

SUMMARY
Somatic stem cells contribute to tissue ontogenesis, homeostasis, and regeneration through sequential processes. Systematic molecular analysis of stem cell behavior is challenging because classic approaches cannot resolve cellular heterogeneity or capture developmental dynamics. Here we provide a comprehensive resource of single-cell transcriptomes of adult hippocampal quiescent neural stem cells (qNSCs) and their immediate progeny. We further developed Waterfall, a bioinformatic suite, to statistically quantify singe-cell gene expression along de novo reconstructed continuous developmental trajectory. Our study reveals molecular signatures of adult qNSCs, characterized by active niche signaling integration and low protein translation capacity. Our analyses further delineate molecular cascades underlying qNSC activation and neurogenesis initiation, exemplified by decreased extrinsic signaling capacity, primed translational machinery, and regulatory switches in transcription factors, metabolism, and energy sources. Our study reveals the molecular continuum underlying adult neurogenesis and illustrates how Waterfall can be used for single-cell omics analyses of various continuous biological processes.
INTRODUCTION
In discrete regions of the adult mammalian brain, quiescent neural stem cells (qNSCs) continuously generate new neurons through a recurrent process involving quiescent to active state transitions, cell cycle entry and neuronal fate specification (Ming and Song, 2011). Understanding molecular mechanisms underlying adult NSC regulation and neurogenesis will advance our knowledge of neural development and plasticity and enable new approaches for regenerative medicine and treatment of brain disorders. Mechanistic analysis of stem cell biology requires comprehensive quantification of molecular properties, such as gene expression. In contrast to traditional approaches targeting individual candidate genes, transcriptome profiling through RNA-sequencing (RNA-seq) provides an unbiased and quantitative proxy for molecular features of cellular states. Such a blueprint may reveal unexpected features of NSC biology, generate hypotheses for functional analysis, and lead to novel strategies to manipulate neurogenesis processes.
Classic approaches for molecular characterization of somatic stem cell behavior use population-based readouts at a few time points along development, which faces two major challenges: resolving cellular heterogeneity and capturing developmental dynamics. Adult stem cells constitute a minor population within complex tissues, intermingled with their progeny at different developmental stages and supporting cells. They switch among different states, such as quiescence and activation (Li and Clevers, 2010), and thus exhibit significant cellular and molecular differences even upon prospective isolation via fluorescence-activated cell sorting or genetic labeling (Codega et al., 2014; Lu et al., 2011). Furthermore, snapshots of molecular composition at selected time points are not sufficient to understand the dynamic nature of stem cell development.
Single-cell RNA-seq generates gene expression profiles at the resolution of an individual cell and has thus far revealed molecular profiles of cell types that were not previously recognized at the population level (Stegle et al., 2015). Single-cell RNA-seq has not yet been widely adopted for adult somatic stem cell studies due to technical difficulties in obtaining individual stem cells from complex tissues. Further, the stochastic nature of gene expression in individual cells (Muramoto et al., 2012; Novick and Weiner, 1957; Raj et al., 2006) may lead to overestimation of cellular heterogeneity and requires a new approach for statistical quantification. And for biological systems with only a few known markers, current approaches are not sufficient to map out a developmental trajectory at high resolution from single-cell datasets (Bendall et al., 2014; Trapnell et al., 2014).
Despite recent advances in acquiring snapshots of transcriptomes, epigenomes, and proteomes from individual cells, a remaining hurdle is the lack of methodology to identify molecular state transitions over a developmental continuum. Cells are destroyed during acquisition of omic data, so the same cell can’t be tracked over time. Here we developed a conceptually different approach, analogous to the “shot-gun” method used in the human genome project characterized by parallel sequencing and bioinformatic reconstruction. We focused on the narrow time window of adult qNSC activation and neurogenesis initiation. Using the Nestin-CFPnuc transgenic genetic labeling system, we produced single-cell transcriptomes from a mixed population of precursor cells at different developmental stages. We then developed a bioinformatic pipeline named Waterfall to reconstruct continuous biological processes at single-cell resolution using adult neurogenesis as our model, and applied this methodology to other stem cell datasets.
RESULTS
Single-cell RNA-seq of Neural Precursor Cells from the Adult Mouse Dentate Gyrus
In the adult dentate gyrus, radial glia-like qNSCs give rise to new neurons via a sequential process of activation, proliferation and generation of intermediate precursor cells (IPCs; Figure 1A) (Ming and Song, 2011). To elucidate detailed molecular dynamics during initial phases of adult neurogenesis in vivo, we used a transgenic mouse line that expresses nucleus-localized cyan fluorescent protein (CFP) under the Nestin regulatory elements (Nes-CFPnuc) (Encinas et al., 2006), in which CFP proteins carry over from adult qNSCs to their immediate progeny (collectively named NPCs; Figure S1A). The SMART-seq protocol (Ramskold et al., 2012) was modified by adding DNase I treatment step to remove genomic DNA for single-cell cDNA amplification (Figure S1A). In total, we performed single-cell RNA-seq for 142 CFPnuc+ and 26 CFPnuc– single cells (Table S1). Total RNA from wild-type adult mouse dentate gyri was serially diluted to 3 pg and processed in parallel for comparison.
Figure 1. Single-cell transcriptomes of adult neural stem cells and their immediate progeny.
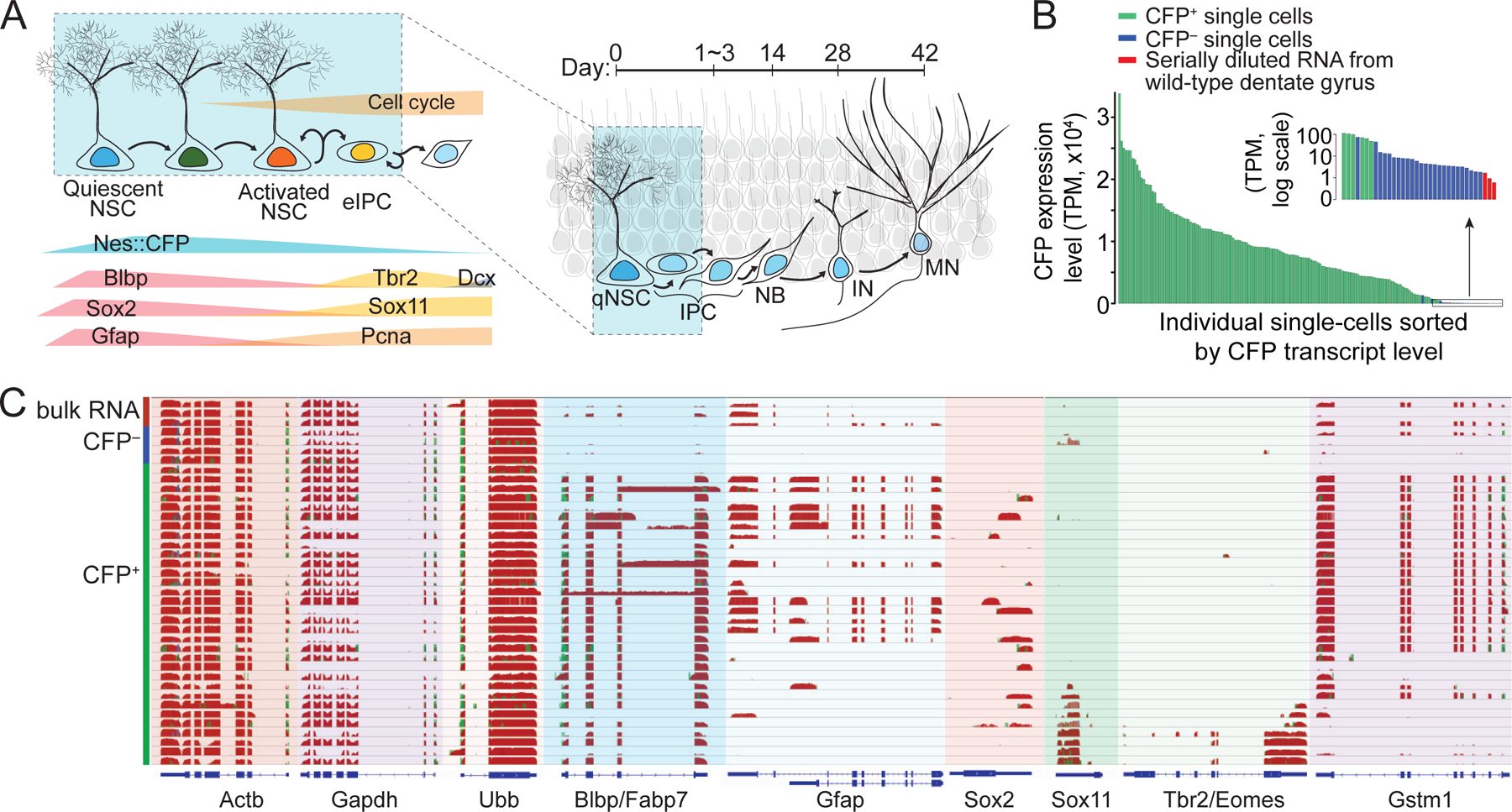
(A) A schematic diagram of the process of adult neurogenesis in the dentate gyrus of the mouse hippocampus. Once quiescent neural stem cells (qNSCs) become activated (aNSCs), they enter cell cycle and generate early intermediate progenitor cells (eIPCs), which in turn give rise to neuroblasts (NB), immature neurons (IN) and then mature neurons (MN). Area highlighted with blue background indicates cell types fluorescently labeled in adult Nestin-CFPnuc animals.
(B) Expression levels of transcript encoding CFP in each single cell and diluted whole dentate RNA samples (TPM, transcripts per million). Inset: enlarged view of CFP transcript levels in logarithmic scale of samples with low abundance of CFP transcript.
(C) Representative coverage profile of diluted total RNA from the whole dentate gyrus, CFP– individual cells, and CFP+ individual cells at selected genomic loci, including house-keeping genes (β-actin/Actb, Gapdh, ubiquitin B/Ubb), known NSC markers (Blbp/Fabp7, Gfap, Sox2), known IPC markers (Sox11, Tbr2/Eomes), and potential new NPC markers (Gstm1).
See also Figure S1.
We achieved, on average, 87% mapping onto annotated genes (Table S1). Sequencing reads were evenly distributed throughout the whole span of transcripts with 3’ bias comparable to recent studies (See Supplementary Methods). Correlation analyses of RNA samples from different batches indicated minimal technical fluctuation during cDNA amplification or across batches compared to significant biological heterogeneity among single-cell transcriptomes (Figure S1B; Table S1). Universally expressed genes, such as β-Actin/Actb, Gapdh, or Ubiquitin B/Ubb, showed even expression patterns across all individual cells, whereas known NSC markers Gfap and Sox2, or early IPC (eIPC) markers Tbr2/Eomes and Sox11, were expressed in subsets of cells (Figure 1C; Table S2).
Nes-CFPnuc reporting system also labeled a small percentage of non-NPCs in the adult dentate gyrus (Figure S1) (Encinas et al., 2011). CFP transcript levels were multiple orders of magnitude higher in CFPnuc+ cells compared to CFPnuc– cells or diluted dentate RNA (Figure 1B). We excluded cells that exhibited transcriptomic profiles markedly different from the majority of the CFPnuc+ population or were clearly identifiable as non-NPCs, such as oligodendrocyte progenitor cells or pericytes (Figure S1C). By differential expression analysis, we identified the top 35 genes enriched in CFPnuc+ cells, which included known NSC markers, Blbp, Spot14/Thrsp, Sox9 and GLAST/Slc1a3 (Tables S2 and S3). In total, 31 out of 35 top genes exhibited SGZ-enriched expression patterns and/or were known NPC genes (p = 6.1× 10−40; hypergeometric test; Table S3). These results provided initial validation of our approach.
Waterfall: Analyzing Single-Cell Datasets from Continuous in vivo Processes
We next examined the whole-transcriptome dataset of individual CFPnuc+ NPCs. Unsupervised hierarchical clustering analysis resulted in two super-groups with six sub-groups (Figure 2A). Notably, these six CFPnuc+ groups were not clearly segregated on the plot of principal component analysis (PCA; Figure 2B), which was consistent over different batches of sequencing runs with multiple biological replicates (Figure S2A; Table S1). The continuous trajectory called for a new approach not relying on segmentation into a few groups of cell clusters. We could not use currently available single-cell analysis software, such as Monocle (Trapnell et al., 2010) or Wanderlust (Bendall et al., 2014), for our system due to the lack of sufficient prior information, such as temporal delineators or a robust set of specific markers (See Supplementary Methods). We thus developed a more generally applicable pipeline of algorithms to perform unbiased statistical analyses of multidimensional single-cell datasets from continuous biological processes. We collectively named the suite of algorithms “Waterfall”, which involves three steps: pre-processing, pseudotime reconstruction, and gene expression analysis (Figure 2C; See Experimental Procedures and Supplementary Methods).
Figure 2. Waterfall for analyzing single-cell data from continuous in vivo process.
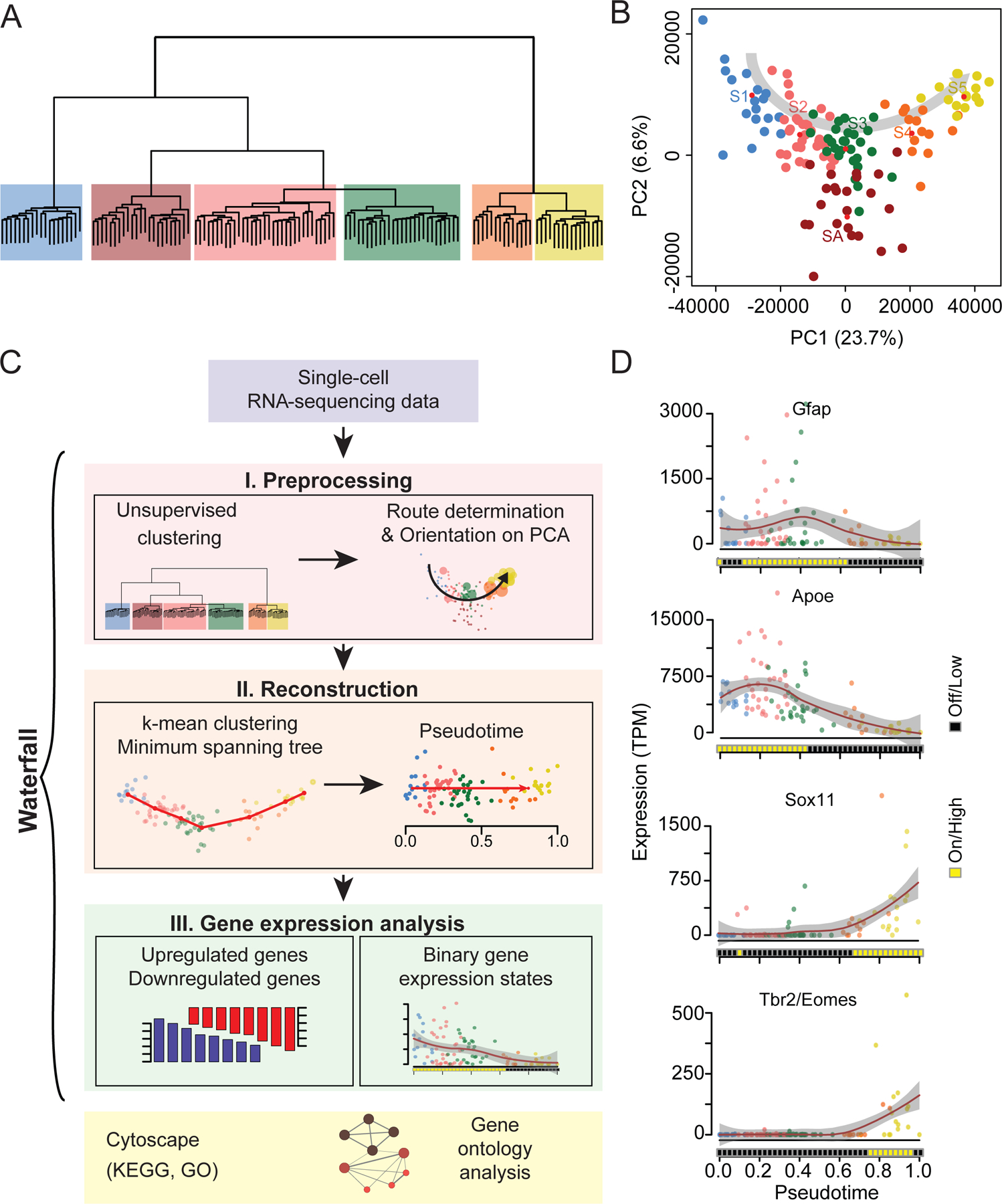
(A) Unsupervised clustering analysis of CFPnuc+ NPCs resulting in two super-groups with six subgroups. Different groups are color coded in the same fashion in this figure and across all other figures.
(B) Principal component analysis (PCA) plot shows one of the possible linear trajectories of different groups with the exception of SA.
(C) A schematic diagram of multiple components and workflow of Waterfall. Waterfall is a full range of algorithms for processing multi-dimensional single-cell datasets derived from continuous biological processes. Please see Supplementary Methods for more information and Waterfall analyses of other biological systems.
(D) Representative expression profiles of marker genes of adult neurogenesis. Each data point represents the gene expression level of a single cell with color scheme following Figure 2B. Data points are fitted with local polynomial regression fitting (red lines) with 95% confidence interval (gray area). HMM-predicted underlying states are represented as black and yellow squares on the bottom of the graphs.
See also Figures S2 and S3
Pre-processing defined the trajectory of interest following dimensionality reduction of the data. Unsupervised learning identified six clusters of cells (Figure 2A), which were then labeled S1–5 and SA based on their relative location in a PCA plot (Figure 2B). SA was recognized as a branch by the minimum spanning tree (MST) algorithm (See Supplementary Methods). Although characterizing SA would be interesting (See Supplementary Methods), we focused on the major neurogenic pathway in the current study. The expression profiles of a few known developmental genes were used to orient the most probable trajectory of interest (Figures 2B and S2B).
To reconstruct the chronology, we first determined the most probable route of transcriptomic progression. We performed k-means clustering of single-cell transcriptomes on the PCA plot after excluding SA, followed by constructing a MST trajectory to connect cluster centers (Figure S3A). We then introduced “pseudotime” (Trapnell et al., 2014) to define the relative location of each cell on the MST trajectory (Figure S3A). Taking the Euclidian distance defined by the whole transcriptomic difference from each cell to the next in pseudotime, we found that the total path length reconstructed by Waterfall was significantly shorter than would result from random ordering of cells (See Supplementary Methods). The pseudotime algorithm reconstructs molecular state transitions of a continuous process by quantifying the gradual divergence of single-cell transcriptomes individually, rather than as members of pre-classified groups.
For gene expression analysis, we developed an algorithm to determine the binary on/high or off/low expression state of each gene along pseudotime in an unbiased fashion using a hidden Markov model (HMM; Figure S3B). Gene expression at the single-cell level is highly stochastic and binary (Muramoto et al., 2012; Novick and Weiner, 1957; Raj et al., 2006). When analyzing continuous processes represented by single-cell transcriptomes in the absence of discrete groups, conventional statistical methods, such as arithmetic mean or t-test, are not appropriate. We adopted HMM to statistically convert stochastic expression patterns of individual genes into binary on/high or off/low states (Figure S3B). The binary gene expression states were then shown as heat maps, which quantified the molecular cascade over time (Figures 2C-D). To discover novel developmentally regulated genes, we correlated gene expression levels with pseudotime and subjected identified genes to gene ontology (GO) analyses (Figure 2C).
Validation for the Reconstructed Adult Neurogenesis Process
We validated molecular dynamics revealed by Waterfall at multiple levels. First, known NSC markers Gfap and Apoe, and eIPC markers Sox11 and Tbr2, showed non-overlapping expression states over developmental pseudotime (Figure 2D; Table S2). Second, we evaluated in vivo expression of Aldoc and Stmn1, which have not been previously studied in adult hippocampal neurogenesis. Over pseudotime, Aldoc was initially highly expressed (on/high state), then downregulated (off/low state), whereas Stmn1 was initially off, then upregulated (Figures 3A and 3C). In the hippocampal dentate subgranular zone (SGZ) of adult Nes-GFPcyto mice (Mignone et al., 2004), Aldoc+GFP+ precursors were almost exclusively PCNA– qNSCs, with very few PCNA+ active NSCs (aNSCs) or IPCs (Figure 3B). In contrast, Stmn1+GFP+ precursors were mostly eIPCs and PCNA+ aNSCs, but not PCNA– qNSCs (Figure 3D). Thus, Waterfall accurately predicted the in vivo expression dynamics of both known and unknown genes. Third, for functional validation, we explored the possibility of genetic labeling of a specific developmental stage based on Waterfall results. Hopx (Hop homeobox) was highly expressed in qNSCs, but downregulated around the transition point from qNPC to aNSC (Figure 3E). Upon a single low-dose tamoxifen injection into a Hopx-CreERT2 mouse line (Takeda et al., 2011) crossed with a mT/mGf/f reporter line for clonal lineage-tracing (Bonaguidi et al., 2011), almost all labeled precursors at three days post injection were nestin+GFAP+ qNSCs in the adult SGZ (Figure 3F). By 7 days, we were able to observe GFP-labeled clones that contained both NSCs and their progeny (Figure 3G), indicating self-renewal and differentiation, two hallmarks of NSCs.
Figure 3. Validation for Waterfall predictions for early adult neurogenesis.
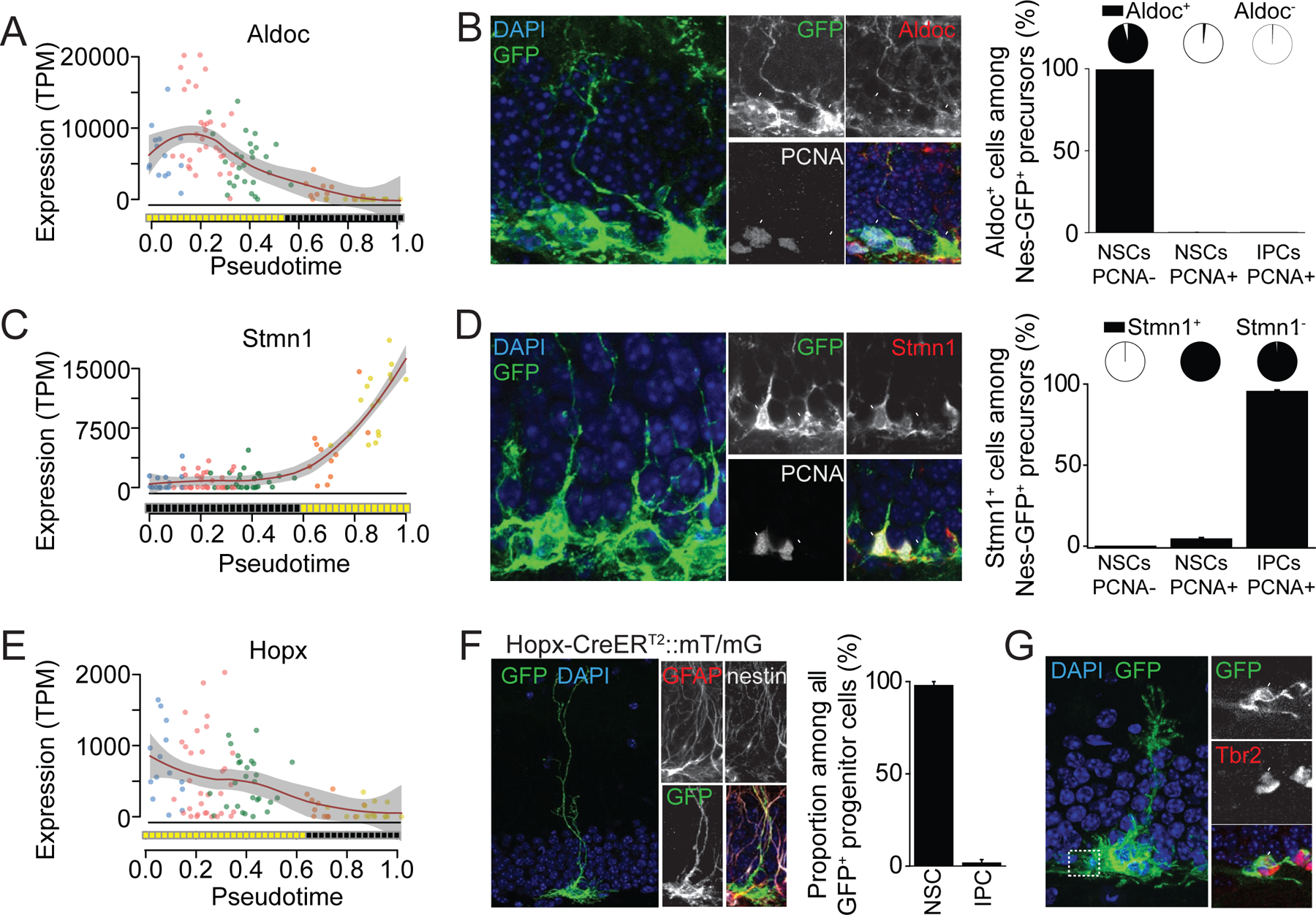
(A-D) Validation of gene expression patterns and on/off binary states of Aldolase C (Aldoc, A) and Stmn1 (C) over pseudotime by immunohistology. Also shown are sample confocal images of GFP, cell proliferation marker PCNA, Aldoc or Stmn1 in the dentate gyrus of adult Nestin-GFPcyco mice (left panels) and quantifications (right panels). Values represent mean ± SEM (n = 3 animals). The pie chart represents the proportion of Aldoc+ or Stmn1+ cells among each category of the GFP+ progenitor population. Scale bars: 20 µm (left) and 10 µm (right).
(E-G) Validation of gene expression patterns and on/off binary states of Hopx over pseudotime (E) by genetic labeling and lineage-tracing. Adult Hopx-CreERT2::mT/mG mice were injected with a single dose of tamoxifen and examined 3 (F) or 7 days (G) later. Shown in (F) are sample confocal images of GFP, GFAP, Nestin and DAPI. Also shown is quantification of percentages of GFP+ cells as NSCs or IPCs. Values represent mean ± SEM (n = 5 dentate gyri). Shown in (G) is an example of a labeled clone containing an NSC and multiple Tbr2+ neuronal progeny. Scale bars: 20 µm (left) and 10 µm (right).
Transcription Factor Expression during Adult NSC Activation and Neurogenesis
Waterfall allows an unbiased prediction of the relative chronological position of each individual cell and distribution of binary gene expression over the developmental trajectory. To delineate molecular cascades underlying adult qNSC activation and neurogenesis, we generated a list of the top 1000 negatively correlated genes with pseudotime (DOWN1000 genes; Spearman correlation coefficient < –0.13; Table S4), which represent qNSC-enriched genes down-regulated during activation and neurogenesis, as well as the top 1000 positively correlated genes with pseudotime (UP1000 genes; Spearman correlation coefficient > 0.20; Table S4), which represent newly activated genes during qNSC activation and early neurogenesis.
Out of these 2000 genes, we initially focused on transcription factors (TFs). Most known TFs involved in adult neurogenesis were discovered by extrapolating findings from embryonic studies. In contrast, our database provides unbiased genome-wide profiles of TF expression. Systematic analyses of our dataset revealed a total of 41 down-regulated TFs and 42 up-regulated TFs during adult hippocampal neurogenesis (Figures 4A and S4; Table S5).
Figure 4. Molecular cascade underlying adult quiescent neural stem cell activation and neurogenesis initiation.
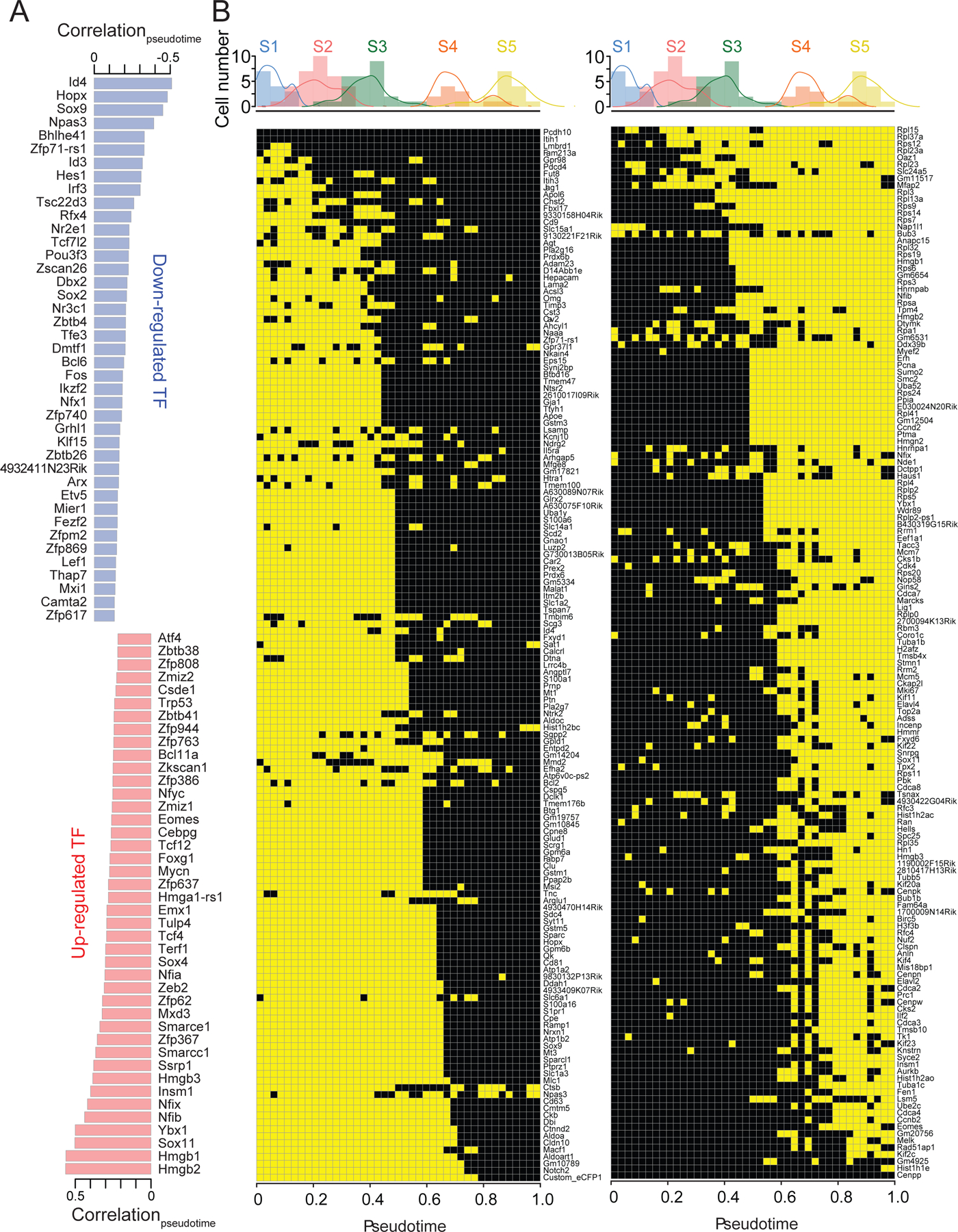
(A) Lists of DOWN and UP TFs and their Spearman correlation coefficient with pseudotime.
(B) ON/HIGH (yellow) or OFF/LOW (black) states of top 150 DOWN (left) and UP (right) genes sorted by the timing of transition points. Shown on the top are histograms of the numbers of individual cells examined along the pseudotime progression. The colors on the histogram follow the color scheme of Figure 2B.
See also Figure S4.
First, the set of dynamic TFs we identified included known regulators of adult NSCs and neurogenesis, which provided additional validation of our approach. Among DOWN TFs, Sox2, Sox9, Id3, nuclear receptor Nr2e1/Tlx, and Hes1 have been shown to regulate adult NSC maintenance and function. Among UP TFs, SoxC (Sox4 and Sox11), Foxg1, Tbr2, Insm1, Tcf12 and Nfib are critical in proliferative adult NPCs (Table S2).
Second, we identified multiple dynamic TFs that are regulators of embryonic neurogenesis but have not yet been studied in adult neurogenesis. DOWN TFs include homeobox protein Dbx2, nuclear glucocorticoid receptor Nr3c1 and Id4. UP TFs included chromatin protein Hmgb1 and proto-oncogene N-myc. During embryonic neurogenesis, these DOWN TFs inhibit cell cycle (Nr3c1) or prevent premature differentiation (Id4), whereas UP TFs regulate progenitor proliferation (Hmgb1, N-myc), suggesting conserved functions during embryonic and adult neurogenesis (Table S2).
Third, more than half of UP TFs and DOWN TFs are largely uncharacterized in the context of neurogenesis, but many of them are close paralogs or binding partners to other neurogenesis-related genes, or have been implicated in other somatic stem cell systems. Examples include Hmgb1 paralogs (Hmgb2, Hmgb3, Hmga1-rs1), SWI/SNF-related Brg1/Smarca4-associated factors (Smarcc1/Baf155, Smarce1/Baf57), and Nfib paralogs (Nfia, Nfix). In addition, Mxd3, Zeb2 and ZT3/Zfp, regulate adipocyte, melanocyte and myogenic differentiation, respectively, whereas Tsc22d3/Gilz inhibits myogenic differentiation (Table S2). Hopx, which we found to mark qNSCs in the adult dentate gyrus (Figures 3E-G), is expressed in intestinal stem cells and a subset of multipotent hair follicle stem cells (Table S2).
Together, our single-cell transcriptome datasets are a rich resource of genome-wide dynamic expression profiles of TFs during adult neurogenesis. Many TFs that were previously uncharacterized in adult neurogenesis are known to be involved in embryonic neurogenesis or regulation of other somatic stem cells, suggesting shared biology among different stem cell systems and the potential utility of our resource for the general stem cell field.
Molecular Cascades underlying Adult qNSC Activation and Neurogenesis Initiation
The vast majority of UP1000 and DOWN1000 genes in our dataset were not TFs (Table S4). We investigated their characteristics from three perspectives: transition patterns along the developmental trajectory, cellular location of gene products, and biological function.
For transition patterns, plots of the top 150 genes each from UP1000 and DOWN1000 lists showed a wave of molecular activation or inactivation events over time, highlighting the sequential transition of gene expression during qNSC activation and neurogenesis (Figure 4B). To obtain biological insight into these transition patterns, we performed multiple gene ontology analyses. Strikingly, the predicted cellular localizations of protein products of UP1000 genes and DOWN1000 genes were drastically different. 51% of DOWN1000 genes, as opposed to 20% of UP1000 genes, encode proteins associated with the membrane (Figure 5A, p = 2.6 × 10−29). On the other hand, 58% of UP1000 genes, as opposed to 20% of DOWN1000 genes, encode proteins associated with the nucleus (Figure 5A, p = 1.2 × 10−36). Similar results were obtained using different thresholds for generating lists of UP and DOWN genes (Figure S5A).
Figure 5. Functional characterization of UP1000 genes and DOWN1000 genes.
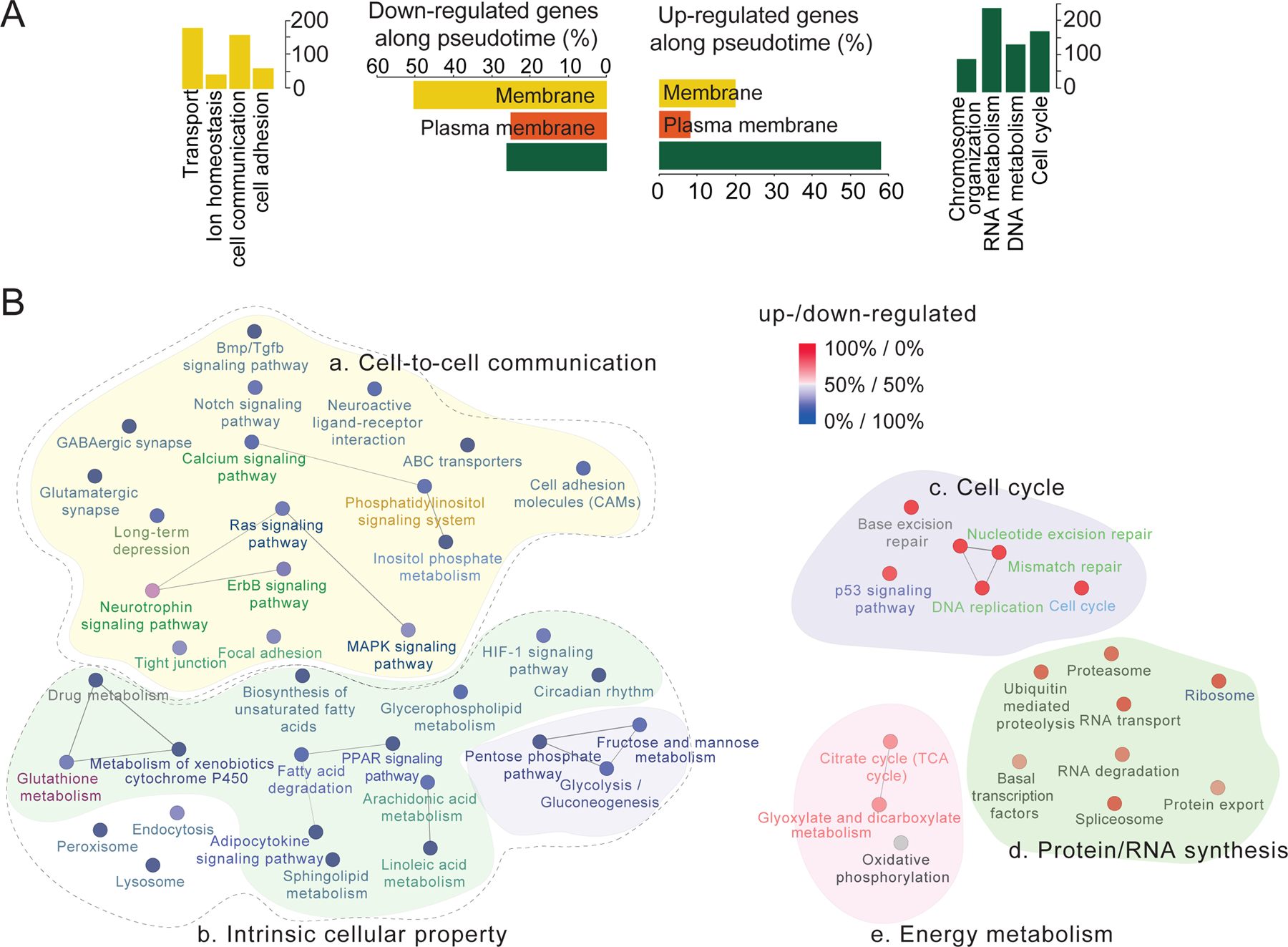
(A) Quantification of predicted cellular location of gene products of UP1000 genes and DOWN1000 genes (two middle panels). Also shown are numbers of genes with indicated functions for membrane-associated DOWN genes (left panel) and those for nucleus-associated UP genes (right panel).
(B) Functional GO analysis for DOWN1000 and UP1000 genes. Color of each functional entity represents proportion of UP genes (blue) and DOWN genes (red). Connections between each pair of data points represent sharing more than 5 genes between the pair. Functionally similar entities are grouped with same background colors. Broken lines represent two categories of DOWN genes: genes encoding proteins involved in the intra- or extra-cellular communication and genes encoding proteins defining intrinsic stem cell properties. The P values are from hypergeometric tests, and corrected by Holm–Bonferroni method.
See also Figures S5, S6 and S7.
Molecular Signatures of Adult qNSCs Revealed by DOWN1000 Genes
Functional annotation of DOWN1000 membrane genes revealed enrichment for ion or protein transport, cell communication and cell adhesion (Figure 5B). Further classification identified proteins specific to the plasma membrane, endoplasmic reticulum, Golgi apparatus, and cytoplasmic vesicles (Figure S5B). KEGG pathway analysis revealed diverse functional entities involved in intra- and inter-cellular communication (Figure 5B). Specifically, Notch signaling, GABAergic synapses, glutamatergic synapses, BMP pathways, MAPK pathway, calcium and cell adhesion-related genes were down-regulated upon qNSC exit from quiescence (Figures 5B, 6A-B and S6A). Electrophysiological recordings of Nestin-GFPcyto+ NSCs in acute slices from adult animals showed responses to both AMPA and NMDA, suggesting expression of functional receptors (Figure 6B). Each functional signaling pathway entity contained key genes that encode receptors, subunits or downstream mediators (Figures 6A-B). Notably, many ligands for these receptors, including glutamate, GABA, Wnts, BDNF/neurotrophin, Jagged1, BMPs, FGFs and Insulin/IGF2, are known to be present in the adult SGZ niche (Table S2), suggesting active signaling in qNSCs. While previous studies have examined each of these ligands and receptors in regulation of adult neurogenesis in isolation, our systematic genome-wide analyses unified disparate information and suggested a novel model that quiescent adult NSCs are not passive or dormant, but instead are actively integrating various niche signals. More surprisingly, qNSC activation was associated with decreased expression of genes involved in transducing local environment cues and pervasive down-regulation of various signaling pathway-related genes (Figure 6A). These results suggest that, once activated, adult NSCs shunt their capacity to respond to external regulation.
Figure 6. Sequential molecular dynamics during adult neural stem cell activation and neurogenesis.
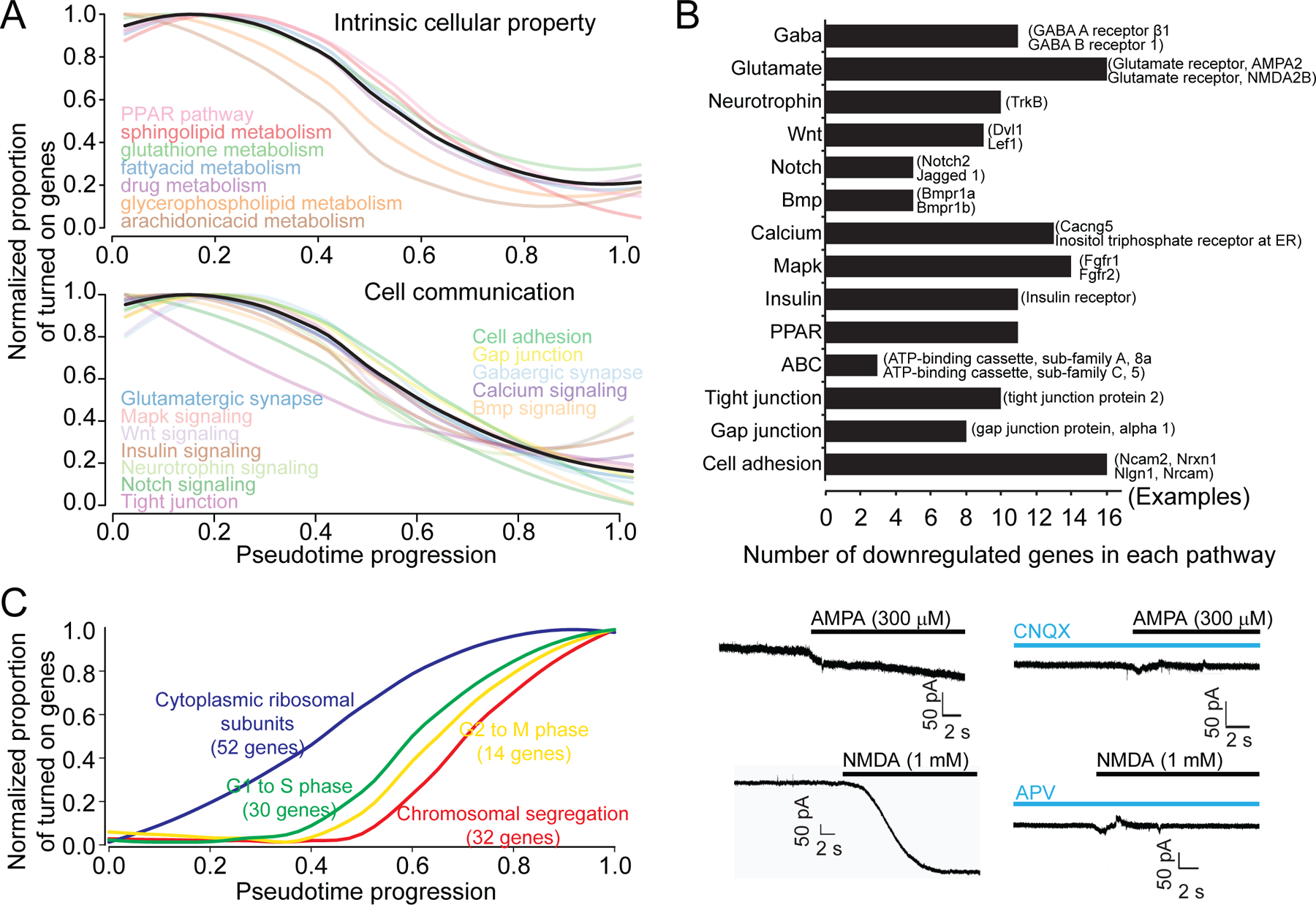
(A) Gradual down-regulation of averaged binary states of each functional entity defining intrinsic stem cell properties (top) and those defining intra- or extracellular communication (bottom). On/off binary states of expressed genes within each functional entity were averaged and normalized to show the timing of the transition.
(B) Number of genes in different DOWN gene ontology groups and representative example genes. Expression patterns of representative genes over developmental pseudotime are shown in Figure S6A. Electrophysiological recording of Nestin-GFPcyto+ NSCs in acute hippocampal slices showed responses to both AMPA and NMDA (bottom).
(C) Gradual up-regulation of averaged binary states of each functional entity defining cell cycle checkpoints and cytoplasmic ribosomal subunits. On/off binary states of up-regulated genes within each functional entity are averaged and normalized to exemplify the timing of the transition. Cell cycle checkpoint genes were up-regulated in the sequence of the cell cycle progression. The up-regulation of cytoplasmic ribosomal subunits preceded up-regulation of the earliest cell cycle checkpoint gene.
See also Figure S6.
KEGG analysis of DOWN1000 genes also revealed a shift in energy source and metabolism. First, multiple lipid metabolism-related functional entities, including fatty acid degradation and sphingolipid metabolism, were enriched in qNSCs, but down-regulated upon activation (Figure 5B). As previously reported (Knobloch et al., 2013), qNSCs exhibited the highest level of Spot14 (Figure S6B), which regulates lipid metabolism. qNSCs also maintained an active fatty acid degradation pathway (Acsl3, Acsl6, and Acsbg1; Figure S6B; Table S2). Second, pathway analysis consistently indicated glutathione metabolism and glycolysis as an adult qNSC characteristic, which was lost upon activation. Among glycolysis genes, aldolase A, aldolase C, and Ldhb decreased significantly, whereas most other glycolysis genes including Gapdh did not change during initiation of neurogenesis (Figure S6C; Table S2).
To validate results from analyses of the top 1000 significantly down-regulated genes, which contained a limited number of genes in each particular pathway, we performed analysis using all expressed genes and an independent functional annotation database wikipathway (Pico et al., 2008). Virtually identical results were obtained (Figure S7; Table S7). Together, analyses of down-regulated genes provided novel insight into molecular signatures of adult qNSCs, including both intrinsic properties and regulation of intra- or inter-cellular signaling pathways.
Sequential Molecular Dynamics during Adult Neurogenesis Revealed by UP1000 Genes
We next analyzed UP1000 genes, which were nucleus-associated and/or related to cell cycle, DNA/RNA metabolism and chromosome organization (Figure 5A). Detailed analysis revealed pervasive activation of cell cycle-related genes, ranging from cell cycle supporting genes, such as nucleotide synthesis, protein/RNA synthesis, and DNA fidelity controls (DNA repair and p53 signaling pathways), to genes directly involved in cell cycle, such as DNA replication, kinetochore complex, cyclin/cyclin-dependent kinases, or cytosolic mitotic spindle genes (Figure 5B).
As opposed to down-regulation of glycolysis-related genes, oxidative phosphorylation-related genes were up-regulated (Figure S6C). Specifically, in contrast to stable expression of earlier mitochondrial respiratory chain complexes (complex I, II, III and IV), expression of subsequent complexes (complex V) increased over pseudotime, implying a gradual completion of the full electron transport chain during neurogenesis (Figure S6C).
The high resolution of Waterfall analyses revealed temporal relationships among genes in different functional groups. Cell cycle checkpoint genes were sequentially activated following the known biological sequence of cell cycles: G1 to S transition, followed by G2 to M transition and then chromosomal segregation, indicating that our pseudotime accurately reconstructs sequential biological events (Figure S6D). Initiation of cell cycle preceded the major transcriptomic shift (Figure 6C). Notably, up-regulation of genes encoding ribosomal subunits preceded the appearance of any cell cycle checkpoint genes (Figures 6C and S6D), suggesting that priming of protein synthesis machinery may mark the G0 to G1 transition ahead of cell cycle entry during adult qNSC activation.
Together, analyses of UP1000 genes suggested that molecular dynamics of qNSC activation and initiation of neurogenesis are largely defined by priming of protein synthesis machinery, cell cycle entry, activation of RNA and protein biogenesis, and a shift in energy metabolism from glycolysis to oxidative phosphorylation. An independent approach using a different functional annotation database showed similar results (Figure S7; Table S7).
Holistic Picture of Molecular Cascades underlying Adult Neurogenesis Initiation
Based on the molecular dynamics from qNSCs to aNSCs and then eIPCs, we have reconstructed sequential waves of biological events from single-cell RNA-seq data and Waterfall (Figure 7). The process begins with adult qNSCs down-regulating transcription factors defining quiescence and decreasing competence for cell signaling (RTKs, GPCRs, neurotransmitter receptors, cytokines, calcium). Concurrently, glycolysis, glutathione and fatty acid metabolism begins to wane, while up-regulation of protein translation capacity is the first marker of a pre-activation stage. As NSCs enter cell cycle, oxidative phosphorylation becomes the primary energy source. Progression through cell cycle accompanies a major decline in NSC metabolism (glutathione, fatty acid, drug metabolism) and an increase in eIPC transcription factors. Finally, kinetochore and chromosomal segregation occurs in the first neurogenic progeny. Overall, the developmental trajectory is defined by a coordinated switch from a membrane-targeted to a nuclear-targeted transcriptome, suggesting a transition from qNSCs dominated by extrinsic signaling to eIPCs dominated by a pre-programmed intrinsic molecular cascade.
Figure 7. Schematic summary of molecular signatures of quiescent adult neural stem cells and molecular cascades underlying their activation and neurogenesis.
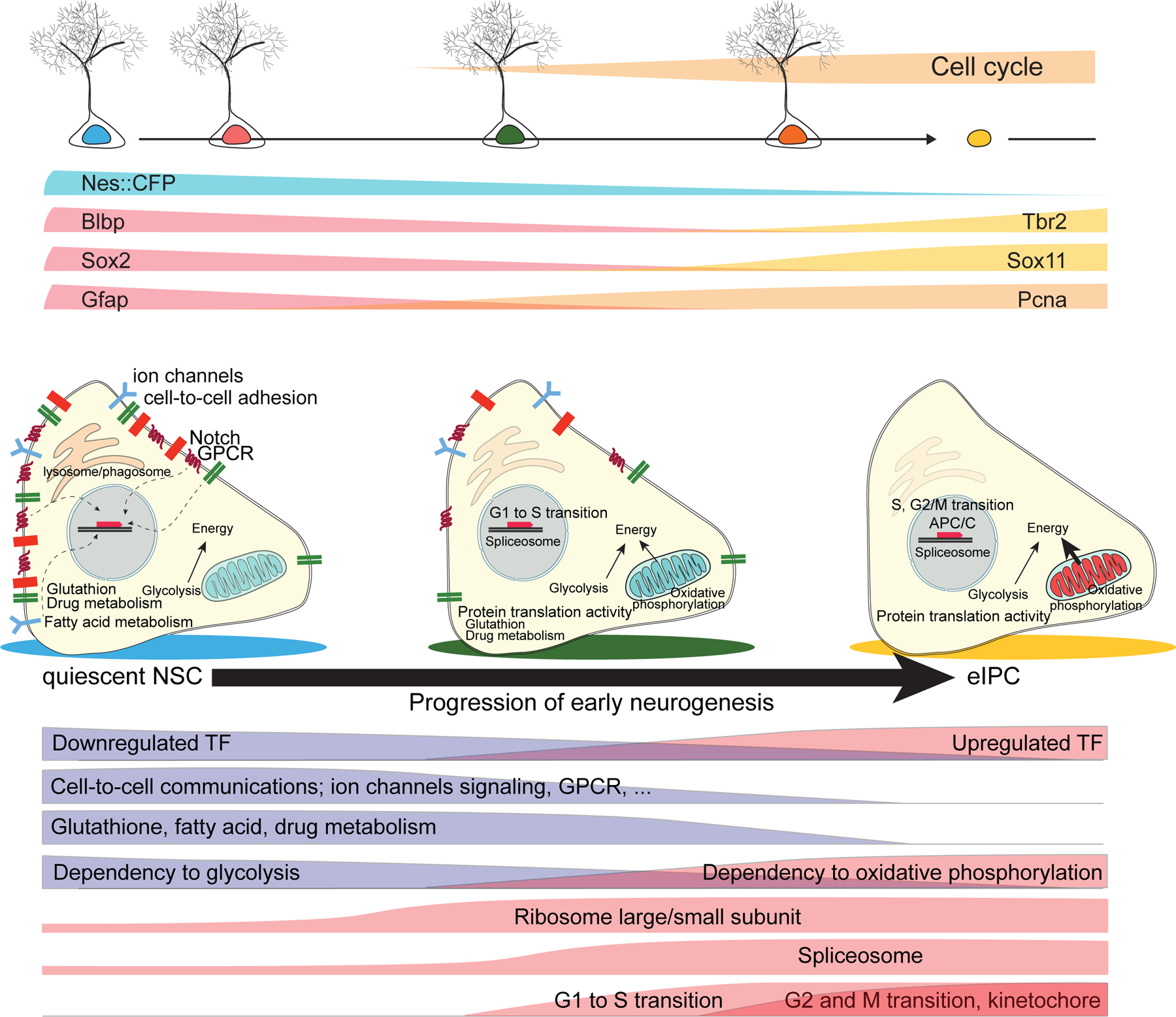
Shown on top is an illustration of known marker expression and cell cycle activation during adult hippocampal neurogenesis. Shown in the middle is an illustration of molecular signatures of adult qNSCs and their immediate progeny. Shown at the bottom are functional categories of genes that show a clear shift during adult qNSC activation and generation of eIPCs. qNSCs exhibit intra- and inter-cellular signaling to actively sense the local niche, rely mostly on glycolysis for energy, and have highly active fatty acid, glutathione, and drug metabolism. Upon activation, NSCs increase translational capacity, followed by cell cycle entry with G1 to S transition. Oxidative phosphorylation starts to be active and stem cell specific properties are down-regulated. eIPCs maintain active cell cycle genes, ribosomal activity and fully active oxidative phosphorylation for energy generation. The color scheme on the top and the middle illustration follows the colors from Figure 2B.
DISCUSSION
Understanding adult NSC behavior and neurogenesis requires quantification of molecular states along a continuous developmental process. In the current study we generated three major resources. First, we provide a comprehensive dataset of single-cell transcriptomes of qNSCs and their immediate progeny in the adult mouse hippocampus in vivo. Second, we provide Waterfall, an unsupervised bioinformatic suite for in silico reconstruction of molecular trajectories based on snapshots of single-cell transcriptomes and statistical gene expression analysis over continuous developmental processes. Third, we provide a holistic picture of adult qNSC molecular signatures and dynamic molecular cascades underlying initial phases of adult neurogenesis at unprecedented temporal resolution (Figure 7). Our study provides an example of how to resolve cellular heterogeneity and reveal developmental dynamics for systematic molecular characterization of stem cells and their differentiation in vivo. Our approach can be adapted for various single-cell omics analyses (transcriptomics, proteomics, epigenomics, lipidomics, and metabolomics) of many continuous biological processes, such as development, physiological and pharmacological stimulation, and disease progression (See examples in Supplementary Methods).
A Resource of in vivo Single-cell Transcriptomes of qNSCs and their Immediate Progeny
Our study provides a single-cell RNA-seq dataset and the first comprehensive view of transcriptome dynamics underlying adult qNSC behavior in vivo. Currently, there is no published dataset for transcriptome dynamics during stem cell development in any somatic system in vivo. We performed multiple levels of validation of our dataset and approach, including comparison with an in situ database, confirmation with known and unknown marker expression during adult neurogenesis in vivo, and functional validation via clonal lineage tracing and electrophysiology (Figures 3 and 6B; Table S3).
Our resource of holistic molecular profiles during early neurogenesishas three unique features that increase its versatility. First, our whole-transcriptome information includes unannotated transcripts, isoforms, and retrotransposon-derived transcripts, as opposed to multiplexed qPCR or microarray-based studies which can only provide limited annotated transcripts (Hoppe et al., 2014). Second, we animated static single-cell transcriptomes over the in vivo neurogenesis trajectory, allowing queries of molecular dynamics for each gene over the continuous biological process and generation of novel hypotheses during specific phases of adult qNSC maintenance, activation and neurogenesis initiation. For example, expression of nuclear glucocorticoid receptor Nr3c1 in adult qNSCs but not in eIPCs (Figure 4A) suggests a cellular target of glucocorticoids during adult hippocampal neurogenesis and a means to manipulate qNSCs in vivo. Our resource also reveals potential prospective markers of adult neurogenesis, such as using Hopx-CreERT2 to target adult qNSCs in vivo (Figures 3E-G). Third, each transcriptome in our dataset represents a biological state within a single cell. This modular construction allows for flexible reorganization of the dataset to probe different questions as our understanding of the biology evolves. For example, instead of generating reporter lines for individual genes or using different surface markers for physical sorting of specific cell populations, investigators can perform unlimited in silico cell sorting with any individual gene, or multiple genes in combination, to obtain a selected cell population to probe their gene expression characteristics at the genome-wide level using our single-cell datasets.
Developmental Dynamics of Adult Neurogenesis at the System Level
Recent genome-wide studies have begun to provide a system-level understanding of in vivo adult NSC biology using marker-defined NPC populations (Bracko et al., 2012; Codega et al., 2014; Kriegstein and Alvarez-Buylla, 2009). Yet previous studies have only acquired snapshots of transciptomes, which limits investigation of developmental dynamics among different cellular states. We co-opted the imperfection of the Nestin-CFPnuc genetic labeling system to collect individual qNSCs and their immediate neuronal progeny concurrently. Aligning cells along the developmental trajectory yielded, for the first time, a molecular continuum with sequential progression of the individual transcriptome from qNSC to aNSC and then eIPC. Importantly, this novel approach does not divide developmental processes into discrete stages that are defined a priori by capturing populations sharing specific markers.
Our resources provide unparalleled temporal resolution to identify new mechanisms underlying adult NSC biology. For example, we showed that Acyl-CoA synthetases (Acsl3, Acsl6 and Acsbg1), the enzymes for the first step of fatty acid β-oxidation, were highly expressed only in qNSCs (Figure S6B), suggesting a novel role for active fatty acid β-oxidation in qNSCs and thereby extending previous findings on the role of fatty acid metabolism in adult neurogenesis (Knobloch et al., 2013) (Table S2). We also found that ribosomal subunits were the first genes up-regulated upon adult qNSC activation and early differentiation (Figures 6C and S6D), suggesting a possible demarcation of G0 to G1 transition and providing the timing for switches in protein synthesis, the regulation of which is important for somatic stem cell function (Signer et al., 2014). The holistic picture we obtained unifies disparate information and illuminates novel biological themes in stem cell biology. For example, our analysis suggests that qNSCs actively respond to local environmental cues through various signaling pathways, but gradually and globally shut off signaling capacity upon activation. These observations support the concept of a niche wherein mammalian somatic stem cells are tightly controlled by a regulatory microenvironment (Schofield, 1978), and predict that eIPCs are less responsive to environmental input (Berg et al., 2015). This novel biological insight may be applicable to many somatic stem systems defined by stochastic behavior (Simons and Clevers, 2011).
Waterfall Analysis of Single-cell Transcriptomes within a Continuum
Waterfall has three key differences from previously methodologies. First, it requires very little prior information to generate a highly accurate temporal trajectory at single-cell resolution. Previous methods have been able to reconstruct accurate trajectories by relying on a robust set of known markers to establish cell order and validate cell alignment at numerous points along the timeline. For many biological systems and processes, we have much less information. Second, in contrast to Monocle (Trapnell et al., 2014) or Wanderlust (Bendall et al., 2014), Waterfall uses k-means clustering to build a trajectory and assign an individual cell a pseudotime based on each cell’s proximity to the cluster-derived trajectory, rather than constructing a trajectory by directly connecting each cell to the next. Third, in order to analyze stochastic gene expressions, we adopted HMM to predict consecutive binary states in gene expression activity over pseudotime. HMM permits the interpretation of highly variable data without logarithmic transformation, normalization, or the input of any arbitrary parameters, such as threshold for gene expression noise or Markovian parameters (transition probability, initial probability and emission probability). Our validation for known and unknown genes in adult neurogenesis indicated that HMM correctly predicted temporal dynamics of in vivo biology.
There is no conceptual restriction of our approach to transcriptome studies of adult neurogenesis. Indeed, in Supplementary Methods we provide examples of how Waterfall could be broadly applicable for single-cell RNA seq datasets such as in vitro myogenesis, in vivo embryonic lung development, single-cell mass-cytometry dataset from in vivo B cell development, and synthetic datasets. We expect that Waterfall algorithms can be adopted for diverse single-cell multi-dimensional datasets, including single-cell transcriptomes, epigenomes, proteomes, and metabolomes, of various continuous biological processes.
EXPERIMENTAL PROCEDURES
Preparation of Individual Cells from Adult Mouse Dentate Gyrus
Homozygous transgenic mice expressing nuclear localized CFP (CFPnuc) driven by the Nestin regulatory elements (Encinas et al., 2006) were used for all single-cell RNA-seq experiments. Mice were euthanized by cervical dislocation, and brains were immediately immersed into ice cold Dulbecco’s Phosphate-Buffered Saline (DPBS, Corning). All procedures were performed with approved protocols in accordance with institutional animal guidelines.
The dissected dentate gyrus was incubated in Hibernate A (BrainBits) containing papain (100 U; Sigma) and RNase-free DNase I (100 units; NEB) at 37°C for 18 minutes with intermittent flicking. The tissue was triturated into individual cell suspension by 1 ml pipette (Denville Scientific). Enzymes and cellular debris were removed with multiple rounds (~4–5 times) of mild centrifugation at 200g and washing with Hibernate A minus Ca2+ and Mg2+ (BrainBits). The individual cell suspension was plated onto a glass bottom plate (MatTek) and picked up using glass pipettes (World Precision Instruments) under a fluorescent microscope. The glass tip was broken into the bottom of each PCR tube containing water (2.4 µl) with RNase-free DNase I (0.2 µl; NEB) and Murine origin RNase inhibitor (0.25 µl; NEB). Importantly, the addition of DNase I significantly improved the quality of the data by removing contamination from the random amplification of genomic DNA (See Supplementary Methods Figure M4B).
Library Preparation and Sequencing
cDNA amplification followed the previously published SMART protocol (Ramskold et al., 2012). Briefly, the DNase I was first inactivated by increasing the temperature (75°C for 10 minutes) and samples were then stored on ice. Custom designed 2A oligo 1 µl (12 µM, Integrated DNA Technologies, sequence shown in Figure S1A) was added and annealed to the polyadenylated RNA by increasing temperature (75°C for 3 minutes) and quenching on ice. A mixture of 2 µl Superscript II First-Strand Buffer (5X, Invitrogen), 1 µl custom designed TS oligo (12 µM, Integrated DNA Technologies, Figure S1A), 0.3 µl MgCl2 (200 mM, Sigma), 0.5 µl RNase inhibitor (Neb), 1 µl dNTP (10 mM each, Thermo), 0.25 μl DTT (100 mM, Invitrogen), and 1 µl Superscript II (200 U/μl, Invitrogen), were incubated at 42°C for 90 minutes, followed by enzyme inactivation at 75°C for 10 minutes. A mixture of 29 µl Water, 5 µl Advantage2 taq polymerase buffer, 2 µl dNTP (10 mM each, Thermo), 2 µl custom designed PCR primer (12 µM, Integrated DNA Technologies, Figure S1A), 2 µl Advantage2 taq polymerase was directly added to the reverse transcription product and the amplification was performed for 19 cycles. The amplification product was purified using Ampure XP beads (Beckman-Coulter). Library preparation was performed using Ovation Ultralow library systems (Nugen inc). Libraries were multiplexed and sequenced using Illumina Hiseq 2500 (Illumina Inc) (Table S1).
Bioinformatic Analyses
Mapping and calculating gene expression levels:
Raw reads were trimmed for the Illumina adapter sequences using Trimmomatic (Bolger et al., 2014), and for the 5’ TS oligo sequences and the 3’ primer sequences using custom R codes. RSEM (Li and Dewey, 2011) was used to map and calculate gene expression levels represented as transcripts per million (TPM). The reference genome was modified to include chrC, which contained the sequence of part of Nestin enhancer followed by eCFP transcripts, and reconstructed from sequencing reads. We used following parameters: rsem-calculate-expression -p 12--fragment-length-mean 500 $input.fastq $rsem_ref $cell_id. For downstream analyses, we used a table with single cells at the columns and the genes at the rows (Table S6).
Waterfall 1–pre-processing:
Waterfall input is an expression matrix from RSEM after eliminating outliers (Figure S1C). Unsupervised clustering was performed using a distance matrix based on Pearson correlation between each pair of single cells (Figure 2A). We defined the neurogenic trajectory on the PCA plot and determined the direction using known markers such as Sox11, Tbr2, Blbp, and Gfap.
Waterfall 2 - Building an in vivo trajectory:
We used custom R codes (included in Supplementary Data) to determine pseudotime for each single cell on the trajectory (Figure S3A; Also see Supplementary Methods). Briefly, we performed parametric PCA, and extracted k-means from the distribution of single-cell transcriptomes. We generate an unbiased trajectory by connecting k-means centers using a minimum spanning tree (MST) algorithm (Paradis et al., 2004). First, we set zero for the origin of the continuous trajectory, determined by pre-processing. Second, we assigned locations to individual cell data points on the trajectory. We assigned each cell to the closest MST segment (lines between k means) or vertex (k-mean) with a single perpendicular projection. Third, we straightened all the segments into one horizontal line, and determined the relative order of the assigned locations of single cell data points on the trajectory. Pseudotime values ranged from 0 (at the origin) to 1 (at the end).
Waterfall 3 - Gene expression analysis by Hidden Markov model:
We used custom R codes to apply a Hidden Markov model (HMM) to predict gene expression states throughout pseudotime. Briefly, we divided pseudotime into 40 bins, each of which contained an average of 2.5 single cells. We averaged the expression level within each bin and assigned the expression values to observed variables for HMM. We used Baum-Welch algorithm to extract the most probable emission probabilities and transition probabilities. Using the output from Baum-Welch algorithm along with observed variables, we applied the Viterbi algorithm to predict binary gene expression states (Figure S3B).
Functional gene expression analysis:
We calculated the Spearman correlation coefficient between pseudotime points and each gene’s expression TPM values. Genes with relatively high Spearman correlations were defined as UP genes and genes with relatively low correlations were DOWN genes and the highest and lowest 1,000 genes defined as UP1000 and DOWN1000, respectively (Table S4). A small subset of the UP1000 and DOWN1000 genes with low average expression values (< 50 TPM) and low coefficient of variation (< 1.95) were from repeat elements within their exons and excluded from downstream analyses. Raw mapping profiles of all genes shown in pseudotime figures were closely inspected to rule out false positives. We identified transcription factors using public databases (Zhang et al., 2012). We used GO (Ashburner et al., 2000), Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa and Goto, 2000) and wikipathway (Pico et al., 2008) for functional gene ontology analyses, along with the R bioconductor package (Gentleman et al., 2004) or Cytoscape software (Shannon et al., 2003).
For alternative functional gene expression analysis (Figure S7), we first divided the entire transcriptome dataset into three equal groups based on their Spearman correlation to pseudotime: positively correlated, uncorrelated, and negatively correlated (Table S7). We then evaluated the proportion of positively correlated genes versus negatively correlated genes within each functional entity from an independent functional annotation database wikipathway rather than the KEGG pathway database. If a functional entity contained a disproportionally larger number of up-regulated genes than down-regulated genes, we considered the functional entity to be generally activated, and, conversely, a disproportionally larger number of down-regulated genes indicated that the pathway was generally inactivated over time.
Validation with in situ Database Comparison, Immunohistology, Genetic Labeling and Electrophysiology
We validated our NPC-enriched genes by the Allen mouse brain atlas in situ hybridization dataset (Lein et al., 2007) (Table S3). We inspected the gene expression patterns within the adult dentate gyrus at sagittal views. Genes with clear and relatively even distribution within the SGZ were determined to be “SGZ enriched”, whereas genes with subtle or scattered enrichment within the SGZ were determined to be “ambiguous”. Genes without any enrichment at the SGZ were determined to be “not SGZ enriched”.
Adult Nestin-GFPcyto animals were used for immunohistochemical validation. Hopx-CreERT2 f/+;;mT/mGf/+mice were generated by crossing Hopx-CreERT2 f/+ (Takeda et al., 2011) (Strain: Hopxtm2.1(cre/ERT2)Joe/J, Jackson Labs Stock: 017606) with the mT/mGf/f reporter line (Strain: B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J; Jackson Labs Stock: 007676). Tamoxifen (62 mg/ml; Sigma; T5648) was prepared in a 5:1 ratio of corn oil/ethanol and heated to 37°C and mixed. Eight week-old HopX-CreERT2 f/+;;mT/mGf/+ animals were injected intraperitoneally with 124 mg/kg tamoxifen and analyzed by immunohistology as previously described (Bonaguidi et al., 2011). The following antibodies were used: Aldoc (1:200, goat; Cat#SC12065; Santa Cruz), GFAP (1:2000, rabbit; Cat#Z0334; DAKO), GFP (1:1000, chicken; Cat#GFP-1020; Aves), GFP (1:1000, goat; Cat#600–101-215; Rockland), Nestin (1:500, chicken; Cat#NES; Aves), PCNA (1:2000, rabbit; Cat#ab18197; Abcam), PCNA (1:500, goat; Cat#SC9857; Santa Cruz), Stmn1 (1:200, rabbit; Cat#ab24445; Abcam), Tbr2 (1:1000, rabbit Cat#Ab23345; Abcam). GFP cells were identified with an Axiovert 200M microscope (Zeiss) and then acquired as z-stacks on Zeiss 710 single-photon confocal microscope using 40X or 63X objectives. For quantification of Stmn1, Aldoc and PCNA expression in Nestin-GFPcyto mice, Z-stacks were acquired from 3 animals. Images were analyzed using Imaris 7.1.1 (Bitplane). RGLs were identified by their radial process and soma situated in the SGZ and IPCs were identified by their small soma and tangential process as previously described (Bonaguidi et al., 2011).
Adult nestin-GFPcyto transgenic mice were used to validate expression of functional glutamate receptors on NSCs. GFP+ radial glia like NSCs in slices prepared acutely from adult animals were recorded by whole-cell patch-clamp upon puffing of AMPA or NMDA in the presence or absence of antagonists as previously described (Song et al., 2012).
Supplementary Material
ACKNOWLEDGEMENTS.
We thank S.L. Salzberg, H.H. Kazazian, and B. Langmead for suggestions, members of Song and Ming laboratories for discussion, Y. Cai and L. Liu for technical support. This work was supported by MSCRF to H.S. and G.L.M., fellowship from Samsung to J. Shin, and EMBO long-term postdoctoral fellowship and Swedish Research Council to D.A.B.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature genetics 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall SC, Davis KL, Amir el AD, Tadmor MD, Simonds EF, Chen TJ, Shenfeld DK, Nolan GP, and Pe’er D (2014). Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell 157, 714–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg DA, Yoon K-J, Will B, Xiao AY, Kim N-S, Christian KM, Song H, and Ming G. l. (2015). Tbr2-expressing intermediate progenitor cells in the adult mouse hippocampus are unipotent neuronal precursors with limited amplification capacity under homeostasis. Frontiers in Biology 10, 262–271. [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaguidi MA, Wheeler MA, Shapiro JS, Stadel RP, Sun GJ, Ming GL, and Song H (2011). In vivo clonal analysis reveals self-renewing and multipotent adult neural stem cell characteristics. Cell 145, 1142–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracko O, Singer T, Aigner S, Knobloch M, Winner B, Ray J, Clemenson GD Jr., Suh H, Couillard-Despres S, Aigner L, et al. (2012). Gene expression profiling of neural stem cells and their neuronal progeny reveals IGF2 as a regulator of adult hippocampal neurogenesis. J Neurosci 32, 3376–3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codega P, Silva-Vargas V, Paul A, Maldonado-Soto AR, Deleo AM, Pastrana E, and Doetsch F (2014). Prospective identification and purification of quiescent adult neural stem cells from their in vivo niche. Neuron 82, 545–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encinas JM, Michurina TV, Peunova N, Park JH, Tordo J, Peterson DA, Fishell G, Koulakov A, and Enikolopov G (2011). Division-coupled astrocytic differentiation and age-related depletion of neural stem cells in the adult hippocampus. Cell Stem Cell 8, 566–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encinas JM, Vaahtokari A, and Enikolopov G (2006). Fluoxetine targets early progenitor cells in the adult brain. Proc Natl Acad Sci U S A 103, 8233–8238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. (2004). Bioconductor: open software development for computational biology and bioinformatics. Genome biology 5, R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe PS, Coutu DL, and Schroeder T (2014). Single-cell technologies sharpen up mammalian stem cell research. Nature cell biology 16, 919–927. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, and Goto S (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic acids research 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knobloch M, Braun SM, Zurkirchen L, von Schoultz C, Zamboni N, Arauzo-Bravo MJ, Kovacs WJ, Karalay O, Suter U, Machado RA, et al. (2013). Metabolic control of adult neural stem cell activity by Fasn-dependent lipogenesis. Nature 493, 226–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegstein A, and Alvarez-Buylla A (2009). The glial nature of embryonic and adult neural stem cells. Annual review of neuroscience 32, 149–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, et al. (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176. [DOI] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, and Clevers H (2010). Coexistence of quiescent and active adult stem cells in mammals. Science 327, 542–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, Neff NF, Quake SR, and Weissman IL (2011). Tracking single hematopoietic stem cells in vivo using high-throughput sequencing in conjunction with viral genetic barcoding. Nature biotechnology 29, 928–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignone JL, Kukekov V, Chiang AS, Steindler D, and Enikolopov G (2004). Neural stem and progenitor cells in nestin-GFP transgenic mice. The Journal of comparative neurology 469, 311–324. [DOI] [PubMed] [Google Scholar]
- Ming GL, and Song H (2011). Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron 70, 687–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramoto T, Cannon D, Gierlinski M, Corrigan A, Barton GJ, and Chubb JR (2012). Live imaging of nascent RNA dynamics reveals distinct types of transcriptional pulse regulation. Proceedings of the National Academy of Sciences of the United States of America 109, 7350–7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick A, and Weiner M (1957). Enzyme Induction as an All-or-None Phenomenon. Proceedings of the National Academy of Sciences of the United States of America 43, 553–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E, Claude J, and Strimmer K (2004). APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics 20, 289–290. [DOI] [PubMed] [Google Scholar]
- Pico AR, Kelder T, van Iersel MP, Hanspers K, Conklin BR, and Evelo C (2008). WikiPathways: pathway editing for the people. PLoS biology 6, e184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, Peskin CS, Tranchina D, Vargas DY, and Tyagi S (2006). Stochastic mRNA synthesis in mammalian cells. PLoS biology 4, e309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramskold D, Luo S, Wang YC, Li R, Deng Q, Faridani OR, Daniels GA, Khrebtukova I, Loring JF, Laurent LC, et al. (2012). Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nature biotechnology 30, 777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield R (1978). The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells 4, 7–25. [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signer RA, Magee JA, Salic A, and Morrison SJ (2014). Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 509, 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons BD, and Clevers H (2011). Strategies for homeostatic stem cell self-renewal in adult tissues. Cell 145, 851–862. [DOI] [PubMed] [Google Scholar]
- Song J, Zhong C, Bonaguidi MA, Sun GJ, Hsu D, Gu Y, Meletis K, Huang ZJ, Ge S, Enikolopov G, et al. (2012). Neuronal circuitry mechanism regulating adult quiescent neural stem-cell fate decision. Nature 489, 150–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegle O, Teichmann SA, and Marioni JC (2015). Computational and analytical challenges in single-cell transcriptomics. Nature reviews Genetics 16, 133–145. [DOI] [PubMed] [Google Scholar]
- Takeda N, Jain R, LeBoeuf MR, Wang Q, Lu MM, and Epstein JA (2011). Interconversion between intestinal stem cell populations in distinct niches. Science 334, 1420–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nature biotechnology 32, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, and Pachter L (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HM, Chen H, Liu W, Liu H, Gong J, Wang H, and Guo AY (2012). AnimalTFDB: a comprehensive animal transcription factor database. Nucleic acids research 40, D144–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.