

Abstract
Amplifications of oncogenic genes are often considered actionable. However, not all patients respond. Questions have therefore arisen regarding the degree to which amplifications, especially non-focal ones, mediate overexpression. We found that a subset of high-level gene amplifications (≥ 6 copies) (from The Cancer Genome Atlas database) was not over-expressed at the RNA level. Unexpectedly, focal amplifications were more frequently silenced than non-focal amplifications. Most non-focal amplifications were not silenced; therefore, non-focal amplifications, if over-expressed, may be therapeutically tractable. Furthermore, specific silencing of high-level focal or non-focal gene amplifications may explain resistance to drugs that target the relevant gene product.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13045-021-01211-1.
Keywords: Gene amplification, mRNA expression, Therapeutic actionability
To the Editor,
Targeted therapy resistance affects a subset of cancer patients [1, 2]. Indeed, ~ 13% of somatic mutations are not expressed at the RNA level [3]. Little is known regarding gene amplifications. We examined The Cancer Genome Atlas (TCGA) to determine RNA expression of high-level amplifications, including in cancer-related genes. We observed that a minority of high-level amplifications are silenced. Silencing is, unexpectedly, more frequent in focal than non-focal amplifications. However, most focal and non-focal amplifications are over-expressed. Therefore, our observations suggest important points, which require further clinical validation: (1) high-level amplifications can be silenced and therefore may not be amenable to therapeutic targeting: and (2) non-focal amplifications are sometimes not considered druggable; however, they are frequently overexpressed, suggesting that they could be pharmacologically tractable.
Data from 675 TCGA samples (23 tumor types) with differential RNA expression was retrieved (Additional file 1: Methods ; Additional file 2: Table S1). A total of 166,707 amplifications were reviewed; there was an average of 304 [268–340] (mean [95% confidence interval (CI)]) high-level amplifications (≥ 6 copies) per sample. Amplifications were categorized as “non-focal” when ≥ 1 other amplification was observed within a 0.1 megabase (Mb) genomic window. Non-focal amplifications result from large genome rearrangements encompassing > 1 gene. Using the aforementioned threshold, 137,819 (83%) amplifications were considered non-focal; 28,888 (17%) amplifications, focal; respectively, 5612 non-focal (68%) and 2599 focal (32%) amplifications in cancer-related genes (Table 1).
Table 1.
Silencing distribution between focal and non-focal high-level amplifications (data from TCGA)
| Total number of high-level amplifications* | Tumor-to-normal differential RNA expression | Number of silenced** amplifications | Number of non-silenced amplifications | Chi-square p value | ||
|---|---|---|---|---|---|---|
| N (%) | Mean [95% confidence interval] | T test p value | N (%) | N (%) | ||
| All genes (N = 18,870) | ||||||
| All amplifications | 166,707 (100%) | + 548 [+ 500 to + 595] % | – | 9534 (6%) | 157,173 (94%) | – |
| Focal amplifications | 137,819 (83%) | + 559 [+ 505 to + 613] % | 0.296 | 6973 (4%) | 130,846 (78%) | < 0.00001 |
| Non-focal amplifications*** | 28,888 (17%) | + 493 [+ 400 to + 585] % | 2561 (2%) | 26,328 (16%) | ||
| Cancer-related genes**** (N = 832) | ||||||
| All amplifications | 8211 (100%) | + 1084 [+ 742 to + 1426] % | – | 485 (6%) | 7726 (94%) | – |
| Focal amplifications | 5612 (68%) | + 1207 [+ 723 to + 1691] % | 0.300 | 233 (3%) | 5379 (66%) | < 0.00001 |
| Non-focal amplifications | 2599 (32%) | + 818 [+ 549 to + 1088] % | 252 (3%) | 2347 (29%) | ||
Numbers in bold represent statistically significant Chi-square p-values at the alpha level of 0.05, as well as the criteria that have the largest contribution to the Chi-square statistic
*Gene level amplification included those genes with ≥ 6 copies
**RNA silencing was defined by an 80% decrease of expression in the tumor sample compared to the normal sample, including only tumor samples that presented a high-level amplification for that gene (≥ 6 copies of the gene)
***Non-focal amplifications are co-amplification of genes that are located in the same 0.1 megabase genomic window
****Cancer-related genes are listed in Additional file 2: Table S2. The list of cancer-related genes was defined as the union of genes curated by the Cancer Gene Census (CGC) from the Catalogue of Somatic Mutations in Cancer (COSMIC) and genes analyzed by Foundation Medicine Inc. in their commercial panels Foundation One and Foundation One Heme (N = 946 distinct genes)
High-level amplifications correlated with a + 548 [+ 500 to + 595] % (mean, 95% CI) increase of expression for the corresponding mRNA in the tumor compared to the adjacent tissue. When considering only cancer-related genes, the overall expression increase was + 1084 [+ 742 to + 1426]% (mean, 95% CI). There was no difference between the focal and the non-focal groups (Table 1).
A subset of 9534 (6%) amplifications were silenced (i.e., presenting a decrease of expression > 80% in the tumor compared to normal adjacent tissue). This proportion was similar when only considering cancer-related genes (N = 485, 6%) (Table 1).
A Chi-square test was performed to examine the relationship between gene silencing and amplification type. Interestingly, gene amplifications were either consistently focal or non-focal. Amongst the 832 amplified cancer-related genes, 243 were always focally amplified and 589 were uniformly non-focally amplified. Focal amplifications were more likely to be silenced (9% vs 5% silenced amplifications; odds-ratio (OR) [95% CI] = 1.83 [1.74–1.91]; X2 (1, N = 166,707) = 641.4, p < 0.00001), and this held true when considering only cancer-related genes (10% versus 4% silenced amplifications; OR = 2.48 [2.06–2.98]; X2 (1, N = 8211) = 98.2, p < 0.00001) (Table 1). Interestingly, many cancer-related genes that are druggable/activate druggable pathways were less frequently silenced than cancer-related genes without active therapies (Fig. 1).
Fig. 1.
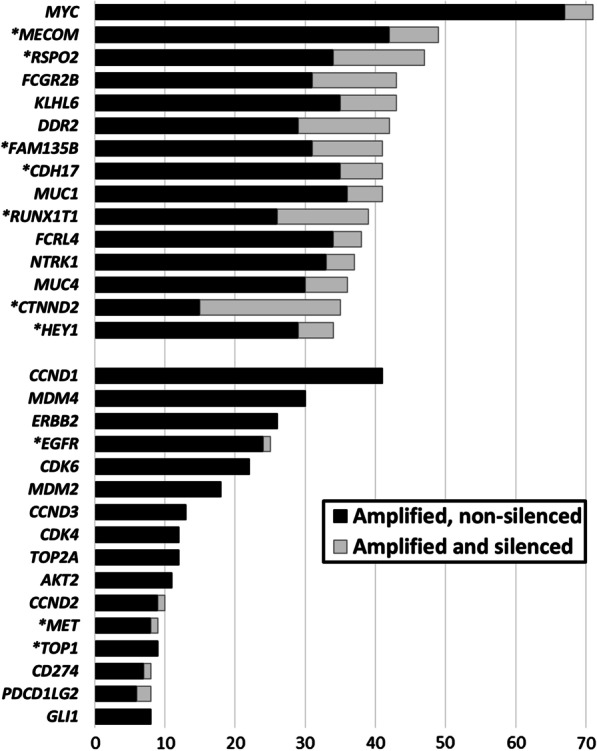
Frequently silenced cancer-related gene amplifications (TCGA). Top panel: cancer-related genes that are amplified in ≥ 5% (N ≥ 34) of the tumor samples (N = 675) and silenced in ≥ 10% of those cases (N ≥ 4). Black = amplified, not silenced; grey = amplified and silenced. Bottom panel (starting with CCND1): sampling of common potentially actionable oncogenes; they are infrequently silenced. *Genes preceded by an asterisk were exclusively found to be focally amplified. Gene level amplification included those genes with ≥ 6 copies; RNA silencing was defined by ≥ 80% expression decrease in the tumor sample compared to the normal sample, including only tumor samples that had high-level amplification for that gene (≥ 6 copies of the gene); non-focal amplifications referred to co-amplification of genes located in the same 0.1 megabase genomic window
The mechanism of amplification silencing was not elucidated. Prior studies suggest potential mechanisms such as epigenetic modifications [4], miRNA regulation [5], and/or factors that influence RNA-decay such as RNA-binding proteins [6].
Overall, our findings indicate that high-level amplifications are prevalent in tumors. Further, as expected, gene copy-number amplifications correlated with an overall increase in mRNA expression. Even so, high-level amplifications may be associated with gene-specific silenced RNA expression. Surprisingly, focal amplifications were more likely to be silenced than non-focal amplifications, and this held true when considering only cancer-related genes. Furthermore, most non-focal amplifications were not silenced, suggesting that such amplifications may still be actionable. Of interest, cancer genes that were more likely to be considered druggable and/or have established prognostic or predictive attributes were usually not silenced, while cancer-related genes that are often considered therapeutically intractable were more often silenced. Since silencing of amplifications would nullify tumorigenic impact, and also lead to resistance (if the gene product was the treatment target), it is conceivable that more frequently silenced amplifications would be less likely to be associated with therapeutic or prognostic/predictive impact. These findings echo those previously published in gliomas where the authors suggested that, even when amplified, genes that are normally silent in a given cell type may play no role in tumor progression [7].
In conclusion, our study indicates that the consequences of silencing on response versus resistance after targeted therapies matched to oncogenic amplifications requires in vitro verification and prospective clinical studies. Taken together with the existing literature [3, 8, 9], we suggest that gene silencing may be an important mechanism of therapeutic resistance, and that optimal pharmacologic intervention in cancer may demand transcriptomic in addition to genomic interrogation and considerations for epigenetic modulation.
Supplementary Information
Additional file 2. Supplementary Tables 1 and 2.
Acknowledgements
Not applicable.
Abbreviations
- CGC
Cancer Gene Census
- CI
Confidence interval
- CNV
Copy number variation
- COSMIC
Catalogue Of Somatic Mutation In Cancer
- GISTIC
Genomic Identification of Significant Targets In Cancer
- Mb
Megabase
- N
Number
- OR
Odds ratio
- RNA/mRNA
Ribonucleic acid/messenger ribonucleic acid
- RSEM
RNA-seq by expectation–maximisation
- TCGA
The Cancer Genome Atlas
- UCSC
University of California Santa Cruz
Authors' contributions
Study conception and design (AB, RK), data acquisition and analysis (AB), result interpretation (AB, SML, RK), manuscript writing and revision (AB, SML, RK). All authors read and approved the final manuscript.
Authors' information
Amelie Boichard, PharmD PhD: Clinical Molecular Geneticist—Department of Molecular Cancer Genetics, University of Strasbourg Hospitals, France (amelie.boichard@chru-strasbourg.fr).
Scott M. Lippman, MD: Distinguished Professor of Medicine and Director of Moores Cancer Center at UC San Diego Health.
Razelle Kurzrock, MD: WIN Consortium for Precision Medicine (teoam2011@gmail.com).
Funding
RK was funded in part by the Joan and Irwin Jacobs Fund. RK and SML received funding from NIH P30 CA023100.
Availability of data and materials
The data that support the findings of this study derived from The Cancer Genome Atlas (TCGA) project (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga).
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
RK receives research funding from Genentech, Merck Serono, Pfizer, Boehringer Ingelheim, TopAlliance, Takeda, Incyte, Debiopharm, Medimmune, Sequenom, Foundation Medicine, Konica Minolta, Grifols, Omniseq, and Guardant, as well as consultant and/or speaker fees and/or advisory board for X-Biotech, Neomed, Pfizer, Actuate Therapeutics, Roche, Turning Point Therapeutics, TD2/Volastra, Bicara Therapeutics, Inc., has an equity interest in IDbyDNA and CureMatch Inc, serves on the Board of CureMatch and CureMetrix, and is a co-founder of CureMatch. S.M.L. is a co-founder of io9 and member of Biological Dynamics scientific advisory board.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Gerlinger M, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sicklick JK, et al. Molecular profiling of cancer patients enables personalized combination therapy: the I-PREDICT study. Nat Med. 2019;25:744–750. doi: 10.1038/s41591-019-0407-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adashek JJ, et al. Transcriptomic silencing as a potential mechanism of treatment resistance. JCI Insight. 2020;5:e134824. doi: 10.1172/jci.insight.134824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tycko B. Epigenetic gene silencing in cancer. J Clin Invest. 2000;105:401–407. doi: 10.1172/JCI9462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet. 2011;12:99–110. doi: 10.1038/nrg2936. [DOI] [PubMed] [Google Scholar]
- 6.Oliveira C, Faoro H, Alves LR, Goldenberg S. RNA-binding proteins and their role in the regulation of gene expression in Trypanosoma cruzi and Saccharomyces cerevisiae. Genet Mol Biol. 2017;40:22–30. doi: 10.1590/1678-4685-gmb-2016-0258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vogt N, et al. Relationships linking amplification level to gene over-expression in gliomas. PLoS ONE. 2010;5(12):e14249. doi: 10.1371/journal.pone.0014249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rodon J, et al. Genomic and transcriptomic profiling expands precision cancer medicine: the WINTHER trial. Nat Med. 2019;25:751–758. doi: 10.1038/s41591-019-0424-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Uhlen M, et al. A pathology atlas of the human cancer transcriptome. Science. 2017;357(eaan2507):1–11. doi: 10.1126/science.aan2507. [DOI] [PubMed] [Google Scholar]
- 10.Mermel CH, et al. GISTIC20 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12(4):R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vivian J, et al. Toil enables reproducible, open source, big biomedical data analyses. Nat Biotechnol. 2017;35(4):314–316. doi: 10.1038/nbt.3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eijkelenboom A, et al. Recommendations for the clinical interpretation and reporting of copy number gains using gene panel NGS analysis in routine diagnostics. Virchows Arch. 2019;474:673–680. doi: 10.1007/s00428-019-02555-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 2. Supplementary Tables 1 and 2.
Data Availability Statement
The data that support the findings of this study derived from The Cancer Genome Atlas (TCGA) project (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga).