

Abstract
Metabolic reprogramming confers cancer cells plasticity and viability under harsh conditions. Such active alterations lead to cell metabolic dependency, which can be exploited as an attractive target in development of effective antitumor therapies. Similar to cancer cells, activated T cells also execute global metabolic reprogramming for their proliferation and effector functions when recruited to the tumor microenvironment (TME). However, the high metabolic activity of rapidly proliferating cancer cells can compete for nutrients with immune cells in the TME, and consequently, suppressing their anti-tumor functions. Thus, therapeutic strategies could aim to restore T cell metabolism and anti-tumor responses in TME by targeting the metabolic dependence of cancer cells. In this review, we highlight current research progress on metabolic reprogramming and the interplay between cancer cells and immune cells. We also discuss potential therapeutic intervention strategies for targeting metabolic pathways to improve tumor immunotherapy efficacy.
Keywords: Metabolic reprogramming, nutrients competing, tumor microenvironment, tumor-infiltrating lymphocytes
1. Introduction
The tumor microenvironment (TME) is comprised of components such as stromal cells, immune cells (together with their secretory factors,) vascular endothelial cells and extracellular matrix (ECM) [1]. Immune cells and related matrix components recruited and activated by tumor cells, clustering a tumor-suppressing inflammatory microenvironment during the early stages of tumor colonization or growth, thus hindering tumorigenesis and development. However, after persistent tumor antigen stimulation and immune activation, the associated effector cells in the microenvironment are either depleted or remodeled, resulting in an immunosuppressive microenvironment [2]. Cancer cells eventually get advanced progression due to the failure of the immune system surveillance and elimination. Since the major cellular components that maintain the immunosuppressive microenvironment also play an antitumor role early in tumor progression, it is possible to stimulate or restore the inherent tumor suppressive ability of the immune system, remodel the active immune microenvironment, and produce a comprehensive response effect through immunotherapeutic strategies targeting TME [3].
Cancer cells reprogram their metabolic pathways to fulfill a selective advantage for rapid proliferation and survival in TEM. This reprogramming of energy metabolism has been considered a key hallmark of cancer [4]. The high rate of aerobic glycolysis [5] and glutamine addiction [6] are the two most prominent features in cancer cell metabolic reprogramming during its initiation and progression. Therefore, this leads to cell metabolic dependency became an attractive target in designing effective antitumor therapy. Immune cells share similarities with cancer cells of engaging metabolic regulation to sustain proliferation and survival. The reprogramming of metabolic pathways plays an important role in immune cell activation [7]. When activated, T cells shift glucose metabolism from oxidative phosphorylation (OXPHOS) to aerobic glycolysis, which promotes glycolytic flux and lactate production [8]. Accordingly, blocking T cells from engaging glycolysis compromises their ability to produce IFN-γ [9]. Therefore, remodeling T cell metabolism and nutrient availability to enhance T cell activation and function could be a promising therapeutic strategy for cancer [8].
Cancer cells modulate the metabolite profiles in the TME (Figure 1). The “metabolic competition” within TME might allow cancer cells to effectively suppress antitumor immunity and drive cancer progression [10, 11]. Cancer cells consume most of the glucose within the TME to produce lactate. Lactate-induced acidification diminishes T and NK cell activation and lead to immune evasion [12]. The compromised effector function of immune cells may contribute to the restricted response to immune checkpoint blockade therapy [10, 11, 13]. Thus, re-balancing the utilization of nutrients within TME by targeting metabolic pathways in cancer cells could restore T cell function, thereby enhancing cancer immunotherapy efficacy [14].
Figure 1. The impacts of cancer cells on the tumor microenvironment and TILs.
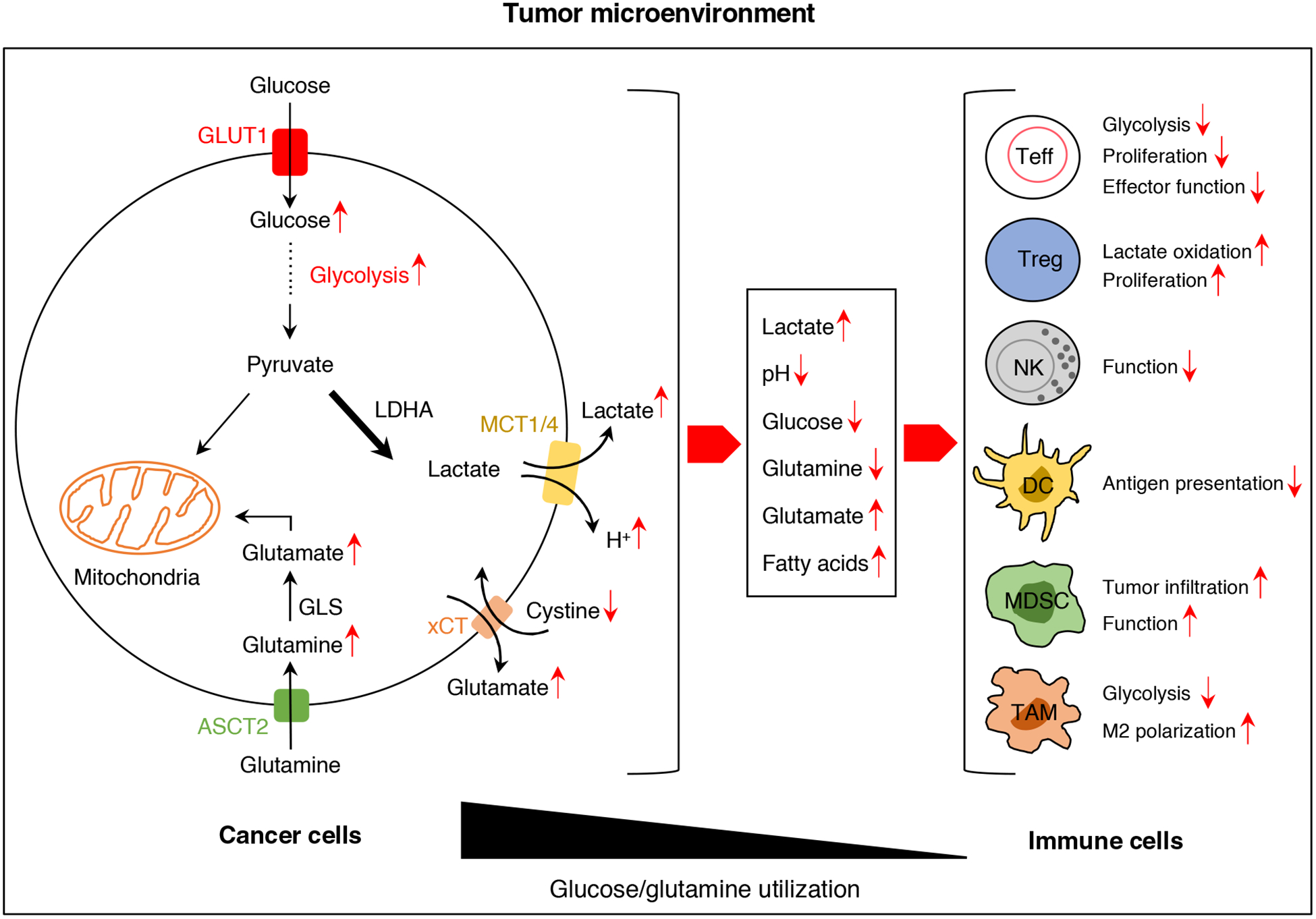
Cancer cells modulate the metabolite profiles in the tumor microenvironment. The “metabolic competition” within tumor microenvironment and tumor-derived lactate might allow cancer cells to effectively suppress antitumor immunity and drive cancer progression.
Abbreviations: TILs: tumor-infiltrating lymphocytes; GLUT1: Glucose transporter 1; LDHA: Lactate dehydrogenase A; MCT1/4: Monocarboxylate transporter 1/4; xCT: cystine/glutamate transporter xCT; ASCT2: alanine-serine-cysteine transporter 2; Teff: T effector cell; Treg: regulatory T cell; NK: natural killer cell; DC: dendritic cell; MDSC: myeloid-derived suppressor cell; TAM: tumor-associated macrophage.
Here, we briefly review the interplay between cancer cells and immune cells from the metabolic perspectives and discuss the potential feasibility for enhancing cancer immunotherapy efficacy via metabolic interventions.
2. Metabolic reprogramming in cancer cells
Cancer cells reprogram metabolic pathways to fulfill rapid and continuous proliferation (Figure 1). Even in the presence of sufficient oxygen, cancer cells selectively undergo high rate of glycolysis instead of oxidative phosphorylation (OXPHOS) to generate adenosine triphosphate (ATP). This common feature of cancer cell aerobic glycolysis is referred to as the “Warburg effect” [15]. Although the causal relationship between aerobic glycolysis and cancer progression still remains controversial, some findings suggested that high rate of glycolysis not only generate the energy that is required to support rapid cancer cell division but also provide building blocks to fulfill biosynthesis needs and maintain redox balance [16].
The metabolic reprogramming in cancer cells is regulated by different signaling pathways and transcriptional factors [16]. Some studies suggested that the enhanced glycolysis in cancer cells was linked to defects in mitochondrial respiration. However, current data indicate that most cancer cells do not have such defects [5, 17]. Beyond the Warburg effect, the reduced nicotinamide adenine dinucleotide phosphate (NADPH) is rapidly produced through pentose phosphate pathway (PPP) to allow cancer cells to counteract the detrimental effects of reactive oxygen species (ROS) during rapid proliferation [18].
Moreover, aerobic glycolysis promotes lactate production in cancer cells. The key enzyme in lactate production is lactate dehydrogenase A (LDH-A), which is regulated by oncogenes and overexpressed in many cancers [19]. The conversion of pyruvate to lactate via LDH-A regenerates NAD+, which is required for conversion of glyceraldehyde-3-phosphate to 1,3-bisphosphoglycerate. Therefore, the NAD+/NADH redox balance maintained by NAD+ regeneration is necessary for continued flux through glycolysis [5]. In addition, lactate has been found to serve as a primary TCA substrate in tumors [20, 21] and also play an important role in suppressing antitumor immunity [12].
The amino acids also experience reprogrammed metabolism in cancer cells [22]. Glutamine is the major amino acid avidly consumed by cancer cells to support their proliferation and survival. Glutamine is one of the primary mitochondrial substrates and most cancer cell lines require constant supply of exogenous glutamine in vitro to fuel the tricarboxylic acid (TCA) cycle, maintain mitochondrial membrane potential and integrity, as well as provide nitrogen for protein and nucleotide synthesis [6]. Once glutamine enters the cells through solute carrier family 1 member 5 (SLC1A5, also known as ASCT2), glutaminase convert it to glutamate. Glutamate then can be converted either to glutathione (GSH) by glutathione cysteine ligase (GCl) or to α-ketoglutarate (α-KG) and enters the TCA cycle [16]. Cancer cells either upregulate glutaminase or increase glutamine flux for amino acid, nucleotide or fatty acid biosynthesis. Glutamine-addicted cancer cells are dependent on glutamine for viability and are sensitive to glutamine withdrawal and glutaminase blockade [23–25]. Glutamine regulates the activation of STAT3, which mediates signaling pathways in invasive ovarian cancer cells [26] and the proliferative effects on glycolytic and oxidative cancer cells [27].
In addition to amino acids, cancer cells need to produce massive amounts of fatty acids via de novo fatty acid synthesis to support the high demand for membrane production and signaling molecules during rapid proliferation [28]. As a result, the increased fatty acids levels in the TME may also regulate the function of immune cells [29].
3. Metabolic reprogramming in immune cells
Metabolic reprogramming is not limited to cancer cells, it is also observed in highly proliferating cells and immune cells, such as activated T cells, activated B cells, activated natural killer (NK) cells, lipopolysaccharide (LPS)-activated macrophages and dendritic cells [30]. Given the similarities of metabolic requirements between cancer cells and immune cells, they compete for nutrients within TME, and this competition sets the stage for tumor progression [31].
In general, naïve T cells require low metabolite needs. However, once the T cell receptor (TCR) recognizes an antigen along with receiving co-stimulatory signals from antigen presenting cells (APCs), T cells undergo changes in metabolic profiles due to the high demand for biomolecule generations to expand and exert effector functions [8, 32]. Glucose consumption is significantly elevated in activated T cells. Activation rapidly switches T cell metabolism from fatty acid β-oxidation and pyruvate oxidation via the TCA cycle to aerobic glycolysis, PPP and glutamine oxidation [33]. Glucose typically enters the cell via solute carrier family 2 (facilitated glucose transporter) [7]. It has been found that the solute carrier family 2 member 1 (SLC2A1) is rapidly upregulated upon T cells and macrophage activation [34, 35]. The increased glucose intake facilitates activated T cells to engage high rate of aerobic glycolysis for proliferation and effector functions [36].
TCR signaling promotes activation of pyruvate dehydrogenase kinase 1 (PDHK1) and PDHK1-mediated aerobic glycolysis integrates glucose availability and lactate dehydrogenase (LDH) activity to control cytokine synthesis at the post-transcriptional level in T cells [37]. The glycolysis enzyme GAPDH can also regulate interferon-γ (IFN-γ) production in CD4+ T cells via binding to the AU-rich elements within the 3’ UTR of IFN-γ mRNA, making glycolysis an essential regulator for T cell effector functions [9]. T cell activation induces many transcriptional changes and mTOR activation. The activation of mTOR and glycolysis induces HIF-1α, which in turn, reprograms transcriptional profiles [22]. Limiting glucose availability or inhibiting glycolysis activates AMPK and represses mTOR and HIF-1α activity, favoring immunosuppressive regulatory T cells (Tregs) and anti-inflammatory M2 microphages, and impairing dendritic cell functions [7].
In addition to glucose, glutamine also serves as an important source for biosynthetic precursors in active T cells. Deprivation of glutamine compromises activation-induced T cell growth and proliferation [33]. It has been demonstrated that the transcription factor Myc acts a dominant role in the regulation of metabolic reprogramming in activated T cells by promoting glycolysis and glutaminolysis and suppressing fatty acid β-oxidation [33]. The balance between fatty acid oxidation and synthesis influences the development of Tregs, macrophages and memory T cells. The immunosuppressive Tregs and anti-inflammatory M2 microphages have much higher fatty acid oxidation than effector T cells and inflammatory M1 macrophages. Inhibiting fatty acid oxidation in Tregs and M2 macrophages impairs their functions and lineage development [7]. The increased immunosuppressive characteristics of myeloid-derived suppressor cells (MDSCs) are also associated with elevated expression of fatty acid oxidation genes [38]. Therefore, metabolic status of immune cells can determine immune responses.
4. Metabolic interplay between cancer cells and immune cells
4.1. Glucose and glycolysis
Cancer cells exploit elevated aerobic glycolysis and limit extracellular glucose level within the TME (Figure 1). T cells are outcompeted by tumor cells for glucose uptake; this metabolic imbalance diminishes T cell effector functions. Most tumor-associated macrophages (TAMs) are the M2 phenotype, which secrete IL-6 to induce PDPK1-dependent PGK1 T243 phosphorylation in cancer cells and promote cancer cell glycolysis and tumor growth [39]. Glucose consumption by tumors metabolically restricts T cells, impairing their mTOR activity, glycolytic capacity, IFN-γ production, and supporting tumor progression [10]. It has been reported that tumor infiltrated CD4+ and CD8+ T cells rewire their metabolic activities in response to the reduced aerobic glycolysis induced by glucose deficient TME via increasing the phosphoenolpyruvate (PEP) level. The PEP produced by phosphoenolpyruvate carboxykinase 1 (PCK1) acts as a metabolic checkpoint to strengthen T cell receptor-mediated Ca2+-NFAT pathway and improves antitumor responses in glucose-poor conditions [11]. Moreover, activated CD4+ T cells utilize different metabolic profiles based on their differentiation goals. Inflammatory CD4+ T cells were shown to be dependent on glycolysis, while the regulatory cells depend on mitochondrial electron transport [40]. T cells and M1 macrophages depend on aerobic glycolysis to be functionally activated [41]. Nevertheless, high glucose utilization by cancer cells and endothelial cells out compete immune cells, resulting in reduced INF-ɣ production, Ca2+ dependent pathways, T-cell motility and cytotoxicity and macrophage proinflammatory functions [9, 10, 34, 42].
Cancer cells employ different mechanisms to access more glucose. Increased expression level of glycolysis enzyme hexokinase 2 (HK2) is one approach which allows cancer cells to exhibit increased rates of glycolysis and diminishes T cell glucose uptake and INF-ɣ production [10, 11]. Methyltransferase EZH2 is involved in cytokine expression and survival of T-cells. The negative regulation of EZH2 expression by miRNAs in CD8+ T cells is among other immunosuppressive approaches, being reinforced by cancer cells [43]. Insufficient glucose induces sustained PD-1 expression on T cells that may recruit more CD4+ Tregs by diminishing TCR, mTOR, PI3K signaling, glycolysis, and activating of fatty acid oxidation [44–46]. T cell glycolysis is prohibited by PD-1 and PD-L1 interaction, which promotes fatty acid oxidation, repressing T cell antitumor functions. Data obtained from blockade of these checkpoints confirm this findings by replenishing glucose in the TME and increasing T cell glycolysis and activity [31]. Checkpoint blockade antibodies against CTLA-4, PD-1, and PD-L1 have been shown to restore glucose in the TME, permitting T cell glycolysis and IFN-γ production [10].
4.2. Lactate and extracellular acidity
The cancer cell monocarboxylate transporters (MCT1 & 4) co-transport lactate and protons to the extracellular matrix within TME and create a high lactate concentration and acidic environment (Figure 1). The physiological concentration range of lactate in human blood is 1.5–3.0 mM but can reach up to 30 mM in a given tumor tissue [47]. Lactate produced from cancer cells has been demonstrated to play a key role in inhibiting T cells and NK cells from attacking cancer cells [12]. High level of lactate in the TME blocks T cells lactic acid export, thereby disturbing their metabolism and functions. The lactate produced by cancer cells lowers the extracellular pH [47, 48]. It is reported that acidity limits T cell glycolysis and blocks T cell activation in vitro, and neutralizing acidity within TEM has been shown to improve antitumor responses to immunotherapy [49]. The proliferation and cytokine production of human cytotoxic T lymphocytes (CTLs) can be suppressed up to 95% and lead to a 50% decrease in cytotoxic activity by lactate [50]. Also, lactate produced by tumor cells has been shown to promote M2-like polarization of tumor-associated macrophages via mono-carboxylate transporters (MCT1, MCT2, MCT3 and MCT4)-mediated lactic acid uptake and induction of vascular endothelial growth factor (vegf) [51]. Another study reported that lactate-induced acidity leads to mitochondria malfunctioning and subsequent apoptosis of NK cells infiltrated to TME [52]. Lactate prevents NKT cells function through mTOR inhibition, which in turn, prohibits INF-ɣ and IL-4 production [53]. The elevated levels of lactate within the TME has also been revealed to regulate PD-L1 expression and LDH-A silencing in melanoma boosts anti PD-1 immune responses. It has been demonstrated that LDH-A inhibition strengthened CD8+ T cells and NK cells tumor infiltration in addition to IFN-γ and granzyme B production [54]. In patients, the lactate level is associated with the metastatic spread of tumor [47]. LDH-A expression is negatively correlated with the levels of T cell activation markers in human melanoma patients [12]. The impacts of tumor-derived lactate on tumor infiltrating lymphocytes in the TEM are summarized and listed in Table 1. Therefore, lactate and its regulating molecules could be promising targets in combination with tumor immunotherapy.
Table 1.
The impacts of lactate and acidification on immune cells in the TEM
| Immune cells | Effects |
|---|---|
| CD8 T cells | Inhibition of proliferation [48] |
| Inhibition of cytokine production/cytotoxicity [12, 48, 201] | |
| Induction of apoptosis [12, 48] | |
| CD4 T cells | Reduction of the antitumoral Th1 subset [202] |
| Inhibition of motility [201] | |
| Regulatory T cells | Increase of polarization/proliferation [202] |
| NK cells | Inhibition of cytokine production/effector functions [12, 203] |
| Induction of apoptosis [12] | |
| Dendritic cells | Inhibition of antigen presentation [204] |
| Inhibition of activation [205] | |
| MDSCs | Increase of development/tumor infiltration [203] |
| Enhancement of immunosuppressive effect [203] | |
| TAMs | Induction of M2 polarization [51, 206] |
4.3. Glutamine and glutaminolysis
Glutamine is an essential amino acid in functional activity, differentiation and expansion of tumor-infiltrating lymphocytes (TILs). Depletion of glutamine in the culture medium blocks T cell proliferation and cytokine production, and supplying biosynthetic precursors of glutamine cannot rescue such effects [55]. The essential demand of a high rate of glutaminolysis coerces TILs and tumor cells to compete for glutamine in the TME [41]. It is likely that the upregulation of glutaminolysis in cancer cells might decrease extracellular glutamine level and limit T cell antitumor functions [56]. Glutamine fuels TCA intermediates, mediates reduced glutathione formation, and sustains mitochondrial membrane integrity in the proliferating cells [57]. Nitrogen is an essential constituent for the synthesis of nitrogen-containing biomolecules in the cell. Glutamine and glutamate are among the major resources of nitrogen in the cell. Activated T cells require more nitrogen and carbon to generate the biomolecules needed for proliferation and effector functions, so they up-regulate glutaminolysis to fulfill their demands [8]. Glutaminolysis deaminases glutamine into α-KG an intermediate of TCA [58]. Glutamate is mainly produced from glutamine by glutaminase and less commonly in some cancers from α-KG [59]. To overcome the consequences of glutamate deprivation and proliferation, some cancer cells convert the ammonia, a by-product of metabolism, and α-KG to glutamate [60]. This conversion is mediated by glutamate dehydrogenase (GDH) to generate glutamate followed by elevated activity of glutamine synthetase (GS) to form glutamine. Silencing both GDH and GS diminishes tumor growth [58, 59, 61]. While T cells are getting activated, limited glutamine availability bolsters CD4+ T cells with augmented FOXP3 expression level. This effect is reversed by silencing GS enzyme [62]. CD4+ T cells use α-KG generated from glutamine to induce a transcription factor supporting their differentiation to T-helper1 (Th1) cell [63]. c-Myc induces glutaminolysis enzymes to synthesize biomolecules required for T cell expansion [64].
By upregulating glutaminolysis, cancer cells secrete large amounts of glutamate, which inhibits cystine uptake by the xCT glutamate-cystine antiporter and leads to intracellular cystine depletion [65]. T cell receptor stimulation lead to upregulation of the xCT and increased cystine uptake in CD4+ and CD8+ human T cells [66]. Although xCT is dispensable for T cell proliferation in vivo [67]. Cancer cell-derived glutamate in the TME may compromise T cell antitumor immunity by impairing the import of cystine and leading to dysregulation of ROS [66]. Moreover, it has been reported that dendritic cells-derived glutamate stimulates the constitutively expressed metabotropic glutamate receptor 5 (mGlu5R) and impairs T cell activation [68]. These findings indicate that the glutamate in the TME is a negative regulator of antitumor immunity.
4.4. Amino acids metabolism
Tumor associated myeloid cells (TAMCs) upregulate arginase 1 (arg1) expression to hydrolyze arginine leading to arginine inadequacy for T cells. TAMCs also cripple expansion and function of cytotoxic T cells by nitrogen oxide generation from arginine [57]. Arginase 1 expression in M2 macrophages, MDSCs and cancer cells breaks down extracellular arginine levels [69]. Transporter of L-arginine has been proposed to be accountable for different immunosuppressive characteristics of MDSCs both in tumor and spleen [38]. Alanine is another amino acid required for T cell activation and re-stimulation of memory T cell. However, these cells do not express the required transaminases to synthesize alanine as needed; they, then, mainly depend on extracellular alanine. Therefore, competitive uptake of alanine by cancer cells may adversely affect theses T-cells [59].
Increased tryptophan catabolism is observed in cancer cells, macrophages, and dendritic cells. Lack of tryptophan and the accumulated kynurenine, a tryptophan catabolite, hamper immune response [70]. Tryptophan is used as a precursor for proteins and various other biomolecules in the body. Lack of tryptophan contributes to tumorigenesis by suppressing immunity through favoring Treg formation, decreasing TCR ζ-chain and stimulating ILT3 and ILT4 inhibitory receptors on dendritic cells [71]. Indoleamine-2,3-dioxygenase (IDO) is a rate-limiting enzyme in tryptophan metabolism that contributes to immune regulation [71, 72]. IDO expression level is increased in both cancer cells and APCs [69]. In many tumor types, IDO1 expression is shown to be associated with unfavorable clinical outcomes. It is also correlated with higher levels of FOXP3+ Tregs, MDSCs, decreased levels of CD57+ NK, CD8+, CD3+ T cells and B cells. Downregulated expansion and effector functions of T cell and NK cells are linked to IDO1 expression [71, 72]. Cytokines secreted from tumor-reactive immune cells and dysregulated molecular mechanisms in cancer cells can stimulate IDO1 expression [73]. An IDO1 inhibitor is under study in clinical trials to explore if it can boost the efficiency of antitumor vaccines [72]. IDO2 also impinges on CD4+ and CD8+ T cell proliferation [71]. The general control nonderepressible 2 (GCN2), a stress sensing kinase, is an important downstream player of IDO-1. IDO1 expression in macrophages results in elevated IL-10 and TGF-β levels and dampens IL-12 production in a GCN2 dependent manner [74]. In another scenario, tryptophan insufficiency is translated to T cell activation prohibition, mTOR1 pathway inhibition and FOXP3 Treg formation [75]. GCN2, regulating a stress response, is a principal player to suppress T cell functions in response to lack of tryptophan [76]. Tryptophan insufficiency mediated by IDO enzyme impedes T cell proliferation by inducing G1 arrest [77]. In lung cancer, cisplatin resistant cells relying on glutamine and tryptophan show elevated expression levels of IDO1 to tackle the ROS. IDO1 inhibition in cisplatin resistant lung cancer cells raises ROS level and diminishes cell growth while cisplatin sensitive cells do not respond to IDO1 inhibition [78]. Immunosuppressive effects of tryptophan metabolism are related to tryptophan insufficiency and the production of its metabolite, kynurenine; however, the latter is the main mechanism of its immunosuppressive effects [72]. Kynurenine concentration in tumor and blood samples of breast and colon cancer patients are associated with augmented levels of PD-1 on CD8+ T cells [71]. Immunosuppressive features of accumulated tryptophan catabolites such as kynurenine mediated by aryl hydrocarbon receptor (AHR) are as important as lack of tryptophan to induce T cell anergy and apoptosis [76]. Cancer associated fibroblasts contribute to tumor immune escape by producing kynurenine and generating tryptophan-deficient environments for immune cells [79]. Increased tryptophan catabolism leading to tryptophan depletion and elevated kynurenine is mediated by both IDO and tryptophan-2,3-dioxygenase (TDO) enzymes [76]. Another metabolite generated from tryptophan is 3-hydroxyanthranilate (3-HAA) that represses T cell activation and proliferation and induces Treg differentiation [63].
Serine is an important amino acid that contributes to T cell induced immune response. During the activation process, T cells rely on the extracellular serine as a major source to produce glycine and one-carbon units to generate de-novo nucleic acids for rapid proliferation and growth [80]. In addition, many cancer cells are highly dependent on serine for growth and survival [81]. Therefore, extracellular serine is an important resource for the competition between tumor cells and immune cells.
mTOR pathway is a principal pathway in regulating immune cells fate and is affected by environmental alterations. Branched chain amino acids (BCAAs) such as leucine are shown to be involved in mTOR regulation [57, 82]. Leucine antagonists block mTOR augmentation in T cells, resulting in T cell tolerance [83]. However, an evident relationship between the BCAAs metabolism in TME and mTOR pathway is yet to be clarified [57].
4.5. Lipid metabolism
The competition between the tumor cells and the immune cells for nutrients and metabolites affects various signaling pathways, gene expression patterns and metabolic activity of the cells [41]. In this setting, lipidomic remodeling is a well-known characteristic of cancer [84]. It was shown that cancer cells acquire their needed fatty acids by elevating the lipolysis of surrounding adipocytes [41]. It was also reported that T-helper 17 (Th17) cells rely on acetyl-CoA carboxylase 1 (ACC1) to synthesize fatty acids whereas Tregs utilize the extracellular fatty acid to provide the required cell membrane phospholipids, and ACC1 blockade attenuates Th17 cells while it strengthens Foxp3+ Tregs formation [85]. The increased fatty acid levels in the TME induced by de novo fatty acid synthesis in cancer cells are proposed to weaken T cell effector functions and give rise to Tregs and M2-like macrophage polarization [31, 69]. MDSCs and dendritic cells containing high lipids negatively regulate immunity by more potent immunosuppressive functions and less antigen presentation, respectively [69]. Cancer cells release eicosanoids, significant bioactive lipid molecules, to the environment and promote cell proliferation. These molecules act as a hurdle for CD8+ T cells co-stimulation and full activation by mediating the binding of cancer cell ICAM-1 and lymphocyte receptor LFA-1. They also inhibit migration of conventional type 1 dendritic cells, required initiators of antitumor response, to the TEM [84]. Other antitumor immunity roles have been attributed to lipids. Endoplasmic reticulum stress response factor XBP1 activation, dependent on lipid peroxidation, promotes ovarian cancer development. It exerts its effects by prompting triglyceride biosynthesis in tumor associated dendritic cells (tDCs) and lipid buildup, which in turn diminishes tDCs to activate T cell. Silencing XBP1 reinstated tDC-induced T cell activation [86]. PD-L1 is found in macrophages, dendritic cells and cancer cells in the TME. PD-1 and PD-L1 interaction supports T cell lipolysis and fatty acid oxidation, hampering T cell antitumor functions [31].
5. Targeting metabolism to improve immune checkpoint blockade efficacy
It has long been recognized that immune evasion is a major hallmark of cancer cells [4]. Generating tumor-specific T effector cells is a major goal of current immunotherapy development. T cell cytotoxicity could be impaired by the metabolic reprogramming of both tumor and immune cells. This phenomenon makes cell metabolic dependency an attractive target in designing effective antitumor therapy. Meanwhile, considering the overlapping of metabolic changes between cancer cells and immunes cells, interventions on metabolic dependency could work synergistically with immune checkpoint blockade. Table 2 lists the tentative therapeutic targets for the integration of metabolism and immunotherapy and will be further discussed below.
Table 2.
Tentative metabolism targeting approaches for tumor immunotherapy
| Drugs | Targets | Consequences | Effects |
|---|---|---|---|
| 2-Deoxyglucose | Hexokinase | Inhibits glycolysis | Increase CD8+ T cell memory and cytotoxic activity [95] |
| PD-1 blocking Antibodies | PD-1 | Inhibits glycolysis | Reinvigorating Exhausted T Cells [207] |
| Rapamycin | mTOR | Inhibits glycolysis | Promote memory CD8+ T cell differentiation [174] |
| Metformin | AMPK | Promote ATP production | Increase tumor CD8+ T cell infiltration and differentiation [180] |
| Diclofenac | MCT1 & MCT4 | Reduce lactate efflux | Delay tumor growth, reverse tumor acidification, preserve T cell function and improve immune checkpoint therapy [14] |
| AZD3965 | MCT1 | Reduce lactate export into TME | Increase tumor immune cell infiltration [122] |
| Lenalidomide | MCT1-CD147 ligation | Reduce lactate export into TME | Increase IL-2 and IFN-γ production in T cells [123] |
| 6-diazo-5-oxo-L-norleucine (DON) | Glutamine-requiring enzymes | Decrease glutamine supply | Increase longevity and activity of effector T cells [139] |
| INCB024360 | IDO1 | Inhibit tryptophan catabolism | Promote T and NK cells proliferation and IFN-γ production, reduce conversion to Tregs [208] |
| GDC-0919 | IDO1 | Inhibit tryptophan catabolism | Restore CD8+ T cell activity [209] |
| Fenofibrate | PPAR | Promote T cell FAO | Increase tumor CD8+ T cell infiltration [170] |
5.1. Targeting glucose metabolism
The glycolytic activity of tumor cells is negatively correlated with their response to immune checkpoint blockade therapy [87]. Tumor cells prefer to use aerobic glycolysis and lactate fermentation as main modes for glycose metabolism, which causes the TME to become acidic and hypoxic. Due to the low efficiency of aerobic glycolysis to generate ATP, tumor cells increase the uptake of glucose from TME. Therefore, blocking glucose transporter could be an effective therapeutic strategy to dampen tumor cell metabolic reprogramming [88]. Some drugs have been developed to target Glut1 on tumor cells. STF-31 is a selective glucose transporter Glut1 inhibitor. It impairs the growth of cells which lack the von Hippel-Lindau (VHL) tumor suppressor protein and depend on Glut1 and aerobic glycolysis for live [89]. WZB117 is another compound that inhibiting Glut1-mediated glucose transport. Studies have found that WZB117 can inhibit the generation and expansion of T cells with high Glut1 expression [90]. The effect of combined treatment with GLut1 inhibitors and immune checkpoint blockade needs further verification.
Several drugs have been reported to target glycolytic enzymes in cancer cells. 2-Deoxyglucose (2DG), a glucose analog, can be taken up by glucose transporter of cancer cells and phosphorylated by hexokinase to produce competitive inhibitor of glycolysis [91]. Phosphorylated 2DG also becomes trapped inside the cell and inhibits hexokinase [92]. It was reported that 2DG arrests cancer cell growth and/or induces apoptosis [93] and stimulates tumor cells into immunogenic ones when combine with chemotherapy [94]. 2DG has also been shown to enhance the antitumor function and generation of CD8+ T memory cells [95]. Hexokinase also can be inhibited by 3-bromopyruvate (3-BrPA). Cell death occurs when intracellular ATP levels deplete with 3-BrPA treatment [96]. 3-BrPA also could overcome chemoresistance, but so far, no report on its effect on T cell activation [97].
PKM2 is an isoenzyme of the pyruvate kinase and plays an important role in cancer cell metabolic reprogramming and in regulating gene expression [98, 99]. Small molecule inhibitors of PKM2 have been identified and showed anticancer effects [100]. What is more, PKM2 has been shown to play an important role in Th1 and Th17 differentiation and dimeric PKM2 is critical to inflammatory macrophage activation [101, 102]. A study recently reported that a small molecule PKM2 activator, TEPP-46, could induce PKM2 tetramerization and block its nuclear translocation, resulting in reduced T cell activation and Th1 and Th17 induction [103]. From this perspective, targeting PKM2 with either inhibitors or activators could be a therapeutic approach in T cell-mediated inflammation and autoimmunity. Phosphofructokinase 2 (PFK2) is another glycolytic enzyme. Its FB3 isoform (PFKFB3) is expressed in many cancers and has been reported to be an essential enzyme for T cell proliferation [104]. Small molecular inhibitors of PFKFB3 showed cytostatic effects on tumor cells and suppression on T cell activation [104, 105]. Dichloroacetate (DCA) is a generic drug that targeting pyruvate dehydrogenase kinase (PDHK). This drug promotes the shift from glycolysis to glucose oxidation in cancer cells and inhibiting tumor growth both in vivo and in vitro [106, 107]. However, like other drugs targeting glycolytic enzyme, DCA also induces Treg cells differentiation and could result in decreased immunosurveillance in case of its use as an anticancer drug [108].
Glycolysis is required for T cells effector functions. Therefore, it is a challenge to find a way to block the metabolism of tumor cells while improving the nutrient uptake of T cells. The expression of PD-L1 on cancer cells is associated with enhanced glycolysis in cancer cells via PI3K/Akt pathway and mTOR [109]. T cells with PD-1 activation have altered metabolism with inhibited glycolysis and increased lipolysis and fatty acid oxidation [110]. It was also reported that hypoxia-induced PD-L1 expression contributed to suppression on T cell cytotoxicity [111]. This suggests that using antibodies that block PD-1/PD-L1 checkpoint may remodel glucose level in the TME and promote T cell cytotoxicity by regulating T cell metabolism. Some reports have highlighted the potential of using cytokines to modulate T cell metabolism. For example, interleukin-2 (IL-2) promote glycolysis in T cells by inducing PI3K/AKT pathway [112]. A recombinant IL-2 immunocytokine was reported to promote tumor infiltrating cytotoxic T lymphocytes and increase the antitumor effect [113].
5.2. Target lactate accumulation
As a consequence of increased glycolysis, cancer cells augment a significant amount of lactate production. Lactate dehydrogenase (LDH) mediates bidirectional conversion between pyruvate and lactate. It is the rate-limiting enzyme for lactate production. A high rate of lactate production leads to intracellular lactate accumulation, which is toxic to cells and then released into the extracellular matrix. The family of monocarboxylate transporters (MCTs) is responsible for export lactate as well as protons [19]. This subsequently results in the acidification of the milieu, which provides growth advantage to cancer cells at the expense of human T cells [50]. The high level of lactate does not only degrade extracellular matrix to promote tumor migration and metastasis, it also upregulates the expression of pro-tumor molecules and angiogenesis factors [114]. Therefore, targeting lactate production or lactate transportation to tamper the acidic TME becomes a strategy for targeting cancer metabolism to improve immunotherapy.
LDHA is the predominant isoform of LDH and has a higher binding affinity to pyruvate. It is dispensable for most non-tumor tissues, making LDHA a selective target for drug discovery [115]. Several LDHA inhibitors were developed and tested their anticancer activity both in vivo and in vitro, such as gossypol and its derivative FX-11 [115]. Gossypol is a natural product found in cotton seeds. It blocks LDH non selectively and now being used in clinical oncology trials as monotherapy or combination with other drugs [115]. FX-11 has selectivity for LDH-A and was shown to inhibit LDH-A in the pancreatic cancer model. Although the treatment of FX-11 alone did not affect tumor growth, its combination with the anti-PD-1 antibody showed significant inhibition on tumor growth and metastasis [116]. Target LDHA directly using siRNA is another strategy and has been assessed in a gastric cancer model [117].
Besides inhibiting the enzyme LDH, the transporter MCTs family also can work as a target for reducing acidification of TME. MCTs family includes 14 different subtypes; only MCT1–4 were identified as lactate : proton symporters [118]. Not only did MCT function to export lactate, but it also necessary for lactate import into cells. The expression patterns and related functions of MCT vary in different tissues. While MCT2 and MCT3 have restricted distribution pattern, MCT1 is ubiquitously expressed at low levels as well as MCT4 is expressed in high glycolytic tissues, particularly [115]. The elevated expression level of MCT1 and/or MCT4 was observed in several human malignancies, including breast, colorectal, renal cell carcinoma, and prostate cancer [115]. Meanwhile, the cell surface glycoprotein CD147 is critical to the activity of MCT1 and MCT4. CD147 facilitates the cell surface expression and stable localization of MCT1 and MCT4 [119]. Thus, either targeting transporters directly or interrupting the ligation between transporter and CD147 could work as potential anticancer therapies. MCT1 is predominantly function to uptake catabolites. To date, several MCT1 inhibitors have been designed and reported to have the antitumor effect, such as AR-C155858, SR138009, AZD3965, as well as lenalidomide and pomalidomide [120–124]. AZD3965 is currently in phase I clinical trials for advanced tumors and diffuse large B cell lymphoma. A recent study reported that the MCT1 blockade by AZD3965 inhibits lipid biosynthesis and increases tumor immune cell infiltration, involving dendritic and natural killer cells [122]. Lenalidomide and pomalidomide are drugs that impair the MCT1-CD147 ligation. The treatment with lenalidomide can enhance IL-2 and IFN-γ secretion in T cells [123]. MCT4 mainly functions to facilitate lactate efflux from the high glycolytic cells. AZD0095 is a highly selective and potent inhibitor of MCT4. Beyond the modulation of lactate transport, a combination of AZD0095 with checkpoint inhibitors increases the tumor infiltrating lymphocytes and the activity of cytotoxic T cells, which reversing the lactate driven immunosuppression [125]. Furthermore, a non-steroidal anti-inflammatory drug, diclofenac, was investigated on its inhibition of MCT1 and MCT4. Diclofenac blocks the lactate transporters MCT1 and MCT4 and diminishes lactate efflux, delays tumor growth, and improves immune checkpoint therapy [87]. These findings collectively highlight that targeting the lactate transporter MCTs provides an efficient anticancer strategy that may improve cancer immunotherapy.
Since the lactate accumulation in the extracellular matrix results in the acidification of TME, drugs that can alkalinize the medium were tested for their anti-cancer capabilities. Proton pump inhibitors (PPIs) are commonly prescribed for acid related disorders. PPIs can inhibit the vacuolar-ATPase on cancer cells, a proton pump involved in the TME acidification. The inhibition of ATPase by PPIs decreases the proton extrusion from cells and thus reverse the extracellular acidity [114]. PPIs have been studies in several tumor models. It was reported that PPIs against melanoma both in vitro and in vivo by modifying the tumor PH environment [126]. A completed phase 2 clinical trial showed that the administration of PPIs improved the antitumor effect of docetaxel and cisplatin chemotherapy in metastatic breast cancer [127]. The impact of PPIs on the immunotherapy is controversial. PPIs can increase the extracellular pH, which subsequently reverts T cell energy and increase immunotherapy efficacy [128]. Nevertheless, recent studies found that PPIs may blunt the effect of immune checkpoint inhibitors in NSCLC patients [129]. Meanwhile, long-term use of PPIs is associated with an increased risk for gastric cancer, although PPIs can reduce the chemo-resistance by targeting the STAT3 pathway in gastric cancer [130]. These suggest more researches need to be done to identify biomarkers for cancer patient who might benefit from the combination of PPIs with immunotherapy.
5.3. Target glutamine metabolism
Glutamine is an essential nitrogen source for nucleotides and nonessential amino acid synthesis. One strategy to starve cancer cells of glutamine is to block the glutamine uptake. Alanine-serine-cysteine transporter 2 (ASCT2, also name as SLC1A5) is the major glutamine transporter, and it is upregulated in several solid tumors such as melanoma, colorectal cancer, and breast cancer [131]. Compounds have been designed to target ASCT2, which inhibits glutamine uptake and cellular proliferation of cancer cells [132]. However, ASCT2 is required for naïve CD4+ T cell differentiation. It was reported that ASCT2 deficiency could impair the induction of Th1 and Th17 cells and attenuate inflammatory T cell responses [133]. Glutamine is also used to replenish intermediates that are depleted during the TCA cycle. Through intracellular glutaminolysis, glutamine is converted to glutamate by glutaminase and subsequently converted to α-ketoglutarate, representing a pathway for glutamine carbons to enter the TCA cycle [134]. Glutaminase has two major isoforms, glutaminase 1 (GLS1) and GLS2. Several agents have been developed to inhibit GLS and showed promising effects as cancer treatments by single or combined with other drugs [134]. Several small molecule inhibitors against GLS have been developed and tested, such as BPTES, CB-839, and compound 968 [135]. CB-839 is one of the most potent and selective GSL1 inhibitor. It has been shown to suppress growth in various cancer cell types [25, 136], synergize with carfilzomib in resistant multiple myeloma cells [137] and overcome the resistance to catalytic mTOR kinase inhibitor MLN128 in lung squamous cell carcinoma [138]. Instead of targeting GLS itself, a glutamine antagonist 6-diazo-5-oxo-L-norleucine (DON) inhibits a broad range of enzymes that require glutamine as a substrate, have been shown dismantled the immunosuppressive TME. The glutamine blockage by DON causes decreased hypoxia, acidosis, and nutrient depletion in the TME, which conditions effector T cells toward a long-lived, highly activated phenotype [139]. This finding points to the potential of exploring glutamine antagonists as “metabolic checkpoints” for tumor immunotherapy.
5.4. Target amino acids metabolism
Cancer cells exhibit increased demand for energy and can redirect nutrients for their advantage. This creates a nutrient deprived TME for immune cells, especially T cells. Besides increased glucose consumption, amino acids also essential to support high metabolic demands for both cancer and immune cells. Several common characteristics have been revealed for the amino acid metabolism in cancer cells, such as increased uptake of nonessential amino acids from exogenous sources and altered enzyme levels for amino acid synthesis and catabolism [140]. Therefore, targeting amino acids in cancer therapy becomes a promising strategy and potential for benefit antitumor immune response.
Several amino acids have been studied as targets for anticancer drug development, including asparagine, glutamine, l-arginine, and l-tryptophan. Asparagine is an important regulator for amino acid homeostasis and anabolic metabolism. Some cancer cells have decreased expression levels of asparagine synthetase (ASNS), making them rely on asparagine supply from blood serum [141]. L-asparaginase is an FDA approved drug for several cancers, especially acute lymphocytic leukemia. It catalyzes the deamidation of asparagine, which results in amino acid depletion in blood serum [142]. Although l-asparagine administration is associated with immunosuppression as a major side effect, researchers found that antibody against PD-1 can be used to treat NK/T cell lymphoma failing l-asparaginase [143]. Similar approaches have been investigated for targeting arginine metabolism. Many cancers, such as hepatocellular carcinoma, bladder, and breast cancer, have deficient arginine metabolism and rely on extracellular arginine supply [144]. Arginine deprivation by deiminase or arginase can inhibit cancer proliferation both in vitro and in vivo. The combination of arginase inhibitor and immune checkpoint inhibitor is currently tested in clinical [145].
L-tryptophan is another essential amino acid that both itself and its metabolites have an important role in several physiological processes, including immune cell response and neuronal excitability [146]. Indoleamine-2,3-dioxygenase (IDO) is the rate-limiting enzyme that catalyzes the tryptophan catabolism in the kynurenine pathway. IDO is encoded by the two genes, IDO1 as well as IDO2. It is overexpressed in tumor cells and tumor-related immune cells and endothelial cells [114]. The function of IDO is associated with multiple levels of tumorigenesis, including immune evasion, tumor invasion, and metastasis [147]. Overexpressed IDO also mediates tumor resistance to both chemotherapy and immune checkpoint inhibitors in a preclinical tumor model [148]. There are two central mechanisms by which the overexpressed IDO causes reduced tumor immune surveillance through tryptophan metabolism: tryptophan deprivation in TME and the release of toxic tryptophan catabolites into the plasma. Both mechanisms inhibit the proliferation and activity of cytotoxic T cells. Furthermore, tryptophan deprivation also induces impaired phenotype on antigen-presentation cells and promotes the generation of some immunosuppressive cells, including Tregs and MDSCs [114]. Accordingly, IDO is an attractive target for drug discovery in cancer immunotherapy. Different strategies have been tested to block IDO or to target its signaling pathway. Epacadostat (INCB024360) is one of the clinical-stage IDO1 inhibitors. Its combination with immune checkpoint inhibitor pembrolizumab showed encouraging data in phase I/II study of melanoma patients but failed to meet their endpoint in the phase III trial [149]. Meanwhile, several ongoing clinical trials are testing the combination of IDO1 inhibitor with antitumor vaccines. All these motivate researchers to learn more about IDO1 inhibition and tryptophan metabolism in the tumor, to perform a better patient selection for cancer immunotherapy.
5.5. Target fatty acid metabolism
In contrast to the normal cells, which take up lipids for new membranes, cancer cell proliferation relies on de novo fatty acid (FA) synthesis to provide building blocks for new membrane assemble. This indicates altered FA metabolism is crucial to tumorigenesis. Recent studies have found that not only de novo FA synthesis, but also FA uptake from extracellular, FA modification and degradation, all play distinctive roles during different stages of tumor development [150].
The sterol regulatory element-binding protein 1 (SREBP-1) is the major transcription factor that activates FA biosynthetic genes expression, including ACLY, ACC, and FASN. Although targeting SREBP-1 represents an efficient way to inhibit the expression of lipogenic genes, no inhibitor was reported since SREBP-1 is widely considered undruggable. Another strategy was developed to inhibit the maturation of SREBPs through block its association with SREBP-cleavage activating protein (SCAP) [151]. Fatostatin is such an inhibitor and displayed antitumor activities in different cancer models, including prostate, NSCLC, and endometrial carcinoma [152–154]. Several other compounds have been reported to target the SCAP/SREBPs pathway and inhibit tumor growth, such as botulin, xanthohumol, BF175, and nelfinavir [155]. This suggests that targeting SCAP/SREBPs interaction is a promising strategy and may combine with immunotherapy for cancer treatment.
Directly target enzymes involved in FA synthesis might be a therapeutic interest. Fatty acid synthase (FASN) is a crucial enzyme involved in de novo lipogenesis. FASN is overexpressed in many cancers, and its expression correlates with poor prognosis. Accumulating evidence has suggested FASN is a metabolic oncogene for its important role in tumor growth and metastasis in several human tumor studies, including prostate, breast, and lung adenocarcinoma [156–158]. Inhibition of FASN has numerous anticancer effects, including induction of apoptosis, inhibition of DNA replication, and disruption on cellular membrane functions [159]. There are extensive preclinical studies on FASN inhibitors, but many limited by their off-target effects and pharmacologic properties. TVB-2640 is the only FASN inhibitor that has moved to the clinical trials. Meanwhile, it was reported that elevated FASN expression in ovarian cancer correlates with immunosuppressive status. Researchers found that constitutive FASN activation leads to abnormal lipid accumulation, which blunts DCs activity, resulting in the failure to initiate T cell cytotoxicity in antitumor immunity [160]. This indicates that targeting FASN could lead to improved cancer immunotherapy with FASN overexpression. Acetyl-CoA carboxylase 1 (ACC1) is another rate-limiting enzyme in FA synthesis. ACC1 inhibitors, such as ND-654, showed anticancer activities in preclinical models [161]. However, T cells are highly dependent on ACC1 to generate long-chain FAs [162]. ACC1-mediated de novo lipogenesis is essential for CD8+ T cell expansion, ACC1 deficient CD8+ T cells show low survival rate during antigen-specific response [162]. Although this can be rescued by exogenous FA, agents need to be considered carefully before combining with immunotherapies.
Synthesized FAs are broken down through fatty acid oxidation (FAO) in the mitochondria to produce ATP and NADPH. FAO is important for the development of CD8+ T cell memory cells, CD4+ Treg cells, and the MDSC-mediated T cell suppressive function [163]. Carnitine palmitoyltransferase 1 (CPT1) is the rate-limiting enzyme in FA transportation into mitochondria. It also plays a critical role in the generation of CD8+ memory T cells and Treg cells [164]. Etomoxir has been a well-studied CPT1 inhibitor. It showed antitumor effects in prostate cancer, glioblastoma, and leukemia models [165–167]. However, studies found that Etomoxir lacks specificity for CPT1 at high concentration and induces oxidative stress as off-target effects [168]. Another study using CPT1A-deficient T cells reported that CPT1 mediated FA oxidation may not be necessary for Etomoxir actions on regulatory and memory T cells [169]. Since targeting FAO may affects several immune cell populations, the usage of CPT1 inhibitors for anticancer treatments may have unpredictable outcomes. Alternatively, it was reported that enhancing the FAO of CD8+ T cells improved their cytotoxicity in cancer treatment. Researchers used a peroxisome proliferator-activated receptor (PPAR) agonist, Fenofibrate, to modify CD8+ T cells FA metabolism and subsequent improved CD8+ T cell tumor infiltrating and enhanced the therapeutic effects of PD-1 blockade in melanoma [170]. This study demonstrated that promoting the FAO in T cells might be a therapeutic option when there is T cell exhaustion and FA as an alternative energy source for T cell activity.
5.6. Target other metabolism-regulating molecules
Considering the effects of TME metabolic profile on tumor immune evasion, several metabolism-regulating molecules, such as AMPK, mTOR, and HIF-1α, were investigated as targets for cancer immunotherapy.
The PI3K-AKT-mTOR pathway is a vital transduction cascade that is critical in both cancer and immune cell metabolism. PI3K-AKT-mTOR pathway coordinates the uptake and metabolism of several nutrients, including glucose, glutamine, and lipids, to support the proliferation and survival of cancer cells [171]. Therapies that target this pathway were developed based on rapamycin, which is a well-known mTOR inhibitor. Treatment with rapamycin alone or combine with AKT inhibitors showed antitumor activity in various tumor study models, including breast, renal and pancreatic cancers [172]. The PI3K-AKT-mTOR pathway is also important for T cell fate decisions. T cells need mTOR signaling to sense and integrate immune signals, including antigenic, co-stimulatory, and cytokine signals [173]. The administration of rapamycin can improve the quantity and quality of CD8+ T cells [174]. However, rapamycin also can affect the differentiation of other immune cells, including T effector cells, Tregs, and macrophages [163], which makes the outcome of rapamycin treatment unpredictable. A study in glioblastoma showed that the combination of rapamycin with immunotherapy enhanced cytotoxic and memory T cell functions [175]. In sum, mTOR is an attractive metabolite target, but more work needs to be done in grouping patients base on the benefit from the treatment.
AMP-activated protein kinase (AMPK) is a well-known regulator in maintaining cellular energy homeostasis. It is activated response to energy shortage, and its activation can promote ATP production. In the past, AMPK was mainly studied in metabolic syndrome and type-2 diabetes. Recently, AMPK gained attention in cancer study due to its ability to oppose the metabolic reprogramming involved in the Warburg effect [176]. AMPK is the upstream regulator of several critical mediators involved in tumor development, such as mTOR, COX-2, p53, and ACC [177]. It also participates in mediating DNA damage repair and the cytotoxicity effect of both radiation and chemotherapy [176]. Preclinical studies have demonstrated the tumor suppression function of AMPK in lung, prostate, and colorectal cancer models [177]. Regulation of AMPK activity using drugs such as metformin has been tested for antitumor activity in clinical trials [178]. Beyond effects on cancer cells, AMPK also has an important role in the T cell metabolic adaption [179]. Activation of AMPK by metformin leads to increased CD8+ tumor-infiltrating lymphocytes [180], suggesting metformin treatment might benefit the effect of immune checkpoint inhibitors. AMPK can also promote the formation of Tregs and reduce the differentiation of Th1 and Th17 cells, which might result in an unwanted immune modulation in the TME [163]. Thus, the synergistic effect of targeting AMPK with immunotherapy might vary in different cancers considering the properties of immune cells dominate the TME.
HIF-1α plays a major role in the cellular response to hypoxia, a common feature of tumor TME. HIF-1α is overexpressed in most cancer types. It is involved in different cancer cell phenotypes, including immortalization, EMT, cancer stem cell maintenance, and angiogenesis [181]. It also affects the performance of immune cells, including modulating the pro-tumor effect of TAMs and supper effective T cell function by producing metabolites such as ROS [114]. Several compounds were designed to inhibit HIF-1α at different levels. PX-478 is a molecule derived from melphalan, and it can decrease the expression level of HIF-1α while inhibiting HIF-1α deubiquitination [182]. It was reported that, in a PDAC study model, a combination of PX-478 with chemotherapy gemcitabine shown inhibition on tumor growth while favors immune response by inducing DCs maturation and cytokines secretion from T cells [183]. Many other HIF-1α targeting compounds are under investigation now, and it is wise to test their effect on immune cells and potential on immunotherapy.
6. Outlook
In 2011, the Food and Drug Administration (FDA) approved the first anti-CTLA-4 monoclonal antibody drug Ipilimumab for metastatic melanoma [184]. Three years later, the first anti-PD-1 monoclonal antibody drugs Nivolumab and Pembrolizumab were approved in Japan and the U.S., respectively, for treating high-level metastatic melanoma in which other treatments were ineffective. Patients treated with Pembrolizumab had a more prolonged progression-free survival and fewer toxic side effects than Ipilimumab [185–191]. Following the FDA approval of the anti-PD-1 treatments, the indications of such monotherapy were gradually expanding to treat non-small cell lung cancer, kidney cancer, bladder cancer, gastric cancer, head and neck cancer, and other malignancies [192–196]. More so, the combination of PD-1 and CTLA-4 blockades rendered as a more effective synergistic treatment option comparing to monotherapy [197]. However, most patients have not shown stable remission after PD1 therapy. Thus, further studies are needed to determine the intrinsic and extrinsic factors that contribute to the variability in patient responses to immune checkpoint blockade [198].
In this review, we have summarized the metabolic reprogramming and the interplay between cancer cells and immune cells. Cancer cells can reshape their nutrient profiles and induce acidification in TME, dampening activated immune cells survival, differentiation, and effector functions. Immunometabolism, therefore, becomes an intriguing topic in anti-tumor immunity and immunotherapy. However, recent discoveries have pointed out the complex regulation of immunometabolism. Specifically, glutamine metabolism was found to be an essential metabolic pathway for effector T cell survival and functions [33, 55], but recent report has shown that glutamine antagonism supports the activation, proliferation and long-lived phenotype of effector T cells [139]. It has been proposed that glutamine-restricted metabolism promoted the differentiation into memory precursors and increased the antitumor activity, thereby reducing T-cell exhaustion [199]. Nonetheless, the role of glutamine metabolism in regulating T cell proliferation and effector functions still need to be further elucidated. Another newly published paper showed that inosine can be used as an alternative carbon source by CD8+ T cells to achieve effector functions under glucose restriction, and supplementation with inosine improved effector T cell-mediated antitumor activities in animal models [200]. This finding provides a new strategy to improve the efficacy of cancer immunotherapy by modulating T cell metabolism.
Above all, the balance of utilization of nutrients between cancer cells and immune cells within TME is a crucial regulator and may determine antitumor immunity and immunotherapy efficacy. Pharmacological interventions targeting the metabolic properties of cancer cells or immune cells can have potential synergic effects with current immune and targeted therapies.
Acknowledgement
This work was supported in part by US National Institutes of Health grants R01CA203737 (X.L.), R01CA222251 (X.L.), and T32GM077229 (X.W), by US Department of Defense Prostate Cancer Research Program Idea Development Award PC190042 (X.Z.), by Indiana University Strategic Research Initiative fund (X.L.) and Vera Bradley Foundation for Breast Cancer Research (X.L. and X.Z.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
Authors declare no conflict of interest.
References
- [1].Hanahan D, Coussens LM, Accessories to the crime: functions of cells recruited to the tumor microenvironment, Cancer Cell 21(3) (2012) 309–22. [DOI] [PubMed] [Google Scholar]
- [2].Turley SJ, Cremasco V, Astarita JL, Immunological hallmarks of stromal cells in the tumour microenvironment, Nature reviews. Immunology 15(11) (2015) 669–82. [DOI] [PubMed] [Google Scholar]
- [3].Clara JA, Monge C, Yang Y, Takebe N, Targeting signalling pathways and the immune microenvironment of cancer stem cells - a clinical update, Nat Rev Clin Oncol 17(4) (2020) 204–232. [DOI] [PubMed] [Google Scholar]
- [4].Hanahan D, Weinberg RA, Hallmarks of cancer: the next generation, Cell 144(5) (2011) 646–74. [DOI] [PubMed] [Google Scholar]
- [5].Lunt SY, Vander Heiden MG, Aerobic glycolysis: meeting the metabolic requirements of cell proliferation, Annu Rev Cell Dev Biol 27 (2011) 441–64. [DOI] [PubMed] [Google Scholar]
- [6].Wise DR, Thompson CB, Glutamine addiction: a new therapeutic target in cancer, Trends in biochemical sciences 35(8) (2010) 427–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Andrejeva G, Rathmell JC, Similarities and Distinctions of Cancer and Immune Metabolism in Inflammation and Tumors, Cell metabolism 26(1) (2017) 49–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Maciolek JA, Pasternak JA, Wilson HL, Metabolism of activated T lymphocytes, Curr Opin Immunol 27 (2014) 60–74. [DOI] [PubMed] [Google Scholar]
- [9].Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O’Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J, Weber JD, Pearce EJ, Jones RG, Pearce EL, Posttranscriptional control of T cell effector function by aerobic glycolysis, Cell 153(6) (2013) 1239–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJ, Tonc E, Schreiber RD, Pearce EJ, Pearce EL, Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression, Cell 162(6) (2015) 1229–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, Tsui YC, Cui G, Micevic G, Perales JC, Kleinstein SH, Abel ED, Insogna KL, Feske S, Locasale JW, Bosenberg MW, Rathmell JC, Kaech SM, Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses, Cell 162(6) (2015) 1217–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, Matos C, Bruss C, Klobuch S, Peter K, Kastenberger M, Bogdan C, Schleicher U, Mackensen A, Ullrich E, Fichtner-Feigl S, Kesselring R, Mack M, Ritter U, Schmid M, Blank C, Dettmer K, Oefner PJ, Hoffmann P, Walenta S, Geissler EK, Pouyssegur J, Villunger A, Steven A, Seliger B, Schreml S, Haferkamp S, Kohl E, Karrer S, Berneburg M, Herr W, Mueller-Klieser W, Renner K, Kreutz M, LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells, Cell metabolism 24(5) (2016) 657–671. [DOI] [PubMed] [Google Scholar]
- [13].Anderson KG, Stromnes IM, Greenberg PD, Obstacles Posed by the Tumor Microenvironment to T cell Activity: A Case for Synergistic Therapies, Cancer Cell 31(3) (2017) 311–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Renner K, Bruss C, Schnell A, Koehl G, Becker HM, Fante M, Menevse AN, Kauer N, Blazquez R, Hacker L, Decking SM, Bohn T, Faerber S, Evert K, Aigle L, Amslinger S, Landa M, Krijgsman O, Rozeman EA, Brummer C, Siska PJ, Singer K, Pektor S, Miederer M, Peter K, Gottfried E, Herr W, Marchiq I, Pouyssegur J, Roush WR, Ong S, Warren S, Pukrop T, Beckhove P, Lang SA, Bopp T, Blank CU, Cleveland JL, Oefner PJ, Dettmer K, Selby M, Kreutz M, Restricting Glycolysis Preserves T Cell Effector Functions and Augments Checkpoint Therapy, Cell reports 29(1) (2019) 135–150 e9. [DOI] [PubMed] [Google Scholar]
- [15].Vander Heiden MG, Cantley LC, Thompson CB, Understanding the Warburg effect: the metabolic requirements of cell proliferation, Science 324(5930) (2009) 1029–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cairns RA, Harris IS, Mak TW, Regulation of cancer cell metabolism, Nature reviews. Cancer 11(2) (2011) 85–95. [DOI] [PubMed] [Google Scholar]
- [17].Moreno-Sanchez R, Rodriguez-Enriquez S, Marin-Hernandez A, Saavedra E, Energy metabolism in tumor cells, FEBS J 274(6) (2007) 1393–418. [DOI] [PubMed] [Google Scholar]
- [18].Nathan C, Ding A, SnapShot: Reactive Oxygen Intermediates (ROI), Cell 140(6) (2010) 951–951 e2. [DOI] [PubMed] [Google Scholar]
- [19].Doherty JR, Cleveland JL, Targeting lactate metabolism for cancer therapeutics, The Journal of clinical investigation 123(9) (2013) 3685–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, Esparza LA, Reya T, Le Z, Yanxiang Guo J, White E, Rabinowitz JD, Glucose feeds the TCA cycle via circulating lactate, Nature 551(7678) (2017) 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, Yang C, Do QN, Doucette S, Burguete D, Li H, Huet G, Yuan Q, Wigal T, Butt Y, Ni M, Torrealba J, Oliver D, Lenkinski RE, Malloy CR, Wachsmann JW, Young JD, Kernstine K, DeBerardinis RJ, Lactate Metabolism in Human Lung Tumors, Cell 171(2) (2017) 358–371 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lieu EL, Nguyen T, Rhyne S, Kim J, Amino acids in cancer, Exp Mol Med 52(1) (2020) 15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Jin L, Alesi GN, Kang S, Glutaminolysis as a target for cancer therapy, Oncogene 35(28) (2016) 3619–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Korangath P, Teo WW, Sadik H, Han L, Mori N, Huijts CM, Wildes F, Bharti S, Zhang Z, Santa-Maria CA, Tsai H, Dang CV, Stearns V, Bhujwalla ZM, Sukumar S, Targeting Glutamine Metabolism in Breast Cancer with Aminooxyacetate, Clin Cancer Res 21(14) (2015) 3263–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, Janes JR, Laidig GJ, Lewis ER, Li J, Mackinnon AL, Parlati F, Rodriguez ML, Shwonek PJ, Sjogren EB, Stanton TF, Wang T, Yang J, Zhao F, Bennett MK, Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer, Mol Cancer Ther 13(4) (2014) 890–901. [DOI] [PubMed] [Google Scholar]
- [26].Yang L, Moss T, Mangala LS, Marini J, Zhao H, Wahlig S, Armaiz-Pena G, Jiang D, Achreja A, Win J, Roopaimoole R, Rodriguez-Aguayo C, Mercado-Uribe I, Lopez-Berestein G, Liu J, Tsukamoto T, Sood AK, Ram PT, Nagrath D, Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer, Mol Syst Biol 10 (2014) 728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cacace A, Sboarina M, Vazeille T, Sonveaux P, Glutamine activates STAT3 to control cancer cell proliferation independently of glutamine metabolism, Oncogene 36(15) (2017) 2074–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Currie E, Schulze A, Zechner R, Walther TC, Farese RV Jr., Cellular fatty acid metabolism and cancer, Cell metabolism 18(2) (2013) 153–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ho PC, Liu PS, Metabolic communication in tumors: a new layer of immunoregulation for immune evasion, J Immunother Cancer 4 (2016) 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].O’Neill LA, Kishton RJ, Rathmell J, A guide to immunometabolism for immunologists, Nature reviews. Immunology 16(9) (2016) 553–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kim J, Regulation of Immune Cell Functions by Metabolic Reprogramming, Journal of immunology research 2018 (2018) 8605471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Buck MD, O’Sullivan D, Pearce EL, T cell metabolism drives immunity, The Journal of experimental medicine 212(9) (2015) 1345–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, Green DR, The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation, Immunity 35(6) (2011) 871–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, Anderson SM, Abel ED, Chen BJ, Hale LP, Rathmell JC, The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function, Cell metabolism 20(1) (2014) 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, Macintyre AN, Goraksha-Hicks P, Rathmell JC, Makowski L, Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype, The Journal of biological chemistry 289(11) (2014) 7884–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Fox CJ, Hammerman PS, Thompson CB, Fuel feeds function: energy metabolism and the T-cell response, Nature reviews. Immunology 5(11) (2005) 844–52. [DOI] [PubMed] [Google Scholar]
- [37].Menk AV, Scharping NE, Moreci RS, Zeng X, Guy C, Salvatore S, Bae H, Xie J, Young HA, Wendell SG, Delgoffe GM, Early TCR Signaling Induces Rapid Aerobic Glycolysis Enabling Distinct Acute T Cell Effector Functions, Cell reports 22(6) (2018) 1509–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kumar V, Patel S, Tcyganov E, Gabrilovich DI, The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment, Trends Immunol 37(3) (2016) 208–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhang Y, Yu G, Chu H, Wang X, Xiong L, Cai G, Liu R, Gao H, Tao B, Li W, Li G, Liang J, Yang W, Macrophage-Associated PGK1 Phosphorylation Promotes Aerobic Glycolysis and Tumorigenesis, Molecular cell 71(2) (2018) 201–215.e7. [DOI] [PubMed] [Google Scholar]
- [40].Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, Winter PS, Liu X, Priyadharshini B, Slawinska ME, Haeberli L, Huck C, Turka LA, Wood KC, Hale LP, Smith PA, Schneider MA, MacIver NJ, Locasale JW, Newgard CB, Shinohara ML, Rathmell JC, Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation, The Journal of clinical investigation 125(1) (2015) 194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Buck MD, Sowell RT, Kaech SM, Pearce EL, Metabolic Instruction of Immunity, Cell 169(4) (2017) 570–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Cham CM, Driessens G, O’Keefe JP, Gajewski TF, Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells, European journal of immunology 38(9) (2008) 2438–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhao E, Maj T, Kryczek I, Li W, Wu K, Zhao L, Wei S, Crespo J, Wan S, Vatan L, Szeliga W, Shao I, Wang Y, Liu Y, Varambally S, Chinnaiyan AM, Welling TH, Marquez V, Kotarski J, Wang H, Wang Z, Zhang Y, Liu R, Wang G, Zou W, Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction, Nature immunology 17(1) (2016) 95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, Stelekati E, McLane LM, Paley MA, Delgoffe GM, Wherry EJ, Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8(+) T Cell Exhaustion, Immunity 45(2) (2016) 358–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, Linsley PS, Thompson CB, Riley JL, CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms, Molecular and cellular biology 25(21) (2005) 9543–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, Karoly ED, Freeman GJ, Petkova V, Seth P, Li L, Boussiotis VA, PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation, Nat Commun 6 (2015) 6692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].de la Cruz-Lopez KG, Castro-Munoz LJ, Reyes-Hernandez DO, Garcia-Carranca A, Manzo-Merino J, Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches, Front Oncol 9 (2019) 1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, Gottfried E, Schwarz S, Rothe G, Hoves S, Renner K, Timischl B, Mackensen A, Kunz-Schughart L, Andreesen R, Krause SW, Kreutz M, Inhibitory effect of tumor cell-derived lactic acid on human T cells, Blood 109(9) (2007) 3812–9. [DOI] [PubMed] [Google Scholar]
- [49].Pilon-Thomas S, Kodumudi KN, El-Kenawi AE, Russell S, Weber AM, Luddy K, Damaghi M, Wojtkowiak JW, Mule JJ, Ibrahim-Hashim A, Gillies RJ, Neutralization of Tumor Acidity Improves Antitumor Responses to Immunotherapy, Cancer Res 76(6) (2016) 1381–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, Gottfried E, Schwarz S, Rothe G, Hoves S, Inhibitory effect of tumor cell–derived lactic acid on human T cells, Blood 109(9) (2007) 3812–3819. [DOI] [PubMed] [Google Scholar]
- [51].Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM, Cline GW, Phillips AJ, Medzhitov R, Functional polarization of tumour-associated macrophages by tumour-derived lactic acid, Nature 513(7519) (2014) 559–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Harmon C, Robinson MW, Hand F, Almuaili D, Mentor K, Houlihan DD, Hoti E, Lynch L, Geoghegan J, O’Farrelly C, Lactate-Mediated Acidification of Tumor Microenvironment Induces Apoptosis of Liver-Resident NK Cells in Colorectal Liver Metastasis, Cancer Immunol Res 7(2) (2019) 335–346. [DOI] [PubMed] [Google Scholar]
- [53].Xie D, Zhu S, Bai L, Lactic acid in tumor microenvironments causes dysfunction of NKT cells by interfering with mTOR signaling, Sci China Life Sci 59(12) (2016) 1290–1296. [DOI] [PubMed] [Google Scholar]
- [54].Daneshmandi S, Wegiel B, Seth P, Blockade of Lactate Dehydrogenase-A (LDH-A) Improves Efficacy of Anti-Programmed Cell Death-1 (PD-1) Therapy in Melanoma, Cancers 11(4) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, Turay AM, Frauwirth KA, Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation, Journal of immunology 185(2) (2010) 1037–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Cluntun AA, Lukey MJ, Cerione RA, Locasale JW, Glutamine Metabolism in Cancer: Understanding the Heterogeneity, Trends Cancer 3(3) (2017) 169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ananieva E, Targeting amino acid metabolism in cancer growth and anti-tumor immune response, World journal of biological chemistry 6(4) (2015) 281–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Bott AJ, Shen J, Tonelli C, Zhan L, Sivaram N, Jiang YP, Yu X, Bhatt V, Chiles E, Zhong H, Maimouni S, Dai W, Velasquez S, Pan JA, Muthalagu N, Morton J, Anthony TG, Feng H, Lamers WH, Murphy DJ, Guo JY, Jin J, Crawford HC, Zhang L, White E, Lin RZ, Su X, Tuveson DA, Zong WX, Glutamine Anabolism Plays a Critical Role in Pancreatic Cancer by Coupling Carbon and Nitrogen Metabolism, Cell reports 29(5) (2019) 1287–1298 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kurmi K, Haigis MC, Nitrogen Metabolism in Cancer and Immunity, Trends Cell Biol 30(5) (2020) 408–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Takeuchi Y, Nakayama Y, Fukusaki E, Irino Y, Glutamate production from ammonia via glutamate dehydrogenase 2 activity supports cancer cell proliferation under glutamine depletion, Biochemical and biophysical research communications 495(1) (2018) 761–767. [DOI] [PubMed] [Google Scholar]
- [61].Spinelli JB, Yoon H, Ringel AE, Jeanfavre S, Clish CB, Haigis MC, Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass, Science 358(6365) (2017) 941–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Metzler B, Gfeller P, Guinet E, Restricting Glutamine or Glutamine-Dependent Purine and Pyrimidine Syntheses Promotes Human T Cells with High FOXP3 Expression and Regulatory Properties, Journal of immunology (Baltimore, Md. : 1950) 196(9) (2016) 3618–30. [DOI] [PubMed] [Google Scholar]
- [63].Fahrmann JF, Vykoukal JV, Ostrin EJ, Amino Acid Oncometabolism and Immunomodulation of the Tumor Microenvironment in Lung Cancer, Frontiers in oncology 10 (2020) 276–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Chang CH, Pearce EL, Emerging concepts of T cell metabolism as a target of immunotherapy, Nature immunology 17(4) (2016) 364–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Briggs KJ, Koivunen P, Cao S, Backus KM, Olenchock BA, Patel H, Zhang Q, Signoretti S, Gerfen GJ, Richardson AL, Witkiewicz AK, Cravatt BF, Clardy J, Kaelin WG Jr., Paracrine Induction of HIF by Glutamate in Breast Cancer: EglN1 Senses Cysteine, Cell 166(1) (2016) 126–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Siska PJ, Kim B, Ji X, Hoeksema MD, Massion PP, Beckermann KE, Wu J, Chi JT, Hong J, Rathmell JC, Fluorescence-based measurement of cystine uptake through xCT shows requirement for ROS detoxification in activated lymphocytes, J Immunol Methods 438 (2016) 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Arensman MD, Yang XS, Leahy DM, Toral-Barza L, Mileski M, Rosfjord EC, Wang F, Deng S, Myers JS, Abraham RT, Eng CH, Cystine-glutamate antiporter xCT deficiency suppresses tumor growth while preserving antitumor immunity, Proceedings of the National Academy of Sciences of the United States of America 116(19) (2019) 9533–9542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Pacheco R, Oliva H, Martinez-Navio JM, Climent N, Ciruela F, Gatell JM, Gallart T, Mallol J, Lluis C, Franco R, Glutamate released by dendritic cells as a novel modulator of T cell activation, Journal of immunology 177(10) (2006) 6695–704. [DOI] [PubMed] [Google Scholar]
- [69].Ho P-C, Liu P-S, Metabolic communication in tumors: a new layer of immunoregulation for immune evasion, J Immunother Cancer 4 (2016) 4–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Wang R, Green DR, Metabolic checkpoints in activated T cells, Nature immunology 13(10) (2012) 907–15. [DOI] [PubMed] [Google Scholar]
- [71].Opitz CA, Somarribas Patterson LF, Mohapatra SR, Dewi DL, Sadik A, Platten M, Trump S, The therapeutic potential of targeting tryptophan catabolism in cancer, British journal of cancer 122(1) (2020) 30–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Platten M, Nollen EAA, Rohrig UF, Fallarino F, Opitz CA, Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond, Nature reviews. Drug discovery 18(5) (2019) 379–401. [DOI] [PubMed] [Google Scholar]
- [73].Mellor AL, Lemos H, Huang L, Indoleamine 2,3-Dioxygenase and Tolerance: Where Are We Now?, Front Immunol 8 (2017) 1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Ravishankar B, Liu H, Shinde R, Chaudhary K, Xiao W, Bradley J, Koritzinsky M, Madaio MP, McGaha TL, The amino acid sensor GCN2 inhibits inflammatory responses to apoptotic cells promoting tolerance and suppressing systemic autoimmunity, Proceedings of the National Academy of Sciences of the United States of America 112(34) (2015) 10774–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Cobbold SP, Adams E, Farquhar CA, Nolan KF, Howie D, Lui KO, Fairchild PJ, Mellor AL, Ron D, Waldmann H, Infectious tolerance via the consumption of essential amino acids and mTOR signaling, Proceedings of the National Academy of Sciences of the United States of America 106(29) (2009) 12055–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Platten M, Wick W, Van den Eynde BJ, Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion, Cancer research 72(21) (2012) 5435–40. [DOI] [PubMed] [Google Scholar]
- [77].Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, Boon T, Van den Eynde BJ, Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase, Nature medicine 9(10) (2003) 1269–74. [DOI] [PubMed] [Google Scholar]
- [78].Nguyen DJM, Theodoropoulos G, Li YY, Wu C, Sha W, Feun LG, Lampidis TJ, Savaraj N, Wangpaichitr M, Targeting the Kynurenine Pathway for the Treatment of Cisplatin-Resistant Lung Cancer, Molecular cancer research : MCR 18(1) (2020) 105–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Hsu YL, Hung JY, Chiang SY, Jian SF, Wu CY, Lin YS, Tsai YM, Chou SH, Tsai MJ, Kuo PL, Lung cancer-derived galectin-1 contributes to cancer associated fibroblast-mediated cancer progression and immune suppression through TDO2/kynurenine axis, Oncotarget 7(19) (2016) 27584–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Ma EH, Bantug G, Griss T, Condotta S, Johnson RM, Samborska B, Mainolfi N, Suri V, Guak H, Balmer ML, Verway MJ, Raissi TC, Tsui H, Boukhaled G, Henriques da Costa S, Frezza C, Krawczyk CM, Friedman A, Manfredi M, Richer MJ, Hess C, Jones RG, Serine Is an Essential Metabolite for Effector T Cell Expansion, Cell metabolism 25(2) (2017) 345–357. [DOI] [PubMed] [Google Scholar]
- [81].Yang M, Vousden KH, Serine and one-carbon metabolism in cancer, Nature reviews. Cancer 16(10) (2016) 650–62. [DOI] [PubMed] [Google Scholar]
- [82].Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, Myer VE, MacKeigan JP, Porter JA, Wang YK, Cantley LC, Finan PM, Murphy LO, Bidirectional transport of amino acids regulates mTOR and autophagy, Cell 136(3) (2009) 521–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Zheng Y, Delgoffe GM, Meyer CF, Chan W, Powell JD, Anergic T cells are metabolically anergic, Journal of immunology 183(10) (2009) 6095–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Koundouros N, Poulogiannis G, Reprogramming of fatty acid metabolism in cancer, Br J Cancer 122(1) (2020) 4–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, Sandouk A, Hesse C, Castro CN, Bahre H, Tschirner SK, Gorinski N, Gohmert M, Mayer CT, Huehn J, Ponimaskin E, Abraham WR, Muller R, Lochner M, Sparwasser T, De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells, Nature medicine 20(11) (2014) 1327–33. [DOI] [PubMed] [Google Scholar]
- [86].Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, Zhang S, Bettigole SE, Gupta D, Holcomb K, Ellenson LH, Caputo T, Lee AH, Conejo-Garcia JR, Glimcher LH, ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis, Cell 161(7) (2015) 1527–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Renner K, Bruss C, Schnell A, Koehl G, Becker HM, Fante M, Menevse A-N, Kauer N, Blazquez R, Hacker L, Restricting glycolysis preserves T Cell effector functions and augments checkpoint therapy, Cell reports 29(1) (2019) 135–150. e9. [DOI] [PubMed] [Google Scholar]
- [88].Macheda ML, Rogers S, Best JD, Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer, Journal of cellular physiology 202(3) (2005) 654–662. [DOI] [PubMed] [Google Scholar]
- [89].Chan DA, Sutphin PD, Nguyen P, Turcotte S, Lai EW, Banh A, Reynolds GE, Chi JT, Wu J, Solow-Cordero DE, Bonnet M, Flanagan JU, Bouley DM, Graves EE, Denny WA, Hay MP, Giaccia AJ, Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality, Sci Transl Med 3(94) (2011) 94ra70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Vignali D, Cantarelli E, Bordignon C, Canu A, Citro A, Annoni A, Piemonti L, Monti P, Detection and Characterization of CD8(+) Autoreactive Memory Stem T Cells in Patients With Type 1 Diabetes, Diabetes 67(5) (2018) 936–945. [DOI] [PubMed] [Google Scholar]
- [91].Ralser M, Wamelink MM, Struys EA, Joppich C, Krobitsch S, Jakobs C, Lehrach H, A catabolic block does not sufficiently explain how 2-deoxy-D-glucose inhibits cell growth, Proceedings of the National Academy of Sciences 105(46) (2008) 17807–17811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Parniak M, Kalant N, Incorporation of glucose into glycogen in primary cultures of rat hepatocytes, Canadian journal of biochemistry and cell biology 63(5) (1985) 333–340. [DOI] [PubMed] [Google Scholar]
- [93].Aft RL, Zhang F, Gius D, Evaluation of 2-deoxy-D-glucose as a chemotherapeutic agent: mechanism of cell death, British journal of cancer 87(7) (2002) 805–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Bénéteau M, Zunino B, Jacquin MA, Meynet O, Chiche J, Pradelli LA, Marchetti S, Cornille A, Carles M, Ricci J-E, Combination of glycolysis inhibition with chemotherapy results in an antitumor immune response, Proceedings of the National Academy of Sciences 109(49) (2012) 20071–20076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, Roychoudhuri R, Palmer DC, Muranski P, Karoly ED, Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function, The Journal of clinical investigation 123(10) (2013) 4479–4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Ganapathy-Kanniappan S, Vali M, Kunjithapatham R, Buijs M, Syed L, Rao P, Ota S, Kwak B, Loffroy R, Geschwind J, 3-bromopyruvate: a new targeted antiglycolytic agent and a promise for cancer therapy, Current pharmaceutical biotechnology 11(5) (2010) 510–517. [DOI] [PubMed] [Google Scholar]
- [97].Dell’Antone P, Targets of 3-bromopyruvate, a new, energy depleting, anticancer agent, Medicinal chemistry 5(6) (2009) 491–496. [DOI] [PubMed] [Google Scholar]
- [98].Israelsen WJ, Vander Heiden MG, Pyruvate kinase: function, regulation and role in cancer, Seminars in cell & developmental biology, Elsevier, 2015, pp. 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Dong G, Mao Q, Xia W, Xu Y, Wang J, Xu L, Jiang F, PKM2 and cancer: The function of PKM2 beyond glycolysis, Oncology letters 11(3) (2016) 1980–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Vander Heiden MG, Christofk HR, Schuman E, Subtelny AO, Sharfi H, Harlow EE, Xian J, Cantley LC, Identification of small molecule inhibitors of pyruvate kinase M2, Biochemical pharmacology 79(8) (2010) 1118–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Kono M, Maeda K, Stocton-Gavanescu I, Pan W, Umeda M, Katsuyama E, Burbano C, Orite SYK, Vukelic M, Tsokos MG, Pyruvate kinase M2 is requisite for Th1 and Th17 differentiation, JCI insight 4(12) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, van den Bosch MW, Quinn SR, Domingo-Fernandez R, Johnston DG, Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages, Cell metabolism 21(1) (2015) 65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Angiari S, Runtsch MC, Sutton CE, Palsson-McDermott EM, Kelly B, Rana N, Kane H, Papadopoulou G, Pearce EL, Mills KH, Pharmacological activation of pyruvate kinase M2 inhibits CD4+ T cell pathogenicity and suppresses autoimmunity, Cell metabolism 31(2) (2020) 391–405. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Telang S, Clem BF, Klarer AC, Clem AL, Trent JO, Bucala R, Chesney J, Small molecule inhibition of 6-phosphofructo-2-kinase suppresses t cell activation, Journal of translational medicine 10(1) (2012) 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Clem B, Telang S, Clem A, Yalcin A, Meier J, Simmons A, Rasku MA, Arumugam S, Dean WL, Eaton J, Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth, Molecular cancer therapeutics 7(1) (2008) 110–120. [DOI] [PubMed] [Google Scholar]
- [106].Vella S, Conti M, Tasso R, Cancedda R, Pagano A, Dichloroacetate inhibits neuroblastoma growth by specifically acting against malignant undifferentiated cells, International journal of cancer 130(7) (2012) 1484–1493. [DOI] [PubMed] [Google Scholar]
- [107].Zheng M.-f., Shen S.-y., DCA increases the antitumor effects of capecitabine in a mouse B16 melanoma allograft and a human non-small cell lung cancer A549 xenograft, Cancer chemotherapy and pharmacology 72(5) (2013) 1031–1041. [DOI] [PubMed] [Google Scholar]
- [108].Eleftheriadis T, Pissas G, Karioti A, Antoniadi G, Antoniadis N, Liakopoulos V, Stefanidis I, Dichloroacetate at therapeutic concentration alters glucose metabolism and induces regulatory T-cell differentiation in alloreactive human lymphocytes, Journal of basic and clinical physiology and pharmacology 24(4) (2013) 271–276. [DOI] [PubMed] [Google Scholar]
- [109].Chang C-H, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, Van Der Windt GJ, Metabolic competition in the tumor microenvironment is a driver of cancer progression, Cell 162(6) (2015) 1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, Karoly ED, Freeman GJ, Petkova V, Seth P, PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation, Nature communications 6(1) (2015) 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].McDonald PC, Chafe SC, Dedhar S, Overcoming hypoxia-mediated tumor progression: combinatorial approaches targeting pH regulation, angiogenesis and immune dysfunction, Frontiers in cell and developmental biology 4 (2016) 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Wieman HL, Wofford JA, Rathmell JC, Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking, Molecular biology of the cell 18(4) (2007) 1437–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Sun Z, Ren Z, Yang K, Liu Z, Cao S, Deng S, Xu L, Liang Y, Guo J, Bian Y, A next-generation tumor-targeting IL-2 preferentially promotes tumor-infiltrating CD8+ T-cell response and effective tumor control, Nature communications 10(1) (2019) 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Domblides C, Lartigue L, Faustin B, Control of the antitumor immune response by cancer metabolism, Cells 8(2) (2019) 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Doherty JR, Cleveland JL, Targeting lactate metabolism for cancer therapeutics, The Journal of clinical investigation 123(9) (2013) 3685–3692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Hashim AAI, Abrahams D, Xu L, Centeno B, Sunassee E, Abddelgader R, Dubois L, Lambin P, Gatenby RA, Gillies RJ, Targeting tumor acidity with the LDHA inhibitor (FX11) and CAIX inhibitor (DH348) overcomes resistance to PD-1 blockade and inhibits metastasis in a pancreatic cancer model, AACR, 2017. [Google Scholar]
- [117].Zhang Y, Zhang X, Wang X, Gan L, Yu G, Chen Y, Liu K, Li P, Pan J, Wang J, Inhibition of LDH-A by lentivirus-mediated small interfering RNA suppresses intestinal-type gastric cancer tumorigenicity through the downregulation of Oct4, Cancer letters 321(1) (2012) 45–54. [DOI] [PubMed] [Google Scholar]
- [118].Halestrap AP, The SLC16 gene family–structure, role and regulation in health and disease, Molecular aspects of medicine 34(2–3) (2013) 337–349. [DOI] [PubMed] [Google Scholar]
- [119].Kirk á., Wilson M, Heddle C, Brown M, Barclay A, Halestrap A, CD147 is tightly associated with lactate transporters MCT1 and MCT4 and facilitates their cell surface expression, The EMBO journal 19(15) (2000) 3896–3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Ovens MJ, Davies AJ, Wilson MC, Murray CM, Halestrap AP, AR-C155858 is a potent inhibitor of monocarboxylate transporters MCT1 and MCT2 that binds to an intracellular site involving transmembrane helices 7–10, Biochemical Journal 425(3) (2010) 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Doherty JR, Yang C, Scott KE, Cameron MD, Fallahi M, Li W, Hall MA, Amelio AL, Mishra JK, Li F, Blocking lactate export by inhibiting the Myc target MCT1 Disables glycolysis and glutathione synthesis, Cancer research 74(3) (2014) 908–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Beloueche-Babari M, Galobart TC, Delgado-Goni T, Wantuch S, Parkes HG, Tandy D, Harker JA, Leach MO, Monocarboxylate transporter 1 blockade with AZD3965 inhibits lipid biosynthesis and increases tumour immune cell infiltration, British journal of cancer (2020) 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Görgün G, Calabrese E, Soydan E, Hideshima T, Perrone G, Bandi M, Cirstea D, Santo L, Hu Y, Tai Y-T, Immunomodulatory effects of lenalidomide and pomalidomide on interaction of tumor and bone marrow accessory cells in multiple myeloma, Blood, The Journal of the American Society of Hematology 116(17) (2010) 3227–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Eichner R, Heider M, Fernández-Sáiz V, van Bebber F, Garz A-K, Lemeer S, Rudelius M, Targosz B-S, Jacobs L, Knorn A-M, Immunomodulatory drugs disrupt the cereblon–CD147–MCT1 axis to exert antitumor activity and teratogenicity, Nature medicine 22(7) (2016) 735. [DOI] [PubMed] [Google Scholar]
- [125].Critchlow SE, Hughes G, Staniszewska A, King M, Michopoulos F, Hopcroft L, Brown MR, Bradley J, Hammond B, MorentinGutierrez P, Reversing lactate-driven immunosuppression using the novel, potent and selective MCT4 inhibitor AZD0095, AACR, 2019. [Google Scholar]
- [126].De Milito A, Canese R, Marino ML, Borghi M, Iero M, Villa A, Venturi G, Lozupone F, Iessi E, Logozzi M, pH-dependent antitumor activity of proton pump inhibitors against human melanoma is mediated by inhibition of tumor acidity, International journal of cancer 127(1) (2010) 207–219. [DOI] [PubMed] [Google Scholar]
- [127].Wang B-Y, Zhang J, Wang J-L, Sun S, Wang Z-H, Wang L-P, Zhang Q-L, Lv F-F, Cao E-Y, Shao Z-M, Intermittent high dose proton pump inhibitor enhances the antitumor effects of chemotherapy in metastatic breast cancer, Journal of Experimental & Clinical Cancer Research 34(1) (2015) 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Bellone M, Calcinotto A, Filipazzi P, De Milito A, Fais S, Rivoltini L, The acidity of the tumor microenvironment is a mechanism of immune escape that can be overcome by proton pump inhibitors, Oncoimmunology 2(1) (2013) e22058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Chalabi M, Cardona A, Nagarkar D, Scala AD, Gandara D, Rittmeyer A, Albert M, Powles T, Kok M, Herrera F, Efficacy of chemotherapy and atezolizumab in patients with non-small cell lung cancer receiving antibiotics and proton pump inhibitors: pooled post-hoc analyses of the OAK and POPLAR trials, Annals of Oncology (2020). [DOI] [PubMed] [Google Scholar]
- [130].Cheung KS, Leung WK, Long-term use of proton-pump inhibitors and risk of gastric cancer: a review of the current evidence, Therapeutic advances in gastroenterology 12 (2019) 1756284819834511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Punekar S, Cho DC, Novel Therapeutics Affecting Metabolic Pathways, American Society of Clinical Oncology Educational Book 39 (2019) e79–e87. [DOI] [PubMed] [Google Scholar]
- [132].van Geldermalsen M, Quek L-E, Turner N, Freidman N, Pang A, Guan YF, Krycer JR, Ryan R, Wang Q, Holst J, Benzylserine inhibits breast cancer cell growth by disrupting intracellular amino acid homeostasis and triggering amino acid response pathways, BMC cancer 18(1) (2018) 689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Nakaya M, Xiao Y, Zhou X, Chang J-H, Chang M, Cheng X, Blonska M, Lin X, Sun S-C, Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation, Immunity 40(5) (2014) 692–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Altman BJ, Stine ZE, Dang CV, From Krebs to clinic: glutamine metabolism to cancer therapy, Nature Reviews Cancer 16(10) (2016) 619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Chen L, Cui H, Targeting Glutamine Induces Apoptosis: A Cancer Therapy Approach, Int J Mol Sci 16(9) (2015) 22830–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Zacharias NM, Baran N, Shanmugavelandy SS, Lee J, Lujan JV, Dutta P, Millward SW, Cai T, Wood CG, Piwnica-Worms D, Konopleva M, Bhattacharya PK, Assessing Metabolic Intervention with a Glutaminase Inhibitor in Real-Time by Hyperpolarized Magnetic Resonance in Acute Myeloid Leukemia, Mol Cancer Ther 18(11) (2019) 1937–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Thompson RM, Dytfeld D, Reyes L, Robinson RM, Smith B, Manevich Y, Jakubowiak A, Komarnicki M, Przybylowicz-Chalecka A, Szczepaniak T, Mitra AK, Van Ness BG, Luczak M, Dolloff NG, Glutaminase inhibitor CB-839 synergizes with carfilzomib in resistant multiple myeloma cells, Oncotarget 8(22) (2017) 35863–35876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Momcilovic M, Bailey ST, Lee JT, Fishbein MC, Braas D, Go J, Graeber TG, Parlati F, Demo S, Li R, Walser TC, Gricowski M, Shuman R, Ibarra J, Fridman D, Phelps ME, Badran K, St John M, Bernthal NM, Federman N, Yanagawa J, Dubinett SM, Sadeghi S, Christofk HR, Shackelford DB, The GSK3 Signaling Axis Regulates Adaptive Glutamine Metabolism in Lung Squamous Cell Carcinoma, Cancer Cell 33(5) (2018) 905–921 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Leone RD, Zhao L, Englert JM, Sun IM, Oh MH, Sun IH, Arwood ML, Bettencourt IA, Patel CH, Wen J, Tam A, Blosser RL, Prchalova E, Alt J, Rais R, Slusher BS, Powell JD, Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion, Science 366(6468) (2019) 1013–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Weinberg SE, Chandel NS, Targeting mitochondria metabolism for cancer therapy, Nature chemical biology 11(1) (2015) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Krall AS, Xu S, Graeber TG, Braas D, Christofk HR, Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor, Nature communications 7(1) (2016) 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Covini D, Maggi M, Tardito S, Bussolati O, Chiarelli LR, Pasquetto MV, Vecchia L, Valentini G, Scotti C, Expanding targets for a metabolic therapy of cancer: L-asparaginase, Topics in anti-cancer research 3 (2015) 418–445. [DOI] [PubMed] [Google Scholar]
- [143].Kwong Y-L, Chan TS, Tan D, Kim SJ, Poon L-M, Mow B, Khong P-L, Loong F, Au-Yeung R, Iqbal J, PD1 blockade with pembrolizumab is highly effective in relapsed or refractory NK/T-cell lymphoma failing l-asparaginase, Blood, The Journal of the American Society of Hematology 129(17) (2017) 2437–2442. [DOI] [PubMed] [Google Scholar]
- [144].Qiu F, Huang J, Sui M, Targeting arginine metabolism pathway to treat arginine-dependent cancers, Cancer letters 364(1) (2015) 1–7. [DOI] [PubMed] [Google Scholar]
- [145].Arginase inhibitor INCB001158 as a single agent and in combination with immune checkpoint therapy in patients with advanced/metastatic solid tumors, 2017.
- [146].Cervenka I, Agudelo LZ, Ruas JL, Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health, Science 357(6349) (2017) eaaf9794. [DOI] [PubMed] [Google Scholar]
- [147].Prendergast GC, Smith C, Thomas S, Mandik-Nayak L, Laury-Kleintop L, Metz R, Muller AJ, Indoleamine 2, 3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer, Cancer immunology, immunotherapy 63(7) (2014) 721–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP, Indoleamine 2, 3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4, Journal of Experimental Medicine 210(7) (2013) 1389–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Platten M, Nollen EA, Röhrig UF, Fallarino F, Opitz CA, Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond, Nature Reviews Drug Discovery 18(5) (2019) 379–401. [DOI] [PubMed] [Google Scholar]
- [150].Chen M, Huang J, The expanded role of fatty acid metabolism in cancer: new aspects and targets, Precision clinical medicine 2(3) (2019) 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Tang J-J, Li J-G, Qi W, Qiu W-W, Li P-S, Li B-L, Song B-L, Inhibition of SREBP by a small molecule, betulin, improves hyperlipidemia and insulin resistance and reduces atherosclerotic plaques, Cell metabolism 13(1) (2011) 44–56. [DOI] [PubMed] [Google Scholar]
- [152].Li X, Chen Y-T, Hu P, Huang W-C, Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling, Molecular cancer therapeutics 13(4) (2014) 855–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Gao S, Shi Z, Li X, Li W, Wang Y, Liu Z, Jiang J, Fatostatin suppresses growth and enhances apoptosis by blocking SREBP-regulated metabolic pathways in endometrial carcinoma, Oncology reports 39(4) (2018) 1919–1929. [DOI] [PubMed] [Google Scholar]
- [154].Li J, Yan H, Zhao L, Jia W, Yang H, Liu L, Zhou X, Miao P, Sun X, Song S, Inhibition of SREBP increases gefitinib sensitivity in non-small cell lung cancer cells, Oncotarget 7(32) (2016) 52392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Cheng X, Li J, Guo D, SCAP/SREBPs are central players in lipid metabolism and novel metabolic targets in Cancer therapy, Current topics in medicinal chemistry 18(6) (2018) 484–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Migita T, Ruiz S, Fornari A, Fiorentino M, Priolo C, Zadra G, Inazuka F, Grisanzio C, Palescandolo E, Shin E, Fatty acid synthase: a metabolic enzyme and candidate oncogene in prostate cancer, Journal of the National Cancer Institute 101(7) (2009) 519–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Menendez J, Lupu R, Fatty acid synthase regulates estrogen receptor-α signaling in breast cancer cells, Oncogenesis 6(2) (2017) e299–e299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Gouw AM, Eberlin LS, Margulis K, Sullivan DK, Toal GG, Tong L, Zare RN, Felsher DW, Oncogene KRAS activates fatty acid synthase, resulting in specific ERK and lipid signatures associated with lung adenocarcinoma, Proceedings of the National Academy of Sciences 114(17) (2017) 4300–4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Lupu R, Menendez JA, Pharmacological inhibitors of fatty acid synthase (FASN)-catalyzed endogenous fatty acid biogenesis: a new family of anti-cancer agents?, Current pharmaceutical biotechnology 7(6) (2006) 483–494. [DOI] [PubMed] [Google Scholar]
- [160].Jiang L, Fang X, Wang H, Li D, Wang X, Ovarian cancer-intrinsic fatty acid synthase prevents anti-tumor immunity by disrupting tumor-infiltrating dendritic cells, Frontiers in immunology 9 (2018) 2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Lally JS, Ghoshal S, DePeralta DK, Moaven O, Wei L, Masia R, Erstad DJ, Fujiwara N, Leong V, Houde VP, Inhibition of acetyl-CoA carboxylase by phosphorylation or the inhibitor ND-654 suppresses lipogenesis and hepatocellular carcinoma, Cell metabolism 29(1) (2019) 174–182. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Lee J, Walsh MC, Hoehn KL, James DE, Wherry EJ, Choi Y, Regulator of fatty acid metabolism, acetyl coenzyme a carboxylase 1, controls T cell immunity, The Journal of Immunology 192(7) (2014) 3190–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Le Bourgeois T, Strauss L, Aksoylar H-I, Daneshmandi S, Seth P, Patsoukis N, Boussiotis VA, Targeting T cell metabolism for improvement of cancer immunotherapy, Frontiers in oncology 8 (2018) 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang L-S, Jones RG, Choi Y, Enhancing CD8 T-cell memory by modulating fatty acid metabolism, Nature 460(7251) (2009) 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Pike LS, Smift AL, Croteau NJ, Ferrick DA, Wu M, Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells, Biochimica et Biophysica Acta (BBA)-Bioenergetics 1807(6) (2011) 726–734. [DOI] [PubMed] [Google Scholar]
- [166].Schlaepfer IR, Rider L, Rodrigues LU, Gijón MA, Pac CT, Romero L, Cimic A, Sirintrapun SJ, Glodé LM, Eckel RH, Lipid catabolism via CPT1 as a therapeutic target for prostate cancer, Molecular cancer therapeutics 13(10) (2014) 2361–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Ricciardi MR, Mirabilii S, Allegretti M, Licchetta R, Calarco A, Torrisi MR, Foa R, Nicolai R, Peluso G, Tafuri A, Targeting the leukemia cell metabolism by the CPT1a inhibition: functional preclinical effects in leukemias, Blood, The Journal of the American Society of Hematology 126(16) (2015) 1925–1929. [DOI] [PubMed] [Google Scholar]
- [168].O’Connor RS, Guo L, Ghassemi S, Snyder NW, Worth AJ, Weng L, Kam Y, Philipson B, Trefely S, Nunez-Cruz S, The CPT1a inhibitor, etomoxir induces severe oxidative stress at commonly used concentrations, Scientific reports 8(1) (2018) 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Raud B, Roy DG, Divakaruni AS, Tarasenko TN, Franke R, Ma EH, Samborska B, Hsieh WY, Wong AH, Stüve P, Etomoxir actions on regulatory and memory T cells are independent of Cpt1a-mediated fatty acid oxidation, Cell metabolism 28(3) (2018) 504–515. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, Filisio F, Giles-Davis W, Xu X, Karakousis GC, Enhancing CD8+ T cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy, Cancer cell 32(3) (2017) 377–391. e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Lien EC, Lyssiotis CA, Cantley LC, Metabolic reprogramming by the PI3K-Akt-mTOR pathway in cancer, Metabolism in Cancer, Springer; 2016, pp. 39–72. [DOI] [PubMed] [Google Scholar]
- [172].Kouidhi S, Ben Ayed F, Benammar Elgaaied A, Targeting tumor metabolism: a new challenge to improve immunotherapy, Frontiers in immunology 9 (2018) 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Chi H, Regulation and function of mTOR signalling in T cell fate decisions, Nature reviews immunology 12(5) (2012) 325–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Araki K, Youngblood B, Ahmed R, The role of mTOR in memory CD8+ T-cell differentiation, Immunological reviews 235(1) (2010) 234–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Mineharu Y, Kamran N, Lowenstein PR, Castro MG, Blockade of mTOR signaling via rapamycin combined with immunotherapy augments antiglioma cytotoxic and memory T-cell functions, Molecular cancer therapeutics 13(12) (2014) 3024–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Sanli T, Steinberg GR, Singh G, Tsakiridis T, AMP-activated protein kinase (AMPK) beyond metabolism: a novel genomic stress sensor participating in the DNA damage response pathway, Cancer biology & therapy 15(2) (2014) 156–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Li W, Saud SM, Young MR, Chen G, Hua B, Targeting AMPK for cancer prevention and treatment, Oncotarget 6(10) (2015) 7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Crawley D, Chandra A, Loda M, Gillett C, Cathcart P, Challacombe B, Cook G, Cahill D, Santa Olalla A, Cahill F, Metformin and longevity (METAL): a window of opportunity study investigating the biological effects of metformin in localised prostate cancer, BMC cancer 17(1) (2017) 494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia-Vázquez G, Yurchenko E, Raissi TC, van der Windt GJ, Viollet B, Pearce EL, The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo, Immunity 42(1) (2015) 41–54. [DOI] [PubMed] [Google Scholar]
- [180].Eikawa S, Nishida M, Mizukami S, Yamazaki C, Nakayama E, Udono H, Immune-mediated antitumor effect by type 2 diabetes drug, metformin, Proceedings of the National Academy of Sciences 112(6) (2015) 1809–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Semenza GL, Hypoxia-inducible factors in physiology and medicine, Cell 148(3) (2012) 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Koh MY, Spivak-Kroizman T, Venturini S, Welsh S, Williams RR, Kirkpatrick DL, Powis G, Molecular mechanisms for the activity of PX-478, an antitumor inhibitor of the hypoxia-inducible factor-1α, Molecular cancer therapeutics 7(1) (2008) 90–100. [DOI] [PubMed] [Google Scholar]
- [183].Zhao T, Ren H, Jia L, Chen J, Xin W, Yan F, Li J, Wang X, Gao S, Qian D, Inhibition of HIF-1α by PX-478 enhances the anti-tumor effect of gemcitabine by inducing immunogenic cell death in pancreatic ductal adenocarcinoma, Oncotarget 6(4) (2015) 2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Graziani G, Tentori L, Navarra P, Ipilimumab: a novel immunostimulatory monoclonal antibody for the treatment of cancer, Pharmacol Res 65(1) (2012) 9–22. [DOI] [PubMed] [Google Scholar]
- [185].Joseph RW, Shillington AC, Lee TA, Macahilig CP, Diede SJ, Dave V, Harshaw Q, Scherrer E, Liu FX, Hospitalization and emergency department utilization in patients with advanced melanoma receiving pembrolizumab versus ipilimumab plus nivolumab in US academic centers, J Med Econ 23(2) (2020) 132–138. [DOI] [PubMed] [Google Scholar]
- [186].Robert C, Ribas A, Schachter J, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil CM, Lotem M, Larkin JMG, Lorigan P, Neyns B, Blank CU, Petrella TM, Hamid O, Su SC, Krepler C, Ibrahim N, Long GV, Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study, Lancet Oncol 20(9) (2019) 1239–1251. [DOI] [PubMed] [Google Scholar]
- [187].Petrella TM, Robert C, Richtig E, Miller WH Jr., Masucci GV, Walpole E, Lebbe C, Steven N, Middleton MR, Hille D, Zhou W, Ibrahim N, Cebon J, Patient-reported outcomes in KEYNOTE-006, a randomised study of pembrolizumab versus ipilimumab in patients with advanced melanoma, Eur J Cancer 86 (2017) 115–124. [DOI] [PubMed] [Google Scholar]
- [188].Hamid O, Puzanov I, Dummer R, Schachter J, Daud A, Schadendorf D, Blank C, Cranmer LD, Robert C, Pavlick AC, Gonzalez R, Hodi FS, Ascierto PA, Salama AKS, Margolin KA, Gangadhar TC, Wei Z, Ebbinghaus S, Ibrahim N, Ribas A, Final analysis of a randomised trial comparing pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory advanced melanoma, Eur J Cancer 86 (2017) 37–45. [DOI] [PubMed] [Google Scholar]
- [189].Schachter J, Ribas A, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, Larkin J, Lorigan P, Neyns B, Blank C, Petrella TM, Hamid O, Zhou H, Ebbinghaus S, Ibrahim N, Robert C, Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006), Lancet 390(10105) (2017) 1853–1862. [DOI] [PubMed] [Google Scholar]
- [190].Wang J, Chmielowski B, Pellissier J, Xu R, Stevinson K, Liu FX, Cost-Effectiveness of Pembrolizumab Versus Ipilimumab in Ipilimumab-Naive Patients with Advanced Melanoma in the United States, J Manag Care Spec Pharm 23(2) (2017) 184–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, Larkin J, Lorigan P, Neyns B, Blank CU, Hamid O, Mateus C, Shapira-Frommer R, Kosh M, Zhou H, Ibrahim N, Ebbinghaus S, Ribas A, K.−. investigators, Pembrolizumab versus Ipilimumab in Advanced Melanoma, N Engl J Med 372(26) (2015) 2521–32. [DOI] [PubMed] [Google Scholar]
- [192].Shaverdian N, Lisberg AE, Bornazyan K, Veruttipong D, Goldman JW, Formenti SC, Garon EB, Lee P, Previous radiotherapy and the clinical activity and toxicity of pembrolizumab in the treatment of non-small-cell lung cancer: a secondary analysis of the KEYNOTE-001 phase 1 trial, Lancet Oncol 18(7) (2017) 895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Chouaid C, Bensimon L, Clay E, Millier A, Levy-Bachelot L, Huang M, Levy P, Cost-effectiveness analysis of pembrolizumab versus standard-of-care chemotherapy for first-line treatment of PD-L1 positive (>50%) metastatic squamous and non-squamous non-small cell lung cancer in France, Lung Cancer 127 (2019) 44–52. [DOI] [PubMed] [Google Scholar]
- [194].She L, Hu H, Liao M, Xia X, Shi Y, Yao L, Ding D, Zhu Y, Zeng S, Shen L, Huang J, Carbone DP, Cost-effectiveness analysis of pembrolizumab versus chemotherapy as first-line treatment in locally advanced or metastatic non-small cell lung cancer with PD-L1 tumor proportion score 1% or greater, Lung Cancer 138 (2019) 88–94. [DOI] [PubMed] [Google Scholar]
- [195].Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, Carcereny E, Ahn MJ, Felip E, Lee JS, Hellmann MD, Hamid O, Goldman JW, Soria JC, Dolled-Filhart M, Rutledge RZ, Zhang J, Lunceford JK, Rangwala R, Lubiniecki GM, Roach C, Emancipator K, Gandhi L, K.−. Investigators, Pembrolizumab for the treatment of non-small-cell lung cancer, N Engl J Med 372(21) (2015) 2018–28. [DOI] [PubMed] [Google Scholar]
- [196].Weng X, Luo S, Lin S, Zhong L, Li M, Xin R, Huang P, Xu X, Cost-Utility Analysis of Pembrolizumab Versus Chemotherapy as First-Line Treatment for Metastatic Non-Small Cell Lung Cancer With Different PD-L1 Expression Levels, Oncol Res 28(2) (2020) 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF, Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment, J Immunother Cancer 2 (2014) 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Sharpe AH, Pauken KE, The diverse functions of the PD1 inhibitory pathway, Nature reviews. Immunology 18(3) (2018) 153–167. [DOI] [PubMed] [Google Scholar]
- [199].Nabe S, Yamada T, Suzuki J, Toriyama K, Yasuoka T, Kuwahara M, Shiraishi A, Takenaka K, Yasukawa M, Yamashita M, Reinforce the antitumor activity of CD8(+) T cells via glutamine restriction, Cancer Sci 109(12) (2018) 3737–3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Wang T, Gnanaprakasam J, Chen X, Kang S, Xu X, Sun H, Liu L, Rodgers H, Miller E, Cassel T, Sun Q, Vicente-Muñoz S, Warmoes M, Lin P, Piedra-Quintero Z, Guerau-de-Arellano M, Cassady K, Zheng S, Yang J, Lane A, Song X, Fan T, Wang R, Inosine is an alternative carbon source for CD8+-T-cell function under glucose restriction, Nat Metab (2020) 10.1038/s42255-020-0219-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Haas R, Smith J, Rocher-Ros V, Nadkarni S, Montero-Melendez T, D’Acquisto F, Bland EJ, Bombardieri M, Pitzalis C, Perretti M, Marelli-Berg FM, Mauro C, Lactate Regulates Metabolic and Pro-inflammatory Circuits in Control of T Cell Migration and Effector Functions, PLoS Biol 13(7) (2015) e1002202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Comito G, Iscaro A, Bacci M, Morandi A, Ippolito L, Parri M, Montagnani I, Raspollini MR, Serni S, Simeoni L, Giannoni E, Chiarugi P, Lactate modulates CD4(+) T-cell polarization and induces an immunosuppressive environment, which sustains prostate carcinoma progression via TLR8/miR21 axis, Oncogene 38(19) (2019) 3681–3695. [DOI] [PubMed] [Google Scholar]
- [203].Husain Z, Huang Y, Seth P, Sukhatme VP, Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells, Journal of immunology 191(3) (2013) 1486–95. [DOI] [PubMed] [Google Scholar]
- [204].Caronni N, Simoncello F, Stafetta F, Guarnaccia C, Ruiz-Moreno JS, Opitz B, Galli T, Proux-Gillardeaux V, Benvenuti F, Downregulation of Membrane Trafficking Proteins and Lactate Conditioning Determine Loss of Dendritic Cell Function in Lung Cancer, Cancer Res 78(7) (2018) 1685–1699. [DOI] [PubMed] [Google Scholar]
- [205].Gottfried E, Kunz-Schughart LA, Ebner S, Mueller-Klieser W, Hoves S, Andreesen R, Mackensen A, Kreutz M, Tumor-derived lactic acid modulates dendritic cell activation and antigen expression, Blood 107(5) (2006) 2013–21. [DOI] [PubMed] [Google Scholar]
- [206].Ohashi T, Aoki M, Tomita H, Akazawa T, Sato K, Kuze B, Mizuta K, Hara A, Nagaoka H, Inoue N, Ito Y, M2-like macrophage polarization in high lactic acid-producing head and neck cancer, Cancer Sci 108(6) (2017) 1128–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Lee J, Ahn E, Kissick HT, Ahmed R, Reinvigorating exhausted T cells by blockade of the PD-1 pathway, Forum on immunopathological diseases and therapeutics, Begel House Inc., 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Liu X, Shin N, Koblish HK, Yang G, Wang Q, Wang K, Leffet L, Hansbury MJ, Thomas B, Rupar M, Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity, Blood, The Journal of the American Society of Hematology 115(17) (2010) 3520–3530. [DOI] [PubMed] [Google Scholar]
- [209].Prendergast GC, Malachowski WJ, Mondal A, Scherle P, Muller AJ, Indoleamine 2, 3-dioxygenase and its therapeutic inhibition in cancer, International review of cell and molecular biology, Elsevier; 2018, pp. 175–203. [DOI] [PMC free article] [PubMed] [Google Scholar]