

Abstract
Objective:
To evaluate the clinical features and etiology of precocious puberty (PP) in Chinese boys.
Methods:
In this study, data from boys who were referred for evaluation of PP from 2015 to 2020 at a tertiary hospital in Northern China were retrospectively analyzed.
Results:
Eighty-two boys were diagnosed with PP from 2015 to 2020. Sixty-two patients (75.6%) were diagnosed with central PP (CPP), and twenty patients (24.4%) were diagnosed with peripheral PP (PPP). In the CPP group, forty-nine cases were classified as idiopathic CPP, and thirteen patients had pathogenic CPP. The top three causes of PPP were congenital adrenal hyperplasia, germ cell tumors and familial male-limited PP.
Conclusion:
The etiology of PP in males is diverse. The majority of CPP cases in Chinese boys are idiopathic rather than organic.
Keywords: Male, precocious puberty, etiology
What is already known on this topic?
The etiology of precocious puberty (PP) in boys is diverse.
What this study adds?
Although previous studies have shown that the majority of central PP (CPP) cases in boys are pathological, our findings showed a high prevalence of idiopathic CPP in Northern China.
Introduction
Precocious puberty (PP) in boys refers to the development of secondary sexual characteristics before nine years of age. The prevalence of PP is approximately 10 times lower in boys than in girls (1). Female PP is mostly idiopathic, while male PP may be due to pathological factors. However, there are fewer studies about male than female PP, and the male studies have tended to include fewer patients. The proportion of organic central PP (CPP) in males is 26~64% (2,3,4). Over 15 years in South Korea, Lee et al (3) reported that 38% (27/71) of male PP cases were organic CPP, and the majority were due to intracranial diseases or tumors. Topor et al (4) reported all cases of male CPP seen at a US pediatric hospital for 10 years; almost two-thirds of the boys were overweight/obese, and more than 60% had neurogenic CPP. Neurofibromatosis type 1 (NF1) was the most common diagnosis. In one Polish study, 16 boys were referred for hospital evaluation because of pubertal signs; 50% were diagnosed with precocious adrenarche, four with central nervous system (CNS) tumors, two with congenital adrenal hyperplasia (CAH), and one each with a Leydig cell tumor and idiopathic CPP (5).
Few studies have analyzed peripheral PP (PPP) and CPP together, and most studies have focused solely on either male CPP or male PPP. We did not focus exclusively on CPP or PPP because central and peripheral manifestations sometimes overlap. In fact, males with PPP can present with CPP because of complexity. Studies from different countries and regions seem to support the role of pathological factors in males with PP, but the etiology is not the same. Data on male PP are still insufficient. Therefore, this retrospective study of male PP diagnosed at tertiary hospitals in northern China was conducted and the clinical manifestations and etiology were analyzed.
Methods
Subjects and Clinical Assessment
This retrospective study included patients who were evaluated for PP between 2015 and 2020 at the Department of Endocrinology, Children’s Hospital Capital Institute of Pediatrics, Beijing, China. PP was defined as the onset of secondary sexual characteristics before 9 years of age, and the medical records of each patient were complete (6). Patients who had previously been diagnosed at other hospitals and those with variants of PP, such as premature adrenarche, were excluded from the analysis.
The clinical, laboratory, radiological, and molecular study data (if needed) were evaluated for all patients. Chinese growth charts for children were applied to interpret growth data, which were described as standard deviation (SD) score values (7). The diagnosis of CPP was made based on a clinical evaluation, which included taking a detailed history of the patient and his caregivers, followed by a physical examination and Tanner staging. Testicular volume was assessed using an orchidometer. All boys included in this study underwent blood collection for the measurement of luteinizing hormone (LH), follicle stimulating hormone (FSH), and testosterone (T). Bone age was assessed by a single pediatric endocrinologist and a radiologist, using the comparative method of Greulich and Pyle. Cranial/pituitary magnetic resonance imaging (MRI) was mandatory in all cases, and cerebrospinal fluid (CSF) sampling was required when a germ cell tumor (GCT) was strongly suspected. A diagnosis of CPP was confirmed via elevated LH response in a gonadotropin-releasing hormone (GnRH) analog stimulation test using subcutaneous triptorelin at a dose of 0.1 mg/m2, to a maximum of 0.1 mg (Ferring, Hoechst, Frankfurt, Germany). CPP was defined as peak LH >7 IU/L and bone age at least 1 year greater than chronological age (8).
CAH was diagnosed based on elevated 17-hydroxyprogesterone (17-OHP), progesterone and adrenocorticotropic hormone; decreased cortisone, with or without hyperkalemia and hyponatremia; and a computed tomography (CT) or ultrasound scan suggesting adrenal hyperplasia (9). Patients with CNS GCTs were diagnosed through histopathology and/or elevated CSF human chorionic gonadotropin (hCG) levels (normal: <0.1 IU/L) and the appearance of an intracranial mass on MRI (10). Patients with hypothalamic hamartomas (HH) were diagnosed with CPP, either isolated or in combination with seizures, and their MRI scans showed a mass either located adjacent to hypothalamic structures (i.e., in a parahypothalamic position) or infiltrating the hypothalamus (11). Familial male-limited PP (FMPP) was diagnosed in boys who had a family history of PP, normal hCG levels and a constitutively activated mutant LH receptor (LHCGR) leading to elevated T synthesis (12). Leydig cell tumors, optic pathway tumors (OPT) and choriocarcinomas were diagnosed using histopathological proof. The study protocol was approved by the Children’s Hospital Capital Institute of Pediatrics Ethics Committee (SHERLL2020003, date: 14.01.2020).
The boys were divided into two groups: the CPP (gonadotropin-dependent) group and the PPP (gonadotropin-independent) group.
Molecular Analysis
Genomic analysis of LHCGR was performed by direct sequencing. Genomic DNA was extracted from peripheral blood lymphocytes. The coding regions and intron-exon boundaries of LHCGR were amplified by polymerase chain reaction (PCR). The pathogenicity of each variant was interpreted according to American College of Medical Genetics and Genomics standards and categorized (13). PolyPhen-2 and SIFT were used in silico prediction analysis.
Statistical Analysis
Statistical analyses were conducted using Statistical Package for the Social Sciences, version 22.0 (IBM Inc., Armonk, NY, USA). The results were expressed as the mean±SD, and t-tests for independent samples were applied when normally distributed. Non-parametric data were presented as median and the interquartile range, analyzed by the Mann-Whitney U test. P values below 0.05 were regarded as statistically significant.
Results
Eighty-seven boys were admitted to our hospital for the evaluation of pubertal development. After clinical and laboratory evaluation of all patients except those excluded with PA (n=5), a total of 82 (94%) boys were diagnosed with PP. The patients’ age at the time of referral ranged from 0.92 to 10.5 years; 73.5% were over 7 years old at the time of their first visit, 20.7% were 3 to 6 years old, and 7% were under 3 years old (Figure 1). The main complaints included penis and/or testis enlargement (n=45, 54.8%), growth acceleration (n=45, 54.8%), the development of pubic and/or axillary hair (n=39, 47.6%), voice change (n=10, 12.2%), breast mass (n=5, 6.1%), acne (n=4, 4.9%), and erection/ejaculation (n=2, 2.4%).
Figure 1.
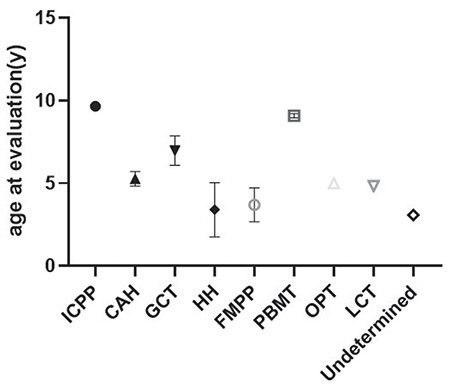
The age at evaluation by etiology
ICPP: idiopathic central precocious puberty, CAH: congenital adrenal hyperplasia, GCT: germ cell tumor, HH: hypothalamic hamartoma, FMPP: familial male limited precocious puberty, PBMT: post bone marrow transplant, OPT: optic pathway tumor, LCT: Leydig cell tumor
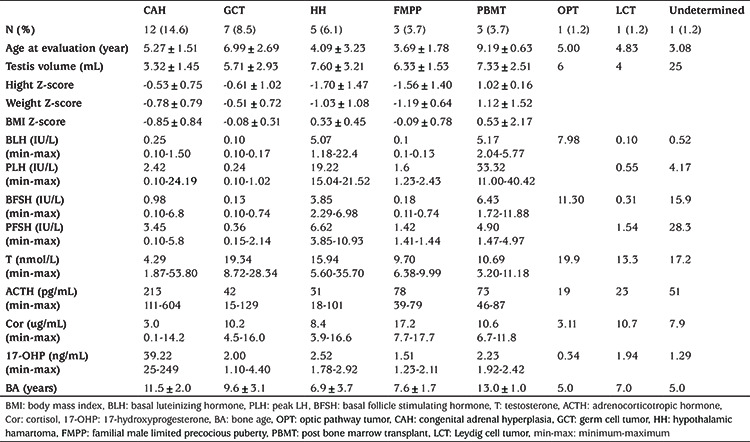
Sixty-two boys (75.6%) were diagnosed with CPP, and 20 boys were diagnosed with PPP (24.4%). The average age at evaluation of the PPP group was much lower than that of the CPP group (5.50±2.23 years vs 8.83±2.19 years, p<0.001), while the T levels did not differ significantly (median: 8.03 nmol/L vs 8.21 nmol/L, p=0.781). The difference between bone age and chronological age in PPP patients was significantly greater than that in CPP patients (4.44±2.41 years vs 2.26±1.31 years, p=0.001). Descriptive data are listed in Table 1 and Table 2.
Table 1. Comparison between CPP and PPP groups. The values in brackets indicate the 25th and 75th percentiles.
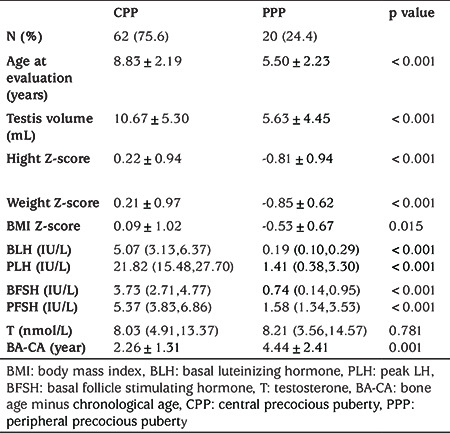
Table 2. Comparison between different subgroups of pathogenic precocious puberty.
In the CPP group, 49 boys were diagnosed with idiopathic CPP (ICPP, 79%). Thirteen boys were diagnosed with pathogenic CPP (21%), including all cases of HH and post bone marrow transplant (PBMT), three cases of CAH, one case of OPT and one undetermined case (Figure 2).
Figure 2.
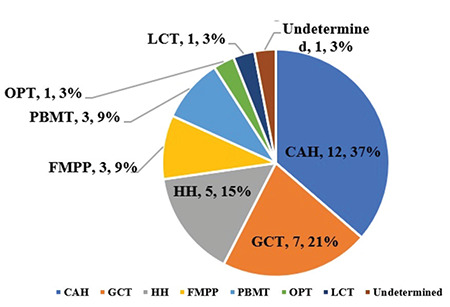
Etiologic spectrum of boys with pathologic precocious puberty
ICPP: idiopathic central precocious puberty, CAH: congenital adrenal hyperplasia, GCT: germ cell tumor, HH: hypothalamic hamartoma, FMPP: familial male limited precocious puberty, PBMT: post bone marrow transplant, OPT: optic pathway tumor, LCT: Leydig cell tumor
Hypothalamic Hamartomas
Five patients were diagnosed with HH (0.92~8.17 years). The imaging findings were assessed by two radiologists (Figure 3A). The patients’ basal LH levels (median: 5.07 IU/L, range:1.18~22.40 IU/L) were consistent with the diagnosis of CPP. Four cases were of the “parahypothalamic type”, in which the hamartoma is attached only to the floor of the third ventricle or tethered to the floor by a peduncle, and two of the patients presented with gelastic seizures. One case was of the “intrahypothalamic type”, in which the hamartoma is involved or enveloped by the hypothalamus and the tumor distorts the third ventricle; this patient also presented with gelastic seizures.
Figure 3.
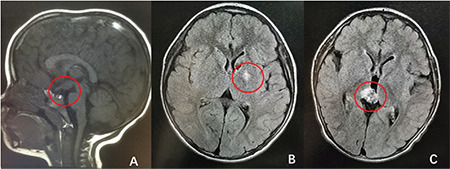
Pathological brain magnetic resonance imaging from three representative cases: Hypothalamic hamartoma (A), basal ganglia germ cell tumor (B), and choriocarcinoma (C)
Post Bone Marrow Transplant
Three patients (one each of Fanconi anemia, myelodysplastic syndrome and aplastic anemia) received chemotherapy and bone marrow transplantation. Busulfan/cyclophosphamide (BU/CY) was used as the conditioning regimen for myelodysplastic syndrome and aplastic anemia. The patient with aplastic anemia took Epimedium for at least 6~7 months after transplantation. The patient with Fanconi anemia had a conditioning regimen including BU/fludarabine+ antithymocyte globulin +TBI. PP occurred 1~3 years after allogeneic hematopoietic stem cell transplantation. All three patients presented with CPP.
The etiology of CPP in one 3-year-old boy remains unknown. He had enlarged testes shortly after birth, and he was admitted to our department because of extremely large, symmetrical testes (25 mL). His blood test results showed an LH peak of 4.17 IU/L, an FSH peak of 28.3 IU/L and extremely high T (17.15 nmol/L). He had a long face, large ears, white skin color and mild mental retardation. A testicular biopsy revealed normal findings. The MRI findings and hCG and 17-OHP results were normal. The results of a fragile X test were negative. Whole-exome sequencing or microarray examination showed no expected typical mutations in the KISS1, KISS1R, MKRN3 or DLK1 genes.
In the PPP group, CAH (n=9), CNS GCT (germinomas n=6 and choriocarcinoma n=1) and FMPP (n=3) were the most common causes (45%, 35% and 15%, respectively). We also diagnosed a boy with a Leydig cell tumor (n=1, 5%) (Figure 2).
Tumor Causes
In all, nine boys had tumors (aged 2.25 to 10.0 years), including seven boys with CNS GCTs, one boy with a Leydig cell tumor (chief complaint: testis mass on the left side; laboratory examinations consistent with PPP) and one boy with an OPT (chief complaints: blurred vision and growth acceleration; laboratory examinations consistent with CPP), all of which were diagnosed by histopathology. Among patients with GCTs (Figure 3B), six showed moderately elevated hCG in serum (0.1~40.6 IU/L) and/or CSF (20.5~32.3 IU/L); their tumor sites were suprasellar (n=3) or in the basal ganglia (n=3). One 2.5-year-old boy presented with rapid pubertal development; testing showed remarkably elevated hCG (10,000 IU/L). hCG in patients with GCTs was significantly elevated, and the other patients had hCG levels that were below the lower limit of detection. MRI of one boy revealed an approximately 5.6×6.7×12.8 mm mass in the pineal gland (Figure 3C), and biopsy showed choriocarcinoma. CT scans of the abdomen, pelvis and chest were normal in all of the GCT patients. In the seven GCT patients, although their testicular volumes varied from 4 mL to 8 mL, all of the GnRH analog stimulation tests revealed a prepubertal response to gonadotropin.
Familial Male Limited Precocious Puberty (OMIM #176410)
LHCGR gene screening was performed in three unrelated boys (patients coded A, B and C) (1.67 to 4.41 years old) (Figure 4). In patients A and B, sequencing analysis of the LHCGR gene revealed a heterozygous C-to-T transition at codon 577 (c.1730C>T), and this transition led to the substitution of threonine with isoleucine at codon 577 (p.T577I). The two patients’ mothers were heterozygous for the p.T577I mutation, while neither patient’s father carried the mutation. Patient C had a heterozygous T-to-C transition at codon 576 (c.1726T>C) of the LHCGR gene, and this transition led to the substitution of phenylalanine with leucine at codon 576 (p.F576L). Patient C’s father (who was also a patient) was heterozygous for the p.F576L mutation, while the mother did not carry the mutation. These two variants were predicted to be deleterious/damaging by both PolyPhen-2 and SIFT.
Figure 4.
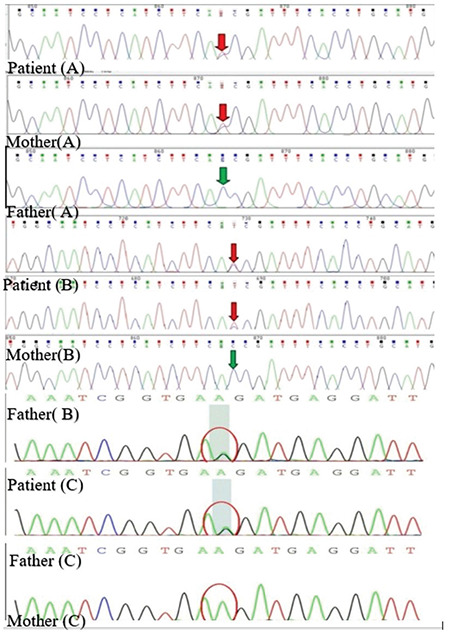
Mutation analysis of the LHCGR gene. In patient A and B, sequencing analysis of the LHCGR gene revealed a heterozygous C to T transition at the codon 577 (c.1730C>T), both from the mother. Patient C had a heterozygous T to C transition at the codon 576 (c.1726T>C) of the LHCGR gene, from his father (who was also a patient)
Congenital Adrenal Hyperplasia (PPP combined with CPP)
All 12 boys (aged 3.25 to 7.67 years) were diagnosed with a 21-hydroxylase deficiency. All presented with penis enlargement and three also presented with premature pubarche. Nine boys presented with PPP, and three patients presented with CPP. The evaluation age of the CAH-PPP group was slightly lower than that of the CAH-CPP group (5.03±1.42 vs 6.00±1.86 years, p=0.360). The 17-OHP levels were higher than the rest of groups. but showed no significant difference. In the CAH-CPP group, one patient presented with CPP before the administration of hydrocortisone, and the other two patients presented with CPP at six months and one year after treatment. The testicular volumes of the latter two were both 2 mL before treatment but increased to 4 mL and 5 mL, respectively, after treatment. The basal value of LH increased from 0.1 IU/L to 0.4 IU/L and 5.0 IU/L, respectively.
Discussion
Of the 62 boys who were diagnosed with CPP in our department, 79% were diagnosed with ICPP. No pathological factors were found during the follow-up. Such a high proportion of ICPP is quite different from what has been reported in other countries. Studies have shown that 50-70% of boys with CPP have identifiable pathological changes (6). The majority (64%) of boys with CPP in a ten-year study in Boston had organic CPP (4). A study from Saudi Arabia reported that organic CPP accounted for 50% of CPP cases in boys (14). This discrepancy may be caused by several factors. Previous studies have suggested that there is a downward trend in the age of puberty onset in boys (15,16). The same trend may have occurred in our patients. Increased awareness may be another cause of the increased number of ICPP diagnoses. At the same time, genetic background could play a role, as suggested by the difference in neurofibromatosis rates: neurofibromatosis was the main cause of CPP in the Boston study but was not found in a single case in our study.
Other than ICPP, HH was the most common etiology of CPP in our cohort. Lesions or malformations of the CNS are usually considered first in the differential diagnosis of organic CPP. The main risk factors for a physical CNS etiology include young age and male sex (17,18). The most common CNS lesion associated with pathogenic CPP is HH, which is a rare congenital non-neoplastic CNS lesion containing mature tissue in a heterotopic location (19). For most symptomatic patients with HH associated with CPP and/or gelastic seizures, the relationship between the neuroradiological findings and clinical presentation has not been fully investigated (20,21). In our study, 41.6% of patients with pathogenic CPP had HH. However, in the US, 64% of boys with CNS-CPP had NF I (4). This suggests that the etiology of CPP differs among different races.
We also found that in addition to the usual etiology, a relatively rare but not negligible cause of male CPP is post BMT. Most studies suggest that gonadal impairment is an important side effect in transplanted patients, and busulfan conditioning regimens and TBI are associated with gonad damage (22,23). Interestingly, three patients developed CPP after BMT. A Korean study suggested that the application of cyclophosphamide may be related to PP (3). In our study, only two patients’ conditioning regimens included cyclophosphamide. One of our patients had a long history of using Epimedium, which has the effect of enhancing sexual function in mice (24). In addition to chemotherapy and herbal remedies, cranial irradiation might cause damage to gamma-aminobutyric acid–secreting neurons, which could lead to premature activation of GnRH neurons (25). Further studies are needed to determine whether BMT and irradiation could be associated with male CPP.
The spectrum of male PPP is complex. In our study, CAH, GCT and FMPP were the three most common etiologies of PPP. CAH is the predominant etiology in boys with PPP (26). In a six-year retrospective study of PP cases at a tertiary hospital in Southern China, 38% were due to CAH (27). CAH was also the most common etiology of organic PP in our study, occurring in 12 patients. Three patients with CAH developed CPP. Secondary CPP may complicate CAH. First of all, chronic hyperandrogenemia may trigger the activation of the hypothalamic-pituitary axis, leading to CPP (28,29) and hydrocortisone treatment reduces androgen levels and activates gonadotropins. The decrease in sex steroids during treatment of the primary underlying disorder causes activation of the precociously matured hypothalamic GnRH pulse generator via feedback mechanisms, resulting in secondary CPP (30). CPP can occur either before or after treatment. This combination of PPP and CPP in patients with CAH is worthy of wider clinical recognition and attention, and better clinical management of patients is needed.
Usually, patients with hCG-secreting GCTs present with signs of increased intracranial pressure and diabetes insipidus, and PPP is less common than other manifestations (31). In a study by Atay et al (32) that excluded CAH, hCG-secreting tumors, Leydig cell tumors and adrenocortical tumors played equally important roles in the etiology of PPP. All of our patients had a significant increase in blood and/or CSF hCG. hCG-secreting GCTs can cause PP in boys by stimulating Leydig cells to secrete T, consistent with previous studies almost exclusively in boys (33). Although seven of our patients had PPP, it has been reported that some boys with hCG-secreting GCTs near the hypothalamus may actually develop CPP (34).
FMPP is an autosomal-dominant form of PPP in males. FMPP is a very rare disorder, affecting approximately nine individuals per million (35). The LHR gene, located on human chromosome 2p21, encodes a transmembrane receptor expressed on the cell membranes of testicular Leydig cells (36). Affected males usually begin pubertal development by 1~4 years of age, with rapid growth and progressive virilization, and the same characteristics were observed in the present study (37). The most common mutation reported in the literature in the LHCGR gene is a D578G missense mutation (38). In our study, PCR product sequencing revealed a heterozygous adenosine-guanine transition at nucleotide 1730 in codon 577 in two boys; this mutation in LHCGR has been identified before (39). One patient was found to have a heterozygous mutation in LHCGR, causing an F576L substitution within Transmembrane region 6 of LHCGR. Although this mutation has not been previously reported, given that it occurs in a mutation hotspot in a patient with a family history, and the variants were predicted to be damaging, we suspect that it is also pathogenic. All three boys presented with PPP, but as seen with CAH, if the diagnosis was delayed, CPP could also be present at the first visit.
Different studies have shown different disease spectra. Based on these five years of data and a review of the literature, we propose a diagnostic algorithm (summarized in Figure 5) (32). Zou et al (27) reported that 23% of boys with PPP in Southern China had a history of exogenous hormone intake, such as contraceptives, whereas not a single case due to exogenous hormone intake was found among our patients. Testicular tumors were also common in the study by Zou et al (27) (19%) but very rare in our study. Genetic and dietary differences may be among the reasons for these discrepancies. We did not find any cases of McCune-Albright syndrome or hypothyroidism, which have been reported in other studies (40). In addition to untreated primary hypothyroidism, very rare syndromes, such as Prader-Willi syndrome and Sotos syndrome, can also result in PP (41). In addition, some etiologies remain undetermined despite complex and detailed investigation.
Figure 5.
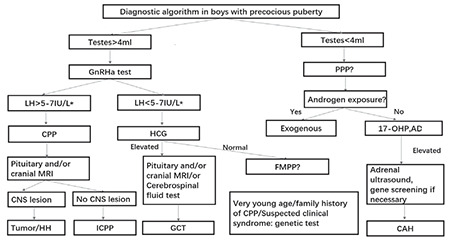
Algorithm for etiological spectrum
CAH: congenital adrenal hyperplasia, GCT: germ cell tumor, HH: hypothalamic hamartoma, FMPP: familial male limited precocious puberty, MRI: magnetic resonance imaging
Study Limitations
This study had several limitations. The enrollees were treated at a single medical center, and the data on male PP could vary from that found in other centers. In the ICPP group, further studies are needed to examine the possible contribution of genetic factors. Sequencing and genetic studies are undertaken increasingly often in affected individuals, thereby resulting in a declining number of “idiopathic” cases.
Conclusion
The etiology of PP in boys is diverse. In our cohort of Chinese males, CPP was mainly idiopathic, and HH was the most common etiology of pathogenic CPP. CAH and GCTs accounted for the etiology of 80% of PPP cases. It should be borne in mind that PPP patients may become CPP patients, and the diagnosis of some cases remains challenging.
Footnotes
Ethics
Ethics Committee Approval: The study protocol was approved by the Children’s Hospital Capital Institute of Pediatrics Ethics Committee (SHERLL2020003, date: 14.01.2020).
Informed Consent: Retrospective study.
Peer-review: Externally peer-reviewed.
Authorship Contributions
Concept – Design - Data Collection or Processing - Analysis or Interpretation - Literature Search - Writing: Liu Ziqin, Li Xiaohui, Chen Xiaobo.
Financial Disclosure: Supported by the Beijing Municipal Administration of Hospitals Incubating Program (PX2020055).
References
- 1.Kalantaridou SN, Chrousos GP. Clinical review 148: monogenic disorders of puberty. J Clin Endocrinol Metab. 2002;87:2481–2894. doi: 10.1210/jcem.87.6.8668. [DOI] [PubMed] [Google Scholar]
- 2.Alikasifoglu A, Vuralli D, Gonc EN, Ozon A, Kandemir N. Changing etiological trends in male precocious puberty: evaluation of 100 cases with central precocious puberty over the last decade. Horm Res Paediatr. 2015;83:340–344. doi: 10.1159/000377678. [DOI] [PubMed] [Google Scholar]
- 3.Lee J, Kim J, Yang A, Cho SY, Jin DK. Etiological trends in male central precocious puberty. Ann Pediatr Endocrinol Metab. 2018;23:75–80. doi: 10.6065/apem.2018.23.2.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Topor LS, Bowerman K, Machan JT, Gilbert CL, Kangarloo T, Shaw ND. Central precocious puberty in Boston boys: A 10-year single center experience. PLoS One. 2018;13:e0199019. doi: 10.1371/journal.pone.0199019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Januszek-Trzciakowska A, Kalina-Faska B, Kalina M, Zachurzok-Buczynska A, Gawlik A, Malecka-Tendera E. Przyczyny przedwczesnego dojrzewania dzieci kierowanych do dianostyki w warunkach szpitalnych [Causes of precocious puberty in children referred for evaluation in hospital conditions] Endokrynol Pol. 2007;58:291–296. [PubMed] [Google Scholar]
- 6.Latronico AC, Brito VN, Carel JC. Causes, diagnosis, and treatment of central precocious puberty. Lancet Diabetes Endocrinol. 2016;4:265–274. doi: 10.1016/S2213-8587(15)00380-0. [DOI] [PubMed] [Google Scholar]
- 7.Li H, Ji CY, Zong XN, Zhang YQ. [Height and weight standardized growth charts for Chinese children and adolescents aged 0 to 18 years] Zhonghua Er Ke Za Zhi. 2009;47:487–492. [PubMed] [Google Scholar]
- 8.Freire AV, Escobar ME, Gryngarten MG, Arcari AJ, Ballerini MG, Bergadá I, Ropelato MG. High diagnostic accuracy of subcutaneous Triptorelin test compared with GnRH test for diagnosing central precocious puberty in girls. Clin Endocrinol (Oxf) 2013;78:398–404. doi: 10.1111/j.1365-2265.2012.04517.x. [DOI] [PubMed] [Google Scholar]
- 9.El-Maouche D, Arlt W, Merke DP. Congenital adrenal hyperplasia. Lancet. 2017;390:2194–2210. doi: 10.1016/S0140-6736(17)31431-9. [DOI] [PubMed] [Google Scholar]
- 10.Göbel U, Schneider DT, Calaminus G, Haas RJ, Schmidt P, Harms D. Germ-cell tumors in childhood and adolescence. GPOH MAKEI and the MAHO study groups. Ann Oncol. 2000;11:263–271. doi: 10.1023/a:1008360523160. [DOI] [PubMed] [Google Scholar]
- 11.Luo S, Li C, Ma Z. [The diagnosis and treatment of hypothalamic hamartoma in children] Zhonghua Yi Xue Za Zhi. 2001;81:212–215. [PubMed] [Google Scholar]
- 12.Shenker A, Laue L, Kosugi S, Merendino JJ Jr, Minegishi T, Cutler GB Jr. A constitutively activating mutation of the luteinizing hormone receptor in familial male precocious puberty. Nature. 1993;365:652–654. doi: 10.1038/365652a0. [DOI] [PubMed] [Google Scholar]
- 13.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Osman HA, Al-Jurayyan NAM, Babiker AMI, Al-Otaibi HMN, AlKhalifah RDH, Al Issa SDA, Mohamed S. Precocious puberty: an experience from a major teaching hospital in Central Saudi Arabia. Sudan J Paediatr. 2017;17:19–24. [PMC free article] [PubMed] [Google Scholar]
- 15.Herman-Giddens ME, Steffes J, Harris D, Slora E, Hussey M, Dowshen SA, Wasserman R, Serwint JR, Smitherman L, Reiter EO. Secondary sexual characteristics in boys: data from the Pediatric Research in Office Settings Network. Pediatrics. 2012;130:e1058–e1068. doi: 10.1542/peds.2011-3291. [DOI] [PubMed] [Google Scholar]
- 16.Sørensen K, Aksglaede L, Petersen JH, Juul A. Recent changes in pubertal timing in healthy Danish boys: associations with body mass index. J Clin Endocrinol Metab. 2010;95:263–270. doi: 10.1210/jc.2009-1478. [DOI] [PubMed] [Google Scholar]
- 17.Soriano-Guillén L, Corripio R, Labarta JI, Cañete R, Castro-Feijóo L, Espino R, Argente J. Central precocious puberty in children living in Spain: incidence, prevalence, and influence of adoption and immigration. J Clin Endocrinol Metab. 2010;95:4305–4313. doi: 10.1210/jc.2010-1025. [DOI] [PubMed] [Google Scholar]
- 18.Kim SH, Huh K, Won S, Lee KW, Park MJ. A significant increase in the incidence of central precocious puberty among Korean girls from 2004 to 2010. PLoS One. 2015;10:e0141844. doi: 10.1371/journal.pone.0141844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jung H, Parent AS, Ojeda SR. Hypothalamic hamartoma: a paradigm/model for studying the onset of puberty. Endocr Dev. 2005;8:81–93. doi: 10.1159/000084095. [DOI] [PubMed] [Google Scholar]
- 20.Mong JA, Glaser E, McCarthy MM. Gonadal steroids promote glial differentiation and alter neuronal morphology in the developing hypothalamus in a regionally specific manner. J Neurosci. 1999;19:1464–1472. doi: 10.1523/JNEUROSCI.19-04-01464.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arita K, Ikawa F, Kurisu K, Sumida M, Harada K, Uozumi T, Monden S, Yoshida J, Nishi Y. The relationship between magnetic resonance imaging findings and clinical manifestations of hypothalamic hamartoma. J Neurosurg. 1999;91:212–220. doi: 10.3171/jns.1999.91.2.0212. [DOI] [PubMed] [Google Scholar]
- 22.Wei C, Albanese A. Endocrine disorders in childhood cancer survivors treated with haemopoietic stem cell transplantation. Children (Basel) 2014;1:48–62. doi: 10.3390/children1010048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shalitin S, Phillip M, Stein J, Goshen Y, Carmi D, Yaniv I. Endocrine dysfunction and parameters of the metabolic syndrome after bone marrow transplantation during childhood and adolescence. Bone Marrow Transplant. 2006;37:1109–1117. doi: 10.1038/sj.bmt.1705374. [DOI] [PubMed] [Google Scholar]
- 24.Gu S, Zhou R, Wang X. Comparison of enhanced male mice sexual function among three medicinal materials. Andrologia. 2018;50:e13087. doi: 10.1111/and.13087. [DOI] [PubMed] [Google Scholar]
- 25.Rose SR, Horne VE, Howell J, Lawson SA, Rutter MM, Trotman GE, Corathers SD. Late endocrine effects of childhood cancer. Nat Rev Endocrinol. 2016;12:319–336. doi: 10.1038/nrendo.2016.45. [DOI] [PubMed] [Google Scholar]
- 26.Haddad NG, Eugster EA. Peripheral precocious puberty including congenital adrenal hyperplasia: causes, consequences, management and outcomes. Best Pract Res Clin Endocrinol Metab. 2019;33:101273. doi: 10.1016/j.beem.2019.04.007. [DOI] [PubMed] [Google Scholar]
- 27.Zou CC, Liang L, Dong GP, Zhao ZY. Peripheral precocious puberty: a retrospective study for six years in Hangzhou, China. J Paediatr Child Health. 2008;44:415–418. doi: 10.1111/j.1440-1754.2008.01320.x. [DOI] [PubMed] [Google Scholar]
- 28.Sahana PK, Gopal Sankar KS, Sengupta N, Chattopadhyay K. Boy with central precocious puberty probably due to a peripheral cause. BMJ Case Rep. 2016;2016:bcr2016214554. doi: 10.1136/bcr-2016-214554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Güven A, Nurcan Cebeci A, Hancili S. Gonadotropin releasing hormone analog treatment in children with congenital adrenal hyperplasia complicated by central precocious puberty. Hormones (Athens) 2015;14:265–271. doi: 10.14310/horm.2002.1555. [DOI] [PubMed] [Google Scholar]
- 30.Lim YJ, Batch JA, Warne GL. Adrenal 21-hydroxylase deficiency in childhood: 25 years’ experience. J Paediatr Child Health. 1995;31:222–227. doi: 10.1111/j.1440-1754.1995.tb00790.x. [DOI] [PubMed] [Google Scholar]
- 31.Kakkar A, Biswas A, Kalyani N, Chatterjee U, Suri V, Sharma MC, Goyal N, Sharma BS, Mallick S, Julka PK, Chinnaswamy G, Arora B, Sridhar E, Chatterjee S, Jalali R, Sarkar C. Intracranial germ cell tumors: a multi-institutional experience from three tertiary care centers in India. Childs Nerv Syst. 2016;32:2173–2180. doi: 10.1007/s00381-016-3167-2. [DOI] [PubMed] [Google Scholar]
- 32.Atay Z, Yesilkaya E, Erdeve SS, Turan S, Akin L, Eren E, Goyal N, Sharma BS, Mallick S, Julka PK, Chinnaswamy G, Arora B, Sridhar E, Chatterjee S, Jalali R, Sarkar C. The etiology and clinical features of non-CAH gonadotropin-independent precocious puberty: a multicenter study. J Clin Endocrinol Metab. 2016;101:1980–1988. doi: 10.1210/jc.2015-3500. [DOI] [PubMed] [Google Scholar]
- 33.Calaminus G, Frappaz D, Kortmann RD, Krefeld B, Saran F, Pietsch T, Vasiljevic A, Garre ML, Ricardi U, Mann JR, Göbel U, Alapetite C, Murray MJ, Nicholson JC. Outcome of patients with intracranial non-germinomatous germ cell tumors-lessons from the SIOP-CNS-GCT-96 trial. Neuro Oncol. 2017;19:1661–1672. doi: 10.1093/neuonc/nox122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berberoğlu M. Precocious puberty and normal variant puberty: definition, etiology, diagnosis and current management. J Clin Res Pediatr Endocrinol. 2009;1:164–174. doi: 10.4274/jcrpe.v1i4.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lim YY, Chan RM, Loke KY, Ho CW, Lee YS. Familial male-limited precocious puberty in neurofibromatosis type I. Eur J Pediatr. 2014;173:219–222. doi: 10.1007/s00431-013-2141-1. [DOI] [PubMed] [Google Scholar]
- 36.Schedewie HK, Reiter EO, Beitins IZ, Seyed S, Wooten VD, Jimenez JF, Aiman EJ, DeVane GW, Redman JF, Elders MJ. Testicular leydig cell hyperplasia as a cause of familial sexual precocity. J Clin Endocrinol Metab. 1981;52:271–278. doi: 10.1210/jcem-52-2-271. [DOI] [PubMed] [Google Scholar]
- 37.Schoelwer M, Eugster EA. Treatment of peripheral precocious puberty. Endocr Dev. 2016;29:230–239. doi: 10.1159/000438895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yano K, Okuno A. [Constitutively activating mutations in the luteinizing hormone receptor gene in cases of male-limited precocious puberty] Nihon Rinsho. 1998;56:1843–1847. [PubMed] [Google Scholar]
- 39.Chan WY. Molecular genetic, biochemical, and clinical implications of gonadotropin receptor mutations. Mol Genet Metab. 1998;63:75–84. doi: 10.1006/mgme.1997.2650. [DOI] [PubMed] [Google Scholar]
- 40.Lumbroso S, Paris F, Sultan C. McCune-Albright syndrome: molecular genetics. J Pediatr Endocrinol Metab. 2002;15(Suppl 3):875–882. [PubMed] [Google Scholar]
- 41.Setian NS. Hypothyroidism in children: diagnosis and treatment. J Pediatr (Rio J) 2007;83(Suppl 5):S209–S216. doi: 10.2223/JPED.1716. [DOI] [PubMed] [Google Scholar]