

Abstract
Homeostatic plasticity maintains network stability by adjusting excitation, inhibition, or the intrinsic excitability of neurons, but the developmental regulation and coordination of these distinct forms of homeostatic plasticity remains poorly understood. A major contributor to this information gap is the lack of a uniform paradigm for chronically manipulating activity at different developmental stages. To overcome this limitation, we used designer receptors exclusively activated by designer drugs (DREADDs) to directly suppress neuronal activity in layer2/3 (L2/3) of mouse primary visual cortex of either sex at two important developmental timepoints: the classic visual system critical period [CP; postnatal day 24 (P24) to P29], and adulthood (P45 to P55). We show that 24 h of DREADD-mediated activity suppression simultaneously induces excitatory synaptic scaling up and intrinsic homeostatic plasticity in L2/3 pyramidal neurons during the CP, consistent with previous observations using prolonged visual deprivation. Importantly, manipulations known to block these forms of homeostatic plasticity when induced pharmacologically or via visual deprivation also prevented DREADD-induced homeostatic plasticity. We next used the same paradigm to suppress activity in adult animals. Surprisingly, while excitatory synaptic scaling persisted into adulthood, intrinsic homeostatic plasticity was completely absent. Finally, we found that homeostatic changes in quantal inhibitory input onto L2/3 pyramidal neurons were absent during the CP but were present in adults. Thus, the same population of neurons can express distinct sets of homeostatic plasticity mechanisms at different development stages. Our findings suggest that homeostatic forms of plasticity can be recruited in a modular manner according to the evolving needs of a developing neural circuit.
SIGNIFICANCE STATEMENT Developing brain circuits are subject to dramatic changes in inputs that could destabilize activity if left uncompensated. This compensation is achieved through a set of homeostatic plasticity mechanisms that provide slow, negative feedback adjustments to excitability. Given that circuits are subject to very different destabilizing forces during distinct developmental stages, the forms of homeostatic plasticity present in the network must be tuned to these evolving needs. Here we developed a method to induce comparable homeostatic compensation during distinct developmental windows and found that neurons in the juvenile and mature brain engage strikingly different forms of homeostatic plasticity. Thus, homeostatic mechanisms can be recruited in a modular manner according to the developmental needs of the circuit.
Keywords: critical periods, DREADDs, homeostatic plasticity, intrinsic homeostatic plasticity, synaptic scaling, visual cortex
Introduction
Homeostatic plasticity ensures the proper development of neocortical circuitry by maintaining network stability (Turrigiano and Nelson, 2004). Past work shows that neocortical pyramidal neurons use the following three main routes of homeostatic control: modulation of excitation, inhibition, and intrinsic excitability (Turrigiano, 2011). Evidence suggests that these mechanisms are sometimes simultaneously engaged (Lambo and Turrigiano, 2013; Wu et al., 2020), but at other times are not (Maffei and Turrigiano, 2008b; Barnes et al., 2015). Therefore, it remains unclear how they are coordinated within the network. Furthermore, our understandings of homeostatic mechanisms come primarily from the malleable juvenile brain, and information about how these mechanisms evolve as neural circuits mature is scarce. A major impediment to answering these questions has been the lack of a uniform activity manipulation method to induce homeostatic plasticity across development. Here we report a reliable chemogenetic approach that allows us to manipulate the same population of neurons in vivo at distinct developmental stages and use this method to characterize the developmental regulation of homeostatic plasticity within layer2/3 (L2/3) of mouse primary visual cortex (V1).
Many forms of homeostatic compensation have been described in a variety of neuronal circuits and cell types (Zhang and Linden, 2003; Marder and Goaillard, 2006; Turrigiano, 2012; Davis, 2013). In neocortical networks, synaptic and intrinsic homeostatic plasticity are widely expressed; the former adjusts synaptic inputs in the appropriate direction to compensate for changes in activity, while the latter modulates intrinsic excitability to alter the input–output relationship of the neuron (Turrigiano, 2011). These mechanisms were first identified in dissociated neuronal cultures, where activity can be easily perturbed but developmental changes are difficult to assess (O'Brien et al., 1998; Turrigiano et al., 1998; Desai et al., 1999; Burrone et al., 2002). Subsequent studies in sensory cortex used an “ex vivo” approach in which sensory deprivation was followed by acute slice recordings (Maffei and Turrigiano, 2008a; Gainey and Feldman, 2017). This allows assessments of homeostatic plasticity in intact networks, but several challenges remain. First, sensory deprivation paradigms generally induce both Hebbian and homeostatic plasticity in parallel, so disentangling one mechanism from another can be challenging. Second, the same paradigm can have fundamentally different effects on the patterns of activity reaching cortex at different developmental stages, making it difficult to directly compare homeostatic compensation across development. Recently, designer receptors exclusively activated by designer drugs (DREADDs) have emerged as an alternative for activity manipulation in vivo (Roth, 2016). Using this toolset, we can directly suppress activity in a specific cell type at particular developmental stages, and thus assess how homeostatic mechanisms are developmentally regulated.
In this study, we unilaterally expressed inhibitory DREADD hM4Di in L2/3 pyramidal neurons in monocular mouse V1 (V1m) and delivered clozapine dihydrochloride (CNO) to the animal to suppress the activity of DREADD-expressing neurons for 24 h. We recorded miniature EPSCs (mEPSCs) from both hM4Di+ neurons in the injected hemisphere and hM4Di– neurons in the control hemisphere. During the classical visual system critical period (CP), we found that hM4Di activation scaled up mEPSC amplitude and increased intrinsic excitability. Notably, DREADD-induced synaptic scaling was blocked by the C-terminal tail (C-tail) of GluA2 AMPA receptor subunit, as is pharmacologically and sensory deprivation-induced synaptic scaling (Gainey et al., 2009; Lambo and Turrigiano, 2013). Further, this same paradigm failed to induce synaptic scaling and intrinsic homeostatic plasticity in Shank3 knock-out (KO) mice, which are known to have deficits in both forms of homeostatic plasticity (Tatavarty et al., 2020). Interestingly, although activity suppression in adult V1 continued to elicit robust synaptic scaling, intrinsic homeostatic plasticity was absent. Recordings of miniature IPSCs (mIPSCs) from adult but not CP neurons revealed a reduction in frequency, suggesting that the mature cortex uses inhibitory mechanisms in lieu of intrinsic homeostatic plasticity. Our data show that the same population of neurons can engage different sets of homeostatic mechanisms at distinct developmental stages. We propose that excitatory, inhibitory, and intrinsic homeostatic plasticity mechanisms subserve modular functions, and can be turned on and off to suit distinct developmental needs.
Materials and Methods
Animals
Experiments were performed on C57BL/6J mice [both wild-type (WT) and transgenic mouse lines]. For all experiments, both males and females were used for slice physiology, either between postnatal day 24 (P24) and P29 (for juveniles) or between P45 and P55 (for adults). All animals were treated in accordance with Brandeis Institutional Biosafety Committee and Institutional Animal Care and Use Committee protocols. Specifically, they were housed on a 12 h light/dark cycle with ad libitum access to food and water except when experiments dictated otherwise (see below for details). Pups were weaned between P19 and P21 and housed with at least one littermate except when experiments required single housing. For most experiments, genetic manipulations (e.g., virus injection) were performed unilaterally so that the other hemisphere could serve as a within-animal control. The number of animals and neurons for each experiment are given in the figure legends, and individual data points represent neurons unless indicated otherwise.
Drug administration
For subcutaneous and intraperitoneal injections, CNO (Hello Bio) was dissolved in 0.9% sterile saline to reach the desired concentration (1 mg/ml). For acute DREADD activation, CNO was administered via subcutaneous injections at a dose of 5 mg/kg. For chronic DREADD activation, CNO was administered via intraperitoneal injections every 12 h, starting 24 h before slice physiology (two injections in total) at a dose of 5 mg/kg. For drinking water (DW) administrations, CNO was dissolved in water to 0.05 mg/ml, and 10 mm saccharine chloride was added to the solution before giving to the animals. All animals that underwent the drinking water paradigm were water deprived for 16 h before switching the regular water to the CNO-containing water. They were killed for slice physiology after 24 h of CNO administration unless indicated otherwise.
Virus injection
Most virus injections were performed between P14 and P19 on a stereotaxic apparatus under ketamine/xylazine/acepromazine anesthesia. For adult slice physiology experiments, virus injections were performed between P20 and P22. V1m was targeted unilaterally using stereotaxic coordinates (Allen Brain Atlas) that were proportionally adjusted according to the age-dependent bregma–lambda distance difference. Unless noted otherwise, 200–300 nl of virus were delivered into the targeted area via a micropipette. Animals that underwent surgery were allowed to recover in their home cages for a week before slice physiology experiments.
Ex vivo brain slice preparation
Animals were anesthetized with isoflurane. After a toe-pinch check, the animal was decapitated and coronal slices (300 µm) containing V1m from both hemispheres were obtained using a vibratome (model VT1000S, Leica Biosystems). Slices were first transferred to an oxygenated chamber filled with choline solution (in mm: 110 choline-Cl, 25 NaHCO3, 11.6 Na-ascorbate, 7 MgCl2, 3.1 Na-pyruvate, 2.5 KCl, 1.25NaH2PO4, and 0.5 CaCl2, with osmolarity adjusted to 310 mOsm with dextrose, pH 7.4) for recovery, and then transferred back to oxygenated standard artificial CSF (ACSF; in mm: 126 NaCl, 25 NaHCO3, 3 KCl, 2 CaCl2, 2 MgSO4, 1 NaH2PO4, and 0.5 Na-ascorbate, with osmolarity adjusted to 310 mOsm with dextrose, pH 7.4) and incubated for 40 min. Slices were used for electrophysiology 1–5 h post-slicing.
Electrophysiology
Slices containing V1 were placed on an upright epifluorescence microscope (model BX51WI, Olympus) equipped with infrared-differential interference contrast optics. To identify V1m, white matter morphology of the slice was compared with those from coronal sections illustrated in the Allen Reference Atlas (https://mouse.brain-map.org/static/atlas). Slices located between −4.5 and −3.5 mm (anterior–posterior distance from bregma) were used for recording, where the white matter generally showed a triangle-shaped morphology toward the rostral-medial area, and V1m was identified as the region above the medial tip of the triangle. L2/3 was identified as the region between 120 and 500 µm (depth from pia mater), and cells at the borders were avoided. Pyramidal neurons were visually targeted for whole-cell recordings using a 40× water-immersion objective; visual identification was based on the teardrop-shaped somata and the presence of an apical dendrite, and morphology was confirmed post hoc from biocytin fill reconstructions. DREADD+ neurons were identified by the expression of fluorescent markers. Borosilicate glass pipettes were pulled to achieve pipette resistances between 4 and 6 MΩ and were filled with K+ gluconate-based internal solution (in mm: 100 K-gluconate, 10 KCl, 10 HEPES, 5.37 biocytin, 0.5 EGTA, 10 Na-phosphocreatine, 4 Mg-ATP, and 0.3 Na-GTP, with osmolarity adjusted to 295 mOsm with sucrose and pH adjusted to 7.4 with KOH) unless otherwise noted. All recordings were performed in slices that were superfused in oxygenated standard ACSF at 34°C. Data were acquired at 10 kHz with Multiclamp 700B amplifiers and CV-7B headstages (Molecular Devices); mEPSC/IPSC data were low-pass filtered at 5 kHz, and intrinsic excitability data were not filtered. Data were acquired using the open-source MATLAB-based software WaveSurfer (Janelia Research Campus, Howard Hughes Medical Institute), and data analysis was performed using in-house scripts written in MATLAB.
mEPSC recordings.
For spontaneous mEPSC recordings, slices were superfused with standard ASCF containing a drug cocktail of tetrodotoxin (TTX; 0.1 μm), aminophosphonopentanoic acid (AP-5; 50 μm), and picrotoxin (25 μm) to isolate mEPSCs. L2/3 pyramidal neurons were targeted and held at −70 mV in whole-cell voltage clamp. Each neuron was recorded for 3–5 min in a series of 30 s segments, and a 500 ms 5 mV hyperpolarizing voltage step was administered at the beginning of each segment so that passive properties could be monitored throughout the recording. Neurons were excluded if access resistance was >20 MΩ, input resistance was <100 MΩ, membrane potential was greater than −55 mV, or these properties changed by >15% during the recording.
Intrinsic excitability measurement.
For intrinsic excitability measurements, slices were superfused with standard ACSF containing AP-5 (50 μm), picrotoxin (25 μm), and 6,7-dinitroquinoxaline-2,3-dione (DNQX; 25 μm) to block synaptic currents. L2/3 pyramidal neurons were held in current clamp with a small DC injection to maintain the resting membrane potential at −70 mV. Frequency–current (f–I) curves were obtained by delivering a series of 20 1-s-long current injections in amplitude increments of 20 pA. Passive properties were monitored in voltage clamp before and after current steps, and neurons that did not meet the criteria listed above for mEPSC recordings were excluded; in addition, neurons were excluded if the deviation of baseline potential from −70 mV was >5%.
mIPSC recordings.
For spontaneous mIPSC recordings, slices were superfused with standard ASCF containing TTX (0.1 μm), AP-5 (50 μm), and DNQX (25 μm) to isolate mIPSCs. L2/3 pyramidal neurons were held at −70 mV under voltage clamp. The internal solutions for these experiments were adjusted so that the reversal potential for Cl– was 0 mV (in mm: 120 KCl, 10 HEPES, 5.37 biocytin, 0.5 EGTA, 10 Na-phosphocreatine, 4 Mg-ATP, and 0.3 Na-GTP, with osmolarity adjusted to 300 mOsm with sucrose and pH adjusted to 7.4 with KOH), and mIPSCs were recorded as inward currents. Inclusion criteria were adjusted for these following recording conditions: neurons were excluded if access resistance was >20 MΩ, input resistance was <90 MΩ, membrane potential was greater than −60 mV, or passive properties changed during the recording by >15%.
Spontaneous firing in active slices.
To measure spontaneous firing, slices were superfused with a modified active ACSF with ionic concentrations that more closely resemble CSF and encourage network activity (in mm: 126 NaCl, 25 NaHCO3, 3.5 KCl, 1 CaCl2, 0.5 MgCl2, 0.5 NaH2PO4, and 0.5 Na-ascorbate, with osmolarity adjusted to 310 mOsm with dextrose, pH 7.4; Maffei et al., 2004). Whole-cell recordings from DREADD-expressing L2/3 pyramidal neurons were obtained in current clamp. After 5 min of recording, active ACSF containing 500 nm CNO was washed in. Passive properties were monitored before and after wash-in, as described above. Neurons were excluded if access resistance was >20 MΩ, input resistance was <100 MΩ, membrane potential was greater than −55 mV, or any property during the recording changed by >15%.
Immunostaining
For acute slices, following slice physiology recordings, slices were postfixed in 4% paraformaldehyde (PFA) overnight and transferred to 0.01 m PBS for storage before staining. For all other immunostaining experiments, animals were anesthetized with a ketamine/xylazine/acepromazine cocktail and transcardially perfused with 4% PFA. Brains were then removed and postfixed in PFA overnight before sectioning V1 into 60 µm slices. Slices were washed in 0.01 m PBS for at least 30 min (6 × 5 min) before being incubated in a blocking solution (0.1% Triton X-100, 0.05% NaN3, and 1% BSA in 0.01 m PBS) for 1 h. Blocked slices were then incubated in the same blocking solution with primary antibodies added (1:1000) at 4°C for 24 h. The following day, slices were washed for 30 min in PBS and then incubated in a solution (0.05% NaN3 and 1% BSA in 0.01 m PBS) containing secondary antibodies (1:500). Acute slices were incubated at 4°C overnight, all other slices were incubated at room temperature for 3 h. Finally, slices were washed for another 30 min before being mounted in Fluoromount G Mounting Medium (Southern Biotech), and images were obtained using a confocal microscope (model LSM880, Zeiss).
Quantification and statistical analysis
Data were analyzed using in-house scripts written in MATLAB. For each experiment, unless otherwise noted, results are reported as the mean ± SEM, and effect sizes are reported as a percentage of control; sample sizes (both the number of neurons and the number of animals), statistical tests used, and p values are given either in the corresponding Results section or in the figure legends.
mEPSC/IPSC recordings.
Mean amplitude and frequency were first calculated for each 30 s of recording, which were then averaged to give the mean value for each neuron. Rise time and decay time constants were calculated from the waveform average traces for each neuron. Rise time is defined as the time for the current to increase from 10% to 90% of the peak amplitude. For mEPSC recordings, the decay time constant (τ) is derived from a first-order exponential fit of the decay phase. For mIPSC recordings, the decay phase is fitted to a second-order exponential function, yielding a fast decay τ (decay_fast) and a slow decay τ (decay_slow), respectively. The percentage of the fast component (fast%) is calculated at the peak. To generate the cumulative distribution function (CDF) of mean amplitudes for each condition, 100 events were randomly selected from each neuron and pooled.
Intrinsic excitability recordings.
Definitions of neuronal properties and firing parameters were primarily adapted from the electrophysiology technical white paper published by the Allen Institute (https://celltypes.brain-map.org/). Briefly, mean instantaneous firing rate (IFR) is defined as the mean reciprocal of the first two interspike intervals. Spike adaptation index is defined as the sum of interspike intervals during a 300 pA current step; higher values indicate more adaptation. Latency is defined as the time difference between the current injection onset and the first spike. Spike width is defined as the width at half-maximum height for the first action potential evoked at the rheobase. Action potential voltage threshold is defined as the membrane potential when dV/dt (rate of rise) reaches 5% of the maximum dV/dt during the depolarizing phase of the first action potential evoked at rheobase; this measurement can produce more consistent estimates of threshold across cells of the same type compared with the one based on an absolute dV/dt value (Jackson et al., 2004). Afterhyperpolarization (AHP) amplitude is calculated for the first action potential evoked at rheobase, defined as the difference between the minimum membrane potential reached after the action potential and the average membrane potential between the current injection onset and the firing threshold; AHP amplitudes from different cells are normalized to the peak amplitude of the same cell.
Immunohistochemistry quantification.
To quantify the fraction of pyramidal neurons expressing virally injected hM4Di in the L2/3 of V1, we stained against NeuN and mCherry (fluorescence marker for hM4Di) for slices that showed mCherry signals. For each image, a region of interest (ROI) of 200 × 150 µm in L2/3 was selected, which was then background subtracted and thresholded, and cell somas were outlined in the NeuN channel. Subsequently, mCherry signals were measured in each identified cell soma, and cells with nonzero values of mCherry intensity were considered as hM4Di+. The fraction of hM4Di-expressing cells was calculated by taking the ratio of the number of hM4Di+ cells and the total number of cells identified. Three ROIs were selected from each animal, corresponding to regions of high expression close to the injection site, similar to those targeted for electrophysiology. To quantify the c-Fos+ cells following acute hM4Di activation, images were similarly processed as described above, and cell somas were outlined in the c-Fos channel. Eight ROIs at similar positions from both hemispheres were selected, and the number of c-Fos+ cells was reported for each ROI.
Statistical analysis.
All datasets were subjected to a normality test (Shapiro–Wilk test). For normally distributed data, an unpaired two-sample t test was used for pairwise comparison, a one-way ANOVA followed by Tukey's post hoc correction was used for multiple comparisons (n > 2 groups). For other data, a Mann–Whitney U test was used for pairwise comparison, and a Kruskal–Wallis test followed by Tukey's post hoc correction was used for multiple comparison. For distribution comparisons, a two-sample Kolmogorov–Smirnov test was used. Results were considered significant if p < 0.05 Significance symbols used in figures are defined as such: *p < 0.05; **p < 0.01; ***p < 0.001; n.s., not significant.
Data availability
All data generated in this study are included in this article and are available on request. All MATLAB scripts used in this study have been deposited at https://github.com/weiwen107/DREADD2021.
Key resource availability Key resources used in this study are summarized in Table 4.
Table 4.
Key resources
| Reagent or resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Rat anti-mCherry | Thermo Fisher Scientific | Catalog #M11217 |
| Rabbit anti-GFP | Thermo Fisher Scientific | Catalog #A-11122 |
| Rabbit anti-NeuN | Thermo Fisher Scientific | Catalog #702022 |
| Rabbit anti-c-Fos | Cell Signaling Technology | Catalog #2250 |
| Goat anti-rabbit Alexa Fluor 488 | Thermo Fisher Scientific | Catalog #A-11070 |
| Goat anti-rat Alexa Fluor 594 | Thermo Fisher Scientific | Catalog #A-11007 |
| Anti-streptavidin Alexa Fluor 488 | Thermo Fisher Scientific | Catalog #S11223 |
| Anti-streptavidin Alexa Fluor 594 | Thermo Fisher Scientific | Catalog #S11227 |
| Recombinant DNA | ||
| AAV-hSyn-DIO-hM4D(Gi)-mCherry | Addgene | Catalog #44362 |
| AAV-CaMKIIa-hM4D(Gi)-mCherry | Addgene | Catalog #50477 |
| AAV-CMV-GluA2(CT)-GFP | Lambo and Turrigiano (2013) | N/A |
| AAV-CMV-GFP | Lambo and Turrigiano (2013) | N/A |
| Experimental models: organisms/strains | ||
| Mouse: WT C57BL/6J | The Jackson Laboratory | RRID:IMSR_JAX:000664 |
| Mouse: Emx1-Cre C57BL/6J | The Jackson Laboratory | B6.129S2-Emx1tm1(cre)Krj/J RRID:IMSR_JAX:005628 |
| Mouse: Shank3B- C57BL/6J | The Jackson Laboratory | B6.129-Shank3tm2Gfng/J RRID:IMSR_JAX:017688 |
| Software | ||
| MATLAB | MathWorks | https://www.mathworks.com |
| Zen Lite | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen-lite.html |
| ImageJ | National Institutes of Health | https://imagej.nih.gov/ij/ |
| Illustrator | Adobe | https://www.adobe.com/products/illustrator.html |
| Photoshop | Adobe | https://www.adobe.com/products/photoshop.html |
N/A, Not applicable.
Results
Chronic chemogenetic activity suppression induces synaptic scaling in L2/3 pyramidal neurons during the CP
To study the developmental regulation of homeostatic plasticity mechanisms, we needed a paradigm that would allow us to induce comparable activity deprivation at different developmental stages. DREADD-mediated inhibition in vivo has been shown to induce synaptic scaling up in insular cortex (Wu and Ramos et al., 2021), so we adapted this approach for use in L2/3 pyramidal neurons in mouse V1, where we know that homeostatic and intrinsic plasticity are coinduced by visual deprivation during the classic visual system CP (Lambo and Turrigiano, 2013).
We began by validating the effectiveness of the inhibitory DREADD, hM4Di, under our experimental conditions. To target DREADDs to excitatory neurons, an adeno-associated virus (AAV) vector for Cre-dependent expression of hM4Di (AAV-hSyn-DIO-hM4Di-mCherry) was stereotaxically delivered into one hemisphere of Emx1-Cre mouse V1 between P14 and P16 (see Fig. 2A). After 7–10 d to allow the virus to express, we observed intense expression of hM4Di in L2/3 pyramidal neurons (Fig. 1A). The majority (67.3 ± 1.5%, n = 3 animals) of NeuN-labeled neurons close to the site of injection also showed hM4Di expressions (Fig. 1A); for electrophysiological recordings, we targeted neurons in the middle of the hM4Di-expressing region. To confirm that these exogenously expressed hM4Di receptors can be activated by CNO and lead to reduced neuronal firing, we used an active slice preparation (in which the Ca2+/Mg2+ ratio is adjusted to enhance spontaneous synaptic activity; see Materials and Methods) to record spontaneous firing in L2/3 pyramidal neurons before and after perfusion with active ACSF containing 500 nm CNO. As expected, CNO wash-in triggered a dramatic reduction in firing rate and a mild hyperpolarization in hM4Di+ neurons from the infected hemisphere, whereas neurons from the uninfected control hemisphere were not affected by CNO wash-in (Fig. 2C). Furthermore, to confirm that hM4Di expression in V1 reduces network activity in vivo, we administered CNO 1 h before mice were killed and transcardially perfused, V1 was then sectioned and stained against both hM4Di and c-Fos, and the number of neurons that showed detectable c-Fos in L2/3 of the hM4Di-injected (L) and the control (R) hemispheres was quantified. Consistent with our in vitro data, 1 h of DREADDs activation led to a significant reduction in the number of c-Fos+ cells in the infected hemisphere (Fig. 1B; reduction from control, 41.2 ± 7.6%; n = 4 animals).
Figure 2.
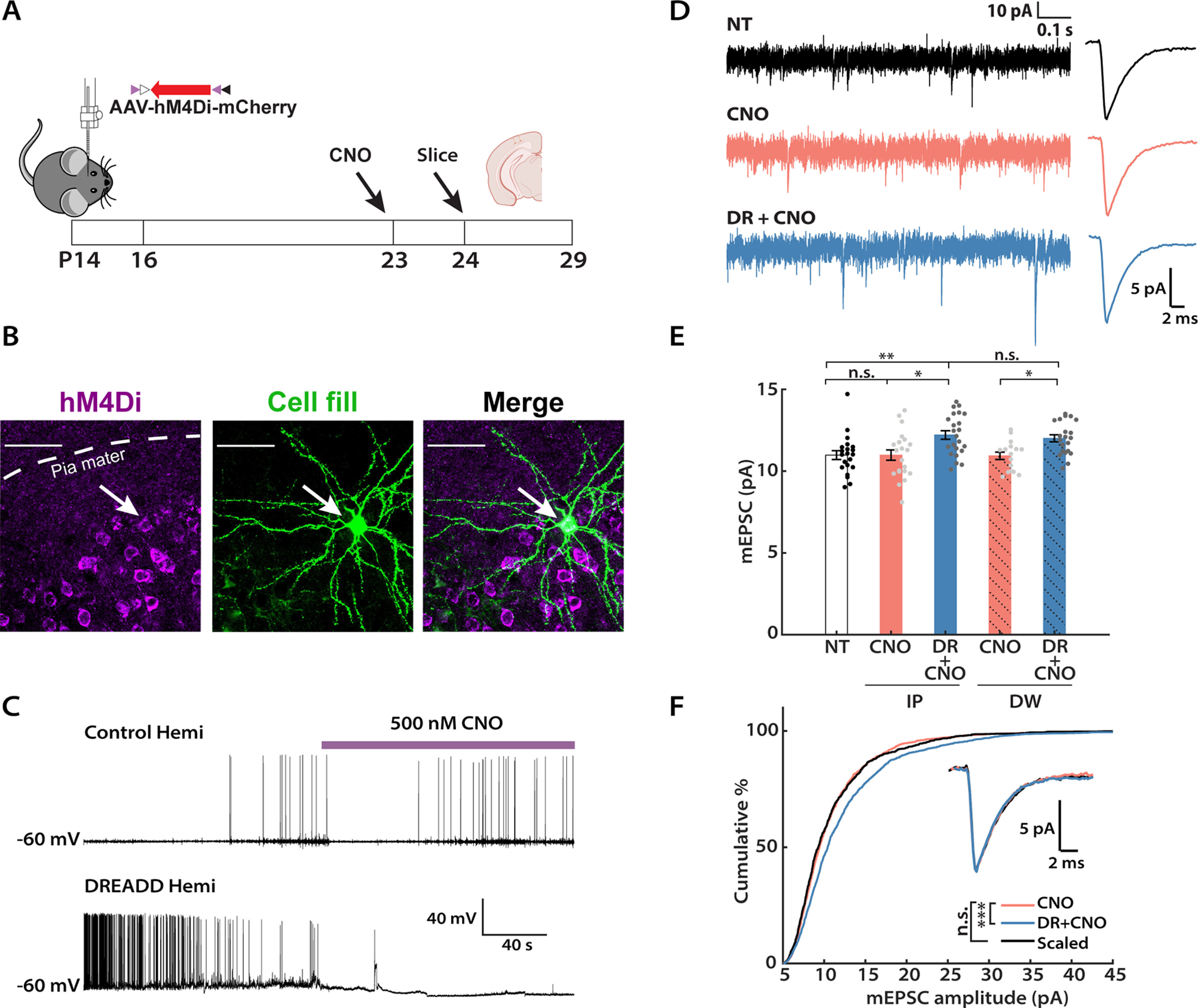
Chronic activity suppression by the inhibitory DREADD induces synaptic scaling up during the classical visual system critical period. A, Experimental timeline. Viruses carrying hM4Di were stereotaxically delivered into V1 between P14 and P16. CNO was administered by intraperitoneal injections (IP) or orally via DW for 24 h before mice were killed. Slice electrophysiology was performed between P24 and P29. Mouse and brain slice icons were adapted from BioRender.com. B, Representative images of DREADD expression and biocytin cell fill in L2/3 of mouse V1. Left, DREADD+ neurons (magenta); middle, a cell filled with biocytin during whole-cell recording (green); right, colocalization of DREADD and biocytin signals. Scale bar, 50 µm. C, Validation of hM4Di function via acute CNO perfusion. Representative current-clamp recordings of spontaneous firing from the infected hemisphere (DREADD hemi) and the uninfected hemisphere (control hemi), respectively, before and after CNO wash-in. Note the decrease in firing rate and hyperpolarization in the DREADD+ cell after CNO onset. D, Representative raw mEPSC traces and corresponding waveform averages under different conditions. CNO, Uninfected hemisphere from animals that received CNO; DR + CNO, the DREADD-expressing hemisphere from the same animal. E, Comparison of the mean mEPSC amplitude following two different methods of CNO delivery: IP (solid color) or DW (solid color + shadow). NT and two conditions from each delivery method were considered as a group, and a Kruskal–Wallis test was performed separately on each group, followed by Tukey's post hoc correction (IP: NT vs CNO, p = 0.9653; NT vs DR + CNO, p = 0.0069; CNO vs DR + CNO, p = 0.0154; DW: NT vs CNO, p = 0.9942; NT vs DR + CNO, p = 0.0084; CNO vs DR + CNO, p = 0.0214). An unpaired t test was performed on the two DR + CNO groups (p = 0.1441). F, Cumulative distributions of mEPSC amplitudes from the CNO and DR + CNO condition, respectively. Events from the DR + CNO condition were scaled according to the linear function y = 1.26× – 1.30 (black trace). Note that events from these two conditions belong to distinct distributions before scaling but are not significantly different after scaling (Kolmogorov–Smirnov test: CNO vs DR + CNO, p = 2.50E-12; CNO vs Scaled, p = 0.1550). Inset, Overlay of peak-scaled waveform average traces from all three conditions to illustrate mEPSC kinetics. Sample sizes: NT: n = 21, 5 animals; IP-CNO: n = 21, 7 animals; IP-DR + CNO: n = 23, 7 animals; DW-CNO: n = 16, 5 animals; DW-DR + CNO: n = 21, 5 animals.
Figure 1.

DREADD-mediated acute activity suppression in mouse V1. A, Representative images of DREADD expression pattern in the L2/3 of mouse V1. Left, DREADD+ neurons (violet); middle, NeuN (teal); right, overlay. Scale bar, 50 µm. B, C, Acute effects (1 h) of DREADD-mediated network activity suppression were comparable in juvenile (B, CP) and adult (C, AD) mice. Left, DREADD+ cells (magenta); middle, c-Fos signals (green); right, overlay. Bar plots, Numbers of c-Fos+ cells detected in L2/3 for both hemispheres, each data point indicates cells detected in an ROI from a single slice (unpaired t test: CP, p = 1.09E-5; AD, p = 2.05E-6). Scale bar, 50 µm. Sample sizes: CP: n = 30, 4 animals; AD, n = 28, 3 animals.
We next designed an experimental paradigm to chronically perturb neuronal activity in vivo (Fig. 2A). Inhibitory DREADDs were expressed in V1 as above, and mice received CNO for 24 h before slice electrophysiology. Slice recordings were obtained between P24 and P29, well within the classical visual system CP (Espinosa and Stryker, 2012). We compared the following two methods of CNO delivery: intraperitoneal injections (5 mg/kg; every 12 h), as described previously (Wu et al., 2021); and by inclusion of CNO in the DW (available ad libitum; 0.05 mg/ml). Under both delivery methods, the average mEPSC amplitude of hM4Di+ L2/3 pyramidal neurons [DR (DREADD) + CNO] increased significantly relative to hM4Di– neurons from the uninfected hemisphere (Fig. 2D,E; intraperitoneal injection, 111.3 ± 3.7%; DW, 109.8 ± 2.9% of control). Importantly, there was no difference between the no-treatment (NT) group (representing neurons from mice that experienced neither virus injection nor CNO administration) and the CNO group (neurons from the uninfected hemisphere of mice that received CNO), indicating that CNO administration itself did not affect mEPSC amplitude. Furthermore, when we examined the CDF of mEPSC amplitudes for the CNO and DR + CNO group, respectively, the latter showed a rightward shift toward larger amplitudes (Fig. 2F, blue vs orange). When scaled down by a multiplicative factor (Turrigiano et al., 1998; Torrado Pacheco et al., 2021), this distribution was indistinguishable from the CNO-alone distribution (Fig. 2F, black vs orange). No significant changes were observed in mEPSC frequency, kinetics, or passive properties of the recorded neurons across any conditions (Table 1). Because administration of CNO in drinking water is less invasive, we used this delivery method for all subsequent experiments unless otherwise indicated. In summary, these data demonstrate that 24 h of activity suppression by in vivo hM4Di activation can robustly induce synaptic scaling up in L2/3 pyramidal neurons during the CP.
Table 1.
Passive neuronal properties and kinetics for mEPSC experiments
| Experimental condition | Frequency (Hz) | Rin (MΩ) | Vm (mV) | Rise time (ms) | Decay τ (ms) |
|---|---|---|---|---|---|
| Figure 2 a | |||||
| NT | 4.13 ± 0.45 | 151 ± 7 | −74 ± 2 | 0.56 ± 0.02 | 2.97 ± 0.12 |
| IP: CNO | 3.32 ± 0.47 | 172 ± 12 | −77 ± 2 | 0.56 ± 0.01 | 2.72 ± 0.11 |
| IP: DR + CNO | 4.59 ± 0.41 | 176 ± 10 | −73 ± 2 | 0.56 ± 0.02 | 2.72 ± 0.09 |
| DW: CNO | 4.13 ± 0.45 | 189 ± 7 | −81 ± 1 | 0.53 ± 0.01 | 2.51 ± 0.07 |
| DW: DR + CNO | 4.19 ± 0.46 | 188 ± 7 | −79 ± 2 | 0.54 ± 0.01 | 2.42 ± 0.06 |
| Figure 3 b | |||||
| CT+ | 1.54 ± 0.18 | 184 ± 10 | −79 ± 1 | 0.57 ± 0.02 | 3.26 ± 0.16 |
| CT– | 2.33 ± 0.24 | 202 ± 7 | −82 ± 1 | 0.56 ± 0.01 | 2.79 ± 0.10 |
| GFP | 1.99 ± 0.28 | 171 ± 8 | −81 ± 1 | 0.59 ± 0.02 | 3.00 ± 0.11 |
| Figure 5 c | |||||
| Shk3 KO: NT | 4.09 ± 0.41 | 192 ± 9 | −79 ± 2 | 0.53 ± 0.01 | 2.59 ± 0.08 |
| Shk3 KO: CNO | 4.30 ± 0.37 | 206 ± 6 | −83 ± 1 | 0.51 ± 0.01 | 2.51 ± 0.06 |
| Shk3 KO: DR + CNO | 4.95 ± 0.47 | 209 ± 10 | −82 ± 2 | 0.56 ± 0.01 | 2.60 ± 0.08 |
| Figure 7 d | |||||
| NT | 2.98 ± 0.25 | 146 ± 7 | −80 ± 2 | 0.57 ± 0.01 | 3.09 ± 0.08 |
| 24 h | 3.37 ± 0.27 | 152 ± 8 | −78 ± 2 | 0.59 ± 0.01 | 3.04 ± 0.07 |
| 48 h | 1.90 ± 0.10 | 157 ± 8 | −83 ± 1 | 0.56 ± 0.02 | 2.80 ± 0.10 |
Rin, input resistance; Vm, resting membrane potential; IP, intraperitoneal injection; Shk3, Shank3.
aMultigroup comparison (NT; IP: CNO; IP: DR + CNO): frequency, p = 0.054; Rin, p = 0.2016; Vm, p = 0.4854; rise time, p = 0.8648; decay τ, p = 0.1773. Two-group comparison (DW: CNO; DW: DR + CNO): frequency, p = 0.2366; Rin, p = 0.9982; Vm, p = 0.5476; rise time, p = 0.4422; decay τ, p = 0.7535.
bMultigroup comparison (CT+, CT–, GFP): frequency, p = 0.0510; Rin, p = 0.3211; Vm, p = 0.1993; rise time, p = 0.3189; decay τ, p = 0.2015.
cMultigroup comparison (Shk3 KO: NT; Shk3 KO: CNO; Shk3 KO: DR + CNO): frequency, p = 0.4776; Rin, p = 0.345; Vm, p = 0.1637; rise time, p = 0.1724; decay τ, p = 0.8728.
dMultigroup comparison (NT, 24 h, 48 h): frequency, p = 0.053; Rin, p = 0.1849; Vm, p = 0.1780; rise time, p = 0.0615; decay τ, p = 0.057.
The GluA2 C-tail is required for DREADD-induced synaptic scaling
One of the molecular signatures of deprivation-induced synaptic scaling in neocortical neurons is a reliance on protein interactions involving the GluA2 subunit of the AMPA receptors (Wierenga et al., 2005; Gainey et al., 2009; Goold and Nicoll, 2010). Synaptic scaling induced by TTX in culture and visual deprivation in V1 can be blocked by the expression of GluA2 C-tail fragments (Gainey et al., 2009; Lambo and Turrigiano, 2013). To verify that DREADD-induced scaling in L2/3 neurons operates through the same molecular pathways as classical synaptic scaling, we expressed hM4Di (AAV-hSyn-DIO-hM4Di-mCherry) and GluA2 C-tail (AAV-CMV-GluA2CT-GFP) together (Fig. 3C, CT+) in the left hemisphere of the Emx1-Cre mouse, and hM4Di alone (Fig. 3C, CT–) in the right hemisphere to serve as a within-animal control (Fig. 3A), and recoded mEPSCs after 24 h of CNO delivery. As an additional control, we coexpressed hM4Di and GFP (AAV-CMV-GFP) in a vector that shares the same backbone as the C-tail-carrying AAV construct, in a different group of mice (Fig. 3C, GFP). In line with our expectation, neurons that expressed both hM4Di and GluA2 C-tail showed similar mean mEPSC amplitudes (Fig. 3D) to the NT and CNO groups (Fig. 2E) following 24 h of CNO treatment, indicating that these neurons failed to engage synaptic scaling to combat the DREADD-induced chronic silencing. Concomitantly, we observed normal scaling up of mEPSC amplitudes in both the CT– and GFP groups (Fig. 3D: CT–, 113.0 ± 3.2%; GFP, 110.0 ± 3.6% of control). We further compared the CDFs of mEPSC amplitude for the CT+ and CT– groups. The distribution of the CT– group was significantly shifted to the right compared with the CT+ group (Fig. 3E, pink vs blue), but, once scaled down, the distributions were indistinguishable (Fig. 3E, pink vs black). There were no significant differences in mEPSC frequency across these three groups (Table 1), and no substantial differences in input resistance, resting membrane potential, or mEPSC kinetics (Table 1). In conclusion, the DREADD-induced global increase in synaptic strength requires GluA2 C-tail interactions, clearly exhibiting a hallmark characteristic of synaptic scaling.
Figure 3.
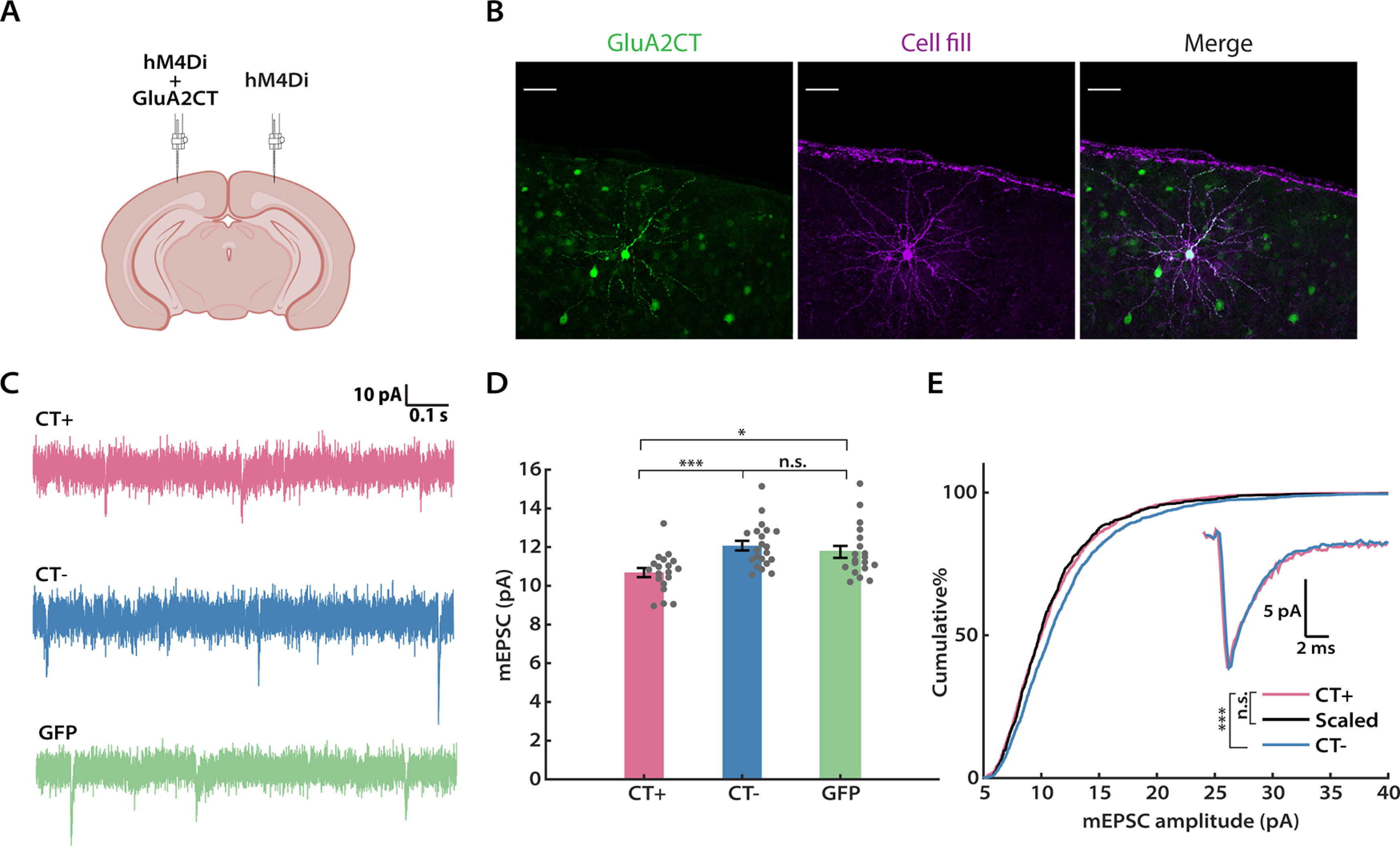
DREADD-induced synaptic scaling is blocked by the GluA2 C-tail. A, A schematic of the experimental design (adapted from icons on BioRender.com). Viruses expressing hM4Di and the GluA2 C-tail, or hM4Di alone, were delivered to the left and right hemispheres of V1, respectively. B, Representative images of GluA2 C-tail expression and biocytin cell fill from a L2/3 pyramidal neuron. C, Representation mEPSC traces from hM4Di+ neurons after CNO administration, with or without the GluA2 CT (CT+ and CT–), or GFP. Scale bar, 50 µm. D, Comparison of the mean mEPSC amplitude from each condition (Kruskal–Wallis test followed by Tukey's post hoc correction, CT+ vs CT–, p = 0.0008; CT+ vs GFP, p = 0.0388; CT– vs GFP: p = 0.4924). E, Cumulative distributions of mEPSC amplitudes for the indicated conditions. Events from the CT– condition were scaled according to the linear function y = 1.21× – 1.10 (black trace). Similarly, two distributions are not significantly different after scaling (Kolmogorov–Smirnov test: CT+ vs CT–, p = 5.28E-9; CT+ vs Scaled, p = 0.4355). Inset, Overlay of peak-scaled waveform average traces from all three conditions, to illustrate mEPSC kinetics. Sample sizes: CT+: n = 19, 7 animals; CT–, n = 21, 7 animals; GFP: n = 19, 6 animals.
DREADD-induced activity suppression increases intrinsic excitability of L2/3 pyramidal neurons during the CP
Following prolonged visual deprivation during the CP, L2/3 pyramidal neurons simultaneously engage synaptic scaling and intrinsic homeostatic plasticity to restore overall activity (Hengen et al., 2013; Lambo and Turrigiano, 2013). We therefore wished to know whether hM4Di silencing in vivo during the CP was also able to induce homeostatic changes in intrinsic excitability. To investigate this, we expressed hM4Di in Emx1-Cre mice as described above, administered CNO for 24 h (via intraperitoneal injection), then cut acute slices and obtained whole-cell current-clamp recordings from L2/3 pyramidal neurons, and generated f–I curves in the presence of synaptic blockers (Fig. 4A). Indeed, we found that the hM4Di+ neurons exhibited an upward- and leftward-shifted f–I curve, characterized by a higher mean IFR at all current steps (Fig. 4B, left). Quantification of the area under the f–I curve (Trojanowski et al., 2021) for each neuron demonstrated that this measure increased significantly following hM4Di-mediated inhibition (158.1 ± 14.4% of control), while CNO alone had no impact (Fig. 4B, right). To dissect the underlying cellular properties that could lead to such an increase, we further analyzed the firing pattern and the action potential shape of neurons from each group, respectively. In line with a higher intrinsic excitability, when compared with neurons from the other two groups, hM4Di+ neurons exhibited a lower rheobase (Fig. 4C; 81.1 ± 7.8% of control), less spike frequency adaptation (Fig. 4D; 56.5 ± 16.4% of control), and shorter latency to the first spike (Fig. 4E; 75.0 ± 6.3% of control). In contrast, we did not see any change in the threshold voltage, the amplitude of the afterhyperpolarization, or the action potential width at half-maximum (Fig. 4G–I). We further generated the waveform average trace of the first action potentials evoked at the rheobase for each group. Consistent with the above findings, action potential waveforms from CNO and DR + CNO groups were indistinguishable (Fig. 4F). Under our mEPSC recording conditions, input resistance was slightly but not significantly increased by activity suppression (Table 1); however, under these f–I recording conditions input resistance did increase significantly (Table 2), similar to our previous observations following monocular deprivation (Lambo and Turrigiano, 2013). Together, these results demonstrate that the two major forms of homeostatic plasticity, synaptic scaling and intrinsic homeostatic plasticity, work in parallel to restore neuronal excitability of L2/3 pyramidal neurons following hM4Di-induced activity suppression during the CP.
Figure 4.
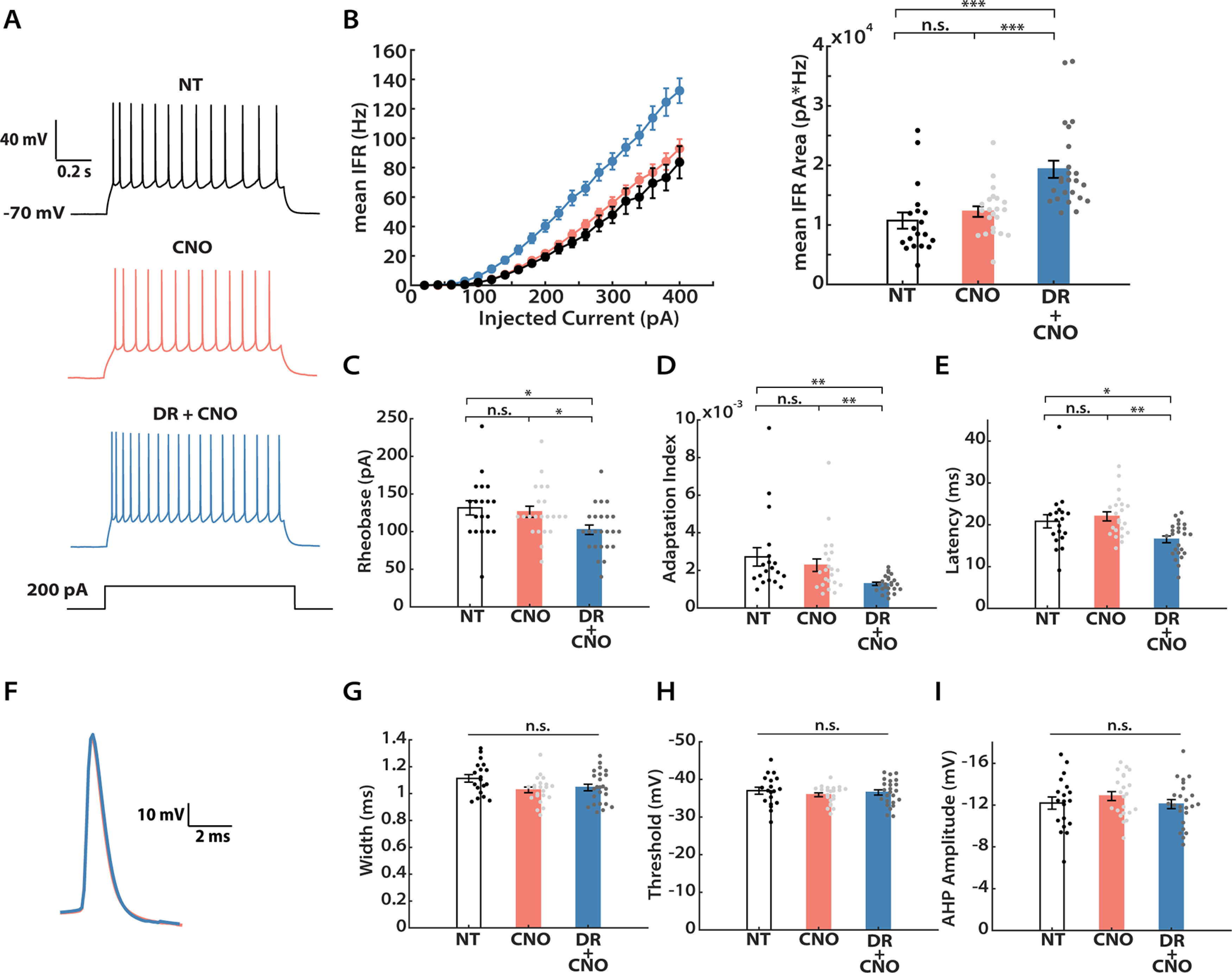
DREADD-induced chronic activity suppression increases intrinsic excitability. A, Representative recordings from L2/3 pyramidal neurons under three conditions (NT, CNO, DR + CNO) evoked by a 200 pA current injection. B, Left, f–I curves for the three conditions. The y-axis indicates mean IFR. Right, Quantification of the area under each f–I curve, calculated individually for each neuron and then averaged for each condition. A Kruskal–Wallis test followed by Tukey's post hoc correction was performed (NT vs CNO, p = 0.2063; NT vs DR + CNO, p = 1E-6; CNO vs DR + CNO, p = 1E-4). C, Mean rheobase for each condition (Kruskal–Wallis test followed by Tukey's post hoc correction: NT vs CNO, p = 0.8902; NT vs DR + CNO, p = 0.0103; CNO vs DR + CNO, p = 0.0306). D, Mean adaptation index (Kruskal–Wallis test followed by Tukey's post hoc correction: NT vs CNO, p = 0.7401; NT vs DR + CNO, p = 0.0011; CNO vs DR + CNO, p = 0.0099). E, Mean latency to the first spike for 300 pA current injection (Kruskal–Wallis test followed by Tukey's post hoc correction: NT vs CNO, p = 0.6692; NT vs DR + CNO, p = 0.0362; CNO vs DR + CNO, p = 0.0015). F, Overlay of waveform average trace for the first evoked action potentials at rheobase, from CNO and DR + CNO conditions, indicating no change in spike waveform. G, Spike widths at half-maximum height (Kruskal–Wallis test followed by Tukey's post hoc correction, p = 0.0651). H, Action potential voltage thresholds (Kruskal–Wallis test followed by Tukey's post hoc correction, p = 0.6681). I, Afterhyperpolarization (AHP) amplitudes (Kruskal–Wallis test followed by Tukey's post hoc correction, p = 0.5062). Sample sizes: NT: n = 19, 3 animals; CNO: n = 22, 7 animals; DR + CNO: n = 24, 7 animals.
Table 2.
Passive neuronal properties for intrinsic excitability experiments
| Experimental condition | Rin (MΩ) | Vm (mV) |
|---|---|---|
| Figure 4 a | ||
| NT | 141 ± 6 | −76 ± 1 |
| CNO | 148 ± 7 | −75 ± 1 |
| DR + CNO | 181 ± 10 | −74 ± 1 |
| Figure 6 b | ||
| Shk3 KO: CNO | 193 ± 12 | −80 ± 1 |
| Shk3 KO: DR + CNO | 203 ± 10 | −82 ± 1 |
| Figure 7 c | ||
| CNO (pooled control) | 143 ± 6 | −82 ± 1 |
| DR + 24 h | 165 ± 10 | −80 ± 1 |
| DR + 48 h | 162 ± 10 | −82 ± 1 |
Rin, input resistance; Vm, resting membrane potential; Shk3, Shank3.
aMultigroup comparison (NT, CNO, DR + CNO): Rin: NT versus CNO, p = 0.7491; NT versus DR + CNO, p = 0.0032; CNO versus DR + CNO, p = 0.0234; Vm: p = 0.7837.
bTwo-group comparison: Rin, p = 0.4647; Vm, p = 0.3315.
cMultigroup comparison (CNO, DR + 24 h, DR + 48 h): Rin, CNO versus DR + 24 h, p = 0.0372; CNO versus DR + 48 h, p = 0.0290; DR + 24 h versus DR + 48 h, p = 0.9966; Vm: p = 0.3465.
Shank3 knock-out mice show deficits in DREADD-induced homeostatic plasticity
We recently reported that acute knockdown of Shank3 in young cultured neocortical neurons results in simultaneous loss of synaptic scaling and intrinsic homeostatic plasticity, as well as loss of firing rate homeostasis in Shank3 KO mice (Tatavarty et al., 2020). We therefore wished to assess whether the hM4Di-mediated induction of synaptic scaling and intrinsic homeostatic plasticity are absent in these mice. To examine this possibility, we expressed hM4Di (AAV-CaMKIIa-hM4Di-mCherry) in Shank3B−/− KO animals and WT littermates, and repeated the in vivo hM4Di silencing paradigm (Peça et al., 2011; Tatavarty et al., 2020). We first compared the ability of the two groups to express synaptic scaling during the CP (Fig. 5A). CNO was administered via drinking water, and the average CNO consumption did not differ between the two groups (WT, 0.28 ± 0.01 mg; Shank3 KO, 0.29 ± 0.03 mg; unpaired t test, p = 0.8067). Analyses of the mEPSC recordings revealed that while mEPSC amplitudes of L2/3 pyramidal neurons were scaled up as expected in WT littermates (Fig. 5B, left three columns, D), there was no significant increase in Shank3 KO animals (Fig. 5B, right three columns, E). There were no significant differences in mEPSC frequency, kinetics, or passive neuronal properties in Shank3 KO animals (Fig. 5C, Table 1). Therefore, DREADD-induced synaptic scaling is absent in these animals.
Figure 5.

DREADD-induced synaptic scaling is impaired in Shank3 knock-out mice. A, Representative mEPSC traces and their waveform averages from Shank3 knockouts (Shk3 KO; right) and WT littermates (left). B, Mean mEPSC amplitude following CNO administration from WT (solid color) and Shk3 KO (solid color + shadow) littermates. Both groups underwent a Kruskal–Wallis test followed by Tukey's correction (for WT: NT vs CNO, p = 0.9942; NT vs DR + CNO, p = 0.0084; CNO vs DR + CNO, p = 0.0214; for Shank3 KO, p = 0.3720). C, Comparisons of the mean mEPSC frequency from each group (one-way ANOVA followed by Tukey's correction: WT, p = 0.2366; Shk3 KO, p = 0.4776). D, Cumulative distributions of mEPSC amplitudes from WT littermates (Kolmogorov–Smirnov test: CNO vs DR + CNO, p = 2.28E-4). E, Same as D, but for Shk3 KO animals (Kolmogorov–Smirnov test: CNO vs DR + CNO, p = 0.5456). Sample sizes: WT-NT: n = 21, 5 animals; WT-CNO: n = 16, 5 animals; WT-DR + CNO: n = 21, 5 animals; Shk3 KO-NT: n = 21, 3 animals; Shk3 KO-CNO: n = 23, 6 animals; Shk3 KO-DR + CNO: n = 23, 6 animals.
Next, we assessed intrinsic excitability (Fig. 6). Examination of the f–I curves from WT and KO littermates revealed two clear effects (Fig. 6A,B, left). First, KO neurons showed greater intrinsic excitability at baseline than WT neurons. Second, while DREADD-mediated inhibition increased the intrinsic excitability of WT neurons as expected, neurons from the KO group showed no change in intrinsic excitability. A similar pattern was observed for peak instantaneous firing rate (Fig. 6C). Furthermore, KO neurons were similar to the WT DREADD group in possessing lower rheobase (Fig. 6D; 68.6 ± 8.0% of control), shorter latency to the first spike (Fig. 6E; 67.2 ± 7.1% of control), and less spike adaptation (Fig. 6F; 63.4 ± 16.6% of control). Thus, Shank3 loss has rendered these L2/3 pyramidal neurons more excitable and has possibly occluded the normal activity-dependent change in intrinsic excitability observed in WT animals. In summary, both the global increase in mEPSC amplitudes and the shift in f–I curves that are normally induced by DREADD-mediated inhibition are absent in Shank3 KO animals.
Figure 6.
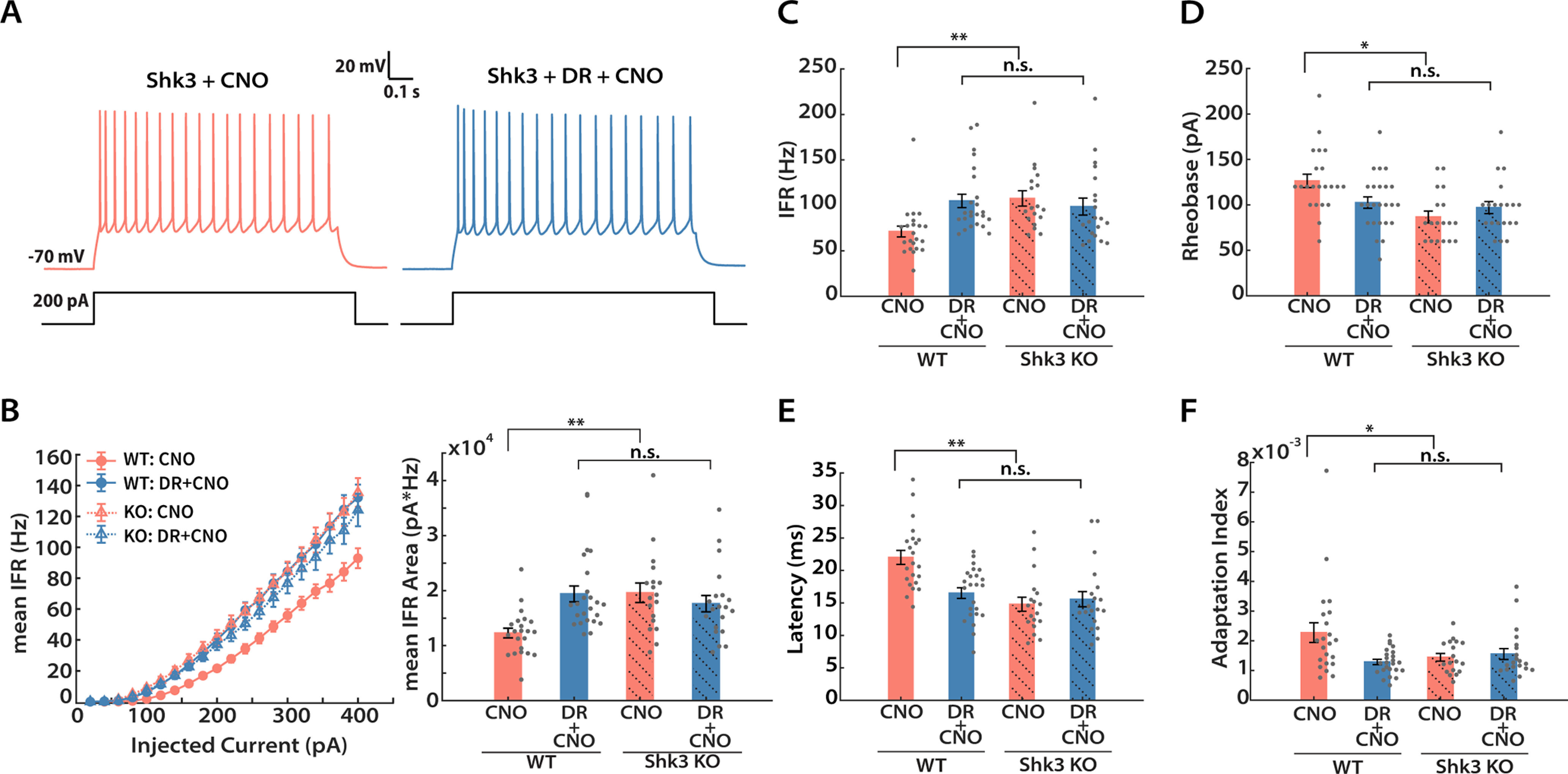
DREADD-induced intrinsic homeostatic plasticity is impaired in Shank3 knock-out mice. A, Representative recordings from Shk3 KO neurons under the indicated conditions. B, Mean IFR from WT (solid circle, solid line) and Shk3 KO (hollow triangle, dashed line) animals under DREADD manipulation (left) and quantification of areas under the curve (right). Kruskal–Wallis test followed by Tukey's correction was performed (WT vs WT + DR, p = 0.0003; WT vs Shk3, p = 0.0004; WT vs Shk3 + DR, p = 0.0113; WT + DR vs Shk3, p = 0.9934; WT + DR vs Shk3 + DR, p = 0.8657; Shk3 vs Shk3 + DR, p = 0.7619). C, Peak instantaneous firing rate at 300 pA current injection (Kruskal–Wallis test followed by Tukey's post hoc correction: WT vs WT + DR, p = 0.0008; WT vs Shk3, p = 0.0006; WT vs Shk3 + DR, p = 0.0458; WT + DR vs Shk3, p = 0.9875; WT + DR vs Shk3 + DR, p = 0.7086; Shk3 vs Shk3 + DR, p = 0.5544). D, Mean rheobase (Kruskal–Wallis test followed by Tukey's post hoc correction: WT vs WT + DR, p = 0.011; WT vs Shk3, p = 0.0008; WT vs Shk3 + DR, p = 0.0184; WT + DR vs Shk3, p = 0.2951; WT + DR vs Shk3 + DR, p = 0.8609; Shk3 vs Shk3 + DR, p = 0.7765). E, Mean latency to first spike at 300 pA current injection (Kruskal–Wallis test followed by Tukey's post hoc correction: WT vs WT + DR, p = 0.01; WT vs Shk3, p = 0.0001; WT vs Shk3 + DR, p = 0.0004; WT + DR vs Shk3, p = 0.4851; WT + DR vs Shk3 + DR, p = 0.7495; Shk3 vs Shk3 + DR, p = 0.9727). F, Mean adaptation index (at 300 pA current injection; Kruskal–Wallis test followed by Tukey's post hoc correction: WT vs WT + DR, p = 0.0244; WT vs Shk3, p = 0.0318; WT vs Shk3 + DR, p = 0.0205; WT + DR vs Shk3, p = 0.9187; WT + DR vs Shk3 + DR, p = 0.8727; Shk3 vs Shk3 + DR, p = 0.9997). Sample sizes: WT-CNO and DR + CNO: same as indicated in Figure 3; Shk3 KO-CNO: n = 20, 4 animals; Shk3 KO-DR + CNO: n = 18, 4 animals.
Developmental regulation of intrinsic homeostatic plasticity
To determine whether homeostatic plasticity is maintained into adulthood (AD), we used the same DREADD-induced inhibition paradigm in adult Emx1-Cre animals that we used previously in juveniles (Fig. 2). We again administered CNO for 24 h, then performed slice electrophysiology experiments between P45 and P55, well after the end of the classical rodent visual system CP (Espinosa and Stryker, 2012). Interestingly, we still observed a significant increase in the mean mEPSC amplitude after 24 h of CNO administration (Fig. 7A,B; 24 h, 110.1 ± 2.7% of control). We next wondered whether longer deprivation would induce stronger synaptic scaling in adult animals as it does during the CP (Lambo and Turrigiano, 2013). To investigate this possibility, we repeated these mEPSC measurements after an additional 24 h of activity suppression. Longer deprivation indeed induced a further increase in the mean amplitude (Fig. 7A,B; 48 h, 117.7 ± 3.0% of control). Similar to their younger counterparts, passive neuronal properties and mEPSC kinetics were not significantly different across conditions (Table 1). Thus, robust synaptic scaling is still present in adult V1.
Figure 7.
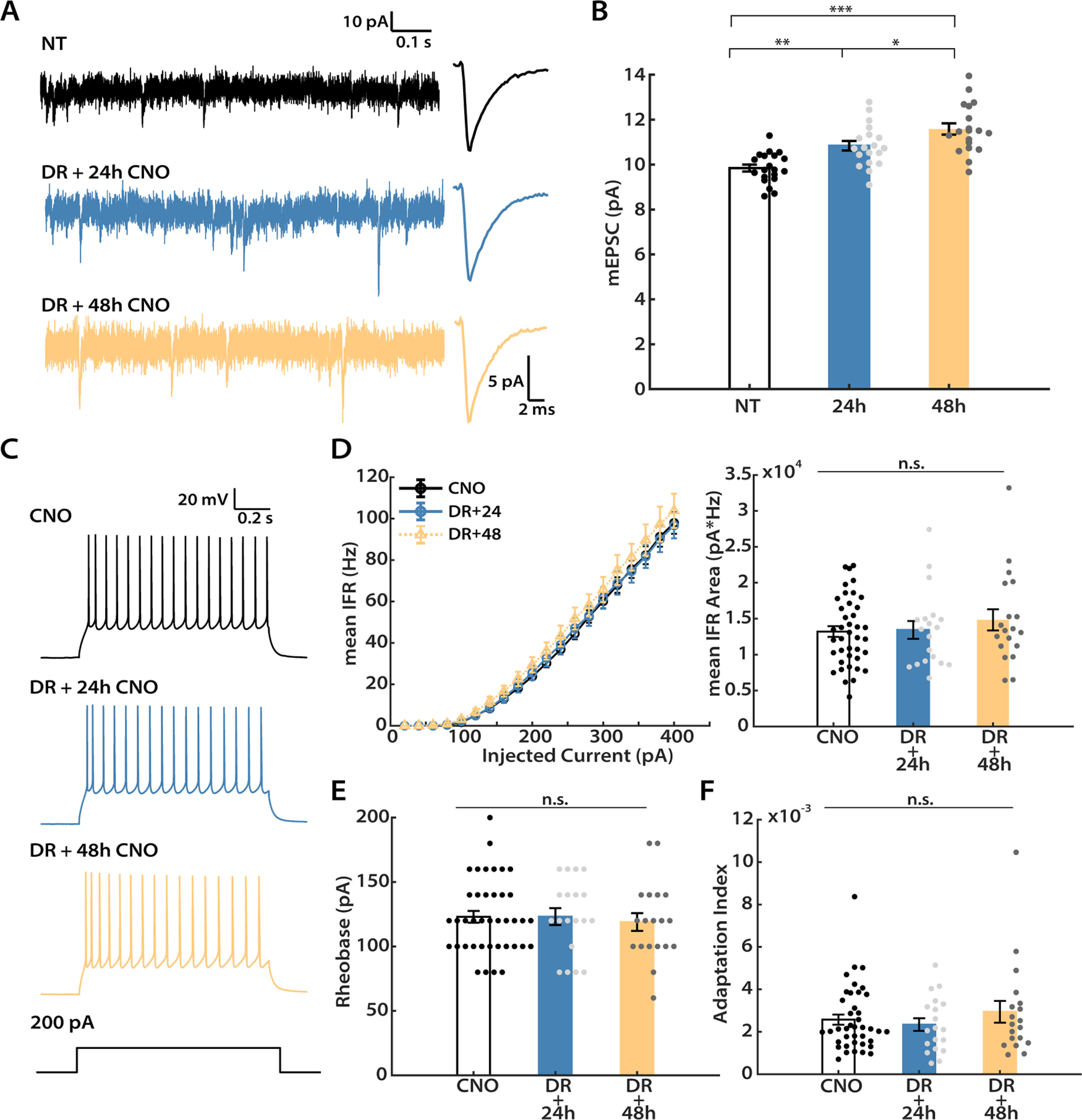
DREADD-induced synaptic scaling persists into adulthood, while intrinsic homeostatic plasticity does not. Recordings were performed on adult mice (P45 to P55) after hM4Di expression in one hemisphere, and either 24 or 48 h of CNO administration. A, Representative mEPSC traces and corresponding waveform averages from three conditions: NT (no treatment); DR + 24 h (24 h of CNO); and DR + 48 h (48 h of CNO). B, Mean mEPSC amplitude for the three conditions (one-way ANOVA followed by Tukey's post hoc correction: NT vs 24 h, p = 0.0036; NT vs 48 h, p = 6.0E-7; 24 h vs 48 h, p = 0.0393). C, Representative evoked firings from all three conditions. D, Left, f–I curves generated from neurons under each condition. The y-axis indicates the mean IFR. Right, Quantification of the area under each f–I curve (Kruskal–Wallis test followed by Tukey's post hoc correction, p = 0.7341). Data from the uninfected hemisphere after 24 and 48 h CNO were not significantly different and were pooled for the CNO condition. E, Mean rheobase (Kruskal–Wallis test followed by Tukey's post hoc correction, p = 0.8205). F, mean adaptation index evoked by 300 pA current injection (Kruskal–Wallis test followed by Tukey's post hoc correction, p = 0.8148). Sample sizes for mEPSC experiments: CNO: n = 39, 10 animals; DR + 24 h: n = 19, 6 animals; DR + 48 h: n = 19, 4 animals. Sample sizes for intrinsic excitability experiments: NT: n = 20, 3 animals; DR + 24 h: n = 19, 6 animals; DR + 48 h: n = 19, 4 animals.
Next, we probed for intrinsic homeostatic plasticity in adult animals (Fig. 7C). To our great surprise, there was no discernable change in intrinsic excitability of hM4Di+ neurons following either 24 or 48 h of activity suppression, as indicated by both the almost superimposable f–I curves (Fig. 7D, left), and the lack of change in mean area under the f–I curves (Fig. 7D, right). Cell properties that contributed to the elevated intrinsic excitability in juvenile animals, such as rheobase and spike adaptation, were also not different in adult animals at either time point (Fig. 7E,F). Interestingly, consistent with our observation in CP animals, there was also a small (∼13%) increase in input resistance under these recording conditions (Table 2). However, these changes were not sufficient to alter f–I curves or other measures of excitability in adult animals. To confirm that hM4Di expression indeed reduces V1 network activity in adults as it does in juveniles, we quantified c-Fos expression after 1 h of CNO administration as for juveniles; there was a significant and comparable reduction in the number of c-Fos+ neurons at both ages (Fig. 1B,C). Together, our data show that, while synaptic scaling is intact, intrinsic homeostatic plasticity is absent in L2/3 pyramidal neurons in adult animals.
Developmental regulation of inhibitory homeostatic plasticity
Plasticity of inhibition can play a prominent role in the adult visual cortex (Kameyama et al., 2010; Ribic, 2020). We therefore wondered whether neurons in the adult visual cortex might engage inhibitory homeostatic mechanisms in place of intrinsic homeostatic plasticity. To investigate this possibility, we recorded mIPSCs from both DREADD+ and control L2/3 pyramidal neurons following 24 h of DREADD-induced activity suppression, from both CP and adult animals (Fig. 8A,B). mIPSCs were recorded as inward currents from a holding potential of −70 mV in a symmetrical chloride solution. Interestingly, mIPSC amplitude was not affected by activity suppression at either age (Fig. 8C). In CP animals, mIPSC frequency was also unaffected by activity suppression (Fig. 8D, left two columns). In marked contrast, there was a 52% reduction in mean mIPSC frequency in adult L2/3 pyramidal neurons (Fig. 8C, right two columns), indicating that spontaneous release events are dramatically reduced following chronic activity suppression. We observed no major changes in passive neuronal properties or mIPSC kinetics for any condition (Tables 3). Together, the above results show that, following the same activity perturbation, L2/3 pyramidal neurons engage different sets of homeostatic plasticity mechanisms at distinct developmental stages. After CP closure, these neurons no longer homeostatically adjust intrinsic excitability, but instead adjust inhibition.
Figure 8.
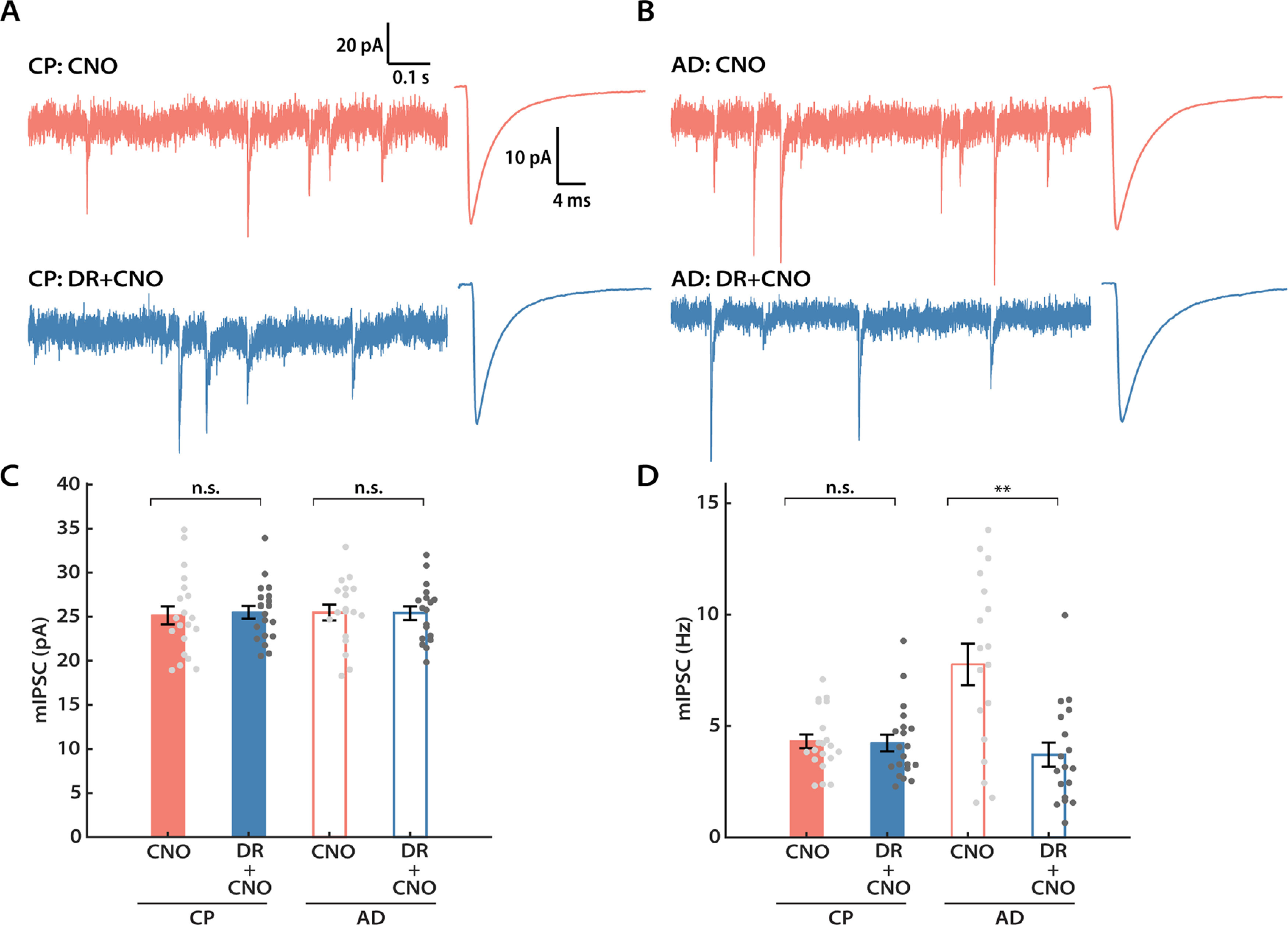
L2/3 pyramidal neurons engage inhibitory plasticity following DREADD-induced chronic silencing in AD but not during the CP. A, Representative mIPSC traces and corresponding waveform averages from the control (CNO) and DREADD (DR + CNO) conditions during CP. B, Same as A, but for adult animals. C, Mean mIPSC amplitude from both conditions for CP and AD, respectively (CP: unpaired t test, p = 0.4351; AD: unpaired t test, p = 0.8868). D, Same as C, but comparing the mean mIPSC frequency (CP: Mann–Whitney U test, p = 0.5978; AD: Mann–Whitney U test, p = 0.0029). Sample sizes: for AD experiments, both conditions, n = 18, 5 animals; for CP experiments, both conditions, n = 20, 5 animals.
Table 3.
Passive neuronal properties and kinetics for mIPSC experiments
| Experimental condition | Rin (MΩ) | Vm (mV) | Rise time (ms) | decay_fast (ms) | decay_slow (ms) | Fast (%) |
|---|---|---|---|---|---|---|
| Figure 8 a | ||||||
| Adult: CNO | 171 ± 8 | −81 ± 1 | 0.43 ± 0.01 | 3.38 ± 0.18 | 53.59 ± 8.09 | 80.2 ± 4.3 |
| Adult: DR + CNO | 152 ± 8 | −81 ± 1 | 0.45 ± 0.02 | 3.34 ± 0.16 | 21.77 ± 5.30 | 70.9 ± 3.7 |
| CP: CNO | 135 ± 8 | −82 ± 1 | 0.44 ± 0.02 | 3.61 ± 0.13 | 20.80 ± 2.10 | 74.5 ± 3.3 |
| CP: DR + CNO | 154 ± 10 | −81 ± 1 | 0.44 ± 0.01 | 3.56 ± 0.17 | 27.15 ± 5.43 | 76.6 ± 2.8 |
Rin, input resistance; Vm, resting membrane potential.
aTwo-group comparison (Adult, CP): Rin: adult, p = 0.054; CP, p = 0.1263; Vm: adult, p = 0.6647; CP, p = 0.8804; Rise time: adult, p = 0.7544; CP, p = 0.4351; decay_fast: adult, p = 0.7637; CP, p = 0.5792; decay_slow: adult, p = 0.0028; CP, p = 0.9031; Fast: adult, p = 0.6554, CP, p = 0.1080.
Discussion
While homeostatic compensation has been widely observed within sensory cortex, the developmental regulation of homeostatic plasticity remains poorly understood. Here we developed an in vivo approach (DREADD-mediated activity suppression) that allowed us to manipulate activity similarly in young and adult V1. This paradigm induced classical excitatory synaptic scaling up in L2/3 pyramidal neurons, which depended on GluA2-mediated trafficking mechanisms and the synaptic scaffold protein Shank3, as it does in vitro (Gainey et al., 2015; Tatavarty et al., 2020). We found that homeostatic compensation within L2/3 pyramidal neurons is dramatically different in CP and adult animals (Fig. 9). While excitatory synaptic scaling and intrinsic homeostatic plasticity cooperate to restore excitability during the CP, intrinsic homeostatic plasticity is absent in adults. Further, the plasticity of quantal inhibitory transmission in L2/3 neurons is absent during the CP, but is robustly recruited in adults. Our data suggest that, rather than being redundant, individual homeostatic mechanisms subserve distinct aspects of excitability maintenance, and can be turned on or off depending on the current needs of neurons and circuits.
Figure 9.

Summary illustration of the distinct homeostatic responses of L2/3 pyramidal neurons during different developmental stages. Left, During the classical visual system CP (Juvenile), neurons engage excitatory synaptic scaling and intrinsic homeostatic plasticity in response to DREADD-induced activity suppression. Right, However, while excitatory synaptic scaling persisted into adulthood, intrinsic mechanisms are replaced by inhibitory homeostatic compensations.
When compared with other in vivo homeostatic plasticity induction methods, DREADDs have several advantages. Most work on neocortical homeostatic plasticity has been limited to sensory cortex following sensory deprivation (Turrigiano, 2011; Gainey and Feldman, 2017). However, the complexity of activity changes during sensory deprivation makes it difficult to disentangle direct from indirect compensations. First, sensory deprivation induces changes along the entire sensory pathway that depend on the deprivation method, and, for many paradigms, the induction of homeostatic plasticity requires the prior induction of Hebbian plasticity (Espinosa and Stryker, 2012; Hengen et al., 2016; Torrado Pacheco et al., 2021). Since identical manipulations can produce distinct forms of Hebbian plasticity in juvenile and adult animals (Cooke and Bear, 2014; Hübener and Bonhoeffer, 2014), their impact on neocortical activity at different developmental stages is rarely equivalent, and thus the sites and time courses of homeostatic compensation may differ. DREADDs allow us to circumvent both limitations. With the ability to restrict DREADD expression to specific neuronal types in desired regions, and the direct action of DREADDs on these neurons, we can induce comparable activity changes in the same subset of neurons at different developmental timepoints. Second, the timeline of DREADD-induced homeostatic plasticity is significantly shorter. DREADD activation robustly induces homeostatic changes within 24 h; in contrast, sensory perturbations generally take longer to induce commensurate changes (Lambo and Turrigiano, 2013; Greenhill et al., 2015; Teichert et al., 2017), mainly because activity suppression in neocortex itself takes time to develop (Espinosa and Stryker, 2012; Hengen et al., 2013, 2016; Pacheco et al., 2019). Last, the flexibility of viral-mediated DREADDs expression allows the direct activity manipulation of targeted cells in nonsensory cortical and subcortical areas (Sternson and Roth, 2014), potentially expanding the scope of homeostatic plasticity research in vivo.
There are a number of technical issues to consider when adopting DREADDs for chronic activity manipulations. First, the efficacy of DREADDs depends critically on the local availability of the activating ligand. This is especially a challenge for long-term manipulations in the CNS, where ligand concentration diminishes with time after administration (Guettier et al., 2009), although some behavioral phenotypes can last for as long as 8 h (Alexander et al., 2009). Drinking water delivery has an advantage over bolus administration for chronic manipulations, as animals have a continuous intake of the ligand, but there may still be variations in ligand concentration depending on when animals drink. Despite these caveats, we found that in vivo DREADD activation via both delivery methods induced an increase in mEPSC amplitude that was comparable to that observed following visual deprivation (Desai et al., 2002; Hengen et al., 2013; Lambo and Turrigiano, 2013). Second, off-target effects of CNO or other ligands have been reported after acute CNO application (MacLaren et al., 2016; Jendryka et al., 2019), although CNO concentrations in the CSF following doses as high as 10 mg/kg are still low enough to keep the off-target effects minimal (Jendryka et al., 2019). Here we found that synaptic and intrinsic properties from our within-animal controls from the uninfected hemisphere (exposed to CNO but without DREADD expression) were not different from those of unmanipulated controls, indicating an absence of major off-target effects. Last, one might worry that activity suppression via DREADDs is nonphysiological and thus might not activate the same homeostatic mechanisms that are induced by sensory manipulations. This concern is mitigated by the observation that DREADD-induced synaptic and intrinsic homeostatic plasticity are phenotypically similar and rely on the same molecular pathways as homeostatic changes induced by pharmacological and sensory deprivation (Lambo and Turrigiano, 2013; Gainey et al., 2015; Tatavarty et al., 2020).
Ample evidence shows that sensory deprivation can induce many forms of homeostatic plasticity in a cell type-specific and layer-specific manner (Feldman, 2009; Sanes and Kotak, 2011; Turrigiano, 2011). Despite these past efforts, our understanding of the developmental regulation of homeostatic plasticity remains nascent. One major reason is that different studies have used disparate paradigms (optic nerve block, lid suture, retinal lesions, enucleation) across ages and cell types, which precludes direct comparison of results from different studies. For example, synaptic scaling induced by optic nerve block is absent in L2/3 until CP onset (Desai et al., 2002), but whether it persists into adulthood has been difficult to test, mainly because visual deprivation has fundamentally different effects on V1 activity after the CP ends (Sawtell et al., 2003; Sato and Stryker, 2008). Interestingly, monocular enucleation in adult mice induces synaptic scaling in L5, but not in L2/3, pyramidal neurons (Barnes et al., 2015). In contrast, we found robust synaptic scaling in adult L2/3 pyramidal neurons following DREADD-mediated activity suppression. This demonstrates that the ability to express synaptic scaling persists in these neurons into adulthood and suggests that enucleation either modulates the activity of L2/3 pyramidal neurons insufficiently to induce scaling, or possibly induces additional forms of plasticity that mask the homeostatic response.
Lid suture induces synaptic scaling and intrinsic homeostatic plasticity in tandem in L2/3 pyramidal neurons during the CP (Maffei and Turrigiano, 2008b; Lambo and Turrigiano, 2013), but neither L2/3 nor L5 adult pyramidal neurons express intrinsic homeostatic plasticity following bilateral retinal lesioning or monocular enucleation (Keck et al., 2013; Barnes et al., 2015). Here we could directly assess whether L2/3 pyramidal neurons lose the ability to express intrinsic homeostatic plasticity after the CP ends. While DREADD-mediated activity suppression induced robust intrinsic plasticity during the CP, it was completely absent in adult neurons, even after an extended period of activity suppression. Our data suggest that, unlike synaptic scaling, intrinsic homeostatic plasticity in L2/3 is tightly coupled to the CP. This critical window for intrinsic plasticity has also been reported in the auditory cortex (Rao et al., 2010). Therefore, intrinsic homeostatic plasticity and synaptic scaling are subject to starkly different developmental forces.
Like excitatory synapses, inhibitory synapses are also subject to homeostatic regulation. In the adult mouse V1, pyramidal neurons in both L2/3 and L5 show reduced mIPSC frequency following visual deprivation (Keck et al., 2013; Barnes et al., 2015; Gao et al., 2017), suggesting that excitatory neurons receive less overall inhibitory input. Intriguingly, we also observed a substantial decrease in the mean mIPSC frequency in adult L2/3 pyramidal neurons following DREADD-mediated activity suppression. This reduction in frequency could reflect inhibitory synapse loss, as reported in previous studies (Chen et al., 2011; Keck et al., 2011; van Versendaal et al., 2012). During the CP, visual deprivation-induced disinhibition in V1 is transient, and precedes and possibly creates a permissive environment for subsequent excitatory plasticity (Hengen et al., 2013; Kuhlman et al., 2013). In contrast, here in adult V1 we found that changes in excitation and inhibition occurred in parallel. The fact that this disinhibition is still present when excitatory scaling up has already been elicited suggests that it serves a purpose other than facilitating excitatory plasticity. Finally, we found that the homeostatic regulation of inhibition was also developmentally regulated. It was only observed in adults, when intrinsic plasticity was missing, but not in juveniles, when excitatory synaptic scaling and intrinsic plasticity were both present. These results support the idea that individual homeostatic mechanisms are modular and can be turned on and off in the same cell type at different developmental stages.
Neural circuits are endowed with a diverse repertoire of homeostatic plasticity mechanisms to maintain stability during learning and development (Turrigiano, 2011). A fundamental challenge is to resolve how these mechanisms are regulated across different developmental stages to achieve the appropriate homeostatic outcome. Past efforts to answer this question have been hindered by the lack of a consistent activity manipulation method that can induce comparable changes in the same type of neurons at different ages. Here, we report a DREADD-mediated approach to bypass this limitation. Our data show that L2/3 pyramidal neurons engage strikingly distinct sets of homeostatic mechanisms in juvenile and adult mice. These mechanisms exhibit unique developmental profiles, supporting the hypothesis that individual homeostatic mechanisms subserve specific functions and will be recruited appropriately to meet changing developmental demands.
Footnotes
This work was supported by National Institutes of Health Grant R35-NS-111562 (G.G.T.). We thank Zhe Meng for technical support, and all members of the Turrigiano laboratory for helpful discussions.
The authors declare no competing financial interests.
References
- Alexander GM, Rogan SC, Abbas AI, Armbruster BN, Pei Y, Allen JA, Nonneman RJ, Hartmann J, Moy SS, Nicolelis MA, McNamara JO, Roth BL (2009) Remote control of neuronal activity in transgenic mice expressing evolved G protein-coupled receptors. Neuron 63:27–39. 10.1016/j.neuron.2009.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes SJ, Sammons RP, Jacobsen RI, Mackie J, Keller GB, Keck T (2015) Subnetwork-specific homeostatic plasticity in mouse visual cortex in vivo. Neuron 86:1290–1303. 10.1016/j.neuron.2015.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrone J, O'Byrne M, Murthy VN (2002) Multiple forms of synaptic plasticity triggered by selective suppression of activity in individual neurons. Nature 420:414–418. 10.1038/nature01242 [DOI] [PubMed] [Google Scholar]
- Chen JL, Lin WC, Cha JW, So PT, Kubota Y, Nedivi E (2011) Structural basis for the role of inhibition in facilitating adult brain plasticity. Nat Neurosci 14:587–594. 10.1038/nn.2799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke SF, Bear MF (2014) How the mechanisms of long-term synaptic potentiation and depression serve experience-dependent plasticity in primary visual cortex. Philos Trans R Soc Lond B Biol Sci 369:20130284. 10.1098/rstb.2013.0284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW (2013) Homeostatic signaling and the stabilization of neural function. Neuron 80:718–728. 10.1016/j.neuron.2013.09.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai NS, Rutherford LC, Turrigiano GG (1999) Plasticity in the intrinsic excitability of cortical pyramidal neurons. Nat Neurosci 2:515–520. 10.1038/9165 [DOI] [PubMed] [Google Scholar]
- Desai NS, Cudmore RH, Nelson SB, Turrigiano GG (2002) Critical periods for experience-dependent synaptic scaling in visual cortex. Nat Neurosci 5:783–789. 10.1038/nn878 [DOI] [PubMed] [Google Scholar]
- Espinosa JS, Stryker MP (2012) Development and plasticity of the primary visual cortex. Neuron 75:230–249. 10.1016/j.neuron.2012.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman DE (2009) Synaptic mechanisms for plasticity in neocortex. Annu Rev Neurosci 32:33–55. 10.1146/annurev.neuro.051508.135516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainey MA, Feldman DE (2017) Multiple shared mechanisms for homeostatic plasticity in rodent somatosensory and visual cortex. Philos Trans R Soc Lond B Biol Sci 372:20160157. 10.1098/rstb.2016.0157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainey MA, Hurvitz-Wolff JR, Lambo ME, Turrigiano GG (2009) Synaptic scaling requires the GluR2 subunit of the AMPA receptor. J Neurosci 29:6479–6489. 10.1523/JNEUROSCI.3753-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainey MA, Tatavarty V, Nahmani M, Lin H, Turrigiano GG (2015) Activity-dependent synaptic GRIP1 accumulation drives synaptic scaling up in response to action potential blockade. Proc Natl Acad Sci U S A 112:E3590–E3599. 10.1073/pnas.1510754112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Whitt JL, Huang S, Lee A, Mihalas S, Kirkwood A, Lee H-K (2017) Experience-dependent homeostasis of “noise” at inhibitory synapses preserves information coding in adult visual cortex. Phil Trans R Soc B Biol Sci 372:20160156. 10.1098/rstb.2016.0156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goold CP, Nicoll RA (2010) Single-cell optogenetic excitation drives homeostatic synaptic depression. Neuron 68:512–528. 10.1016/j.neuron.2010.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenhill SD, Ranson A, Fox K (2015) Hebbian and homeostatic plasticity mechanisms in regular spiking and intrinsic bursting cells of cortical layer 5. Neuron 88:539–552. 10.1016/j.neuron.2015.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guettier J-M, Gautam D, Scarselli M, Ruiz de Azua I, Li JH, Rosemond E, Ma X, Gonzalez FJ, Armbruster BN, Lu H, Roth BL, Wess J (2009) A chemical-genetic approach to study G protein regulation of β cell function in vivo. Proc Natl Acad Sci U S A 106:19197–19202. 10.1073/pnas.0906593106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengen KB, Lambo ME, Van Hooser SD, Katz DB, Turrigiano GG (2013) Firing rate homeostasis in visual cortex of freely behaving rodents. Neuron 80:335–342. 10.1016/j.neuron.2013.08.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengen KB, Torrado Pacheco A, McGregor JN, Van Hooser SD, Turrigiano GG (2016) Neuronal firing rate homeostasis is inhibited by sleep and promoted by wake. Cell 165:180–191. 10.1016/j.cell.2016.01.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hübener M, Bonhoeffer T (2014) Neuronal plasticity: beyond the critical period. Cell 159:727–737. 10.1016/j.cell.2014.10.035 [DOI] [PubMed] [Google Scholar]
- Jackson AC, Yao GL, Bean BP (2004) Mechanism of spontaneous firing in dorsomedial suprachiasmatic nucleus neurons. J Neurosci 24:7985–7998. 10.1523/JNEUROSCI.2146-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jendryka M, Palchaudhuri M, Ursu D, van der Veen B, Liss B, Kätzel D, Nissen W, Pekcec A (2019) Pharmacokinetic and pharmacodynamic actions of clozapine-N-oxide, clozapine, and compound 21 in DREADD-based chemogenetics in mice. Sci Rep 9:4522. 10.1038/s41598-019-41088-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameyama K, Sohya K, Ebina T, Fukuda A, Yanagawa Y, Tsumoto T (2010) Difference in binocularity and ocular dominance plasticity between GABAergic and excitatory cortical neurons. J Neurosci 30:1551–1559. 10.1523/JNEUROSCI.5025-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck T, Scheuss V, Jacobsen RI, Wierenga CJ, Eysel UT, Bonhoeffer T, Hübener M (2011) Loss of sensory input causes rapid structural changes of inhibitory neurons in adult mouse visual cortex. Neuron 71:869–882. 10.1016/j.neuron.2011.06.034 [DOI] [PubMed] [Google Scholar]
- Keck T, Keller GB, Jacobsen RI, Eysel UT, Bonhoeffer T, Hübener M (2013) Synaptic scaling and homeostatic plasticity in the mouse visual cortex in vivo. Neuron 80:327–334. 10.1016/j.neuron.2013.08.018 [DOI] [PubMed] [Google Scholar]
- Kuhlman SJ, Olivas ND, Tring E, Ikrar T, Xu X, Trachtenberg JT (2013) A disinhibitory microcircuit initiates critical-period plasticity in the visual cortex. Nature 501:543–546. 10.1038/nature12485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambo ME, Turrigiano GG (2013) Synaptic and intrinsic homeostatic mechanisms cooperate to increase L2/3 pyramidal neuron excitability during a late phase of critical period plasticity. J Neurosci 33:8810–8819. 10.1523/JNEUROSCI.4502-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLaren DAA, Browne RW, Shaw JK, Radhakrishnan SK, Khare P, España RA, Clark SD (2016) Clozapine N-oxide administration produces behavioral effects in Long–Evans rats: implications for designing DREADD experiments. eNeuro 3:ENEURO.0219-16.2016. 10.1523/ENEURO.0219-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffei A, Turrigiano G (2008a) The age of plasticity: developmental regulation of synaptic plasticity in neocortical microcircuits. Prog Brain Res 169:211–223. [DOI] [PubMed] [Google Scholar]
- Maffei A, Turrigiano GG (2008b) Multiple modes of network homeostasis in visual cortical layer 2/3. J Neurosci 28:4377–4384. 10.1523/JNEUROSCI.5298-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffei A, Nelson SB, Turrigiano GG (2004) Selective reconfiguration of layer 4 visual cortical circuitry by visual deprivation. Nat Neurosci 7:1353–1359. 10.1038/nn1351 [DOI] [PubMed] [Google Scholar]
- Marder E, Goaillard J-M (2006) Variability, compensation and homeostasis in neuron and network function. Nat Rev Neurosci 7:563–574. 10.1038/nrn1949 [DOI] [PubMed] [Google Scholar]
- O'Brien RJ, Kamboj S, Ehlers MD, Rosen KR, Fischbach GD, Huganir RL (1998) Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron 21:1067–1078. 10.1016/S0896-6273(00)80624-8 [DOI] [PubMed] [Google Scholar]
- Pacheco AT, Tilden EI, Grutzner SM, Lane BJ, Wu Y, Hengen KB, Gjorgjieva J, Turrigiano GG (2019) Rapid and active stabilization of visual cortical firing rates across light–dark transitions. Proc Natl Acad Sci U S A 116:18068–18077. 10.1073/pnas.1906595116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peça J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, Lascola CD, Fu Z, Feng G (2011) Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 472:437–442. 10.1038/nature09965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao D, Basura GJ, Roche J, Daniels S, Mancilla JG, Manis PB (2010) Hearing loss alters serotonergic modulation of intrinsic excitability in auditory cortex. J Neurophysiol 104:2693–2703. 10.1152/jn.01092.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribic A (2020) Stability in the face of change: lifelong experience-dependent plasticity in the sensory cortex. Front Cell Neurosci 14:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth BL (2016) DREADDs for neuroscientists. Neuron 89:683–694. 10.1016/j.neuron.2016.01.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes DH, Kotak VC (2011) Developmental plasticity of auditory cortical inhibitory synapses. Hear Res 279:140–148. 10.1016/j.heares.2011.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Stryker MP (2008) Distinctive features of adult ocular dominance plasticity. J Neurosci 28:10278–10286. 10.1523/JNEUROSCI.2451-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawtell NB, Frenkel MY, Philpot BD, Nakazawa K, Tonegawa S, Bear MF (2003) NMDA receptor-dependent ocular dominance plasticity in adult visual cortex. Neuron 38:977–985. 10.1016/s0896-6273(03)00323-4 [DOI] [PubMed] [Google Scholar]
- Sternson SM, Roth BL (2014) Chemogenetic tools to interrogate brain functions. Annu Rev Neurosci 37:387–407. 10.1146/annurev-neuro-071013-014048 [DOI] [PubMed] [Google Scholar]
- Tatavarty V, Torrado Pacheco A, Groves Kuhnle C, Lin H, Koundinya P, Miska NJ, Hengen KB, Wagner FF, Van Hooser SD, Turrigiano GG (2020) Autism-associated Shank3 is essential for homeostatic compensation in rodent V1. Neuron 106:769–777.e4. 10.1016/j.neuron.2020.02.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teichert M, Liebmann L, Hübner CA, Bolz J (2017) Homeostatic plasticity and synaptic scaling in the adult mouse auditory cortex. Sci Rep 7:17423. 10.1038/s41598-017-17711-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrado Pacheco A, Bottorff J, Gao Y, Turrigiano GG (2021) Sleep promotes downward firing rate homeostasis. Neuron 109:530–544.e6. 10.1016/j.neuron.2020.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojanowski NF, Bottorff J, Turrigiano GG (2021) Activity labeling in vivo using CaMPARI2 reveals intrinsic and synaptic differences between neurons with high and low firing rate set points. Neuron 109:663–676.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G (2011) Too many cooks? Intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Annu Rev Neurosci 34:89–103. 10.1146/annurev-neuro-060909-153238 [DOI] [PubMed] [Google Scholar]
- Turrigiano G (2012) Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb Perspect Biol 4:a005736. 10.1101/cshperspect.a005736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB (2004) Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci 5:97–107. 10.1038/nrn1327 [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB (1998) Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391:892–896. 10.1038/36103 [DOI] [PubMed] [Google Scholar]
- van Versendaal D, Rajendran R, Saiepour MH, Klooster J, Smit-Rigter L, Sommeijer J-P, De Zeeuw CI, Hofer SB, Heimel JA, Levelt CN (2012) Elimination of inhibitory synapses is a major component of adult ocular dominance plasticity. Neuron 74:374–383. 10.1016/j.neuron.2012.03.015 [DOI] [PubMed] [Google Scholar]
- Wierenga CJ, Ibata K, Turrigiano GG (2005) Postsynaptic expression of homeostatic plasticity at neocortical synapses. J Neurosci 25:2895–2905. 10.1523/JNEUROSCI.5217-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C-H, Ramos R, Katz DB, Turrigiano GG (2021) Homeostatic synaptic scaling establishes the specificity of an associative memory. Curr Biol 31:2274–2285.e5. 10.1016/j.cub.2021.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YK, Hengen KB, Turrigiano GG, Gjorgjieva J (2020) Homeostatic mechanisms regulate distinct aspects of cortical circuit dynamics. Proc Natl Acad Sci U S A 117:24514–24525. 10.1073/pnas.1918368117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Linden DJ (2003) The other side of the engram: experience-driven changes in neuronal intrinsic excitability. Nat Rev Neurosci 4:885–900. 10.1038/nrn1248 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated in this study are included in this article and are available on request. All MATLAB scripts used in this study have been deposited at https://github.com/weiwen107/DREADD2021.
Key resource availability Key resources used in this study are summarized in Table 4.
Table 4.
Key resources
| Reagent or resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Rat anti-mCherry | Thermo Fisher Scientific | Catalog #M11217 |
| Rabbit anti-GFP | Thermo Fisher Scientific | Catalog #A-11122 |
| Rabbit anti-NeuN | Thermo Fisher Scientific | Catalog #702022 |
| Rabbit anti-c-Fos | Cell Signaling Technology | Catalog #2250 |
| Goat anti-rabbit Alexa Fluor 488 | Thermo Fisher Scientific | Catalog #A-11070 |
| Goat anti-rat Alexa Fluor 594 | Thermo Fisher Scientific | Catalog #A-11007 |
| Anti-streptavidin Alexa Fluor 488 | Thermo Fisher Scientific | Catalog #S11223 |
| Anti-streptavidin Alexa Fluor 594 | Thermo Fisher Scientific | Catalog #S11227 |
| Recombinant DNA | ||
| AAV-hSyn-DIO-hM4D(Gi)-mCherry | Addgene | Catalog #44362 |
| AAV-CaMKIIa-hM4D(Gi)-mCherry | Addgene | Catalog #50477 |
| AAV-CMV-GluA2(CT)-GFP | Lambo and Turrigiano (2013) | N/A |
| AAV-CMV-GFP | Lambo and Turrigiano (2013) | N/A |
| Experimental models: organisms/strains | ||
| Mouse: WT C57BL/6J | The Jackson Laboratory | RRID:IMSR_JAX:000664 |
| Mouse: Emx1-Cre C57BL/6J | The Jackson Laboratory | B6.129S2-Emx1tm1(cre)Krj/J RRID:IMSR_JAX:005628 |
| Mouse: Shank3B- C57BL/6J | The Jackson Laboratory | B6.129-Shank3tm2Gfng/J RRID:IMSR_JAX:017688 |
| Software | ||
| MATLAB | MathWorks | https://www.mathworks.com |
| Zen Lite | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen-lite.html |
| ImageJ | National Institutes of Health | https://imagej.nih.gov/ij/ |
| Illustrator | Adobe | https://www.adobe.com/products/illustrator.html |
| Photoshop | Adobe | https://www.adobe.com/products/photoshop.html |
N/A, Not applicable.