

Abstract
Kabuki syndrome (KS) is a disease characterized by distinctive facial features, skeletal anomalies and delay in neuromotor development. KS 1 is an autosomal dominant condition caused by mutations in the KMT2D gene, whereas KS 2 is an X-linked disorder caused by mutations in the KDM6A gene. In the majority of KS patients who present with hypoglycemia, KDM6A is the defective gene. A 9-month old girl was admitted to our emergency department due to a seizure. On physical examination, hypotonia, mild facial dysmorphism, brachydactyly of the 5th finger, prominent finger pads and pansystolic murmur were detected. A fasting glucose tolerance test was performed the next day due to her history of hypoglycemia, but she had convulsions at the fifth hour of the test. Her serum glucose was 24 mg/dL, insulin 1.94 mIU/L, C-peptide 0.94 ng/mL, growth hormone 11 ng/mL, anti-insulin antibody 4.2 IU/mL, cortisol 19.8 μg/dL, and adrenocorticotropic hormone 9.3 pg/mL. A diagnosis of hyperinsulinemic hypoglycemia was considered. Given the abnormalities, genetic analysis for congenital hyperinsulinism, including the genes causing KS was performed. A heterozygous frameshift mutation (c.2579del, p.Leu860Argfs*70) was detected in the KMT2D gene. Epilepsy and other neurological symptoms may be seen in KS patients and in some of these the neurological symptoms are the result of hypoglycemia. In such cases, the detection and prevention of hypoglycemia can help prevent the progression of neurological symptoms. We suggest considering the diagnosis of KS for patients with hypoglycemia and dysmorphic features, even if the patient does not manifest all features of KS.
Keywords: Diazoxide, hyperinsulinemic hypoglycemia, Kabuki syndrome, KMT2, KDM6A
What is already known on this topic?
Kabuki syndrome (KS) patients with the KMT2D gene mutations develop symptoms like facial dysmorphism, neonatal feeding problems, kidney anomalies, and skeletal malformations, whereas KS patients with the KDM6A mutations have a higher risk of hyperinsulinemic hypoglycemia (HH).
What this study adds?
The presented case is very rare because the infant had a mutation in KMT2D but presented with HH.
Introduction
Kabuki syndrome (KS) is a rare congenital syndrome first described by Niikawa et al (1) and Kuroki et al (2) in 1981. Its prevalence is 1:32,000 in Japan, and 1:86,000 in Australia and New Zealand (3,4).In approximately 85% of KS patients, mutations in either KMT2D gene (previously known as MLL2) located at 12q13.13 (autosomal dominant) or KDMGA gene located at Xp11.3 (X-linked dominant) were identified (5,6,7,8,9). There are five cardinal criteria for the diagnosis of KS: postnatal short stature, mild to moderate developmental delay/intellectual disability, distinctive facial features, skeletal abnormalities, and persistent fetal fingertip pads (3,10,11).Other symptoms, including hypoglycemia, congenital heart defects, congenital hypothyroidism, seizures, hypotonia, and gastrointestinal problems, might be seen infrequently (12).Here, we present a Turkish female infant with KS who also exhibited hyperinsulinemic hypoglycemia (HH).
Case Report
The patient was a female infant, born as the second child of healthy, non-consanguineous parents at 32 weeks of gestation by an uncomplicated cesarean section. Her birth weight was 2410 grams [+2.20 standard deviation score (SDS)]. She was hospitalized because of prematurity, respiratory distress syndrome and indirect hyperbilirubinemia. Hypoglycemia developed within the first few days of life due to hyperinsulinism (serum insulin 181.5 mIU/L and concurrent glucose 37 mg/dL). She was initially treated by intravenous glucose infusion (up to 15 mg/kg/min) and oral diazoxide. After achieving normoglycemia with oral feeding, diazoxide was eventually discontinued. Subsequently, neonatal transient hyperinsulinism was considered and no other treatment was initiated. She also had a ventricular septal defect (VSD) along with patent ductus arteriosus. The karyotype analysis was found to be 46,XX. After 45 days, she was discharged and given furosemide and captopril to address cardiac problems.
At the age of nine months, she was admitted to the pediatric emergency department because of somnolence after a seizure. Weight, height, and head circumference were 6760 grams (-1.93 SDS), 71 cm (-0.07 SDS), and 39.8 cm (-3.59 SDS), respectively. Hypotonia, pansystolic murmur, and facial dysmorphic features including wide forehead, arched and sparse eyebrows, ptosis, eversion of lateral third of inferior eyelids, and a short columella were noted. In addition, persistent fetal pads, and brachydactyly of fifth fingers were observed in the physical examination (Figure 1). Biochemical tests and electroencephalogram were found to be normal.
Figure 1.
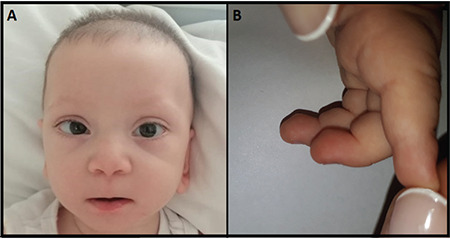
A) Facial appearance (wide forehead, arched and sparse eyebrows, ptosis, eversion of lateral third of inferior eyelids, short columella), B) Persistent fetal pads
A fasting glucose tolerance test was performed due to the history of hypoglycemia. She developed hypoglycemic convulsion at the fifth hour of the fasting test. The capillary blood glucose was 32 mg/dL. Critical blood and urine samples were collected. Hypoglycemia was treated by intravenous bolus injection of 0.2 g/kg dextrose, followed by an infusion at the rate of 8 mg/kg/min. The patient improved rapidly with this treatment. The samples collected at the time of hypoglycemia revealed normal blood count, electrolytes, liver, and kidney functions. Serum glucose was 24 mg/dL, insulin 1.94 mIU/L, C-peptide 0.94 ng/mL, growth hormone 11 ng/mL, anti-insulin antibody 4.2 IU/mL, cortisol 19.8 µg/dL, and adrenocorticotropic hormone 9.3 pg/mL. The levels of blood carnitine and acylcarnitines were normal and no ketone bodies were observed in the urinalysis. Skeletal radiographs, and abdominal ultrasound were normal. Based on the fasting test, HH was considered and diazoxide treatment was initiated at a dose of 3 mg/kg/day and that the dose was gradually increased to 15 mg/kg/day while tapering down dextrose. Feeding intervals of four hours, and dietary supplementation with uncooked cornstarch were recommended. The patient did not suffer from hypoglycemia under diazoxide therapy. The dysmorphic features, along with VSD and HH, suggested a possible diagnosis of KS. Genetic analysis was performed using a next-generation sequencing panel, including KMT2D and KDM6A genes, which revealed a heterozygous frameshift mutation in KMT2D gene (c.2579del, p.Leu860Argfs*70). The mutation had previously been reported to be associated with KS in a single case in ClinVar database, confirming the clinical diagnosis (13). The mutation was considered to be de novo, as the variant was not detected in the molecular genetic analysis of the parents. Informed consent was obtained from the parents for reporting genetic testing and publication of related data.
Discussion
KMT2D encodes a lysine-specific histone methyltransferase and is responsible for over 75% of KS cases (5,6,7,8). KDM6A encodes a histone demethylase and accounts for 5-8% of KS cases (9,14). KS patients with KMT2D gene mutations develop symptoms such as facial dysmorphism, neonatal feeding problems, kidney anomalies, and skeletal malformations (8), whereas KS patients with KDM6A mutations have a higher risk of HH (15).
Hyperinsulinism may manifest in various syndromes, especially Beckwith-Wiedemann, but it has rarely been observed in Kabuki, Sotos, Costello, Turner, Simpson-Golabi-Behmel, Ondine, Usher, Perlman and Timothy syndromes, and congenital disorders of glycosylation (16,17,18,19,20,21,22). Although the frequency of neonatal hypoglycemia is 6.7%, HH is extremely rare (0.3%) in KS (10). A small cohort study indicated that the incidence of KS in neonates with HH may be around 1%. Moreover, this cohort reported that 45.5% of KS patients, who presented with HH, had KDM6A mutations. However, KDM6A mutations were detected in only 5-8% of all KS patients. These data support the fact that there is a higher risk of HH in KS patients with KDM6A mutations compared to those with KMT2D mutations (15). This makes the presented case unusual because the infant had a mutation in KMT2D but presented with HH, which was probably the cause of the seizure.
KS may not be easily identified in neonates and infants because the characteristic facial features may not yet have become distinct. Similar to our 9-month old patient, KS is usually diagnosed between 6 and 18 months of age. Although KS is very rare, it is important to consider KS in patients with HH to provide genetic counseling and determine the prognosis of hypoglycemia. In fact, in the case of our 9-month old patient, failure to diagnose KS during the neonatal period caused the clinicians to assume that her hyperinsulinism was transient. Therefore, diazoxide treatment was discontinued, which might have contributed to the severe re-occurrence of hypoglycemia, accompanied by seizures, loss of consciousness and worsening of neurological damage and of developmental problems. As seen in our case, it is not safe to completely stop diazoxide treatment before excluding any underlying genetic etiology. In the literature, it has been reported that diazoxide treatment may be needed until five years of age to maintain normoglycemia in KS patients with HH (11,23).Our patient is currently being continued on diazoxide with proper maintenance of glycemia. In addition, the 4-hour interval feeding has been maintained, including nocturnal feeds, to prevent hypoglycemia.
Conclusion
In conclusion, early detection and proper management of hypoglycemia would help to prevent progression of neurological symptoms and permanent sequelae in KS. KS should be considered in the differential diagnosis of infants with hypoglycemia and dysmorphic features, even if the patient does not manifest with all features of the syndrome.
Acknowledgments
Genetic testing was performed by the Department of Molecular Genetics at the University of Exeter Medical School, UK.
Footnotes
Ethics
Informed Consent: Our patient’s mother and father gave informed consent for the genetic testing reported in this paper and for the publication of related data.
Peer-review: Externally peer-reviewed.
Authorship Contributions
Surgical and Medical Practices: Onur Akın, Yılmaz Yıldız, Mina Mısırlıgil, Concept: Onur Akın, Yılmaz Yıldız, Mina Mısırlıgil, Design: Onur Akın, Mutluay Arslan, Data Collection or Processing: Sevinç Odabaşı Güneş, Mina Mısırlıgil, Analysis or Interpretation: Bülent Ünay, Literature Search: Mina Mısırlıgil, Writing: Yılmaz Yıldız, Mina Mısırlıgil.
Financial Disclosure: The authors declared that this study received no financial support.
References
- 1.Niikawa N, Matsuura N, Fukushima Y, Ohsawa T, Kajii T. Kabuki make-up syndrome: a syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. J Pediatr. 1981;99:565–569. doi: 10.1016/s0022-3476(81)80255-7. [DOI] [PubMed] [Google Scholar]
- 2.Kuroki Y, Suzuki Y, Chyo H, Hata A, Matsui I. A new malformation syndrome of long palpebral fissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal dwarfism and mental retardation. J Pediatr. 1981;99:570–573. doi: 10.1016/s0022-3476(81)80256-9. [DOI] [PubMed] [Google Scholar]
- 3.Niikawa N, Kuroki Y, Kajii T, Matsuura N, Ishikiriyama S, Tonoki H, Ishikawa N, Yamada Y, Fujita M, Umemoto H, Iwama Y, Kondoh I, Fukushima Y, Nako Y, Matsui I, Urakami T, Aritaki S, Hara M, Suzuki Y, Chyo H, Sugio Y, Hasegawa T, Yamanaka T, Tsukino R, Yoshida A, Nomoto N, Kawahito S, Aihara R, Toyota S, Ieshima A, Funaki H, Ishitobi K, Ogura S, Furumae T, Yoshino M, Tsuji Y, Kondoh T, Matsumoto T, Abe K, Harada N, Miike T, Ohdo S, Naritomi K, Abushwereb AK, Braun OH, Schmid E, Opitz JM, Reynolds JF. Kabuki make-up (Niikawa-Kuroki) syndrome: a study of 62 patients. Am J Med Genet. 1988;31:565–589. doi: 10.1002/ajmg.1320310312. [DOI] [PubMed] [Google Scholar]
- 4.White SM, Thompson EM, Kidd A, Savarirayan R, Turner A, Amor D, Delatycki MB, Fahey M, Baxendale A, White S, Haan E, Gibson K, Halliday JL, Bankier A. Growth, behavior, and clinical findings in 27 patients with Kabuki (Niikawa-Kuroki) syndrome. Am J Med Genet A. 2004;127A:118–127. doi: 10.1002/ajmg.a.20674. [DOI] [PubMed] [Google Scholar]
- 5.Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, Beck AE, Tabor HK, Cooper GM, Mefford HC, Lee C, Turner EH, Smith JD, Rieder MJ, Yoshiura K, Matsumoto N, Ohta T, Niikawa N, Nickerson DA, Bamshad MJ, Shendure J. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010;42:790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Y, Bögershausen N, Alanay Y, Simsek Kiper PO, Plume N, Keupp K, Pohl E, Pawlik B, Rachwalski M, Milz E, Thoenes M, Albrecht B, Prott EC, Lehmkühler M, Demuth S, Utine GE, Boduroglu K, Frankenbusch K, Borck G, Gillessen-Kaesbach G, Yigit G, Wieczorek D, Wollnik B. A mutation screen in patients with Kabuki syndrome. Hum Genet. 2011;130:715–724. doi: 10.1007/s00439-011-1004-y. [DOI] [PubMed] [Google Scholar]
- 7.Paulussen AD, Stegmann AP, Blok MJ, Tserpelis D, Posma-Velter C, Detisch Y, Smeets EE, Wagemans A, Schrander JJ, van den Boogaard MJ, van der Smagt J, van Haeringen A, Stolte-Dijkstra I, Kerstjens-Frederikse WS, Mancini GM, Wessels MW, Hennekam RC, Vreeburg M, Geraedts J, de Ravel T, Fryns JP, Smeets HJ, Devriendt K, Schrander-Stumpel CT. MLL2 mutation spectrum in 45 patients with Kabuki syndrome. Hum Mutat. 2011;32:E2018–E2025. doi: 10.1002/humu.21416. [DOI] [PubMed] [Google Scholar]
- 8.Banka S, Veeramachaneni R, Reardon W, Howard E, Bunstone S, Ragge N, Parker MJ, Crow YJ, Kerr B, Kingston H, Metcalfe K, Chandler K, Magee A, Stewart F, McConnell VP, Donnelly DE, Berland S, Houge G, Morton JE, Oley C, Revencu N, Park SM, Davies SJ, Fry AE, Lynch SA, Gill H, Schweiger S, Lam WW, Tolmie J, Mohammed SN, Hobson E, Smith A, Blyth M, Bennett C, Vasudevan PC, García-Miñaúr S, Henderson A, Goodship J, Wright MJ, Fisher R, Gibbons R, Price SM, C de Silva D, Temple IK, Collins AL, Lachlan K, Elmslie F, McEntagart M, Castle B, Clayton-Smith J, Black GC, Donnai D. How genetically heterogeneous is Kabuki syndrome?: MLL2 testing in 116 patients, review and analyses of mutation and phenotypic spectrum. Eur J Hum Genet. 2012;20:381–388. doi: 10.1038/ejhg.2011.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lederer D, Grisart B, Digilio MC, Benoit V, Crespin M, Ghariani SC, Dallapiccola B, Verellen-Dumoulin C. Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with Kabuki syndrome. Am J Hum Genet. 2012;90:119–124. doi: 10.1016/j.ajhg.2011.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geneviève D, Amiel J, Viot G, Le Merrer M, Sanlaville D, Urtizberea A, Gérard M, Munnich A, Cormier-Daire V, Lyonnet S. Atypical findings in Kabuki syndrome: report of 8 patients in a series of 20 and review of the literature. Am J Med Genet A. 2004;129A:64–68. doi: 10.1002/ajmg.a.30144. [DOI] [PubMed] [Google Scholar]
- 11.Zarate YA, Zhan H, Jones JR. Infrequent Manifestations of Kabuki syndrome in a patient with novel MLL2 mutation. Mol Syndromol. 2012;3:180–184. doi: 10.1159/000342253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bögershausen N, Wollnik B. Unmasking Kabuki syndrome. Clin Genet. 2013;83:201–211. doi: 10.1111/cge.12051. [DOI] [PubMed] [Google Scholar]
- 13.Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, Karapetyan K, Katz K, Liu C, Maddipatla Z, Malheiro A, McDaniel K, Ovetsky M, Riley G, Zhou G, Holmes JB, Kattman BL, Maglott DR. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46:D1062–D1067. doi: 10.1093/nar/gkx1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miyake N, Mizuno S, Okamoto N, Ohashi H, Shiina M, Ogata K, surusaki Y, Nakashima M, Saitsu H, Niikawa N, Matsumoto N. KDM6A point mutations cause Kabuki syndrome. Hum Mutat. 2013;34:108–110. doi: 10.1002/humu.22229. [DOI] [PubMed] [Google Scholar]
- 15.Yap KL, Johnson AEK, Fischer D, Kandikatla P, Deml J, Nelakuditi V, Halbach S, Jeha GS, Burrage LC, Bodamer O, Benavides VC, Lewis AM, Ellard S, Shah P, Cody D, Diaz A, Devarajan A, Truong L, Greeley SAW, De Leon DD, Edmondson AC, Das S, Thornton P, Waggoner D, Del Gaudio D. Correction: “Congenital hyperinsulinism as the presenting feature of Kabuki syndrome: clinical and molecular characterization of 10 affected individuals”. Genet Med. 2019;21:262–265. doi: 10.1038/s41436-018-0126-1. [DOI] [PubMed] [Google Scholar]
- 16.Alexander S, Ramadan D, Alkhayyat H, Al-Sharkawi I, Backer KC, El-Sabban F, Hussain K. Costello syndrome and hyperinsulinemic hypoglycemia. Am J Med Genet A. 2005;139:227–230. doi: 10.1002/ajmg.a.31011. [DOI] [PubMed] [Google Scholar]
- 17.Kapoor RR, James C, Hussain K. Hyperinsulinism in developmental syndromes. Endocr Dev. 2009;14:95–113. doi: 10.1159/000207480. [DOI] [PubMed] [Google Scholar]
- 18.Arnoux JB, Verkarre V, Saint-Martin C, Montravers F, Brassier A, Valayannopoulos V, Brunelle F, Fournet JC, Robert JJ, Aigrain Y, Bellanné-Chantelot C, de Lonlay P. Congenital hyperinsulinism: current trends in diagnosis and therapy. Orphanet J Rare Dis. 2011;6:63. doi: 10.1186/1750-1172-6-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cappella M, Graziani V, Pragliola A, Sensi A, Hussain K, Muratori C, Marchetti F. Hyperinsulinemic hypoglycaemia in a turner syndrome with ring (X) Case Rep Pediatr. 2015;2015:561974. doi: 10.1155/2015/561974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nessa A, Rahman SA, Hussain K. Hyperinsulinemic hypoglycemia - the molecular mechanisms. Front Endocrinol (Lausanne). 2016;7:29. doi: 10.3389/fendo.2016.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Toda N, Ihara K, Kojima-Ishii K, Ochiai M, Ohkubo K, Kawamoto Y, Kohno Y, Kumasaka S, Kawase A, Ueno Y, Futatani T, Miyazawa T, Nagaoki Y, Nakata S, Misaki M, Arai H, Kawai M, Sato M, Yada Y, Takahashi N, Komatsu A, Maki K, Watabe S, Sumida Y, Kuwashima M, Mizumoto H, Sato K, Hara T. Hyperinsulinemic hypoglycemia in Beckwith-Wiedemann, Sotos, and Kabuki syndromes: a nationwide survey in Japan. Am J Med Genet A. 2017;173:360–367. doi: 10.1002/ajmg.a.38011. [DOI] [PubMed] [Google Scholar]
- 22.Péanne R, de Lonlay P, Foulquier F, Kornak U, Lefeber DJ, Morava E, Pérez B, Seta N, Thiel C, Van Schaftingen E, Matthijs G, Jaeken J. Congenital disorders of glycosylation (CDG): Quo vadis. Eur J Med Genet. 2018;61:643–663. doi: 10.1016/j.ejmg.2017.10.012. [DOI] [PubMed] [Google Scholar]
- 23.Subbarayan A, Hussain K. Hypoglycemia in Kabuki syndrome. Am J Med Genet A. 2014;164A:467–471. doi: 10.1002/ajmg.a.36256. [DOI] [PubMed] [Google Scholar]