

Abstract
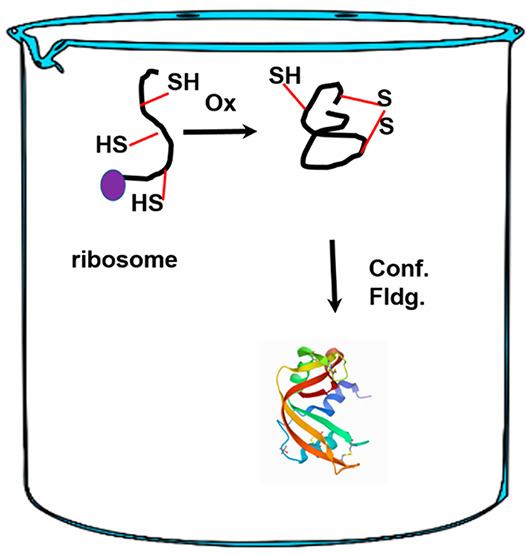
Disulfide bonds play an important role in physiology and are the mainstay of proteins that reside in the plasma membrane and of those that are secreted outside the cell. Disulfide-bond-containing proteins comprise ∼30% of all eukaryotic proteins. Using bovine pancreatic ribonuclease A (RNase A) as an exemplar, we review the regeneration (oxidative folding) of disulfide-bond-containing proteins from their fully reduced state to the biologically active form. We discuss the key aspects of the oxidative folding landscape w.r.t. the acquisition and retention of native disulfide bonds which is an essential requirement for the polypeptide to be biologically functional. By re-examining the regeneration trajectory in light of the symbiotic relationship between native disulfide bonds and a protective structure, we describe the elements that compete with the processes that secure native disulfide bonds in disulfide-coupled protein folding. The impact of native-disulfide-bond formation on protein stability, trafficking, protein misfolding, and neurodegenerative onset is elaborated upon.
Introduction
Proteins that reside in the relatively harsh environments of the plasma membrane (outer membrane in bacteria) or are secreted outside the cell (in higher eukaryotes) have evolved to form disulfide bonds. As a result, disulfide bonds are not only present in a large fraction of the proteome but also play important roles in organismal physiology by providing critical structural support to constitutive proteins and enzymes.1 In bacteria, the disulfide-bond-containing proteins constitute 11% of the total proteins expressed. The number rises to 14 and 29% in archaea and eukaryotes, respectively.2
These intramolecular covalent linkages between cysteines provide added stability over the noncovalent interactions typified by van der Waal’s forces, salt bridges, the hydrophobic effect, and hydrogen bonds. However, the introduction of disulfide bonds to the structure increases the complexity of the folding trajectory.3−5 Proteins that possess disulfide bonds acquire their biologically active conformation through a process called oxidative folding. Oxidative protein folding couples disulfide-bond-forming chemical reactions with a physical (conformational) folding event. The latter is the only reaction found in proteins that do not possess disulfide bonds.1,6
The culmination of the oxidative folding process produces the native protein with a unique complement of native disulfide bonds and is considered “native” as it is the set found in the biologically active form of the protein.7 Using the well-studied regeneration pathway of bovine pancreatic ribonuclease A as an exemplar, we first discuss a typical oxidative folding trajectory that transforms the nascent polypeptide chain to the native disulfide-bond-containing protein.8 Next, we examine the role of elements that either facilitate or retard the process along the path toward securing native disulfide bonds.
Regeneration of Bovine Pancreatic Ribonuclease A (RNase A)
Postribosomal synthesis, RNase A, is threaded into the endoplasmic reticulum (ER) in its fully reduced form (R) which includes eight cysteines at positions 26, 40, 58, 65, 72, 84, 90, and 110. Oxidative folding is initiated in the ER where the ratio of oxidized to reduced glutathione (GSSG/GSH) favors the formation of disulfide bonds. To mimic this process in the laboratory, the fully reduced protein (R) was prepared by reducing the disulfide bonds in the native protein (N) using a 6 M urea and a reducing agent such as 10 mM DTTred.9 The presence of the denaturing agent exposes the disulfide bonds to the reducing agent R exposed to an oxidizing environment (pH 8, 100 mM DTTox). The regeneration mixture is periodically sampled for the acquisition of disulfide bonds R by a variety of techniques including NMR spectroscopy, UV–vis analysis, enzyme activity, disulfide-bond sequencing, etc. However, in order to obtain a precise understanding of the intramolecular forces and disulfide pattern(s) that guide the folding of the polypeptide, separation techniques are usually applied prior to analyses of the intermediates using the aforementioned techniques.11−13
Scheme 1 shows the regeneration pathway of RNase A. The rate of oxidation of R to the one-disulfide-containing ensemble (1S) proceeds with a rate that is dependent on the redox potential of the external oxidizing agent.10−14 The 1S ensemble is a mixture of 28 possible 1S species that are in equilibrium with one another via intramolecular thiol–disulfide exchange reactions. The oxidation of the 1S ensemble results in the formation of a 2S ensemble and so on until the 4S ensemble is generated. The distribution of disulfide-bond-containing isomers within each ensemble is governed by the entropic cost of forming a disulfide bond potentiated by enthalpic interactions.15,16
Scheme 1. Regeneration of RNase A (25 °C, pH 8) Proceeds via the Successive Oxidation of Its Cysteines to Form Disulfide Bonds.
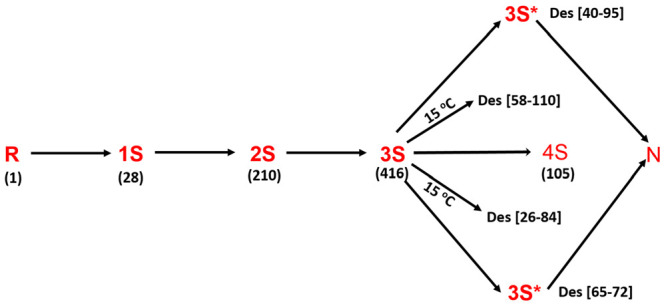
Intermediates (1S–4S) differing in their degree of oxidation (number of disulfide bonds) populate the regeneration chromatogram, and each nS “intermediate” is an ensemble of isomers containing n disulfide bonds. Native protein (N) is formed via the oxidation of two three-native-disulfide-containing 3S* intermediates that accumulate from their 3S isomers by conformational folding. At temperatures <15 °C, two dead-end three-disulfide-bond-containing structured species also populate the oxidative folding landscape of RNase A.
Both secondary and tertiary structures are not detectable in the R–4S species of RNase A. However, there are likely to exist elements of local order via collapse of the polypeptide chain in an aqueous environment leading to a statistical coil. There is also the possibility of flickering secondary structural elements in the unfolded state. It is notable that the loop, which is a key secondary structural element in proteins, is usually indistinguishable from the unfolded structure. As a result, their disulfide bonds and cysteines are exposed and participate in reduction, oxidation, and intramolecular thiol–disulfide exchange reactions. The result of these chemical reactions is the rapid formation of a quasi-equilibrium distribution of species that dominates the regeneration landscape in terms of both dwell time and the diversity of the intermediates. At 25 °C, two structured (native-like) species eventually emerge in the regeneration trajectory of RNase A. Des [65–72] and des [40–95], each lacking the bond in brackets, are formed in a rate-determining step from the 3S ensemble via thiol–disulfide exchange reactions and a conformational folding reaction.13,14 Each des species possesses three native disulfide bonds that are preserved by the tertiary structure of the des species which are alternatively termed as 3S* species, where the * signifies native-like structure.
The formation of both des species can be deemed irreversible since they have melting temperatures of ∼42 °C each. They also possess some enzymatic activity, suggesting the maturation of the catalytic triad. Native RNase A is then rapidly generated by the oxidation of the remaining cysteines in each des species. The formation of N is considered irreversible (Tm = 65 °C). The irreversible formation of the two 3S* species, along with N, causes a gradual decline in the concentration of the R–4S ensemble via the consumption of the unstructured 3S pool (3S → 3S*).
At lower temperatures (<15 °C), two other 3S* species also populate the regeneration landscape of RNase A. Des [58–110] and des [26–84], formed from the 3S ensemble, are kinetically trapped 3S* species. This is because the remaining pair of cysteines in each of these two 3S* species remains buried within native-like structure, making them inaccessible for oxidation to N.17
A survey of the structures of disulfide-bond-containing proteins in conjunction with oxidative protein folding studies on RNase A and other disulfide-bond-containing proteins including hen-egg white lysozyme (HEWL) and bovine pancreatic trypsin inhibitor (BPTI) reveal key finding.7
Biologically active disulfide-bond-containing proteins possess a unique set of disulfide bonds in the native state. The so-called “native disulfide bonds” are only the ones that occur in the native protein. As a corollary, non-native disulfide bonds do not occur in biologically functional disulfide-bond-containing proteins.6
Furthermore, few disulfide-coupled folding trajectories of proteins possess structured intermediates containing non-native disulfide bonds.1 This feature highlights the pivotal role of native disulfides as a precursor to structure/conformational folding.
A minimum number of native disulfide bonds, three in RNase A and HEWL and one in BPTI, is necessary and sufficient to provoke conformational folding. The securing of native disulfide bonds within a structure is essential as it protects them from fruitless reduction or reshuffling (to non-native isomers) reactions.1
These observations underscore the pivotal and “symbiotic” relationship between the formation of native disulfide bonds, a requirement to provoke native/native-like structures, and their preservation through the formed structure.
Based on these observations, we define and discuss the role of elements that either facilitate or compete with (retard) the process of securing native disulfide bonds along the path to acquiring native-like/native structures. For ease of comprehension, we define the trajectory to N as the path along the x-axis heading away from the origin (Figure 1).
Figure 1.
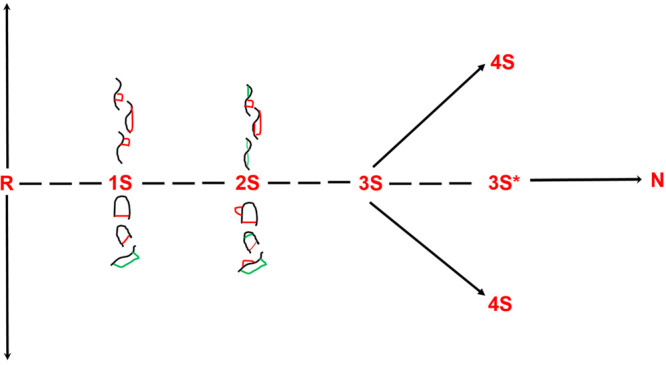
Isomers in every nS ensemble contain native (green) and non-native (red) disulfide bonds containing n disulfide bonds (as shown for the 1S and 2S species). Since the native protein contains only native disulfide bonds, the non-native disulfide bonds must reshuffle to native disulfide bonds. Native protein (N) is formed via the oxidation of two three-native-disulfide-containing 3S* intermediates that accumulate from their 3S isomers by conformational folding.
The origin itself corresponds to R.
Furthermore, native disulfides reside along the pathway Y = 0, and processes that compromise the securing of native disulfide bonds reside along Y = c.
Elements That Compromise the Trajectory to N
Thiol–Disulfide Isomerization Reactions in the Structure-Forming Step of Oxidative Folding
An examination of the rate-determining step in RNase A can provide insight into the impact of thiol–disulfide isomerization on the existing population of native disulfide bonds and its impact on the trajectory to N. As previously stated, native disulfide bonds alone can provoke conformational folding to form native/native-like structures. The ensuing native/native-like structures, in turn, secure the formed native disulfide bonds from becoming reshuffled to their non-native counterparts. To further elaborate, the rate-determining formation of native-like 3S* species from their unstructured 3S counterparts takes place in two steps (Figure 2).18 The first step is chemical in nature and involves intramolecular, thiol–disulfide reshuffling of non-native disulfide-bond-containing unstructured 3S isomers (3SNNU) to form unstructured 3S isomers (3SNU) that contain only native disulfide linkages. It is mandatory for only native disulfide bonds to be present prior to the second, conformational folding, step (as previously discussed, only native disulfide bonds provoke native-like/native structure). In RNase A, a second requirement is that a minimum of three native linkages are necessary to provoke conformational folding. The second step is physical in nature and involves the formation of fold-creating and fold-stabilizing interactions (van der Waals, hydrogen bonding, salt bridges, and hydrophobic interactions). Note: In RNase A, the second step also involves the locking of X–Pro peptide bonds from their equilibrium unfolded-state distribution (70:30 trans:cis) to either a trans-locked or a cis-locked conformation at each X–Pro bond.3−6
Figure 2.
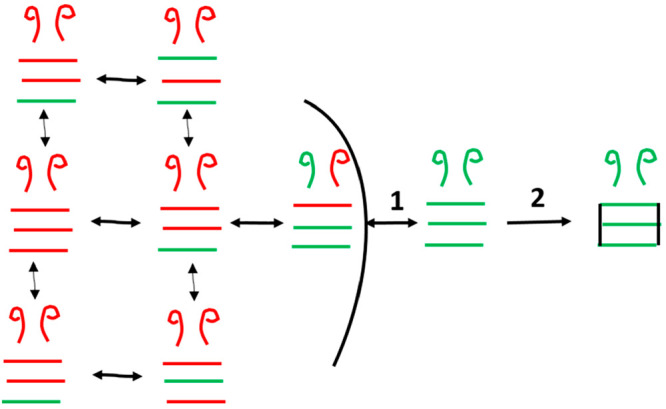
Two-step structure-forming process in RNase A involves a competition between thiol–disulfide isomerization reactions (step 1) between 3SNU and 3SNNU and a conformational folding reaction (step 2; 3SNNU to 3S*) that secures the native disulfide bonds within the structure.
The yield of the forward, physical, reaction depends on the concentration of 3SNU that is available to conformationally fold. However, prior to conformational folding, the 3SNU cluster is transient. I.e., it is vulnerable to intramolecular thiol–disulfide exchange.
To address the role of thiol–disulfide exchange reactions in the rate-determining step of RNase A, oxidative folding let us consider a fixed concentration of isolated 3SNU species at t = 0 and pH 2. A pH jump from 2 to 8 at 25 °C (folding conditions) promotes conformational folding to form des [65–72] and des [40–95]. However, it simultaneously induces intramolecular thiol–disulfide exchange. Essentially, the 3SNU cluster rapidly “decomposes” to 3SNNU. The ensemble (now statistically composed of 420 3S species) starts “oscillating” back and forth while transiently populating the 3SNU cluster, the only subensemble capable of conformational folding (Figure 3). Since only native disulfide-bond-containing intermediates can provoke conformational folding, the reduction in the concentration of native disulfide-bond-containing intermediates will inhibit the folding rate. That is, since only the subcluster with three native disulfides can conformationally fold, any pH-dependent excursions away from the pool will reduce the forward reaction rate.
Figure 3.
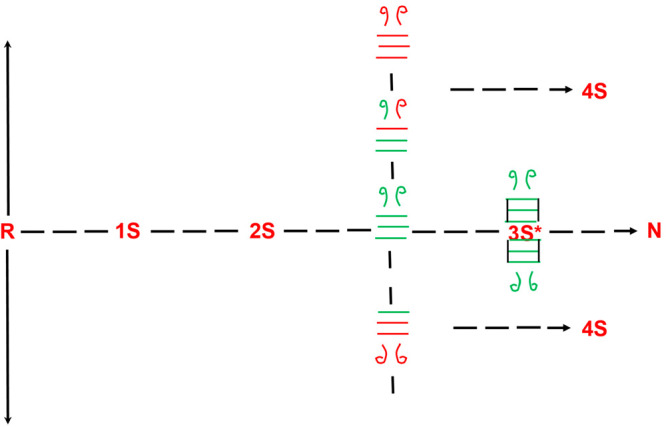
Intramolecular reshuffling reactions within the 3S ensemble compete with the conformational folding reaction in the rate-determining, structure-forming step of RNase A. Reshuffling of native-disulfide-bond-containing 3S species (all green bonds), which is shown to reside orthogonal to the trajectory to N, results in the formation of non-native disulfide-bond-containing species (red ± green bonds). By contrast, 3S species with all native disulfide bonds can conformationally fold to the structured 3S* species that protect the native bonds from undergoing isomerization reactions.
A reduction in the final pH of the pH jump experiment (pH 2 to say pH 6, instead of 8) will reduce the rate of intramolecular thiol–disulfide exchange of the 3SNU pool and, thereby, the loss of native disulfides via excursions away from 3SNU (the formation of 3SNNU isomers). The drop in final pH preserves native disulfides and tilts the competition between thiol–disulfide isomerization and conformational folding to favor the physical (conformational folding) reaction. Conversely, an increase in the final pH of the pH jump experiment, to say pH 9, will lead to a rapid loss in the initial population of native disulfide-bond-containing species (3SNU).
In any final pH scenario (pH 2 to 6, 8, or 9), it is always incumbent upon the “oscillating ensemble” that only transiently acquires native disulfide bonds to conformationally fold when the native set is acquired. The window of opportunity to do so is nothing but the pH-dependent persistence time that 3SNU is populated.
However, there is a special case when it is fruitful for thiol–disulfide exchange isomerization reactions to be rapid and consume the 3SNU pool prior to its conformational folding to form 3S* species. Using RNase A as an example, we recall that only two of the four 3S* species that emerge at low temperatures from the conformational folding of the 3SNU species are on the pathway. Des [40–95] and des [65–72] are the only two 3S* species capable of forming N (13, 14). Des [58–100] and des [26–84] are kinetically trapped and must unfold and reshuffle via 3NNU prior to repopulating on-pathway-forming 3SNU species.19,20 pH-driven excursions from the initial 3SNU pool would prevent kinetic-trap-forming 3SNU species from conformationally folding into kinetically trapped intermediates. The beneficial role of intramolecularly driven excursions away from native disulfides is particularly useful in the case of BPTI which forms two kinetically trapped intermediates, whose reshuffling to on-pathway species is rate determining.1
Native Disulfide Bonds vs Redox Concentration in Oxidative Folding
Akin to thiol–disulfide isomerization reactions that can compromise the pool of unstructured native disulfide-bond-containing intermediates, the redox concentration in the oxidative folding milieu can also adversely impact the class of the aforementioned intermediates.
Consider the forward trajectory: R → 1S → 2S → 3S → 4S. The forward rate in each step depends on the concentration and oxidation potential of the redox reagent. For example, compared to DTTox, GSSG is a strong oxidizing agent. Therefore, for a fixed concentration of either reagent, the “dwell time” of the 3SNU pool will be shorter for GSSG than DTTox as it is oxidized more rapidly to 4S in the presence of the oxidizing agent with a stronger oxidation potential. Furthermore, w.r.t. any oxidizing agent, an increase in the concentration of the oxidizing agent will compete with persistence time of the bonds in every ensemble. Therefore, as the concentration of the oxidizing agent increases, there will be a tendency to reduce the persistence time of each species in an nS ensemble. Statistically, in each ensemble, there are fewer species with all native disulfide bonds than those with one or more non-native disulfide bonds. For example, there are four 1S intermediates that possess native disulfide bonds in RNase A ([40–95], [26–84], [58–110], and [65–72]) and 24 non-native disulfide-bond-containing 1S intermediates. The non-native disulfide-bond-containing intermediates must reshuffle to their native disulfide-bond-containing isomers for conformational folding (to a native/native-like state) to occur. The intramolecular reshuffling rate is in direct competition with the intermolecular oxidation reaction. If the oxidizing potential is too strong, then the non-native disulfide-bond-containing isomers in any nS ensemble will become oxidized to the (n + 1)S ensemble prior to reshuffling to their native disulfide-containing counterparts (Figure 4). The overall outcome would be the terminal oxidation to the 4S ensemble (in RNase A), leading to reduced yields and compromised biological function.
Figure 4.
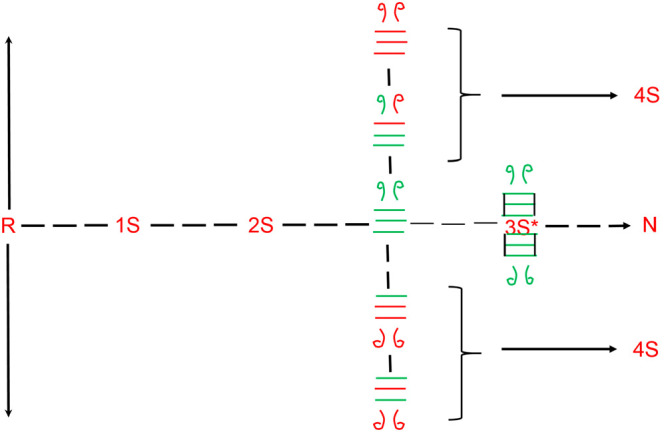
Number of non-native disulfide-bond-containing isomers in any nS ensemble far exceeds the number of native disulfide-bond-containing species. The presence of a strong oxidizing agent rapidly oxidizes unstructured species, such as the 3SNNU species, to 4S prior to it being able to reshuffle to 3SNU, compromising the formation of N.
Therefore, the population of every 3SNU species, in RNase A, is compromised by two chemical reactions: (1) Intramolecular thiol–disulfide exchange reactions that reduce the concentration of 3SNU and (2) intermolecular oxidizing (and, in principal, reducing) reactions that convert 3SNU to 4SNU before it can conformationally fold. It is noted that 4SNU can fold to N, but as the next section discusses, in RNase A the conformational folding is proline-isomerization-dependent and slow.
Role of Proline Isomerization in the Preserving Native Disulfide Bonds
Generally, amino acids across peptide bonds predominantly exist in the trans conformation. However, the X–Pro (where X is any preceding amino acid) peptide bond can exist in both cis and trans conformations in the unfolded state. The conformations are almost isoenergetic. As a result, the equilibrium ratio of the cis to trans configurations of the X–Pro peptide bond is 30:70. Kinetically, the process of isomerization is slow, as the activation energy between the two states is large. Furthermore, once the protein has acquired native-like or native conformation, the X–Pro peptide bond remains locked in either one of the two states (100% cis or 100% trans). The locking of the X–Pro peptide bond is a result of both steric influence and enthalpic interactions.3−5
In RNase A, three of the four prolines are essential to conformational folding. The isomerization of prolines 93, 114, and 117 to their cis, cis, and trans conformations, viz., “cct”, is a precedent to the conformational folding event that ensures that the X–Pro peptides become locked in their aforementioned conformations. Pro isomerization is a rate-determining event in the conformation folding of RNase A.3
To address the role of proline isomerization in preserving any existing set of native disulfide bonds in an nS species, we will consider a box model to depict a 3SNU intermediate in any of the eight possible isomerization states of its essential prolines (Figure 5). The 3SNU state cct possesses, by coincidence, the same proline isomerization state found in 3S* and N. Thus, the conformational folding of 3SNU cct is very fast (milliseconds). 3SNUcct therefore is able to preserve its native disulfides by rapidly acquiring a native-like structure that protects the disulfides from isomerization.
Figure 5.
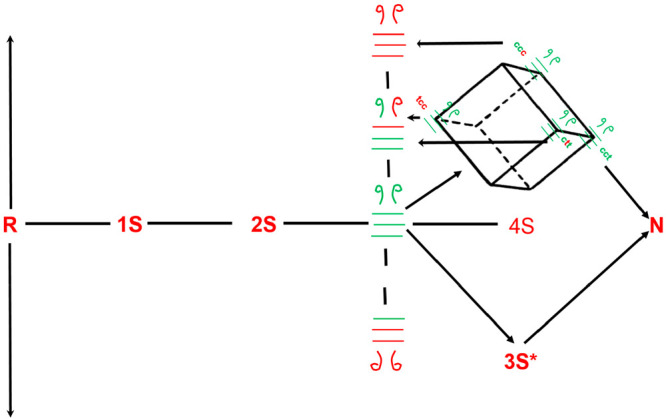
Cis–trans proline isomerization around the X–Pro peptide bond competes with the reshuffling reactions. Only native disulfide-bond-containing 3S species with their X–Pro peptide bonds in the c,c,t conformation can conformationally fold to form the 3S* species. The remaining X–pro isomers (e.g., c,t,c) must isomerize to c,c,t prior to conformational folding. However, since proline isomerization is slow, there is a high likelyhood that the native disulfide-bond-containing 3S intermediate (all green bonds with c,t,c disposition) reshuffles to a non-native disulfide-bond-containing isomer.
By contrast, consider a 3SNU species whose prolines are in a conformation that is one step away from cct. 3SNU ccc is one such conformation along with 3SNU tct and 3SNU ctt. Proline 117 in 3SNU ccc must isomerize from the cis to the trans state (3SNU ccc → 3SNU cct) for RNase to form the 3S* species. However, this kinetically slow step competes with two (previously discussed) chemical reactions: (1) the 3SNU ccc species can undergo intramolecular reshuffling to form 3SNNU ccc prior to forming 3SNU cct, and (2) 3SNU ccc can become oxidized to 4SNU ccc.
The former scenario adversely impacts the formation of 3S* species. Not only is the native disulfide converted to a non-native disulfide but also the ccc proline conformation can convert to other cis–trans combinations (for example, ctc) that are further removed from cct.
Proline isomerization is rate limiting in the conformation folding (3SNU → 3S*) step of RNase A. The slow rate of isomerization is detrimental to the preservation of native disulfide bonds.
Discussion
The oxidative folding of RNase A underscores the role of native disulfide bonds in the trajectory toward acquisition of the native fold (R → N). The findings from RNase regeneration, coupled with those from other disulfide-bond-containing proteins such as BPTI and HEWL, reveal that native disulfide bonds are a necessary denominator for conformational folding of the protein to form native-like/native structures.18,21 Conversely, native or native-like structures secure native disulfide bonds from reshuffling to their non-native isomers or from becoming reduced. Furthermore, while a minimum set of native disulfide bonds is required for an intermediate to acquire a structure, the presence of non-native disulfide bonds precludes conformational folding to a native state. Once it is established that only native disulfide bonds can provoke the formation of native-like/native structure, it is crucial to recognize elements that compete with the formed set of native disulfide bonds prior to the conformational folding reaction that secures them.
Here, we have showcased three distinct elements that compete with the formed set of native disulfides. (1) Intrinsic competitors include thiol–disulfide exchange reactions that lead to an attrition in the concentration of all native disulfide bond species in any intermediate. This has particularly deleterious consequences when the intermediate is poised to conformationally fold. (2) The slow rate of proline isomerization is a second intrinsic feature that contributes to the loss of all native disulfide-bond-containing intermediates. (3) The redox potential in the endoplasmic reticulum milieu also factors into the ability of native disulfide bonds to be preserved.
The innate nature of elements (1) and (2) and the in situ nature of (3) have consequences for homeostasis within the cell.20 Delays in protein folding or the misfolding of proteins lead to the exposure of hydrophobic domains for prolonged periods. Intermolecular interactions due to the hydrophobic effect trigger aggregation and disrupt traffic through the endoplasmic reticulum (ER). The ER-associated protein degradation (ERAD) machinery exports misfolded debris from the ER to the cytoplasm where it is ubiquitinated prior to proteasomal degradation.19,22
The ubiquitination machinery can become overburdened due to misfolded debris exiting the ER. As a consequence, misfolding-prone proteins such as the amyloid family, including amyloid β, α-synuclein, mutant Huntingtin, etc., fail to be ubiquitinated in a timely manner.19,20 The amyloid accumulates peripherally and causes cellular (neuronal) damage due to pore formation, disruption of Ca2+ homeostasis, etc. Amyloid accumulation correlates with oxidative stress and disrupts protein homeostasis in neurons by colocalizing with chaperones and other proteins, further disrupting cellular function. Amyloid seeds can be transmitted across neurons where they continue to corrupt neuronal assemblies, contributing to neuronal injury and neuronal demise and onset neurodegenerative pathologies. A specific example of the role played by disulfide bonds in protein-misfolding-related disorder onset is observed in the role played by the [2–7] disulfide bond of islet amyloid peptide (IAPP) and type 2 diabetes mellitus (T2DM).23 The reduction of this disulfide bond modulates the aggregation of IAPP in T2DM where extracellular amyloid deposits localized to the dysfunctional area of pancreatic cells are found in >90% of patients. The disulfide-intact peptide demonstrates increased stability and a reduced tendency to form a fibril-driving β sheet structure.
Disulfide bonds play a key role in the stability of nonamyloid proteins as well. The only covalent bond besides the peptide backbone can exert a tremendous influence of protein stability compared to the remainder of the stabilizing forces (van der Waals, hydrogen bonding, salt bridges, and the hydrophobic interactions) which are noncovalent in nature. Each of the four disulfide bonds of RNase plays a crucial role in its stability as evidenced from a comparison of the midpoints in thermal transitions of des [40–95], des [65–72], des [58–110], and des [26–84] relative to native RNase A (65 °C).13,14,17 Furthermore, their contributions to stability are not equal. While des [40–95] and des [65–72] have thermal transitions ∼25 °C less than that of native RNase A, both des [58–110] and des [26–84] do not appear in the folding trajectory of RNase A at 25 °C, suggesting that their thermal transitions are 40 °C lower than that of the native protein.
In azurin, reduction of the [3–26] does not affect the spectroscopic signals associated with its structure to the extent that it affects its thermal stability.24 The midpoint of thermal transition of Cys-3Ala/Cys-26Ala azurin is down-shifted by 20 °C compared to the wild-type protein.
In each of the examples mentioned below it is critical to note that while the reduction of individual disulfides resulted in a loss of protein stability the native-like features of the corresponding “des” species were retained. These observations once again reinforce the notion that a critical number of native disulfide bonds provoke native/native-like structure, albeit with compromised stability. The number of disulfides required for engendering structural stability can be a subset of the total number of disulfide bonds. As exhaustively discussed, it is “in the interest” of the protein to develop native-like structure early on in the oxidative folding trajectory to align the remaining cysteines for the rapid oxidation to native disulfide bonds while limiting non-native cysteine oxidation. The former scenario further stabilizes the already formed native-like structure and has a “cooperative” (allosteric) effect on the rate of acquisition of the native, biologically active fold. In sum and substance, a native disulfide-bond-endowed native/native-like structure is an epoch-making event in the oxidative folding trajectory as it removes a productive “native/native-like” species from the equilibrium of unfolded species in different oxidative states.
In conclusion, timely acquisition of native disulfide bonds can contribute to cell health.25 The interplay between native disulfide bonds, native structure, and elements that compete with the tendency to preserve and protect native disulfide bonds can have important consequences not only for protein folding but also for organismal homeostasis and function.
Acknowledgments
The author would like to thank Mr. Gyan M. Narayan for assistance with the references and Giber Fonseca, Jyoti Ahlawat, Zhaobo Li, Rui Dong, Gabriel Henriquez, Dayan Viera, Shima Masoudi Asil, Daniel Von Salzen, Elizabeth Noriega Landa, and Gileydis Guillama Barroso for discussions.
Biography
Mahesh Narayan is currently serving as a professor in the Department of Chemistry and Biochemistry at the University of Texas at El Paso. In summary, he has authored and coauthored over 85 research and review articles and book chapters in the areas of free radical biology, protein–structure function, oxidative folding and protein misfolding, halogen bonding, and in silico drug design for a collective citation count of 3720. Five other manuscripts have been submitted, are under revision, or are being prepared. He retains corresponding authorship of 84% of the papers he has published post-tenure. His work has been recognized through invitations to speaking engagements in over 15 international forums and by recognition in a variety of media outlets. Currently, he serves on the editorial board of PLOS One (Public Library of Science) and the editorial board of Cell Biochemistry and Biophysics (Springer) starting in 2016. He has been invited to deliver lectures at the Institute of NanoChemistry and NanoBiology of Shanghai University.
Author Contributions
M.N. conceived the topic and wrote the manuscript.
M.N. acknowledges support from NIH 1SC3 GM111200 01A1 for this work.
The author declares no competing financial interest.
References
- Narayan M.; Welker E.; Wedemeyer W. J.; Scheraga H. A. Oxidative folding of proteins. Acc. Chem. Res. 2000, 33, 805–812. 10.1021/ar000063m. [DOI] [PubMed] [Google Scholar]
- Bechtel T. J.; Weerapana E. From structure to redox: the diverse functional roles of disulfides and implications in disease. Proteomics 2017, 17, 1600391. 10.1002/pmic.201600391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welker E.; Narayan M.; Wedemeyer W. J.; Scheraga H. A. Structural determinants of oxidative folding in proteins. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 2312–2316. 10.1073/pnas.041615798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedemeyer W. J.; Welker E.; Narayan M.; Scheraga H. A. Disulfide bonds and protein folding. Biochemistry 2000, 39, 4207–4216. 10.1021/bi992922o. [DOI] [PubMed] [Google Scholar]
- Welker E.; Wedemeyer W. J.; Narayan M.; Scheraga H. A. Coupling of conformational folding and disulfide-bond reactions in oxidative folding of proteins. Biochemistry 2001, 40, 9059–9064. 10.1021/bi010409g. [DOI] [PubMed] [Google Scholar]
- Hudson D. A.; Gannon S. A.; Thorpe C. Oxidative protein folding: From thiol-disulfide exchange reactions to the redox poise of the endoplasmic reticulum. Free Radical Biol. Med. 2015, 80, 171–182. 10.1016/j.freeradbiomed.2014.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woycechowsky K. J.; Raines R. T. Native disulfide bond formation in proteins. Curr. Opin. Chem. Biol. 2000, 4, 533–539. 10.1016/S1367-5931(00)00128-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamathambika B. S.; Bardwell J. C. Disulfide-linked protein folding pathways. Annu. Rev. Cell Dev. Biol. 2008, 24, 211–35. 10.1146/annurev.cellbio.24.110707.175333. [DOI] [PubMed] [Google Scholar]
- Rothwarf D. M.; Scheraga H. A. Regeneration of bovine pancreatic ribonuclease A. 1. Steady-state distribution. Biochemistry 1993, 32, 2671–2679. 10.1021/bi00061a027. [DOI] [PubMed] [Google Scholar]
- Rothwarf D. M.; Scheraga H. A. Regeneration of bovine pancreatic ribonuclease A. 2. Kinetics of regeneration. Biochemistry 1993, 32, 2680–2689. 10.1021/bi00061a028. [DOI] [PubMed] [Google Scholar]
- Rothwarf D. M.; Scheraga H. A. Regeneration of bovine pancreatic ribonuclease A. 3. Dependence on the nature of the redox reagent. Biochemistry 1993, 32, 2690–2697. 10.1021/bi00061a029. [DOI] [PubMed] [Google Scholar]
- Rothwarf D. M.; Scheraga H. A. Regeneration of bovine pancreatic ribonuclease A. 4. Temperature dependence of the regeneration rate. Biochemistry 1993, 32, 2698–2703. 10.1021/bi00061a030. [DOI] [PubMed] [Google Scholar]
- Rothwarf D. M.; Li Y. J.; Scheraga H. A. Regeneration of bovine pancreatic ribonuclease A: Identification of two native like three-disulfide intermediates involved in separate pathways. Biochemistry 1998, 37, 3760–3766. 10.1021/bi972822n. [DOI] [PubMed] [Google Scholar]
- Rothwarf D. M.; Li Y. J.; Scheraga H. A. Regeneration of bovine pancreatic ribonuclease A: Detailed kinetic analysis of two independent folding pathways. Biochemistry 1998, 37, 3767–3776. 10.1021/bi972823f. [DOI] [PubMed] [Google Scholar]
- Xu X.; Rothwarf D. M.; Scheraga H. A. Nonrandom distribution of the one-disulfide intermediates in the regeneration of ribonuclease A. Biochemistry 1996, 35, 6406–6417. 10.1021/bi960090d. [DOI] [PubMed] [Google Scholar]
- Volles M. J.; Xu X.; Scheraga H. A. Distribution of disulfide bonds in the two-disulfide intermediates in the regeneration of bovine pancreatic ribonuclease A: Further insights into the folding process. Biochemistry 1999, 38, 7284–7293. 10.1021/bi990570f. [DOI] [PubMed] [Google Scholar]
- Welker E.; Narayan M.; Volles M. J.; Scheraga H. A. Two new structured intermediates in the oxidative folding of RNase A. FEBS Lett. 1999, 460, 477–479. 10.1016/S0014-5793(99)01391-5. [DOI] [PubMed] [Google Scholar]
- Narayan M.; Welker E.; Scheraga H. A. Development of a novel method to study the rate-determining step during protein regeneration: Application to the oxidative folding of RNase A at low temperature reveals BPTI-like kinetic traps. J. Am. Chem. Soc. 2001, 123, 2909–2910. 10.1021/ja003934w. [DOI] [PubMed] [Google Scholar]
- Narayan M. Disulfide bonds: Protein folding and subcellular protein trafficking: Disulfide bonds. FEBS J. 2012, 279, 2272–2282. 10.1111/j.1742-4658.2012.08636.x. [DOI] [PubMed] [Google Scholar]
- Narayan M.The case of oxidative folding of ribonuclease A: Factors impacting fold maturation of ER-processed proteins. In Folding of Disulfide Proteins; Springer: New York, NY, USA, 2011; pp 23–42. [Google Scholar]
- Arai K.; Shibagaki W.; Shinozaki R.; Iwaoka M. Reinvestigation of the oxidative folding pathways of hen egg white lysozyme: Switching of the major pathways by temperature control. Int. J. Mol. Sci. 2013, 14, 13194–13212. 10.3390/ijms140713194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson D. A.; Gannon S. A.; Thorpe C. Oxidative protein folding: From thiol-disulfide exchange reactions to the redox poise of the endoplasmic reticulum. Free Radical Biol. Med. 2015, 80, 171–182. 10.1016/j.freeradbiomed.2014.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milardi D.; Pappalardo M.; Pannuzzo M.; Grasso D. M.; La Rosa C. L. The role of the Cys2-Cys7 disulfide bridge in the early steps of Islet amyloid polypeptide aggregation: A molecular dynamics. Chem. Phys. Lett. 2008, 463, 396–399. 10.1016/j.cplett.2008.07.110. [DOI] [Google Scholar]
- Guzzi R.; Sportelli L.; La Rosa C.; Milardi D.; Grasso D.; Verbeet M. P.; Canters G. W. A spectroscopic and calorimetric investigation on the thermal stability of the Cys3Ala/Cys26Ala azurin mutant. Biophys. J. 1999, 77, 1052–63. 10.1016/S0006-3495(99)76955-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossuto M. F. Disulfide bonding in neurodegenerative misfolding diseases. Int. J. Cell Biol. 2013, 2013, 318319. 10.1155/2013/318319. [DOI] [PMC free article] [PubMed] [Google Scholar]