

Abstract
Iron-Sulfur (Fe-S) clusters function as core prosthetic groups known to modulate the activity of metalloenzymes, act as trafficking vehicles for biological iron and sulfur, and participate in several intersecting metabolic pathways. The formation of these clusters is initiated by a class of enzymes called cysteine desulfurases, whose primary function is to shuttle sulfur from the amino acid L-cysteine to a variety of sulfur transfer proteins involved in Fe-S cluster synthesis as well as in the synthesis of other thiocofactors. Thus, sulfur and Fe-S cluster metabolism are connected through shared enzyme intermediates, and defects in their associated pathways cause a myriad of pleiotropic phenotypes, which are difficult to dissect. Post-transcriptionally modified transfer RNA (tRNA) represents a large class of analytes whose synthesis often requires the coordinated participation of sulfur transfer and Fe-S enzymes. Therefore, these molecules can be used as biologically relevant readouts for cellular Fe and S status. Methods employing LC-MS technology provide a valuable experimental tool to determine the relative levels of tRNA modification in biological samples and, consequently, to assess genetic, nutritional, and environmental factors modulating reactions dependent on Fe-S clusters. Herein, we describe a robust method for extracting RNA and analytically evaluating the degree of Fe-S-dependent and -independent tRNA modifications via an LC-MS platform.
Keywords: tRNA, Bacteria, Iron-sulfur cluster, Sulfur metabolism, LC-MS, RNA extraction, tRNA modification, Thionucleosides
1. Introduction
For nearly six decades, the study of biological Fe-S clusters has captured the interest of the biochemical and bioinorganic scientific community [1]. When associated with proteins, these inorganic cofactors play indispensable roles in central metabolism, gene regulation, DNA repair, respiration, and translation [2, 3]. Due to the essential nature of these molecules, it is therefore not surprising that Fe-S clusters are found throughout all domains of life and remain an active topic of exploration for their functionality and biogenesis [4–6]. Different pathways have been identified for the synthesis of Fe-S clusters. Despite their differences in biosynthetic components, all systems reported thus far include a consortium of proteins dedicated to the recruitment and assembly of individual Fe and S components into clusters, followed by transfer to final acceptor proteins. In bacteria, the iron-sulfur cluster (ISC), sulfur mobilization (SUF), and nitrogen fixation (NIF) systems serve as three distinct biosynthetic routes for Fe-S cluster assembly [7–9]. A conserved feature of these systems is the presence of a cysteine desulfurase involved in the abstraction and passage of sulfur from the amino acid L-cysteine to downstream components involved in sulfur and Fe-S cluster mobilization pathways [10, 11]. Thus, defects during the sulfur mobilization step not only affect the activity of Fe-S enzymes, but also impair the synthesis of several S-containing metabolites [12, 13].
The functionality of sulfur and Fe-S cluster pathways affects the efficiency and accuracy of translational processes through modulation of tRNA structure and function [14, 15]. Post-transcriptional modifications are installed at multiple locations on the tRNA structure by specialized tRNA-modifying enzymes. These modifications expand their reactivity while simultaneously promoting proper protein-RNA recognition events and aminoacylation [16, 17]. To date, over 100 modifications to tRNA have been described [18]. The synthesis and functionalities of modified tRNAs are known to be affected by environmental and nutritional factors [19]. Likewise, alterations to sulfur and Fe-S metabolism are reflected in the overall tRNA epitranscriptome through direct and indirect mechanisms [20].
Relevant to the metabolism of sulfur and Fe-S clusters are the modifications listed in Table 1 and Fig. 1. Analysis of these modifications can serve as an in vivo readout of the functionality of their corresponding pathways. Modifications found in the anticodon domain of tRNA are of particular importance due to their role during translation in preventing frameshifting and maintaining accurate codon-anticodon recognition [21]. An example of this type of chemical alteration is the 2-methylthio-N6-isopentenyladenosine (ms2i6A) s-tRNA modification found on A37. Iron starvation or oxidative stress leads to decreased levels of this modification. Loss of ms2i6A is attributed to defects in Fe-S cluster synthesis caused by adverse conditions that inactivate MiaB, an Fe-S SAM--dependent methylthiotransferase involved in ms2i6A formation [22]. MiaB is a bifunctional enzyme that uses its Fe-S centers along with AdoMet to thiolate and methylate its substrate tRNA, N6-isopentenyladenosine (i6A), through a radical-dependent mechanism [23]. In Salmonella species, MiaB performs identical methylthiolation reactions on the same A37 base, but is able to use the hydroxylated form of i6A (io6A) as a target substrate [24]. This substrate promiscuity sets it apart from its E. coli ortholog in that E. coli does not have the hydroxylated precursor nucleoside. The Bacillus subtilis MiaB ortholog, MtaB, is also a SAM-dependent Fe-S enzyme and performs similar functions to MiaB [25, 26]. However, B. subtilis MtaB uses N6-threonyl-carbamoyladenosine (t6A) as a distinct substrate. Interestingly, t6A’s cyclized form, cyclic-N6-threonyl-carbamoyladenosine (ct6A), and its hypermodified form, 2-methylthiocyclic-N6-threonyl-carbamoyladenosine (ms2ct6A), have also been reported in this organism [27].
Table 1.
tRNA modifications whose synthesis is either dependent on FeS proteins or independent of FeS processes in the Gram-positive model organism, Bacillus subtilis, and Gram-negative Escherichia coli. The latter half of the table entitled “Fe-S-Independent tRNA Modifications” highlights commonly used internal standards comprising highly abundant and stable nucleosides
| Fe-S-Dependent tRNA Modifications | |||
| Position | Modification | Observed Molecular Ion (m/z+) | Biosynthetic Enzyme and Cluster Type |
| 34 | oQ | 426.1620 | QueE (B. subtilis & E. coli); [4Fe-4S] |
| 34 | Q | 410.1670 | QueG (B. subtilis & E. coli); (2) [4Fe-4S] |
| 37 | ms2i6A | 382.1544 | MiaB (B. subtilis & E. coli); (2) [4Fe-4S] |
| 37 | m2Aa | 282.1197 | RlmN (B. subtilis & E. coli); (2) [2Fe-2S] |
| 32 | s2C | 260.0699 | TtcA (E. coli); (2) [4Fe-4S] |
| 37 | ms2t6A/ms2ct6A | 459.1293/440.1235 | MtaB (B. subtilis); (2) [4Fe-4S] |
| Fe-S-Independent tRNA Modifications | |||
| Position | Modification | Observed Molecular Ion (m/z+) | Biosynthetic Enzyme and Cluster Type |
| 55b | ψ | 245.0767 | TruB (B. subtilis & E. coli) |
| 17c | D | 247.0925 | Dus1 or Dus2/DusA (B. subtilis & E. coli) |
| 34 | I | 269.0881 | TadA (B. subtilis & E. coli) |
| 37 | t6A | 413.1416 | TsaBCDE (B. subtilis & E. coli) |
| 37 | i6A | 336.1667 | MiaA (B. subtilis & E. coli) |
| 37 | m6A | 282.1197 | TrmN6 (B. subtilis & E. coli) |
m2A is also found in 23S rRNA and modified by the same biosynthetic enzyme, RlmN
Pseudouridine is also found at positions 32, 38, 39, 40 in B. subtilis and E. coli, and additionally at positions 13 and 35 in E. coli
Dihydrouridine is also found at positions 20 and 20a in B. subtilis and E. coli, at position 47 in B. subtilis only, and at position 16 in E. coli only
Fig. 1.
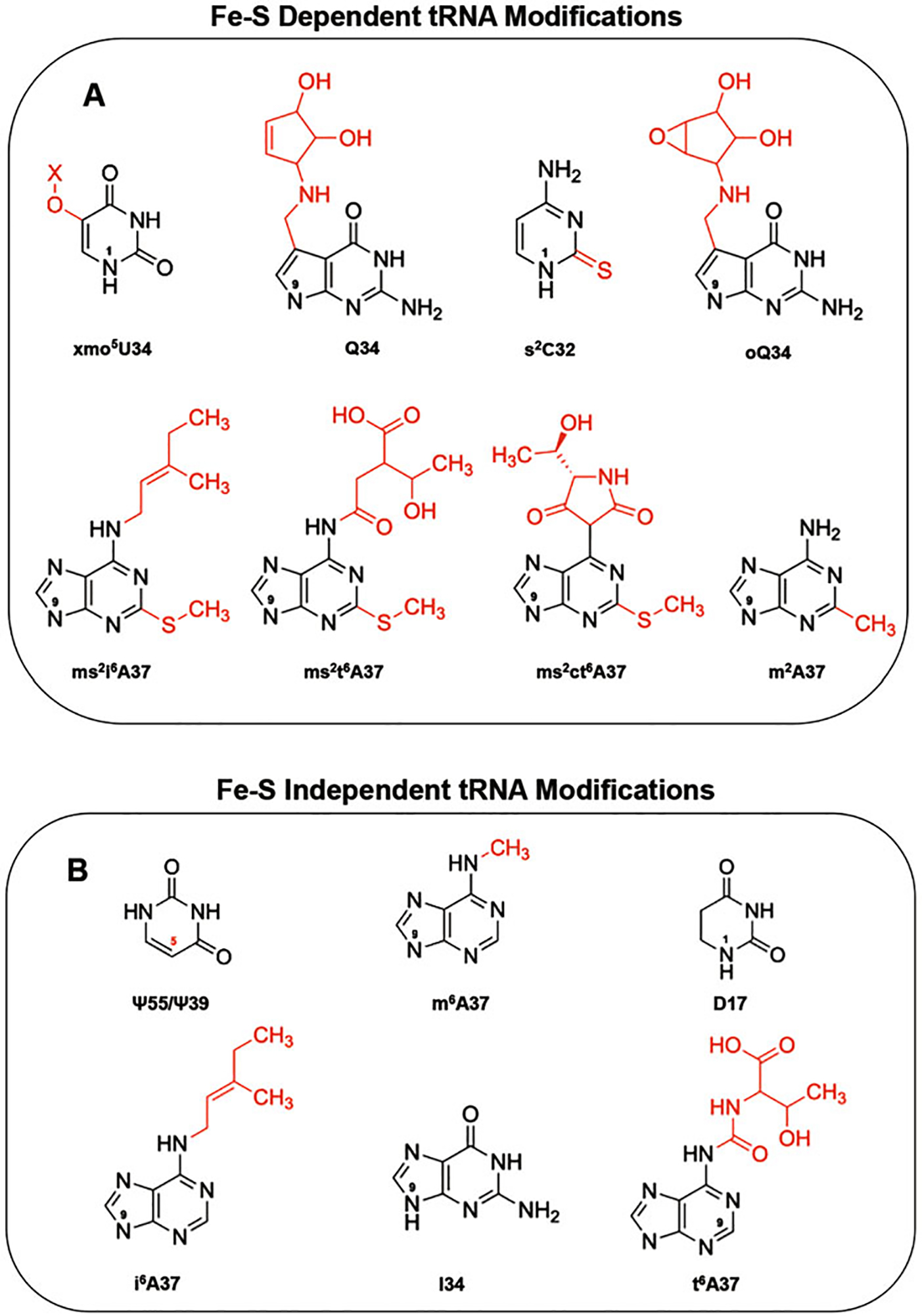
Structures of modified bacterial tRNA and derivatives. (a) Structures of modified tRNAs found at positions 32, 34, and 37 whose synthesis is dependent on Fe-S proteins, and (b) structures of modified tRNAs found at positions 17a, 34, 37, and 55b whose synthesis is independent of Fe-S proteins are shown with their corresponding modified/hypermodified moieties indicated in red. The numbers “1” and “9” found within each base denote the β-glycosidic bond orientation found in the pyrimidine and purine bases, with their adjacent riboses in tRNA. (a) Pseudouridine is also found at positions 32, 38, 39, 40 in B. subtilis and E. coli, and additionally at positions 13 and 35 in E. coli. (b) Dihydrouridine is also found at positions 20 and 20a in B. subtilis and E. coli, at position 47 in B. subtilis only, and at position 16 in E. coli only
2-thiocytidine (s2C) found at position C32 of tRNAArg and tRNASer represents another sulfur-containing modification that depends on an Fe-S tRNA- modifying enzyme [28]. The Gram-negative bacterial species, E. coli and Salmonella enterica serovar typhimurium, use the [4Fe-4S] tRNA thioltransferase TtcA for the synthesis of s2C. This unique enzyme contains a PP-loop known for its ATPase activity and six highly conserved cysteine residues, four of which are bundled into CysXXCys motifs. Possibly one of the most intriguing characteristics of TtcA is that it was the first Fe-S enzyme of its kind demonstrated to thiolate its tRNA substrates in a nonradical catalyzed mechanism [29, 30]. Other thiolating enzymes that share common PP-loop motif characteristics with TtcA include the 4-thiouridine (s4U) modifying enzyme, ThiI, and the 2-thiouridine (s2U) thiouridylase, MnmA [31–35]. In sharp contrast, ThiI’s and MnmA’s functionality is not contingent on the presence of an Fe-S cluster biosynthetic apparatus for thiolation and catalysis and additionally contain no CysXXCys motifs in the majority of their sequences. However, it was recently shown that a few archaeal and extreme thermophilic bacterial species possess an MnmA or a ThiI that requires an Fe-S cluster for their tRNA-modifying activities. Thermus thermophilus MnmA bears conserved cysteines, which are canonically found in Fe-S proteins. These cysteines coordinate an oxygen-sensitive [4Fe-4S] cluster that is required for catalytic activity [36]. Similarly, in Methanococcus maripaludis, ThiI has a redox-active [3Fe-4S] cluster that is also required for the tRNA thiouridylation reaction [37]. Surprisingly, like several methanogenic species, M. maripaludis’ genome does not code for a cysteine desulfurase, and the formation of thionucleosides is thought to use sulfide as a sulfur source [37, 38].
A distinct subset of tRNA modifications are dependent on Fe-S tRNA-modifying enzymes but do not require sulfur to be incorporated into their structures. For instance, in E. coli, the Fe-S SAM-dependent enzyme RlmN implements the 2-methyladenosine (m2A) base modification onto messenger RNA (mRNA) as well as A37 in tRNA, making this enzyme unique for its dual substrate specificity [39]. At the neighboring G34 position of tRNAs, the 7-deaza-guanosine hypermodification known as queuosine (Q) possesses an elaborate and highly coordinated biosynthetic pathway. In both B. subtilis and E. coli, the 7-carboxy-7-deazaguanine synthase, QueE, uses a [4Fe-4S] cluster along with AdoMet to catalyze the heterocyclic, radical-mediated conversion of 6-carboxy-5,6,7,8-tetrahydropterin (CPH4) to 7-carboxy-7-deazaguanine (CDG), a highly conserved biosynthetic step intrinsic to all compounds containing 7-deazapurine [40]. In the same queuosinilation pathway, these organisms also employ the epoxide reductase, QueG, to complete the last Q synthesis step. In this scheme, QueG uses a redox chain of two [4Fe-4S] clusters to reduce the epoxide found on the Q-tRNA precursor, epoxyqueuosine (oQ), and form the cyclopentediol product Q [41, 42]. Lastly, the inventory of Fe-S-dependent modifications has been recently expanded to include the hydrouridine (hoU) tRNA modification in a pathway that depends on a specialized ferredoxin in B. subtilis and E. coli [43].
The type and number of Fe-S clusters vary substantially in these tRNA-modifying Fe-S proteins. Characterization of these proteins has been extensively undertaken using a combination of genetic, biochemical, and spectroscopic techniques, in vitro Fe-S cluster assays, and Fe/sulfide analyses. However, integrating this information into Fe-S protein pathway functionality also requires a broad, in vivo assessment. Analysis of tRNA modifications provides a valuable method for identifying factors that affect Fe-S and/or S metabolism at the organismal level. LC-MS-mediated analysis of total tRNA isolated from an organism of interest allows for the determination of the composition and identity of modified tRNA nucleosides. The quantification of tRNA modifications that depend on Fe-S modifying enzymes provides a unique avenue for monitoring Fe-S cluster metabolism in the cell.
Epitranscriptomic analyses can also provide a readout on the functionality of pathways involving sulfur transfer reactions, specifically those that do not employ Fe-S enzymes in their biosynthetic pathways. Thus, this approach permits deconvolution of thiomodifications that are dependent and independent of Fe-S clusters. The abundance of these modifications can then be normalized to the levels of modifications that are independent of Fe-S and S metabolism, such as dihydrouridine (D), inosine (I), pseudouridine (ψ), and many others [14] (Table 1). Dihydrouridine is particularly useful as a normalizing tool due to its consistency, high abundance, and near exclusivity in bacterial tRNA [44]. The necessity of this modified nucleotide is further highlighted when attempting to quantify low abundance tRNA modifications. For instance, in most organisms, the C34 lysidine (k2C) modification is present in a single tRNAIle isoacceptor bearing the CUA anticodon sequence [45]. Since this modification is exceptionally rare, normalizing k2C abundance to its highly abundant unmodified cytidine precursor base would grossly underrepresent lysidine’s physiological prevalency in tRNA. Additionally, cytidine is also present in ribosomal RNA (rRNA) and mRNA, further diluting the significance of the tRNA-specific k2C when using total RNA during these types of analyses. Factors like these reiterate the importance of using moderately abundant normalizing factors such as dihydrouridine to get an accurate representation of modification levels in the overall pool of cellular tRNA.
Cultures exposed to varying stressors known to affect Fe-S protein competency impact the cellular tRNA pool composition and give insight into interlinked metabolic processes that affect translation and protein synthesis [13, 19, 46, 47]. The methods described in this chapter enable the use of tRNA modifications as a readout of Fe-S protein activity as well as proteins involved in sulfur metabolism. This methodology allows for a comprehensive understanding of how Fe-S proteins, specifically those that modify tRNAs, function at any given time under a range of environmental and nutritional conditions.
2. Materials
2.1. Growth of Cells for Total RNA Extraction
LB broth: 12.5 g of Luria Bertani (LB) medium and 500 mL of water autoclaved. Stored at room temperature (see Note 1).
LB agar: 12.5 g of LB medium and add 7.5 g agar; add 500 mL of water and autoclave. Aseptically, pour roughly 20 mL of autoclaved LB agar into sterile Petri dishes and store them at 4 °C.
Bacillus subtilis PS832 strain (see Note 2).
2.2. Saturation of Phenol with NaOAc pH 4.3
2.3. Total RNA Extraction
Cell resuspension buffer: 300 mM sodium acetate, 190 mM NaCl, pH 4.5, 10 mM ethylenediaminetetraacetic acid (EDTA) (see Notes 4 and 5). NaOAc and EDTA stock solutions should be filtered with 0.45 μM MCE membrane filter before being combined for the final buffer solution.
NaOAc-saturated phenol, pH 4.3.
Chloroform-isoamyl alcohol mix (IAA) (24:1).
Pure ethanol (EtOH), 200 proof, chilled in −20 °C freezer.
70% EtOH, chilled in −20 °C freezer.
High-speed polypropylene copolymer (PPCO) 50 mL centrifuge tubes.
6 M ammonium acetate (NH4OAc), pH 5.3.
Water, Optima LC-MS grade.
2.4. Digestion of Total RNA and LC-MS Sample Preparation
Nuclease P1: 250 U/mL Nuclease P1 from Penicillium citrinum, resuspended from the lyophilized form in Water, Optima LC-MS grade; stored as 50 μL aliquots in −80 °C freezer.
rSAP: 1000 U/mL Shrimp Alkaline phosphatase.
Nuclease P1 digestion buffer: 400 mM NH4OAc (HPLC grade); 200 mM ZnCl2, pH 5.3.
CutSmart Digestion Buffer: 50 mM Potassium Acetate, 20 mM tris (hydroxymethyl) aminomethane (Tris)-acetate, pH 7.9; 10 mM Magnesium Acetate; 100 μg/mL Bovine Serum Albumin (BSA).
100 mM NH4HCO3 (see Note 6).
Formic acid 99.0 + %, Optima LC-MS grade.
Water, Optima LC-MS grade.
Blue screw-top LC-MS sample vials with 200 μL glass inserts.
80:4 MeOH:FA: 80% mixture of methanol (MeOH), Optima, LC-MS grade containing 4% formic acid (FA) 99.0 + % Optima LC-MS grade and 16% water, Optima, LC-MS grade.
2.5. LC-MS Method Run, Mobile Phases, and Data Analysis
Solvent A: Water with 0.1% formic acid, Optima LC-MS grade.
Solvent B: Methanol and 0.1% formic acid, Optima LC-MS grade.
Formic acid 99.0 + %, Optima LC-MS grade.
LC-18 analytical column, 3 μm particle size, 100 mm column length, 4.6 mm internal diameter.
LC-MS: HPLC instrument coupled to a linear ion trap mass spectrometer.
Data analysis software (see Note 7).
3. Methods
Assessing the function of Fe-S proteins in vivo gives an insightful look into cellular redox status, degree of oxidative stress, and bioavailability of core elements such as iron and sulfur at the organismal level [4, 48]. In the past few decades, LC-MS technology has been used to analyze the tRNAome and closely monitor changes in its composition and degree of modifications. These changes can act as signals of depleted nutritional status or markers of oxidative stress and cellular competency. Fluctuating modification levels also serve as direct/indirect readouts of upstream Fe-S protein activity, as many tRNA-modifying enzymes utilize Fe-S clusters for their activities. Challenging growth cultures with varying stressors such as nutrient limitation or peroxide stress allows for the evaluation of tRNA modification levels and the linking of those patterns to Fe-S protein expression levels and activity profiles. These analyses also provide information about Fe-S cluster independent modifications, which serve as internal controls in these biological samples.
The following methods outline the procedures used for cellular growth, total RNA extraction, sample preparation, and LC-MS sample processing/analysis. The levels and types of modification vary across species and growth conditions and are growth phase-dependent. Therefore, it is important to maintain the same experimental conditions and harvest cells at the same cell density to obtain meaningful results. Below we describe the workflow for isolation and analysis of tRNA modified nucleosides from B. subtilis cultures. We have successfully implemented these methods using bacterial species such as B. subtilis, E. coli, Azotobacter vinelandii, and Borrelia burgdorferi. This methodology may be successfully extended to other organisms.
3.1. Growth of Cells for Total RNA Extraction
Using a sterile technique, streak the desired bacterial strain onto an LB agar plate and incubate at 37 °C to obtain isolated colonies.
Inoculate 250 mL of sterile LB liquid medium (see Note 8) with a single colony from overnight LB agar plate and place it in a 37 °C shaking incubator until OD600 ~ 1.0 is reached (mid-exponential growth phase).
Dilute the starting culture in 500 mL fresh LB liquid medium to OD600 0.01, and place in 37 °C shaking incubator until OD600 0.4–0.6 is reached (early exponential growth phase).
Centrifuge 500 mL of culture at 6744 × g for 10 min at 4 °C, decanting supernatant in between spins; store pellet in −80 °C freezer for up to 1 year.
3.2. Saturation of Phenol with NaOAc pH 4.3
Melt 100 g bottle of phenol by loosening cap and placing the bottle in a 65 °C water bath. Phenol should be completely melted in 20–30 min (see Notes 9 and 10).
Transfer melted phenol to separatory funnel in a fume hood while phenol is still hot. Make sure the valve on the funnel is in the horizontal closed position.
Add 100 mL of NaOAc buffer to liquid phenol, close the funnel with the appropriate ground glass stopper, carefully invert the separatory funnel, and shake vigorously to mix phases. Stop and vent every four to five inversions.
Allow phases to separate in a ring clamp. This step may take 6–8 h at room temperature. At that point, the bottom organic phase (phenol) should be clear if full separation is achieved.
Place a clean glass beaker under the opening of the separatory funnel, then open the valve to the vertical open position, and collect the bottom organic layer. Stop collecting when the interphase, or the murky layer found between two phases, is reached. Discard the upper aqueous layer.
Return the bottom organic layer to the funnel, add 100 mL of NaOAc buffer, and repeat steps 4 and 5 for two additional times.
Transfer the bottom organic layer to a brown, light-resistant glass container and store at 4 °C for up to 6 months (see Note 11).
3.3. Total RNA Extraction
The following protocol uses manually presaturated acidic phenol with chloroform/IAA for cell disruption and subsequent RNA extraction and EtOH precipitation (see Note 12). When using any phenol/chloroform/IAA-containing reagents, all experiments must be performed in a well-ventilated fume hood. The appropriate gloves, eye protection, and lab coats should be used.
Thaw a bacterial cell pellet and add 10 mL of cell resuspension buffer. Mix until resuspension is homogenous and transfer to a 50-mL high-speed PPCO centrifuge tube.
Add 3.5 mL of NaOAc-saturated phenol (bottom layer) to the 10 mL resuspension, and vortex vigorously for 60 s, followed by a 30-s rest and resume with a final 90 s of vortexing.
Centrifuge the mixture at 33,745 × g for 15 min at 25 °C.
Carefully remove the spun tubes without disturbing extracted layers. Remove the top layer (aqueous layer) and transfer it to a clean 50-mL high-speed PPCO tube.
Extract again by adding 3.5 mL of NaOAc-saturated phenol to the aqueous layer and vortex for 60 s.
Centrifuge the mixture at 33,745 × g for 15 min at 25 °C.
Carefully remove the spun tubes from the centrifuge, remove the top aqueous layer, and transfer to a clean 50-mL high-speed PPCO tube.
Add 0.5 g of NaCl and 10 mL of 24:1 chloroform/IAA mixture to aqueous layer and vortex for 10 s.
Centrifuge the mixture at 33,745 × g for 3 min at 25 °C.
Remove the top layer (take care to not disturb the grainy, white interphase) and transfer to a clean 50-mL high-speed PPCO tube.
Fill the 50 mL tube containing aqueous layer with ice-cold, 200 proof, pure EtOH.
Allow the RNA to precipitate for 4 h to overnight in a −20 °C freezer.
After precipitation, centrifuge tubes at 33,745 × g for 25 min at 4 °C.
Carefully remove the tubes from the centrifuge and gently decant the liquid EtOH from the tubes. At this point, there should be a distinct pellet at the bottom that can be easily dislodged during decanting, so care should be taken to prevent this occurrence.
Add 10 mL of chilled 70% EtOH to the pellet along with 166 μL of 6 M NH4OAc, pH 5.3 (final concentration of 100 mM), and vortex for 15 s.
Centrifuge tubes at 33,745 × g for 30 min at 4 °C.
Carefully remove the tubes from the centrifuge and gently decant the liquid without disturbing the pellet.
Repeat steps 15 and 16 without the addition of the 100 mM NH4OAc, pH 5.3.
Carefully remove the tubes from the centrifuge and gently decant the liquid without disturbing the pellet.
Invert tubes in a rack over a dry surface and allow them to air-dry for 30–45 min, or until pellets are dry and no residual EtOH exists on the sides of the tubes (see Note 13).
Resuspend RNA pellet in 750 μL of Optima, LC-MS grade water, and store in −80 °C freezer for further use.
3.4. Digestion of Total RNA and LC-MS Sample Preparation
Perform a 1:10 RNA/water dilution and measure the absorbance of the RNA sample at 260 nm. Calculate the concentration of RNA using an extinction coefficient of 0.025 (μg/mL)−1 cm−1.
Aliquot 40 μg of total RNA into a 0.6-mL high-binding microfuge tubes.
Heat denature for 5 min in a 95 °C heating block.
Perform a quick spin to pull denatured RNA to the bottom of the tube and allow it to come to room temperature before adding any digestion components.
Add Optima, LC-MS grade water to cooled RNA up to 150 μL.
Add 4.1 μL of nuclease P1 digestion buffer and 5 μL of 0.25 U/ μL nuclease P1 and incubate at 50 °C for 4 h.
Perform a quick spin to collect evaporated digestion products in the tube.
Add 20 μL of CutSmart buffer, 20 μL of 100 mM NH4HCO3, and 10 μL of the rSAP enzyme, and incubate at 37 °C for 2 h.
Spin digested RNA for 10 min at 16,873 × g at room temperature (see Note 14).
Take 196 μL of RNA supernatant and transfer to the new 0.6 mL microfuge tube.
Add 5 μL of freshly prepared 80:4 MeOH/FA mixture to the supernatant.
Transfer MeOH/FA-spiked mixture to glass 200 μL LC-MS vial insert and place inside LC-MS sample vial for subsequent analysis.
3.5. LC-MS Method Run and Tune Parameters
Perform all nucleoside analyses on an LC-MS using the following column equilibration prior to experimental sample run: 98% Solvent A and 2% Solvent B for 15 column volumes (45 min) at 300 μL min−1 flow rate.
Adjust the tune file to the following settings: Voltage (kV): 4.01; Sheath Gas Flow Rate (arb): 47.00; Auxiliary Gas Flow Rate (arb): 30.00; Sweep Gas Flow Rate (arb): 0.00; Capillary Voltage (V): 2.00; Capillary Temperature (C): 350.00; Tube Lens Voltage (V): 49.89.
Over the course of a 39-min run, set up the gradient of solvent B as follows: 0–4 min, hold at 2% B (v/v); 4–25 min, increase B from 2% to 100%; 25–33 min, hold at 100% B; 33–39 min switch from 100% B to 2% B to allow column reequilibration at 2% B.
3.6. LC-MS Data Analysis Using MestReNova
The Fourier transformed m/z data from each experimental run will be saved in a .raw file format; transfer these files from the computer that controls the OrbiTrap to your personal/shared device with a copy of MestReNova data analysis software along with the appropriate license files.
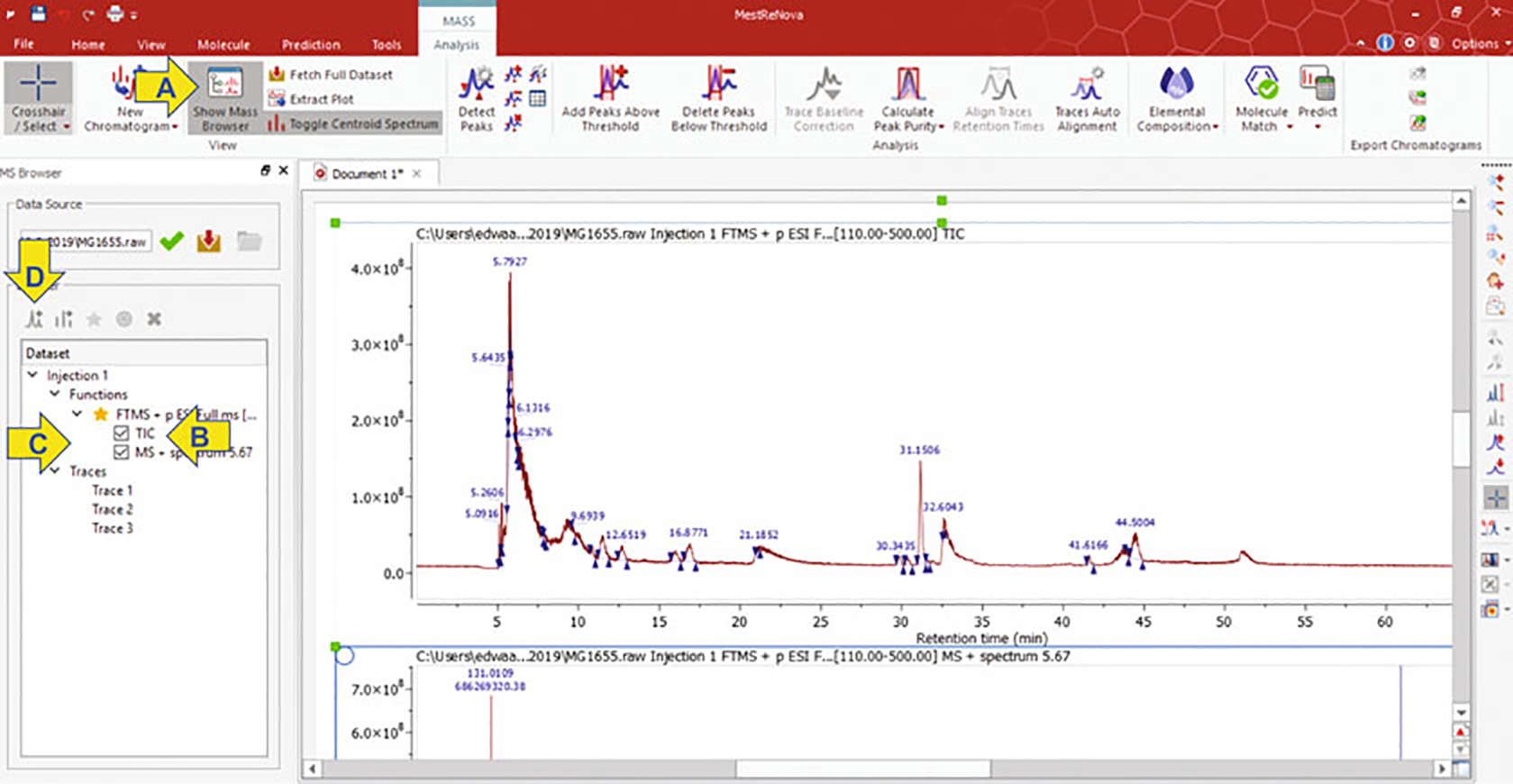
Open the MestReNova application, then open your .raw file. This command will open with two screens; one is a total ion chromatogram (TIC), and the other is a mass spectrum (MS+ Spectrum).
In the top panel of the MestReNova application, click on the “Show Mass Browser” icon (Fig. 2a arrow), and a mass browser panel will be displayed on the left side of the screen. Uncheck the “TIC” panel (Fig. 2b arrow).
In the mass browser, click in the white space next to the dropdown boxes, and then select the “open new chromatograph” icon, shown as a red chromatogram (Fig. 2c, d arrows).
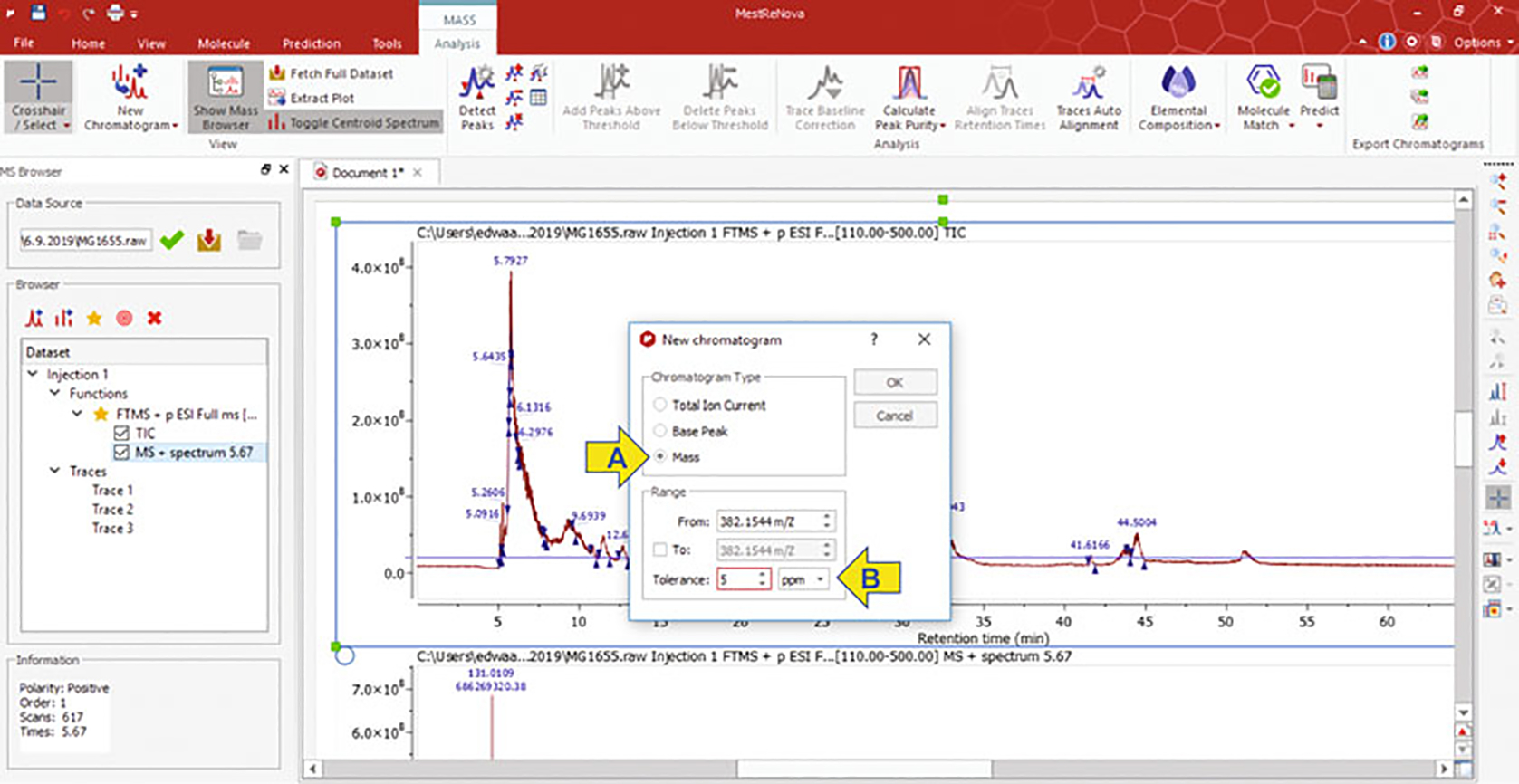
In the new window, select the “Mass” option. In the dropdown box, add the mass over charge ratio (m/z) calculated from the monoisotopic mass of the targeted modification of interest with four decimal places. Lastly, change the “tolerance” from “Da” to “PPM” in the dropdown menu, and set the threshold to five parts per million (PPM) (Fig. 3a, b arrows).
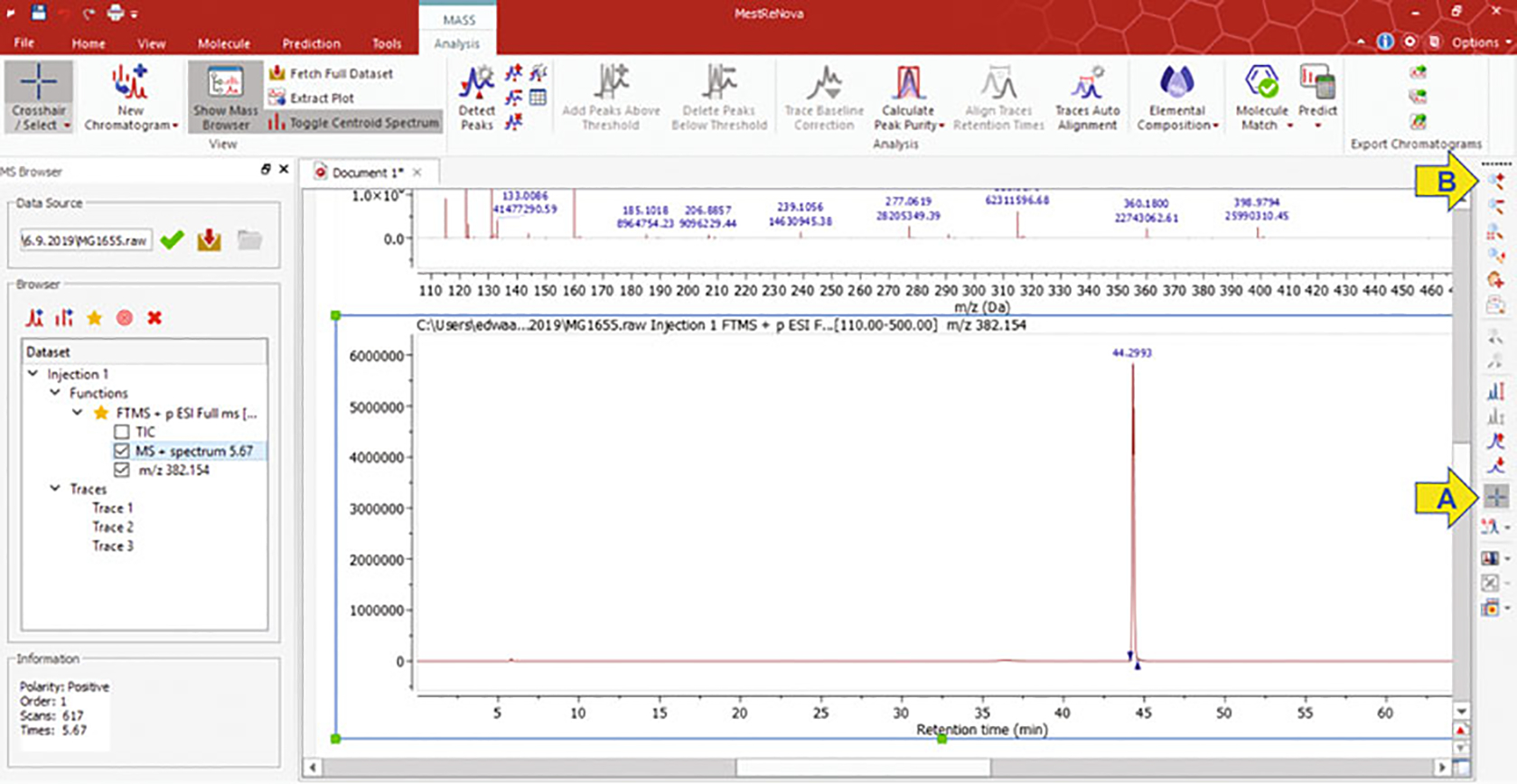
The new panel will display the extracted ion chromatogram (EIC) (Fig. 4). This chromatogram shows the profile of species containing the selected m/z within five PPMs with associated retention times.
Select the blue crosshair icon located on the right panel of icons and drag the crosshair from one side of the peak of interest to the other side, indicated by up/down faced blue triangles on EIC (Fig. 4a arrow). The “MS + Spectrum” of the highlighted peak will be displayed in the top panel with the corresponding range of masses.
From the right-side panel, click on the magnifying glass with a red plus sign icon for “Zoom In,” and then zoom in on the specific mass of interest (Fig. 4b arrow). The zoom in feature allows visualization of the spectral counts associated with your m/z of interest (Fig. 5).
After obtaining the quantitative value associated with your nucleoside of interest, steps 3–8 should be repeated for an internal normalizing factor that is highly abundant, robust, and chemically stable (dihydrouridine, pseudouridine, etc.).
After gathering the values corresponding to all other analytes of interest, use the value associated with your chosen normalizing factor as a divisor and obtain the ratio of your modification in relation to dihydrouridine or pseudouridine (see Note 15).
Fig. 2.
The layout of MestreNova application displaying a total ion chromatograph of a 70-min LC-MS run. Arrows indicate the application commands for selecting the mass browser (a), TIC panel (b), new chromatogram (c), and a new window (d)
Fig. 3.
Layout of MestreNova application when mass browsing to produce an extracted ion chromatogram (EIC). Arrows indicate commands for selecting type of chromatogram (a) and tolerance range (b)
Fig. 4.
Extracted ion chromatogram of 382.1544 m/z and the corresponding retention times of all analytes that fall within 5 PPM of this m/z of interest. Arrows indicate commands for selecting the range (a) and zoom in (b)
Fig. 5.
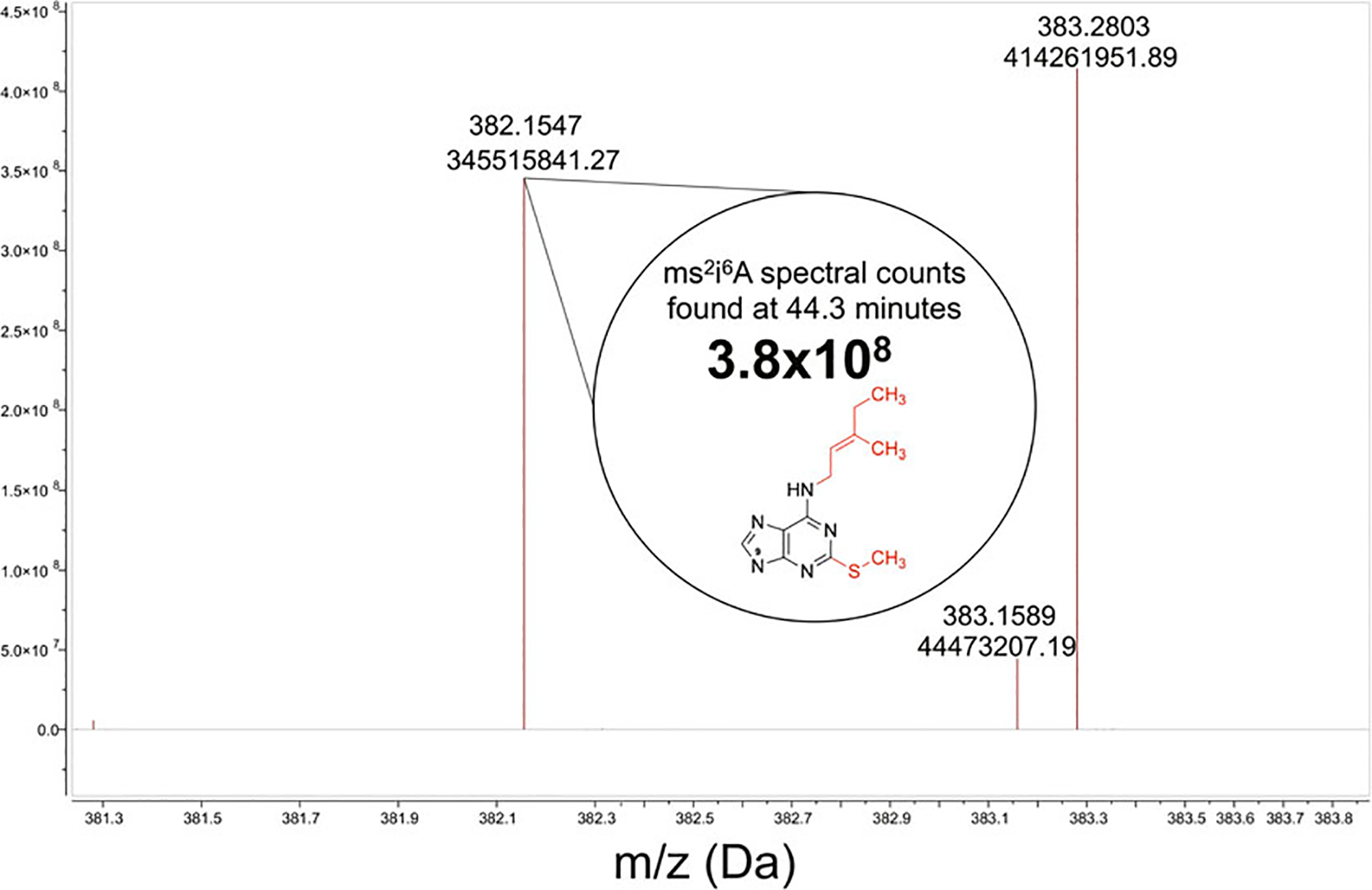
Mass spectrum of the 43.7- to 44.9-min retention time, the m/z values found within this time range, and the spectral count associated with the ms2i6A37 modified nucleoside
Acknowledgments
The research and methodology developed in the PDS laboratory was supported by the National Science Foundation (1716535). A. M.E was a recipient of a T32 NIH predoctoral fellowship (GM 95440-9).
Footnotes
Our lab uses 500 mL cultures for these analyses, but successful extraction and LC-MS experiments have been performed using culture volumes as small as 5 mL.
Our lab chose to use B. subtilis PS832 to perform the majority of our analyses. However, this method can be applied to other bacterial species and produce comparable results.
Solid phenol is the preferred state in this method. Liquid phenol and crystallized phenol yield different extraction efficiencies in our hands.
A final concentration of 1 M NaOAc salt dissolved in water and pH is adjusted with concentrated HCl. At equilibrium, the solution will contain NaOAc and NaCl as indicated.
The NaOAc portion of the buffer should be made using NaOAc salt. The EDTA should be made as a liquid stock solution at 0.5 M EDTA, pH 8.0. The stock EDTA can then be added to the NaOAc buffer, and finally, pH adjusted to pH 4.3 using glacial acetic acid.
All enzymatic digestion buffers should use compounds that are at least 99% assay purity, preferably Optima or HPLC-grade purity. If not, the impurities lead to a background signal and increased signal/noise ratio of MS analyses.
Our lab uses MestReNova analytical software, but there are multiple different data analysis software available.
The growth medium can be modified to test the effects of different stressors/nutritional conditions on the relative abundance of tRNA modifications. For example, when Saccharomyces cerevisiae is grown in sulfur amino acid limited-standard glucose minimal media, a sharp decrease in tRNA thiolation is observed in a dose-dependent manner [49].
If 100 g phenol is not in the bottle, saturate the phenol with NaOAc buffer at a 1:1 v/v ratio.
Saturation of phenol should be performed in a chemical fume hood with gloves, safety eyewear, and lab coat.
If the phenol turns brown/pink, the phenol has become oxidized and should be disposed appropriately.
The extraction of total RNA can be performed using any standard RNA extraction kit (i.e., Trizol).
Do not over-dry pellets and do not use speed-vac to speed up the drying process. This prevents the resolubilization of RNA in your resuspending buffer of choice or water.
Do not disturb the pellet of digest products; this can clog the column and produce a high background signal on MS.
Each sample within an LC-MS run will have its own internal normalizing factors, which are the levels of modification that are not fluctuating with experimental conditions. These internal standards normalize the desired analyte quantification and account for variations on extraction, digestion, and MS quantification across samples.
References
- 1.Nicholas DJ, Wilson PW, Heinen W et al. (1962) Use of electron paramagnetic resonance spectroscopy in investigations of functional metal components in micro-organisms. Nature 196:433–436 [DOI] [PubMed] [Google Scholar]
- 2.Kiley PJ, Beinert H (2003) The role of Fe-S proteins in sensing and regulation in bacteria. Curr Opin Microbiol 6:181–185 [DOI] [PubMed] [Google Scholar]
- 3.Py B, Barras F (2010) Building Fe-S proteins: bacterial strategies. Nat Rev Microbiol 8:436–446 [DOI] [PubMed] [Google Scholar]
- 4.Lill R, Freibert SA (2020) Mechanisms of mitochondrial iron-sulfur protein biogenesis. Annu Rev Biochem 89:471–499 [DOI] [PubMed] [Google Scholar]
- 5.Blahut M, Sanchez E, Fisher CE et al. (2020) Fe-S cluster biogenesis by the bacterial Suf pathway. Biochim Biophys Acta, Mol Cell Res 1867:118829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zheng C, Dos Santos PC (2018) Metallocluster transactions: dynamic protein interactions guide the biosynthesis of Fe-S clusters in bacteria. Biochem Soc Trans 46:1593–1603 [DOI] [PubMed] [Google Scholar]
- 7.Takahashi Y, Tokumoto U (2002) A third bacterial system for the assembly of iron-sulfur clusters with homologs in archaea and plastids. J Biol Chem 277:28380–28383 [DOI] [PubMed] [Google Scholar]
- 8.Zheng L, Cash VL, Flint DH et al. (1998) Assembly of iron-sulfur clusters. Identification of an iscSUA-hscBA-fdx gene cluster from Azotobacter vinelandii. J Biol Chem 273:13264–13272 [DOI] [PubMed] [Google Scholar]
- 9.Dos Santos PC, Dean DR (2014) A retrospective on the discovery of [Fe-S] cluster biosynthetic machineries in Azotobacter vinelandii. In: Rouault T (ed) Iron-sulfur clusters in chemistry and biology. Verlag Walter de Gruyter, Berlin, pp 267–296 [Google Scholar]
- 10.Dos Santos PC (2017) B. subtilis as a model for studying the assembly of Fe-S clusters in grampositive bacteria. Methods Enzymol 595:185–212 [DOI] [PubMed] [Google Scholar]
- 11.Mihara H, Kurihara T, Yoshimura T et al. (2000) Kinetic and mutational studies of three NifS homologs from Escherichia coli: mechanistic difference between L-cysteine desulfurase and L- selenocysteine lyase reactions. J Biochem (Tokyo) 127:559–567 [DOI] [PubMed] [Google Scholar]
- 12.Lauhon CT, Kambampati R (2000) The iscS gene in Escherichia coli is required for the biosynthesis of 4-thiouridine, thiamin, and NAD. J Biol Chem 275:20096–20103 [DOI] [PubMed] [Google Scholar]
- 13.Lauhon CT (2002) Requirement for IscS in biosynthesis of all thionucleosides in Escherichia coli. J Bacteriol 184:6820–6829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zheng C, Black KA, Dos Santos PC (2017) Diverse mechanisms of sulfur decoration in bacterial tRNA and their cellular functions. Biomol Ther 7:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cavuzic M, Liu Y (2017) Biosynthesis of sulfur-containing tRNA modifications: a comparison of bacterial, archaeal, and eukaryotic pathways. Biomol Ther 7:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agris PF, Vendeix FA, Graham WD (2007) tRNA’s wobble decoding of the genome: 40 years of modification. J Mol Biol 366:1–13 [DOI] [PubMed] [Google Scholar]
- 17.Motorin Y, Helm M (2010) tRNA stabilization by modified nucleotides. Biochemistry 49:4934–4944 [DOI] [PubMed] [Google Scholar]
- 18.Boccaletto P, Machnicka MA, Purta E et al. (2018) MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res 46:D303–D307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edwards AM, Addo MA, Dos Santos PC (2020) Extracurricular functions of tRNA modifications in microorganisms. Genes (Basel) 11(8):907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alings F, Sarin LP, Fufezan C et al. (2015) An evolutionary approach uncovers a diverse response of tRNA 2-thiolation to elevated temperatures in yeast. RNA 21:202–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yarian C, Townsend H, Czestkowski W et al. (2002) Accurate translation of the genetic code depends on tRNA modified nucleosides. J Biol Chem 277:16391–16395 [DOI] [PubMed] [Google Scholar]
- 22.Buck M, Griffiths E (1982) Iron mediated methylthiolation of tRNA as a regulator of operon expression in Escherichia coli. Nucleic Acids Res 10:2609–2624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang B, Arcinas AJ, Radle MI et al. (2020) First step in catalysis of the radical S-Adenosylmethionine Methylthiotransferase MiaB yields an intermediate with a [3Fe-4S] (0)-like auxiliary cluster. J Am Chem Soc 142:1911–1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Esberg B, Bjork GR (1995) The methylthio group (ms2) of N6-(4-hydroxyisopentenyl)-2-methylthioadenosine (ms2io6A) present next to the anticodon contributes to the decoding efficiency of the tRNA. J Bacteriol 177:1967–1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arragain S, Handelman SK, Forouhar F et al. (2010) Identification of eukaryotic and prokaryotic methylthiotransferase for biosynthesis of 2-methylthio-N6-threonylcarbamoyladenosine in tRNA. J Biol Chem 285:28425–28433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anton BP, Russell SP, Vertrees J et al. (2010) Functional characterization of the YmcB and YqeV tRNA methylthiotransferases of Bacillus subtilis. Nucleic Acids Res 45:2124–2136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kang BI, Miyauchi K, Matuszewski M et al. (2017) Identification of 2-methylthio cyclic N6-threonylcarbamoyladenosine (ms2ct6A) as a novel RNA modification at position 37 of tRNAs. Nucleic Acids Res 45:2124–2136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reichle VF, Petrov DP, Weber V et al. (2019) NAIL-MS reveals the repair of 2-methylthiocytidine by AlkB in E. coli. Nat Commun 10:5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jager G, Leipuviene R, Pollard MG et al. (2004) The conserved Cys-X1-X2-Cys motif present in the TtcA protein is required for the thiolation of cytidine in position 32 of tRNA from Salmonella enterica serovar typhimurium. J Bacteriol 186:750–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bouvier D, Labessan N, Clemancey M et al. (2014) TtcA a new tRNA-thioltransferase with an Fe-S cluster. Nucleic Acids Res 42:7960–7970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rajakovich LJ, Tomlinson J, Dos Santos PC (2012) Functional analysis of Bacillus subtilis genes involved in the biosynthesis of 4-Thiouridine in tRNA. J Bacteriol 194:4933–4940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Black KA, Dos Santos PC (2015) Abbreviated pathway for biosynthesis of 2-Thiouridine in Bacillus subtilis. J Bacteriol 197:1952–1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palenchar PM, Buck CJ, Cheng H et al. (2000) Evidence that ThiI, an enzyme shared between thiamin and 4-thiouridine biosynthesis, may be a sulfurtransferase that proceeds through a persulfide intermediate. J Biol Chem 275:8283–8286 [DOI] [PubMed] [Google Scholar]
- 34.Numata T, Ikeuchi Y, Fukai S et al. (2006) Snapshots of tRNA sulphuration via an adenylated intermediate. Nature 442:419–424 [DOI] [PubMed] [Google Scholar]
- 35.Bimai O, Arragain S, Golinelli-Pimpaneau B (2020) Structure-based mechanistic insights into catalysis by tRNA thiolation enzymes. Curr Opin Struct Biol 65:69–78 [DOI] [PubMed] [Google Scholar]
- 36.Shigi N, Horitani M, Miyauchi K et al. (2020) An ancient type of MnmA protein is an iron-sulfur cluster-dependent sulfurtransferase for tRNA anticodons. RNA 26:240–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y, Zhu X, Nakamura A et al. (2012) Biosynthesis of 4-thiouridine in tRNA in the methanogenic archaeon Methanococcus maripaludis. J Biol Chem 287:36683–36692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu Y, Vinyard DJ, Reesbeck ME et al. (2016) A [3Fe-4S] cluster is required for tRNA thiolation in archaea and eukaryotes. Proc Natl Acad Sci U S A 113:12703–12708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schwalm EL, Grove TL, Booker SJ et al. (2016) Crystallographic capture of a radical S-adenosylmethionine enzyme in the act of modifying tRNA. Science 352:309–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mccarty RM, Krebs C, Bandarian V (2013) Spectroscopic, steady-state kinetic, and mechanistic characterization of the radical SAM enzyme QueE, which catalyzes a complex cyclization reaction in the biosynthesis of 7-deazapurines. Biochemistry 52:188–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Phillipson DW, Edmonds CG, Crain PF et al. (1987) Isolation and structure elucidation of an epoxide derivative of the hypermodified nucleoside queuosine from Escherichia coli transfer RNA. J Biol Chem 262:3462–3471 [PubMed] [Google Scholar]
- 42.Payne KA, Fisher K, Sjuts H et al. (2015) Epoxyqueuosine reductase structure suggests a mechanism for cobalamin-dependent tRNA modification. J Biol Chem 290:27572–27581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lauhon CT (2019) Identification and characterization of genes required for 5-Hydroxyuridine synthesis in Bacillus subtilis and Escherichia coli tRNA. J Bacteriol 201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dalluge JJ, Hashizume T, Mccloskey JA (1996) Quantitative measurement of dihydrouridine in RNA using isotope dilution liquid chromatography-mass spectrometry (LC/MS). Nucleic Acids Res 24:3242–3245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Soma A, Ikeuchi Y, Kanemasa S et al. (2003) An RNA-modifying enzyme that governs both the codon and amino acid specificities of isoleucine tRNA. Mol Cell 12:689–698 [DOI] [PubMed] [Google Scholar]
- 46.Schwartz CJ, Djaman O, Imlay JA et al. (2000) The cysteine desulfurase, IscS, has a major role in in vivo Fe-S cluster formation in Escherichia coli. Proc Natl Acad Sci U S A 97:9009–9014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Skovran E, Lauhon CT, Downs DM (2004) Lack of YggX results in chronic oxidative stress and uncovers subtle defects in Fe-S cluster metabolism in Salmonella enterica. J Bacteriol 186:7626–7634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Imlay JA (2006) Iron-Sulphur clusters and the problem with oxygen. Mol Microbiol 59:1073–1082 [DOI] [PubMed] [Google Scholar]
- 49.Gupta R, Walvekar AS, Liang S et al. (2019) A tRNA modification balances carbon and nitrogen metabolism by regulating phosphate homeostasis. Elife 8:e44795. [DOI] [PMC free article] [PubMed] [Google Scholar]