

Abstract
Mitochondria, the powerhouse of a cell, are closely linked to the pathophysiology of various common as well as not so uncommon disorders of the liver and beyond. Evolution supports a prokaryotic descent, and, unsurprisingly, the organelle is worthy of being labeled an organism in itself. Since highly metabolically active organs require a continuous feed of energy, any dysfunction in the structure and function of mitochondria can have variable impact, with the worse end of the spectrum producing catastrophic consequences with a multisystem predisposition. Though categorized a hepatopathy, mitochondrial respiratory chain defects are not limited to the liver in time and space. The liver involvement is also variable in clinical presentation as well as in age of onset, from acute liver failure, cholestasis, or chronic liver disease. Other organs like eye, muscle, central and peripheral nervous system, gastrointestinal tract, hematological, endocrine, and renal systems are also variably involved. Diagnosis hinges on recognition of subtle clinical clues, screening metabolic investigations, evaluation of the extra-hepatic involvement, and role of genetics and tissue diagnosis. Treatment is aimed at both circumventing the acute metabolic crisis and long-term management including nutritional rehabilitation. This review lists and discusses the burden of mitochondrial respiratory chain defects, including various settings when to suspect, their evolution with time, including certain specific disorders, their tiered evaluation with diagnostic algorithms, management dilemmas, role of liver transplantation, and the future research tools.
Keywords: Mitochondrial hepatopathy, Respiratory chain defects, Maternal inheritance, Neonatal liver failure, DNA depletion syndrome, Pearson syndrome
Core Tip: Liver disease with multi-system involvement should arouse the suspicion for mitochondrial respiratory chain hepatopathies. These disorders are predominantly autosomal recessive with some having a maternal inheritance. Presence of lactic acidosis without hypoglycemia is an important clue. A tiered evaluation yields the most data, with the final step being a genetic and enzyme analysis from tissue of interest. Treatment is largely supportive with blood transfusions, correction of acidosis and shock, providing cofactors and salvage therapies, with liver transplantation in a select group. A periodic follow-up is mandatory for monitoring evolution of disease including “migration” to other systems.
INTRODUCTION
Mitochondria are intracellular organelles, with a double lamellar covering outer membrane serving as a corset that holds the highly convoluted inner membrane in place (Figure 1). The inter-membrane space is the first of two liquid components within the mitochondria mainly participating in the exchange of lipids, proteins, and metal ions and also signaling cascades[1]. The second space is the soluble matrix, lying within the inner membrane and the hub of various metabolically active processes, most notably the tricarboxylic acid cycle, fatty acid oxidation, and urea synthesis. The inner membrane is folded into multiple cristae, which are shelf like projections into the matrix. The number of cristae are reflective of a metabolically active state, with a more active tissue having numerous mitochondria with many cristae, an ideal comparison being striated muscle tissue against adipocytes. This inner membrane of the mitochondria houses the respiratory chain comprised of electron carriers (complexes I, II, III, and IV, cytochrome c, coenzyme Q) and complex V, which is the hydrogen adenosine triphosphatases complex (Figure 2). All metabolic processes within the matrix generate reducing equivalents in the form of electrons (carried as NADPH2), which pass through these complexes, entering it at various points. While doing so, from one complex to another, it also results in proton (H+) flow from matrix to intermembrane space leading to its pooling up and a chemical gradient that then flows down the potential via the complex V, which utilizes the energy to generate adenosine triphosphate from adenosine diphosphate, the ultimate objective of this intricately woven complex process called oxidative phosphorylation[2].
Figure 1.

Diagrammatic representation of structure of mitochondria. mtDNA: Mitochondrial DNA.
Figure 2.

The electron transport chain formed by the respiratory chain complexes and process of oxidative phosphorylation. ADP: Adenosine diphosphate; ATP: Adenosine triphosphate; CoQ: Coenzyme Q; Cyt c: Cytochrome c; FAD: Flavin adenine dinucleotide; FADH2: Reduced form of FAD; NAD: Nicotinamide adenine dinucleotide; NADH: Reduced form of NAD.
MITOCHONDRIAL GENOME AND ITS IMPLICATIONS
From the perspective of evolution, the classical endosymbiont theory proposes that mitochondria are actually prokaryotes within eukaryotic cells and hence have a genome of their own[3]. Mitochondrial genome consists of a circular double stranded DNA made of 16569 base pairs organized to make up 37 genes. Of these, 13 genes are exclusively for synthesis of proteins that are part of the respiratory chain. The other 24 genes are required for mitochondrial DNA (mtDNA) translation process (22 genes for an equal number of transfer RNA and two for ribosomal RNA synthesis). There are three major differences between mitochondrial and Mendelian inheritance: Maternal inheritance, heteroplasmy and threshold effect, and mitotic segregation. Maternal inheritance in simple terms means that the mtDNA and its aberrations are transferred from mother (ovum) to its offspring (zygote), as there is hardly any mitochondria left in the sperm, which concentrates itself to fill its entire cytoplasm with the energy dense nucleus. However, there are a few exceptions, as reported in skeletal muscle defects linked to mitochondrial inheritance that are transmitted by father to offspring[4]. It is essential to understand that all characteristics encoded by mtDNA are maternally inherited but all mitochondrial diseases are not maternally inherited.
Nuclear DNA (nuDNA) encodes most of the metabolic processes occurring in the mitochondria. NuDNA also encodes many enzymes and cofactors required for maintenance of mtDNA as well as approximately 70 respiratory chain subunits[5]. Figures 3 and 4 describe the way of inheritance and the mathematics of genetics in mitochondrial diseases. In normal persons, all mtDNA are identical, a state known as homoplasmy. Presence of both mutated and non-mutated wild type mtDNA containing mitochondria together in a cell is cellular heteroplasmy, while having 2 types of mtDNA within a single mitochondrion is organellar heteroplasmy. A particular number of abnormal mtDNA burden should exist for disease phenotype to manifest, a phenomenon known as threshold effect. This effect is seen at different levels of mutated mtDNA in various organs, the lowest threshold (and hence maximum susceptibility) being in organs dependent highly on oxidative metabolism like brain, heart, skeletal muscle, retina, and endocrine organs. Another interesting phenomenon is “skewed heteroplasmy” where some organs selectively have a higher burden of abnormal mitochondria, exemplified by mitochondrial diabetes, cardiomyopathies, and deafness[6-8]. Mitotic segregation effect refers to the random distribution of mitochondria at end of cell division, which can segregate mutated and non-mutated mtDNA in a variable manner into the two daughter cells. This may result in a daughter cell phenotype that is diseased (due to presence of more abnormal mitochondria), i.e. more abnormal than the originator cell in the subsequent divisions. With age, abnormal cells may predominate, explaining age related unmasking of diseases.
Figure 3.

Various modes of inheritance of mitochondrial disease. mtDNA: Mitochondrial DNA.
Figure 4.

Comparison of mitochondrial and nuclear DNA influence in genetics of mitochondria. mtDNA: Mitochondrial DNA; rRNA: Ribosomal RNA; tRNA: Transfer RNA.
THE PROBLEM STATEMENT: EPIDEMIOLOGY
Prevalence of respiratory chain defects is variable across geographical lines as well as across eras. A large study examining birth prevalence of mitochondrial respiratory chain disorders (RCDs) up to 16 years of age puts the figure at 5/100000 births[9]. This would mean that for every 20000 births in a particular time period, 1 child has the probability of getting affected by a respiratory chain defect of any type till he or she reaches the age of 16. The same study extrapolated the prevalence as 13.1/100000 births with onset at any age when seen together in the light of another study by Chinnery et al[10]. According to the Swedish registry, in a population study identifying mitochondrial encephalomyopathies, 20% had liver involvement[11]. In a 5 year French study of 1041 children, 22 (10%) of the 234 patients with respiratory chain defects had hepatopathy[12]. We would, however, add a word of caution that these figures can be an underrepresentation of true values in view of the heterogeneity of presentation and difficulty in diagnosis of mitochondrial respiratory chain defects.
CLASSIFICATION OF MITOCHONDRIAL HEPATOPATHIES AND STATUS OF RESPIRATORY CHAIN DISORDERS
Mitochondrial disorders are characterized by their variability in presentation and predilection for more than one organ system simultaneously or separated in time.
Sokol and Treem proposed classifying these disorders as primary and secondary depending on whether defect is inherently present in the mitochondria and leads to liver dysfunction or there is secondary involvement of mitochondria in the form of injury or alteration in non-mitochondrial genetics. There are two broad types of mitochondrial hepatopathies, one which affects the respiratory chain present on the inner mitochondrial membrane and the other includes fatty acid oxidation defects, which are related to the process within the mitochondrial matrix. The RCDs can also be divided into those arising due to defective mtDNA and those due to defect/ mutation in nuDNA. Among the diseases affecting mtDNA, the affliction can be in the form of either mutations or an overall depletion of quantity of mtDNA compared to nuDNA in a cell/tissue. Figure 5 shows a simplified way of classification of mitochondrial hepatopathies (MH), and a tabular representation of primary MH individual disorders is shown in Table 1. It is worthwhile to note that usually mitochondrial disorders with primary myopathic involvement have mutations in mtDNA, while those with primary hepatic involvement have mutations in nuDNA affecting mitochondrial processes, with some exceptions[13]. Since we are discussing respiratory chain disorders not confined to liver but to include the gastrointestinal tract, we will include one prototype non-hepatic RCD affecting the gastrointestinal (GI) tract, mitochondrial neurogastrointestinal encephalomyopathy (MNGIE), in our review. This review does not cover non-RCD mitochondrial hepatopathies (fatty acid oxidation disorders and others) and GI manifestations of non-RCD, non-hepatic mitochondrial disorders.
Figure 5.

Simplified way of classification of mitochondrial hepatopathies based on location of defect. IMS: Intermembrane space; mtDNA: Mitochondrial DNA; PEP: Phosphoenolpyruvate.
Table 1.
Various mitochondrial primary respiratory chain disorders
|
Disorder
|
Mutation/defective gene
|
Location of defect
|
Affected proteins/consequence
|
| Neonatal liver failure: (1) Complex I deficiency; (2) Complex III deficiency; (3) Complex IV deficiency; and (4) Multiple complex deficiencies | ACAD9; BCS1L; SCO1 | nuDNA | Respective complexes deficiency as per name |
| Delayed onset liver failure: Alper’s Huttenlocher syndrome | POLG mutation | nuDNA | Defective mtDNA polymerase; mtDNA depletion |
| MtDNA depletion syndrome | DGUOK; TK-2; MPV 17; POLG | All nuDNA | Decreased deoxyribonucleotide concentrations within mitochondria |
| Mitochondrial neuro-gastrointestinal encephalomyelopathy | TYMP | nuDNA | Markedly low levels of thymidine phosphorylase activity |
| Pearson marrow pancreas syndrome | 4000-5000 bp deletions in mtDNA; tRNA gene of mtDNA | Both mtDNA | Complex I, IV, V |
| Navajo neurohepatopathy | MPV 17 mutations | nuDNA | mtDNA depletion |
| Villous atrophy with hepatic involvement | Rearrangement defect/deletion-duplications in mtDNA | mtDNA | Complex III deficiency |
nuDNA: Nuclear DNA; mtDNA: Mitochondrial DNA.
CLINICAL PRESENTATION
Mitochondrial disorders are often called mitochondrial multiorgan disorder syndrome (MIMODS) in view of their heterogenous presentation affecting the nervous system (central and peripheral), eyes, ears, endocrine system, kidneys, heart and blood vessels, bone marrow, lungs, and also the intestinal tract, apart from affecting the liver (hepatopathy). The liver involvement is also variable in clinical presentation as well as in age of onset, from acute liver failure, cholestasis, or as chronic liver disease. A graphical summary of all mitochondrial RCDs affecting the liver is represented in Figure 6. Each of the individual disorders is briefly discussed.
Figure 6.

Graphical summary of various respiratory chain disorders involving liver on a timeline with key features. CNS: Central nervous system; IDDM: Insulin dependent diabetes mellitus; mtDNA: Mitochondrial DNA; PNS: Peripheral nervous system.
Neonatal liver failure
Neonatal liver failure is a catastrophic event, and there are few disorders that present in the first few months as liver failure. Neonatal acute liver failure (ALF) is distinct from pediatric and adult liver failures in that it can include causes that have underlying cirrhosis. Also, the cut-off for coagulopathy is proposed as an international normalized ratio (INR) of ≥ 3 for newborns, as normal INR can be up to 2 in this age[14]. The four main causes of neonatal liver failure are: (1) Gestational alloimmune liver disease (neonatal hemochromatosis); (2) Viral infections (herpes simplex); (3) Hemophagocytic lymphohistiocytosis (primary-familial/secondary to infections); and (4) Mitochondrial hepatopathies (respiratory chain defects).
Apart from these, galactosemia, tyrosinemia, and hereditary fructose intolerance can present as ALF in early infantile period and rarely in neonatal age[15]. The key here is to keep mitochondrial hepatopathy (RCD and non-RCD) as one of the differentials of acute liver failure in a newborn/early infantile period, though it accounts for < 5% in neonatal ALF series[14]. Multi-system involvement, especially with neurological symptoms in form of lethargy, floppy tone, vomiting, poor suck, and seizures, are diagnostic clues. Some patients are apparently normal until a viral illness or an unknown inciting event seems to trigger a downhill course either hepatic or neurological or both. Infants with mitochondrial hepatopathies are seen to have a low birth weight in up to 23%, and associated intrauterine growth retardation is seen in 16%, likely due to insult beginning from intrauterine period[16]. Laboratory findings of metabolic acidosis, elevated lactate levels, high lactate to pyruvate ratio often more than 30 mol/mol, elevated ketone bodies betahydroxybutyrate, and betahydroxybutyrate to acetoacetate ratio > 2 mol/mol are corroborative, but absence does not rule out the diagnosis. Liver biopsy findings may yield micro or macrovesicular steatosis, which reflects impaired energy metabolism. Liver or muscle tissue respiratory chain analysis shows decreased levels of complexes I, III, or IV. Liver biopsy is often done post-mortem due to the inability to do so percutaneously in view of coagulopathy. Treatment including liver transplant is discussed subsequently.
Delayed onset liver disease: Alpers Huttenlocher syndrome
This syndrome presents anywhere from 2 mo to 8 years of age, predominantly in late infancy to childhood (Figure 7 graphical summary). The diagnostic criteria include[17]: (1) Presence of refractory seizures including focal seizures; (2) Infection triggered psychomotor regression that is episodic in nature; and (3) Liver dysfunction with or without liver failure. Liver involvement is in the form of hepatomegaly, jaundice, coagulopathy, and episodes of hypoglycemia. Gastrointestinal involvement mainly due to the muscle impairment results in progressive feeding difficulty and gastroesophageal reflux, progressing to intractable vomiting. One series of 5 patients with Alpers Huttenlocher syndrome (AHS) showed mean age of liver disease presentation of 35 mo, and all died over a mean 4.6 wk period, due to progressive liver failure[18]. Autopsy findings across series show macrovesicular steatosis, massive hepatocyte dropout, proliferating bile ductular elements replacing hepatocytes, and often cirrhosis[17,18]. Valproate is known to precipitate liver failure in these patients when given for the frequently associated seizure disorder, which often demands use of more than one anticonvulsant. This is possibly because of depletion of respiratory chain enzyme activity by the drug and inability to increase metabolic rate by the DNA polymerase subunit gamma (POLG) deficient cells[19]. Valproate increases glycolysis, likely an indirect clue of impaired mitochondrial function as shown in yeast and mouse liver models[20]. POLG mutation subtype and zygosity influence outcome, with worst outcomes shown in compound heterozygous mutations for A467T and W748S[21].
Figure 7.

Graphical summary of Alpers Huttenlocher syndrome and its natural history. BiPAP: Bilevel positive airway pressure; C/I: Contraindicated; CPAP: Continuous positive airway pressure; FTT: Failure to thrive; LTx: Liver transplantation; Mx: Management; PEG: Percutaneous endoscopic gastrostomy; Vx: Vomiting.
Liver failure management, addressing feeding issues often mandating percutaneous endoscopic gastrostomy tube insertion, seizure control, and use of respiratory aids like continuous positive airway pressure in view of progressive motor impairment are cornerstones of management. Liver transplantation is often contraindicated in view of the multisystem involvement.
MtDNA depletion syndrome
DNA depletion is distinct from DNA deletion. MtDNA depletion refers to a state when a cell contains less than normal mtDNA per unit nuDNA. Depletion diseases are much more severe and earlier in onset compared to deletion diseases[22]. NuDNA encodes for processes within the mitochondria including production and stability of mtDNA. Mutations in nuDNA may result in low levels of or increased destruction of DNA pool essential for mtDNA synthesis[23], thereby reducing the concentration of mtDNA in the cell or tissue as a whole. The end result is suboptimal mitochondrial function. DGUOK mutations lead to predominant neuro-hepatopathy, while TK2 mutations lead to predominant myopathy[23]. As an illustration to above statements, TK2 induced depletions present early in the first few years with myopathy, feeding difficulty, hypotonia, and respiratory failure as a terminal event. However, multiple TK2 deletions present as proximal myopathy and chronic progressive external ophthalmoplegia later in life[24]. Two additional genes, POLG coding for mtDNA polymerase and MPV17, have been described in hepatocerebral form of MDS. POLG mutations in older children have been associated with AHS as already described. It should be understood that MDS and AHS both have mtDNA depletion. AHS got its name earlier and was later found to have its molecular basis as mtDNA depletion, and it characteristically refers to a comparatively delayed onset (> 2 mo age), compared to MDS, which has its onset in the first few weeks of life. The other mutation in nuclear gene MPV17 leads to decreased synthesis of an unknown inner mitochondrial membrane protein that possibly has a role in oxidative phosphorylation, and knockout mice (-/-) have shown impaired oxidative phosphorylation and also mtDNA depletion[25].
Liver failure in infancy is the common presentation of the hepatopathic form. There is notably an overlap between the hepatopathic form of MDS and neonatal liver failure presentation of RCD. The difference exists in the fact that the former has mtDNA quantity that is < 10% of nuDNA, and there is no sequence alteration in mtDNA.
Pearson syndrome
Pearson syndrome is one among three mitochondrial diseases (Kearne Sayre syndrome and chronic progressive external ophthalmoplegia being the other two) associated with a single large deletion in mtDNA[26]. This is a multi-systemic fatal disorder with involvement of exocrine pancreas, eyes, skin, hematological system, liver, and kidneys[22]. MtDNA rearrangements form the etiological basis, and it is associated with large 4-5 kbp deletions in a large proportion of cases. All respiratory chain complexes can suffer a decreased synthesis, with complex I most severely affected. Refractory anemia with ring sideroblasts occurs in infancy with vacuolization in bone marrow of myeloid and erythroid precursors[27]. Elevated plasma alanine and fumaric acid levels are discriminating from other non-mitochondrial bone marrow failure syndromes[28], though neither specific for Pearson syndrome nor distinguishing it from other mitochondrial disorders. Hematological manifestations may occur alone or in combination with renal tubular dysfunction (Fanconi syndrome) and hepatic failure. If the patient survives this phase of hematological symptoms, its intensity begins to decrease[29], and symptoms change from hematological to a phenotype of severe pancreatic insufficiency in late infancy to early childhood, during the same time which villous atrophy is found to appear. Eye involvement is in form of pigmentary retinopathy and external ophthalmoplegia and appears in early to late childhood. Liver involvement is in form of hepatomegaly with cirrhosis, cholestatic jaundice, elevated liver enzymes, and progressive liver failure leading to death in early childhood similar to what is seen in MDS[30]. Recent series have shown age of death ranging from 5 to 11 years, and mortality is worse in Pearson syndrome compared to other single large mitochondrial deletions[28,31]. A graphical summary outlining the natural history is shown in Figure 8. Supportive therapy with packed red cell transfusions for anemia, granulocyte colony stimulated factor for neutropenia, and bicarbonate for metabolic acidosis forms the basis of care.
Figure 8.

Graphical summary of Pearson marrow pancreas syndrome and its natural history. GCSF: Granulocyte colony stimulation factor; insuff: Insufficiency; KSS: Kearns Sayre syndrome; Pancr: Pancreatic; Plt: Platelets; PRBC: Packed red blood cells.
Navajo neurohepatopathy
This is an autosomal recessive disease prevalent in southwestern United States. The genetic defect is a nuclear gene MPV17 (chromosome 2p24)[32], whose product is located on the inner mitochondrial membrane and is responsible for mtDNA maintenance and regulation of oxidative phosphorylation. Hence, there is impaired pool of mtDNA and disrupted oxidative phosphorylation. While earlier only neurological manifestations were known and this entity was called Navajo neuropathy, liver manifestations in form of jaundice, failure to thrive, and liver failure were recognized to be part of the same disease spectrum prompting a change in name to Navajo neurohepatopathy[33]. Clinical features are outlined in Figure 9 graphical summary. All the three subtypes have occurred in same kindred, underscoring the pattern of mitochondrial inheritance.
Figure 9.

Graphical summary of Navajo neurohepatopathy. Bx: Biopsy; FTT: Failure to thrive.
Villous atrophy syndrome
This disorder was described in 1994 by Cormier-Daire et al[34] in 2 unrelated children presenting as chronic diarrhea in infancy with villous atrophy. The defect was identified as mtDNA rearrangements in the form of deletion-duplications. Hepatomegaly and steatosis on biopsy with mildly deranged transaminases was the liver manifestation. Both children survived the diarrheal phase, which subsided by early childhood, including a reversal in histology (Figure 10: Graphical summary). However, the phenotype then changed to neuromuscular and ophthalmic involvement and death by the end of first decade. Complex III defect was detected on muscle biopsy after the advent of neuromuscular symptoms and was normal in lymphocytes. Intravenous dextrose for resuscitation should not be used in high rates as it may lead to worsening of metabolic acidosis.
Figure 10.

Graphical summary of villous atrophy syndrome and its natural history. HPE: Histopathological examination; SNHL: Sensorineural hearing loss; Vx: Vomiting.
Mitochondrial neurogastrointestinal encephalomyopathy
This entity is discussed purely as a prototype for GI (non-hepatic) manifestations of mitochondrial disorders, and also since it is a respiratory chain disorder, though not classically a “hepatopathy”, and additionally as it is rewarding to diagnose in view of available therapy[35]. It is to be understood that RCD and non RCD mitochondrial diseases can have some or the other GI manifestation (Table 2). MNGIE was earlier known as polyneuropathy, ophthalmoplegia, leukoencephalopathy and intestinal pseudo-obstruction, oculogastrointestinal encephalopathy syndrome, or oculogastrointestinal muscular distrophy[36]. The current nomenclature was given by Hirano et al[37].
Table 2.
Gastrointestinal manifestations of mitochondrial respiratory chain defects
|
Site
|
Manifestation
|
| Oral cavity and esophagus | Sicca syndrome; Dry mouth; Dysphagia |
| Stomach | Vomiting; Reflux; Pseudo-obstruction |
| Small bowel and large bowel | Pseudo-obstruction; Diarrhea; Megacolon; Constipation |
| Extra-luminal/miscellaneous | Poor appetite; Pancreatitis; Pancreatic cysts |
MNGIE occurs due to mutation in a nuclear gene encoding TYMP, encoding thymidine phosphorylase, deficiency of which leads to toxic accumulation of pyrimidine nucleosides thymidine and deoxyuridine. This impairs mtDNA synthesis thereby leading to a mtDNA depletion state. Clinical symptoms of MNGIE usually begin between the first and fifth decades of life and before 20 years of age in approximately 60%. GI dysmotility is one of the most important features in form of dysphagia, gastroparesis, and pseudo-obstruction leading to consequences like small bowel bacterial overgrowth, nutritional deficiencies, and severe weight loss[38]. Hepatic steatosis, hepatomegaly, elevated transaminases, and cirrhosis have also been described[38,39].
A diagnostic delay of about 5 to 10 years can occur in view of multisystem and complex clinical presentation[40,41]. Often there are unnecessary exploratory surgeries for the GI symptoms before being diagnosed as pseudo-obstruction[36]. Neurological involvement is mainly in form of peripheral neuropathy (demyelination with or without axonal neuropathy)[38], oculoparesis, with subtle central nervous system manifestations due to subcortical white matter involvement, and magnetic resonance imaging changes showing leukoencephalopathy. Muscle biopsies may show ragged red fibers due to proliferation of abnormal mitochondria. Current diagnostic methods employ testing for plasma thymidine and deoxyuridine levels (> 3 μmol/L and > 5 μmol/L, respectively)[42] or elevated urinary concentrations[43] and thymidine phosphorylase activity in leucocytes (< 10% of healthy controls)[43]. TYMP gene (nuDNA) mutations and also consequent mtDNA abnormalities can be identified on Sanger sequencing and Southern blot assays[44]. A graphical summary is as shown in Figure 11.
Figure 11.

Graphical summary of mitochondrial neurogastrointestinal encephalomyopathy. BMT: Bone marrow transplantation; dThd: Thymidine; dUrd: Deoxy uridine levels; MRI: Magnetic resonance imaging; TP: Thymidine phosphorylase. Vx: Vomiting.
Symptomatic management remains the cornerstone. Experimental therapies include hemodialysis and peritoneal dialysis[43], platelet transfusions, hematopoietic stem cell transplant, enzyme replacement, and liver transplant[45,46]. All above therapies concentrate on 2 aspects: To reduce the toxic load of nucleosides and to replace the enzyme thymidine phosphorylase.
SETTINGS TO SUSPECT RCD AND DIAGNOSTIC EVALUATION
The settings of when to suspect a mitochondrial hepatopathy are shown in Figure 12.
Figure 12.
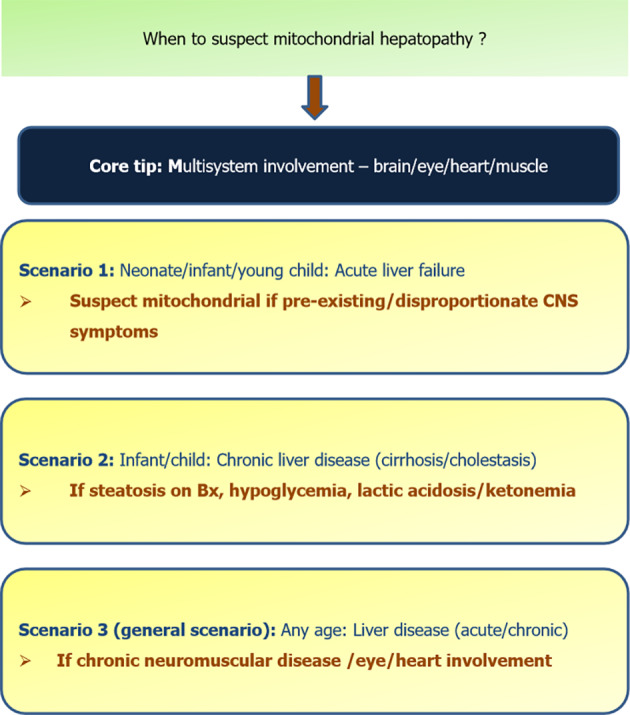
Scenarios when to suspect mitochondrial hepatopathy. Bx: Biopsy; CNS: Central nervous system.
Individual disorders discussed above and their graphical summaries outlined give specific information. The diagnostic evaluation of mitochondrial disorders follows once a clinical suspicion is raised, and in this section we highlight general steps towards approaching to diagnose a mitochondrial RCD[30]. Parallel evaluation of extra-hepatic and extra-GI symptoms, if present, need to be carried out for “mapping” the disease and for aid in management and improving quality of life. Table 3 elucidates a stepwise evaluation algorithm[30]. Diagnostic steps proceed from non-invasive, easily available, and less expensive investigations to more complex elaborate tests, some of which are available in the research setting only. Level-1 entails workup for basic metabolic causes including checking for hypoglycemia and, if present, whether it is ketotic or non-ketotic. Fatty acid oxidation defects (but not RCDs) are known to have non-ketotic hypoglycemic episodes. Lactate levels more than 2.1 mmol/L (mmol) are significant, and this may often not be observed when not in a metabolic crisis. Notably, lactate may not be elevated much in POLG1 mutations[47]. Normal lactate to pyruvate ratio is less than 20 mol/mol. However, the value often rises above 30 and is typical though not exclusive to RCDs and is discriminatory from pyruvate metabolism defects[48]. Similarly, 3-hydroxy butyrate to acetoacetate ratio is normally less than 4, and values above this should arouse a suspicion of mitochondrial dysfunction. Urine organic acids like lactate, succinate, fumarate, malate, and 3-methyl-glutaconic are seen elevated in Pearson syndrome. Serum alanine elevation is also a clue; however, it is often more elevated in pyruvate dehydrogenase deficiency than in RCDs[48]. Creatine kinase elevation and concomitant low levels of phosphocreatine in brain and muscle tissue are seen in RCDs[49]. Branched chain amino acid to glutamine ratios were highest in RCDs and lowest in pyruvate dehydrogenase deficiency compared to controls, according to one study[48].
Table 3.
Stepwise evaluation of mitochondrial hepatopathies (respiratory chain disorder/non- respiratory chain disorders)
|
Steps
|
Description
|
Additional action
|
| Level-1 (body fluids) | Basic: CBC, INR, AFP, CPK, NH3, sugars, phosphorous, urine ketones. Advanced: Lactate: Pyruvate (1 h post feeds); Ketone Body ratio, 3OH-butyrate: Acetoacetate; Serum acylcarnitine profile; Urine organic acidogram; Serum aminoacidogram; 3 Methyl Glutaconic acid in serum/urine; CSF lactate: Pyruvate, CSF alanine, protein; Plasma thymidine (MNGIE); Leucocyte CoQ levels | Parallel level-1: Evaluate other involved systems: CNS: MRI/MR-Spectroscopy, EEG; Eye: Fundus evaluation, clinical evaluation for ophthalmoplegias; Hearing screen; Heart: 2D-Echo, ECG; Renal: urine electrolytes, proteins, amino acids; Muscle: Muscle biopsy (Level-1 in case of primary muscle involvement, level-3 otherwise); Endocrine: HbA1c, 8 AM cortisol; Pancreas: Fecal elastase |
| Level-2 (genetics) | Common genes genotyping: POLG-1; DGUOK; MPV-17; SUCLG-1; TRMU; C10ORF2/Twinkle; CPT-1; mtDNA point mutations | Alternative level-2: Next generation sequencing/clinical exome sequencing for simultaneous evaluation of all mitochondrial DNA and nuclear DNA |
| Level-3 (invasive) | Tissue diagnosis: (1) Liver biopsy: Light microscopy including oil red O stain for steatosis; Electron microscopy for structural mitochondrial alterations; Frozen tissue analysis for respiratory chain enzymes, DNA quantification. (2) Muscle biopsy: Frozen tissue analysis as above; Blue native page analysis. (3) Skin biopsy: Same as muscle biopsy | Key points to note during level-3 evaluation: Biopsy specimens for electron microscopy need to be preserved in glutaraldehyde and not formalin; It is possible that one invasive test may not give a clue and one has to proceed for an additional invasive test. This is usually because of heteroplasmy. Often liver biopsy molecular analysis provides a final definitive answer; Combination of level-1, level-2 and level-3 studies are sometimes needed to provide comprehensive management and for prognostication |
2D Echo: Two-dimensional echocardiography; AFP: Alpha-fetoprotein; CBC: Complete blood count; CNS: Central nervous system; CoQ: Coenzyme Q; CPK: Creatine phosphokinase; CSF: Cerebrospinal fluid; EEG: Electroencephalogram; HbA1c: Glycosylated hemoglobin; INR: International normalized ratio; MRI: Magnetic resonance imaging; NH3: Serum ammonia levels; POLG: DNA polymerase subunit gamma; RCD: Respiratory chain disorders.
Table 4 helps differentiate the common metabolic disorders encountered in the pediatric patient and how to filter out RCD.
Table 4.
Biochemical differentiation between various metabolic hepatopathies (respiratory chain disorder vs non respiratory chain disorder comparison)
|
|
Acidosis
|
Urine ketones
|
Blood sugar
|
Serum lactate
|
Serum ammonia
|
| RCD | ++ | ++ | Normal | ++++ | ± |
| FAOD | ++ | Nil (non-ketotic) | Low (hypoglycemia) | + | + |
| OA | +++ (persistent) | ++/+++ | Low/normal/high | Normal | ++ |
| UCD | Normal | Normal | Normal | Normal | ++++ |
FAOD: Fatty acid oxidation defects; OA: Organic acidemias; RCD: Respiratory chain defects; UCD: Urea cycle defects.
Role of genetic testing
Genetic studies are confirmatory but have a high turnaround time of 4-6 wk. They may not also be available freely at all centers or in resource poor settings. Timely referral to tertiary care centers for management is advisable. A major limitation is selection of the gene panel testing for the phenotypic presentation. In products of consanguineous union and multiple affected siblings, genetic evaluation is better guided, and it is possible to identify the index patient’s chromosomal region containing the abnormality by linkage analysis. Targeted gene analysis is performed if the phenotype matches the previous cases in a family and there is an already identified mutation responsible for the clinical features in those particular kindred. Whole exome analysis for nuDNA and mtDNA is preferred otherwise as an alternative step in case there are no previously affected siblings or if the phenotype does not classically match previously described entities[50]. Targeted analysis can be performed using Sanger method of few genes, while whole exome sequencing refers to a massive parallel sequencing technique of multiple genes or the entire exome using next generation sequencing[51]. Another method to short-list genes for analysis is to study the expression profiles of RNA or specific proteins or polypeptides levels encoded by the gene(s) of interest, especially after a biochemical diagnosis is made. As an example, complex I deficiency can be caused by any of the multiple mtDNA and nuDNA responsible for each of its subunits. To identify a particular gene (of the multiple encoding ones) responsible for causing overall complex I deficiency in an index patient, analyzing the distribution or expression of proteins or RNA and its deviation from healthy controls or known standards can help pinpoint which gene may be defective. Once a specific change is identified, either in RNA expression, which can be detected by microarray assays, or in enzyme levels or stress protein expression, which can be identified by immunoblotting using an antibody panel, indirectly “reverse identification” of the causative gene(s) is facilitated[52]. This of course is only possible when, with time, all genes encoding each subunit of large proteins are identified so that such indirect, simpler, and time saving methods may be employed. Tables 1 and 3 list genes implicated in mitochondrial RCDs. Figure 13 suggests a two-step strategy of genetic evaluation in mitochondrial RCDs and also a few phenotypes for which specific genes should be tested.
Figure 13.

Step-wise strategy of genetic evaluation in mitochondrial respiratory chain defects. MRCD: Mitochondrial respiratory chain disorders; ALF: Acute liver failure; MMA: Methylmalonic acid.
It is pertinent to note that mitochondrial hepatopathies, unlike other metabolic disorders, require analysis of mtDNA in addition to nuDNA defects. Hence, when screening genetics do not yield the diagnosis and the next step of whole exome sequencing is being undertaken, it is essential to specify to the testing lab that mtDNA analysis be included in addition to the wide gamut of nuDNA being tested. While for nuDNA analysis, the tissue of interest may not be specific and whole blood sample may serve the purpose, for mtDNA molecular analysis, specific tissues (liver, muscle) may be required, mainly because of the phenomenon of heteroplasmy.
MtDNA depletions are diagnosed by first isolating the DNA of the tissue biopsied, which is then subjected to electrophoresis and blotting followed by hybridization with probes specific for mtDNA and nuDNA both. The relative levels of autoradiographic signals emitted post hybridization are detected for mtDNA and nuDNA, and this helps in diagnosing mtDNA depletions. MtDNA deletions and point mutations on the other hand can be detected by single strand conformational polymorphisms[53,54].
Next generation sequencing by parallel exome sequencing undertaken with blood or any tissue is limited by its inability to detect mutations in the non-exonic region, like untranslated regions or intronic splice sites. It is also not adept in diagnosing trinucleotide repeat sequences, complex genetic inheritance like synergistic contribution of nuDNA and mtDNA to cause a particular disease, and epigenetic effects[51].
Role of tissue biopsies
Tissue biopsies are important despite having readily evident biochemical abnormalities; only one-third to one-half of mitochondrial disorders have identifiable mutations despite extensive exome sequencing of known genetic defects[51,55]. That is to say, all genes related to mitochondrial disorders have not yet been identified. Biopsies from the most involved site are more likely to yield the diagnosis[56]. Respiratory chain enzymes can be analyzed and activity quantified on tissue biopsy specimens. Quantitative Southern blot analysis or real-time quantitative polymerase chain reaction to detect mtDNA depletion can be done in liver biopsy specimens. Skin biopsy for cultured skin fibroblasts can be stored indefinitely and retrieved for re-culture once newer diagnostic modalities are available. It is simpler to perform and less invasive compared to muscle biopsy. However, the downside is that not all diseases are detectable on skin fibroblast analysis[47].
How to select which tissue to test is an important question that the clinician must be aware. Most mitochondrial disorders involve the muscle, and hence muscle is one of the most useful sites for analysis of enzymes, metabolites, and even molecular DNA studies. While earlier 1-5 g of muscle tissue was required for respiratory chain enzyme assays, now even 100-200 mg of skeletal muscle tissue (usually quadriceps or soleus) is sufficient especially in young children, which then yields a mitochondrial enriched fraction of 400-500 μg of protein, enough to characterize the respiratory chain enzyme deficiencies[57]. Muscle biopsies may be analyzed either as frozen or fresh samples. Samples once collected should be snap frozen immediately bedside or in the procedure room at -80 °C till analysis of mitochondrial enzymes[53]. Fresh muscle samples should not be frozen and transported in cool buffer solution, which offers the advantage of analysis of the entire mitochondrial energy generation system in addition to mitochondrial enzymes being studied in frozen samples[58].
Those diseases that have primary liver involvement and no apparent muscle involvement, especially the ones with liver failure phenotype, liver tissue of up to 10 mg can be more yielding than muscle. Cardiac tissue requirement, when indicated, is even less, about 1-2 mg, obtained by endomyocardial biopsy[53]. These invasive techniques may be gradually substituted by molecular DNA techniques done on whole blood and cater to only research purpose over time as cost and availability of next generation sequencing is eased.
MANAGEMENT OF MITOCHONDRIAL RESPIRATORY CHAIN DEFECTS
There are three aspects to management: Firstly, acute management of crisis, second is general management of children with metabolic liver disease, followed by specific treatment if available including the role of liver transplant. An additional important component relates to parental counseling and to bust myths and avoid patients to resort to non-scientific therapies and refraining from standard of care.
Acute crisis management
Treatment of acute liver failure and progressive liver disease remains unsatisfactory. The aim of therapy is mainly mitigating, postponing, or circumventing damage to the respiratory chain. The basic steps and precautions are outlined as in Table 5. Bicarbonate infusions when used for a longer time may itself worsen cerebral function. An alternative is dichloroacetate, which inhibits pyruvate dehydrogenase kinase, hence favoring persistent levels of active pyruvate dehydrogenase and hence preventing pyruvate accumulation and pyruvate to lactate conversion.
Table 5.
Management during evaluation in acute phase
|
Following thumb rules while attending to a patient with suspected mitochondrial disorder
|
| Monitor closely for hypoglycemia and acidosis |
| Avoid lactated ringer’s solution for fluid administration: Worsens acidosis |
| Bicarbonate infusions as 1st line of defense |
| Avoid propofol for sedation/anesthesia |
| Avoid fasting > 12 h; avoid high rate glucose only infusions |
| Avoid drugs that are toxic to mitochondria: Chloramphenicol, valproate, aminoglycosides, phenytoin, carbamazapine, phenobarbital, statins, linezolid |
| Avoid drugs precipitating hepatopathy/liver dysfunction |
Other supportive therapy is to give packed red cell and platelet transfusions for anemia and thrombocytopenia and pancreatic enzyme replacement in case of insufficiency.
General considerations for managing a child with mitochondrial hepatopathy
Once the acute crisis is settled, it is important to improve the nutrition of the child as malnutrition itself can lead to secondary mitochondrial dysfunction[59]. These children often have increased caloric needs and an inability to maintain it owing to either repeated sickness bouts or a general anorexia associated with liver disorders. The issues of swallowing difficulties, impaired gut motility, and gastro-esophageal reflux need to be addressed often to the extent of placement of feeding tubes (orogastric or nasogastric), percutaneous endoscopic feeding gastrostomy, or using parenteral nutrition therapy. Nutritional improvement has led to improved quality of life and an increase in developmental quotients in these children[60]. Ketogenic diet may be useful in some mitochondrial disorders but may worsen fatty acid oxidation defects and should be avoided in them. Exercise helps in reducing the burden of abnormal mitochondria[61]. It is useful to do so regularly, under supervision in a graded manner, and to have a meal prior to exercise[62].
Pharmacotherapy
A combination of drugs is often empirically administered to suspected mitochondrial disorders and comprises: Coenzyme Q, carnitine, thiamine, riboflavin, vitamins C and E, and creatine. Of all, Coenzyme Q shows promise and along with B vitamins remains the most common combination as part of cocktail therapy[63]. The various drugs and their pediatric dosages are outlined in Table 6[13,63,63].
Table 6.
Pharmacotherapy used for mitochondrial diseases
|
Drug
|
Pediatric dose
|
Remark
|
| Coenzyme Q: (1) Ubiquinol form; (2) Ubiquinone form | 2-8 mg/kg/d in BD dosing; 10-30 mg/kg/d BD dosing | Preferably had after meals; Most effective and most used therapy; Free radical scavenger; Bypasses complex I |
| Idebenone | 5 mg/kg/d | Synthetic form of CoQ; Penetrates blood-brain barrier |
| L-carnitine | 10-100 mg/kg/d IV or oral divided 3 times/d | Avoid in long chain FAO-Ds: May lead to cardiac arrhythmias |
| Creatine | 0.1 g/kg PO, OD | Used for repletion of muscle phosphocreatine levels |
| L-arginine | 500 mg/kg IV per day for 1-3 d followed by 150-300 mg/kg oral daily in BD dosing | Used for acute stroke; Watch for hypotension while infusion; Evidence is anecdotal |
| Thiamine | 100 mg/d | Cofactor of PDH; useful for thiamine responsive PDH deficiency; Helpful in leigh disease |
| Riboflavin | 50-400 mg/d | Give at night time before sleep; Shown to be useful in ACAD9 mutations; Flavin precursor for complex I & II |
| Vitamin C | 5 mg/kg/d OD | Antioxidant; Artificial electron acceptor |
| Vitamin E | Variable dosing, up to 25 IU/kg/d OD (avoid > 400 IU/d) | Absorption better when taken with meals |
| Dichloroacetate | 25-50 mg/kg/d | Improves lactic acidosis |
BD: Twice daily; CoQ: Coenzyme Q; FAO-D: Fatty acid oxidation defects; IV: Intravenous; PDH: Pyruvate dehydrogenase; PO: Per oral; OD: Once daily.
Role of organ transplant
Multisystem involvement in mitochondrial hepatopathies often precludes performing a liver transplant. However, in hepatocerebral form of DGUOK defects when detected in infancy without neurological involvement, liver transplant has shown to be effective. Those with neurological involvement do not benefit from liver transplant[65]. Overall post-transplant survival is less with RCDs than non-RCDs. Sokal et al[66] reported 8 cases with a survival of 50% post transplantation for RCDs. In an elaborate compilation of 40 cases with mitochondrial RCDs across various centers at different time points, it was noted that 22 (55%) patients died within 24 mo post-transplant[67]. Early postoperative multi-organ failure and neuro-degeneration followed by respiratory complications and severe pulmonary hypertension were the cause of death in these patients. The same group recognized that those diagnosed pre-transplant had a higher survival (58%) than those recognized to have RCD after transplant (29%). Thus, the emphasis is on early recognition of the diagnosis and a thorough evaluation for extra-hepatic manifestations, adding investigations like magnetic resonance imaging of the brain and echocardiography.
MNGIE stands out as the single mitochondrial disorder for which replacement of the missing enzyme thymidine phosphorylase by stem cell transplantation can be curative and lead to improvement in long term outcomes. While earlier enzyme levels were artificially increased using repeated platelet transfusions[40], stem cell transplant has come up as a definitive modality[68,69].
Myths in mitochondrial disorders
Immunizations are not contraindicated in children with mitochondrial diseases. This is to be emphasized because of certain misconceptions that immunization may lead to autism in children with mitochondrial diseases for which there is no evidence[64]. The other important aspect that needs to be clarified is that there is no role of hyperbaric therapy in treatment of MH and in fact may lead to oxygen toxicity. Vagus nerve stimulation may not be very helpful in controlling refractory seizures in children with MH[63].
CONCLUSION
In a nutshell: (1) Liver along with other system involvement may not be just sepsis – think of mitochondrial respiratory chain hepatopathy; (2) Lactic acidosis without hypoglycemia is an important clue, avoid ringer lactate and drugs causing hepatopathy; (3) Evaluation should be done in a tiered manner – genetic evaluation and enzyme analysis from tissue of interest; (4) Treatment is largely supportive with transfusions, correction of acidosis, shock, and providing cofactors/salvage therapies; (5) Liver transplantation needs to be considered in only a select group and may worsen disease despite adequate precautions; and (6) Periodic follow-up is mandatory for monitoring evolution of disease including “migration” to other organ systems.
Footnotes
Conflict-of-interest statement: Gopan A and Sarma MS declare no conflict of interest related to this publication.
Provenance and peer review: Invited article; Externally peer reviewed.
Peer-review started: May 24, 2021
First decision: June 15, 2021
Article in press: August 18, 2021
Specialty type: Gastroenterology and hepatology
Country/Territory of origin: India
Peer-review report’s scientific quality classification
Grade A (Excellent): A
Grade B (Very good): 0
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
P-Reviewer: El-Karaksy H, Xia JK S-Editor: Yan JP L-Editor: Filipodia P-Editor: Guo X
Contributor Information
Amrit Gopan, Department of Gastroenterology, Seth G.S Medical College and K.E.M Hospital, Mumbai 400012, India.
Moinak Sen Sarma, Department of Pediatric Gastroenterology, Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow 226014, India. moinaksen@gmail.com.
References
- 1.Herrmann JM, Riemer J. The intermembrane space of mitochondria. Antioxid Redox Signal. 2010;13:1341–1358. doi: 10.1089/ars.2009.3063. [DOI] [PubMed] [Google Scholar]
- 2.Lee WS, Sokol RJ. Mitochondrial hepatopathies: advances in genetics and pathogenesis. Hepatology. 2007;45:1555–1565. doi: 10.1002/hep.21710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gray MW. Mitochondrial evolution. Cold Spring Harb Perspect Biol. 2012;4:a011403. doi: 10.1101/cshperspect.a011403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwartz M, Vissing J. Paternal inheritance of mitochondrial DNA. N Engl J Med. 2002;347:576–580. doi: 10.1056/NEJMoa020350. [DOI] [PubMed] [Google Scholar]
- 5.Di Mauro S, Schon EA. Mitochondrial Respiratory-Chain Diseases. N Engl J Med. 2003;348:2656–2668. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- 6.Kadowaki T, Kadowaki H, Mori Y, Tobe K, Sakuta R, Suzuki Y, Tanabe Y, Sakura H, Awata T, Goto Y. A subtype of diabetes mellitus associated with a mutation of mitochondrial DNA. N Engl J Med. 1994;330:962–968. doi: 10.1056/NEJM199404073301403. [DOI] [PubMed] [Google Scholar]
- 7.Meyers DE, Basha HI, Koenig MK. Mitochondrial cardiomyopathy: pathophysiology, diagnosis, and management. Tex Heart Inst J. 2013;40:385–394. [PMC free article] [PubMed] [Google Scholar]
- 8.Fischel-Ghodsian N. Mitochondrial mutations and hearing loss: paradigm for mitochondrial genetics. Am J Hum Genet. 1998;62:15–19. doi: 10.1086/301695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skladal D, Halliday J, Thorburn DR. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain. 2003;126:1905–1912. doi: 10.1093/brain/awg170. [DOI] [PubMed] [Google Scholar]
- 10.Chinnery PF, Johnson MA, Wardell TM, Singh-Kler R, Hayes C, Brown DT, Taylor RW, Bindoff LA, Turnbull DM. The epidemiology of pathogenic mitochondrial DNA mutations. Ann Neurol. 2000;48:188–193. [PubMed] [Google Scholar]
- 11.Darin N, Oldfors A, Moslemi AR, Holme E, Tulinius M. The incidence of mitochondrial encephalomyopathies in childhood: clinical features and morphological, biochemical, and DNA abnormalities. Ann Neurol. 2001;49:377–383. [PubMed] [Google Scholar]
- 12.Cormier-Daire V, Chretien D, Rustin P, Rötig A, Dubuisson C, Jacquemin E, Hadchouel M, Bernard O, Munnich A. Neonatal and delayed-onset liver involvement in disorders of oxidative phosphorylation. J Pediatr. 1997;130:817–822. doi: 10.1016/s0022-3476(97)80027-3. [DOI] [PubMed] [Google Scholar]
- 13.Lee WS, Sokol RJ. Liver disease in mitochondrial disorders. Semin Liver Dis. 2007;27:259–273. doi: 10.1055/s-2007-985071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taylor SA, Whitington PF. Neonatal acute liver failure. Liver Transpl. 2016;22:677–685. doi: 10.1002/lt.24433. [DOI] [PubMed] [Google Scholar]
- 15.Durand P, Debray D, Mandel R, Baujard C, Branchereau S, Gauthier F, Jacquemin E, Devictor D. Acute liver failure in infancy: a 14-year experience of a pediatric liver transplantation center. J Pediatr. 2001;139:871–876. doi: 10.1067/mpd.2001.119989. [DOI] [PubMed] [Google Scholar]
- 16.von Kleist-Retzow JC, Cormier-Daire V, Viot G, Goldenberg A, Mardach B, Amiel J, Saada P, Dumez Y, Brunelle F, Saudubray JM, Chrétien D, Rötig A, Rustin P, Munnich A, De Lonlay P. Antenatal manifestations of mitochondrial respiratory chain deficiency. J Pediatr. 2003;143:208–212. doi: 10.1067/S0022-3476(03)00130-6. [DOI] [PubMed] [Google Scholar]
- 17.Harding BN. Progressive neuronal degeneration of childhood with liver disease (Alpers-Huttenlocher syndrome): a personal review. J Child Neurol. 1990;5:273–287. doi: 10.1177/088307389000500402. [DOI] [PubMed] [Google Scholar]
- 18.Narkewicz MR, Sokol RJ, Beckwith B, Sondheimer J, Silverman A. Liver involvement in Alpers disease. J Pediatr. 1991;119:260–267. doi: 10.1016/s0022-3476(05)80736-x. [DOI] [PubMed] [Google Scholar]
- 19.Sitarz KS, Elliott HR, Karaman BS, Relton C, Chinnery PF, Horvath R. Valproic acid triggers increased mitochondrial biogenesis in POLG-deficient fibroblasts. Mol Genet Metab. 2014;112:57–63. doi: 10.1016/j.ymgme.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salsaa M, Pereira B, Liu J, Yu W, Jadhav S, Hüttemann M, Greenberg ML. Valproate inhibits mitochondrial bioenergetics and increases glycolysis in Saccharomyces cerevisiae. Sci Rep. 2020;10:11785. doi: 10.1038/s41598-020-68725-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tzoulis C, Engelsen BA, Telstad W, Aasly J, Zeviani M, Winterthun S, Ferrari G, Aarseth JH, Bindoff LA. The spectrum of clinical disease caused by the A467T and W748S POLG mutations: a study of 26 cases. Brain. 2006;129:1685–1692. doi: 10.1093/brain/awl097. [DOI] [PubMed] [Google Scholar]
- 22.Rötig A, Cormier V, Blanche S, Bonnefont JP, Ledeist F, Romero N, Schmitz J, Rustin P, Fischer A, Saudubray JM. Pearson's marrow-pancreas syndrome. A multisystem mitochondrial disorder in infancy. J Clin Invest. 1990;86:1601–1608. doi: 10.1172/JCI114881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mandel H, Szargel R, Labay V, Elpeleg O, Saada A, Shalata A, Anbinder Y, Berkowitz D, Hartman C, Barak M, Eriksson S, Cohen N. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat Genet. 2001;29:337–341. doi: 10.1038/ng746. [DOI] [PubMed] [Google Scholar]
- 24.Tyynismaa H, Sun R, Ahola-Erkkilä S, Almusa H, Pöyhönen R, Korpela M, Honkaniemi J, Isohanni P, Paetau A, Wang L, Suomalainen A. Thymidine kinase 2 mutations in autosomal recessive progressive external ophthalmoplegia with multiple mitochondrial DNA deletions. Hum Mol Genet. 2012;21:66–75. doi: 10.1093/hmg/ddr438. [DOI] [PubMed] [Google Scholar]
- 25.Spinazzola A, Viscomi C, Fernandez-Vizarra E, Carrara F, D'Adamo P, Calvo S, Marsano RM, Donnini C, Weiher H, Strisciuglio P, Parini R, Sarzi E, Chan A, DiMauro S, Rötig A, Gasparini P, Ferrero I, Mootha VK, Tiranti V, Zeviani M. MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat Genet. 2006;38:570–575. doi: 10.1038/ng1765. [DOI] [PubMed] [Google Scholar]
- 26.Saneto RP. Mitochondrial diseases: expanding the diagnosis in the era of genetic testing. J Transl Genet Genom. 2020;4:384–428. doi: 10.20517/jtgg.2020.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dror Y. Inherited Bone Marrow Failure Syndromes. In: Hoffman R, Benz Jr E, Silberstein L, Weitz J, Heslop H, Anastasi J, Salama M, Abutalib SA, editors. Hematology: Basic Principles and Practice. Elsevier Science, 2017: 350-393. [Google Scholar]
- 28.Broomfield A, Sweeney MG, Woodward CE, Fratter C, Morris AM, Leonard JV, Abulhoul L, Grunewald S, Clayton PT, Hanna MG, Poulton J, Rahman S. Paediatric single mitochondrial DNA deletion disorders: an overlapping spectrum of disease. J Inherit Metab Dis. 2015;38:445–457. doi: 10.1007/s10545-014-9778-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muraki K, Nishimura S, Goto Y, Nonaka I, Sakura N, Ueda K. The association between haematological manifestation and mtDNA deletions in Pearson syndrome. J Inherit Metab Dis. 1997;20:697–703. doi: 10.1023/a:1005378527077. [DOI] [PubMed] [Google Scholar]
- 30.Molleston JP, Sokol RJ, Karnsakul W, Miethke A, Horslen S, Magee JC, Romero R, Squires RH, Van Hove JL Childhood Liver Disease Research Education Network (ChiLDREN) Evaluation of the child with suspected mitochondrial liver disease. J Pediatr Gastroenterol Nutr. 2013;57:269–276. doi: 10.1097/MPG.0b013e31829ef67a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Farruggia P, Di Cataldo A, Pinto RM, Palmisani E, Macaluso A, Valvo LL, Cantarini ME, Tornesello A, Corti P, Fioredda F, Varotto S, Martire B, Moroni I, Puccio G, Russo G, Dufour C, Pillon M. Pearson Syndrome: A Retrospective Cohort Study from the Marrow Failure Study Group of A.I.E.O.P. (Associazione Italiana Emato-Oncologia Pediatrica) JIMD Rep. 2016;26:37–43. doi: 10.1007/8904_2015_470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karadimas CL, Vu TH, Holve SA, Chronopoulou P, Quinzii C, Johnsen SD, Kurth J, Eggers E, Palenzuela L, Tanji K, Bonilla E, De Vivo DC, DiMauro S, Hirano M. Navajo neurohepatopathy is caused by a mutation in the MPV17 gene. Am J Hum Genet. 2006;79:544–548. doi: 10.1086/506913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holve S, Hu D, Shub M, Tyson RW, Sokol RJ. Liver disease in Navajo neuropathy. J Pediatr. 1999;135:482–493. doi: 10.1016/s0022-3476(99)70172-1. [DOI] [PubMed] [Google Scholar]
- 34.Cormier-Daire V, Bonnefont JP, Rustin P, Maurage C, Ogler H, Schmitz J, Ricour C, Saudubray JM, Munnich A, Rötig A. Mitochondrial DNA rearrangements with onset as chronic diarrhea with villous atrophy. J Pediatr. 1994;124:63–70. doi: 10.1016/s0022-3476(94)70255-1. [DOI] [PubMed] [Google Scholar]
- 35.Finsterer J, Frank M. Gastrointestinal manifestations of mitochondrial disorders: a systematic review. Therap Adv Gastroenterol. 2017;10:142–154. doi: 10.1177/1756283X16666806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pacitti D, Levene M, Garone C, Nirmalananthan N, Bax BE. Mitochondrial Neurogastrointestinal Encephalomyopathy: Into the Fourth Decade, What We Have Learned So Far. Front Genet. 2018;9:669. doi: 10.3389/fgene.2018.00669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hirano M, Silvestri G, Blake DM, Lombes A, Minetti C, Bonilla E, Hays AP, Lovelace RE, Butler I, Bertorini TE. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): clinical, biochemical, and genetic features of an autosomal recessive mitochondrial disorder. Neurology. 1994;44:721–727. doi: 10.1212/wnl.44.4.721. [DOI] [PubMed] [Google Scholar]
- 38.Garone C, Tadesse S, Hirano M. Clinical and genetic spectrum of mitochondrial neurogastrointestinal encephalomyopathy. Brain. 2011;134:3326–3332. doi: 10.1093/brain/awr245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Finkenstedt A, Schranz M, Bösch S, Karall D, Bürgi SS, Ensinger C, Drach M, Mayr JA, Janecke AR, Vogel W, Nachbaur D, Zoller H. MNGIE Syndrome: Liver Cirrhosis Should Be Ruled Out Prior to Bone Marrow Transplantation. JIMD Rep. 2013;10:41–44. doi: 10.1007/8904_2012_199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lara MC, Weiss B, Illa I, Madoz P, Massuet L, Andreu AL, Valentino ML, Anikster Y, Hirano M, Martí R. Infusion of platelets transiently reduces nucleoside overload in MNGIE. Neurology. 2006;67:1461–1463. doi: 10.1212/01.wnl.0000239824.95411.52. [DOI] [PubMed] [Google Scholar]
- 41.Taanman JW, Daras M, Albrecht J, Davie CA, Mallam EA, Muddle JR, Weatherall M, Warner TT, Schapira AH, Ginsberg L. Characterization of a novel TYMP splice site mutation associated with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) Neuromuscul Disord. 2009;19:151–154. doi: 10.1016/j.nmd.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 42.Martí R, Nishigaki Y, Hirano M. Elevated plasma deoxyuridine in patients with thymidine phosphorylase deficiency. Biochem Biophys Res Commun. 2003;303:14–18. doi: 10.1016/s0006-291x(03)00294-8. [DOI] [PubMed] [Google Scholar]
- 43.Spinazzola A, Marti R, Nishino I, Andreu AL, Naini A, Tadesse S, Pela I, Zammarchi E, Donati MA, Oliver JA, Hirano M. Altered thymidine metabolism due to defects of thymidine phosphorylase. J Biol Chem. 2002;277:4128–4133. doi: 10.1074/jbc.M111028200. [DOI] [PubMed] [Google Scholar]
- 44.Nishino I, Spinazzola A, Hirano M. MNGIE: from nuclear DNA to mitochondrial DNA. Neuromuscul Disord. 2001;11:7–10. doi: 10.1016/s0960-8966(00)00159-0. [DOI] [PubMed] [Google Scholar]
- 45.De Giorgio R, Pironi L, Rinaldi R, Boschetti E, Caporali L, Capristo M, Casali C, Cenacchi G, Contin M, D'Angelo R, D'Errico A, Gramegna LL, Lodi R, Maresca A, Mohamed S, Morelli MC, Papa V, Tonon C, Tugnoli V, Carelli V, D'Alessandro R, Pinna AD. Liver transplantation for mitochondrial neurogastrointestinal encephalomyopathy. Ann Neurol. 2016;80:448–455. doi: 10.1002/ana.24724. [DOI] [PubMed] [Google Scholar]
- 46.D'Angelo R, Rinaldi R, Pironi L, Dotti MT, Pinna AD, Boschetti E, Capristo M, Mohamed S, Contin M, Caporali L, Carelli V, De Giorgio R. Liver transplant reverses biochemical imbalance in mitochondrial neurogastrointestinal encephalomyopathy. Mitochondrion. 2017;34:101–102. doi: 10.1016/j.mito.2017.02.006. [DOI] [PubMed] [Google Scholar]
- 47.Mitochondrial Medicine Society's Committee on Diagnosis. Haas RH, Parikh S, Falk MJ, Saneto RP, Wolf NI, Darin N, Wong LJ, Cohen BH, Naviaux RK. The in-depth evaluation of suspected mitochondrial disease. Mol Genet Metab. 2008;94:16–37. doi: 10.1016/j.ymgme.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clarke C, Xiao R, Place E, Zhang Z, Sondheimer N, Bennett M, Yudkoff M, Falk MJ. Mitochondrial respiratory chain disease discrimination by retrospective cohort analysis of blood metabolites. Mol Genet Metab. 2013;110:145–152. doi: 10.1016/j.ymgme.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Atkuri KR, Cowan TM, Kwan T, Ng A, Herzenberg LA, Enns GM. Inherited disorders affecting mitochondrial function are associated with glutathione deficiency and hypocitrullinemia. Proc Natl Acad Sci U S A. 2009;106:3941–3945. doi: 10.1073/pnas.0813409106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.ACMG Board of Directors. Points to consider in the clinical application of genomic sequencing. Genet Med. 2012;14:759–761. doi: 10.1038/gim.2012.74. [DOI] [PubMed] [Google Scholar]
- 51.McCormick E, Place E, Falk MJ. Molecular genetic testing for mitochondrial disease: from one generation to the next. Neurotherapeutics. 2013;10:251–261. doi: 10.1007/s13311-012-0174-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thorburn DR, Sugiana C, Salemi R, Kirby DM, Worgan L, Ohtake A, Ryan MT. Biochemical and molecular diagnosis of mitochondrial respiratory chain disorders. Biochim Biophys Acta. 2004;1659:121–128. doi: 10.1016/j.bbabio.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 53.Rustin P, Chretien D, Bourgeron T, Gérard B, Rötig A, Saudubray JM, Munnich A. Biochemical and molecular investigations in respiratory chain deficiencies. Clin Chim Acta. 1994;228:35–51. doi: 10.1016/0009-8981(94)90055-8. [DOI] [PubMed] [Google Scholar]
- 54.Munnich A, Rötig A, Chretien D, Saudubray JM, Cormier V, Rustin P. Clinical presentations and laboratory investigations in respiratory chain deficiency. Eur J Pediatr. 1996;155:262–274. doi: 10.1007/BF02002711. [DOI] [PubMed] [Google Scholar]
- 55.Calvo SE, Compton AG, Hershman SG, Lim SC, Lieber DS, Tucker EJ, Laskowski A, Garone C, Liu S, Jaffe DB, Christodoulou J, Fletcher JM, Bruno DL, Goldblatt J, Dimauro S, Thorburn DR, Mootha VK. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci Transl Med. 2012;4:118ra10. doi: 10.1126/scitranslmed.3003310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Munnich A, Rustin P. Clinical spectrum and diagnosis of mitochondrial disorders. Am J Med Genet. 2001;106:4–17. doi: 10.1002/ajmg.1391. [DOI] [PubMed] [Google Scholar]
- 57.Janssen AJ, Smeitink JA, van den Heuvel LP. Some practical aspects of providing a diagnostic service for respiratory chain defects. Ann Clin Biochem. 2003;40:3–8. doi: 10.1258/000456303321016114. [DOI] [PubMed] [Google Scholar]
- 58.Lian C, Cao S, Zeng W, Li Y, Su J, Li J, Zhao S, Wu L, Tao J, Zhou J, Chen X, Peng C. RJT-101, a novel camptothecin derivative, is highly effective in the treatment of melanoma through DNA damage by targeting topoisomerase 1. Biochem Pharmacol. 2020;171:113716. doi: 10.1016/j.bcp.2019.113716. [DOI] [PubMed] [Google Scholar]
- 59.Wortmann SB, Zweers-van Essen H, Rodenburg RJ, van den Heuvel LP, de Vries MC, Rasmussen-Conrad E, Smeitink JA, Morava E. Mitochondrial energy production correlates with the age-related BMI. Pediatr Res. 2009;65:103–108. doi: 10.1203/PDR.0b013e31818d1c8a. [DOI] [PubMed] [Google Scholar]
- 60.Choi HS, Lee YM. Enteral Tube Feeding in Paediatric Mitochondrial Diseases. Sci Rep. 2017;7:16909. doi: 10.1038/s41598-017-17256-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tarnopolsky MA. Mitochondrial DNA shifting in older adults following resistance exercise training. Appl Physiol Nutr Metab. 2009;34:348–354. doi: 10.1139/H09-022. [DOI] [PubMed] [Google Scholar]
- 62.Jeppesen TD, Schwartz M, Olsen DB, Wibrand F, Krag T, Dunø M, Hauerslev S, Vissing J. Aerobic training is safe and improves exercise capacity in patients with mitochondrial myopathy. Brain. 2006;129:3402–3412. doi: 10.1093/brain/awl149. [DOI] [PubMed] [Google Scholar]
- 63.Parikh S, Saneto R, Falk MJ, Anselm I, Cohen BH, Haas R, Medicine Society TM. A modern approach to the treatment of mitochondrial disease. Curr Treat Options Neurol. 2009;11:414–430. doi: 10.1007/s11940-009-0046-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dimauro S, Rustin P. A critical approach to the therapy of mitochondrial respiratory chain and oxidative phosphorylation diseases. Biochim Biophys Acta. 2009;1792:1159–1167. doi: 10.1016/j.bbadis.2008.10.015. [DOI] [PubMed] [Google Scholar]
- 65.Dimmock DP, Dunn JK, Feigenbaum A, Rupar A, Horvath R, Freisinger P, Mousson de Camaret B, Wong LJ, Scaglia F. Abnormal neurological features predict poor survival and should preclude liver transplantation in patients with deoxyguanosine kinase deficiency. Liver Transpl. 2008;14:1480–1485. doi: 10.1002/lt.21556. [DOI] [PubMed] [Google Scholar]
- 66.Sokal EM, Sokol R, Cormier V, Lacaille F, McKiernan P, Van Spronsen FJ, Bernard O, Saudubray JM. Liver transplantation in mitochondrial respiratory chain disorders. Eur J Pediatr. 1999;158 Suppl 2:S81–S84. doi: 10.1007/pl00014328. [DOI] [PubMed] [Google Scholar]
- 67.De Greef E, Christodoulou J, Alexander IE, Shun A, O'Loughlin EV, Thorburn DR, Jermyn V, Stormon MO. Mitochondrial respiratory chain hepatopathies: role of liver transplantation. A case series of five patients. JIMD Rep. 2012;4:5–11. doi: 10.1007/8904_2011_29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hirano M, Martí R, Casali C, Tadesse S, Uldrick T, Fine B, Escolar DM, Valentino ML, Nishino I, Hesdorffer C, Schwartz J, Hawks RG, Martone DL, Cairo MS, DiMauro S, Stanzani M, Garvin JH Jr, Savage DG. Allogeneic stem cell transplantation corrects biochemical derangements in MNGIE. Neurology. 2006;67:1458–1460. doi: 10.1212/01.wnl.0000240853.97716.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rahman S, Hargreaves IP. Allogeneic stem cell transplantation corrects biochemical derangements in MNGIE. Neurology. 2007;68:1872; author reply 1872; discussion 1872–1872; author reply 1872; discussion 1873. doi: 10.1212/01.wnl.0000265356.98295.be. [DOI] [PubMed] [Google Scholar]