

Abstract
This is the second part of a two-part review on pheochromocytoma and paragangliomas (PPGLs). In this part, perioperative management, including preoperative preparation, intraoperative, and postoperative interventions are reviewed. Current data on outcomes following resection are presented, including outcomes after cortical-sparing adrenalectomy for bilateral adrenal disease. In addition, pathological features of malignancy, surveillance considerations, and the management of advanced disease are also discussed.
PERIOPERATIVE CONSIDERATIONS
Patients who undergo PPGL resection may exhibit labile blood pressure (BP), arrhythmias, and tachycardia in the perioperative period. Relatively small retrospective observational studies of patients undergoing surgery for PPGL have not reported intraoperative deaths but have reported arrhythmias, sustained hypertension and/or hypotension, postoperative myocardial infarction, stroke, and prolonged intubation.1-3 Significant risk factors include large tumor size, increased levels of preoperative catecholamines, preexisting catecholamine-induced heart failure, and lengthy complicated surgery.2,3 Therefore, management of PPGL should be multidisciplinary, including experienced surgeons, anesthesiologists, and endocrinologists.
PREOPERATIVE BLOCKADE
The goals of preoperative management include control of hypertension and normalization of intravascular volume. Clinical institutional practices vary with no universally accepted algorithm. Preoperative readiness is assessed by orthostatic BP measurements, aiming for a seated blood pressure of 120–130/80 mmHg, a standing systolic blood pressure (SBP) ≥ 90 mmHg, and a heart rate between 60 and 70 beats/min (bpm) seated and 70 to 80 bpm standing. Patients should also be well hydrated and have a high-sodium diet prior to surgery.3 No data exist on optimal duration of preoperative treatment, but a minimum of 7 days is advised.
Alpha-selective or nonselective blockade is the most common approach to preoperative preparation of patients with PPGL and is always initiated first, with the addition of beta blockade as needed for tachycardia after at least 3–4 days of alpha blockade administration because unopposed alpha-adrenergic stimulation can lead to worsening of hypertension. Calcium channel blockers (CCBs) are used to supplement alpha and beta blockade or as an alternative. Metyrosine is typically reserved for patients who cannot tolerate alpha blockers or have alpha receptor and CCB-refractory hypertension,4 and recent data suggest that its use may be associated with improved cardiovascular outcomes in combination with a nonselective alpha blockade5 (Table 1).
TABLE 1.
Medications used in preoperative management of pheochromocytoma and paraganglioma
| Drug | Class | Starting dose |
Final dose | Side effects |
|---|---|---|---|---|
| Phenoxybenzaminea | Alpha-blocker | 10 mg bid | 1 mg/kg/day | Tachycardia, postural hypotension, nasal congestion, miosis, inhibition of ejaculation |
| Doxazosina | Alpha-blocker | 2 mg/day | 32 mg/day | Dizziness, fatigue, somnolence, edema, dyspnea, hypotension, dry mouth |
| Propranolol | Beta-blocker | 20 mg tid | 40 mg tid | Fatigue, nausea, drowsiness, diarrhea, bronchitis, bradycardia, impotence, dry eyes |
| Atenolol | Beta-blocker | 25 mg/day | 50 mg/day | Fatigue, nausea, dizziness, bradycardia, postural hypotension, vertigo |
| Nifedipine | Calcium channel blocker | 30 mg/day | 60 mg/day | Dizziness, flushing, headache, weakness, nausea, muscle cramps, peripheral edema, anxiety, palpitation, dyspnea, nasal congestion |
| Amlodipine | Calcium channel blocker | 5 mg/day | 10 mg/day | Edema, dizziness, flushing, palpitation, fatigue, nausea, abdominal pain, somnolence |
| Metyrosine | Tyrosine hydroxylase inhibitor | 250 mg/qid | 4 g/day | Sedation, diarrhea, extrapyramidal signs such as drooling, speech difficulty, and tremor, anxiety, depression, confusion |
Alpha blockade should start 714 days prior to surgery. Beta blockade should be initiated 34 days after alpha-blockade when necessary. Calcium channel blockers should be added as needed post alpha and beta blockade. Metyrosine should be offered as a fourth-line agent for patients who are refractory to alpha and calcium channel blockade
Alpha blockade is most commonly achieved with phenoxybenzamine, a nonselective irreversible blocker of alpha receptors; or a selective reversible alpha-1 antagonist such as doxazosin, prazosin, and others. Several retrospective papers have addressed the superiority of one strategy versus the other, which have not shown a difference in increased complications.6,7 A recent prospective open-label randomized trial comparing selective and nonselective alpha blockade did show phenoxybenzamine as more effective at preventing intraoperative hemodynamic instability but no difference in clinical outcome.8 Phenoxybenzamine is currently extremely expensive in the USA and has a high side-effect profile. In practice and depending on local preference, the surgeon, the endocrinologist, or the hypertension expert initiates the oral alpha blockade (Table 1). The patient should have access to a blood pressure cuff at home and record a minimum of twice daily blood pressure and heart rate measurements. After 2 or 3 days, the alpha blocker dose is then increased to target a systolic blood pressure consistently at or below < 130 mmHg. Beta blockade is added if the pulse rate is consistently > 100 bpm. Tachycardia is less common when a selective reversible alpha blocker is used,9 but it is often necessary when the nonselective alpha blocker phenoxybenzamine is utilized. If the patient does not have the diagnosis of hypertension, the patient should still be blocked starting with a low-dose alpha blocker.
THE PREGNANT PATIENT
In a gravid patient, the goal is to prevent extreme hemodynamic fluctuations. No data exist on optimal blockade drug or its duration,10,11 but phenoxybenzamine, betablockers, CCBs, and doxazosin have all been used safely.12,13 The importance of balance between hypertension control and adequate uterine perfusion is vital.14,15 Maternal deaths have been reported as high as 48%, but with earlier diagnosis and appropriate perioperative care, mortality is as low as 2%.12,14 The optimal timing of PPGL resection is the second trimester, with minimally invasive techniques if feasible. Alternatively, a concomitant planned c-section and PPGL resection can be performed, or resection can be performed in the medically well-controlled patient in the postpartum period.16
MANAGEMENT BY ANESTHESIA
Intraoperative communication between surgeons and anesthesiologist is vital. Patients require adequate IV access and an arterial catheter for continuous blood pressure monitoring. A discussion of the anesthetic management of PPGL is beyond the scope of this review, but the anesthesia team should ideally be experienced with the management of such complex cases.
PERSONALIZED SURGICAL MANAGEMENT
Surgical approaches for adrenalectomy include both minimally invasive (laparoscopic or robotic) and open techniques (Table 2). The guiding principles include early identification and ligation of the adrenal vein, and minimal manipulation of the tumor to prevent tumor rupture and intraoperative release of catecholamines with subsequent blood pressure lability.17-19 Studies have shown improved outcomes when resection is performed by advanced laparoscopic surgeons and high-volume adrenal surgeons (> 4–6 adrenalectomies/year).20-23
TABLE 2.
Excerpts from the Endocrine Society Guidelines for surgical management of pheochromocytomas and paragangliomas
| Surgical approaches |
| Minimally invasive adrenalectomy for most adrenal pheochromocytomas (recommendation; low-quality evidence) |
| Open resection for large (> 6 cm) or invasive pheochromocytomas (recommendation; very low-quality evidence) |
| Open resection for paragangliomas, with laparoscopic resection for small, noninvasive paragangliomas in surgically favorable locations (suggestion; very low-quality evidence) |
| Extent of adrenalectomy |
| Partial adrenalectomy for selected patients, such as those with hereditary pheochromocytoma, with small tumors who have already undergone a contralateral complete adrenalectomy to spare adrenal cortex and prevent permanent hypocortisolism (suggestion; very low-quality evidence) |
| Postoperative management |
| Postoperatively patients should have monitoring of blood pressure, heart rate and blood glucose levels with adjustment of associated therapies (strong recommendation; low-quality evidence)19 |
Minimally invasive (laparoscopic or robotic) adrenalectomy has become the standard of care for the majority of patients with pheochromocytomas. This approach may be performed with either the transabdominal (TA) or posterior retroperitoneoscopic (PRA) approach.19,24-29 The choice of approach depends on surgeon preference and familiarity with the techniques, as well as factors such as patient body habitus/body mass index (BMI), indications for surgery, tumor characteristics, size and location, and history of prior abdominal or retroperitoneal procedures that may influence the decision.24-29
The minimally invasive TA approach is familiar to most surgeons and may allow for easier conversion to open if necessary. TA can be performed in patients with prior upper abdominal surgery (ipsilateral or contralateral to the adrenal). However, adhesions may add to the complexity and length of the case.30 The PRA approach provides direct access to the adrenal gland, without the need for mobilization or adhesiolysis.24-27 In a study of 46 patients with similar demographics and tumor characteristics undergoing adrenalectomy for pheochromocytoma (23 TA and 23 PRA), PRA was associated with shorter operative times (99.9 vs. 144.8 min; p > 0.001), estimated blood loss (8.4 vs. 123.8 mL; p = 0.02), and postoperative length-of-stay (1.9 vs. 3.1 nights; p < 0.01).26 The study suffered from selection bias, since the PRA approach was associated with smaller tumors. PRA has been shown to be safe in children as well.31 Other benefits of PRA include the ability to perform bilateral adrenalectomy without repositioning. However, this must be balanced by potential limitations, including a “cognitive reorientation” of anatomical landmarks and appropriate patient selection based on body habitus and tumor size/location in relation to the kidney and its hilum. Typically, it is best for tumors < 6 cm located posterior and superior to the kidney in patients with little retroperitoneal fat24-26,28 (Fig. 1).
FIG. 1.

a Diagram outlining port placement for robotic retroperitoneoscopic approach to an adrenalectomy. b Patient prepped, draped, and ports placed for robotic retroperitoneal total adrenalectomy
Robotic-assisted adrenalectomy, using both TA and PRA approaches, has been described for benign tumors, including pheochromocytoma.32-35 Advantages of robotic adrenalectomy include ergonomics, three-dimensional visualization, and improved wrist articulation and degrees of freedom allowing for greater surgical precision. Relative disadvantages include cost, a possible learning curve, and the lack of haptic feedback.32-35 Although a recent report showed similar costs and slightly faster procedure times for RA versus LA in a high-volume center.36 The robotic PRA can be hampered by the small access window which can inhibit robotic arm movement with currently available systems.
Open resection remains the standard of care when there is clinical concern for malignant/invasive PPGLs or with large tumors at risk for rupture. The prevention of tumor rupture is important to avoid local recurrence or pheochromocytomatosis, especially given the few effective systemic therapies for metastatic pheochromocytoma.19 Furthermore, open adrenalectomy may be preferred in patients with SDHB, TMEM127, or FH germline mutations, since these mutations are associated with a higher risk of extraadrenal disease, metastatic disease, and recurrence compared with germline mutations in NF1, RET, or VHL.37
Cortical-sparing (partial) adrenalectomy consists in resection of the adrenal pheochromocytoma with preservation of as much normal adrenal as possible to preserve a functioning gland. Cortical-sparing adrenalectomy is offered to patients with syndromic diseases such as multiple endocrine neoplasia type 2 (MEN2) or von Hippel–Lindau disease (VHL) (Fig. 2). This approach has been demonstrated to prevent postoperative adrenal insufficiency in up to 90% of patients,19,37-39 although the exact amount of remnant adrenal gland required is unknown.
FIG. 2.

Eleven-year-old male with VHL syndrome who underwent laparoscopic bilateral partial adrenalectomies guided with intraoperative ultrasound for bilateral pheochromocytomas. a Right adrenal tumor measuring 1.8 cm. b Post right partial adrenalectomy with preservation of the right adrenal vein. c Left adrenal tumor measuring 3.0 cm. d Post left partial adrenalectomy with preservation of the left adrenal vein. *Left and right adrenal tumors, respectively, ^Left and right adrenal veins, respectively, IVC inferior vena cava
IMMEDIATE POSTOPERATIVE PERIOD
After monitoring in the recovery room, most patients, if they have an uneventful intraoperative course and are hemodynamically stable, can be admitted to a floor bed. A monitored bed or intensive care unit stay may be required if there is a need for telemetry, hemodynamic monitoring, or the need for ongoing pharmacologic hemodynamic support.
Two potential postoperative complications are hypotension and rebound hypoglycemia.40,41 Transient episodes of postoperative hypotension are common. Postoperative hypotension is attributed to a combination of: (1) residual preoperative alpha blockade with an abrupt fall in circulating catecholamines, (2) residual effects of long-acting non-alpha-blocker antihypertensive medications, and (3) hypovolemia due to preoperative volume contraction and intraoperative blood loss. Treatment includes aggressive IV fluid boluses for volume resuscitation. If postoperative hypotension persists with adequate fluid resuscitation, about 10% of patients may transiently require IV vasopressors (e.g., phenylephrine, ephedrine, norepinephrine).9,17,42
A relatively rare event after PPGL resection includes rebound insulinemia and hypoglycemia, which typically occurs within the first 4 h postoperatively.40 The high preoperative levels of catecholamines causes suppression of alpha and beta cell pancreatic function, which results in insulin resistance and increased liver glycogenolysis.43-45 In addition, preoperative glycogenolysis is no longer present, the hepatic glycogen stores are depleted, and glucagon release is impaired.40,46 Finally, improved peripheral insulin sensitivity with increased uptake of glucose by skeletal muscle contributes to hypoglycemia.47 Reported incidence rates of 5–15% are largely limited to case reports and a few retrospective studies.45,46,48 Patients with higher preoperative urinary metanephrines/catecholamine levels are at higher risk.45,46 Patients should have regular glucose monitoring (at least every 6 h) and receive 5% dextrose-containing fluids until tolerating oral intake.45,49 Insulin requirements should be readjusted for those with preoperative diabetes mellitus.4
OUTCOMES
Adrenal Remnant Recurrence in the Setting of Familial Syndromes (MEN and VHL)
Patients with familial syndromes such as MEN 2A, MEN 2B, and VHL may benefit from cortical-sparing adrenalectomies to prevent steroid dependency (Fig. 2). This benefit must be balanced with the risk of tumor recurrence in the remnant adrenal gland; for VHL patients, the estimated risk of recurrence is 10–15% over 10 years with a cortical-sparing approach,50 while it has been reported to be as high as 39% for MEN2 patients.51 A recent series of 96 patients with hereditary pheochromocytomas reported a 3-year recurrence rate of 7% and steroid independence rate of 78%.39
In a large retrospective observational study encompassing 30 medical centers and 563 patients with MEN2 associated pheochromocytomas, 21% of patients (n = 114) underwent cortical sparing adrenalectomy, whereas 79% (n = 438) underwent total adrenalectomy.52 When recurrence was analyzed by operated gland, 4 (3%) out of 153 cortically spared glands had an ipsilateral recurrence with a mean follow-up of 10 years. In comparison, 11 (2%) of 717 total adrenalectomy glands recurred within the adrenal bed with a mean follow-up of 14.2 years. The rate of steroid dependency for patients who underwent either unilateral or bilateral cortical-sparing adrenalectomy was 43%. Of note, patients with at least one-third of an adrenal gland remnant were not steroid dependent.
Outcomes for patients with VHL undergoing cortical-sparing adrenalectomy are equally encouraging. Twentysix patients with VHL seen at the National Institutes of Health (NIH) over an 8-year period underwent 36 partial adrenalectomies. With a median follow-up of 9.25 years (5–46 years), no patients developed metastatic pheochromocytoma, 11% of patients developed recurrences within the ipsilateral adrenal gland remnant, and 11% of patients developed a pheochromocytoma within the contralateral adrenal gland, requiring partial adrenalectomy, and only 11% of patients became steroid dependent.5
RESOLUTION OF HYPERTENSION/CARDIAC MORBIDITY AND DIABETES
Previous studies report that pheochromocytomas increase cardiovascular morbidity and mortality. A retrospective case–control study examining cardiovascular events (myocardial infarction, stroke, or angina pectoris) 5 years prior to diagnosis between patients with pheochromocytomas (n = 109) matched to a control group of patients with essential hypertension (n = 183) showed a 14-fold increased risk for patients with pheochromocytomas.53 Timmers and colleagues studied the outcomes of 64 patients undergoing surgery for presumably benign pheochromocytoma. Most patients (64%) showed a decrease in blood pressure, but only one-third of patients became normotensive.54 Although hypertension may not be cured by surgical resection, surgical resection likely provides cardiovascular benefit. However, long-term data evaluating the outcome of patients with surgically treated PPGL have shown that such patients have a higher mortality compared with the general population. Khorram-Manesh and colleagues followed 121 patients after adrenalectomy for an average of 15 years and found 8 patients to have a recurrence at a mean follow-up of 8.5 years. Of those eight patients, two had a local recurrence, five had a distant recurrence, and one patient had both local and distant recurrences. Half of those patients eventually succumbed to their disease. When comparing the mortality of patients with pheochromocytoma with the general Swedish population, 42 patients with pheochromocytoma died compared with an estimated 23.6 expected deaths. Preoperatively, 86% of the cohort had hypertension, and although there was improvement at 1 year after resection, half of all patients with pheochromocytoma were still hypertensive. Although patients did not have a high recurrence rate and mortality was low, patients had a long-term increased risk of death compared with the general population.55 An estimated 15–35% of patients with PPGLs have diabetes and/or impaired glucose tolerance. Beninanto and colleagues reported the outcomes of 153 patients with PPGL, of whom 36 patients had diabetes. After surgery, 78.6% of patients had complete resolution and 93.0% had improvement of their diabetes.56
RISK OF OVERALL RECURRENCE AND MALIGNANCY
After diagnosis and treatment of a PPGL, patients require lifelong follow-up due to the risk of recurrence and malignancy. A retrospective study by Schovanek and colleagues showed that patients with PPGLs greater than 4.5 cm were at a significantly higher risk of developing metastatic disease. In addition, patients with tumors greater than 5.5 cm had an overall worse survival compared with smaller tumors. The age of initial diagnosis was also found to be an independent predictor of survival.57 Recurrence rates have been reported from 2.0 to 6.5% in patients with presumably benign PPGLs.58-60 These recurrences can happen decades later, and therefore, these patients require lifelong biochemical follow-up.
In a large cohort of patients spanning 55 years seen at the Mayo Clinic, 272 out of 3280 patients (8.3%) were found to have malignant PPGLs. Nearly two-thirds of those patients developed metachronous metastases at a median of 5.5 years (range 0.3–53.4 years), and the 5-year overall survival rate for patients with malignant PPGLs was 85.4%. Hamidi and colleagues reported genetic mutations as well in this series, but given the long time span, it was difficult to draw conclusions.61 Patients that were initially considered with benign sporadic disease may develop malignancy. Goldstein and colleagues reported an overall malignancy rate of 13%, which reemphasizes the need for lifelong follow-up.1 Dhir and colleagues assessed clinical factors associated with malignancy in patients with PPGLs and identified patients who are younger, have larger tumors, paragangliomas, and positive genetic testing to have a higher risk of malignancy.62
The outcome of patients with hereditary PPGLs who have a germline mutation in the succinate dehydrogenase subunit B (SDHB) (see Part 1 of this review) differs from those patients with sporadic PPGLs. Patients with SDHB mutation are at higher risk for metastatic disease and disease-specific mortality. Gimenez-Togueplo and colleagues reported a rate of 87% (7 out of 8) malignancy rate with SDHB compared with a rate of 26% (20 out of 76) in non-SDHB PPGLs.63 In a larger study by Neumann and colleagues, the rate of malignancy was 34.3% (11 out of 34) for patients with SDHB.64 Disease-specific survival is also reduced when comparing patients with SDHB mutations and non-SDHB mutations. Amar and colleagues reported the 5-year survival after the diagnosis of metastases was 36% for those with SDHB mutation compared with 67% for those without SDHB mutations. The risk of death was 2.7 times higher by multivariate analysis in patients with SDHB mutation.65 Therefore, these patients require biochemical testing as well as imaging to detect metastatic and/or early recurrence in the hopes that earlier detection can lead to improved treatment and survival.
PATHOLOGIC FEATURES
Malignant PPGLs are defined by the presence of chromaffin cells in tissues and sites that normally do not contain these cells. To predict their clinical behavior, Thompson and colleagues reported 100 patients with PPGLs. Half of the patients were deemed benign, while the other half was defined as malignant by histology with or without the presence of metastases. A histologically weighted pheochromocytoma of the adrenal gland scaled score (PASS) that assessed 12 histological factors was created. A PASS score ≥ 4 identified all tumors that were biologically aggressive.66 Strong and colleagues validated the PASS score in a comparison of 43 patients with benign tumors and 5 patients with malignant tumors with a median follow-up of 43 months (range 6.7–220.6 months). Tumor necrosis, tumor cell spindling, and higher mitotic rate were the significant factors that differentiated malignant from benign behavior. A PASS score < 4 predicted benign pheochromocytomas, and therefore the authors recommended for close surveillance of any patients with a PASS score ≥ 4.67 However, the largest impediment to wide adoption of the PASS score relates to the inter- and intra-observer variation and scoring. Wu and colleagues assessed scoring by five experienced endocrine pathologists and found the scores to be widely variable. Therefore, the PASS score is not recommended for clinical prognostication at this time.68
SURVEILLANCE
Current consensus guidelines recommend surveillance for the detection of recurrent disease postsurgical resection.19,69,70 However, the true incidence of recurrence as well as the optimal frequency, methods, and duration of follow-up are not well defined. In a systematic review and metaanalysis, Amar and colleagues71 evaluated the occurrence of “new events” following resection PPGLs. A pooled incidence of 0.98 events/100 person-years (95% CI 0.71, 1.25) was calculated, which corresponds to an estimated 5 events per 100 patients followed-up for 5 years. Across the studies analyzed, paraganglioma (PGL) and familial disease were independent predictors of a higher risk of recurrence. An analysis of 701 patients from the European Network for the Study of Adrenal Tumors database, found that the overall risk of recurrences (local recurrence, distant metastases, and new tumors) was 10% within 5 years of follow-up.70 In this study, PGL (versus pheochromocytoma), age < 20 years, hereditary disease, and tumor size > 15 cm were each associated with a recurrence risk of > 15%. In addition, patients with PGL and hereditary syndromes have the risk of late disease relapse (> 5 years).70
Following resection for localized PPGL, early postoperative history, physical, and biochemical evaluation will establish completeness of resection and provide a postoperative baseline. Postoperative biochemical evaluation is ideally performed using the same sampling conditions and methods used preoperatively.19,70,72 Frequency and duration of subsequent surveillance are best assessed using an individualized approach that considers concern for persistent disease, history of tumor spillage, age and genetic status of the patient, and location and size of the primary tumor. Although a risk-stratified algorithm for surveillance is not available, a personalized approach should be utilized19,70; For instance, in a 60-year-old patient with a small isolated sporadic PHEO, one may consider annual history and physical with biochemical evaluation for up to 10 years. However, a 20-year-old with an SDHB mutation and a history of resected multifocal PGL is best served with lifelong surveillance. Imaging should generally be reserved for symptomatic patients and positive screening laboratory values. Imaging may be required for surveillance in the rare patient with a biochemically inactive PPGL or in a patient who did not have any preoperative biochemical evaluation.
MANAGEMENT OF ADVANCED DISEASE
PPGLs are considered malignant when there is direct invasion into adjacent organs or when chromaffin tissue is present in areas normally lacking chromaffin cells. Common sites of distant metastases include liver, lung, bone, and lymph nodes. The treatment options for advanced disease are limited and include 131I MIBG, peptide receptor radionuclide therapy (PRRT), surgical debulking/cytoreduction, chemotherapy, and targeted therapies.
131I-MIBG
Since 1983, specialized centers around the world have used 131I-MIBG to treat patients with metastatic PPGLs. In a metaanalysis of 116 patients, Loh and colleagues73 found that 76% of patients had symptomatic improvement, 30% of patients had tumor responses, and 45% of patients had a biochemical response with 131I-MIBG treatment. Gonias and colleagues evaluated high-dose 131I-MIBG treatment in 49 patients with metastatic PPGLs and found the overall response rate to be 22%. The estimated 5-year survival was 64% with a median follow-up time of 2 years (0.1–15 years).74 The optimal dosing and the selection of patients who can benefit continue to be active areas of research.
PEPTIDE RECEPTOR RADIONUCLIDE THERAPY
Peptide receptor radionuclide therapy (PRRT) is an emerging therapy utilizing radiolabeled somatostatin analogues to treat metastatic neuroendocrine tumors, including PPGLs. In a series of 22 patients with metastatic PPGL and either disease progression and/or failure to control symptoms of catecholamine excess, Nastos and colleagues reviewed outcomes for patients allocated to either 123I-MIBG, 90Y-DOTATATE, or 177Lu-DOTATATE treatment based on MIBG or DOTATATE uptake. Although an improvement in progression-free survival was observed, two patients had received a combination of 123I-MIBG treatment and PRRT, rendering it difficult to conclude the superior modality. Identifying the optimal radionuclide analogue for progressive metastatic PPGLs is an active area of research.75 The National Institutes of Health is currently enrolling and treating patients with progressive inoperable metastatic PPGLs with PRRT (NCT03206060) (Fig. 3).
FIG. 3.
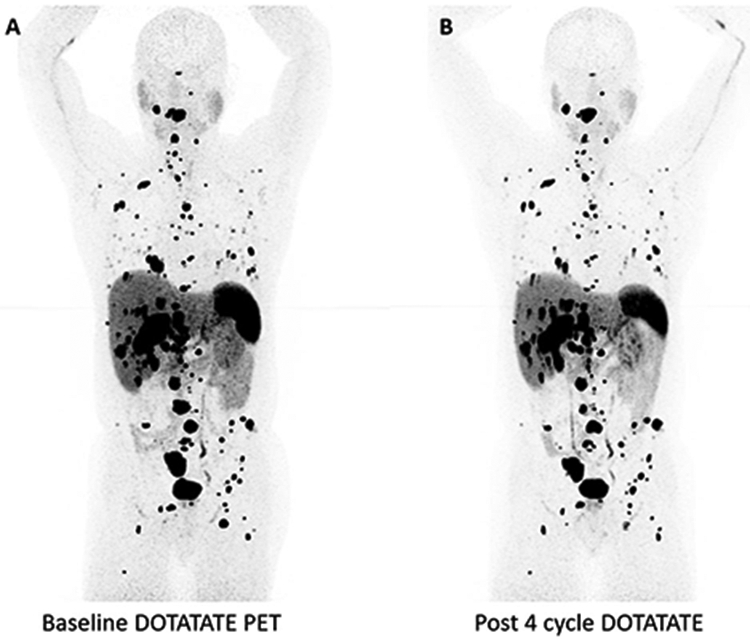
A 59-year-old male with SDHB who underwent PRRT for progressive metastatic PPGL showing mild response after four cycles of treatment. a Baseline 68-gallium DOTATATE PET/CT scan showing widely metastatic disease. b 68-Gallium DOTATATE PET/CT scan showing mild tumor response after four cycles of Lutathera therapy
SURGICAL DEBULKING/CYTOREDUCTION
The role of surgical debulking and cytoreduction of malignant PPGLs is not well defined. In a review of 30 patients who underwent surgical resection for metastatic PPGL, Ellis and colleagues found that complete resection of all gross disease (R0/R1) was critical to achieving a durable biochemical response and pharmacologic independence.76 In contrast, R2 resection was almost invariably associated with either no response or partial response, and very few patients were able to discontinue alpha blockade for any meaningful period of time. Resection of metastatic PPGL should be reserved for patients in whom complete resection is anticipated or in situations where surgical debulking and cytoreduction will remove all the gross disease. However, selected patients who require surgical palliation may benefit and must be based on expert clinical and multidisciplinary judgement.
CHEMOTHERAPY AND TARGETED THERAPIES
Combination chemotherapy with cyclophosphamide, vincristine, and dacarbazine (CVD) has been the standard chemotherapy regimen for patients with malignant PPGLs. Averbuch and colleagues initial report in 1988 was a nonrandomized single-arm trial showing a complete and partial response rate of 57% (median duration of 21 months) and a complete and partial biochemical response rate of 79% (median duration greater than 22 months) in 14 patients with malignant PPGLs.77 Tanabe and colleagues reported a nonrandomized single-arm trial of 17 patients with metastatic PPGL treated with CVD (n = 17). In this study, nearly 50% of patients responded to treatment, defined as either complete or partial biochemical response and/or partial tumor response (no patients in this study had complete tumor regression). The patients who responded to treatment had improvements in hypertension and impaired glucose tolerance. Moreover, the median survival was 6 years for responders, 4 years for nonresponders, and 3 years for those with progressive disease (median follow-up of 60 months, range 12–192 months).78
Given the limited options of effective cytotoxic therapy for advanced PPGL, there is interest in molecular target therapies. In a small series of patients with progressive metastatic PPGL, Ayala-Ramirez and colleagues reported modest benefit with the multiple tyrosine kinase receptor inhibitor, sunitinib. In this study, 8 of 14 patients with metastatic PPGLs and hypertension became normotensive, with 2 of those 8 patients discontinuing all antihypertensives. Despite promising results, the progression-free survival was only 4.1 months, indicating that better treatments are needed.79 Further prospective multiinstitutional studies are needed to identify and define the best treatment for patients with malignant PPGLs.
CONCLUSIONS
Expert and experienced surgeons, endocrinologists, and anesthesiologists are required for the preoperative blood pressure and intravascular management of patients with PPGLs. After resection, patients with PPGLs have reasonable outcomes with an improved biochemical response and blood pressure control. Cortical-sparing adrenalectomy is the ideal approach for patients with germline mutations such as VHL and RET, whose adrenal tumors have a low malignancy potential to prevent steroid dependency. Pathological features of PPGLs have not been clearly delineated and will require further study. All patients with PPGLs require biochemical and potentially radiological surveillance. Advanced disease continues to be difficult to manage and is an important area of research.
ACKNOWLEDGMENT
This manuscript was reviewed by the SSO Endocrine and Head and Neck Disease Site Working Group, and all members expressed support for its publication.
DISCLOSURES
Dr. Yang’s research is supported by the National Heart Lung and Blood Institute (NHLBI) of the National Institutes of Health (K08HL145139).
REFERENCES
- 1.Goldstein RE, O’Neill JA Jr., Holcomb GW, et al. Clinical experience over 48 years with pheochromocytoma. Ann Surg. 1999;229(6):755–64 (discussion 764–756). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kinney MA, Warner ME, van Heerden JA, et al. Perianesthetic risks and outcomes of pheochromocytoma and paraganglioma resection. Anesth Analg. 2000;91(5):1118–1123. [DOI] [PubMed] [Google Scholar]
- 3.Kercher KW, Novitsky YW, Park A, Matthews BD, Litwin DE, Heniford BT. Laparoscopic curative resection of pheochromocytomas. Ann Surg. 2005;241(6):919–26 (discussion 926–918). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steinsapir J, Carr AA, Prisant LM, Bransome ED Jr. Metyrosine and pheochromocytoma. Arch Intern Med. 1997;157(8):901–906. [PubMed] [Google Scholar]
- 5.Wachtel H, Kennedy EH, Zaheer S, et al. Preoperative metyrosine improves cardiovascular outcomes for patients undergoing surgery for pheochromocytoma and paraganglioma. Ann Surg Oncol. 2015;22 Suppl 3:S646–54. [DOI] [PubMed] [Google Scholar]
- 6.van der Zee PA, de Boer A. Pheochromocytoma: a review on preoperative treatment with phenoxybenzamine or doxazosin. Neth J Med. 2014;72(4):190–201. [PubMed] [Google Scholar]
- 7.Randle RW, Balentine CJ, Pitt SC, Schneider DF, Sippel RS. Selective versus non-selective alpha-blockade prior to laparoscopic adrenalectomy for pheochromocytoma. Ann Surg Oncol. 2017;24(1):244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buitenwerf E, Osinga TE, Timmers H, et al. Efficacy of alpha-blockers on hemodynamic control during pheochromocytoma resection—a randomized controlled trial. J Clin Endocrinol Metab. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kiernan CM, Solorzano CC. Pheochromocytoma and paraganglioma: diagnosis, genetics, and treatment. Surg Oncol Clin N Am. 2016;25(1):119–138. [DOI] [PubMed] [Google Scholar]
- 10.Witteles RM, Kaplan EL, Roizen MF. Safe and cost-effective preoperative preparation of patients with pheochromocytoma. Anesth Analg. 2000;91(2):302–304. [DOI] [PubMed] [Google Scholar]
- 11.Kalra JK, Jain V, Bagga R, et al. Pheochromocytoma associated with pregnancy. J Obstet Gynaecol Res. 2003;29(5):305–8. [DOI] [PubMed] [Google Scholar]
- 12.Lenders JW. Pheochromocytoma and pregnancy: a deceptive connection. Eur J Endocrinol. 2012;166(2):143–50. [DOI] [PubMed] [Google Scholar]
- 13.James MF, Cronje L. Pheochromocytoma crisis: the use of magnesium sulfate. Anesth Analg. 2004;99(3):680–6 (table of contents). [DOI] [PubMed] [Google Scholar]
- 14.Ahlawat SK, Jain S, Kumari S, Varma S, Sharma BK. Pheochromocytoma associated with pregnancy: case report and review of the literature. Obstet Gynecol Surv. 1999;54(11):728–37. [DOI] [PubMed] [Google Scholar]
- 15.Dahia PL, Hayashida CY, Strunz C, Abelin N, Toledo SP. Low cord blood levels of catecholamine from a newborn of a pheochromocytoma patient. Eur J Endocrinol. 1994;130(3):217–9. [DOI] [PubMed] [Google Scholar]
- 16.van der Weerd K, van Noord C, Loeve M, et al. Endocrinology in pregnancy: Pheochromocytoma in pregnancy: case series and review of literature. Eur J Endocrinol. 2017;177(2):R49–58. [DOI] [PubMed] [Google Scholar]
- 17.Kiernan CM, Du L, Chen X, et al. Predictors of hemodynamic instability during surgery for pheochromocytoma. Ann Surg Oncol. 2014;21(12):3865–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Livingstone M, Duttchen K, Thompson J, et al. Hemodynamic stability during pheochromocytoma resection: lessons learned over the last two decades. Ann Surg Oncol. 2015;22(13):4175–80. [DOI] [PubMed] [Google Scholar]
- 19.Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915–42. [DOI] [PubMed] [Google Scholar]
- 20.Hauch A, Al-Qurayshi Z, Kandil E. Factors associated with higher risk of complications after adrenal surgery. Ann Surg Oncol. 2015;22(1):103–10. [DOI] [PubMed] [Google Scholar]
- 21.Park HS, Roman SA, Sosa JA. Outcomes from 3144 adrenalectomies in the United States: which matters more, surgeon volume or specialty? Arch Surg. 2009;144(11):1060–7. [DOI] [PubMed] [Google Scholar]
- 22.Anderson KL Jr., Thomas SM, Adam MA, et al. Each procedure matters: threshold for surgeon volume to minimize complications and decrease cost associated with adrenalectomy. Surgery. 2018;163(1):157–64. [DOI] [PubMed] [Google Scholar]
- 23.Lindeman B, Hashimoto DA, Bababekov YJ, et al. Fifteen years of adrenalectomies: impact of specialty training and operative volume. Surgery. 2018;163(1):150–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walz MK, Alesina PF, Wenger FA, et al. Posterior retroperitoneoscopic adrenalectomy—results of 560 procedures in 520 patients. Surgery. 2006;140(6):943–8 (discussion 948–950). [DOI] [PubMed] [Google Scholar]
- 25.Perrier ND, Kennamer DL, Bao R, et al. Posterior retroperitoneoscopic adrenalectomy: preferred technique for removal of benign tumors and isolated metastases. Ann Surg. 2008;248(4):666–74. [DOI] [PubMed] [Google Scholar]
- 26.Dickson PV, Alex GC, Grubbs EG, et al. Posterior retroperitoneoscopic adrenalectomy is a safe and effective alternative to transabdominal laparoscopic adrenalectomy for pheochromocytoma. Surgery. 2011;150(3):452–8. [DOI] [PubMed] [Google Scholar]
- 27.Carr AA, Wang TS. Minimally Invasive Adrenalectomy. Surg Oncol Clin N Am. 2016;25(1):139–52. [DOI] [PubMed] [Google Scholar]
- 28.Agcaoglu O, Sahin DA, Siperstein A, Berber E. Selection algorithm for posterior versus lateral approach in laparoscopic adrenalectomy. Surgery. 2012;151(5):731–5. [DOI] [PubMed] [Google Scholar]
- 29.Lee CR, Walz MK, Park S, et al. A comparative study of the transperitoneal and posterior retroperitoneal approaches for laparoscopic adrenalectomy for adrenal tumors. Ann Surg Oncol. 2012;19(8):2629–34. [DOI] [PubMed] [Google Scholar]
- 30.Mazeh H, Froyshteter AB, Wang TS, et al. Is previous same quadrant surgery a contraindication to laparoscopic adrenalectomy? Surgery. 2012;152(6):1211–7. [DOI] [PubMed] [Google Scholar]
- 31.Walz MK, Iova LD, Deimel J, et al. Minimally invasive surgery (MIS) in children and adolescents with pheochromocytomas and retroperitoneal paragangliomas: experiences in 42 patients. World J Surg. 2018;42(4):1024–30. [DOI] [PubMed] [Google Scholar]
- 32.Taskin HE, Berber E. Robotic adrenalectomy. J Surg Oncol. 2012;106(5):622–5. [DOI] [PubMed] [Google Scholar]
- 33.Aliyev S, Karabulut K, Agcaoglu O, et al. Robotic versus laparoscopic adrenalectomy for pheochromocytoma. Ann Surg Oncol. 2013;20(13):4190–4. [DOI] [PubMed] [Google Scholar]
- 34.Economopoulos KP, Mylonas KS, Stamou AA, et al. Laparoscopic versus robotic adrenalectomy: a comprehensive meta-analysis. Int J Surg. 2017;38:95–104. [DOI] [PubMed] [Google Scholar]
- 35.Feng Z, Feng MP, Levine JW, Solorzano CC. Robotic retroperitoneoscopic adrenalectomy: useful modifications of the described posterior approach. J Robot Surg. 2017;11(4):409–14. [DOI] [PubMed] [Google Scholar]
- 36.Feng Z, Feng MP, Feng DP, Rice MJ, Solorzano CC. A cost-conscious approach to robotic adrenalectomy. J Robot Surg. 2018;12(4):607–11. [DOI] [PubMed] [Google Scholar]
- 37.Nockel P, El Lakis M, Gaitanidis A, et al. Preoperative genetic testing in pheochromocytomas and paragangliomas influences the surgical approach and the extent of adrenal surgery. Surgery. 2018;163(1):191–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee JE, Curley SA, Gagel RF, Evans DB, Hickey RC. Cortical-sparing adrenalectomy for patients with bilateral pheochromocytoma. Surgery. 1996;120(6):1064–70 (discussion 1070–1061). [DOI] [PubMed] [Google Scholar]
- 39.Grubbs EG, Rich TA, Ng C, et al. Long-term outcomes of surgical treatment for hereditary pheochromocytoma. J Am Coll Surg. 2013;216(2):280–9. [DOI] [PubMed] [Google Scholar]
- 40.Hull CJ. Phaeochromocytoma. Diagnosis, preoperative preparation and anaesthetic management. Br J Anaesth. 1986;58(12):1453–68. [DOI] [PubMed] [Google Scholar]
- 41.Houston M The role of magnesium in hypertension and cardiovascular disease. J Clin Hypertens (Greenwich). 2011;13(11):843–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet. 2005;366(9486):665–75. [DOI] [PubMed] [Google Scholar]
- 43.Hamaji M Pancreatic alpha- and beta-cell function in pheochromocytoma. J Clin Endocrinol Metab. 1979;49(3):322–5. [DOI] [PubMed] [Google Scholar]
- 44.La Batide-Alanore A, Chatellier G, Plouin PF. Diabetes as a marker of pheochromocytoma in hypertensive patients. J Hypertens. 2003;21(9):1703–7. [DOI] [PubMed] [Google Scholar]
- 45.Chen Y, Hodin RA, Pandolfi C, Ruan DT, McKenzie TJ. Hypoglycemia after resection of pheochromocytoma. Surgery. 2014;156(6):1404–8 (discussion 1408–1409). [DOI] [PubMed] [Google Scholar]
- 46.Akiba M, Kodama T, Ito Y, Obara T, Fujimoto Y. Hypoglycemia induced by excessive rebound secretion of insulin after removal of pheochromocytoma. World J Surg. 1990;14(3):317–24. [DOI] [PubMed] [Google Scholar]
- 47.Wiesner TD, Bluher M, Windgassen M, Paschke R. Improvement of insulin sensitivity after adrenalectomy in patients with pheochromocytoma. J Clin Endocrinol Metab. 2003;88(8):3632–36. [DOI] [PubMed] [Google Scholar]
- 48.Plouin PF, Duclos JM, Soppelsa F, Boublil G, Chatellier G. Factors associated with perioperative morbidity and mortality in patients with pheochromocytoma: analysis of 165 operations at a single center. J Clin Endocrinol Metab. 2001;86(4):1480–6. [DOI] [PubMed] [Google Scholar]
- 49.Graham JB. Pheochromocytoma and hypertension; an analysis of 207 cases. Int Abstr Surg. 1951;92(2):105–21. [PubMed] [Google Scholar]
- 50.Benhammou JN, Boris RS, Pacak K, Pinto PA, Linehan WM, Bratslavsky G. Functional and oncologic outcomes of partial adrenalectomy for pheochromocytoma in patients with von Hippel-Lindau syndrome after at least 5 years of followup. J Urol. 2010;184(5):1855–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Asari R, Scheuba C, Kaczirek K, Niederle B. Estimated risk of pheochromocytoma recurrence after adrenal-sparing surgery in patients with multiple endocrine neoplasia type 2A. Arch Surg. 2006;141(12):1199–205 (discussion 1205). [DOI] [PubMed] [Google Scholar]
- 52.Castinetti F, Qi XP, Walz MK, et al. Outcomes of adrenal-sparing surgery or total adrenalectomy in phaeochromocytoma associated with multiple endocrine neoplasia type 2: an international retrospective population-based study. Lancet Oncol. 2014;15(6):648–55. [DOI] [PubMed] [Google Scholar]
- 53.Stolk RF, Bakx C, Mulder J, Timmers HJ, Lenders JW. Is the excess cardiovascular morbidity in pheochromocytoma related to blood pressure or to catecholamines? J Clin Endocrinol Metab. 2013;98(3):1100–6. [DOI] [PubMed] [Google Scholar]
- 54.Timmers HJ, Brouwers FM, Hermus AR, et al. Metastases but not cardiovascular mortality reduces life expectancy following surgical resection of apparently benign pheochromocytoma. Endocr Relat Cancer. 2008;15(4):1127–33. [DOI] [PubMed] [Google Scholar]
- 55.Khorram-Manesh A, Ahlman H, Nilsson O, et al. Long-term outcome of a large series of patients surgically treated for pheochromocytoma. J Intern Med. 2005;258(1):55–66. [DOI] [PubMed] [Google Scholar]
- 56.Beninato T, Kluijfhout WP, Drake FT, et al. Resection of pheochromocytoma improves diabetes mellitus in the majority of patients. Ann Surg Oncol. 2017;24(5):1208–13. [DOI] [PubMed] [Google Scholar]
- 57.Schovanek J, Martucci V, Wesley R, et al. The size of the primary tumor and age at initial diagnosis are independent predictors of the metastatic behavior and survival of patients with SDHB-related pheochromocytoma and paraganglioma: a retrospective cohort study. BMC Cancer. 2014;14:523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Heerden JA, Roland CF, Carney JA, Sheps SG, Grant CS. Long-term evaluation following resection of apparently benign pheochromocytoma(s)/paraganglioma(s). World J Surg. 1990;14(3):325–9. [DOI] [PubMed] [Google Scholar]
- 59.Pruszczyk P, Januszewicz W, Feltynowski T, et al. Long term follow-up after surgical removal of pheochromocytoma—observations in 61 patients. Clin Exp Hypertens A. 1991;13(6–7):1179–94. [DOI] [PubMed] [Google Scholar]
- 60.Favia G, Lumachi F, Polistina F, D’Amico DF. Pheochromocytoma, a rare cause of hypertension: long-term follow-up of 55 surgically treated patients. World J Surg. 1998;22(7):689–93 (discussion 694). [DOI] [PubMed] [Google Scholar]
- 61.Hamidi O, Young WF Jr., Iniguez-Ariza NM, et al. Malignant pheochromocytoma and paraganglioma: 272 patients over 55 years. J Clin Endocrinol Metab. 2017;102(9):3296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dhir M, Li W, Hogg ME, et al. Clinical predictors of malignancy in patients with pheochromocytoma and paraganglioma. Ann Surg Oncol. 2017;24(12):3624–630. [DOI] [PubMed] [Google Scholar]
- 63.Gimenez-Roqueplo AP, Favier J, Rustin P, et al. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res. 2003;63(17):5615–21. [PubMed] [Google Scholar]
- 64.Neumann HP, Pawlu C, Peczkowska M, et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA. 2004;292(8):943–51. [DOI] [PubMed] [Google Scholar]
- 65.Amar L, Baudin E, Burnichon N, et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J Clin Endocrinol Metab. 2007;92(10):3822–28. [DOI] [PubMed] [Google Scholar]
- 66.Thompson LD. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol. 2002;26(5):551–66. [DOI] [PubMed] [Google Scholar]
- 67.Strong VE, Kennedy T, Al-Ahmadie H, et al. Prognostic indicators of malignancy in adrenal pheochromocytomas: clinical, histopathologic, and cell cycle/apoptosis gene expression analysis. Surgery. 2008;143(6):759–68. [DOI] [PubMed] [Google Scholar]
- 68.Wu D, Tischler AS, Lloyd RV, et al. Observer variation in the application of the Pheochromocytoma of the Adrenal Gland Scaled Score. Am J Surg Pathol. 2009;33(4):599–608. [DOI] [PubMed] [Google Scholar]
- 69.Shah MH, Goldner WS, Halfdanarson TR, et al. NCCN guidelines insights: neuroendocrine and adrenal tumors, Version 2.2018. J Natl Compr Canc Netw. 2018;16(6):693–702. [DOI] [PubMed] [Google Scholar]
- 70.Plouin PF, Amar L, Dekkers OM, et al. European Society of Endocrinology Clinical Practice Guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. Eur J Endocrinol. 2016;174(5):G1–10. [DOI] [PubMed] [Google Scholar]
- 71.Amar L, Lussey-Lepoutre C, Lenders JW, Djadi-Prat J, Plouin PF, Steichen O. Management of endocrine disease: recurrence or new tumors after complete resection of pheochromocytomas and paragangliomas: a systematic review and meta-analysis. Eur J Endocrinol. 2016;175(4):R135–145. [DOI] [PubMed] [Google Scholar]
- 72.Eisenhofer G, Peitzsch M. Laboratory evaluation of pheochromocytoma and paraganglioma. Clin Chem. 2014;60(12):1486–99. [DOI] [PubMed] [Google Scholar]
- 73.Loh KC, Fitzgerald PA, Matthay KK, Yeo PP, Price DC. The treatment of malignant pheochromocytoma with iodine-131 metaiodobenzylguanidine (131I-MIBG): a comprehensive review of 116 reported patients. J Endocrinol Invest. 1997;20(11):648–658. [DOI] [PubMed] [Google Scholar]
- 74.Gonias S, Goldsby R, Matthay KK, et al. Phase II study of high-dose [131I]metaiodobenzylguanidine therapy for patients with metastatic pheochromocytoma and paraganglioma. J Clin Oncol. 2009;27(25):4162–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nastos K, Cheung VTF, Toumpanakis C, et al. Peptide receptor radionuclide treatment and (131)I-MIBG in the management of patients with metastatic/progressive phaeochromocytomas and paragangliomas. J Surg Oncol. 2017;115(4):425–34. [DOI] [PubMed] [Google Scholar]
- 76.Ellis RJ, Patel D, Prodanov T, et al. Response after surgical resection of metastatic pheochromocytoma and paraganglioma: can postoperative biochemical remission be predicted? J Am Coll Surg. 2013;217(3):489–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Averbuch SD, Steakley CS, Young RC, et al. Malignant pheochromocytoma: effective treatment with a combination of cyclophosphamide, vincristine, and dacarbazine. Ann Intern Med. 1988;109(4):267–73. [DOI] [PubMed] [Google Scholar]
- 78.Tanabe A, Naruse M, Nomura K, Tsuiki M, Tsumagari A, Ichihara A. Combination chemotherapy with cyclophosphamide, vincristine, and dacarbazine in patients with malignant pheochromocytoma and paraganglioma. Horm Cancer. 2013;4(2):103–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ayala-Ramirez M, Chougnet CN, Habra MA, et al. Treatment with sunitinib for patients with progressive metastatic pheochromocytomas and sympathetic paragangliomas. J Clin Endocrinol Metab. 2012;97(11):4040–50. [DOI] [PMC free article] [PubMed] [Google Scholar]