

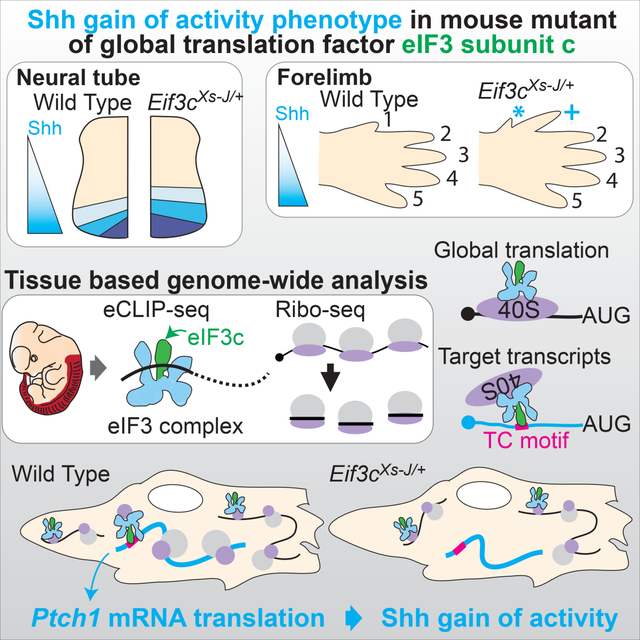
Summary:
Although gene expression is tightly regulated during embryonic development, the impact of translational control has received less experimental attention. Here, we find that eukaryotic translation initiation factor-3 (eIF3) is required for Shh-mediated tissue patterning. Analysis of loss-of-function eIF3 subunit c (Eif3c) mice reveal a unique sensitivity to the Shh receptor Patched 1 (Ptch1) dosage. Genome-wide in vivo enhanced cross-linking immunoprecipitation sequence (eCLIP-seq) shows unexpected specificity for eIF3 binding to a pyrimidine rich motif present in subsets of 5’-UTRs and a corresponding change in the translation of these transcripts by ribosome profiling in Eif3c loss-of-function embryos. We further find that while Eif3c loss-of-function embryos do not show a global decrease in protein synthesis, translation of Ptch1 through this pyrimidine rich motif is specifically sensitive to eIF3 amount. Altogether, this work uncovers hidden specificity of housekeeping translation initiation machinery for the translation of key developmental signaling transcripts.
eTOC Blurb
Using tissue-based eCLIP- and Ribo-seq in mouse embryos, Fujii et al. demonstrate that translation initiation factor 3 (eIF3) directly associates with cell signaling transcripts and regulates their translation. Furthermore, they observed a Shh-gain-of-function phenotype in eIF3c mutant mice associated with reduced translation of the eIF3 direct target, Ptch1 mRNA.
Graphical Abstract
Introduction
Dynamic spatiotemporal gene regulation is critical to specify and differentiate cells at the right time and place during embryonic development. Our current understanding of embryonic development is largely based on studies of mRNA and not on direct assessment of proteins that are ultimately required for tissue patterning. The development of new methods such as ribosome profiling has allowed us to observe the impact of genome-wide translational control (Brar and Weissman, 2015). We have previously applied these methods directly in mammalian embryos and discovered that mRNA translation further diversifies tissue-specific gene expression. In particular, we have shown that multiple cell signaling components belonging to the Shh (Sonic hedgehog), Wnt, and Hippo pathways are all under pervasive translational control (Fujii et al., 2017). This regulation is encoded in elements embedded in the mRNA, including upstream open reading frames (uORFs) located in 5’ untranslated regions (5’-UTRs), which compete with the main ORF for ribosome occupancy and thereby act to repress translation (Bazzini et al., 2014; Chew et al., 2016; Fujii et al., 2017; Johnstone et al., 2016; Tsutsumi et al., 2013). While our knowledge of the prevalence of translation control and regulation through cis-regulatory elements are expanding, the trans-acting factors that modulate spatiotemporal control of these translational programs still remains in its infancy.
Among the largest initiation factors is eIF3, a 13 subunit complex, that binds to the small 40S ribosomal subunit to recruit other translation initiation factors and the large 60S subunit of ribosome in order to stimulate translation initiation (Sonenberg and Hinnebusch, 2009). Reconstitution analysis revealed that the functional core comprises six subunits that are essential for global translation initiation (Masutani et al., 2007). The additional subunits that are dispensable for basic eIF3 function may either serve more regulatory functions in translation initiation, such as by controlling transcript-specific translation and/or involved in additional biological processes. Interestingly, one eIF3 subunit, eIF3h, specifically regulates development of the brain, heart, vasculature, and lateral line during zebrafish embryonic development (Choudhuri et al., 2010, 2013), suggesting that eIF3 may regulate specific developmental programs during vertebrate embryogenesis, however the molecular mechanisms remain poorly understood. Recent cross-linking immunoprecipitation sequence (CLIP-seq) analysis has revealed the direct binding of several eIF3 subunits primarily to the 5’-UTRs of specific mRNAs, which contain short stem loop elements, to both activate or repress translation of transcripts important for control of cell proliferation (Lee et al., 2015; Pulos-Holmes et al., 2019).
In this study, we address the mechanisms of translational regulation of key developmental signaling transcripts. Interestingly, two distinct spontaneous mouse mutants, extra-toes spotting (Xs-J and Xs-L), exhibit a very selective limb phenotype with the presence of an additional anterior digit in heterozygosity, and both of these mutants have been surprisingly mapped to Eif3c, a subunit of the eIF3 complex (Gildea et al., 2011), although the underlying mechanisms for such a tissue selective phenotype remains unknown. In vertebrates, extra digit phenotypes are frequently associated with activation of Shh signaling (Litingtung et al., 2002). Here, we characterize Eif3cXs-J/+ heterozygous mutant mice, and show a gain of function Shh phenotype associated with a specific reduction in the translation of SHH receptor Patched 1 (Ptch1) mRNA, which is a negative regulator of the Shh pathway, that explains the Shh activation phenotype in these mice. Moreover, using tissue-based enhanced CLIP-seq (eCLIP-seq) and ribosome profiling analysis, our findings demonstrate that eIF3 binds to a pyrimidine rich motif within a select subset of 5’UTRs and more broadly controls transcript-specific translation which fine tunes cell signaling activity required for intricate tissue patterning during embryonic development.
Results
Global translation is not reduced in Eif3cXs-J/+ mutant mice
The Eif3cXs-J allele harbors a point mutation from G to T in the CDS that produces a premature stop codon that is predicted to result in nonsense mediated decay (NMD), leading to reduced Eif3c mRNA (Figure 1A and S1A and B) (Gildea et al., 2011). Consistent with this, we also observe that Eif3cXs-J/+ embryos exhibit an ~30 % reduction of eIF3c protein levels which may destabilize the eIF3 complex as we observe a 20% decrease of eIF3d, eIF3e, and eIF3k, but not a reduction in eIF3b, eIF3f, eIF3g, and eIF3h (Figure S1C). Given that eIF3c is a core subunit of the general translation initiation factor eIF3 (Figure 1A) (Masutani et al., 2007), we next monitored global translation. The translation activity of the cell was measured by the distribution of free subunits (40S or 60S), 80S monosome-a single ribosome on an mRNA, and polysomes-multiple ribosomes on an mRNA. Surprisingly, we did not observe a reduction of global translation in Eif3cXs-J/+ mutant embryos, particularly within the most translationally active heavy polysome fractions (Figure 1A and S1D). In agreement with this result, we also did not see a reduction of global protein synthesis in O-propargyl-puromycin (OP-Puro) incorporation in nascent peptides (Figure S1E) and, if anything, we observe a subtle increase. We further investigated translation initiation by monitoring the amount of the eIF3 complex bound to the 40S ribosome (Herrmannová et al., 2020), which is an important event in the initiation step of translation. The eIF3 complex in each fraction was monitored by the eIF3b subunit whose amount did not change in Eif3cXt-J/+ embryos (Figure S1C). We observed around a 20% reduction in eIF3b signal from the fraction containing the eIF3 complex and correspondingly a slight increase in eIF3b in the free fraction not part of the translation initiation complex (Figure S1F), suggesting that the missing eIF3c subunit de-stabilizes the eIF3 complex. However, in good agreement with the normal global protein synthesis in Eif3cXt-J/+ embryos (Figure 1A and S1D–E), we did not see a change in the eIF3–40S initiation complex when compared to WT (Figure S1F). Together, these results strongly suggest that Eif3cXs-J/+ mutant embryos do not exhibit a reduction in global translation nor initiation.
Figure 1. Mice heterozygous for the translation initiation factor Eif3c show tissue specific phenotypes without a reduction in general translation.

(A) The eif3cXt-J allele creates a nonsense stop codon. Eif3c encodes a Yellow subunit of eIF3 complex as illustrated. Global translation of neural tube and somite at E11.5 was monitored by polysome analysis. (B) SHH secreted from the zone of polarizing activity (ZPA) creates signaling gradient in the limb, which controls digit identity. (C-F) Skeletal staining and quantification of digit patterning phenotypes of forelimb (C-F) and hindlimb (C’-F’) at E18.5 in wildtype (n=19) (C, C’), Eif3cXs-J/+ (n=9) (D, D’), and Eif3cXt-J/+; Ptch1+/− (n=9) (E, E’). * indicates polydactyly, + marks posteriorized first digit phenotype. (G) Shh signaling gradient across the ventral-to-dorsal axis of the developing neural tube dictates differentiation of FOXA2+ floor plate (FP), NKX2.2+ (p3), and OLIG2+ (pMN) motor neurons. (H-K) Shown are E9.5 forelimb-level immunofluorescent staining and quantification of cell numbers normalized by neural tube area (1- way ANOVA, n≥3), FOXA2 (H, I, J, K); NKX2.2 (H’, I’, J’, K’); OLIG2 (H”, I”, J”) in wildtype (H, H’, H”), Eif3cXs-J/+ (I, I’, I”), and Eif3cXs-J/+, Ptch1+/− (J, J’, J”) embryos. Yellow brackets indicate shift in OLIG2+ domain.
Eif3c exhibit a Shh signaling specific phenotype with a strong genetic interaction with the Shh receptor, Ptch1
To understand whether haploinsufficiency of Eif3c may affect transcript-specific translational regulation, we next sought to characterize the detailed phenotype of Eif3c mutant embryos. While Eif3cXs-J/Xs-J homozygotes are embryonic lethal prior to E3.5, Eif3cXs-J/+ heterozygote have been reported to be largely normal albeit exhibiting mild forelimb patterning defects when observed in the adult animal (Gildea et al., 2011). We therefore next fully characterized the eIF3c loss of function phenotype. We observe mild to severe polydactyly (extra digits) in 44 % of Eif3cXs-J/+ forelimbs and anterior to posterior transformations of digit 1, evident from increased digit length, in 28 % of Eif3cXs-J/+ forelimbs suggesting a de-repression of Shh signaling in the anterior of the limb bud (Figures 1B–D, and F). A number of previous studies have established the importance of Ptch1 in tissue patterning and development (Goodrich, 1997; Marigo and Tabin, 1996). As the PTCH1 protein is a potent negative regulator of the pathway, we hypothesized that if Eif3c were required for regulation of Shh signaling, Eif3cXs-J/+ heterozygotes would be acutely sensitive to Ptch1 dosage. Importantly, while Ptch1+/− mice exhibit normal digit patterning (n=6) (Feng et al., 2013), we observed a striking increase in the penetrance and severity of limb defects in Eif3cXs-J/+;Ptch1+/− double heterozygous mice (Figures 1E and F). In particular, while the vast majority (89 %) of Eif3cXs-J/+ animals do not display any hindlimb phenotype, we observed that 94% of Eif3cXs-J/+;Ptch1+/− hindlimbs show severe polydactyly or A-P transformations (Figures 1C’-F’). This shift in phenotype severity and distribution indicates a strong genetic interaction between Eif3c and Ptch1 and suggests that Eif3c may be critically required for Shh signaling.
To further understand the role of Eif3c in Shh signaling, we next examined the phenotype in the neural tube, where Shh signaling has an important role in dorsal-ventral patterning of motor neurons, where we did not observe any global translation defect (Figure 1A). Unlike the limb bud, precise quantification of the Shh signaling gradient in the neural tube is greatly facilitated by known molecular markers downstream of the Shh pathway. Shh is secreted from the notochord and ventral region of the neural tube, floor plate (FP), establishing a signaling gradient that specifies neuronal identity. This is readily quantified with known markers FOXA2, NKX2.2, and OLIG2 across the ventral-dorsal axis (Figure 1G). Our analysis revealed that both Eif3cXs-J/+ and Eif3cXs-J/+;Ptch1+/− embryos have a significant increase in the number of both FOXA2+ floor plate (FP) (1.8-fold) (Figures 1H–K) and NKX2.2+ p3 neurons (1.6-fold) (Figures 1H’-K’) but not OLIG2+ motor neurons (p=0.1) (Figures 1H’’-J’’) and a concomitant, statistically significant, dorsal shift of the OLIG2+ pMN domain (p=0.01, 1-way ANOVA) (Figures 1H’’-J’’, yellow bracket). This data further indicates a specific role of Eif3c in negatively regulating Shh pathway activity and suggests that there could be Eif3c-dependent translational regulation critical for control of this pathway.
Identification of a distinct eIF3 binding site using embryonic tissues
To understand the molecular mechanisms underlying the Shh signaling pathway phenotype due to haploinsufficiency of Eif3c, we performed in vivo tissue-based eIF3c-eCLIP-seq analysis using neural tube and somites (Figure 2A) where Eif3c-dependent Shh activation phenotype was observed. We reproducibly observed strong and widespread enrichment signals in the eIF3c-eCLIP samples relative to the input, and peak calling analysis identified 14,310 regions with significant eIF3c binding (FDR<0.05, n=2) across 5,406 transcripts out of 8,496 detectably expressed transcripts (Figure 2B and S2A, Table S1). eIF3c binding was exceedingly overrepresented in the 5’-UTRs, making up 87.2% of the enriched windows which is 4.78 fold (p<2.2×10−16) above 18.3% for the input library (Figure 2C). The binding patterns in the 5’-UTRs were typically very broad, reflecting passive interactions during ribosome scanning (Figure 2D and S2B). However, an alternative binding pattern composed of a distinct, sharp peak was also frequently observed, which may reflect more direct binding of eIF3 to specific sequence or structural elements (Figure 2D and S2C). To more systematically classify these sharp or broad enrichment patterns, we additionally quantified unevenness in the distribution of the sequencing reads along the transcripts. We calculated Gini coefficients in sliding windows across the transcripts: sharp peaks have more unequal distributions and thus high Gini coefficients, while broadly enriched regions have low coefficients (Figure 2D). Significantly enriched regions that are both the local maximum in read counts and above top 30 percentile in windowed Gini coefficients are defined as sharp binding sites (1,899 number total, Figure 2E, Table S1). We next asked whether the translation of transcripts that have sharp eIF3 binding peaks in their 5’-UTRs are also more sensitive to the amount of eIF3c. We carried out ribosome profiling from both the wildtype and Eif3cXs-J/+ neural tube at E11.5 to obtain sufficient embryonic tissue for these experiments (Figure S3A–C). Although we did not see a change in the translation efficiency of Shh transcripts likely because Shh activity is low at E11.5 (Cohen et al., 2015), transcripts that contain sharp peaks in their 5’-UTR tend to have lower translation efficiency in Eif3cXs-J/+ embryos genome-wide (p=5.95×10−15, Figure 2F).
Figure 2. Tissue-based eIF3c eCLIP-Seq and ribosome profiling shows transcripts having distinct sharp peaks of eIF3 in 5’-UTRs tend to have lower translation efficiency in the Eif3cXs-J/+ mouse.

(A) Experimental scheme of tissue-based eIF3c eCLIP-Seq at E11.5 in Neural tube and somite. (B) Reads distribution in the eIF3c eCLIP and input library. (C) The distribution of normalized log2 read count in eIF3c eCLIP library over the input library. Reads are counted within 10nt windows. The red dot indicates 5’-UTR windows significantly enriched in the eCLIP-seq library (FDR<0.05, n=2). (D) Schematic of the sharpness of eCLIP peaks using the Gini Index. (E) The distribution of the Gini index calculated in 100 nt windows for each significantly enriched 5’-UTR over the normalized eIF3c eCLIP log2 read counts. (F) The plot shows that the transcripts having sharp peaks in the 5’-UTR tend to show reduced translation efficiency (TE) in Eif3cXs-J/+ embryos by ribosome profiling (n=3, p=5.95×10−15).
Strikingly, multiple expectation maximization for motif elicitation (MEME) analysis (Bailey et al., 2009) on the sharp binding sites (1,899 sites across 1,342 genes) identified a 20 nt long U and C pyrimidine rich motif (UC motif) with E-value=2.9×10−76 that appeared in 919 peaks (Figure 3A, S4A, Table S2). Interestingly, transcripts containing at least one UC motif overlapping the sharp eIF3 binding peak enriched for Gene Ontology (GO) terms related to tissue morphogenesis and cell migration (Figure 3B, Table S3). These results demonstrate that beyond the general association with mRNA broadly across their 5’-UTRs, the eIF3 complex also more specifically recognizes a UC motif to produce a highly localized RNA binding landscape which can impact translation in a transcript-specific manner that may explain the phenotype of Eif3c haploinsufficiency.
Figure 3. eIF3c associates with a pyrimidine rich (UC) motif that has important roles in translation of the Ptch1 5’-UTR.

(A) MEME motif analysis for the 75 quantile of high Gini Index peaks reveals a 20 nt UC motif. (B) 15 Gene Ontology (GO) term categories with lowest FDR enriched for transcripts having distinct sharp peaks with a UC motif over expressed transcripts in the input library was shown with the % value of transcripts over expected. The number of transcripts having the UC motif in each GO category is indicated. (C-D) eIF3c eCLIP and input plots of the Ptch1 (C) or Gli3 (D) transcript. Orange highlights significantly enriched domains. Green or Red indicate the UC or GC motif respectively. (E-G) Wildtype (WT) or mutant Ptch1 (E and G) or Gli3 (F) 5’-UTRs with Firefly luciferase (Fluc) reporter RNAs were transfected and each Fluc reporter activity was normalized to mRNA and Renilla luciferase (Rluc), shown relative to WT 5’-UTRs constructs. Dashed line indicates deletion of domains. In (G), reporter RNA was transfected in eIF3d knock-down or control siRNA cells. Each luciferase activity was normalized by corresponding RNA amount. (Error bars represent s.d. AU, arbitrary unit; t-test, n≥4, **P<0.01; NS, not significant)
Distinct eIF3 binding in the 5’-UTR is required for Ptch1 mRNA translation
We identified Shh signaling related transcripts, Gli3 and Ptch1 mRNAs, contain distinct sharp peaks with UC motifs within their 5’-UTRs at relatively well conserved regions (Figure 3C–D and S4B–C). We therefore sought to understand the role of the marked association of eIF3 complex and the UC motif in translation. We transfected Firefly luciferase (Fluc) RNA reporters harboring the Ptch1 and Gli3 5’-UTR into C3H10T1/2 mesenchymal cells, which efficiently respond to Shh signaling. Interestingly, the deletion of the entire 75 nt eIF3 binding site (ΔBS) or only the 20 nt UC motif (ΔUC) from Ptch1 5’-UTRs significantly reduced translation of Fluc (Figure 3E). Adding back only the UC motif to the larger ΔBS deletion (ΔBS+UC) rescued Fluc production (Figure 3E). Moreover, other motifs identified by MEME in Ptch1 5’-UTR (Figure 3C and S4A–B) do not have a significant impact (Figure 3E). Similar result was observed in the Gli3 5’-UTR, although the magnitude in the translation of Fluc was much more modest than in the Ptch1 5’-UTR (Figure 3F). To further confirm that the UC motif is acting directly through eIF3, we attempted knock-down of the eIF3c subunit in culture cell. However, we were not successful in downregulating eIF3c to this same extent as observed in vivo (~20–30%) and more pronounced downregulation perturbed global protein synthesis. We therefore turned our attention to additional eIF3 subunits and in particular eIF3d which has been shown to directly interface with specific 5’-UTRs (Lee et al., 2016). As shown in Figure 3G, knock-down of Eif3d reduced translation from the WT Ptch1 5’-UTR, but not Ptch1 ΔBS 5’-UTR and control Renilla luciferase (Rluc) mRNA, revealing the selectivity for the translation of specific transcripts (Figure 3G and S4D). Importantly, these findings show a direct connection between the UC motif and sensitivity to the translation of particular transcripts for the eIF3 complex.
We next sought to further address translational regulation of Shh components in vivo. We detected a marked decrease in PTCH1 protein but not GLI3 repressor (GLI3-R) in Eif3cXs-J/+ neural tube lysates at E11.5 without a reduction in mRNA (Figures 4A–C). Moreover, we further analyzed translation using polysome fractionation followed by qPCR analysis within E9.5 whole embryos. Strikingly the Ptch1 mRNA, but not Actb or Eno1 mRNA, was reduced in the highly translated heavy polysome fractions of Eif3cXs-J/+ embryos (p=0.02, t-test) (Figures 4D–F). We further investigated the unique regulatory role of Eif3c in facilitating the recruitment of the eIF3 complex to the Ptch1 mRNA. To this end, we immunoprecipitated the eIF3 complex in vivo from the neural tube and somites of E11.5 wildtype and Eif3cXs-J/+ embryos using an antibody against eIF3b, which is a core component of the eIF3 complex and its level do not change in Eif3cXs-J/+ mice (Figure S1C). Strikingly, the association of eIF3 complex with Ptch1 mRNA but not other mRNAs, such as Jun and Cdk12, which are known to bind the eIF3 complex (Lee et al., 2015), was selectively reduced in the Eif3cXs-J/+ mutant (Figure 4G). Given that the PTCH1 receptor negatively regulates Shh signaling, reducing PTCH1 protein could explain the Shh gain-of-function phenotypes (Butterfield et al., 2009; Holtz et al., 2013; Makino et al., 2001; Milenkovic et al., 1999; Zhulyn et al., 2014). Together, these data genetically reveal the importance of Eif3c, a perceived housekeeping pre-initiation complex component, in Shh signaling and transcript-specific translation required for normal tissue patterning.
Figure 4. Eif3cXs-J/+ mice specifically show reduced translation of Ptch1 mRNA and eIF3 association.

(A-B) Western blot in neural tube lysates from wildtype and Eif3cXs-J/+ embryos at E11.5 and quantification was shown in (B) (t-test, n=4). (C) Quantification of each mRNA by RT-qPCR (t-test, n=3). (D-F) Relative amounts of Ptch1 (D), Actb (E) and Eno1 (F) mRNA in polysome fractions of E9.5 wildtype (n=4) and Eif3cXs-J/+ (n=8) embryos. Each fraction was normalized by total mRNA amount in the gradient. (G) Neural tube and somites were micro-dissected from wildtype and Eif3cXs-J/+ embryos at E11.5 followed by eIF3 immunoprecipitation (IP) using eIF3b antibody. The relative IP efficiency was calculated by amount of mRNA in eIF3b IP normalized by input and further normalized by positive control Jun mRNA in the wildtype (t-test, n≥3). (Error bars represent s.d.; * p<0.05; NS, not significant).
Discussion
Here, we demonstrate that eIF3 plays a critical role in translation regulation of Shh signaling during embryonic development. Eif3cXs-J/+ embryos show a reduction not only in eIF3c, but also eIF3d, eIF3e, and eIF3k subunits, affecting the eIF3 complex stability. Future studies are required to determine whether the phenotype evident in Eif3cXs-J/+ embryos is directly due to the reduction of the eIF3c subunit or more broadly the disruption observed in additional eIF3 subunits. In this context, given that Eif3d in culture cell regulates mRNA translation from the Ptch1 5’-UTR (Figure 3G), the phenotype of the Eif3c mouse might also stem from reductions in the eIF3d subunit. However, it is intriguing that other eIF3 subunit mutant mice have never been reported to have Shh related phenotypes including Eif3b (Koyanagi-Katsuta et al., 2002), Eif3e (Lin et al., 2020; Sadato et al., 2018), Eif3f (Docquier et al., 2019), Eif3h (Daxinger et al., 2012), and Eif3m (Zeng et al., 2013). The amount of the eIF3d subunit was not reduced in heterozygous mutant mice for Eif3e (Sadato et al., 2018) and Eif3m (Zeng et al., 2013). Interestingly, while Eif3e mutant mice have a progressive decline in muscle strength (Lin et al., 2020) Eif3f mutant mice reduce muscle mass (Docquier et al., 2019). Therefore, the eIF3 subunit specific phenotypes is suggestive of subunit specific functions.
A key feature of translational regulation is the ability to quickly remodel gene expression in response to highly dynamic circumstances. Dynamic spatiotemporal translational regulation, especially for cell signaling transcripts, might fine-tune signaling activity in different developmental stages and tissues (Fujii et al., 2017). Tissue-based eIF3c eCLIP-seq analysis demonstrated unexpected specificity of a global translation initiation factor for binding to a UC motif, which is present in other components of key signaling pathways including Bmp (Bmpr2, Smad1, Smad6, Smad7), Hippo (Yap1, Wwtr1/Taz, Tead1), Wnt (Fzd2, Sfrp1, Lrp6), Notch (Jag1, Dll1), and Semaphorin (Nrp1, Nrp2, Sema5a) signaling pathways (Figure S2C, Table S2). Bmp, Wnt, Notch, and Semaphorin signaling have important roles for neural crest differentiation and migration. Indeed, GO term analysis identified enriched categories related neural crest migration such as ‘embryonic cranial skeleton morphogenesis’ and ‘regulation of chemotaxis’ (Figure 3B and Table S3). Interestingly, most Eif3c mutant mice have a white belly spot, suggesting defects in neural crest cell migration (Gildea et al., 2011). Thus, Eif3c-dependent translation might have a further role in regulating the translation of additional signaling transcripts.
An outstanding question in the field has been how translation of selective transcripts is modulated. Recent genome wide analysis has revealed specificity of other eIFs, for example the eIF4G1 homolog eIF4G2 (known as p97, NAT1, and DAP5) for translation of specific transcripts that are required for cell differentiation (Sugiyama et al., 2017). Also the cytosine enriched regulator of translation (CERT) motif provides differential sensitivity to the amount of eIF4E (Truitt et al., 2015). Such cis-regulatory elements might change the affinity of eIFs for unique transcripts and provide differential sensitivity to the activity and amount of each eIF. Moreover, recent analysis showed that phosphorylation of the eIF3d could influence the translation of specific transcripts (Lamper et al., 2020). There are at least 29 phosphorylation sites in the eIF3 complex (Andaya et al., 2014; Damoc et al., 2007). Future studies should uncover the dynamics of the spatiotemporal stoichiometry and posttranslational modifications of each eIF and address how their activities are fine-tuned between cells and tissues to contribute to tissue- and stage-specific translational programs.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Maria Barna (mbarna@stanford.edu)
Materials availability
All plasmids and DNA constructs generated in this study are available upon request. All antibodies, chemicals, cell lines, and most mouse lines used in this study are commercially available. All other unique materials are also available upon request.
Data and code availability
Data that support the findings of this study have been deposited in the Gene Expression Omnibus under accession number GSE183472.
Code generated in this study has been deposited on Github DOI: 10.5281/zenodo.5555442.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All mice used in the study were housed at Stanford University or University of Florida. All animal work done in Stanford University was reviewed and approved by the Stanford Administrative Panel on Laboratory Animal Care (APLAC). All animal work done in the University of Florida was reviewed and approved by the University of Florida Animal Care Services (ACS). The Stanford APLAC and University of Florida ACS are accredited by the American Association for the Accreditation of Laboratory Animal Care (AAALAC). Mice were housed under a 12 hr light/dark cycle with free access to food and water. Eif3cXs-J/+ (Stock# 006045), Ptch1+/− (Ptch1tm1Mps/+ (Stock# 003081)), C3HeB/FeJ (Stock# 000658), and FVB/NJ (Stock# 001800) lines were purchased from the Jackson Laboratory (Bar Harbor, ME, USA).
Cell line
C3H/10T1/2 cells were purchased from the American Type Culture Collection and grown under standard conditions in DMEM supplemented with 10% FBS and 2 mM L-glutamine. Cells were passaged 1:6 roughly every 2–3 days. All cell lines used in this study were mycoplasma-free. Cells were grown in humidified CO2 incubators at 37°C and 5% CO2.
METHOD DETAILS
OP-Puromycin incorporation assay
E9.5 embryos were dissected in filming media (DMEM/F12, no phenol red, 10 % fetal bovine serum (FBS)). Whole embryos were dissociated in Dissociation Buffer (1 % trypsin in HBSS without Ca2+ or Mg2+) for 15 min at 37 °C. Resuspended cells were used for downstream labeling and analysis. For OP-Puro incorporation, cells were labeled with 20 μM of OPP in DMEM plus drug for 30 min at 37 °C. Following metabolic labeling, cells were washed with twice 1×PBS. Cell pellets were resuspended in Zombie Violet Live-Dead Stain (1:500 in PBS; BioLegend, 423113) and incubated for 15 min in the dark. Cells were then washed with Cell Staining Buffer (0.1 % NaN3, 2 % FBS in HBSS) before being fixed in 1 % PFA for 15 min on ice. Subsequently, cells were permeabilized overnight at 4 °C in Perm Buffer (0.1 % Saponin, 0.1 % NaN3, 3 % FBS in PBS). The next day, cells were washed twice with Cell Staining Buffer (without 0.1 % NaN3), labeled with an Alexa Fluor 555 Picolyl Azide dye (Thermo Fisher, C10642) and incubated for 30 min at room temperature in the dark. Labeled cells were washed and resuspended in Cell Staining Buffer before being analyzed on Novocyte Quanteon flow cytometer (Agilent Technologies) using software packages CellQuest and FlowJo v10.
Polysome analysis by sucrose gradient fractionation
E11.5 neural tubes or forelimb samples were dissected under the 100 μg/ml cycloheximide containing filming medium and dissociated with the Papain dissociation kit as previously described. Neural tube or forelimb samples were lysed on ice in 200 μl of lysis buffer (10 mM Tris pH 7.5, 150 mM KOAc, 15 mM Mg(OAc)2, 1 mM DTT, 8 % glycerol, 0.5 % NP-40, 0.2 % Na-deoxycholate 100 μg/ml cycloheximide, 200 U/ml SUPERase In (Ambion, AM2694)). After lysis, nuclei and membrane debris was removed by centrifugation (1800 g, 5 min, at 4 °C and then 14000 rpm, 5 min, at 4 °C). The supernatant was layered onto a linear sucrose gradient (10–40 % sucrose (w/v), 10 mM Tris, pH 7.5, 100 mM NaCl, 15 mM MgCl2, 100 μg/ml cycloheximide) and centrifuged in an SW41Ti rotor (Beckman) for 2.5 hr at 40000 rpm at 4 °C. Fractions were collected by Density Gradient Fraction System (Brandel) and 3 fg in vitro transcribed Fluc RNA were mixed with each fractions to normalize RNA purification efficiency. RNA was purified by acid phenol/chloroform (Ambion, AM9720) followed by isopropanol precipitation for RT-qPCR analysis.
Translation initiation complex (TIC) analysis by Sucrose gradient fractionation
Formaldehyde cross-linking sucrose density gradient was performed following the procedures previously described (Herrmannová et al., 2020). E11.5 whole embryos were dissected under 100 μg/ml cycloheximide containing filming medium and dissociated with the 1% Trypsin with 100 μg/ml Cycloheximide. Dissociated cells were lysed on ice in 200 μl of lysis buffer (10 mM Hepes pH 7.5, 62.5 mM KCl, 2.5 mM MgCl2, 1 mM DTT, 1 % Triton X-100, 100 μg/ml cycloheximide, RNaseOut, Halt Protease and Phosphatase Inhibitor Cocktail). After lysis, nuclei and membrane debris was removed by centrifugation (1800 g, 5 min, at 4 °C and then 14000 rpm, 5 min, at 4 °C). The supernatant was layered onto a 7–30 % linear sucrose gradient containing a top-to bottom increasing concentration of formaldehyde. Such gradient was prepared by mixing 30% sucrose (w/v) buffer containing 0.05% formaldehyde (10 mM Hepes pH 7.5, 62.5 mM KCl, 2.5 mM MgCl2, 1mM DTT, 100 μg/ml cycloheximide) and 7% sucrose (w/v) buffer without formaldehyde (10 mM Hepes pH 7.5, 62.5 mM KCl, 2.5 mM MgCl2, 1mM DTT, 100 μg/ml cycloheximide). SW41Ti rotor (Beckman) was centrifuged for 5 hr 15min at 40000 rpm at 4 °C. Fractions were collected by Density Gradient Fraction System (Brandel). Each fraction was run on the SDS-PAGE followed by western blotting using anti-eIF3b (1:5000, Santa Cruz, sc-16377), and anti-RPS6/eS6 (Cell Signaling, 5G10). Signal intensity of each band was quantified by ImageJ and percentages of eIF3b subunit in each fraction was calculated.
Skeletal Staining
Skeletal staining was performed on E18.5 embryos following established protocols as described in Xue et al. (2015). Skeletons were cleared in 20 % Glycerol 1 % KOH for 2 days with daily changes and then equilibrated and imaged in 50 % Glycerol: 50 % EtOH. Multiple embryos (n=6–19) were examined for each genotype and the phenotype of the limbs was scored by inspection under a light microscope. Formation of additional phalange on digit 1 was reported as a posteriorized digit 1 phenotype. Digit 1 bifurcation or nub-like outgrowths were reported as mild polydactyly. Full digit duplications were reported as severe polydactyly.
Immunofluorescence
Immunofluorescence was performed following established protocols described in Xue et al., (2015). E9.5 embryos (24.5+/−2.5 somites) and fixed for 1 hr in 4 % PFA in PBS at 4 °C and embedded in O.C.T. Compound (Tissue-Tek, 1S-LB-4583-EA). Embedded embryos were sectioned at 12 μm; sections were stored at −80 °C. The sections were then incubated with SHH (1:50, DSHB, 5E1), FOXA2 (1:100, DSHB, 4C7c), NKX2.2 (1:100, DSHB, 74.5A5) or OLIG2 (1:100, EMD Millipore, AB9610) primary antibodies in blocking buffer overnight at 4 °C, and after washing were incubated with appropriate secondary antibodies (Goat anti-mouse or goat anti-rabbit Alexa fluor 488 or 574 at 1:1000 dilution and DAPI, Invitrogen) for 1 hr at RT, protected from light. Slides were mounted with Fluoromount-G (SouthernBiotech) and examined using a Spinning Disc confocal microscope. At least two forelimb level sections were imaged for each embryo and 4–8 embryos were analyzed for each genotype (evenly spread between 22–27 somites). FOXA2+, NKX2.2+ and OLIG2+ cells were manually quantified using the ‘Cell counter’ feature of Fuji/ImageJ. For each section, ventral neuron number was normalized by neural tube area, determined by examining the DAPI channel to identify and outline the outer perimeter of the neural tube for measurement with ImageJ. Statistical analysis was performed using Excel 2013 and GraphPad Prism6. For the three genotypes, NKX2.2+ and OLIG2+ neuron numbers (normalized by neural tube area) were compared using 1-way ANOVA. Means were deemed statistically significant at p < 0.05. The mean number of FOXA2+ neurons (normalized to neural tube area) across three genotypes were compared using 1-way ANOVA and Welch’s ANOVA with differences deemed statistically significant at p < 0.05. Standard deviations were assessed with the Brown-Forsythe test and Bartlett’s test and deemed to be different at P < 0.05. Graphs plot mean neuron number and 95 % confidence intervals.
Dissection of neural tube and somites
Pregnant FVB females, 3–8 months of age, were euthanized at E11.5, the uterus was dissected, and embryos were taken out and placed into PBS. Microdissections for neural tube & somite were performed in filming media (DMEM F12 1:1, 10 % FBS) containing in a Sylgard dissection dish (Sylgard 184 Silicone Elastomer Kit; Dow Corning). For some experiments, Neural tube & somites were snap frozen in liquid nitrogen and stored at −80°C. Otherwise, they were dissociated at 37 °C for 30 min using Papain tissue dissociation kit (Washington, LK003150). After incubation, cells were washed using manufacturer’s protocol, rinsed in PBS and then UV crosslinked (400 mJ/cm2) for eCLIP-seq library production.
The eCLIP-seq library production
For CLIP-Seq, dissected neural tube and somites from 1 litter of E11.5 embryos (20–23 somite number from tail to hind limb) were used for CLIP-seq. The cell pellet was re-suspended in 0.5 ml of cold lysis buffer (50 mM Tris pH 7.4, 100 mM NaCl, 1 % NP-40, 0.5 % SDS, 0.5 % Sodium deoxycholate, 1x cOmplete, EDTA-free Protease Inhibitor Cocktail (Sigma, 11873580001)) and incubated for 30 min at 4 °C with occasional vortexing. The lysate was clarified by sequential centrifugation for 5 min at 1,800 g and 10,000 g at 4 °C to remove nuclei and mitochondria. The lysate was then treated with RNase I (Ambion, AM2294) for 5 min at 37 °C (1 Unit of RNase1 for 45 μg of RNA in the lysate). The digestion was stopped by adding 16 μl of SUPERaseIn RNase Inhibitor (20 U/μl, Ambion AM2696). 5 % of lysate was taken for input sample. Lysate was then mixed with magnetic beads (Invitrogen, 10002D) 1hr at 4 °C for pre-clear then mixed with 40 μl of beads conjugated with eIF3c antibody (Novus, NB100–511) and incubated for 2 hr at 4 °C. Beads were washed three times by High wash buffer (50 mM Tris pH 7.4, 1 M NaCl, 1 mM EDTA, 1 % NP-40, 0.5 % SDS, 0.5 % Sodium deoxycholate) and twice by low wash buffer (20 mM Tris pH 7.4, 10 mM MgCl2, 0.2 % Tween-20). RNA on the beads was treated with Fast AP (LifeTech, EF0652) and T4 PNK (NEB, M0201L) for dephosphorylation, ligated with 3’ adaptor (NEB, S1315S, Sequence: 5’ rAppCTGTAGGCACCATCAAT-NH2 3’) by T4 RNA ligase 2 truncated (NEB, M0242S), and then radiolabeled by gamma 32P-ATP Kination treating with OptiKinase (Affymetrix, 78334X). After radioactive labeling of RNA, CLIP and Input samples were denatured and run on NuPAGE and transferred to nitrocellulose membrane for the size selection. The strong radioactive band at the size of eIF3c (105 kDa) was cut out from membrane as well as from the size matched input lane. RNA on the membrane was recovered by protease K treatment followed by Phenol/chloroform and Zymo column (Zymo, R1015) RNA purification. Input RNA was processed for dephosphorylation and 3’ linker ligation. To take out the remaining free linker, Input RNA was run on the 10 % Urea Gel and recovered ligated input RNA. The cDNA for eCLIP sample and Input was produced by Super Script III (Invitrogen, 18080093) and RNA was digested by 1 N NaOH. Cleaned cDNAs were circulized by CircLigase (Epicentre, CL4111K) at 60 °C overnight. Library was produced by 2 times of PCR reactions by Phusion polymerase (NEB, M0530S). Primers used in preparation were listed in the Key Resources Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-eIF3c | Novus | Cat# NB100-511 |
| Rabbit polyclonal anti-eIF3d | Proteintech | Cat# 10219-1-AP |
| Rabbit polyclonal anti-eIF3e | BETHYL | Cat# A302-985A-T |
| Rabbit polyclonal anti-eIF3k | Abcome | Cat# ab85968 |
| Goat polyclonal anti-eIF3b | Santa Cruz | Cat# sc-16377 |
| Rabbit polyclonal anti-eIF3g | BETHYL | Cat# A301-757A-T |
| Rabbit polyclonal anti-eIF3f | BETHYL | Cat# A303-005A-T |
| Rabbit monoclonal anti-eIF3h | Cell Signaling | Cat# 3413S |
| Mouse monoclonal anti-GAPDH | Ambion | Cat# AM4300 |
| Rabbit polyclonal anti-RPL23A/uL23 | BETHYL | Cat# A303-932A-M |
| Rabbit polyclonal anti-RPS24/eS24 | BETHYL | Cat# A303-842A-M |
| Rabbit polyclonal anti-RPS19/eS19 | BETHYL | Cat# A304-002A-M |
| Rabbit polyclonal anti-PTCH1 | (Rohatgi et al., 2007) | N/A |
| Goat polyclonal anti-GLI3 | R&D | Cat# AF3690 |
| Mouse monoclonal anti-ACTB | Sigma-Aldrich | Cat# SAB1403520 |
| Rabbit monoclonal anti-RPS6/eS6 | Cell Signaling | Cat# 2217 |
| donkey anti-mouse HRP | GE Healthcare | Cat# NA931-1ML |
| donkey anti-rabbit HRP | GE Healthcare | Cat# NA934-1ML |
| Chicken anti-goat HRP | R&D systems | Cat# HAF019 |
| Goat IgG control antibody | Santa Cruz | Cat# sc-2028 |
| Rabbit IgG control antibody | Thermo Fisher | Cat# 02-6102 |
| Mouse monoclonal anti-FOXA2 | DSHB | Cat# 4C7c |
| Mouse monoclonal anti-NKX2.2 | DSHB | Cat# 74.5A5 |
| Rabbit polyclonal anti-OLIG2 | EMD Millipore | Cat# AB9610 |
| Goat anti-mouse Alexa fluor 488 | Thermo Fisher | Cat# A11017 |
| goat anti-rabbit Alexa fluor 647 | Thermo Fisher | Cat# A27040 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| TRIzol | Invitrogen | Cat# 15596 |
| Acid-Phenol: Chloroform, pH 4.5 (with IAA, 125:24:1) | Thermo Fisher | Cat# AM9722 |
| Chloroform (Ethanol as Preservative/Certified ACS) | Fisher Chemical | Cat# C298-500 |
| iScript Reverse Transcription Supermix kit | Bio-Rad | Cat# 1708841 |
| SsoAdvanced Universal SYBR Green Supermix | Bio-Rad | Cat# 1725274 |
| Trans-Blot Turbo RTA Midi 0.2 μm PVDF Transfer Kit | Bio-Rad | Cat# 170-4273 |
| Clarity Western ECL Substrate | Bio-Rad | Cat# 170-5061 |
| Dulbecco’s Modified Eagle’s Medium | GIBCO | Cat# 11965–118 |
| Fetal calf serum | EMD Millipore Thermo | Cat# TMS-013-B |
| DMEM/F-12, HEPES, no phenol red | Thermo Fisher | Cat# 11039021 |
| Penicillin-Streptomycin Solution 100X | Thermo Fisher | Cat# 15140163 |
| Opti-MEM | GIBCO | Cat# 11058-021 |
| Lipofectamine 2000 | Thermo Fisher | Cat# 11668-019 |
| TransIT-mRNA Transfection Kit | Mirus | Cat# MIR 2225 |
| MISSION siRNA Universal Negative Control 2 | Sigma-Aldrich | Cat# SIC002 |
| Zombie Violet Live-Dead Stain | BioLegend | Cat# 423113 |
| O-propargyl-puromycin | Medchem Source LLP | Cat# JA-1024 |
| Alexa Fluor 555 Picolyl Azide dye | Thermo Fisher | Cat# C10642 |
| Halt™ Protease and Phosphatase Inhibitor Cocktail (EDTA-free) | Thermo Fisher | Cat# 78443 |
| cOmplete, EDTA-free Protease Inhibitor Cocktail | Sigma | Cat# 11873580001 |
| RNaseOUT™ Recombinant Ribonuclease Inhibitor | Invitrogen | Cat# 10777019 |
| SUPERase In | Ambion | Cat# AM2696 |
| TURBO DNase | Ambion | Cat# AM2238 |
| cycloheximide | Sigma | Cat# C7698 |
| RNase I | Ambion | Cat# AM2294 |
| Protein G Dynabeads | Invitrogen | Cat# 10003D |
| Protein A Dynabeads | Invitrogen | Cat# 10001D |
| MyOne Streptavidin C1 DynaBeads | Invitrogen | Cat# 65001 |
| Fast AP | LifeTech | Cat# EF0652 |
| T4 PNK | NEB | Cat# M0201L |
| T4 RNA ligase 2, truncated KQ | NEB | Cat# M0242S |
| Universal miRNA Cloning Linker | NEB | Cat# S1315S |
| EasyTides® ATP, [gamma-32P]-, 250μCi (9.25MBq) | Perkin Elmer | Cat# NEG502A250UC |
| OptiKinase | Affymetrix | Cat# 78334X |
| Super Script III | Invitrogen | Cat# 18080093 |
| CircLigase | Epicentre | Cat# CL4111K |
| Phusion polymerase | NEB | Cat# M0530S |
| Critical commercial assays | ||
| Papain tissue dissociation kit | Washington | Cat# LK003150 |
| RNA Clean and Concentrator-5 columns | Zymo Research | Cat# R1016 |
| PureLink RNA Mini Kit | ThermoFisher | Cat# 12183018 |
| mMESSAGE mMACHINE T7 Transcription Kit | ThermoFisher | Cat# AM1344 |
| Poly(A) Polymerase Tailing Kit | Lucigen | Cat# PAP5104H |
| Dual Luciferase kit | Promega | Cat# E1980 |
| BCA assay | Pierce | Cat# 23225 |
| Oligotex mRNA Mini Kit | Qiagen | Cat# 70022 |
| NextSeq 500/550 v2.5 Kits | Illumina | Cat# 20024906 |
| Deposited data | ||
| GEO | This paper | GSE183472 |
| Data processing and analysis codes | This paper | https://github.com/Kotaro-UF/eif3; https://doi.org/10.5281/zenodo.5555442 |
| Experimental models: Cell lines | ||
| Mouse Cell line: C3H10T1/2 | ATCC | Cat# CCL-226 |
| Experimental models: Organisms/strains | ||
| Mouse: Eif3cXs-J/+ | (Gildea et al., 2011) Jackson Laboratories | JAX: 006045 |
| Mouse: Ptch1+/− | (Goodrich, 1997) Jackson Laboratories | JAX: 003081 |
| Mouse: C3HeB/FeJ | Jackson Laboratories | JAX: 000658 |
| Mouse: FVB/NJ | Jackson Laboratories | JAX: 001800 |
| Oligonucleotides | ||
| Oligonucleotides for ribosome profiling, RT-qPCR, CLIP-seq, Genotyping, siRNA, see Table S4 | This paper | N/A |
| Recombinant DNA | ||
| SV40_Renilla-luc | Promega | E2231 |
| pK429_pGL1_Ptch1_WT | (Fujii et al., 2017) | N/A |
| pK610_pGL54_Ptch1_ΔBS | This paper | N/A |
| pK626_pGL70_Ptch1_ΔUC | This paper | N/A |
| pK625_pGL69_Ptch1_ΔBS+UC | This paper | N/A |
| pK628_pGL72_Ptch1_ΔGC | This paper | N/A |
| pK627_pGL71_Ptch1_ΔBS+GC | This paper | N/A |
| pK435_pGL3_Gli3_WT | This paper | N/A |
| pK437_pGL3_Gli3_ΔBS | This paper | N/A |
| Software and algorithms | ||
| CellQuest | BD Biosciences | N/A |
| FlowJo v10 | BD Biosciences | N/A |
| Fuji/ImageJ | NIH | https://imagej.nih.gov/ij/ |
| GraphPad Prism6 | Graphpad Software Inc. | Version 8.0 |
| SAMtools | (Li et al., 2009) | https://doi.org/10.1093/bioinformatics/btp352 |
| STAR | (Dobin et al., 2013) | https://doi.org/10.1093/bioinformatics/bts635 |
| UMItools | (Smith et al., 2017) | https://doi.org/10.1101/gr.209601.116 |
| BEDTools | (Quinlan and Hall, 2010) | https://doi.org/10.1093/bioinformatics/btq033 |
| limma | (Ritchie et al., 2015) | https://doi.org/10.1093/nar/gkv007 |
| locfdr | (Efron, 2012) | https://doi.org/10.1198/016214504000000089 |
| topGO | Bioconductor | https://bioconductor.org/packages/release/bioc/html/topGO.html |
| wiggleplotr | Bioconductor | https://bioconductor.org/packages/release/bioc/html/wiggleplotr.html |
| tidyverse | (Wickham et al., 2019) | https://doi.org/10.21105/joss.01686 |
| data.table | CRAN | https://cran.r-project.org/web/packages/data.table/index.html |
| zoo | (Zeileis et al., 2005) | https://doi.org/10.18637/jss.v014.i06 |
| GenomicFeatures | (Lawrence et al., 2013) | https://doi.org/10.1371/journal.pcbi.1003118 |
| ineq | CRAN | https://cran.r-project.org/web/packages/ineq/index.html |
| scales | CRAN | https://cran.r-project.org/web/packages/scales/index.html |
| ggrepel | CRAN | https://cran.r-project.org/web/packages/ggrepel/index.html |
| universalmotif | Bioconductor | https://bioconductor.org/packages/release/bioc/html/universalmotif.html |
| MEME | (Bailey et al., 2015) | https://doi.org/10.1093/nar/gkv416 |
| org.Mm.eg.db | Bioconductor | https://bioconductor.org/packages/release/data/annotation/html/org.Mm.eg.db.html |
| edgeR | (Robinson et al., 2009) | https://doi.org/10.1093/bioinformatics/btp616 |
| cutadapt | (Martin, 2011) | https://doi.org/10.14806/ej.17.1.200 |
| Other | ||
| Density Gradient Fraction System | Brandel | Cat# BR-188 |
| Biocomp Model 108 Gradient Master | BioComp | N/A |
| TLA 120.2 rotor | Beckman | Cat# 357656 |
| SW-41Ti | Beckman | Cat# 331362 |
| CFX384 Touch qPCR machine | Bio-Rad | Cat# 1855485 |
| Trans-Blot Turbo Transfer System | Bio-Rad | N/A |
| ChemiDoc MP | Bio-Rad | Cat# 17001402 |
| Novocyte Quanteon flow cytometer | Agilent Technologies | N/A |
| GloMax-Multi Plate Reader | Promega | Cat# E7081 |
eCLIP-seq data analysis
Read alignment:
For removal of adapter sequences, low quality bases, and short reads, we use cutadapt (Martin, 2011) to trim Illumina adapter sequences and <Q20 bases (parameters -m 18 -a CTGTAGGCACCATCAAT --overlap=5 --trimmed-only --quality-cutoff=20). For splice-aware alignment using STAR (Dobin et al., 2013), we used STAR to align the reads to a reference genome/transcriptome. STAR reference is built using a combination of mouse genome (mm10), mouse rDNA sequence (GenBank GU372691), and mouse transcript annotations (GENCODE vM18). Only uniquely mapped reads were retained (Parameters --outFilterMultimapNmax 1 --alignEndsType EndToEnd --alignIntronMax 1000000 --alignIntronMin 20 --outFilterMismatchNmax 999 --alignSJDBoverhangMin 1 --alignSJoverhangMin 8). For deduplication using UMI, we used umi_tools to deduplicate the alignments. Deduplicated alignments are realigned using STAR and the same parameters as before. For read quantification, we used bedtools (Quinlan and Hall, 2010) to count alignments over 10nt sliding windows with step size of 5nt across the mouse genome.
IP enrichment analysis:
We discarded rows whose sum of counts across all libraries was <10. We used the TMM (Robinson and Oshlack, 2010) method to calculate normalization factors. Counts divided by normalization factors were used for plotting tracks along the transcript. Tracks are plotted using wiggleplotR (Alasoo et al., 2015). Each genomic window is annotated as 5’-UTR, ORF, or 3’-UTR based on overlap with any isoform present in the GENCODE vM18 annotation. For statistical significance of enriched windows, we use voom (Law et al., 2014)-limma (Ritchie et al., 2015) to model mean-variance bias and calculate moderated t-statistics and p-values for the difference in IP versus input samples. We used locfdr (Efron, 2012) approach to estimate local false discovery rates. Locfdr parameters: bre=120, df=10, pct=0, nulltype=1, type=0, mlests=(−2, 0.5).
Enrichment in 5’-UTR/CDS/3’-UTR:
To test overrepresentation of enriched windows across 5’-UTR, CDS, 3’-UTR regions, we performed permutation based chi-square test of independence on the contingency table of regions that the windows overlap versus whether the FDR for enrichment of windows were <=0.05.
Sharp peaks:
Across each annotated transcript with at least one overlapping significant IP-enriched window (FDR <=0.05, log2 fold change >=2, minimum normalized IP signal >=−0.5), Gini indices of 2^(normalized IP signal) values were calculated over rolling windows (minimum normalized IP signal >=−2.5) of size 10. Since Gini index values were biased by IP signal, we hierarchically selected top sharp peaks by binning the IP signal. All windows were ordered by IP signal and split into 10 groups by the order. Windows with within-bin rank of the Gini index higher than 60 percentile were categorically classified as having sharp peaks.
GO term enrichment analysis:
For Gene Ontology (GO) term enrichment, GO terms and gene mappings were obtained from Bioconductor annotation package org.Mm.eg.db (version 3.6.0). We used topGO (Alexa et al., 2006) to perform enrichment analysis. We used a combination of Fisher’s exact test and weight01 algorithm for handling local similarities between GO terms. Genes that have at least one window with FDR <=0.05 and further classified as sharp are used as the positive set. All genes that have at least one window tested are used as the background. For the reported list of GO terms in the manuscript, the following criteria are true: observed/expected ratio >= 2, minimum number of observed genes >= 3, Fisher’s exact test FDR<=0.05, and weight01-conditioned Fisher’s exact test p-value <= 0.05. FDR for Fisher’s exact test is estimated by Benjamini-Hochberg procedure.
Motif analysis:
de novo motif discovery analysis was performed using multiple expectation maximization for motif elicitation (MEME) algorithm (Bailey et al., 2015), using the parameters -objfun de -dna -minw 6 -maxw 20 -nmotifs 20 -evt 0.001 -test mrs -brief 1000000. Genes that have at least one window with FDR <=0.05 and further classified as sharp are used as the positive set. All genes that have at least one window tested are used as the background.
Dissection of neural tube
Microdissections of the neural tube was performed in media (DMEM F12 1:1, 10% FBS) containing 100 μg/ml cycloheximide in a Sylgard dissection dish (Sylgard 184 Silicone Elastomer Kit; Dow Corning). Embryos were pinned to the dish (Austerlitz dissecting pins, FST) with the ventral surface of the embryo facing down. The neural tubes were separated from somites utilizing a tungsten needle (Sharpoint) from the hindlimb, which served as a landmark, to the rhombomeres. Neural tubes were dissociated at 37 °C for 30 min using Papain tissue dissociation kit (Washington, LK003150). After incubation, cells were washed using manufacture’s protocol, rinsed in PBS containing 100 μg/ml cycloheximide and then split into two tubes for Ribo- Seq and RNA-Seq respectively.
Ribosome Profiling library production
Ribosome profiling was performed following the procedures described before (Fujii et al., 2017; Ingolia et al., 2011). For Ribo-Seq, neural tubes from 1 litter of E11.5 embryos (20–23 somite number from tail to hind limb) were used. The Papain dissociated cells were split into two tubes. One was dissolved by Trizol (Invitrogen, 15596) for RNA seq. Another cell pellet was re-suspended in 0.5 ml of cold lysis buffer (20 mM Tris pH 7.5, 150 mM NaCl, 15 mM MgCl2, 1 % Triton X-100, 1 mM DTT, 8 % Glycerol, 20 Unit/ml TURBO DNase, 100μg/ml Cycloheximide) and incubated for 30 min at 4 °C with occasional vortexing. The lysate was clarified by sequential centrifugation for 5 min at 1,800 g and 10,000 g at 4 °C to remove nuclei and mitochondria. The lysate was then treated with RNase I (Ambion, AM2294) for 30 min at RT to digest mRNAs not protected by the ribosome. The digestion was stopped by adding 4.5 μl of SUPERaseIn RNase Inhibitor (20 U/μl, Ambion AM2696). Lysate was then loaded onto a 1 M sucrose cushion. Ribosomes and ribosome protected fragments (RPFs) were pelleted by ultracentrifugation at 70,000 rpm for 4 hr at 4 °C by TLA120.2 rotor. The pellet was re-suspended in Trizol (Invitrogen, 15596) and RNAs were extracted by following manufacturer’s protocol.
RNA-seq:
RNA was extracted from Trizol (Invitrogen, 15596) following manufactuer’s protocol and polyA mRNA was isolated using Oligotex mRNA Mini Kit (Qiagen, 70022) following manufacturer’s protocol. Purified mRNAs were fragmented in alkaline fragmentation buffer (100 mM NaCO3 pH 9.2, 2 mM EDTA).
Ribo-seq/RNA-seq Library production:
RPFs and fragmented RNAs were loaded onto a 15% urea gel. 28–31 nt RPFs and 30–50 nt fragmented RNAs were excised from the gel for Ribo-Seq and RNA-Seq respectively. RNAs were eluted, dephosphorylated by PNK (NEB, M0201S), and ligated to the miRNA Cloning linker (NEB, S1315S) by T4 RNA Ligase2 truncated K227Q (NEB, M0242S). Ligated RNA was gel purified and reverse transcribed by Superscript III (Invitrogen, 18080). Gel purified cDNAs were circularized by Circligase (Epicentra, CL4111K) and rRNA sequence were subtracted using biotinylated oligos. Amplification was done using Phusion High Fidelity DNA Polymerase (NEB, M0530S). PCR amplification was performed for 6–7 cycles and products were loaded onto non-denaturing 8 % PAGE gel. DNA fragment were purified for Illumina sequencing. Primers used in preparation were listed in the Key Resources Table.
Ribo-seq data analysis
Read alignment:
For removal of adapter sequences, low quality bases, and short reads, we use cutadapt (Martin, 2011) to trim Illumina adapter sequences and <Q20 bases (parameters -m 18 -a CTGTAGGCACCATCAAT --quality-cutoff=20). For splice-aware alignment using STAR (Dobin et al., 2013), we used STAR to align the reads to a reference genome/transcriptome. STAR reference is built using a combination of mouse genome (mm10), mouse rDNA sequence (GenBank GU372691), and mouse transcript annotations (GENCODE vM18). Only uniquely mapped reads were retained (Parameters -outFilterMultimapNmax 1 --alignEndsType EndToEnd --alignIntronMax 1000000 --alignIntronMin 20 -outFilterMismatchNmax 999 --alignSJDBoverhangMin 1 --alignSJoverhangMin 8). For read quantification, we used bedtools (Quinlan and Hall, 2010) to count alignments over annotated coding regions excluding the first 45 bases from the start codon and last 15 bases from the stop codon.
Translation efficiency analysis:
We discarded rows whose sum of counts across all libraries was <10. We used the TMM (Robinson and Oshlack, 2010) method to calculate normalization factors. The highest read count transcript isoform was kept for each gene. For calculating statistical significance of differential TE between WT and mutant, we use voom (Law et al., 2014)-limma (Ritchie et al., 2015) to model mean-variance bias and calculate moderated t-statistics and p-values for the difference in TE. We used locfdr (Efron, 2012) approach to estimate local false discovery rates. Locfdr parameters: bre=150, df=7, pct=1e-5, nulltype=1, type=0, mlests=(1, 0.75). For the analysis of the distribution of ΔTEmut-WT for sharp 5’UTR eIF3 binding peaks versus no peak, a control set of similar expression level was chosen by selecting a matching non-eIF3 binding gene with closest wild-type RNA-seq library normalized read counts for each gene with the sharp eIF3 binding peak.
RNA transfection and luciferase assay
C3H10T1/2 cells were cultured in DMEM supplemented with 2 mM L-glutamine and 10 % FBS. RNAs were in vitro transcribed using mMESSAGE mMACHINE T7 Transcription Kit (ThermoFisher, AM1344) followed by Poly(A) Polymerase Tailing Kit (Lucigen, PAP5104H). 1 μg of Fluc RNA and 0.2 μg of Rluc RNA were transfected on 12-well plates using TransIT-mRNA Transfection Kit (Mirus, MIR 2225). Cells were harvested 6 hr after transfection by trypsin treatment followed by PBS wash. Cells were split into two tubes. Half of the cells were re-suspended in TRIzol and RNA was extracted using the PureLink RNA Mini Kit (Thermo Fisher, 12183018). 0.1 μg of RNA was converted to cDNA using iScript Reverse Transcription Supermix for RT-qPCR (BioRad, 1708840). cDNA was diluted two-fold and 2 μl used to run a SYBR green detection RT-qPCR assay (SsoAdvanced Universal SYBR Green supermix (1725270) and CFX384, BioRad). Data was analyzed and converted to relative RNA quantity using CFX manager (BioRad). Primers were used at 300 nM per reaction. The other half of the cells was lysed and assayed using Dual Luciferase kit (Promega, E1980). Firefly luciferase activity was normalized to Renilla luciferase activity and the translation efficiency of Firefly reporter construct was calculated from Firefly luciferase RNA amount normalized to Renilla luciferase relative to the Wildtype construct. Each experiment was performed a minimum of three times, each containing two technical replicates. Statistical analysis was performed using Student’s t-test.
siRNA Knock-down
C3H10T1/2 cells were seeded on a 24 well plate and 25 pmol siRNA with 100 ng of GFP plasmid was transfected twice at 6 and 30 hr after seeding using lipofectamine 2000 in Optimem (Invitrogen, 11668–019; 11058–021). In vitro transcribed RNAs (0.5 μg of Fluc RNA and 0.1 μg of Rluc RNA) were transfected at 48 hr after seeding using TransIT-mRNA Transfection Kit (Mirus, MIR 2225). Cells were harvested 54 hr after seeding for luciferase assay and PCR analysis.
Western blotting and qPCR analysis
Microdissected neural tubes were lysed in 50 μl RIPA buffer (150 mM NaCl, 50 mM Tris-HCl (pH 7.4), 5 mM EDTA (pH 8.0), 1 mM EGTA (pH 8.0), 0.1 % SDS, 1.0 % NP-40, 0.5 % deoxycholate, 1x Combined Protease and Phosphatase Inhibitor (Thermo 78443)) and 10 μl of the lysate was removed and dissolved in Trizol (Invitrogen, 15596) for RNA extraction and RT-qPCR analysis. Lysates were incubated for 5 min on ice and genomic DNAs were sonicated by Bioruptor (Diagenode). Cell lysates were then spun at 14,000 rpm for 5 min at 4°C to remove debris and the supernatant was collected. For Western blot analysis, protein concentration was measured by BCA assay (Pierce, 23225), and 20 μg of protein was loaded onto 4–20 % SDS-PAGE gel. After running, proteins were transferred by semi-dry transfer system using Trans-Blot Turbo (Bio-Rad) following manufacturer’s protocol. PTCH1 and GLI3 proteins were wet transferred to PVDF membrane by Mini Trans-Blot Electrophoretic Transfer Cell (Bio-Rad). The PVDF membranes were blocked in 5% nonfat dry milk in PBST for 1 hr, and incubated overnight at 4 °C with anti-eIF3c (1:3000, Novus, NB100–511), anti-PTCH1 (1:500, kind gift of Matthew Scott lab), anti-Actin (1:5000, Sigma), anti-GAPDH (1:5000, Ambion, AM4300), anti-GLI3 (1:1000, R&D, AF3690), anti-eIF3b (1:5000, Santa Cruz, sc-16377), and anti-RPS5 (Abcam, ab58345), then washed three times for 5 min in PBST, incubated with appropriate secondary antibodies conjugated to horseradish peroxidase (anti-Mouse and anti-Rabbit from GE Healthcare, and anti-Goat from R&D) for 1 hr, and then washed three times for 5 min in PBST. The Western-blot signals were developed using Clarity Western ECL Substrate (Bio-Rad, 1705060) and imaged with ChemiDoc MP (Bio-Rad). Signal intensity of each band was quantified by ImageJ and protein amount was normalized to GAPDH or to RPS5 as indicated in figure legends, and compared between tissues or genotypes.
The eIF3b Immunoprecipitation
Immunoprecipitation was performed following established protocols as described in elsewhere (Lee et al., 2015). E11.5 neural tube and somites were microdissected and flash frozen in liquid nitrogen. We collected 10–12 neural tube & somite for each sample (stage matched somite number +/−2 within sample and between genotypes). Tissues were ground to powder under liquid nitrogen (del Prete et al., 2007) and resuspended in 400 μl of lysis buffer (50 mM HEPES-KOH pH 7.5, 150 mM KCl, 2 mM EDTA, 0.5 % NP-40 alternative, 0.5 mM DTT, 1 Complete EDTA-free Proteinase Inhibitor Cocktail Mini tablet per 10 ml of buffer, 20 U/ml TURBO™ DNase (Ambion, AM1907), 100 U/ml SUPERase RNase inhibitor (Ambion)) and incubated on ice for 10 min. After lysis, nuclei and membrane debris was removed by centrifugation (1,300 g, 5min, 4 °C and then 14,000 rpm, 5 min, 4 °C). Lysate was split for two different antibodies (Goat IgG control antibody (Santa Cruz, sc-2028) and eIF3b antibody (Santa Cruz, sc-16377)) conjugated with Protein G Dynabeads (Invitrogen) and incubated 2 hr at 4 °C. Beads were collected and washed three times in Wash buffer (50 mM HEPES-KOH pH 7.5, 500 mM KCl, 0.5 % NP-40 alternative, 0.5 mM DTT) and bound RNAs were isolated by Trizol (Invitrogen, 15596) following the manufacturer’s protocol, and RT-qPCR was performed for IP and input samples.
QUANTIFICATION AND STATISTICAL ANALYSIS
All measurements were sampled from individual biological replicates. Replicates for mouse embryo experiments consisted of individual neural tube, individual neural tube & somites, individual embryos, or pools of embryos if material was limiting. Experiments consisted of multiple embryos from multiple litters. Unless otherwise stated, when groups of continuous data are compared, two-tailed t-tests with unequal variance were performed; * P< 0.05, ** P< 0.01; for bar plots, error bars = SEM. Otherwise, statistical tests and definitions of significance are described in respective figure legends and methods for each experiment. No statistical methods were used to predetermine sample size.
Supplementary Material
Table S1: The input and eIF3c-eCLIP library profiles of Figure 2 for every 10nt windows in transcripts, Related to Figure 2.
Table S2: Transcripts and sequence of the pyrimidine rich motif in distinct eIF3 binding sites, Related to Figure 3.
Table S3: Enriched GO categories among eIF3 associated pyrimidine rich motif containing transcripts, Related to Figure 3.
Table S4: Oligonucleotide table, Related to Key resources table.
Highlights.
The global translation initiation factor (eIF3c) is required for Shh signaling
eIF3c mutant mice do not show reduced global protein synthesis
Tissue-based eCLIP- and Ribo-seq revealed transcript specific regulation by eIF3
Reduced Ptch1 translation may lead to Shh related phenotypes in eIF3c mutant mice
Acknowledgement
We thank the Cate lab (UC Berkeley) and Barna lab members and D. Ruggero (UCSF) for constructive suggestions and thoughtful critiques of the work. We thank R. Flynn (Stanford University) for generous advice and discussion for CLIP library production. We thank L. Milenkovic and M. Scott (Stanford University) for sharing the PTCH1 antibody. This work was supported by the New York Stem Cell Foundation grant NYSCF-R-I36 (M.B.), NIH grant R01HD086634(M.B.), an Alfred P. Sloan Research Fellowship (M.B.), a Pew Scholars Award (M.B.), and JST, PRESTO grant JPMJPR2049 (K.F.). K.F. was supported by the Human Frontier Science Program Fellowship. O.Z. is a Simons Fellow of the Helen Hay Whitney Foundation. M.B. is a New York Stem Cell Foundation Robertson Investigator.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alasoo K, Martinez FO, Hale C, Gordon S, Powrie F, Dougan G, Mukhopadhyay S, and Gaffney DJ (2015). Transcriptional profiling of macrophages derived from monocytes and iPS cells identifies a conserved response to LPS and novel alternative transcription. Sci. Rep. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexa A, Rahnenführer J, and Lengauer T (2006). Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics. [DOI] [PubMed] [Google Scholar]
- Andaya A, Villa N, Jia W, Fraser CS, and Leary JA (2014). Phosphorylation stoichiometries of human Eukaryotic initiation factors. Int. J. Mol. Sci 15, 11523–11538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, and Noble WS (2009). MEME Suite: Tools for motif discovery and searching. Nucleic Acids Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Johnson J, Grant CE, and Noble WS (2015). The MEME Suite. Nucleic Acids Res. 43, W39–W49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzini AA, Johnstone TG, Christiano R, MacKowiak SD, Obermayer B, Fleming ES, Vejnar CE, Lee MT, Rajewsky N, Walther TC, et al. (2014). Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 33, 981–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brar G. a, and Weissman JS (2015). Ribosome profiling reveals the what, when, where and how of protein synthesis. Nat. Rev. Mol. Cell Biol 16, 651–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield NC, Metzis V, McGlinn E, Bruce SJ, Wainwright BJ, and Wicking C (2009). Patched 1 is a crucial determinant of asymmetry and digit number in the vertebrate limb. Development 136, 3515–3524. [DOI] [PubMed] [Google Scholar]
- Chew G-L, Pauli A, and Schier AF (2016). Conservation of uORF repressiveness and sequence features in mouse, human and zebrafish. Nat. Commun 7, 11663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhuri A, Evans T, and Maitra U (2010). Non-core subunit eIF3h of translation initiation factor eIF3 regulates zebrafish embryonic development. Dev. Dyn 239, 1632–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhuri A, Maitra U, and Evans T (2013). Translation initiation factor eIF3h targets specific transcripts to polysomes during embryogenesis. Proc. Natl. Acad. Sci. U. S. A 110, 9818–9823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M, Kicheva A, Ribeiro A, Blassberg R, Page KM, Barnes CP, and Briscoe J (2015). Ptch1 and Gli regulate Shh signalling dynamics via multiple mechanisms. Nat. Commun 6, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damoc E, Fraser CS, Zhou M, Videler H, Mayeur GL, Hershey JWB, Doudna J. a, Robinson CV, and Leary J. a (2007). Structural characterization of the human eukaryotic initiation factor 3 protein complex by mass spectrometry. Mol. Cell. Proteomics 6, 1135–1146. [DOI] [PubMed] [Google Scholar]
- Daxinger L, Oey H, Apedaile A, Sutton J, Ashe A, and Whitelaw E (2012). A forward genetic screen identifies eukaryotic translation initiation factor 3, subunit H (eIF3h), as an enhancer of variegation in the mouse. G3 (Bethesda). 2, 1393–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: Ultrafast universal RNA-seq aligner. Bioinformatics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docquier A, Pavlin L, Raibon A, Bertrand-Gaday C, Sar C, Leibovitch S, Candau R, and Bernardi H (2019). eIF3f depletion impedes mouse embryonic development, reduces adult skeletal muscle mass and amplifies muscle loss during disuse. J. Physiol 597, 3107–3131. [DOI] [PubMed] [Google Scholar]
- Efron B (2012). Large-scale inference: Empirical Bayes methods for estimation, testing, and prediction.
- Feng W, Choi I, Clouthier DE, Niswander L, and Williams T (2013). The Ptch1 DL mouse: A new model to study lambdoid craniosynostosis and basal cell nevus syndrome-associated skeletal defects. Genesis 51, 677–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii K, Shi Z, Zhulyn O, Denans N, and Barna M (2017). Pervasive translational regulation of the cell signalling circuitry underlies mammalian development. Nat. Commun 8, 14443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gildea DE, Luetkemeier ES, Bao X, Loftus SK, Mackem S, Yang Y, Pavan WJ, and Biesecker LG (2011). The pleiotropic mouse phenotype extra-toes spotting is caused by translation initiation factor Eif3c mutations and is associated with disrupted sonic hedgehog signaling. FASEB J. 25, 1596–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich LV (1997). Altered Neural Cell Fates and Medulloblastoma in Mouse patched Mutants. Science (80-. ). 277, 1109–1113. [DOI] [PubMed] [Google Scholar]
- Herrmannová A, Prilepskaja T, Wagner S, Šikrová D, Zeman J, Poncová K, and Valášek LS (2020). Adapted formaldehyde gradient cross-linking protocol implicates human eIF3d and eIF3c, k and l subunits in the 43S and 48S pre-initiation complex assembly, respectively. Nucleic Acids Res. 48, 1969–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtz AM, Peterson K. a, Nishi Y, Morin S, Song JY, Charron F, McMahon AP, and Allen BL (2013). Essential role for ligand-dependent feedback antagonism of vertebrate hedgehog signaling by PTCH1, PTCH2 and HHIP1 during neural patterning. Development 140, 3423–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Lareau LF, and Weissman JS (2011). Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147, 789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone TG, Bazzini AA, and Giraldez AJ (2016). Upstream ORFs are prevalent translational repressors in vertebrates. 35, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyanagi-Katsuta R, Akimitsu N, Hamamoto H, Arimitsu N, Hatano T, and Sekimizu K (2002). Embryonic lethality of mutant mice deficient in the p116 gene. J. Biochem 131, 833–837. [DOI] [PubMed] [Google Scholar]
- Lamper AM, Fleming RH, Ladd KM, and Lee ASY (2020). to metabolic stress. 856, 853–856. [DOI] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, and Smyth GK (2014). Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, and Carey VJ (2013). Software for Computing and Annotating Genomic Ranges. PLoS Comput. Biol 9, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee ASY, Kranzusch PJ, and Cate JHD (2015). eIF3 targets cell-proliferation messenger RNAs for translational activation or repression. Nature 522, 111–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee ASY, Kranzusch PJ, Doudna JA, and Cate JHD (2016). EIF3d is an mRNA cap-binding protein that is required for specialized translation initiation. Nature 536, 96–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Subgroup, 1000 Genome Project Data Processing (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Li F, Huang L, Polte C, Duan H, Fang J, Sun L, Xing X, Tian G, Cheng Y, et al. (2020). eIF3 Associates with 80S Ribosomes to Promote Translation Elongation, Mitochondrial Homeostasis, and Muscle Health. J. Clean. Prod 79, 575–587.e7. [DOI] [PubMed] [Google Scholar]
- Litingtung Y, Dahn RD, Li Y, Fallon JF, and Chiang C (2002). Shh and Gli3 are dispensable for limb skeleton formation but regulate digit number and identity. Nature 418, 979–983. [DOI] [PubMed] [Google Scholar]
- Makino S, Masuya H, Ishijima J, Yada Y, and Shiroishi T (2001). A spontaneous mouse mutation, mesenchymal dysplasia (mes), is caused by a deletion of the most C-terminal cytoplasmic domain of patched (ptc). Dev. Biol 239, 95–106. [DOI] [PubMed] [Google Scholar]
- Marigo V, and Tabin CJ (1996). Regulation of patched by sonic hedgehog in the developing neural tube. Proc. Natl. Acad. Sci 93, 9346–9351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.Journal 17, 10–12. [Google Scholar]
- Masutani M, Sonenberg N, Yokoyama S, and Imataka H (2007). Reconstitution reveals the functional core of mammalian eIF3. EMBO J. 26, 3373–3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milenkovic L, Goodrich LV, Higgins KM, and Scott MP (1999). Mouse patched1 controls body size determination and limb patterning. Development 126, 4431–4440. [DOI] [PubMed] [Google Scholar]
- del Prete MJ, Vernal R, Dolznig H, Müllner EW, and Garcia-Sanz J a (2007). Isolation of polysome-bound mRNA from solid tissues amenable for RT-PCR and profiling experiments. RNA 13, 414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulos-Holmes MC, Srole DN, Juarez MG, Lee ASY, McSwiggen DT, Ingolia NT, and Cate JH (2019). Repression of ferritin light chain translation by human eIF3. Elife. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, and Oshlack A (2010). A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2009). edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadato D, Ono T, Gotoh-Saito S, Kajiwara N, Nomura N, Ukaji M, Yang L, Sakimura K, Tajima Y, Oboki K, et al. (2018). Eukaryotic translation initiation factor 3 (eIF3) subunit e is essential for embryonic development and cell proliferation. FEBS Open Bio. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T, Heger A, and Sudbery I (2017). UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 27, 491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonenberg N, and Hinnebusch AG (2009). Regulation of Translation Initiation in Eukaryotes: Mechanisms and Biological Targets. Cell 136, 731–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama H, Takahashi K, Yamamoto T, Iwasaki M, Narita M, Nakamura M, Rand TA, Nakagawa M, Watanabe A, and Yamanaka S (2017). Nat1 promotes translation of specific proteins that induce differentiation of mouse embryonic stem cells. Proc. Natl. Acad. Sci 114, 340–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truitt ML, Conn CS, Shi Z, Pang X, Tokuyasu T, Coady AM, Seo Y, Barna M, and Ruggero D (2015). Differential Requirements for eIF4E Dose in Normal Development and Cancer. Cell 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsumi R, Masoudi M, Takahashi A, Fujii Y, Hayashi T, Kikuchi I, Satou Y, Taira M, and Hatakeyama M (2013). YAP and TAZ, Hippo signaling targets, act as a rheostat for nuclear SHP2 function. Dev. Cell 26, 658–665. [DOI] [PubMed] [Google Scholar]
- Wickham H, Averick M, Bryan J, Chang W, McGowan L, François R, Grolemund G, Hayes A, Henry L, Hester J, et al. (2019). Welcome to the Tidyverse. J. Open Source Softw 4, 1686. [Google Scholar]
- Zeileis A, Grothendieck G, and Wien W (2005). zoo: An S3 Class and Methods for Indexed Totally Ordered Observations zoo: An S3 Class and Methods for Indexed Totally Ordered Observations.
- Zeng L, Wan Y, Li D, Wu J, Shao M, Chen J, Hui L, Ji H, and Zhu X (2013). The m subunit of murine translation initiation factor eIF3 maintains the integrity of the eIF3 complex and is required for embryonic development, homeostasis, and organ size control. J. Biol. Chem 288, 30087–30093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhulyn O, Li D, Deimling S, Vakili NA, Mo R, Puviindran V, Chen M-H, Chuang P-T, Hopyan S, and Hui C (2014). A switch from low to high shh activity regulates establishment of limb progenitors and signaling centers. Dev. Cell 29. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: The input and eIF3c-eCLIP library profiles of Figure 2 for every 10nt windows in transcripts, Related to Figure 2.
Table S2: Transcripts and sequence of the pyrimidine rich motif in distinct eIF3 binding sites, Related to Figure 3.
Table S3: Enriched GO categories among eIF3 associated pyrimidine rich motif containing transcripts, Related to Figure 3.
Table S4: Oligonucleotide table, Related to Key resources table.
Data Availability Statement
Data that support the findings of this study have been deposited in the Gene Expression Omnibus under accession number GSE183472.
Code generated in this study has been deposited on Github DOI: 10.5281/zenodo.5555442.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.