

Abstract
Marburg virus disease (MVD) caused by the Marburg virus (MARV) generally appears with flu-like symptoms and leads to severe hemorrhagic fever. It spreads via direct contact with infected individuals or animals. Despite being considered to be less threatening in terms of appearances and the number of infected patients, the high fatality rate of this pathogenic virus is a major concern. Until now, no vaccine has been developed to combat this deadly virus. Therefore, vaccination for this virus is necessary to reduce its mortality. Our current investigation focuses on the design and formulation of a multi-epitope vaccine based on the structural proteins of MARV employing immunoinformatics approaches. The screening of potential T-cell and B-cell epitopes from the seven structural proteins of MARV was carried out through specific selection parameters. Afterward, we compiled the shortlisted epitopes by attaching them to an appropriate adjuvant and linkers. Population coverage analysis, conservancy analysis, and MHC cluster analysis of the shortlisted epitopes were satisfactory. Importantly, physicochemical characteristics, human homology assessment, and structure validation of the vaccine construct delineated convenient outcomes. We implemented disulfide bond engineering to stabilize the tertiary or quaternary interactions. Furthermore, stability and physical movements of the vaccine protein were explored using normal-mode analysis. The immune simulation study of the vaccine complexes also exhibited significant results. Additionally, the protein–protein docking and molecular dynamics simulation of the final construct exhibited a higher affinity toward toll-like receptor-4 (TLR4). From simulation trajectories, multiple descriptors, namely, root mean square deviations (rmsd), radius of gyration (Rg), root mean square fluctuations (RMSF), solvent-accessible surface area (SASA), and hydrogen bonds, have been taken into account to demonstrate the inflexible and rigid nature of receptor molecules and the constructed vaccine. Inclusively, our findings suggested the vaccine constructs’ ability to regulate promising immune responses against MARV pathogenesis.
1. Introduction
The Marburg virus (MARV), a single-stranded RNA virus responsible for causing Marburg virus disease (MVD) in humans, was first identified after simultaneous outbreaks in Marburg and Frankfurt of Germany in 1967. In 1987–88, this virus had serious repercussions in the Soviet Union, Koltsovo, and Kenya, leading to 100% mortality. In the year 2004 in Angola, almost 400 people had been infected with MARV, raising the mortality rate to almost 90%. It triggered outbreaks in the USA, Netherlands, and also Uganda in 2008. Notably, Uganda experienced multiple MARV outbreaks in 2012, 2014, and 2017. Recently, Guinea recorded the first-ever case of MARV on August 9, 2021. The Marburg virus is a negative-sensed, unsegmented enveloped RNA virus that has a filamentous structure and snail-like number 6 or spiral shape and occasionally can be branched.1−4 Being a member of the Filovirus family under the order of Mononegavirales, the Marburg virus (MARV); similar to the Ebola virus (EBOV), is responsible for causing severe MVD, which can be fatal.5 Although this virus has different strains, the Lake Victoria Marburg virus strain was reported as responsible for causing severe Marburg hemorrhagic fever (MHF) or MVD.1
Transmission of MARV takes place through mucosal surfaces, skin abrasions, and also parenteral encounters. Different types of bat species have been suggested as reservoir hosts. There is notable confirmation about Rousettus aegyptiacus, the Egyptian fruit bat, as a reservoir for MARV.6 In the course of an outbreak, the most common source of MARV infection comes into contact with infected individuals or animals, especially under nosocomial conditions where parenteral introduction is the most noxious route of infection.7 MVD generally appears with flu-like symptoms, where the symptoms include headache, fever, chills, joint pain, malaise, and myalgia in 2–21 days after the infection occurs initially. Abdominal pain, watery diarrhea, vomiting, lethargy, and nausea can be observed in the patient within 2–5 days of the first symptoms, and the severity of the infection gets enhanced on days 5–7. Sustained fever, conjunctivitis, maculopapular rash, and finally the symptoms of hemorrhagic fever such as petechiae, mucosal bleeding, and blood in the vomitus, stool, and venipuncture sites are experienced by the patients.6
Seven structural proteins are encoded by MARV genomes, that is, nucleoprotein (NP), envelope glycoprotein (GP), RNA-directed RNA polymerase L (L), matrix protein VP40 (VP40), polymerase cofactor VP35 (VP35), minor nucleoprotein VP30 (VP30), and membrane-associated protein VP24 (VP24). The NP, VP35, VP30, and L are four proteins that associate to form a nucleocapsid complex, which encircles the viral genome.8 Glycoprotein (GP), a single surface protein of MARV, is responsible for facilitating viral entry and assisting in the attachment of the virus to target cells.9 The entry of the MARV and its insertion of replication machinery were found to be associated with intravesicular glycoprotein cleavage by host proteases. Thus, the viral core is released into the cell cytoplasm and then replicates.10 Nucleoprotein (NP), the principal nucleocapsid protein of MARV, is accountable for the encapsidation of the genomic RNA of the virus. NP helps to generate helical nucleocapsid precursors that assemble in intracellular inclusions in infected cells and determines the transcription and replication activity.11 The MARV VP40 protein not only regulates viral replication and transcription but also stimulates virus egress.12 MARV VP35 is necessary for nucleocapsid generation, which has some vital roles in different stages of the viral replication cycle including production of type I interferon (IFN), maturation of dendritic cells, and activation of protein kinase R (PKR).13−16 MARV VP30 is closely associated with the nucleocapsid by uniting with the NP.17 This protein is not a requisite in replication or transcription activity.11,18 Interestingly, a MARV full-length clone can only be rescued in the presence of VP30, which suggests its role in viral amplification.19 MARV VP24 generally has roles in the formation of nucleocapsids and assembly.20 The VP24 protein is also involved in the step between replication and budding.21 The large protein (L) is a multifunctional protein that is necessary for replication and transcription. It pairs with VP35 to form an RNA-dependent RNA polymerase complex. This protein is thought to perform capping, polyadenylation of viral RNAs, and synthesis of RNA.22 Therefore, it can be concluded that the structural proteins of MARV are significantly involved in the viral pathogenesis and life cycle of the virus.
Introduction of the peptide allows B-cells to be stimulated by helper T cells and become plasma cells to produce antibodies. In addition to the neutralization of antibodies, the body also counts on helper CD4+ T cells and CD8+ T cytotoxic cells to completely clear the virus from the body. When a mutant virus causes infection to the host cells by ruling out antibodies, the body relies on immunity-mediated T-cells to fight the virus. Also, T-cell immunity is highly dependent on MHC–peptide complexes, which is reminiscent of antigen–antibody binding. MHC proteins are encoded by the human leukocyte antigen (HLA). Each of the HLA alleles represents only a specific set of peptides on the surface of an infected cell and is recognized by T-cell epitopes.23 Previously, several approaches for designing a vaccine against MARV had been followed. This included vaccines based on viral vectors such as the vesicular stomatitis virus, adenovirus-vectored vaccines, DNA plasmid vaccine, and virus-like particles composed of several MARV proteins. Despite these trials, there is no approved vaccine against MARV in the market.24−27
Identification, deep investigation, and setting up an immunological correlation with MARV are some requisites for the traditional method of commanding vaccine development. The development of a vaccine by an experiment-based method is very difficult, costly, and tedious. It takes quite a number of years with a reasonable rate of failure to make a vaccine commercially successful. This may also exhibit contradiction with the normal complement system function of the human body. Epitope-based peptide vaccine designing and development by employing different visionary tools have become areas of interest for researchers. In silico epitope-based peptide vaccine could become a decent candidate against MARV, as these vaccines lack the aforementioned limitations and possess increased immunogenicity and safety. The selection of antigen molecules mainly determines the efficacy of the vaccines.28,29 The multi-epitope vaccines have been designed for the generation of diverse immune responses for incorporation of the antigens and also to reduce the limitation associated with MHC restriction into one single unit.30 Peptide vaccines based on epitopes have been delineated to be effective against hepatitis B, hepatitis C, influenza A, Leishmania, and Mayaro viruses by elucidating protective immunity.31−35 In this research, we investigated the MARV structural proteins to scrutinize prospective antigenic and immunogenic epitopes that prompts not only humoral (B-cell) immune response but also cell-mediated (T-cell) immune response. We have taken potential epitopes from the selected structural proteins into account and construct a multi-epitope vaccine with the addition of an appropriate adjuvant and linkers. We expect the present study to facilitate MARV vaccine development, and experimental work will be performed further to validate our current findings.
2. Results
2.1. Retrieval and Analysis of Protein Sequences
The entire viral proteome of MARV consists of seven structural proteins, namely, nucleoprotein (NP), envelope glycoprotein (GP), RNA-directed RNA polymerase L (L), matrix protein VP40 (VP40), polymerase cofactor VP35 (VP35), minor nucleoprotein VP30 (VP30), and membrane-associated protein VP24 (VP24). We used the VaxiJen v2.0 online server to assess the retrieved structural protein sequences. The number of TM helices was predicted using the TMHMM v2.0 server. The antigenicity of the protein sequences ranges from 0.4107 to 0.5636, indicating the sequences as antigenic. The proteins were also predicted as non-allergenic using the AllergenFP v1.0 server. The number of transmembrane helices was also suitable, as it ranges from 0 to 1. Therefore, we selected all seven proteins for multi-epitope vaccine construction. The selected protein sequences along with their UniProt entry number, antigenicity, allergenicity, and TM helices are displayed in Table 1.
Table 1. List of the Selected Structural Protein Sequences and Their Selection Parameters.
| UniProt entry | entry name | protein name | abbreviation | antigenicity | allergenecity | no. of TM helices |
|---|---|---|---|---|---|---|
| P35253 | VGP_MABVM | envelope glycoprotein | GP | 0.5481 (probable antigen) | probable non-allergen | 0 |
| P31352 | L_MABVM | RNA-directed RNA polymerase L | L | 0.4518 (probable antigen) | probable non-allergen | 1 |
| P35260 | VP40_MABVM | matrix protein VP40 | VP40 | 0.4107 (probable antigen) | probable non-allergen | 0 |
| P27588 | NCAP_MABVM | nucleoprotein | NP | 0.4761 (probable antigen) | probable non-allergen | 0 |
| P35259 | VP35_MABVM | polymerase cofactor VP35 | VP35 | 0.4360 (probable antigen) | probable non-allergen | 0 |
| P35256 | VP24_MABVM | membrane-associated protein VP24 | VP24 | 0.5423 (probable antigen) | probable non-allergen | 0 |
| P35258 | VP30_MABVM | transcriptional activator VP30 | VP30 | 0.5636 (probable antigen) | probable non-allergen | 0 |
2.2. Prediction and Evaluation of Potential CTL Epitopes
A total of 115 potential CTL epitopes from the selected proteins were determined as antigenic, non-allergenic, non-toxic, and also immunogenic under the specified selection range (Table S1). NetCTL 1.2 was utilized to determine the combinatorial score of the selected epitopes. After appropriate screening, we shortlisted two promising CTL epitopes from each of the NP and L proteins and one from each of the GP, VP40, VP35, VP30, and VP24 proteins for multi-epitope vaccine construction. The shortlisted CTL epitopes for vaccine construction are incorporated in Table 2.
Table 2. Shortlisted CTL Epitopes for the Final Vaccine Construction.
| epitopes | protein | C-score | immunogenicity score | antigenicity | allergenicity | toxicity | epitope conservancy hit (maximum identity %) | human homology | interacting MHC-I allelesa |
|---|---|---|---|---|---|---|---|---|---|
| GSNDTELNY | L | 2.1551 | 0.13913 (positive) | 1.1545 (probable antigen) | probable non-allergen | non-toxin | 100 | non-homologue | HLA-A*01:01, HLA-B*58:01, HLA-A*30:02 |
| WTIGNRAPY | L | 2.4674 | 0.14626 (positive) | 1.2809 (probable antigen) | probable non-allergen | non-toxin | 100 | non-homologue | HLA-A*26:01, HLA-A*30:02, HLA-A*01:01, HLA-C*03:03, HLA-B*15:01, HLA-B*35:01 |
| FDERRHVAM | NP | 2.2254 | 0.22313 (positive) | 0.9841 (probable antigen) | probable non-allergen | non-toxin | 100 | non-homologue | HLA-B*14:02, HLA-B*08:01, HLA-C*07:02 |
| RPMNRPTAL | NP | 1.9018 | 0.03207 (positive) | 0.5629 (probable antigen) | probable non-allergen | non-toxin | 100 | non-homologue | HLA-B*07:02, HLA-B*08:01, HLA-B*39:01, HLA-B*48:01, HLA-B*14:02, HLA-B*53:01, HLA-B*35:01 |
| NPHAQGIAL | GP | 1.5028 | 0.10415 (positive) | 1.0162 (probable antigen) | probable non-allergen | non-toxin | 100 | non-homologue | HLA-B*39:01, HLA-B*35:01, HLA-B*15:02, HLA-B*07:02 |
| FTYNLTNLV | VP40 | 1.2927 | 0.00609 (positive) | 0.7288 (probable antigen) | probable non-allergen | non-toxin | 100 | non-homologue | HLA-A*02:01, HLA-A*68:02, HLA-A*02:06, HLA-A*01:01, HLA-A*26:01, HLA-B*51:01, HLA-C*06:02, HLA-C*15:02, HLA-B*46:01 |
| RTTAPAAAF | VP35 | 1.4688 | 0.13388 (positive) | 0.4463 (probable antigen) | probable non-allergen | non-toxin | 100 | non-homologue | HLA-A*32:01, HLA-B*58:01, HLA-B*57:01, HLA-B*07:02, HLA-C*03:03, HLA-B*15:01, HLA-B*35:01 |
| SLTCAGIRK | VP30 | 1.1697 | 0.17091 (positive) | 1.3530 (probable antigen) | probable non-allergen | non-toxin | 100 | non-homologue | HLA-A*03:01, HLA-A*11:01 |
| ISPNLLGIY | VP24 | 1.4959 | 0.07501 (positive) | 1.0528 (probable antigen) | probable non-allergen | non-toxin | 100 | non-homologue | HLA-A*30:02, HLA-A*01:01, HLA-A*26:01, HLA-B*46:01 |
MHC-I alleles with percentile rank ≤2 were taken.
2.3. Prediction and Evaluation of Potential HTL Epitopes
A total of 442 potential HTL epitopes from the selected proteins were determined as antigenic, non-allergenic, and non-toxic under the specified selection range (Table S2). After evaluating the cytokine-inducing ability of the selected HTL epitopes, we shortlisted two promising epitopes from the L protein and one from each of the GP, NP, VP40, VP35, and VP24 proteins for multi-epitope vaccine construction. The shortlisted HTL epitopes for vaccine construction are incorporated in Table 3.
Table 3. Shortlisted HTL Epitopes for the Final Vaccine Construction.
| epitopes | protein | antigenicity | IL4 | IL10 | IFN-γ | allergenicity | toxicity | epitope conservancy hit (maximum identity %) | human homology | interacting MHC-II allelesa |
|---|---|---|---|---|---|---|---|---|---|---|
| LGFPLGFNIDISCFK | L | 1.1972 (probable antigen) | inducer | inducer | positive | probable non-allergen | non-toxin | 100 | non-homologue | HLA-DRB1*04:26, HLA-DRB1*03:05, HLA-DRB1*04:21 |
| SPLLDLRNHFICSLR | L | 1.4076 (probable antigen) | inducer | inducer | positive | probable non-allergen | non-toxin | 100 | non-homologue | HLA-DRB1*13:01, HLA-DRB1*13:27, HLA-DRB1*13:28, HLA-DRB1*13:04, HLA-DRB1*11:02, HLA-DRB1*11:21, HLA-DRB1*13:22 |
| SQRGRIGLFLSFCSL | NP | 0.8219 (probable antigen) | inducer | inducer | positive | probable non-allergen | non-toxin | 100 | non-homologue | HLA-DRB1*15:01, HLA-DRB1*15:06, HLA-DRB1*04:23 |
| WGAFFLYDRIASTTM | GP | 0.5262 (probable antigen) | inducer | inducer | positive | probable non-allergen | non-toxin | 100 | non-homologue | HLA-DRB1*08:13, HLA-DRB1*15:02, HLA-DRB1*08:17, HLA-DRB1*08:01, HLA-DRB1*04:01, HLA-DRB1*15:06, HLA-DRB1*08:02, HLA-DRB1*11:14, HLA-DRB1*13:23, HLA-DRB3*01:01, HLA-DRB1*08:04 |
| HPAVSIHPNLPPIVL | VP40 | 1.0327 (probable antigen) | non-inducer | inducer | positive | probable non-allergen | non-toxin | 100 | non-homologue | HLA-DRB1*13:02 |
| EITPVLKMGRTLEAI | VP35 | 0.9411 (probable antigen) | inducer | inducer | positive | probable non-allergen | non-toxin | 100 | non-homologue | HLA-DRB1*04:02, HLA-DRB1*03:06, HLA-DRB1*03:07, HLA-DRB1*03:08 |
| HVVNDHGKPSSIEIK | VP24 | 0.8888 (probable antigen) | inducer | non-inducer | positive | probable non-allergen | non-toxin | 100 | non-homologue | HLA-DRB1*03:06, HLA-DRB1*03:07, HLA-DRB1*03:08 |
MHC-II alleles with percentile rank ≤2 were taken.
2.4. Prediction and Evaluation of Potential LBL Epitopes
A total of 151 potential LBL epitopes from the selected proteins were determined as non-allergenic, antigenic, and non-toxic (Table S3). Two promising LBL epitopes from each of the NP and L proteins and one from each of the GP, VP40, VP35, VP30, and VP24 proteins were selected for multi-epitope vaccine construction based on the non-allergen nature, highest antigenicity, toxicity profile, and probability scores. The shortlisted LBL epitopes for vaccine construction are compiled in Table 4.
Table 4. Shortlisted LBL Epitopes for the Final Vaccine Construction.
| epitopes | protein | probability score | antigenicity | allergenicity | toxicity | human homology |
|---|---|---|---|---|---|---|
| ASARSQCEDFSPKENP | L | 0.9560 | 1.2618 (probable antigen) | probable non-allergen | non-toxin | non-homologue |
| DELHDLNLKLQISDNK | L | 0.9560 | 1.2314 (probable antigen) | probable non-allergen | non-toxin | non-homologue |
| LELGTKPTAPHVRNKK | NP | 0.9560 | 1.6432 (probable antigen) | probable non-allergen | non-toxin | non-homologue |
| DWPDMSFDERRHVAMN | NP | 0.9560 | 1.2572 (probable antigen) | probable non-allergen | non-toxin | non-homologue |
| PPLPTARPEIKLTSTP | GP | 0.9560 | 1.1812 (probable antigen) | probable non-allergen | non-toxin | non-homologue |
| FSVKEGMMKKRGENSP | VP40 | 0.9560 | 1.0854 (probable antigen) | probable non-allergen | non-toxin | non-homologue |
| AFLGVVPPVIRVKNFQ | VP35 | 0.9560 | 1.0892 (probable antigen) | probable non-allergen | non-toxin | non-homologue |
| SKPHYTNYHPRARSMS | VP30 | 0.9560 | 1.2354 (probable antigen) | probable non-allergen | non-toxin | non-homologue |
| PSVGGWTVKWGNFVFH | VP24 | 0.9560 | 1.3286 (probable antigen) | probable non-allergen | non-toxin | non-homologue |
2.5. Epitope Conservancy and Human Homology Determination
Each shortlisted epitope from both MHC classes displayed 100% maximum identity for conservancy. Furthermore, selected LBL, CTL, and HTL epitopes were non-homologous to the human proteome, confirming the epitopes as antigens or foreign particles within the human body. Human homology and conservancy analysis of the selected epitopes are incorporated in Tables 2 and 3.
2.6. Molecular Docking Studies of the Alleles and CTL Epitopes
Molecular docking simulation was employed to investigate the binding efficacy of the targeted CTL epitopes with their respective HLA alleles. We have selected the HLA-B*35:01 (PDB ID: 1A1N) allele for RTTAPAAAF, WTIGNRAPY, and NPHAQGIAL epitopes. HLA-A*01:01 (PDB ID: 6AT9) was selected for ISPNLLGIY, FTYNLTNLV, and GSNDTELNY epitopes. HLA-B*14:02 (PDB ID: 3BXN) was selected for both epitopes of the nucleoprotein, that is, FDERRHVAM and RPMNRPTAL. Furthermore, the HLA-A*03:01 (PDB ID: 6O9B) allele was selected for the SLTCAGIRK epitope of the VP30 protein. Afterward, we validated the docking studies by considering co-crystallized ligand epitopes as the positive control. Molecular docking results concluded that the positive controls and the shortlisted CTL epitopes interacted with the HLA alleles. All the positive controls of the respective alleles possessed more binding affinities than the targeted CTL epitopes except the control of HLA-B*35:01, where RTTAPAAAF (−10 kcal/mol) and WTIGNRAPY (−9.4 kcal/mol) epitopes produced more docking scores than the positive control (−9.3 kcal/mol) of the HLA-B*35:01 allele. Although they generated greater docking scores than the positive control, the number of hydrogen bond interactions was less than that for the positive control of HLA-B*35:01. The targeted alleles and the positive controls formed almost similar interactions in comparison with the interactions formed between the CTL epitopes and the targeted receptors. GSNDTELNY formed nine hydrogen bonds with the HLA-A*01:01 allele—the highest amongst all the epitopes, with the control of HLA-A*01:01 allele displaying 11 hydrogen bonds. Although FDERRHVAM (−8.6 kcal/mol) and RPMNRPTAL (−7.8 kcal/mol) epitopes of NP represented a low docking score than the positive control of HLA-B*14:02 (−10.0 kcal/mol), the number of hydrogen bonds formed by these epitopes was greater than that for the control. Furthermore, all the selected CTL epitopes and the positive controls displayed a good number of hydrophobic bond interactions with the receptors (Table 5 and Figures 2–5).
Table 5. Binding Affinities and Interaction between Selected CTL Epitopes and HLA Alleles.
| alleles | epitopes | grid box size (Å) | docking score (kcal mol–1) | hydrogen bond interactions | hydrophobic bond interactions | attractive charges | unfavorable bumps |
|---|---|---|---|---|---|---|---|
| HLA-B*35:01 | RTTAPAAAF | 62.33 × 45.41 × 70.54 | –10.0 | Tyr84, Arg97, Trp147 | Tyr74 (pi-sigma), Lys146(pi-alkyl), Tyr159 (pi-sigma), Leu163 (pi-alkyl), Trp167 (pi–pi stacked) | Arg97, Ser116, Lys146, Trp147 | |
| WTIGNRAPY | –9.4 | Tyr9, Arg62, Ser77, Thr143, Trp147 | Ile95 (pi-alkyl), Trp147 (pi–pi T shaped), Tyr159 (pi-sigma), Trp167 (pi–pi stacked) | Lys146 | Arg62 | ||
| NPHAQGIAL | –8.8 | Arg97, Tyr99, Trp147, Trp167 | Tyr7 (pi-alkyl), Ile66 (pi-alkyl), Phe67 (pi-alkyl), Tyr74 (pi-alkyl), Ile95 (pi-alkyl), Trp147 (pi-alkyl) | Ser77 | |||
| control | –9.3 | Asn70, Asn80, Tyr84, Thr143, Lys146, Trp147, Tyr171 | Tyr59 (pi-alkyl), Asn63 (carbon–hydrogen), Ile95 (pi-alkyl), Trp147 (pi–pi stacked), Tyr159 (pi-alkyl), Trp167 (pi-alkyl) | Tyr7 | |||
| HLA-A*01:01 | ISPNLLGIY | 60.64 × 73.76 × 45.49 | –7.8 | Tyr84, Asp116, Trp147, Arg156 | Leu81 (pi-alkyl), Lys146 (pi-alkyl), Trp147 (pi-sigma), Val158 (alkyl), Arg163 (pi-alkyl) | Lys146, Tyr159 | Arg163 |
| FTYNLTNLV | –7.8 | Asn66, Thr73, Asn77, Tyr84, Lys146, Trp147, Arg156 | Leu81 (pi-alkyl), Ile95 (pi-alkyl), Lys146 (pi-alkyl), Val150 (pi-alkyl), Tyr159 (pi-sigma) | Arg156 | Trp147 | ||
| GSNDTELNY | –7.7 | Ala76, Thr80, Arg82, Tyr84, His93, Asp116, Tyr118, Lys146, Trp147 | Gly79 (carbon–hydrogen), Leu81 (pi-alkyl), Gly83 (carbon–hydrogen), Glu89 (carbon–hydrogen), Thr143 (carbon–hydrogen) | Arg75, Arg82, Lys146 | |||
| Control | –8.0 | Glu63, Asn66, Ala69, Thr73, Asn77, Arg114, Asp116, Lys146, Trp147, Gln155, Arg156 | Ala69 (pi-alkyl), His70 (pi-alkyl), Thr73 (pi-sigma), Ala76 (pi-alkyl), Leu81 (pi-alkyl), Thr143 (carbon–hydrogen), Tyr159 (pi-sigma) | Arg156 | |||
| HLA-B*14:02 | FDERRHVAM | 61.25 × 48.69 × 72.95 | –8.6 | Tyr7, Asn63, Asn70, Asp74, Ser77, Trp147 | Ile66 (carbon–hydrogen), Leu81 (pi-alkyl), Leu95 (pi-alkyl), Trp97 (pi-alkyl), Tyr99 (pi-sigma), Phe116 (pi–pi T shaped), Tyr159 (pi-sigma) | Asp74, Glu152, Trp167 | Glu76, Asn80, Tyr159 |
| RPMNRPTAL | –7.8 | Asn70, Thr73, Ser77, Lys146, Trp147, Glu152 | Ser77 (carbon–hydrogen), Trp97 (pi-sigma), Trp147 (pi-sigma) | Glu152 | Asp74, Ser77 | ||
| control | –10.0 | Glu55, Arg62, Asn63, Asn70 | Leu81 (pi-alkyl), Leu95 (pi-alkyl), Phe116 (pi–pi T shaped), Tyr123 (pi–pi stacked), Trp133 (alkyl),Trp147 (pi-sigma), Leu156 (alkyl), Trp167 (pi-sigma) | Trp167 | Tyr7, Gly56, Asn63 | ||
| HLA-A*03:01 | SLTCAGIRK | 73.20 × 52.65 × 53.24 | –6.5 | Glu63, Asn66, Glu152, Gln155 | Glu63 (carbon–hydrogen), Tyr99 (pi-sigma), Tyr159 (pi-sigma) | Tyr7, Glu63, Trp167 | Tyr99 |
| control | –7.1 | Tyr7, Asn66, Thr73, Tyr99, Trp147, Glu152, Gln155, Tyr159 | Tyr7 (pi-sigma), Ala69 (pi-alkyl), Tyr99 (carbon–hydrogen), Tyr159 (pi-sigma), Trp167 (pi-sigma) | Glu63, Glu152, Trp167 | Arg114 |
Figure 2.
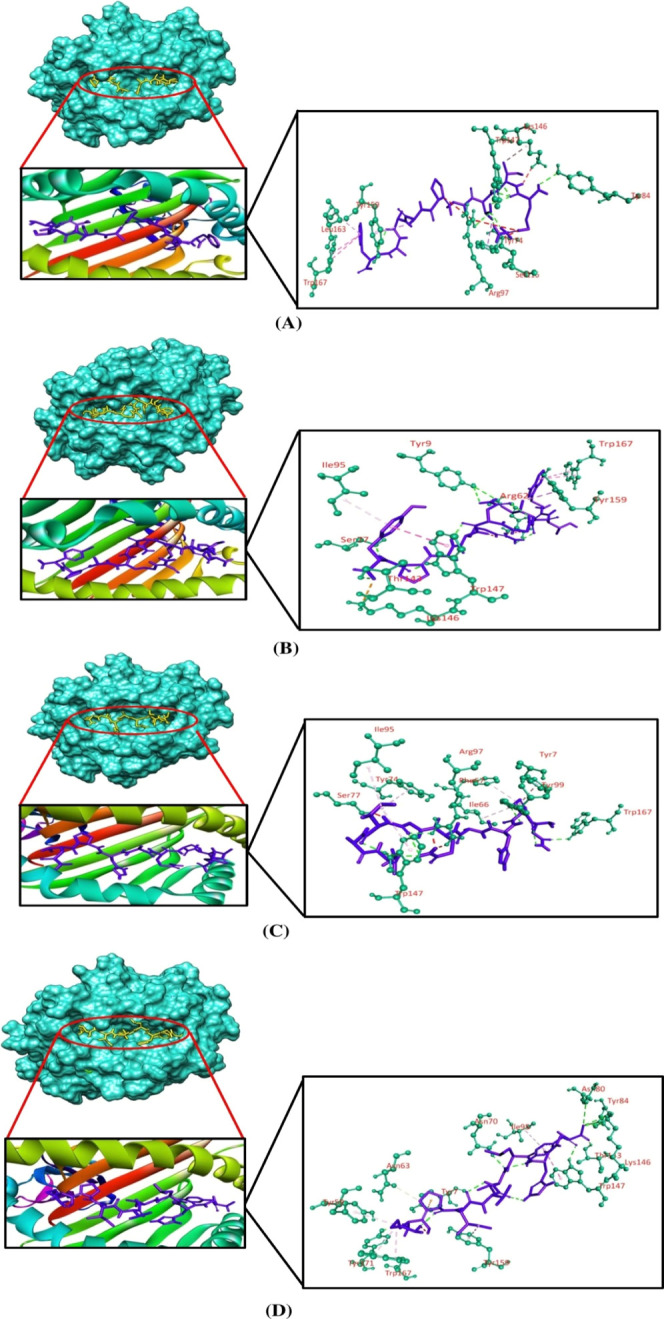
Three-dimensional representation of molecular docking analysis between the predicted epitope (A) RTTAPAAAF, (B) WTIGNRAPY, (C) NPHAQGIAL, and (D) positive control to the groove of the HLA-B*35:01 allele. The interacting residues are displayed as green balls and sticks, conventional hydrogen bonds are displayed as green lines, pi–pi/pi–alkyl stacking bonds are displayed as pink lines, pi–sigma bonds are displayed as violet lines, carbon–hydrogen bonds are displayed as white lines, unfavorable bumps are displayed as red lines, and attractive charges are displayed as gold lines.
Figure 5.

Three-dimensional representation of molecular docking analysis between the predicted epitope of the MARV VP30 protein (A) SLTCAGIRK and (B) positive control to the groove of the HLA-A*03:01 allele. The interacting residues are displayed as green balls and sticks, conventional hydrogen bonds are displayed as green lines, pi–pi/pi–alkyl stacking bonds are displayed as pink lines, pi–sigma bonds are displayed as violet lines, carbon–hydrogen bonds are displayed as white lines, unfavorable bumps are displayed as red lines, and attractive charges are displayed as gold lines.
Figure 3.

Three-dimensional representation of molecular docking analysis between the predicted epitope (A) ISPNLLGIY, (B) FTYNLTNLV, (C) GSNDTELNY, and (D) positive control to the groove of the HLA-A*01:01 allele. The interacting residues are displayed as green balls and sticks, conventional hydrogen bonds are displayed as green lines, pi–pi/pi–alkyl stacking bonds are displayed as pink lines, pi–sigma bonds are displayed as violet lines, carbon–hydrogen bonds are displayed as white lines, unfavorable bumps are displayed as red lines, and attractive charges are displayed as gold lines.
Figure 4.

Three-dimensional representation of molecular docking analysis between the predicted epitope of MARV NP (A) FDERRHVAM, (B) RPMNRPTAL, and (C) positive control to the groove of the HLA-B*14:02 allele. The interacting residues are displayed as green balls and sticks, conventional hydrogen bonds are displayed as green lines, pi–pi/pi–alkyl stacking bonds are displayed as pink lines, pi–sigma bonds are displayed as violet lines, carbon–hydrogen bonds are displayed as white lines, unfavorable bumps are displayed as red lines, and attractive charges are displayed as gold lines.
2.7. Population Coverage Analysis
The population coverage analysis of the MHCI and MHCII epitopes displayed 95.75 and 51.08% coverage for the global population, respectively. As a vaccine protein includes both classes of MHC epitopes, we emphasized their combined population coverage. The combined coverage involved 97.92% of the world’s population. Europe had the highest population coverage for combined MHC class-I and class-II epitopes (99.23%) followed by North America (98.19%), East Asia (96.27%), West Africa (95.89%), West Indies (95.56%), Northeast Asia (95.33%), North Africa (95.05%), South Africa (94.56%), South Asia (94.38%), Southeast Asia (93.71%), East Africa (92.98%), Southwest Asia (92.20%), Central Africa (91.09%), Oceania (89.52%), South America (86.22%), and Central America (40.73%). The comparative population coverages among MHC class-I epitopes, MHC class-II epitopes, and combined MHC epitopes are displayed in Figure 6, Table S4, and Figures S1–S3.
Figure 6.

Worldwide population coverage with MHCI, MHCII, and combined MHC epitopes based on their respective HLA binding alleles.
2.8. MHC Cluster Analysis
The MHCI alleles and MHCII alleles that interacted with the epitopes from the selected structural proteins were clustered using the MHCcluster v2.0 server. A total of 25 alleles from each MHC class were included in the analysis. Figure 7A,C displays the cluster analysis for the MHCI and MHCII alleles, respectively. In addition, Figure 7B,D demonstrates an advanced tree map of MHCI and MHCII cluster analysis. The red regions in the heatmap demonstrated stronger interactions between the clustering HLA alleles, whereas the red regions in the heatmap indicated weaker interactions between the clustering HLA alleles.
Figure 7.

Results of the MHC cluster analysis. Here, (A) heatmap of MHC class-I cluster analysis, (B) advanced tree map of MHC class-I cluster analysis, (C) heatmap of MHC class-II cluster analysis, and (D) advanced tree map of MHC class-II cluster analysis.
2.9. Formulation of the Vaccine Construct
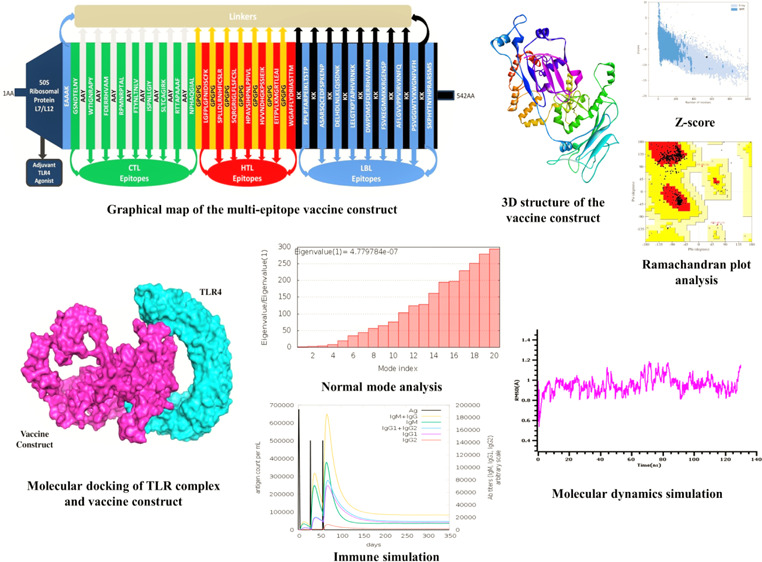
The vaccine construct was formulated by assembling the most promising LBL, CTL, and HTL epitopes with the assistance of KK, AAY, and GPGPG linkers, respectively. The adjuvant 50S ribosomal protein L7/L12 (NCBI ID: P9WHE3) was linked to the front of the vaccine with the EAAAK linker. In the present experiment of multi-epitope vaccine construction, a total of nine CTL epitopes, seven HTL epitopes, and nine LBL epitopes were chosen from the selected MARV structural protein sequences. After proper combination and randomization, we formulated a vaccine construct comprising 542 amino acid residues. The final vaccine construct is MAKLSTDELLDAFKEMTLLELSDFVKKFEETFEVTAAAPVAVAAAGAAPAGAAVEAAEEQSEFDVILEAAGDKKIGVIKVVREIVSGLGLKEAKDLVDGAPKPLLEKVAKEAADEAKAKLEAAGATVTVKEAAAKGSNDTELNYAAYWTIGNRAPYAAYFDERRHVAMAAYRPMNRPTALAAYFTYNLTNLVAAYISPNLLGIYAAYSLTCAGIRKAAYRTTAPAAAFAAYNPHAQGIALGPGPGLGFPLGFNIDISCFKGPGPGSPLLDLRNHFICSLRGPGPGSQRGRIGLFLSFCSLGPGPGHPAVSIHPNLPPIVLGPGPGHVVNDHGKPSSIEIKGPGPGEITPVLKMGRTLEAIGPGPGWGAFFLYDRIASTTMKKPPLPTARPEIKLTSTPKKASARSQCEDFSPKENPKKDELHDLNLKLQISDNKKKLELGTKPTAPHVRNKKKKDWPDMSFDERRHVAMNKKFSVKEGMMKKRGENSPKKAFLGVVPPVIRVKNFQKKPSVGGWTVKWGNFVFHKKSKPHYTNYHPRARSMS (Figure 8).
Figure 8.

Graphical map of the formulated multi-epitope vaccine construct. The vaccine constructs included (left to right) an adjuvant and CTL, HTL, and LBL epitopes, shown in the dark blue, green, red, and blue rectangular boxes, respectively. Herein, the adjuvant and the first CTL epitope were linked by the EAAAK linker (blue), CTL epitopes were added together by AYY linkers (off-white), HTL epitopes were linked by GPGPG linkers (dark yellow), and LBL epitopes were linked by KK linkers (black).
2.10. Antigenicity, Allergenicity, and Physicochemical Profile Evaluation
The evaluation of physicochemical characteristics of the formulated construct was performed. The chemical formula of the vaccine was found to be C2632H4161N721O743S16. The molecular weight was 58306.34 Da, indicating an average weight of the vaccine construct. The theoretical PI property indicated the alkaline nature of the vaccine, as it was 9.56. Additionally, the aliphatic index value was 76.47, and the value of the instability index was found as 33.08. The nature of the construct was hydrophilic given the value (−0.303) of the grand average of hydropathicity (GRAVY). We further utilized multiple servers to assess the allergenicity and antigenicity of the constructed vaccine. In Vaxijen2.0 and ANTIGENPro servers, the antigenicity score was found to be 0.7130 and 0.762192, respectively. Importantly, in all the servers, the vaccine was determined as antigenic and non-allergenic. Furthermore, the vaccine construct was predicted to be soluble by both Solpro and Protein-Sol servers, displaying a score of 0.928259 and 0.509, respectively. Also, the vaccine construct did not display any TM helices and signal peptides in their respective prediction results (Table 6 and Figure S4).
Table 6. Physicochemical, Antigenic, and Allergenic Characteristics of the Vaccine Construct.
| characteristics | findings | remarks |
|---|---|---|
| number of amino acids | 542 | suitable |
| molecular weight | 58306.34 Da | average |
| theoretical pI | 9.56 | basic |
| chemical formula | C2632H4161N721O743S16 | |
| extinction coefficient (at 280 nm in H2O) | 50,100 M–1 cm–1 | |
| estimated half-life (mammalian reticulocytes, in vitro) | 30 h | satisfactory |
| estimated half-life (yeast cells, in vivo) | >20 h | satisfactory |
| estimated half-life (E. coli, in vivo) | >10 h | satisfactory |
| instability index of vaccine | 33.08 | stable |
| aliphatic index of vaccine | 77.34 | thermostable |
| grand average of hydropathicity (GRAVY) | –0.303 | hydrophilic |
| antigenicity | 0.7130 (VaxiJen v2.0) 0.762192 (ANTIGENPro) | antigenic |
| allergenicity | no (AllerTop v2.0) No (AllergenFP v1.0) No (AlgPred) | non-allergen |
| solubility | 0.928259 (SolPro) 0.509 (Protein-Sol) | soluble |
| TM helices | no | suitable |
| signal peptides | no | suitable |
2.11. BLAST Homology Assessment
The sequence homology between the constructed vaccine protein sequence and the proteome sequence of the host revealed that the query coverage of the vaccine construct displayed only 22% homology to Homo Sapiens proteins. The BLAST homology assessment result indicated that the predicted vaccine protein would not cause autoimmune responses to the host.
2.12. Secondary Structure Prediction
The assessment of secondary structural features, including random coils, alpha-helix, and beta-turn, in the constructed vaccine was carried out using two servers. The SOPMA server predicted 36.72% α-helix, 18.08% β-strand, and 45.20% random coils (Table 7 and Figure S4), whereas the PSIPRED server predicted 34.32% α-helix, 14.94% β-strand, and 50.74% random coils in the final vaccine construct (Table 7 and Figure S4).
Table 7. Secondary Structural Features of the Vaccine Construct.
| SOMPA server |
PSIPRED server |
|||
|---|---|---|---|---|
| features | amino acid | percentage (%) | amino acid | percentage (%) |
| alpha-helix | 199 | 36.72 | 186 | 34.32 |
| beta-strand | 98 | 18.08 | 81 | 14.94 |
| random coil | 245 | 45.20 | 275 | 50.74 |
2.13. Homology Modeling, 3D Structure Refinement, and Validation
The Raptor-X server was employed to generate the 3D structure of the final vaccine. The server developed five models for the given construct. Afterward, ProSA and Procheck web servers were used to evaluate these models. In our present study, the pre-refined crude model 4 represented the highest Z-score of −6.66, and 91.7% residues were in the most favored regions (Figure S5). For refinement, model 4 was submitted to the GalaxyRefine server. This server also produces five models for the given crude model. After refinement, all the models showed improved Rama-favored regions compared with the initially submitted crude model. Among the generated models, model 4 was determined as the best refined model. It exhibited good GDT-HA (0.9156), rmsd (0.509), MolProbity (2.086), clash score (21.2), poor rotamers (0.7), and Rama-favored (96.1) scores in the GalaxyRefine server (Table S5). The refined models were further validated using ProSA and Procheck web servers. Here, model 4 possessed the best Z-score (−7.47) and delineated 93.2% amino acid residues in the most favored regions of the Ramachandran plot. Thus, in the present study, we have selected model 4 for further analysis (Figure 9).
Figure 9.

(A) 3D structure of the final vaccine construct, (B) Z-score of the vaccine construct predicted using the PROSA server, and (C) Ramachandran plot analysis of the vaccine construct.
2.14. Disulfide Bond Engineering of the Constructed Vaccine
In disulfide engineering, a total of 45 pairs of amino acid residues were determined as suitable for generating disulfide mutation via the Disulfide by Design v2.12 web server (Table S10). After evaluating the χ3 angle and the energy score, only three pairs of amino acids were finally selected as their value fulfills the following criteria, that is, χ3 angle should range between −87 and +97°, and the energy score should not exceed 2.2 kcal/mol. Thus, six mutations were produced on the residue pairs Leu269-Arg272, Gly283-Gly365, and Asp330-Lys333, for which the χ3 angles were +81.84, −86.62, and +89.66°, while the energy scores were 1.08, 1.49, and 1.57 kcal/mol, respectively (Figure 10 and Table S6).
Figure 10.

Stability of the vaccine construct by disulfide bond engineering in (A) original form and (B) mutant form. Three pairs of amino acids represented in the yellow stick (shown in a black circle).
2.15. Screening for Conformational B-Cell Epitopes
In the vaccine construct sequence, eight conformational B-cell epitopes were identified using the ElliPro tool (Figure 12). The number of amino acid residues ranged from 4 to 86, and the total number of residues was 294, displayed in Table 8. In addition, the predicted conformational B-cell epitope score ranged from 0.518 to 0.921.
Figure 12.

Conformational B-cell epitopes present in the vaccine. The light-yellow spheres showing epitopes containing (A) 4 residues (AA539-542) with 0.921; (B) 42 residues (AA1-32 and AA 34-43) with 0.849; (C) 39 residues (AA64-97 and AA99-103) with 0.77; (D) 13 residues (AA51-63) with 0.734; (E) 86 residues (AA393-427, AA439-460, AA471, AA497-500, AA502-513, AA517-522, AA531, and AA534-538) with 0.696; (F) 30 residues (AA219-239 and AA241-249) with 0.655; (G) 75 residues (AA259-270, AA274, AA297-356, and AA361-362) with 0.616; and (H) 5 residues (AA283-287) with 0.518 scores.
Table 8. Predicted Conformational B-Cell Epitopes of the Constructed Vaccine.
| no. | residues | no. of residues | score |
|---|---|---|---|
| 1 | A:R539, A:S540, A:M541, A:S542 | 4 | 0.921 |
| 2 | A:M1, A:A2, A:K3, A:L4, A:S5, A:T6, A:D7, A:E8, A:L9, A:L10, A:D11, A:A12, A:F13, A:K14, A:E15, A:M16, A:T17, A:L18, A:L19, A:E20, A:L21, A:S22, A:D23, A:F24, A:V25, A:K26, A:K27, A:F28, A:E29, A:E30, A:T31, A:F32, A:V34, A:T35, A:A36, A:A37, A:A38, A:P39, A:V40, A:A41, A:V42, A:A43 | 42 | 0.849 |
| 3 | A:D64, A:V65, A:I66, A:L67, A:E68, A:A69, A:A70, A:G71, A:D72, A:K73, A:K74, A:I75, A:G76, A:V77, A:I78, A:K79, A:V80, A:V81, A:R82, A:E83, A:I84, A:V85, A:S86, A:G87, A:L88, A:G89, A:L90, A:K91, A:E92, A:A93, A:K94, A:D95, A:L96, A:V97, A:G99, A:A100, A:P101, A:K102, A:P103 | 39 | 0.77 |
| 4 | A:G51, A:A52, A:A53, A:V54, A:E55, A:A56, A:A57, A:E58, A:E59, A:Q60, A:S61, A:E62, A:F63 | 13 | 0.734 |
| 5 | A:K393, A:L394, A:T395, A:S396, A:T397, A:P398, A:K399, A:K400, A:A401, A:S402, A:A403, A:R404, A:S405, A:Q406, A:C407, A:E408, A:D409, A:F410, A:S411, A:P412, A:K413, A:E414, A:N415, A:P416, A:K417, A:K418, A:D419, A:E420, A:L421, A:H422, A:D423, A:L424, A:N425, A:L426, A:K427, A:L439, A:G440, A:T441, A:K442, A:P443, A:T444, A:A445, A:P446, A:H447, A:V448, A:R449, A:N450, A:K451, A:K452, A:K453, A:K454, A:D455, A:W456, A:P457, A:D458, A:M459, A:S460, A:K471, A:P497, A:P498, A:V499, A:I500, A:V502, A:K503, A:N504, A:F505, A:Q506, A:K507, A:K508, A:P509, A:S510, A:V511, A:G512, A:G513, A:K517, A:W518, A:G519, A:N520, A:F521, A:V522, A:Y531, A:Y534, A:H535, A:P536, A:R537, A:A538 | 86 | 0.696 |
| 6 | A:Y219, A:R220, A:T221, A:T222, A:A223, A:P224, A:A225, A:A226, A:A227, A:F228, A:A229, A:A230, A:Y231, A:N232, A:P233, A:H234, A:A235, A:Q236, A:G237, A:I238, A:A239, A:G241, A:P242, A:G243, A:P244, A:G245, A:L246, A:G247, A:F248, A:P249 | 30 | 0.655 |
| 7 | A:F259, A:K260, A:G261, A:P262, A:G263, A:P264, A:G265, A:S266, A:P267, A:L268, A:L269, A:D270, A:H274, A:F297, A:C298, A:S299, A:L300, A:G301, A:P302, A:G303, A:P304, A:G305, A:H306, A:P307, A:A308, A:V309, A:S310, A:I311, A:H312, A:P313, A:N314, A:L315, A:P316, A:P317, A:I318, A:V319, A:L320, A:G321, A:P322, A:G323, A:P324, A:G325, A:H326, A:V327, A:V328, A:N329, A:D330, A:H331, A:G332, A:K333, A:P334, A:S335, A:S336, A:I337, A:E338, A:I339, A:K340, A:G341, A:P342, A:G343, A:P344, A:G345, A:E346, A:I347, A:T348, A:P349, A:V350, A:L351, A:K352, A:M353, A:G354, A:R355, A:T356, A:G361, A:P362 | 75 | 0.616 |
| 8 | A:G283, A:P284, A:G285, A:S286, A:Q287 | 5 | 0.518 |
2.16. Molecular Docking of the Vaccine Construct and TLR Complex
For an effective and stable immune response, the interaction between the immune cell and the vaccine construct is required. The Cluspro program prepares 30 different clusters, which have higher interaction energies. The top five clusters had an energy of −1034.9, −859.9, −812, −723, and −716.3 kcal/mol. Based on the energy score, the first cluster was selected, which had better energy (−1034.9 kcal/mol) than other clusters (Figure 11).
Figure 11.
Molecular docking of the TLR complex and vaccine construct.
2.17. Normal-Mode Analysis
The normal-mode analysis of the vaccine complexes was conducted in iMODS tools where multiple descriptors were analyzed to explain the stability and the physical movements of the atoms from the vaccine molecules. From Figure 13B, it can be depicted that the peaks in the graph represent the deformability region in the protein. The B factor graph indicates the visualizations and comparison between the NMA and PDB fields of the complexes (Figure 13C). The eigenvalue graph of the complexes illustrated in Figure 13D showed that the complex had an eigenvalue of 4.779784 × 10–7. Figure 13E illustrates the individual variance as red color, whereas the green color represents the cumulative variance. The covariance map is depicted in Figure 13F, where the red color represents the correlated motion between pairs of residues, white color indicates the uncorrelated motion, and the anticorrelated motion is indicated by blue color. The elastic maps of the complexes represented in Figure 13G represent the connection between atoms, and darker gray indicates the stiffer complex.
Figure 13.

Normal-mode analysis of the vaccine protein, (A) NMA mobility, (B) deformability, (C) B-factor, (D) eigenvalues, (E) variance (red color indicates individual variances, and green color indicates cumulative variances), (F) covariance map (correlated (red), uncorrelated (white) or anti-correlated (blue) motions), and (G) elastic network.
2.18. In Silico Immune Simulation
The immune simulation study was conducted for the vaccine complexes to explore the generation of adaptive immunity and also the immune interactions. The immune simulation study illustrated that after every injection dose, the primary immune response was increased significantly as gradual elevation or decrease rates of the different immunoglobulins were observed. Moreover, the secondary immune response was also increased (Figure 14A). The increasing rate of active B-cells (Figure 14B,C), plasma B-cell (Figure 14D), helper T-cells (Figure 14E,F), and regulatory and cytotoxic T cells (Figure 14G–I) was observed.
Figure 14.

Immune simulation of the predicted vaccine; (A) immunoglobulin and immunocomplex response to the antigen, (B) B lymphocyte total count, (C) B lymphocyte population per entity state, (D) plasma B lymphocyte count, (E) CD4 T-helper lymphocyte count, (F) CD4 T-helper lymphocyte count subdivided per entity state, (G) CD4 T-regulatory lymphocyte count, (H) CD8 T-cytotoxic lymphocyte count, (I) CD8 T-cytotoxic lymphocyte count per entity state, (J) natural killer cell population, (K) dendritic cell per state, and (L) macrophage population per state.
These results indicate the after every injection, strong secondary immune response, increasing clearance of antigens, and strong immune memory generation occur. Moreover, the good antigen presentation was also observed by these antigen-presenting cells from dendritic cell and macrophage cell concentrations (Figure 14J,K). The vaccine protein was also capable of forming a vast number of different types of cytokines (Figure 14L).
2.19. Molecular Dynamics Simulation
The molecular dynamics simulation study was conducted to understand the stable nature of the TLR-4 complexes and the constructed vaccine by analyzing multiple descriptors from the simulation trajectories. The rmsd from the C-alpha atoms of the complexes was analyzed to understand the stable nature of the complexes. From Figure 15A, it can be observed that the rmsd profile was lower at the initial phases, and then, it began to rise till 20 ns. The complexes had a moderate level of fluctuations in the rmsd profile at 40–45 and 70–80 ns simulation times, but the degree of the deviations was little. The whole rmsd profile from the beginning till the last phases of times was below 2.5 Å, which defines the rigidness of the complexes.
Figure 15.

Molecular dynamics simulation of the vaccine and TLR-4 complexes, (A) root mean square deviation, (B) solvent-accessible surface area, (C) radius of gyration, (D) hydrogen bond analysis from the simulation system, and (E) root mean square fluctuations of the amino acid residues.
Moreover, the SASA of the vaccine and TLR-4 complexes was analyzed to understand the change in the protein surface volumes. A higher SASA value defines the expansion of the protein surface, whereas a lower SASA value defines the truncated nature of the proteins. Figure 15B demonstrates that the complexes had a little upper trend from the beginning to 45 ns, which indicates the extension of the protein surface. Then, the vaccine and TLR-4 complexes reached a steady state and maintained a similar trend till the end. Although the SASA trend had some deviations, they are not plentiful.
The radius of gyration profile from the simulation complexes was also analyzed to examine the mobility and overall flexibility of the complexes. The higher Rg profile is linked with the folding or defolding nature of the complexes. Figure 15C illustrates that the complex had higher aberrations from 0 to 40 ns, which indicates the labile nature of the complexes. Therefore, the complexes were stabilized after then and maintain the firm profile. Also, the hydrogen bond of the complexes outlines the constant and steady nature of the complexes. Figure 15D specifies that the vaccine and TLR-4 complexes had stable and lower aberrations in the hydrogen bond during the whole simulation. Besides, the RMSF of the complexes was analyzed to recognize the flexibility across the amino acid residues from the complexes. Figure 15E indicates that maximum residues had a lower RMSF and higher level of stability, as they had an RMSF of below 2.5 Å.
3. Discussion
Conventional or traditional methods for vaccine development include whole organisms that may bring about unwanted exposure to antigens and might cause allergic reactions. Peptide vaccines that contain short fragments of immunogenic peptides create powerful and targeted immune responses are the candidates to solve this problem by escaping the risk of allergic reactions. Vaccinations are now gaining popularity against infectious diseases and prove highly effective against yellow fever, chickenpox, rabies, mumps, Japanese encephalitis, rubella, smallpox, influenza, and hepatitis A and B.36 Both preclinical and clinical study phases of vaccine development involve complex, laborious, and expensive in vivo and in vitro protocols for assuring vaccine efficacy.37 Present-day progress in computational biology and immunoinformatics lessens the degree of in vitro experiments and paves the way to design effective in silico vaccines.38,39 Importantly, designing a multi-epitope vaccine against Mycobacterium tuberculosis was validated experimentally through an in vitro study.40 The main advantage of this process is that it can identify a wide range of vaccine candidates without involving the propagation of pathogenic organisms in the wet laboratory.37 Vaccines against multiple viruses such as Chikungunya, Ebola, SARS-CoV-2, MERS-CoV, HIV, Lassa, Oropouche, Cytomegalovirus, Dengue, Hepatitis C, Zika, Flavivirus, Norovirus, and so on are some of the promising examples that applied vaccinomics to design a multi-epitope-based vaccine model.38,41
Despite MARV being considered to be less intimidating, this thought was needed to be re-evaluated, as MARV reappeared in two substantial outbreaks; one in the Democratic Republic of Congo (DRC) during the period of 1998–2000 and the other in Angola during the period of 2004–2005, where it was identified for the first time.42,43 The high fatality rate of 90% in Angola and 83% in DRC opened a discussion for scientists for considering it as threatening as EBOV.44,45 Recently, in Guinea, a MARV case has been detected on August 9, 2021, indicating the existing threat of the deadly virus.4 Because of its potentiality to offer a serious threat to the safety and public health, the Centre for Disease Control and Prevention (CDC) classified it as a select agent. Additionally, the National Institute of Allergy and Infectious Diseases (NIAID) classified it as a “category A pathogen”, and the World Health Organization (WHO) declared it as a “risk group 4” agent.22
MARV enters the bloodstream or lymphatic system when exposed to mucous membranes or abrasive skin or by needle injections or other penetrating wounds and infects macrophages, monocytes, and dendritic cells. The fast replication in these cells further spreads the virus to hepatocytes, fibroblasts, and endothelial and epithelial cells.46 The virus does not directly infect the lymphocytes, but the apoptosis of T-lymphocyte cells and killer cells induces large-scale lymphoid cell depletion in the liver, spleen, thymus, and lymph nodes, which negatively impacts the induction of adaptive immune response.22,46 The clinical experience of research has shown that no effective treatments such as drugs and vaccines have been developed against the pathogenic MARV. Because no cure exists for this virus, the high mortality rate makes therapeutic identification a top priority. As the Marburg virus is a potential pathogen and outbreaks happen occasionally, it is important to take proper precautionary steps for preventing human death.47 Thereupon, developing vaccines against this pathogenic virus at present is a key challenge.
We retrieved all seven structural proteins of MARV from the UniProt database. The protein sequences were then submitted to the VaxiJen v2.0, AllergenFP v1.0, and TMHMM v2.0 servers to check the antigenicity, allergenicity, and TM helices of the structural proteins, respectively. Generally, proteins displaying >1 TM helices are not considered suitable vaccine targets, as these proteins are difficult to purify and ineffective in cloning and expression. Therefore, we did not utilize these proteins in vaccine design.48,49 Importantly, all the protein sequences were determined as antigenic and non-allergenic and contained a suitable number of TM helices. For each structural protein, all types of epitopes were primarily screened based on their antigenicity, allergenicity, and toxicity profile. Most vaccines nowadays produce immune system stimulation and create allergic reactions. As per the WHO/FAO, a sequence is considered potentially allergenic, if its sequence has at least six contiguous amino acid identities within the range of 80 amino acids (sequence identity of 0.35%) with a known allergen. Therefore, this study allowed only the non-allergenic epitopes from each structural protein for further analysis.50 Based on the C-score provided by the NetCTL v1.2 server and IEDB MHC class-I binding result, two CTL epitopes from each of the NP and L protein and one CTL epitope from each of the remaining five proteins were finally selected. After determining the aforementioned parameters, conservancy was also considered here as a key determinant for the prediction of the best possible epitopes under the selected range. Importantly, all the shortlisted CTL and HTL epitopes displayed 100% maximum identity for conservancy. WTIGNRAPY, NPHAQGIAL, and RTTAPAAAF epitopes interacted with the HLA-B*35:01 allele. Among them, two CTL epitopes, namely, WTIGNRAPY and RTTAPAAAF, strongly interacted with the HLA-B*35:01 allele and displayed better docking scores than the respective positive control. In a previous study of MARV, the WTIGNRAPY epitope of the L protein was reported as a potential CTL epitope for computational vaccine designing.1 ISPNLLGIY, FTYNLTNLV, and GSNDTELNY interacted with the HLA-A*01:01 allele, where all the epitopes displayed almost similar docking scores (7.7–7.8 kcal/mol), and the amino acid residues (Thr73, Asn77, Asp116, Lys146, Trp147, and Arg156) involved in the hydrogen bond formation were almost similar to those of the control. In the case of the NP protein, both FDERRHVAM and RPMNRPTAL epitopes interacted with the HLA-B*14:02 allele. Therefore, we determine the docking interaction of these epitopes toward the HLA-B*14:02 allele. Although the positive control of the HLA-B*14:02 allele surpasses the binding score produced by both FDERRHVAM and RPMNRPTAL epitopes, the number of hydrogen bonds formed by these epitopes was greater compared with that of the positive control. The SLTCAGIRK epitope of the VP30 protein interacted with the HLA-A*03:01 allele and displayed Asn66, Glu152, and Gln155 residues in the hydrogen bond interaction, which is similar to the positive control of the allele. Apart from the control of the HLA-B*14:02 allele, other respective positive controls displayed significant interactions toward the targeted alleles.
In order to elicit a prolonged and significant immune response, the engagement of both B-cells and T-cells is needed to stimulate and attach both humoral and cellular immunity, respectively. There is another reason behind the selection of the two types of episodes, that is, antigens can frequently avoid memory B-cells where their detection is covert. Here, in the prediction of B-cell epitopes, only LBL epitopes were involved in the vaccine construct because of their better stability than conformational epitopes. It is obligatory that the B-cell epitope of a target molecule should combine with a T-cell epitope, so that a peptide vaccine can be considerably immunogenic. In the prediction of T-cells, both CD8+ (MHCI) and CD4+ (MHCII) were predicted to obtain a great number of prospective epitopes that will have the ability to prompt a more significant immune response.28,51
The vaccine protein has some important characteristics like that it must not offer notable homology or sameness to the host proteins as this kind of similarity with the host proteome can lead to autoimmune diseases because there are possibilities of cross-reactivity and molecular mimicry.52−54 For an epitope to become antigenic and elicit a robust immune response, it also must be recognized by MHC complex molecules. In humans, the MHC alleles are reported to be the most polymorphic and present in thousands of HLA combinations. Consequently, high-frequency HLA alleles in most of the world’s population are likely to have immunogenic effects.55 Our constructed vaccine has covered 97.92% of the world’s population, indicating significant global population coverage of the vaccine construct. The vaccine construct in this study showed less similarity (22%) to the human proteins applying the BLASTp tool, offering the vaccine as a suitable candidate with no autoimmunity. Due to the importance of the MHC superfamily in the construction of a vaccine and drug development, MHC cluster analysis has been done. This determined the functional relationship between the MHCI and MHCII clustering variants.50
Vaccine efficacy can be enhanced in multiple ways from increasing the response of the immune system to facilitating vaccine transport and increasing its exposure duration. In silico vaccines possess somewhat low immunogenicity in comparison with live attenuated vaccines, which is a vital demerit for them. Adjuvants are frequently used to eradicate this problem. Therefore, adjuvants have been utilized extensively to enhance the efficacy of vaccines. Adjuvants function by following the principle of creating a deposit of the antigenic compound at the site of the vaccine that is steadily released over time, elongating the robust immune response.56 Linkers were incorporated to assist each epitopes’ functional preservation.57 However, flexibility and structural rigidity are important parameters regulated by the linkers. For multi-epitope vaccines, the use of linkers provides major advantages, causing a reduction in the junctional antigen formation probability and improving the antigen presentation and processing.58 The 50S ribosomal protein (L7/L12) functions as a TLR4 agonist.59 EAAAK, an empirical α-helical linker, has been used to enhance the bi-functional catalytic activity, provide stiffness, and improve fusion protein stability.60 The GPGPG linker was used based on the HTL immune response-inducing ability and was designed as a universal spacer with the ability to break the junctional immunogenicity, leading to individual epitopes’ restoration of immunogenicity.61,62 This was explained experimentally in mouse models by Livingstone and his collaborators.62 In mammalian cells, AAY linkers played their role in the cleavage site for proteasomes and also reduced the junctional immunogenicity.63,64 When linked with T-cell epitopes, these linkers (GPGPG and AAY) allow recognition and separation of each epitope.65 KK, a bi-lysine basic linker, was used to conserve the independent immunogenic function of LBL epitopes and bring the pH value close to the physiological range.29,66 It was reported that the 50S ribosomal protein L7/L12 can induce the maturation of dendritic cells (DC) following the activation of naive T-cells and lead to the formation of CD4+-, CD8+-, and IFN-γ-secreting cells.59
The constructed vaccine was determined to be highly antigenic and non-allergenic by multiple servers, which reveals its capability of triggering robust immune responses without causing any unwanted allergic reaction. Moreover, the molecular weight of the vaccine construct was an average value (58.3 kDa), which further indicated its good antigenic nature.67 Proteins having less than 110 kDa molecular weight are considered as suitable vaccine candidates.68 The theoretical isoelectric point (pI) of the vaccine protein is 9.56 (basic in nature), indicating the stable interaction inside the human body. The short half-life of peptides is considered as one of the major limitations in the therapeutic protein field.69 The vaccine construct displayed a half-life of more than 10 and 20 h in E. coli (in vivo) and yeast cells (in vivo), respectively, whereas 30 h half-life was predicted in mammalian reticulocytes (in vitro). These half-life results are satisfactory based on previous findings.67,70 Thermostability of a protein has a direct correlation with the occupied aliphatic amino acid side chains in the protein structure. The aliphatic index (AI) of the vaccine was 77.08, which ensured the thermostability of the vaccine protein at normal body temperature.71 The instability index was recorded as 33.08 for the final vaccine protein, indicating the stability of the vaccine in biological environments, as an instability index (II) of less than 40 for a compound is considered as stable.72 The negative GRAVY value indicated the hydrophilic nature of the vaccine. The strong affinities for water molecules aid in ease of formulation and purification of the constructed vaccine.73,74 For many functional and biochemical analyses, the solubility of the protein undergoing over-expression in the E. coli host is significant.75 In both Protein-Sol and SOLpro servers, the vaccine protein was found to be soluble upon over-expression in E. coli. This demonstrates better post-production results, as highly soluble proteins during downstream processing demonstrate ease of purification.76 The final vaccine construct also did not present any TM helices and signal peptides, which further validates its ease of purification and expression.77 Six mutations were produced on the residue pairs in structural disulfide engineering, which plays a major role in protein folding and stability. Additionally, disulfide engineering reduces the probable number of conformations, resulting in an increased thermal stability and decreased entropy.75
After 3D structure prediction and refinement of the vaccine model, a significant Z-score of 7.47 was determined, and the Ramachandran plot showed most amino acid residues (93.2%) in the preferable region. To predict the molecular interaction between the constructed vaccine and TLR4, molecular studies were used, and good interactions were found with a global binding score of −1034.9 kcal/mol. TLRs belong to the conserved pattern recognition receptor (PPR) family that are responsible for identifying specific pathogen patterns, including bacteria, viruses, or fungi, and are able to distinguish themselves from foreign substances.78 The cytokine production and MHC molecules are upregulated upon TLR activation by specific ligands, resulting in linking of the adaptive immune response with the innate immune system.79 TLRs have the resemblance of the cytoplasmic region to the IL-1 receptor family; nevertheless, there is a structural difference in extracellular portions. Both the agonist and antagonist of TLRs have been proposed as broad therapeutic potential compounds against respiratory diseases with the association of antiviral agents and vaccine adjuvants.80,81 TLR4 is unique among the TLRs, as it requires myeloid differentiation factor 2 (MD-2), an accessory protein, to respond. It is a major receptor that triggers non-infectious and infectious stimulation, which induces a pro-inflammatory response. Since TLR4 signaling relies on TLR4/MD-2 complex formation, the interface of TLR4 and MD-2 can be targeted to render a new strategy for therapy development.82 TLR4 plays a critical role in inflammatory response amplification and microbial lipopolysaccharide (LPS) recognition.83,84 To enhance the interaction of the constructed vaccine with the TLR4 agonist, a molecular docking study was performed to determine the binding affinity between TLR4 chain A and the final construct. The docking study resulted in a negative value of binding energy, which demonstrated the high binding affinity between the vaccine construct and TLR4 chains. Thus, these interactions with TLR4 are specialized to potentially elicit a protective immune response.
To generate a strong immune response, the vaccine construct should possess a strong immune response within the host. Therefore, after injections of the vaccine, the primary immune response was triggered, and the secondary immune response associated with the constructed vaccine injections was also activated. The increased concentration of multiple immune cells, memory B-cells, plasma B-cells, helper T-cells, cytotoxic T-cells, and multiple types of antibodies indicates the elevated amount of immune response after the vaccine injections.85 Moreover, diverse immune responses can be exhibited from the lower Simpson index, which was found for the constructed vaccine in immune simulation.86 Moreover, the normal-mode analysis from iMODS revealed that the vaccine complex had a higher level of eigenvalues, which defines the lower degree of deformability. Also, the deformability graph indicates that the vaccine construct might have a higher chance of complex rigidity. Furthermore, atomistic simulation can provide information about the stability and conformation variations about the biological systems. Moreover, multiple descriptors from the molecular dynamics simulation trajectories allow the study of the conformational variations and stability more precisely.87 The rmsd, Rg, SASA, and RMSF of the vaccine complex had a steady profile and exhibited a lower degree of fluctuation. These trends in molecular dynamics simulations define the stable nature of the complexes.
4. Conclusions
Multi-epitope vaccines may be regarded as a promising strategy for therapy against viral infections and tumors. The recent advancement of various computational biology and immunoinformatics tools made it plausible to design and develop multi-epitope vaccines in a less-time-consuming and cost-effective manner. This study designed and constructed a multi-epitope vaccine targeting MARV structural proteins. As viruses can trigger the cellular and humoral immune system, stimulation of these particular immune responses might serve to control viral infection. This vaccine was developed with the combination of potential LBL, CTL, and HTL epitopes. At first, reviewing the screening result of antigenicity, toxicity, and allergenicity analysis of these epitopes, we applied an adjuvant and different linkers to attach the shortlisted epitopes. The physicochemical profile and 3D structure validation of our constructed vaccine also represented positive results toward the selected parameters. The final construct exhibited greater binding affinity with the TLR4 receptor in the molecular docking study. Importantly, the descriptors of normal-mode analysis, immune simulation, and molecular dynamics simulation studies were also satisfactory. Although the vaccine showed significant in silico results in our present study, this preliminary-designed vaccine is not exhaustive and requires further in vivo and in vitro experiments to evaluate the constructed vaccine’s effectiveness. We are hopeful that our predicted vaccine model will exhibit positive effects in wet laboratory verification and facilitate the future treatment of MARV infection.
5. Materials and Methods
In the current experiment, the methodologies we applied to design an in silico vaccine candidate against structural proteins of MARV included a minor modification from our previous articles.23,88,89 The schematic representation of the overall workflow applied in the present investigation is displayed in Figure 1.
Figure 1.

Schematic representation of the overall workflow applied in the multi-epitope-based vaccine design from MARV structural proteins.
5.1. Retrieval and Analysis of Protein Sequences
The Marburg virus (Marburg marburgvirus) Musoke-80 strain was selected from the National Center for Biotechnology Information (NCBI) database (https://www.ncbi.nlm.nih.gov/).90 ViralZone, an online resource of the Swiss Institute of Bioinformatics (https://viralzone.expasy.org/), was utilized to further analyze the genus, genome, and proteome of the selected strain.91 The UniProt database (https://www.uniprot.org/) was used to retrieve the entire viral proteome of this strain. UniProt is an established database providing a large amount of biological protein information.92 The selected protein sequences were extracted in the FASTA format. Afterward, the VaxiJen v2.0 web server (http://www.ddg-pharmfac.net/vaxijen/) was employed to find the antigenicity of the proteins using the default threshold value for viruses. This server uses an auto cross-covariance (ACC) transformation method that produces 70–89% accuracy in the prediction result.93,94 Then, allergenic profiles of the selected proteins were determined using the AllergenFP v1.0 (https://ddg-pharmfac.net/AllergenFP/) server. This server utilizes a novel alignment-free descriptor-based fingerprint approach with 88.9% accurate prediction.95 For transmembrane (TM) helix prediction, the TMHMM v2.0 server (http://www.cbs.dtu.dk/services/TMHMM/) based on the hidden Markov model (HMM) was utilized.96 Finally, structural proteins which are antigenic and non-allergenic and display less TM helices were selected for the next step of investigation.
5.2. Prediction and Evaluation of Cytotoxic T-Lymphocyte (CTL) Epitopes
Cytotoxic T-lymphocytes (CTLs) play a significant role in the host defense mechanism. These cells can directly interact and kill the infectious cells in the immune system.97 The NetCTL v1.2 server (http://www.cbs.dtu.dk/services/NetCTL/) is reported to demonstrate high predictive performance, which uses artificial neural networks and weight matrix to predict 9-mer CTL epitopes against 12 supertypes (A1, A2, A3, A24, A26, B7, B8, B27, B39, B44, B58, and B62). This server was utilized to anticipate the CTL epitopes with high combined scores among the selected protein sequences, using a threshold value of 0.90 to maintain a sensitivity of 0.74 and specificity of 0.98.98 The MHC-I binding tool of the IEDB resource (http://tools.iedb.org/mhci/) was used to determine the MHC-I binding alleles for each CTL epitope depending on the CONSENSUS method. A percentile rank score of equal to or less than 2 was considered to narrow down CTL epitopes, as a lower rank indicates higher affinity.99 Then, antigenicity, the allergenic profile, toxicity prediction, and immunogenicity were characterized for individual CTL epitopes using the VaxiJen v2.0 server (http://www.ddg-pharmfac.net/vaxijen/),93 AllerTOP v2.0 server (https://www.ddg-pharmfac.net/AllerTOP/),100 ToxinPred server (http://crdd.osdd.net/raghava/toxinpred/),101 and IEDB Class I Immunogenicity tool (http://tools.iedb.org/immunogenicity/),102 respectively. The AllerTop v2.0 server applies k-nearest neighbor (kNN) methods, amino acid descriptors, and ACC transformation methods to isolate the non-allergens from allergens with 85.3% prediction accuracy at fivefold cross-validation.100 The ToxinPred server evaluates properties of different peptides by applying support-vector machines (SVM), a machine learning approach together with a quantitative matrix for predicting toxicity.101 Immunogenicity prediction was executed to confirm whether a specific epitope will generate an immunogenic response. CTL epitopes that are antigenic, non-allergenic, non-toxic, and immunogenic and display high C-scores are considered for vaccine construction.
5.3. Prediction and Assessment of Helper T-Lymphocyte (HTL) Epitopes
Helper T cells (HTLs) are components of adaptive immunity that can recognize foreign antigens and activate B-cells and CTLs to destroy infectious pathogens.97 The MHC-II binding tool of the IEDB resource (http://tools.iedb.org/mhcii/) was used for the prediction of 15-mer HTL epitopes among the selected protein sequences.103 To predict the respective binding alleles of the proteins, we used the CONSENSUS method where a percentile rank threshold of equal to or less than 2 was taken into consideration to retain consistency.104 Then, allergenicity, antigenicity, and toxicity were predicted for individual HTL epitopes using the AllerTOP v2.0 server (https://www.ddg-pharmfac.net/AllerTOP/),100 VaxiJen v2.0 server (http://www.ddg-pharmfac.net/vaxijen/),93 and ToxinPred server (http://crdd.osdd.net/raghava/toxinpred/), respectively.101 Antigenic, non-allergenic, and non-toxic HTL epitopes were further evaluated by considering their cytokine-inducing abilities. IFN-γ can trigger both specific and native immune responses through macrophages and natural killer cell activation. It also augments MHC response to antigens and plays a major role in detaining viral replication.38,105 We applied the IFNepitope server (http://crdd.osdd.net/raghava/ifnepitope/) that provides 81.39% accuracy for the prediction of interferon-gamma (IFN-γ)106 Also, the interleukin-4 (IL-4)-inducing property was evaluated using the IL4pred server (https://webs.iiitd.edu.in/raghava/il4pred/) with a threshold of 0.2, and for the interleukin-10 (IL-10)-inducing property, the IL10pred server (https://webs.iiitd.edu.in/raghava/il10pred/) was used with a threshold value of −0.3. Both operations were executed by applying SVM-based methods. IL4pred and IL10pred servers provide 75.76 and 81.24% accuracy, respectively.107,108 We prioritized the induction abilities of all three cytokines in HTL epitope selection for vaccine construction. For those proteins, where none of the epitopes displayed all three cytokine-inducing functions, we prioritized IFN-γ induction along with either IL4- or IL10-inducing abilities of the HTL epitopes.41
5.4. Prediction and Assessment of Linear B-Lymphocyte (LBL) Epitopes
B-cell epitopes are key mediators for inducing antibody-mediated or humoral immunity. The ABCpred server (https://webs.iiitd.edu.in/raghava/abcpred/), the first developed server built on a recurrent neural network, was used to predict the LBL epitopes among the selected protein sequences.109,110 The VaxiJen v2.0 server (http://www.ddg-pharmfac.net/vaxijen/),93 AllerTOP v2.0 server (https://www.ddg-pharmfac.net/AllerTOP/),100 and ToxinPred server (http://crdd.osdd.net/raghava/toxinpred/)101 were utilized to evaluate the predicted LBL epitopes’ antigenic, allergenic, and toxic profiles respectively. In addition, the iBCE-EL server (http://www.thegleelab.org/iBCE-EL/) was applied to predict the probability scores of LBLs using default parameters.111
5.5. Epitope Conservancy and Human Homology Determination
The conservancy analysis of the shortlisted MHC-I and MHC-II epitopes was carried out using the “epitope conservancy analysis” tool (https://www.iedb.org/conservancy/) of the IEDB resource. This property indicates the availability of an epitope in a range of different strains. For final vaccine construction, those epitopes found with 100% maximum identity in the conservancy analysis were selected.112 The epitope homology to the human proteome was evaluated to indicate which epitope is homologous to the human proteome. Only non-homologous epitopes were taken since homologous epitopes may lack the ability to elicit a possible immune response. To determine the human homology, the BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) tool’s protein BLAST module was used. In this experiment, the comparison was done with default parameters with Homo sapiens (taxid: 9606), and the threshold e-value was set to 0.05. Epitopes were determined as a non-homologous peptide when no hits under the threshold e-value were found.113,114
5.6. Peptide Modeling and Molecular Docking Studies
The selected CTL epitopes were docked with the HLA binding allele to evaluate the binding capability that becomes part of the vaccine design. The targeted CTL epitopes were modeled using the sOPEP sorting scheme by applying 200 simulations in the PEP-FOLD v3.5 server (https://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-FOLD3/). This server uses a de novo approach to predict the small peptide conformations (5–50 amino acids). It applies the Taboo/Backtract sampling algorithm for predicting conformations.115 Five probable structures were predicted using this server for each peptide sequence. SWISS-PDB VIEWER was used to determine the energy of each structure, and the structure displaying the lowest energy was taken for further analysis.116 The human alleles HLA-A*01:01 (PDB ID: 6AT9), HLA-B*35:01 (PDB ID: 1A1N), HLA-A*03:01 (PDB ID: 6O9B), and HLA-B*14:02 (PDB ID: 3BXN) were selected for CTL epitopes, and the co-crystallized ligand epitopes were selected as positive controls for the validation of the docking approach. The RCSB Protein Data Bank (https://www.rcsb.org/) was used to download the structure of HLA alleles in the pdb format with co-crystallized ligands.117 At first, the protein preparation wizard of UCSF Chimera (version 1.11.2) was used for the preparation of proteins by removing the ligands attached to the structure followed by the addition of hydrogens and Gasteiger–Marsili charges.118 Then, OpenBabel was used to convert this file into the pdbqt format.119 After that, the ligand energy form was minimized and converted to the pdbqt format using the OpenBabel module of PyRx 0.8. The docking parameters were kept default in the present operation. The grid box size for HLA-A*01:01, HLA-B*35:01, HLA-A*03:01, and HLA-B*14:02 in AutoDock Vina was set at 60.64 × 73.76 × 45.49 Å, 62.33 × 45.41 × 70.54 Å, 73.20 × 52.65 × 53.24 Å, and 61.25 × 48.69 × 72.95 Å, for X, Y, and Z axes, respectively. The results of docking simulation are represented as negative scores in kcal mol–1, and lower docking scores present stronger binding affinity.120 The visualization of the molecular docking analyses was done using Discovery Studio (DS) version 4.5, and figures were generated using UCSF Chimera and Microsoft PowerPoint 2019.118,121
5.7. Population Coverage Analysis
The expression and distribution of the HLA allele vary by race and region around the world.122 It may impact the development of a successful epitope-based vaccine. To estimate the populace coverage of the vaccine to be designed, the IEDB population coverage tool (http://tools.iedb.org/population/) was implemented. For this, we examined the shortlisted CTL and HTL epitopes along with their corresponding HLA binding alleles of both MHC class I and MHC class II separately and combined.123 In our present study, we emphasized the overall world coverage of the alleles and parts of different continents including Central America, Central Africa, Europe, East Asia, East Africa, North America, Northeast Asia, North Africa, Oceania, South America, South Africa, South Asia, Southwest Asia, Southeast Asia, West Indies, and West Africa.
5.8. MHC Cluster Analysis
The MHC genomic region is of vast polymorphic nature in most species. The extremely polymorphic human MHC genomic region (HLA) comprises several thousand alleles. In the majority of the cases, the MHC allele’s potentially unique specificity remains uncharacterized.124 Both classes of MHC molecules having similar binding specificities can be identified via cluster analysis of MHC alleles. In this study, the MHCcluster 2.0 web server (http://www.cbs.dtu.dk/services/MHCcluster/) was applied to generate phylogenetic tree-based visualizations and immensely instinctive heatmaps of the functional cluster between MHC variants by keeping the default parameters. In MHC class I cluster analysis, the NetMHCpan-2.8 method was implemented using a HLA-prevalent and -characterized module, while the corresponding DRB allele modules were selected for MHC class II cluster analysis.124,125
5.9. Formulation of the Vaccine Construct
The vaccine construction was designed by applying the targeted LBL, CTL, and HTL epitopes from the selected MARV proteins. Additionally, an adjuvant was attached using a proper linker during vaccine construction.126,127 We employed the TLR4 agonist as an adjuvant because it is found that viral glycoproteins recognize the TLR4 agonist.70,128 Hence, 50S ribosomal protein L7/L12 was taken as an adjuvant (NCBI ID: P9WHE3) and was linked to the vaccine front with a bi-functional linker, EAAAK. On the other hand, the selected LBL, CTL, and HTL were linked with aid of Lys-Lys (KK), Ala-Ala-Tyr (AAY), and Gly-Pro-Gly-Pro-Gly (GPGPG) linkers respectively.126,127 The AAY linker influences protein stability and improves epitope presentation and immunogenicity. The GPGPG or glycine–proline linker restrains “junctional epitope” formation and helps in immune processing, while the KK or bi-lysine linker facilitates the preservation of constructed vaccines’ independent immunogenic activities.129,130
5.10. Physicochemical, Antigenicity, Allergenicity, and Solubility Profile Evaluation
Analysis of physicochemical properties such as the molecular weight (MW), theoretical isoelectric point (pI), aliphatic index (AI), instability index (II), half-life (in vitro and in vivo), and grand average hydropathicity (GRAVY) of the vaccine construct were evaluated using the ProtParam server (https://web.expasy.org/protparam/).131 Furthermore, multiple servers were utilized to analyze the antigenic, allergenic, and solubility profile of the constructed vaccine. The Vaxijen v2.0 server (http://www.ddg-pharmfac.net/vaxijen/)93 and ANTIGENPro tool (http://scratch.proteomics.ics.uci.edu/) of the Scratch protein prediction server were used for antigenicity prediction. ANTIGENPro displayed 76% precision by cross-validation experiments on the combined dataset.94 The allergenic profile of the vaccine construct was predicted using the AllerTop v2.0 (https://www.ddg-pharmfac.net/AllerTOP/) server,100 AllergenFP v1.0 (https://ddg-pharmfac.net/AllergenFP/) server,95 and AlgPred (http://crdd.osdd.net/raghava/algpred/) server, as there should be no allergenic reaction.132 The solubility of the vaccine construct was predicted using the SOLpro online tool (http://scratch.proteomics.ics.uci.edu/) of the Scratch protein prediction server upon overexpression in the E. coli host. The vaccine is expected to be soluble if the prediction score is equal to or more than 0.5.76 Additionally, the Protein-Sol online server (https://protein-sol.manchester.ac.uk/) was also applied for a better understanding of solubility.133 QuerySol (scaled solubility value) is compared with the E. coli proteins’ average solubility from the experimental dataset (PopAvrSol), and the protein is considered soluble if the predicted score exceeds 0.45.134 The TMHMM v2.0 server (http://www.cbs.dtu.dk/services/TMHMM/) was applied to predict the number of transmembrane helices.96 Furthermore, potential signal peptides were searched through the application of the SignalP 4.1 server (http://www.cbs.dtu.dk/services/SignalP-4.1/) in the final vaccine construct.135
5.11. BLAST and Cross-Checking of Host Homology
The NCBI Protein BLAST (BLASTp) module in the PSI-BLAST algorithm was employed to find homology between the human proteome and the final vaccine construct.136,137 The purpose of this cross-checking analysis was to negate the risk of autoimmune response via molecular mimicry. The search in BLASTp was restricted to the records of H. sapiens (taxid: 9606). The query coverage must display no or less than 40% homology to the human proteome.52
5.12. Secondary Structure Prediction
The PSIPRED v4.0 server (http://bioinf.cs.ucl.ac.uk/psipred/) and SOPMA server (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_sopma.html) were used to identify the secondary structural configurations in default parameters.138,139 Both servers were utilized to calculate the percentage of two-dimensional features, namely, the alpha-helix, random coils, and beta-turn in the constructed vaccine. The SOPMA server reported greater than 80% accuracy in the prediction result.138 The PSIPRED server is regarded as one of the most accurate secondary structure generators, displaying 78.1% accuracy in the prediction result.140
5.13. Homology Modeling and 3D Structure Refinement and Validation
The Raptor-X server (http://raptorx.uchicago.edu/), a homology modeling tool, was implemented to produce the tertiary structure (3D) model of the vaccine construct.141 RaptorX predicts the 3D model of the query proteins based on the multiple-template threading method. The 3D model with the lowest p-score is considered as the best-quality model.142 The refinement of the 3D vaccine model was done using the GalaxyRefine module of the GalaxyWEB server (http://galaxy.seoklab.org/). Five refined models are generated in the output result including different parameters such as rmsd, GDT-HA, clash score, poor rotamers, MolProbity, and Rama-favored.143 The predicted models were validated using the ProSA-web server (https://servicesn.mbi.ucla.edu/PROCHECK/). This server estimates the Z-score and analyzes any protein structure’s stereochemical quality by evaluating the residue-by-residue geometry and overall structure geometry.144 The Procheck web server was used to further analyze the Ramachandran plot for determination of the overall quality of the refined 3D vaccine structure.145 The Ramachandran plot visualizes the percentage of amino acid residues in the most favored, disallowed, generously allowed, and additional allowed regions on the basis of dihedral angles psi (ψ) and phi (ϕ) of each amino acid. A model is considered to be of good quality when more than 90% of residues are in the most favored regions.57
5.14. Disulfide Bond Engineering of the Vaccine Protein
Disulfide bonds help to stabilize tertiary or quaternary interactions within a protein.146 After refinement, the vaccine protein was further submitted to the Disulfide by Design v2.12 web server (http://cptweb.cpt.wayne.edu/DbD2/) for disulfide bridging.147 The χ3 value and the Cα-Cβ-Sγ angle were kept as default parameters. As previous studies suggested, the χ3 angle must range between −87 and +97°, and the energy score must not exceed 2.2 kcal/mol for disulfide bridging.147 In the end, to allow bridging between possible residue pairs, they had undergone mutation to cysteine residues applying the server’s “Create/View Mutant” option.
5.15. Screening for Conformational B-Cell Epitopes
The ElliPro tool of the IEDB resource (http://tools.iedb.org/ellipro/) was employed to predict the discontinuous or conformational B-cell epitopes in the constructed vaccine by keeping the default parameters, that is, 6 Å maximum distance and 0.5 minimum score. The results are based on modified Thornton’s method that uses the residue clustering algorithms. The prediction is done by considering the residual protein index (PI), neighbor residue clustering, and protein shape.148
5.16. Molecular Docking of the TLR Complex and Vaccine Construct
The TLR4 complexes (PDB ID: 4G8A) were retrieved from the protein data bank (RCSB) with a resolution of 2.4 Å.117 The heteroatoms and B, C, and D chains were removed in the Discovery Studio software package.121 The protein was energy-minimized in Swiss PDB Viewer software by employing the GROMOS 43B1 force field.116 Then, the vaccine construct and the TLR4 complexes were docked in ClusPro (https://cluspro.bu.edu/) tools.149 This tool represents an automated web-based program for protein–protein or peptide–protein docking. This docking program evaluates the billions of putative complexes with favorable surface complementarities. According to the clustering properties, the program outputs a short list of putative complexes.
5.17. Normal-Mode Analysis
The collective motion of the protein complexes can be explained using the iMODS simulation server through the analysis of normal modes in internal coordinates.150 This process is quicker and cost-efficient compared to the other processes of molecular dynamics simulations.151 This web server predicts the eigenvalues, deformability, B-factors, and covariance. The assessment of motif stiffness was done using eigenvalues, whereas the deformity of the main chain was predicted from the biological targets’ efficacy measurement.152
5.18. In Silico Immune Simulation
To understand and explore the immunogenicity and immune response profile, an immune simulation study was conducted. The C-ImmSim web tool (http://150.146.2.1/CIMMSIM/index.php) was utilized for the immune simulation study. This web tool program has employed machine learning and real-life-like immune interactions and response by applying a position-specific scoring matrix.153 The parameters in CImmSim were set as defaults, and the time step was set as 1, 84, and 170, where time step 1 is injection at time = 0, and each time step is 8 h. A total of three injections were expected with the time duration between two injections set as four weeks, as commercial vaccines recommend an interval between two doses of four weeks.154
5.19. Molecular Dynamics Simulation
The YASARA dynamics software package (version 20.1.1) was utilized to conduct the molecular dynamics simulation of the constructed vaccine and the TLR4 complexes. In this software, the AMBER14 force field was employed.155,156 The complexes were initially cleaned, and hydrogen bond networks were optimized. The TIP3P solvation model157 was used to solvate the protein in a cubic simulation cell with periodic boundary conditions by maintaining a solvent density of 0.997 g L–1. The pKa or the acid dissociation constant value was calculated for the amino acid present in the protein. The SCWRL algorithm was applied to maintain the correct protonation state of each amino acid residue.158 The physiological conditions were maintained at pH 7.4 with the addition of Na and Cl ions at 298 K temperature.159 The energy of the complexes was minimized using the steepest gradient approaches (5000 cycles) by simulated annealing methods. The time step of the simulation system was set as 2.0 fs.160,161 The particle mesh Ewald method was applied for the calculation of the long-range electrostatic interactions with a cutoff radius of 8 Å.162 With a Berendsen thermostat and constant pressure, the simulation was conducted for 100 ns. The simulation trajectories were saved after 100 ps intervals to calculate the root mean square deviations (rmsd), Rg, RMSF, SASA, and hydrogen bonds.163−166
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c04817.
Prediction of potential CTL epitopes from MARV structural proteins, prediction of potential HTL epitopes from MARV structural proteins, prediction of potential LBL epitopes from MARV structural proteins, worldwide population coverage analysis based on MHC restriction data, refined vaccine models generated using the GalaxyRefine server, and list of amino acid residues in disulfide bond engineering (PDF)
Author Contributions
Conceptualization: S.A.S., K.K.S.M., and A.R. Formal analysis: S.A.S., K.K.S.M., and S.M., S.A., A.M.E.-S., A.R., M.M.N.U., M.K.H., and T.B.E. Funding acquisition: T.E.T. and T.B.E. Investigation: S.A.S., K.K.S.M., S.M., M.A.N.K., A.R., A.C., M.M., and T.B.E. Methodology: S.A.S., K.K.S.M., S.M., M.A.N.K., A.R., A.C., M.M, and T.B.E. Project administration: S.A.S. and T.B.E. Resources: K.D., T.E.T., and T.B. E. Software: S.A.S., K.K.S.M., and S.M. Supervision: M.K.H. and T.B.E. Writing—original draft: S.A.S., K.K.S.M., S.M., M.A.N.K., A.C., and M.M. Writing—review and editing: S.A.S., S.A., A.M.E.-S., K.D., M.M.N.U., M.K.H., T.E.T., and T.B.E.
The current work was funded by Taif University Researchers Supporting Project number (TURSP-2020/202), Taif University, Taif, Saudi Arabia.
The authors declare no competing financial interest.
Notes
Availability of data and material: The datasets supporting the conclusions of this study are included within the article (and its additional files).
Supplementary Material
References
- Mahmud S. M. N.; Rahman M.; Kar A.; Jahan N.; Khan A. Designing of an Epitope- Based Universal Peptide Vaccine against Highly Conserved Regions in RNA Dependent RNA Polymerase Protein of Human Marburg Virus: A Computational Assay. Anti-Infect. Agents. 2020, 18, 294–305. 10.2174/2211352517666190717143949. [DOI] [Google Scholar]
- Nyakarahuka L.; Shoemaker T. R.; Balinandi S.; Chemos G.; Kwesiga B.; Mulei S.; Kyondo J.; Tumusiime A.; Kofman A.; Masiira B.; Whitmer S.; Brown S.; Cannon D.; Chiang C.-F.; Graziano J.; Morales-Betoulle M.; Patel K.; Zufan S.; Komakech I.; Natseri N.; Chepkwurui P. M.; Lubwama B.; Okiria J.; Kayiwa J.; Nkonwa I. H.; Eyu P.; Nakiire L.; Okarikod E. C.; Cheptoyek L.; Wangila B. E.; Wanje M.; Tusiime P.; Bulage L.; Mwebesa H. G.; Ario A. R.; Makumbi I.; Nakinsige A.; Muruta A.; Nanyunja M.; Homsy J.; Zhu B.-P.; Nelson L.; Kaleebu P.; Rollin P. E.; Nichol S. T.; Klena J. D.; Lutwama J. J. Marburg virus disease outbreak in Kween District Uganda, 2017: Epidemiological and laboratory findings. PLoS Neglected Trop. Dis. 2019, 13, e0007257 10.1371/journal.pntd.0007257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO . WHO Marburg Virus Disease. https://www.who.int/news-room/fact-sheets/detail/marburg-virus-disease (accessed July 1, 2021).
- WHO Africa . West Africa’s First-Ever Case of Marburg Virus Disease Confirmed in Guinea. https://www.afro.who.int/news/west-africas-first-ever-case-marburg-virus-disease-confirmed-guinea (accessed Aug 14, 2021).
- Bukreyev A. A.; Chandran K.; Dolnik O.; Dye J. M.; Ebihara H.; Leroy E. M.; Mühlberger E.; Netesov S. V.; Patterson J. L.; Paweska J. T.; Saphire E. O.; Smither S. J.; Takada A.; Towner J. S.; Volchkov V. E.; Warren T. K.; Kuhn J. H. Discussions and decisions of the 2012-2014 International Committee on Taxonomy of Viruses (ICTV) Filoviridae Study Group, January 2012-June 2013. Arch. Virol. 2014, 159, 821–830. 10.1007/s00705-013-1846-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shifflett K.; Marzi A. Marburg virus pathogenesis - differences and similarities in humans and animal models. Virol. J. 2019, 16, 165. 10.1186/s12985-019-1272-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehedi M.; Groseth A.; Feldmann H.; Ebihara H. Clinical aspects of Marburg hemorrhagic fever. Future Virol. 2011, 6, 1091–1106. 10.2217/fvl.11.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiCarlo A.; Möller P.; Lander A.; Kolesnikova L.; Becker S. Nucleocapsid formation and RNA synthesis of Marburg virus is dependent on two coiled coil motifs in the nucleoprotein. Virol. J. 2007, 4, 105. 10.1186/1743-422x-4-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will C.; Mühlberger E.; Linder D.; Slenczka W.; Klenk H. D.; Feldmann H. Marburg virus gene 4 encodes the virion membrane protein, a type I transmembrane glycoprotein. J. Virol. 1993, 67, 1203–1210. 10.1128/jvi.67.3.1203-1210.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross R. W.; Mire C. E.; Feldmann H.; Geisbert T. W. Post-exposure treatments for Ebola and Marburg virus infections. Nat. Rev. Drug Discovery 2018, 17, 413–434. 10.1038/nrd.2017.251. [DOI] [PubMed] [Google Scholar]
- Mühlberger E.; Lötfering B.; Klenk H.-D.; Becker S. Three of the four nucleocapsid proteins of Marburg virus, NP, VP35, and L, are sufficient to mediate replication and transcription of Marburg virus-specific monocistronic minigenomes. J. Virol. 1998, 72, 8756–8764. 10.1128/jvi.72.11.8756-8764.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler A.; Pfeiffer S.; Kolesnikova L.; Becker S. Analysis of the multifunctionality of Marburg virus VP40. J. Gen. Virol. 2018, 99, 1614–1620. 10.1099/jgv.0.001169. [DOI] [PubMed] [Google Scholar]
- Albariño C. G.; Guerrero L. W.; Spengler J. R.; Uebelhoer L. S.; Chakrabarti A. K.; Nichol S. T.; Towner J. S. Recombinant Marburg viruses containing mutations in the IID region of VP35 prevent inhibition of Host immune responses. Virology 2015, 476, 85–91. 10.1016/j.virol.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen B. C.; Basler C. F. Effects of filovirus interferon antagonists on responses of human monocyte-derived dendritic cells to RNA virus infection. J. Virol. 2016, 90, 5108–5118. 10.1128/jvi.00191-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanan P.; Edwards M. R.; Shabman R. S.; Leung D. W.; Endlich-Frazier A. C.; Borek D. M.; Otwinowski Z.; Liu G.; Huh J.; Basler C. F.; Amarasinghe G. K. Structural basis for Marburg virus VP35-mediated immune evasion mechanisms. Proc. Natl. Acad. Sci. 2012, 109, 20661–20666. 10.1073/pnas.1213559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume A.; Mühlberger E. Marburg Virus Viral Protein 35 Inhibits Protein Kinase R Activation in a Cell Type-Specific Manner. J. Infect. Dis. 2018, 218, S403–S408. 10.1093/infdis/jiy473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker S.; Rinne C.; Hofsäß U.; Klenk H.-D.; Mühlberger E. Interactions of Marburg virus nucleocapsid proteins. Virology 1998, 249, 406–417. 10.1006/viro.1998.9328. [DOI] [PubMed] [Google Scholar]
- Wenigenrath J.; Kolesnikova L.; Hoenen T.; Mittler E.; Becker S. Establishment and application of an infectious virus-like particle system for Marburg virus. J. Gen. Virol. 2010, 91, 1325–1334. 10.1099/vir.0.018226-0. [DOI] [PubMed] [Google Scholar]
- Enterlein S.; Volchkov V.; Weik M.; Kolesnikova L.; Volchkova V.; Klenk H.-D.; Mühlberger E. Rescue of recombinant Marburg virus from cDNA is dependent on nucleocapsid protein VP30. J. Virol. 2006, 80, 1038–1043. 10.1128/jvi.80.2.1038-1043.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolesnikova L.; Nanbo A.; Becker S.; Kawaoka Y. Inside the cell: assembly of filoviruses. Curr. Top. Microbiol. Immunol. 2017, 411, 353–380. 10.1007/82_2017_15. [DOI] [PubMed] [Google Scholar]
- Bamberg S.; Kolesnikova L.; Möller P.; Klenk H.-D.; Becker S. VP24 of Marburg virus influences formation of infectious particles. J. Virol. 2005, 79, 13421–13433. 10.1128/jvi.79.21.13421-13433.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauburger K.; Hume A. J.; Mühlberger E.; Olejnik J. Forty-five years of Marburg virus research. Viruses 2012, 4, 1878–1927. 10.3390/v4101878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakib A.; Sami S. A.; Islam M. A.; Ahmed S.; Faiz F. B.; Khanam B. H.; Marma K. K. S.; Rahman M.; Uddin M. M. N.; Nainu F.; Emran T. B.; Simal-Gandara J. Epitope-Based Immunoinformatics Approach on Nucleocapsid Protein of Severe Acute Respiratory Syndrome-Coronavirus-2. Molecules 2020, 25, 5088. 10.3390/molecules25215088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daddario-DiCaprio K. M.; Geisbert T. W.; Ströher U.; Geisbert J. B.; Grolla A.; Fritz E. A.; Fernando L.; Kagan E.; Jahrling P. B.; Hensley L. E.; Jones S. M.; Feldmann H. Postexposure protection against Marburg haemorrhagic fever with recombinant vesicular stomatitis virus vectors in non-human primates: an efficacy assessment. Lancet 2006, 367, 1399–1404. 10.1016/s0140-6736(06)68546-2. [DOI] [PubMed] [Google Scholar]
- Callendret B.; Vellinga J.; Wunderlich K.; Rodriguez A.; Steigerwald R.; Dirmeier U.; Cheminay C.; Volkmann A.; Brasel T.; Carrion R.; Giavedoni L. D.; Patterson J. L.; Mire C. E.; Geisbert T. W.; Hooper J. W.; Weijtens M.; Hartkoorn-Pasma J.; Custers J.; Grazia Pau M.; Schuitemaker H.; Zahn R. A prophylactic multivalent vaccine against different filovirus species is immunogenic and provides protection from lethal infections with Ebolavirus and Marburgvirus species in non-human primates. PLoS One 2018, 13, e0192312 10.1371/journal.pone.0192312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant-Klein R. J.; Altamura L. A.; Badger C. V.; Bounds C. E.; Van Deusen N. M.; Kwilas S. A.; Vu H. A.; Warfield K. L.; Hooper J. W.; Hannaman D.; Dupuy L. C.; Schmaljohn C. S. Codon-optimized filovirus DNA vaccines delivered by intramuscular electroporation protect cynomolgus macaques from lethal Ebola and Marburg virus challenges. Hum. Vaccines Immunother. 2015, 11, 1991–2004. 10.1080/21645515.2015.1039757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swenson D. L.; Warfield K. L.; Larsen T.; Alves D. A.; Coberley S. S.; Bavari S. Monovalent virus-like particle vaccine protects guinea pigs and nonhuman primates against infection with multiple Marburg viruses. Expert Rev. Vaccines 2008, 7, 417–429. 10.1586/14760584.7.4.417. [DOI] [PubMed] [Google Scholar]
- Yashvardhini N.; Kumar A.; Jha D. K. Immunoinformatics Identification of B- and T-Cell Epitopes in the RNA-Dependent RNA Polymerase of SARS-CoV-2. Can. J. Infect Dis. Med. Microbiol. 2021, 2021, 6627141. 10.1155/2021/6627141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y.; Sun X.; Li B.; Huang J.; Zhan B.; Zhu X. Vaccination with a paramyosin-based multi-epitope vaccine elicits significant protective immunity against Trichinella spiralis infection in mice. Front. Microbiol. 2017, 8, 1475. 10.3389/fmicb.2017.01475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agallou M.; Athanasiou E.; Koutsoni O.; Dotsika E.; Karagouni E. Experimental Validation of Multi-Epitope Peptides Including Promising MHC Class I- and II-Restricted Epitopes of Four Known Leishmania infantum Proteins. Front. Immunol. 2014, 5, 268. 10.3389/fimmu.2014.00268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staneková Z.; Varečková E. Conserved epitopes of influenza A virus inducing protective immunity and their prospects for universal vaccine development. Virol. J. 2010, 7, 351. 10.1186/1743-422x-7-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sominskaya I.; Skrastina D.; Dislers A.; Vasiljev D.; Mihailova M.; Ose V.; Dreilina D.; Pumpens P. Construction and immunological evaluation of multivalent hepatitis B virus (HBV) core virus-like particles carrying HBV and HCV epitopes. Clin. Vaccine Immunol. 2010, 17, 1027–1033. 10.1128/cvi.00468-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L.; Cheng Y.; Kong L.; Azadnia P.; Giang E.; Kim J.; Wood M. R.; Wilson I. A.; Law M.; Zhu J. Approaching rational epitope vaccine design for hepatitis C virus with meta-server and multivalent scaffolding. Sci. Rep. 2015, 5, 12501. 10.1038/srep12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E Silva R. F.; Ferreira L. F. G. R.; Hernandes M. Z.; de Brito M. E. F.; de Oliveira B. C.; da Silva A. A.; de-Melo-Neto O. P.; Rezende A. M.; Pereira V. R. A. Combination of In Silico Methods in the Search for Potential CD4(+) and CD8(+) T Cell Epitopes in the Proteome of Leishmania braziliensis. Front. Immunol. 2016, 7, 327. 10.3389/fimmu.2016.00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S.; Khan A.; Rehman A. U.; Ahmad I.; Ullah S.; Khan A. A.; Ali S. S.; Afridi S. G.; Wei D.-Q. Immunoinformatics and structural vaccinology driven prediction of multi-epitope vaccine against Mayaro virus and validation through in-silico expression. Infect. Genet. Evol. 2019, 73, 390–400. 10.1016/j.meegid.2019.06.006. [DOI] [PubMed] [Google Scholar]
- Plotkin S. A. Vaccines: past, present and future. Nat. Med. 2005, 11, S5–S11. 10.1038/nm1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugunthan S. P.; Harish M. C. Multi-epitope-Based Vaccine Designed by Targeting Cytoadherence Proteins of Mycoplasma gallisepticum. ACS Omega 2021, 6, 13742–13755. 10.1021/acsomega.1c01032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahir ul Qamar M.; Rehman A.; Tusleem K.; Ashfaq U. A.; Qasim M.; Zhu X.; Fatima I.; Shahid F.; Chen L.-L. Designing of a next generation multiepitope based vaccine (MEV) against SARS-COV-2: Immunoinformatics and in silico approaches. PLoS One 2020, 15, e0244176 10.1371/journal.pone.0244176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar T.; Narsaria U.; Basak S.; Deb D.; Castiglione F.; Mueller D. M.; Srivastava A. P. A candidate multi-epitope vaccine against SARS-CoV-2. Sci. Rep. 2020, 10, 10895. 10.1038/s41598-020-67749-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M. K.; Zaman S.; Chakraborty S.; Chakravorty R.; Alam M. M.; Bhuiyan T. R.; Rahman M. J.; Fernández C.; Qadri F.; Seraj Z. I. In silico predicted mycobacterial epitope elicits in vitro T-cell responses. Mol. Immunol. 2014, 61, 16–22. 10.1016/j.molimm.2014.04.009. [DOI] [PubMed] [Google Scholar]
- Bhuiyan M. A.; Quayum S. T.; Ahammad F.; Alam R.; Samad A.; Nain Z. Discovery of potential immune epitopes and peptide vaccine design - a prophylactic strategy against Rift Valley fever virus. F1000Res. 2020, 9, 999. 10.12688/f1000research.24975.1. [DOI] [Google Scholar]
- Bausch D. G.; Nichol S. T.; Muyembe-Tamfum J. J.; Borchert M.; Rollin P. E.; Sleurs H.; Campbell P.; Tshioko F. K.; Roth C.; Colebunders R.; Pirard P.; Mardel S.; Olinda L. A.; Zeller H.; Tshomba A.; Kulidri A.; Libande M. L.; Mulangu S.; Formenty P.; Grein T.; Leirs H.; Braack L.; Ksiazek T.; Zaki S.; Bowen M. D.; Smit S. B.; Leman P. A.; Burt F. J.; Kemp A.; Swanepoel R. Marburg hemorrhagic fever associated with multiple genetic lineages of virus. N. Engl. J. Med. 2006, 355, 909–919. 10.1056/nejmoa051465. [DOI] [PubMed] [Google Scholar]
- Towner J. S.; Khristova M. L.; Sealy T. K.; Vincent M. J.; Erickson B. R.; Bawiec D. A.; Hartman A. L.; Comer J. A.; Zaki S. R.; Ströher U.; Gomes da Silva F.; del Castillo F.; Rollin P. E.; Ksiazek T. G.; Nichol S. T. Marburgvirus genomics and association with a large hemorrhagic fever outbreak in Angola. J. Virol. 2006, 80, 6497–6516. 10.1128/jvi.00069-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slenczka W.; Klenk H. D. Forty years of Marburg virus. J. Infect. Dis. 2007, 196, S131–S135. 10.1086/520551. [DOI] [PubMed] [Google Scholar]
- Feldmann H. Marburg Hemorrhagic Fever - The Forgotten Cousin Strikes. N. Engl. J. Med. 2006, 355, 866–869. 10.1056/nejmp068160. [DOI] [PubMed] [Google Scholar]
- Rougeron V.; Feldmann H.; Grard G.; Becker S.; Leroy E. M. Ebola and Marburg haemorrhagic fever. J. Clin. Virol. 2015, 64, 111–119. 10.1016/j.jcv.2015.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X.; Smith T. G.; Rupprecht C. E. From brain passage to cell adaptation: the road of human rabies vaccine development. Expert Rev. Vaccines 2011, 10, 1597–1608. 10.1586/erv.11.140. [DOI] [PubMed] [Google Scholar]
- Hassan A.; Naz A.; Obaid A.; Paracha R. Z.; Naz K.; Awan F. M.; Muhmmad S. A.; Janjua H. A.; Ahmad J.; Ali A. Pangenome and immuno-proteomics analysis of Acinetobacter baumannii strains revealed the core peptide vaccine targets. BMC Genom. 2016, 17, 732. 10.1186/s12864-016-2951-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalsass M.; Brozzi A.; Medini D.; Rappuoli R. Comparison of open-source reverse vaccinology programs for bacterial vaccine antigen discovery. Front. Immunol. 2019, 10, 113. 10.3389/fimmu.2019.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan M.; Azim K. F.; Begum A.; Khan N. A.; Shammi T. S.; Imran A. S.; Chowdhury I. M.; Urme S. R. A. Vaccinomics strategy for developing a unique multi-epitope monovalent vaccine against Marburg marburgvirus. Infect. Genet. Evol. 2019, 70, 140–157. 10.1016/j.meegid.2019.03.003. [DOI] [PubMed] [Google Scholar]
- Hossain M. S.; Hossan M. I.; Mizan S.; Moin A. T.; Yasmin F.; Akash A.-S.; Powshi S. N.; Hasan A. K. R.; Chowdhury A. S. Immunoinformatics approach to designing a multi-epitope vaccine against Saint Louis Encephalitis Virus. Inform. Med. Unlocked. 2021, 22, 100500. 10.1016/j.imu.2020.100500. [DOI] [Google Scholar]
- Rojas M.; Restrepo-Jiménez P.; Monsalve D. M.; Pacheco Y.; Acosta-Ampudia Y.; Ramírez-Santana C.; Leung P. S. C.; Ansari A. A.; Gershwin M. E.; Anaya J.-M. Molecular mimicry and autoimmunity. J. Autoimmun. 2018, 95, 100–123. 10.1016/j.jaut.2018.10.012. [DOI] [PubMed] [Google Scholar]
- Kanduc D. Peptide cross-reactivity the original sin of vaccines. Front. Biosci. 2012, S4, 1393–1401. 10.2741/s341. [DOI] [PubMed] [Google Scholar]
- Ojha R.; Pareek A.; Pandey R. K.; Prusty D.; Prajapati V. K. Strategic development of a next-generation multi-epitope vaccine to prevent Nipah virus zoonotic infection. ACS Omega 2019, 4, 13069–13079. 10.1021/acsomega.9b00944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Galarza F. F.; Takeshita L. Y. C.; Santos E. J. M.; Kempson F.; Maia M. H. T.; da Silva A. L. S.; Teles e Silva A. L.; Ghattaoraya G. S.; Alfirevic A.; Jones A. R.; Middleton D. Allele frequency net 2015 update: new features for HLA epitopes, KIR and disease and HLA adverse drug reaction associations. Nucleic Acids Res. 2015, 43, D784–D788. 10.1093/nar/gku1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A.; Rosato A. J.; Cui F. Vaccine candidate designed against carcinoembryonic antigen-related cell adhesion molecules using immunoinformatics tools. J. Biomol. Struct. Dyn. 2021, 39, 6084–6098. 10.1080/07391102.2020.1797539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell S. C.; Davis I. W.; Arendall W. B. 3rd; de Bakker P. I. W.; Word J. M.; Prisant M. G.; Richardson J. S.; Richardson D. C. Structure validation by Cα geometry: ϕ,ψ and Cβ deviation. Proteins 2003, 50, 437–450. 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- Athanasiou E.; Agallou M.; Tastsoglou S.; Kammona O.; Hatzigeorgiou A.; Kiparissides C.; Karagouni E. A Poly(Lactic-co-Glycolic) Acid Nanovaccine Based on Chimeric Peptides from Different Leishmania infantum Proteins Induces Dendritic Cells Maturation and Promotes Peptide-Specific IFNγ-Producing CD8+ T Cells Essential for the Protection against Experimental Visceral Leishmaniasis. Front. Immunol. 2017, 8, 684. 10.3389/fimmu.2017.00684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. J.; Shin S. J.; Lee M. H.; Lee M.-G.; Kang T. H.; Park W. S.; Soh B. Y.; Park J. H.; Shin Y. K.; Kim H. W.; Yun C.-H.; Jung I. D.; Park Y.-M. A potential protein adjuvant derived from Mycobacterium tuberculosis Rv0652 enhances dendritic cells-based tumor immunotherapy. PLoS One 2014, 9, e104351 10.1371/journal.pone.0104351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Zaro J. L.; Shen W.-C. Fusion protein linkers: property, design and functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369. 10.1016/j.addr.2012.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Guo L.; Kong M.; Su X.; Yang D.; Zou M.; Liu Y.; Lu L. Design and evaluation of a multi-epitope peptide of human metapneumovirus. Intervirology 2015, 58, 403–412. 10.1159/000445059. [DOI] [PubMed] [Google Scholar]
- Livingston B.; Crimi C.; Newman M.; Higashimoto Y.; Appella E.; Sidney J.; Sette A. A rational strategy to design multiepitope immunogens based on multiple Th lymphocyte epitopes. J. Immunol. 2002, 168, 5499–5506. 10.4049/jimmunol.168.11.5499. [DOI] [PubMed] [Google Scholar]
- Dolenc I.; Seemüller E.; Baumeister W. Decelerated degradation of short peptides by the 20S proteasome. FEBS Lett. 1998, 434, 357–361. 10.1016/s0014-5793(98)01010-2. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Sun W.; Guo J.; Zhao G.; Sun S.; Yu H.; Guo Y.; Li J.; Jin X.; Du L.; Jiang S.; Kou Z.; Zhou Y. In silicodesign of a DNA-based HIV-1 multi-epitope vaccine for Chinese populations. Hum. Vaccines Immunother. 2015, 11, 795–805. 10.1080/21645515.2015.1012017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velders M. P.; Weijzen S.; Eiben G. L.; Elmishad A. G.; Kloetzel P.-M.; Higgins T.; Ciccarelli R. B.; Evans M.; Man S.; Smith L.; Kast W. M. Defined flanking spacers and enhanced proteolysis is essential for eradication of established tumors by an epitope string DNA vaccine. J. Immunol. 2001, 166, 5366–5373. 10.4049/jimmunol.166.9.5366. [DOI] [PubMed] [Google Scholar]
- Negahdaripour M.; Nezafat N.; Eslami M.; Ghoshoon M. B.; Shoolian E.; Najafipour S.; Morowvat M. H.; Dehshahri A.; Erfani N.; Ghasemi Y. Structural vaccinology considerations for in silico designing of a multi-epitope vaccine. Infect. Genet. Evol. 2018, 58, 96–109. 10.1016/j.meegid.2017.12.008. [DOI] [PubMed] [Google Scholar]
- Khatoon N.; Pandey R. K.; Prajapati V. K. Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci. Rep. 2017, 7, 8285. 10.1038/s41598-017-08842-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naz A.; Awan F. M.; Obaid A.; Muhammad S. A.; Paracha R. Z.; Ahmad J.; Ali A. Identification of putative vaccine candidates against Helicobacter pylori exploiting exoproteome and secretome: a reverse vaccinology based approach. Infect. Genet. Evol. 2015, 32, 280–291. 10.1016/j.meegid.2015.03.027. [DOI] [PubMed] [Google Scholar]
- Mathur D.; Prakash S.; Anand P.; Kaur H.; Agrawal P.; Mehta A.; Kumar R.; Singh S.; Raghava G. P. S. PEPlife: a repository of the half-life of peptides. Sci. Rep. 2016, 6, 36617. 10.1038/srep36617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey R. K.; Bhatt T. K.; Prajapati V. K. Novel immunoinformatics approaches to design multi-epitope subunit vaccine for malaria by investigating anopheles salivary protein. Sci. Rep. 2018, 8, 1125. 10.1038/s41598-018-19456-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhri G.; Quah B. J.; Wang Y.; Tan A. H. Y.; Zhou J.; Karupiah G.; Parish C. R. T cell receptor sharing by cytotoxic T lymphocytes facilitates efficient virus control. Proc. Natl. Acad. Sci. 2009, 106, 14984–14989. 10.1073/pnas.0906554106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guruprasad K.; Reddy B. V. B.; Pandit M. W. Correlation between stability of a protein and its dipeptide composition: a novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng. Des. Sel. 1990, 4, 155–161. 10.1093/protein/4.2.155. [DOI] [PubMed] [Google Scholar]
- Chang K. Y.; Yang J.-R. Analysis and prediction of highly effective antiviral peptides based on random forests. PLoS One 2013, 8, e70166 10.1371/journal.pone.0070166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte J.; Doolittle R. F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Almofti Y. A.; Abd-Elrahman K. A.; Eltilib E. E. M. Vaccinomic approach for novel multi epitopes vaccine against severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). BMC Immunol. 2021, 22, 22–20. 10.1186/s12865-021-00412-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnan C. N.; Randall A.; Baldi P. SOLpro: accurate sequence-based prediction of protein solubility. Bioinformatics 2009, 25, 2200–2207. 10.1093/bioinformatics/btp386. [DOI] [PubMed] [Google Scholar]
- Dar H. A.; Zaheer T.; Shehroz M.; Ullah N.; Naz K.; Muhammad S. A.; Zhang T.; Ali A. Immunoinformatics-aided design and evaluation of a potential multi-epitope vaccine against Klebsiella pneumoniae. Vaccines 2019, 7, 88. 10.3390/vaccines7030088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri A.; Das P.; Chakravortty D. Engagement of TLR signaling as adjuvant: towards smarter vaccine and beyond. Vaccine 2008, 26, 6777–6783. 10.1016/j.vaccine.2008.09.045. [DOI] [PubMed] [Google Scholar]
- Akira S.; Takeda K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- Zheng C.; Chen J.; Chu F.; Zhu J.; Jin T. Inflammatory role of TLR-MyD88 signaling in multiple sclerosis. Front. Mol. Neurosci. 2020, 12, 314. 10.3389/fnmol.2019.00314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onofrio L.; Caraglia M.; Facchini G.; Margherita V.; Placido S. D.; Buonerba C. Toll-like receptors and COVID-19: a two-faced story with an exciting ending. Future Sci. OA 2020, 6, FSO605. 10.2144/fsoa-2020-0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Zhang S.; Li H.; Wang H.; Zhang T.; Hutchinson M. R.; Yin H.; Wang X. Small-molecule modulators of toll-like receptors. Acc. Chem. Res. 2020, 53, 1046–1055. 10.1021/acs.accounts.9b00631. [DOI] [PubMed] [Google Scholar]
- Molteni M.; Gemma S.; Rossetti C. The role of toll-like receptor 4 in infectious and noninfectious inflammation. Mediat. Inflamm. 2016, 2016, 6978936. 10.1155/2016/6978936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K.; Akira S. Toll-like receptors in innate immunity. Int. Immunol. 2005, 17, 1–14. 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- Sarkar B.; Ullah M. A.; Araf Y.; Islam N. N.; Zohora U. S. Immunoinformatics-guided designing and in silico analysis of epitope-based polyvalent vaccines against multiple strains of human coronavirus (HCoV). Expert Rev. Vaccines 2021, 1–21. 10.1080/14760584.2021.1874925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappuoli R. Reverse vaccinology. Curr. Opin. Microbiol. 2000, 3, 445–450. 10.1016/s1369-5274(00)00119-3. [DOI] [PubMed] [Google Scholar]
- Mahmud S.; Paul G. K.; Biswas S.; Afrose S.; Mita M. A.; Hasan M.; Shimu M.; Sultana S.; Hossain A.; Promi M. M. Prospective role of peptide-based antiviral therapy against the main protease of SARS-CoV-2. Front. Mol. Biosci. 2021, 8, 628585. 10.3389/fmolb.2021.628585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obaidullah A. J.; Alanazi M. M.; Alsaif N. A.; Albassam H.; Almehizia A. A.; Alqahtani A. M.; Mahmud S.; Sami S. A.; Emran T. B. Immunoinformatics-guided design of a multi-epitope vaccine based on the structural proteins of severe acute respiratory syndrome coronavirus 2. RSC Adv. 2021, 11, 18103–18121. 10.1039/d1ra02885e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakib A.; Sami S. A.; Mimi N. J.; Chowdhury M. M.; Eva T. A.; Nainu F.; Paul A.; Shahriar A.; Tareq A. M.; Emon N. U.; Chakraborty S.; Shil S.; Mily S. J.; Ben Hadda T.; Almalki F. A.; Emran T. B. Immunoinformatics-guided design of an epitope-based vaccine against severe acute respiratory syndrome coronavirus 2 spike glycoprotein. Comput. Biol. Med. 2020, 124, 103967. 10.1016/j.compbiomed.2020.103967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M.; Zaretskaya I.; Raytselis Y.; Merezhuk Y.; McGinnis S.; Madden T. L. NCBI BLAST: a better web interface. Nucleic Acids Res. 2008, 36, W5–W9. 10.1093/nar/gkn201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulo C.; De Castro E.; Masson P.; Bougueleret L.; Bairoch A.; Xenarios I.; Le Mercier P. ViralZone: a knowledge resource to understand virus diversity. Nucleic Acids Res. 2011, 39, D576–D582. 10.1093/nar/gkq901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. 10.1093/nar/gkw1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doytchinova I. A.; Flower D. R. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinf. 2007, 8, 4. 10.1186/1471-2105-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnan C. N.; Zeller M.; Kayala M. A.; Vigil A.; Randall A.; Felgner P. L.; Baldi P. High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics 2010, 26, 2936–2943. 10.1093/bioinformatics/btq551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrov I.; Naneva L.; Doytchinova I.; Bangov I. AllergenFP: allergenicity prediction by descriptor fingerprints. Bioinformatics 2014, 30, 846–851. 10.1093/bioinformatics/btt619. [DOI] [PubMed] [Google Scholar]
- Krogh A.; Larsson B.; von Heijne G.; Sonnhammer E. L. L. Predicting transmembrane protein topology with a hidden markov model: application to complete genomes11Edited by F. Cohen. J. Mol. Biol. 2001, 305, 567–580. 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Xu X.; Chen P.; Wang J.; Feng J.; Zhou H.; Li X.; Zhong W.; Hao P. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci. China Life Sci. 2020, 63, 457–460. 10.1007/s11427-020-1637-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen M. V.; Lundegaard C.; Lamberth K.; Buus S.; Lund O.; Nielsen M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinf. 2007, 8, 424. 10.1186/1471-2105-8-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moutaftsi M.; Peters B.; Pasquetto V.; Tscharke D. C.; Sidney J.; Bui H.-H.; Grey H.; Sette A. A consensus epitope prediction approach identifies the breadth of murine TCD8+-cell responses to vaccinia virus. Nat. Biotechnol. 2006, 24, 817–819. 10.1038/nbt1215. [DOI] [PubMed] [Google Scholar]
- Dimitrov I.; Flower D. R.; Doytchinova I. AllerTOP - a server for in silico prediction of allergens. BMC Bioinf. 2013, 14, S4. 10.1186/1471-2105-14-s6-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S.; Kapoor P.; Chaudhary K.; Gautam A.; Kumar R.; Raghava G. P. S.; Raghava G. P. In silico approach for predicting toxicity of peptides and proteins. PLoS One 2013, 8, e73957 10.1371/journal.pone.0073957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calis J. J. A.; Maybeno M.; Greenbaum J. A.; Weiskopf D.; De Silva A. D.; Sette A.; Keşmir C.; Peters B. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266 10.1371/journal.pcbi.1003266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P.; Sidney J.; Kim Y.; Sette A.; Lund O.; Nielsen M.; Peters B. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinf. 2010, 11, 568. 10.1186/1471-2105-11-568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P.; Sidney J.; Dow C.; Mothé B.; Sette A.; Peters B. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput. Biol. 2008, 4, e1000048 10.1371/journal.pcbi.1000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Chung H.; Jang Y. Y. High serum IgE level in the children with acute respiratory syncytial virus infection is associated with severe disease. J. Allergy Clin. Immunol. 2016, 137, AB110. 10.1016/j.jaci.2015.12.486. [DOI] [Google Scholar]
- Dhanda S. K.; Gupta S.; Vir P.; Raghava G. P. S. Prediction of IL4 Inducing Peptides. Biol. Direct 2013, 2013, 1–9. 10.1155/2013/263952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanda S. K.; Gupta S.; Vir P.; Raghava G. P. Prediction of IL4 inducing peptides. Clin. Dev. Immunol. 2013, 2013, 263952. 10.1155/2013/263952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagpal G.; Usmani S. S.; Dhanda S. K.; Kaur H.; Singh S.; Sharma M.; Raghava G. P. S. Computer-aided designing of immunosuppressive peptides based on IL-10 inducing potential. Sci. Rep. 2017, 7, 42851. 10.1038/srep42851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S.; Raghava G. P. S. Prediction methods for B-cell epitopes. Methods Mol. Biol. 2007, 409, 387–394. 10.1007/978-1-60327-118-9_29. [DOI] [PubMed] [Google Scholar]
- Saha S.; Raghava G. P. S. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins 2006, 65, 40–48. 10.1002/prot.21078. [DOI] [PubMed] [Google Scholar]
- Manavalan B.; Govindaraj R. G.; Shin T. H.; Kim M. O.; Lee G. iBCE-EL: a new ensemble learning framework for improved linear B-cell epitope prediction. Front. Immunol. 2018, 9, 1695. 10.3389/fimmu.2018.01695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui H.-H.; Sidney J.; Li W.; Fusseder N.; Sette A. Development of an epitope conservancy analysis tool to facilitate the design of epitope-based diagnostics and vaccines. BMC Bioinf. 2007, 8, 361. 10.1186/1471-2105-8-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S. F.; Gish W.; Miller W.; Myers E. W.; Lipman D. J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. 10.1016/s0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Mehla K.; Ramana J. Identification of epitope-based peptide vaccine candidates against enterotoxigenic Escherichia coli: a comparative genomics and immunoinformatics approach. Mol. Biosyst. 2016, 12, 890–901. 10.1039/c5mb00745c. [DOI] [PubMed] [Google Scholar]
- Lamiable A.; Thévenet P.; Rey J.; Vavrusa M.; Derreumaux P.; Tufféry P. PEP-FOLD3: fasterde novostructure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016, 44, W449–W454. 10.1093/nar/gkw329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guex N.; Peitsch M. C. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera?A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- O’Boyle N. M.; Banck M.; James C. A.; Morley C.; Vandermeersch T.; Hutchison G. R. Open Babel: An open chemical toolbox. J. Cheminf. 2011, 3, 33. 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Accelrys Discovery Studio . Discovery Studio Modeling Environment, Release 3.5. Accelrys Discovery Studio; Accelrys Software Inc: San Diego: 2012.
- Adhikari U. K.; Tayebi M.; Rahman M. M. Immunoinformatics approach for epitope-based peptide vaccine design and active site prediction against polyprotein of emerging oropouche virus. J. Immunol. Res. 2018, 2018, 6718083. 10.1155/2018/6718083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui H.-H.; Sidney J.; Dinh K.; Southwood S.; Newman M. J.; Sette A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinf. 2006, 7, 153. 10.1186/1471-2105-7-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen M.; Lundegaard C.; Buus S.; Lund O.; Nielsen M. MHCcluster, a method for functional clustering of MHC molecules. Immunogenetics 2013, 65, 655–665. 10.1007/s00251-013-0714-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan M.; Ghosh P. P.; Azim K. F.; Mukta S.; Abir R. A.; Nahar J.; Hasan Khan M. M. Reverse vaccinology approach to design a novel multi-epitope subunit vaccine against avian influenza A (H7N9) virus. Microb. Pathog. 2019, 130, 19–37. 10.1016/j.micpath.2019.02.023. [DOI] [PubMed] [Google Scholar]
- Dorosti H.; Eslami M.; Negahdaripour M.; Ghoshoon M. B.; Gholami A.; Heidari R.; Dehshahri A.; Erfani N.; Nezafat N.; Ghasemi Y. Vaccinomics approach for developing multi-epitope peptide pneumococcal vaccine. J. Biomol. Struct. Dyn. 2019, 37, 3524–3535. 10.1080/07391102.2018.1519460. [DOI] [PubMed] [Google Scholar]
- Nain Z.; Abdulla F.; Rahman M. M.; Karim M. M.; Khan M. S. A.; Sayed S. B.; Mahmud S.; Rahman S. M. R.; Sheam M. M.; Haque Z.; Adhikari U. K. Proteome-wide screening for designing a multi-epitope vaccine against emerging pathogen Elizabethkingia anophelis using immunoinformatic approaches. J. Biomol. Struct. Dyn. 2020, 38, 4850–4867. 10.1080/07391102.2019.1692072. [DOI] [PubMed] [Google Scholar]
- Olejnik J.; Hume A. J.; Mühlberger E. Toll-like receptor 4 in acute viral infection: too much of a good thing. PLoS Pathog. 2018, 14, e1007390 10.1371/journal.ppat.1007390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdellrazeq G. S.; Fry L. M.; Elnaggar M. M.; Bannantine J. P.; Schneider D. A.; Chamberlin W. M.; Mahmoud A. H. A.; Park K.-T.; Hulubei V.; Davis W. C. Simultaneous cognate epitope recognition by bovine CD4 and CD8 T cells is essential for primary expansion of antigen-specific cytotoxic T-cells following ex vivo stimulation with a candidate Mycobacterium avium subsp. paratuberculosis peptide vaccine. Vaccine 2020, 38, 2016–2025. 10.1016/j.vaccine.2019.12.052. [DOI] [PubMed] [Google Scholar]
- Borthwick N.; Silva-Arrieta S.; Llano A.; Takiguchi M.; Brander C.; Hanke T. Novel nested peptide epitopes recognized by CD4+ T cells induced by HIV-1 conserved-region vaccines. Vaccines 2020, 8, 28. 10.3390/vaccines8010028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E.; Hoogland C.; Gattiker A.; Duvaud S. e.; Wilkins M. R.; Appel R. D.; Bairoch A.. Protein identification and analysis tools on the ExPASy server. The Proteomics Protocols Handbook; Humana Press, 2005; pp 571–607. [Google Scholar]
- Saha S.; Raghava G. P. S. AlgPred: prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res. 2006, 34, W202–W209. 10.1093/nar/gkl343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebditch M.; Carballo-Amador M. A.; Charonis S.; Curtis R.; Warwicker J. Protein-Sol: a web tool for predicting protein solubility from sequence. Bioinformatics 2017, 33, 3098–3100. 10.1093/bioinformatics/btx345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa T.; Ying B.-W.; Saito K.; Jin W.; Takada S.; Ueda T.; Taguchi H. Bimodal protein solubility distribution revealed by an aggregation analysis of the entire ensemble of Escherichia coli proteins. Proc. Natl. Acad. Sci. 2009, 106, 4201–4206. 10.1073/pnas.0811922106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen H. Predicting secretory proteins with SignalP. Methods Mol. Biol. 2017, 1611, 59–73. 10.1007/978-1-4939-7015-5_6. [DOI] [PubMed] [Google Scholar]
- Altschul S.; Madden T. L.; Schäffer A. A.; Zhang J.; Zhang Z.; Miller W.; Lipman D. J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S. F.; Wootton J. C.; Gertz E. M.; Agarwala R.; Morgulis A.; Schaffer A. A.; Yu Y.-K. Protein database searches using compositionally adjusted substitution matrices. FEBS J. 2005, 272, 5101–5109. 10.1111/j.1742-4658.2005.04945.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geourjon C.; Deléage G. SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Bioinformatics 1995, 11, 681–684. 10.1093/bioinformatics/11.6.681. [DOI] [PubMed] [Google Scholar]
- Buchan D. W. A.; Minneci F.; Nugent T. C. O.; Bryson K.; Jones D. T. Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 2013, 41, W349–W357. 10.1093/nar/gkt381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomerie S.; Sundararaj S.; Gallin W. J.; Wishart D. S. Improving the accuracy of protein secondary structure prediction using structural alignment. BMC Bioinf. 2006, 7, 301. 10.1186/1471-2105-7-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Källberg M.; Wang H.; Wang S.; Peng J.; Wang Z.; Lu H.; Xu J. Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 2012, 7, 1511–1522. 10.1038/nprot.2012.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Li W.; Liu S.; Xu J. RaptorX-Property: a web server for protein structure property prediction. Nucleic Acids Res. 2016, 44, W430–W435. 10.1093/nar/gkw306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J.; Park H.; Heo L.; Seok C. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res. 2012, 40, W294–W297. 10.1093/nar/gks493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederstein M.; Sippl M. J. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R. A.; MacArthur M. W.; Moss D. S.; Thornton J. M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. 10.1107/s0021889892009944. [DOI] [Google Scholar]
- Hodder A. N.; Crewther P. E.; Matthew M. L. S. M.; Reid G. E.; Moritz R. L.; Simpson R. J.; Anders R. F. The disulfide bond structure of Plasmodium apical membrane antigen-1. J. Biol. Chem. 1996, 271, 29446–29452. 10.1074/jbc.271.46.29446. [DOI] [PubMed] [Google Scholar]
- Craig D. B.; Dombkowski A. A. Disulfide by Design 2.0: a web-based tool for disulfide engineering in proteins. BMC Bioinf. 2013, 14, 346. 10.1186/1471-2105-14-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarenko J.; Bourne P.; Li W.; Fusseder N.; Bourne P. E.; Sette A.; Peters B. Antibody-protein interactions: benchmark datasets and prediction tools evaluation. BMC Bioinf. 2008, 9, 1–8. 10.4016/5328.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozakov D.; Hall D. R.; Xia B.; Porter K. A.; Padhorny D.; Yueh C.; Beglov D.; Vajda S. The ClusPro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. 10.1038/nprot.2016.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Aalten D. M. F.; De Groot B. L.; Findlay J. B. C.; Berendsen H. J. C.; Amadei A. A comparison of techniques for calculating protein essential dynamics. J. Comput. Chem. 1997, 18, 169–181. . [DOI] [Google Scholar]
- Tama F.; Brooks C. L. III Symmetry, form, and shape: guiding principles for robustness in macromolecular machines. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 115–133. 10.1146/annurev.biophys.35.040405.102010. [DOI] [PubMed] [Google Scholar]
- Prabhakar P. K.; Srivastava A.; Rao K. K.; Balaji P. V. Monomerization alters the dynamics of the lid region inCampylobacter jejuniCstII: an MD simulation study. J. Biomol. Struct. Dyn. 2016, 34, 778–791. 10.1080/07391102.2015.1054430. [DOI] [PubMed] [Google Scholar]
- Del Tordello E.; Rappuoli R.; Delany I. Reverse Vaccinology. Hum. Vaccine 2017, 65–86. 10.1016/b978-0-12-802302-0.00002-9. [DOI] [Google Scholar]
- Gill S. C.; Von Hippel P. H. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989, 182, 319–326. 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- Land H.; Humble M. S. YASARA: a tool to obtain structural guidance in biocatalytic investigations. Methods Mol. Biol. 2018, 1685, 43–67. 10.1007/978-1-4939-7366-8_4. [DOI] [PubMed] [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Harrach M. F.; Drossel B. Structure and dynamics of TIP3P, TIP4P, and TIP5P water near smooth and atomistic walls of different hydroaffinity. J. Chem. Phys. 2014, 140, 174501. 10.1063/1.4872239. [DOI] [PubMed] [Google Scholar]
- Krieger E.; Dunbrack R. L.; Hooft R. W. W.; Krieger B. Assignment of Protonation States in Proteins and Ligands: Combining pKa Prediction with Hydrogen Bonding Network Optimization. Methods Mol. Biol. 2012, 819, 405–421. 10.1007/978-1-61779-465-0_25. [DOI] [PubMed] [Google Scholar]
- Krieger E.; Nielsen J. E.; Spronk C. A. E. M.; Vriend G. Fast empirical pKa prediction by Ewald summation. J. Mol. Graph. Model. 2006, 25, 481–486. 10.1016/j.jmgm.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. 10.1063/1.470117. [DOI] [Google Scholar]
- Krieger E.; Vriend G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. 10.1002/jcc.23899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey M. J.; De Fabritiis G. An implementation of the smooth particle mesh Ewald method on GPU hardware. J. Chem. Theory Comput. 2009, 5, 2371–2377. 10.1021/ct900275y. [DOI] [PubMed] [Google Scholar]
- Mahmud S.; Uddin M. A. R.; Paul G. K.; Shimu M. S. S.; Islam S.; Rahman E.; Islam A.; Islam M. S.; Promi M. M.; Emran T. B.; Saleh M. A. Virtual screening and molecular dynamics simulation study of plant-derived compounds to identify potential inhibitors of main protease from SARS-CoV-2. Briefings Bioinf. 2021, 22, 1402–1414. 10.1093/bib/bbaa428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddin M. Z.; Paul A.; Rakib A.; Sami S. A.; Mahmud S.; Rana M. S.; Hossain S.; Tareq A. M.; Dutta M.; Emran T. B.; Simal-Gandara J. Chemical Profiles and Pharmacological Properties with In Silico Studies on Elatostema papillosum Wedd. Molecules 2021, 26, 809. 10.3390/molecules26040809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta M.; Tareq A. M.; Rakib A.; Mahmud S.; Sami S. A.; Mallick J.; Islam M. N.; Majumder M.; Uddin M. Z.; Alsubaie A.; Almalki A. S. A.; Khandaker M. U.; Bradley D. A.; Rana M. S.; Emran T. B. Phytochemicals from Leucas zeylanica Targeting Main Protease of SARS-CoV-2: Chemical Profiles, Molecular Docking, and Molecular Dynamics Simulations. Biology 2021, 10, 789. 10.3390/biology10080789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakib A.; Nain Z.; Sami S. A.; Mahmud S.; Islam A.; Ahmed S.; Siddiqui A. B. F.; Babu S. M. O. F.; Hossain P.; Shahriar A.; Nainu F.; Emran T. B.; Simal-Gandara J. A molecular modelling approach for identifying antiviral selenium-containing heterocyclic compounds that inhibit the main protease of SARS-CoV-2: an in silico investigation. Briefings Bioinf. 2021, 22, 1476–1498. 10.1093/bib/bbab045. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.