

Abstract
Systemic pseudohypoaldosteronism (PHA) is a rare, salt-wasting syndrome that is caused by inactivating variants in genes encoding epithelial sodium channel subunits. Hyponatremia, hyperkalemia, metabolic acidosis, increased aldosterone and renin levels are expected findings in PHA. Clinical management is challenging due to high dose oral replacement therapy. Furthermore, patients with systemic PHA require life-long therapy. Here we report a patient with systemic PHA due to SCNN1B variant whose hyponatremia and hyperkalemia was detected at the 24th hour of life. Hyperkalemia did not improve with conventional treatments and dialysis was required. He also developed myocarditis and hypertension in follow-up. Challenges for diagnosis and treatment in this patient are discussed herein. In addition, published evidence concerning common features of patients with SCNN1B variant are reviewed.
Keywords: Systemic pseudohypoaldosteronism, hyponatremia, hyperkalemia, metabolic acidosis, epithelial sodium channel, SCNN1B
What is already known on this topic?
Pseudohypoaldosteronism is a life threatening disease due to serious salt loss. Differential diagnosis from other adrenal insufficencies is important because the treatments are different. Patient compliance is difficult due to the need for excessive amounts of oral treatments.
What this study adds?
We present a patient with a difficult diagnostic process due to hypertension. A novel variant resulting in a premature stop codon was detected in the patient. Clinical and laboratory features of all published cases with SCNN1B variant are reviewed.
Introduction
Aldosterone is a mineralocorticoid hormone that provides sodium absorbtion and potassium secretion. Sodium crosses the apical membrane in the principal cells in kidney and enters the epithelial cell through the ion selective epithelial sodium channel (ENaC). Potassium is also secreted into the tight epithelium in the kidney. The ENaC is located in the apical membranes of sensitive tissues, such as the distal nephron, distal colon, salivary and sweat glands and creates a rate-limiting step in sodium reabsorption (1,2). ENaC is a heteromultimeric protein consisting of three subunits, a, b, g (3). ENaC subunits are encoded by the SCNN1A gene on chromosome 12p13, and the SCNN1B and SCNN1G genes on chromosome 16p12.2-p12.1.
Pseudohypoaldosteronism (PHA) is a salt wasting syndrome that develops due to variants in the mineralocorticoid receptor (MR) or ion channels in the kidney tubules. The estimated incidence of this rare disease is between 1/47,000 and 1/80,000, and its prevalence is <1/1,000,000 (4,5,6). PHA1 is divided into renal (PHA1A) and systemic (PHA1B) forms depending on mutation in the NR3C2 gene that codes the MR or in the SCNN1A, SCNN1B and SCNN1G genes that code ENaC subunits, respectively. In the systemic form, there is serious salt loss from the lung, colon, sweat and salivary glands, besides the kidney, and the symptoms begin in the neonatal period. Systemic PHA, which is inherited in an autosomal recessive fashion, results in life-threatening hyponatremia, hyperkalemia and metabolic acidosis. Plasma renin and aldosterone levels increase significantly, indicating end organ resistance. Treatment requires high doses of sodium replacement and potassium-lowering approaches. In this article, the clinical management of a patient with PHA due to an SCNN1B variant and the common features of patients with SCNN1B variants are presented.
Case Report
The male patient was born at term by normal vaginal delivery with a birth weight of 3600 grams and was followed up in the neonatal intensive care unit due to respiratory distress. It was learned that hyponatremia and hyperkalemia were detected (Na 118 mEq/L, K 8 mEq/L) at the 24th hour of hospitalization. He received 6x1 g salt, 4x2 g/kg calcium polystyrene sulfonate, 12x1 mL/kg 8.4% NaHCO3 and 12x4 mL/kg 3% NaCl treatment during this period. He was subsequently referred to our clinic because of lack of response to treatments at the age of 1 month and 15 days. On physical examination, his weight was 4000 grams [-1.32 standard deviation score (SDS)], height was 55 cm (-0.34 SDS), and he did not have hyperpigmentation and any abnormality on genital examination. His daily weight gain was insufficient. His blood pressure was high at 110/80 mmHg (95th percentiles for systolic and diastolic blood pressure for this age group are 94/46 mmHg). There was third degree consanguinity between the parents. His brother had died at the age of seven days with hyponatremia and hyperkalemia. On admission, Na concentration was 123 mEq/L, K 7.1 mEq/L, blood pH 7.12, and HCO3 was 10.8 mmol/L. Echocardiography was normal. He was diagnosed with congenital adrenal hyperplasia. Hydrocortisone and fludrocortisone treatments were started. Calcium gluconate, glucose-insulin infusion, NaHCO3 infusion, and salbutamol inhalation were administered for hyponatremia and hyperkalemia. Despite these interventions, hyponatremia persisted. Anti-hypertensive treatment was started for hypertension (0.1 mg/kg/day amlodipine). Oral 2x0.5 grams of salt was also added to the treatment and the dose was gradually increased. The laboratory findings at admission to our clinic were: 17-hydroxyprogesterone 1.22 ug/L, dehydroepiandrosterone sulfate 241.8 µg/dL, total testosterone 215.6 ng/dL, adrenocorticotropic hormone 255 pg/mL, cortisol 43.8 µg/dL, renin 16.3 ng/mL/hour (NR=2.4-37), aldosterone 6.4 µg/L (NR=0.065-0.86), urine Na 134 mmol/L, urine K 2 mmol/L (when blood Na 123 mEq/L and blood K 7.1 mEq/L). The transtubular potassium gradient (TTKG) was 1.3, indicating very low renal potassium excretion. Based on the laboratory test results, hydrocortisone and fludrocortisone treatments were discontinued. Urinalysis, urine culture and renal ultrasonography were normal. During this process, hypertension continued. The diagnosis of systemic PHA was considered given that the patient was admitted with hyponatremia and hyperkalemia in the neonatal period, with high aldosterone level, increased urinary Na excretion, and decreased K excretion. When hyperkalemia did not respond to conventional treatments, including a trial of calcium polystyrene sulfonate at 1 g/kg/dose in four doses, peritoneal dialysis was required. After three days of peritoneal dialysis, K decreased to 4.18 mEq/L. Electrolyte values of the patient were kept in the normal range with 6x1 g of oral salt and 4x3 g of anti-potassium treatment. The patient had fever during the follow up and despite subsequent normalization of body temperature, tachycardia persisted. The patient was diagnosed with myocarditis due to an increase in acute phase reactants, troponin 1 level and electrocardiographic findings. Myocarditis findings regressed on the tenth day. However, the cause of hypertension could not be explained and was thought to be related to the salt treatment. Then his blood pressure returned to normal ranges and amlodipine and propranalol treatments were discontinued on the fourteenth day. The Sanger sequencing analysis of the SCNN1A gene, which is the most common gene to carry pathogenic variants in systemic PHA type 1B, was found to be normal. In subsequent Illumina MiSeq sequencing, a homozygous c. 978 C>A (p.Tyr326Ter) variant was detected in the sixth exon of the SCNN1B gene (NM_000336). After oral salt (6x1 g) and antipotassium (4x3 g) administration, the patient had normal electrolyte values and was discharged. One month after discharge, during a period of infection, the patient had to be hospitalized again due to the loss of oral intake and salt wasting crisis. At the last follow-up, the patient was seven months old, his weight was 7.3 kg (-1.3 SDS), height was 68 cm (-0.83 SDS) and blood pressure was 80/35 mmHg (50th percentile). His growth and development was appropriate for the age with the current treatments (6x1 g oral salt, 4x3 g calcium polystyrene sulfonate and 4x2 mL NaHCO3). Clinical follow-up continues.
Discussion
Systemic PHA type 1 is a rare life-threatening disease. Clinical manifestations are similar to other adrenal gland insufficiencies, such as congenital adrenal hyperplasia, hypoaldosteronism, and secondary PHA. The clinical presentation is characterized by insufficient weight gain, vomiting, and dehydration (4,6,7,8). Our patient had normal genitalia with hyponatremia and hyperkalemia. Hydrocortisone and fludrocortisone treatments were started until adrenal androgen results were obtained. Since adrenal hormone levels were normal in the follow-up, the patient was diagnosed with PHA1B. In the differential diagnosis of our patient, transient aldosterone resistance secondary to urinary tract infection was also considered (4), and this diagnosis was ruled out when urinalysis, urine culture and renal ultrasonography were normal. Elevated aldosterone level accompanying hyponatremia and hyperkalemia supported resistance to aldosterone in the kidney and directed the treatment of our patient.
To date, more than 40 variants have been reported in the genes encoding ENaC subunits (9), and these variants have most often been found in the gene encoding the alpha subunit. Eleven variants have been reported in the gene encoding the beta subunit (Table 1). Consequently, we first investigated the SCNN1A gene encoding the alpha subunit, but when no pathogenic variant was found, the SCNN1B gene was sequenced. In this patient, a novel c.978 C>A (p.Tyr326Ter) homozygous variant was detected, presumably leading to a premature stop codon in the SCNN1B gene. This variant has not been previously reported in the literature or in the GnomaD database including genome data from healthy individuals, and is anticipated to be a pathogenic change by Variant Taster, one of the in silico assessment tools used to predict pathogenicity of a variant. In addition, this change has been considered pathogenic according to the criteria of American College of Medical Genetics guidelines 2015 (PVS1, PM2, PP3).
Table 1. Clinical and laboratory features of this and other cases with SCNN1B mutation in the literature thoma.
The SCNN1B gene, consisting of 13 exons, encodes a transmembrane protein with two transmembrane segments with 640 amino acids (Figure 1) (7). The detected variant is located in the extracellular region of the protein, and, it is predicted to cause loss of function by creating an early stop codon, presumably resulting in nonsense mediated decay at the mRNA level.
Figure 1.
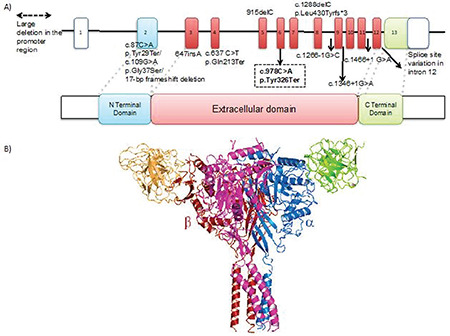
A) 3-D structure of the SCNN1B protein (PDB 6BQN) black arrow indicates the mutated position on the protein. B) Close up view of the tyrosine aminoacid at the 326th position
To date, 11 variants with PHA due to SCNN1B variants have been reported (Table 1). These cases were diagnosed in infancy with classical findings. On follow-up, two case died due to salt wasting crisis (6,8). In four cases, there were skin manifestations, such as dry skin, severe eczema, bullous dermatitis and hidradenitis suppurativa (7,10,11). Recurrent lung infections developed in four cases (7,11,12,13) and gastrostomy was required in four cases due to the salt wasting (8,10,11,13). Pulmonary hypertension developed in one case during follow-up (11). Apart from the case presented here, no other developed viral myocarditis and hypertension was encountered during attacks.
Both urgent and long-term treatments of PHA involve many challenges. Hyperkalemia can be life threatening due to the risk of cardiac arrhythmia. In the literature and similar to our case, there have been previous reports of peritoneal dialysis to correct hyperkalemia (7,14,15,16). Cases requiring gastrostomy due to difficulties in continuing oral treatment have also been reported (4,5,8,10,11,13,17). Patients with PHA are prone to pulmonary infections due to a decrease in sodium-dependent fluid absorption in the lungs. Rapid decompensation may occur during episodes of infection or when oral intake is impaired. After the electrolyte balance was achieved in the follow-up of our patient, salt wasting reccurred due to intervening viral myocarditis and, during this period, treatments were given via nasogastric tube.
Conclusion
Systemic PHA is a challenging disease to manage, with severe salt wasting that starts in the neonatal period. In this disease, life-threatening arrhythmias can be seen due to recurrent salt wasting and severe hyperkalemia. PHA1B can be confused with congenital adrenal hyperplasia. If a patient with hyperkalemia has hyponatremia, elevated urinary sodium excretion and low TTKG, mineralocorticoid resistance/deficiency should be considered. Treatment compliance is difficult due to the need for high dose oral salt and anti-potassium treatment. Long-term follow-up and treatment of these patients should careful, as the patients are frequently non-compliant with treatment and the frequency of rapid decompensation is high, especially during periods of infection.
Footnotes
Ethics
Informed Consent: Written informed consent was obtained from the parents.
Peer-review: Externally peer-reviewed.
Authorship Contributions
Surgical and Medical Practices: Gülin Karacan Küçükali, Semra Çetinkaya, Gaffari Tunç, M. Melek Oğuz, Nurullah Çelik, Kardelen Yağmur Akkaş, Saliha Şenel, Naz Güleray Lafcı, Şenay Savaş Erdeve, Concept: Gülin Karacan Küçükali, Semra Çetinkaya, M.Melek Oğuz, Saliha Şenel, Şenay Savaş Erdeve, Design: Gülin Karacan Küçükali, Naz Güleray Lafcı, Şenay Savaş Erdeve, Data Collection or Processing: Gülin Karacan Küçükali, Semra Çetinkaya, Gaffari Tunç, M. Melek Oğuz, Nurullah Çelik, Kardelen Yağmur Akkaş, Saliha Şenel, Naz Güleray Lafcı, Şenay Savaş Erdeve, Analysis or Interpretation: Gülin Karacan Küçükali, Semra Çetinkaya, M. Melek Oğuz, Saliha Şenel, Naz Güleray Lafcı, Şenay Savaş Erdeve, Literature Search: Gülin Karacan Küçükali, Şenay Savaş Erdeve, Naz Güleray Lafcı, Writing: Gülin Karacan Küçükali, Semra Çetinkaya, Şenay Savaş Erdeve, Naz Güleray Lafcı.
Financial Disclosure: The authors declared that this study received no financial support.
References
- 1.Rossier BC. The epithelial sodium channel (ENaC) and the control of blood pressure. J Am Soc Nephrol. 1997;8:980–992. doi: 10.1681/ASN.V86980. [DOI] [PubMed] [Google Scholar]
- 2.Garty H, Palmer LG. Epithelial sodium channels: function, structure, and regulation. Physiol Rev. 1997;77:359–396. doi: 10.1152/physrev.1997.77.2.359. [DOI] [PubMed] [Google Scholar]
- 3.Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, Rossier BC. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature. 1994;367:463–467. doi: 10.1038/367463a0. [DOI] [PubMed] [Google Scholar]
- 4.Amin N, Alvi NS, Barth JH, Field HP, Finlay E, Tyerman K, Frazer S, Savill G, Wright NP, Makaya T, Mushtag T. Pseudohypoaldosteronism type 1: clinical features and management in infancy. Endocrinol Diabetes Metab Case Rep. 2013;2013:130010. doi: 10.1530/EDM-13-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kala Ahluwalia, Dasouki M, Lennon A. Phenotypic variation of autosomal recessive pseudohypoaldosteronism type 1: a case in point. Clin Case Rep. 2014;2:326–330. doi: 10.1002/ccr3.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cayir A, Demirelli Y, Yıldız D, Kahveci H, Yaralı O, Kurnaz E, Vuralli D, Demirbilek H. Systemic pseudohypoaldosteronism type 1 due to 3 novel mutations in SCNN1A and SCNN1B genes. Horm Res Paediatr. 2019;91:175–185. doi: 10.1159/000498860. [DOI] [PubMed] [Google Scholar]
- 7.Gopal-Kothandapani JS, Doshi AB, Smith K, Christian M, Mushtaq T, Banerjee I, Padidela R, Ramakrishnan R, Owen C, Cheetham T, Dimitri P. Phenotypic diversity and correlation with the genotypes of pseudohypoaldosteronism type 1. J Pediatr Endocrinol Metab. 2019;32:959–967. doi: 10.1515/jpem-2018-0538. [DOI] [PubMed] [Google Scholar]
- 8.Saxena A, Hanukoglu I, Saxena D, Thompson RJ, Gardiner RM, Hanukoglu A. Novel mutations responsible for autosomal recessive multisystem pseudohypoaldosteronism and sequence variants in epithelial sodium channel α-,β-,ϒ- subunit genes. J Clin Endocrinol Metab. 2002;87:3344–3350. doi: 10.1210/jcem.87.7.8674. [DOI] [PubMed] [Google Scholar]
- 9.Nur N, Lang C, Hodax JK, Quintos JB. Systemic pseudohypoaldosteronism type I: A case report and review of the literature. Case Rep Pediatr. 2017;2017:7939854. doi: 10.1155/2017/7939854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Belot A, Ranchin B, Fichtner C, Pujo L, Rossier BC, Liutkus A, Morlat C, Nicolino M, Zennaro MC, Cochat P. Pseudohypoaldosteronisms, report on a 10- patient series. Nephrol Dial Transplant. 2008;23:1636–1641. doi: 10.1093/ndt/gfm862. [DOI] [PubMed] [Google Scholar]
- 11.Nobel YR, Lodish MB, Raygada M, Rivero JD, Faucz FR, Abraham SB, Lyssikatos C, Belyavskaya E, Stratakis CA, Zilbermint M. Pseudohypoaldosteronism type 1 due to novel variants of SCNN1B gene. Endocrinol Diabetes Metab Case Rep. 2016;2016:150104. doi: 10.1530/EDM-15-0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thomas CP, Zhou J, Liu KZ, Mick VE, MacLaughlin E, Knowles M. Systemic pseudohypoaldosteronism from deletion of the promoter region of the human β epithelial Na+ channel subunit. Am J Respir Cell Mol Biol. 2002;27:314–319. doi: 10.1165/rcmb.2002-0029OC. [DOI] [PubMed] [Google Scholar]
- 13.Edelheit O, Hanukoglu I, Gizewska M, Kandemir N, Tenenbaum- Rakover Y, Yurdakök M, Zajaczek S, Hanukoglu A. Novel mutations in epithelial sodium channel (ENaC) subunit genes and phenotypic expression of multisystem pseudohypoaldosteronism. Clin Endocrinol (Oxf) 2005;62:547–553. doi: 10.1111/j.1365-2265.2005.02255.x. [DOI] [PubMed] [Google Scholar]
- 14.Güran T, Degirmenci S, Bulut IK, Say A, Riepe FG, Güran Ö. Critical points in the management of pseudohypoaldosteronism type 1. J Clin Res Pediatr Endocrinol. 2011;3:98–100. doi: 10.4274/jcrpe.v3i2.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saravanapandian N, Paul S, Matthai J. Pseudohypoaldosteronism type 1: a rare cause of severe dyselectrolytemia and cardiovascular collapse in neonates. J Clin Neonatol. 2012;1:224–226. doi: 10.4103/2249-4847.106007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharma R, Pandey M, Kanwal SK, Zennaro MC. Pseudohypoalsteronism type 1: management issues. Indian Pediatr. 2013;50:331–333. doi: 10.1007/s13312-013-0070-8. [DOI] [PubMed] [Google Scholar]
- 17.Önder A, Çetinkaya S, Kara C, Zenciroğlu A, Aycan Z. Multisystemic severe form pseudohypoaldosteronism: can gastrostomy be useful in management? Turkish J Pediatr Dis. 2016;2:134–136. [Google Scholar]
- 18.Chang SS, Grunder S, Hanukoglu A, Rösler A, Mathew PM, Hanukoglu I, Schild L, Lu Y, Shimkets RA, Nelson-Williams C, Rossier BC, Lifton RP. Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkalemic acidosis, pseudohypoaldosteronism type 1. Nat Genet. 1996;12:248–253. doi: 10.1038/ng0396-248. [DOI] [PubMed] [Google Scholar]
- 19.Dogan CS, Parlak M, Akan M, Akcurin S, İffet B, Afig B. A novel splice site mutation of the beta subunit gene of epithelial sodium channel (ENaC) in one Turkish patient with systemic form of pseudohypoaldosteronism type 1. J Pediatr Endocrinol Metab. 2012;25:1035–1039. doi: 10.1515/jpem-2012-0083. [DOI] [PubMed] [Google Scholar]
- 20.Hanukoglu A, Edelheit O, Shriki Y, Gizewska M, Dascal N, Hanukoglu I. Renin-aldosteron response, urinary Na/K ratio and growth in pseudohypoaldosteronism patients with mutations in epithelial sodium channel (ENaC) subunit genes. J Steroid Biochem Mol Biol. 2008;111:268–274. doi: 10.1016/j.jsbmb.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 21.Kerem E, Bistritzer T, Hanukoglu A, Hofmann T, Zhou Z, Bennett W, MacLaughlin E, Barker P, Nash M, Quittell L, Boucher R, Knowles MR. Pulmonary epithelial sodium-channel dysfunction and excess airway liquid in pseudohypoaldosteronism. N Engl J Med. 1999;341:156–162. doi: 10.1056/NEJM199907153410304. [DOI] [PubMed] [Google Scholar]