

Abstract
Objective:
We tested the hypothesis that both chronic and acute inflammatory processes contribute to worse reperfusion injury and stroke outcome in an experimental model of T2DM.
Materials and Methods:
Twelve- to thirteen-week-old male Zucker Diabetic Fatty (ZDF) rats vs. Zucker Lean Controls (ZLC) rats were tested at baseline and after middle cerebral artery occlusion (ischemia) and reperfusion (I–R). Neutrophil adhesion to the cerebral microcirculation, neutrophil expression of CD11b, infarction size, edema, neurologic function, sICAM, and cerebral expression of neutrophil–endothelial inflammatory genes were measured.
Results:
At baseline, CD11b and sICAM were significantly increased in ZDF vs. ZLC animals (p < 0.05). After I–R, significantly more neutrophil adhesion and cell aggregates were observed in ZDF vs. ZLC (p < 0.05); infarction size, edema, and neurologic function were significantly worse in ZDF vs. ZLC (p < 0.05). CD11b and sICAM-1 remained significantly increased in ZDFs (p < 0.05), and cerebral expression of IL-1β, GRO/KC, E-selectin, and sICAM were significantly induced in ZDF, but not ZLC groups (p < 0.05) after 2.5 hours of reperfusion.
Conclusion:
Both sides of the neutrophil–endothelial interface appear to be primed prior to I–R, and remain significantly more activated during I–R in an experimental model of T2DM. Consequently, reperfusion injury appears to play a significant role in poor stroke outcome in T2DM.
Keywords: stroke, type 2 diabetes, Zucker Diabetic Fatty rat, reperfusion injury, neutrophil
INTRODUCTION
Stroke remains a leading cause of death and the leading cause of adult disability in the United States [38]. Individuals with diabetes are at higher risk for stroke occurrence, and experience increased mortality and disability than those without diabetes [1,2]. The incidence of T2DM is rising in the US as obesity increases, leading to projections that one in three adults will have T2DM by 2050 [3]. Thus, increased efforts that examine mechanisms responsible for poor stroke outcomes in T2DM are warranted.
In the absence of diabetes, acute ischemic stroke and reperfusion lead to activation of systemic neutrophils and the vascular endothelium at the site of injury. Upon reperfusion, adherence of activated neutrophils to the injured endothelium initiates a cascade of events leading to additional damage in the microvasculature and surrounding brain tissue [28,34,40,42,43]. It is well documented that soluble inflammatory mediators capable of activating neutrophils and vascular endothelium are chronically increased in T2DM [5–7,15]. Thus, as a result of chronic inflammatory processes occurring at the blood–vascular interface, it seems likely that reperfusion injury following stroke could be significantly more severe in T2DM and may contribute to poor stroke outcome in this population. However, to our knowledge, this postulate has not previously been tested in an experimental model of T2DM, and is the major aim of this study.
Studies indicate that the ZDF rat model of T2DM mimics the metabolic and chronic inflammatory abnormalities observed in humans with T2DM [32,33,44–46]. Chronic systemic inflammation [13,24,32,46] and pro-oxidative and pro-coagulant states [4,10,32,44] have been described in the ZDF. Limited studies in the heart [21,22] and other organs [20] indicate that activation of inflammatory pathways contribute to worse outcomes after I–R in the ZDF model. No studies have examined I–R injury in the brain in the ZDF.
Neutrophil adhesion in the microcirculation is a marker for I–R injury [34,36,37]. To determine whether increased neutrophil adhesion in the cerebral microcirculation, and thus, reperfusion injury is associated with worse stroke outcome in T2DM, male ZDF rats were subjected to MCAO-R. The cerebral microcirculation following MCAO-R was directly observed, so that neutrophil accumulation during early reperfusion could be compared with non-diabetic controls (ZLC). Effects of the diabetic milieu on cellular activation on both sides of the neutrophil–endothelial interface were explored, and downstream effects of diabetes on brain injury were assessed, as measured by edema and infarction volume and neurologic function. Specifically, we postulated that neutrophil accumulation during reperfusion would be significantly worse in animals with T2DM due to increased activation of neutrophils and the vascular endothelium prior to and following ischemia, and that these changes would be associated with increased brain injury.
METHODS
Animal Model of T2DM
All animal procedures were reviewed and approved by the University of Arizona Institutional Animal Care and Use Committee. Male ZDF (fa/fa) rats vs. aged-matched lean littermates, ZLC (fa/−) rats (Charles River Laboratories International, Wilmington, MA, USA), were studied. The ZDF rat is obese and insulin resistant due to a homozygous point mutation in the gene for the leptin receptor, and hyperglycemic due to a genetic defect in the pancreatic beta cell [14]. In contrast, heterozygous ZLC rats are lean and without evidence of metabolic defects [23]. All animals were studied at 12–13 weeks of age, well after the onset of hyperglycemia in the ZDF. At 12–13 weeks of age, ZDF rats are not hypertensive [4,21,45], making this model suitable for studies on inflammation in the absence of this potential confounder. Rats were housed under controlled conditions with 12 hour light/dark cycles and 22–24°C temperature. All rats were placed in standard hanging cages (two per cage), and were fed Purina 5008 (LabDiet), a 6% fat rodent diet, ad libitum. Animal weights and blood glucose levels (mg/dL) (AccuCheck, Roche Diagnostics, Indianapolis, IN, USA) were periodically measured in all animals prior to experimentation. In a subset of ZDF and ZLC animals, blood pressure was measured periodically over a two-week period prior to MCAO-R in non-anesthetized animals via tail cuff (Coda 6; Kent Scientific Corp., Torrington, CT, USA). In addition, immediately after the induction of anesthesia in rats undergoing MCAO-R, non-fasting baseline blood samples were obtained via jugular venipuncture using a 23-gauge needle and syringe containing sodium citrate to assess triglycerides (mg/dL) (CardioCheck, Indianapolis, IN, USA), platelet, and total WBC counts (thousands/mm3) (Beckman Coulter AcT5diff, Brea, CA, USA) and manual determination of differential WBC counts (%). Total neutrophil count was calculated as (total WBC count × % neutrophils)/100 (thousands/mm3).
Middle Cerebral Artery Occlusion and Reperfusion
MCAO/R was surgically induced using the intraluminal filament method as previously described [12,34,40] to compare responses in ZDF vs. ZLC animals. Briefly, animals were anesthetized and a laser doppler flow meter probe (Perimed, North Royalton, OH, USA) was affixed to the skull over the middle cerebral artery territory to measure cerebral blood flow. A 4–0 nylon filament was advanced to the middle cerebral artery. The animal was included in the study if blood flow decreased ≥75% after filament placement. After occlusion was verified, the filament was secured, the neck incision was closed, and the animal was allowed to recover. After two hours of ischemia, the neck incision was reopened and the intraluminal filament was withdrawn to initiate reperfusion.
Direct Observation of the Cerebral Microcirculation
Animals underwent the MCAO-R procedure followed by direct observation of the cerebral microcirculation during reperfusion (n = 3 animals/group) as previously described [12,34,48]. Briefly, anesthetized animals were intubated and mechanically ventilated. Catheters were placed in the tail artery and vein for monitoring arterial pressure and blood gases and for delivering reagents, respectively. The intraluminal filament was then placed to induce focal cerebral ischemia. Fifteen minutes before reperfusion, rhodamine 6G (0.01%) (Sigma-Aldrich, St. Louis, MO, USA) was injected intravenously to label neutrophils. Immediately prior to reperfusion, a craniotomy was performed to expose the microcirculation on the surface of the ischemic cortex. Reperfusion was then initiated by removal of the filament, the dura was opened, and the animal was placed on a microscope stage. The brain surface was continuously superfused with aCSF. Using fluorescence video microscopy (Hamamatsu, Bridgewater, NJ, USA and Zeiss, Thronwood, NY, USA), cell adhesion to cerebral venules was recorded (Mitsubishi U82) during reperfusion. At these time points, at least six postcapillary venules in each animal were randomly selected and videotaped. On video playback, neutrophil adhesion, cell aggregates, and shear rate were quantified by a blinded observer. Neutrophil adhesion (single cells and cells in aggregates) were expressed as the cell number/100 μm venule. To measure cell aggregates, templates of different aggregate sizes were developed based on the area of a single neutrophil. The number of cells in an aggregate was determined by overlaying the template on the video monitor. Aggregates were also further categorized as small (2–5 cells), medium (5–10 cells), or large (>10 cells). Shear rate was calculated as 8 (Vwbc/D), where Vwbc is center neutrophil velocity/1.6, and D is venule diameter.
Neurologic Evaluation
A neurologic evaluation was performed after two hours of ischemia and after four hours of reperfusion. Animals were scored in four categories: level of consciousness, spontaneous circling, front limb symmetry, and front limb paresis. Each category was scored from 0 to 3 (the greater the score, the more severe the impairment). The scores in each category were totaled to yield a total neurologic score. A total score of 6–8 indicates a moderate impairment, and a score of 9–10 or greater indicates a severe impairment [40]. If, after two hours of ischemia, an animal had a total neurologic score of <6 or >10, it was not included in the study.
Histologic Assessment of Brain Injury
Cerebral infarction volume and edema were determined in MCAO-R animals (n = 5 animals/group) as previously described [12,35,40] except that assessment was done at four hours rather than the standard 24 hours of reperfusion. This non-standard reperfusion time point was, of necessity, chosen for comparison with non-diabetic ZLC controls, as no ZDF animal survived beyond four hours of reperfusion in preliminary experiments. After four hours of reperfusion, whole brains were sectioned into seven 2-mm coronal slices and stained with 2% TTC. Infarction volume, corrected for edema, was calculated as the difference between the volume of the contralateral hemisphere and the non-infarcted volume of the ipsilateral hemisphere, and expressed as a percent of the contralateral hemisphere. Edema (%) was calculated as: [(ipsilateral hemisphere volume) − (contralateral hemisphere volume)/(contralateral hemisphere volume)] × 100.
Flow Cytometry
Whole blood was analyzed for CD11b adhesion molecule expression, a measure of neutrophil activation, using methods previously described [16,31,35] (n = 5 animals/group). Briefly, blood was withdrawn from the jugular vein of anesthetized animals prior to MCAO and 15 minutes after reperfusion. Whole blood was diluted with bovine serum albumin (Sigma, St. Louis, MO, USA) in sodium azide staining buffer (BD Biosciences-Pharmingen, San Jose, CA, USA) and an Fc receptor-blocking agent (anti-CD32 monoclonal antibody, 5 mg/mL) to decrease non-specific antibody binding. Whole blood was divided into aliquots and incubated with Phycoerythrin-Cy5-conjugated anti-CD45 antibody (BD Biosciences-Pharmingen), a pan-leukocyte marker, a fluorescein isothiocyanate (FITC)-conjugated anti-CD11b antibody (BD Biosciences-Pharmingen) for detection of activated neutrophils, or a non-fluorescent IgG isotype control antibody to measure background fluorescence. Following incubation, samples were fixed with 1% paraformaldehyde. Data were acquired using a FACS Calibur flow cytometer (BD Biosciences-Pharmingen). Neutrophils, identified by positive fluorescence for CD45 antibody and selected from the total leukocyte population by cell size and granularity, were assessed for CD11b expression. A total of 5000 cells were analyzed. Data are expressed as TFI, which is the product of % gated and the geometric mean of fluorescence, after subtraction of the fluorescence from unlabeled IgG antibody samples.
Real-Time RT-PCR Analysis of Cerebral Gene Expression
Brains were quickly removed from ZLC and ZDF rats (n = 3 animals/group) either at baseline or 2.5 hours after reperfusion in animals subjected to MCAO-R, divided into hemispheres, flash-frozen in liquid nitrogen, and stored at −70°C. Total RNA was extracted from each hemisphere using TRI® Reagent (Sigma-Aldrich, St. Louis, MO, USA) followed by 2.5 M lithium chloride precipitation. RNA was run on a 1% agarose gel to check for purity and integrity. Total RNA (250 ng) was reverse transcribed (iScript; BioRad, Hercules, CA, USA). Changes in expression levels of selected genes were detected by TaqMan real-time RT-PCR analysis using rat-specific primers for IL-10β (Rn00580432_m1), TNF-α (Rn00562055_m1), GRO/KC (Rn00578225_m1), ICAM (Rn00564227_m1), endothelial cell selectin (E-selectin) (Rn00594072_m1), eNOS (Rn02132634_s1), iNOS (Rn00561646_m1), and an 18S primer for use as an internal control (Hs99999901_s1) (Applied Biosystems, Foster City, CA, USA). Data were analyzed using the comparative cycle threshold (Ct) method as a means of relative quantitation of gene expression, normalized to the endogenous reference (18S RNA), and expressed relative to a calibrator (normalized Ct value obtained from an appropriate baseline ZDF or ZLC control brain) and as 2−ΔΔCt, as described by the manufacturer (Applied Biosystems). Changes in gene expression in the ipsilateral (injured) hemisphere in either ZDF or ZLC, is expressed as fold-increase over gene expression in nonischemic (baseline) hemisphere for each strain [11].
Measurements of sICAM
sICAM-1 was determined in ZLC or ZDF rats at baseline and after 2.5 hours of reperfusion in MCAO-R rats using a commercial rat-specific ELISA (R&D Systems, Minneapolis, MN, USA) (n = 5/group). The minimum detectable dose of sICAM ranged from 1.2 to 4.1 pg/mL. Brain sICAM (CD54) immunocytochemistry was performed as previously described [21]. Briefly, brains were harvested from normal ZDF or ZLC animal (n = 1), cut in 2 mm sections, and immediately frozen in tissue matrix (OCT) prior to ) −70°C storage. Five-micron cryosections were fixed in cold acetone, blocked in 3% BSA with 6% H2O2, and incubated overnight with anti-CD54 monoclonal antibody (BD Biosciences-Pharmingen). Antigen–antibody complex was detected by serial incubations with anti-mouse biotinylated anti-Ig antibody, streptavidin–HRP, and DAB as directed by the manufacturer (anti-mouse HRP-DAB Kit, BD Sciences). Brain sICAM expression was quantified using an analysis macro thresholded on HRP-stained pixels, which converted pixels to square micrometers as previously described. sICAM stained area was assessed in 25 randomly selected fields per section, and then averaged per animal. Specificity of staining was determined by comparison with control slides incubated without primary antibody.
Statistical Analysis
All values are expressed as mean ± SEM. Analysis of neutrophil adhesion in the microcirculation and neutrophil CD11b expression between ZLC and ZDF groups over time were performed using ANOVA with post hoc analysis. For all other experiments, comparisons between ZDF and ZLC groups were performed using Student’s t-test (SigmaStat 3.0, San Jose, CA, USA). Differences were considered significant at p ≤ 0.05.
RESULTS
Baseline Physiologic Parameters in ZDF vs. ZLC Rats
At baseline prior to MCAO-R, body weight, glucose, and triglycerides were significantly greater in ZDF animals than in ZLC animals (p ≤ 0.05) (n = 8–13) (Table 1). The number of circulating total WBC, neutrophils, and platelets were each also significantly greater in ZDF animals when compared with ZLC animals (p ≤ 0.05). In contrast, MAP in non-anesthetized animals did not differ between ZLC and ZDF rats (130 ± 5 vs. 129 ± 5 mmHg, respectively).
Table 1.
Physiologic variables at baseline
| ZLC | ZDF | |
|---|---|---|
| Weight (g) | 347.6 ± 12.1 | 467.5 ± 23.1* |
| Glucose (mg/dL) | 140.3 ± 11 | 390.3 ± 24* |
| Triglycerides (mg/dL) | 54.4 ± 4.0 | 452.3 ± 47.7* |
| Platelets (thsn/mm3) | 433.3.± 17.9 | 554.0 ± 8.0* |
| Total WBC (thsn/mm3) | 7.4 ± 0.6 | 9.0 ± 0.8* |
| Neutrophils | ||
| WBC (%) | 20 ± 1.6 | 30 ± 2.5* |
| Total (thsn/mm3) | 1.5 ± 0.7 | 2.9 ± 0.4* |
ZLC, Zucker Lean Control; ZDF, Zucker Diabetic Fatty; WBC, white blood cell. Measure of variance: SEM.
p ≤ 0.05 vs. ZLC (n = 8–13).
Neutrophil Adhesion and Shear Rate in the Cerebral Microcirculation
During the microcirculation experiments, physiologic measures did not differ between groups (ZLC: MAP 92 ± 10.5 mmHg, RT 36.8 ± 1.2C, arterial pO2140.3 ± 20.5 mmHg, pCO2 40.0 ± 6.2 mmHg; ZDF: MAP 96 ± 9.1 mmHg, RT 37.7 ± 0.9C, arterial pO2 156.8 ± 18.7 mmHg, pCO2 39.4 ± 7.1 mmHg). Artificial CSF gases and temperature were within normal ranges, and also did not differ between groups.
Representative still-frame photographs from the video-tape of the cerebral microcirculation are presented in Figure 1. Significantly greater neutrophil adhesion was observed in the cerebral microcirculation in ZDF animals (E,F) compared with ZLC animals (B,C). Extravasated, non-bound fluorescent dye was often observed outside venules in ZDF animals (D), but was rarely observed in ZLC animals (A). Immediately upon reperfusion (0–15 minutes), total neutrophil adhesion was twofold higher in ZDF animals compared with ZLC animals (5.9 ± 0.5 vs. 12.5 ± 0.8 neutrophils adhered/100 μm venule, respectively) (p ≤ 0.05) (Figure 2). This significant increase in neutrophil adhesion in ZDF vs. ZLC animals persisted throughout the first 30 minutes of reperfusion (6.9 ± 0.6 vs. 10.2 ± 1.0 neutrophils adhered/100 μm venule, respectively) (p ≤ 0.05) (Figure 2). In contrast to the single neutrophil adhesion or small aggregates in the ZLC, the cell adhesion in ZDF animals occurred as large neutrophil cell aggregates that also contained platelets (Figure 3). Blood flow velocities (VWBC) were approximately 38% lower in ZDF animals compared with ZLC (p ≤ 0.05) (Table 2). As vessel diameters did not differ between groups, shear rates in ZDF animals were also decreased by more than one-third when compared with non-diabetic ZLC rats (p ≤ 0.05) (Table 2).
Figure 1.
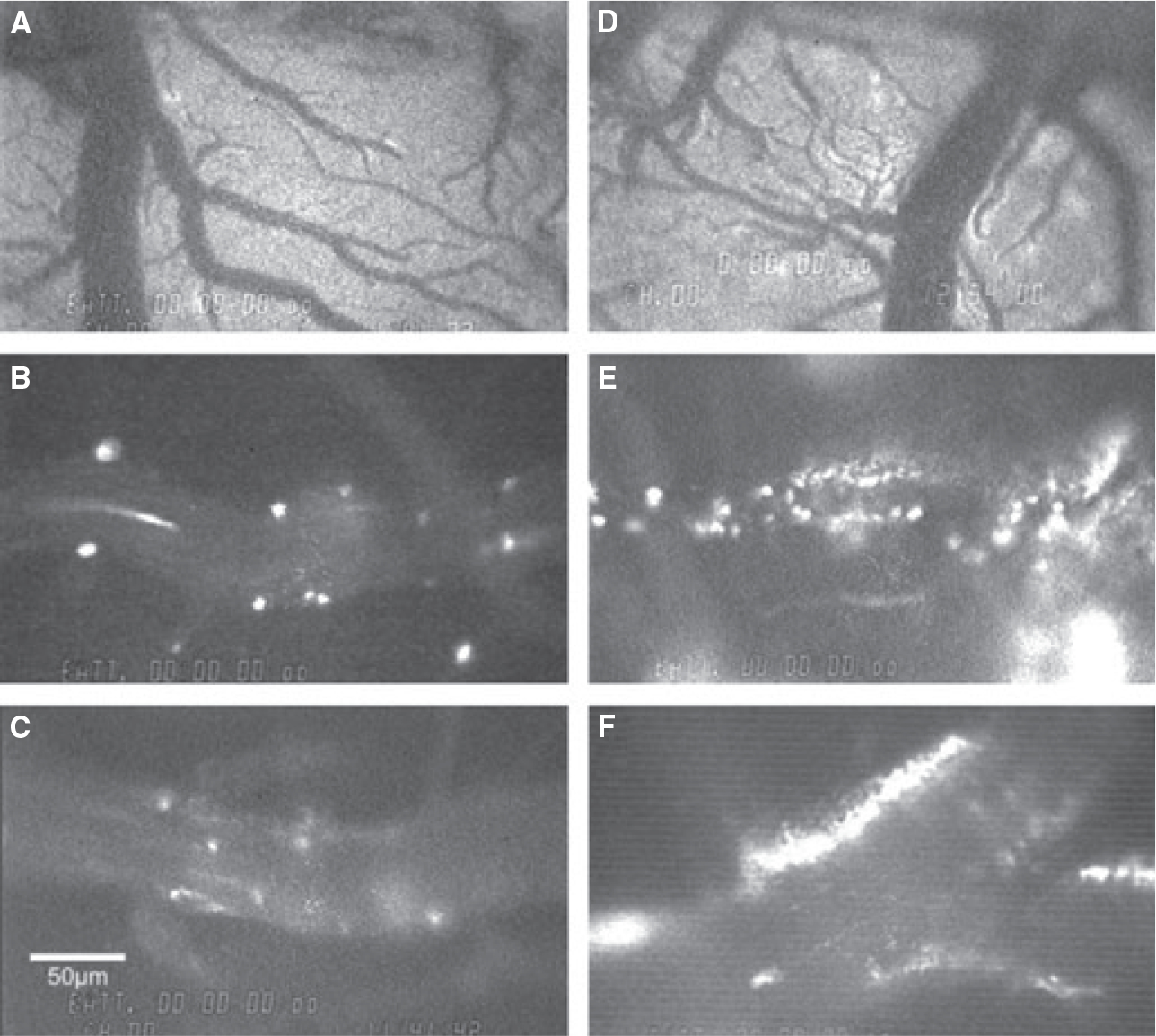
Representative images of the cerebral microcirculation in ZLC (A–C) and ZDF (D–F) animals during reperfusion, and during the first 30 minutes of reperfusion following MCAO-R. (A, D) Low power images of the microcirculation observed within the cranial window. (B–F) High power images of neutrophil adhesion to cerebral venules. Scale bars = 50 μm.
Figure 2.
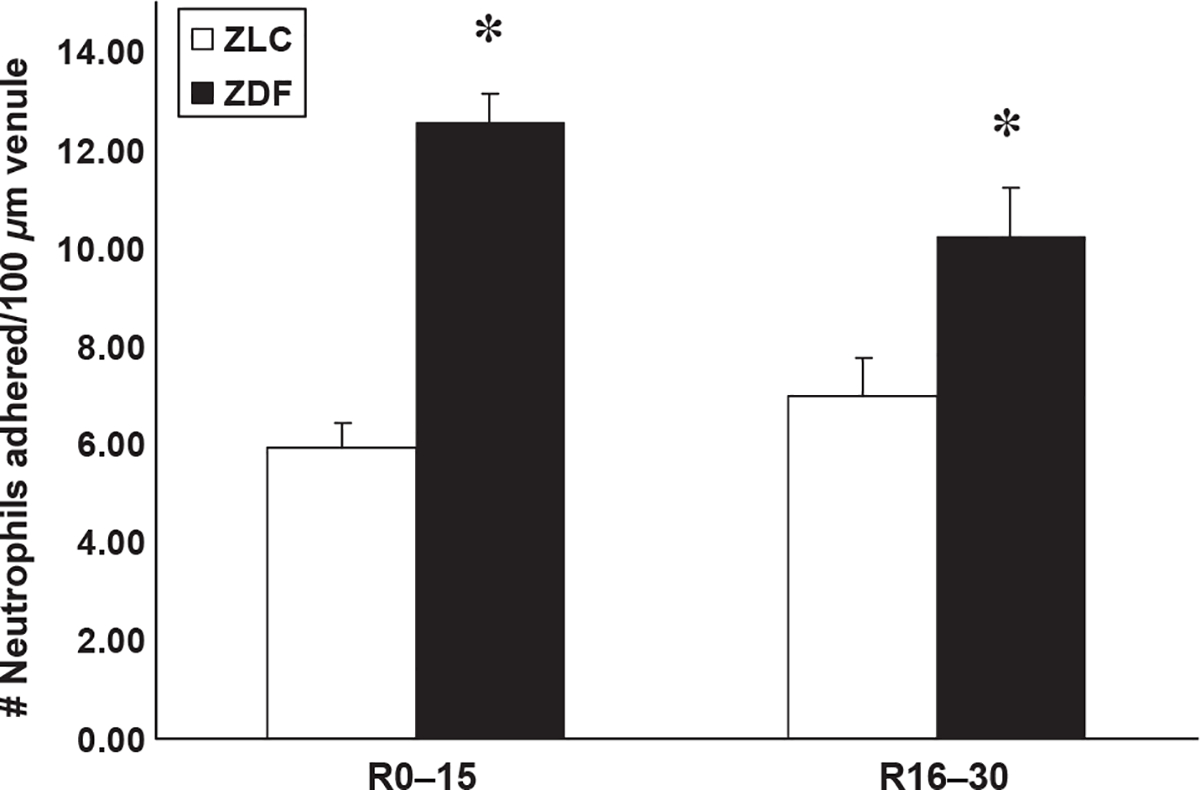
Neutrophil adhesion to venules of ZLC and ZDF at 0–15 and 16–30 minutes of reperfusion after MCAO-R, reperfusion. Measure of variance: SEM. *p ≤ 0.05 compared with ZLC. n = 3 animals/group
Figure 3.
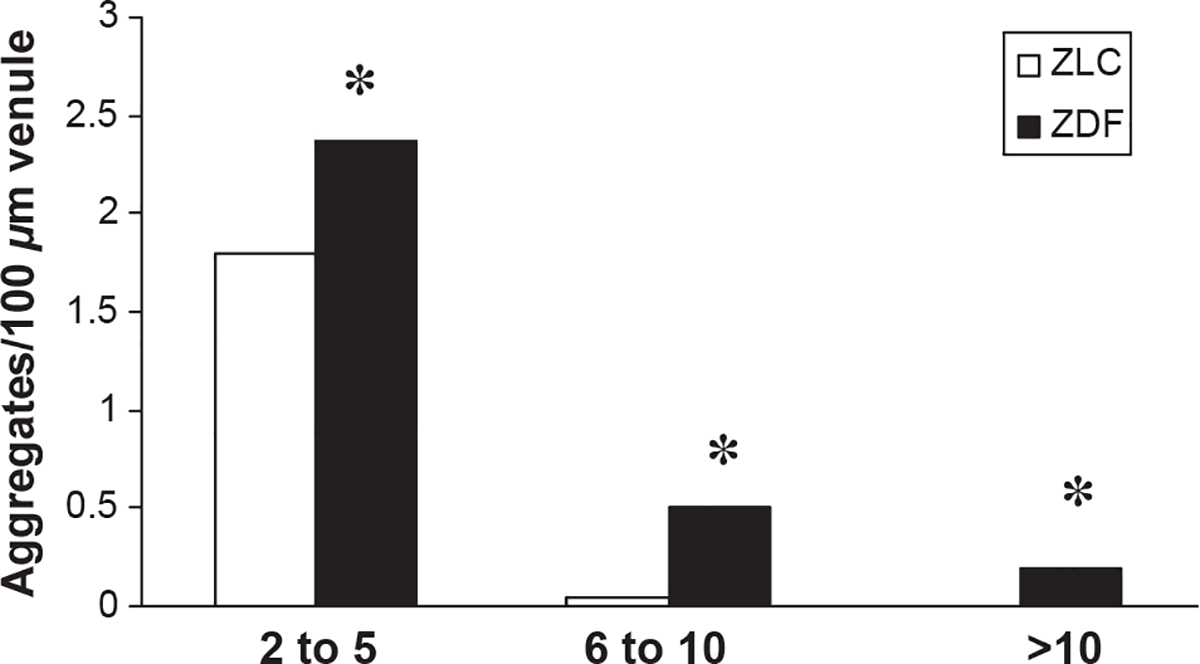
Number and size of neutrophil aggregates in venules of ZLC and ZDF during reperfusion after MCAO. Measure of variance: SEM. *p ≤ 0.05 compared with ZLC. n = 3 animals/group
Table 2.
Hemodynamic characteristics in the cerebral microcirculation during reperfusion after MCAO-R
| Group | N (venules) | Diameter (μm) | Vwbc (μm/sec) | Shear rate (per sec) |
|---|---|---|---|---|
| ZLC | 33 | 68.8 ± 4.2 | 847.1 ± 124.5 | 66.8 ± 9.4 |
| ZDF | 18 | 67.8 ± 5.9 | 521.4 ± 59.5* | 44.2 ± 6.0* |
Measure of variance: SEM.
p ≤ 0.05 vs. ZLC (n = 3 animals/group).
Neurologic Function and Brain Injury
At four hours of reperfusion, neurologic scores were significantly worse in ZDF animals (8.7 ± 0.3) when compared with ZLC animals (6.3 ± 0.3) (p ≤ 0.05) and indicative of severe vs. moderate impairment in ZDF vs. ZLC control animals, respectively. As might be expected, infarction volume in non-diabetic ZLC animals at this early time point was only 7% of the contralateral hemisphere (Figure 4A,B). In contrast, infarct volume in ZDF at this same early time was fivefold greater, and involved one-third of the total hemisphere (Figure 4A,B) (p ≤ 0.05). Cerebral edema after four hours of reperfusion in ZDF animals was also severe (11.2 ± 1.2%) and was significantly (twofold) greater than in ZLC animals (4.6 ± 1.8%) (p ≤ 0.05) (Figure 4C).
Figure 4.
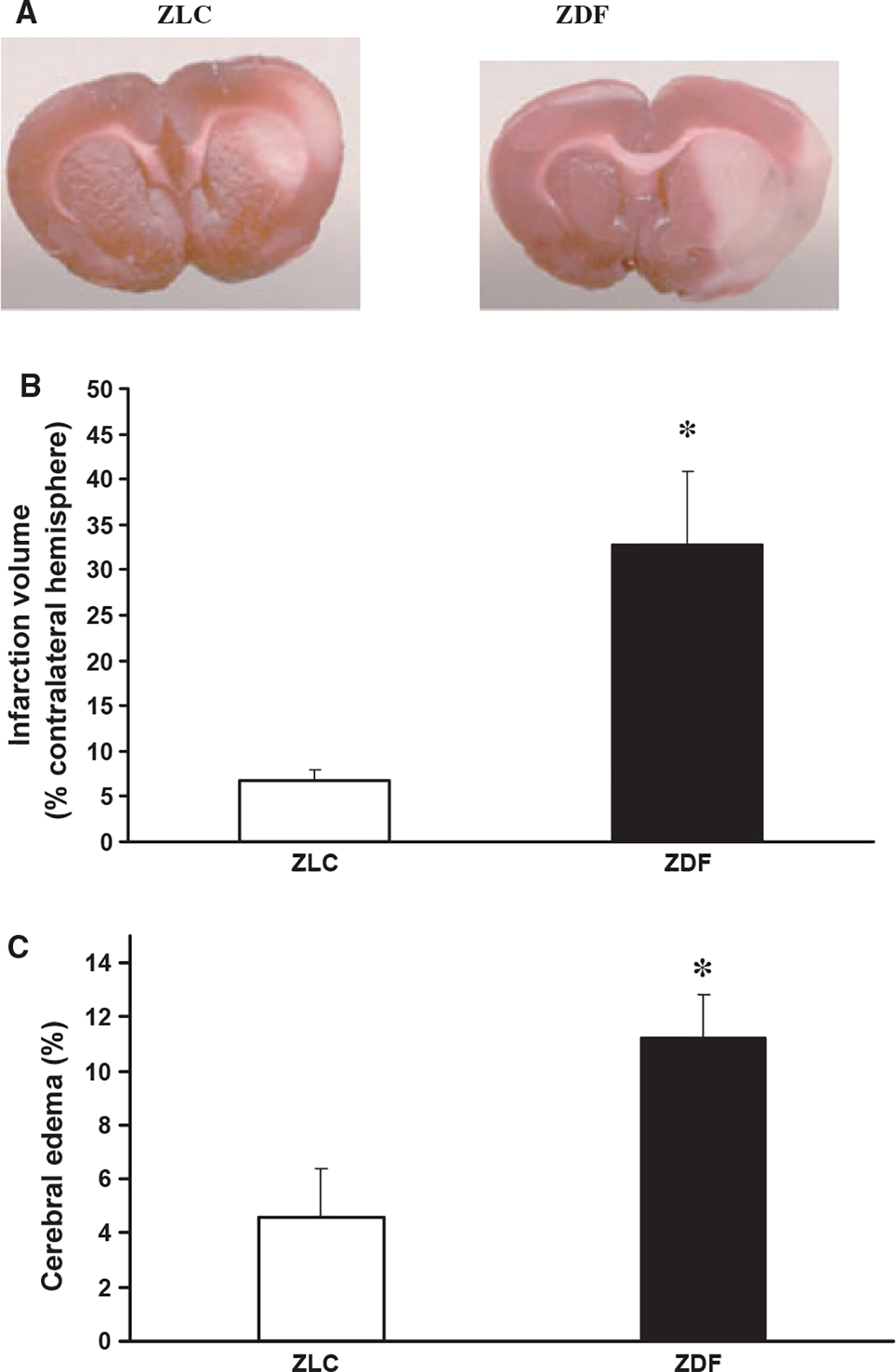
Brain injury in ZLC and ZDF after MCAO-R. (A) Representative images of infarction (light areas) in ipsilateral hemispheres in ZLC (left) and ZDF (right) after four hours of reperfusion. (B) Infarction volume, corrected for edema, after four hours of reperfusion. (C) Cerebral edema at four hours of reperfusion. *p ≤ 0.05 compared with ZLC. n = 5 animals/group.
Neutrophil CD11b Expression
At baseline (pre-ischemia), neutrophil CD11b expression was significantly greater in ZDF animals when compared with ZLCs, being increased twofold (TFI 13.7 ± 1 vs. TFI 6.5 ± 0.8, respectively, p ≤ 0.05) (Figure 5). At 15 minutes of reperfusion after MCAO-R, CD11b expression increased (p ≤ 0.05) in both groups and remained significantly (1.7-fold) greater in diabetic ZDF animals when compared with ZLC animals (TFI 22.4 ± 1 vs. TFI 13.2 ± 1.8, respectively, p ≤ 0.05) (Figure 5)
Figure 5.
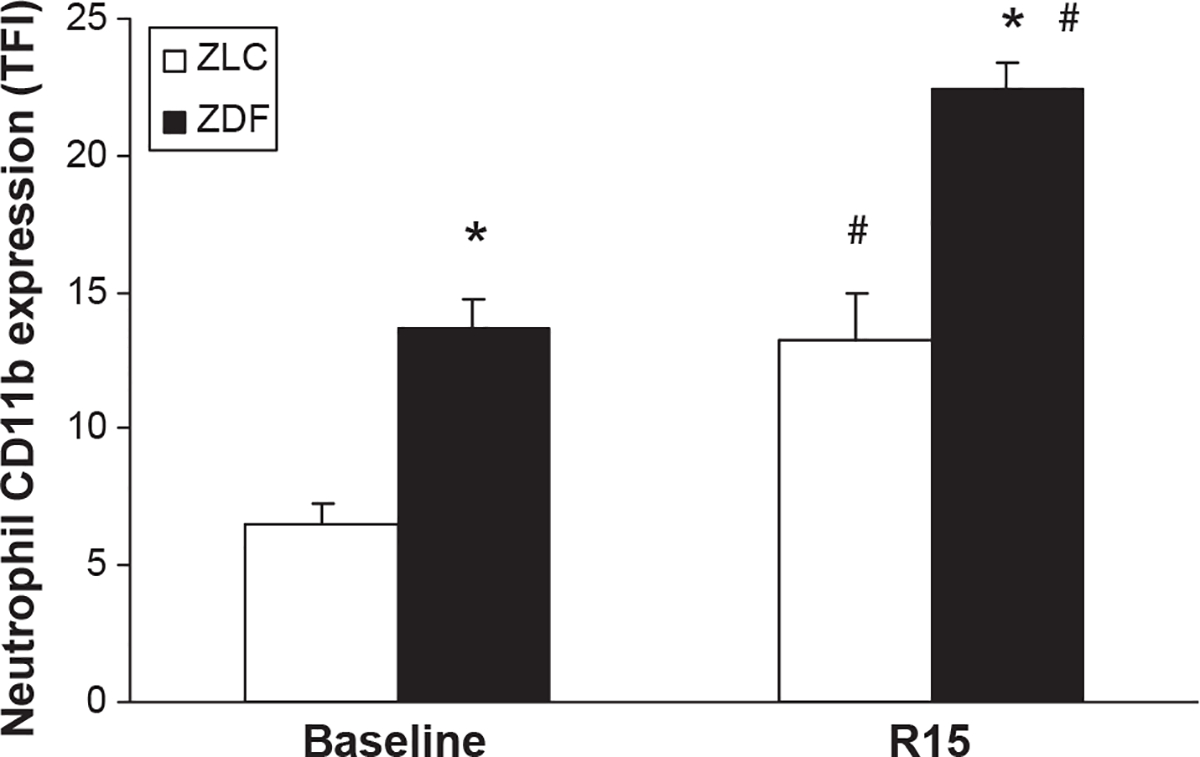
Neutrophil CD11b expression in ZLC and ZDF at baseline and at 15 minutes of reperfusion following MCAO. *p ≤ 0.05 compared with ZLC. #p ≤ 0.05 compared with pre-ischemia. n = 5 animals/group.
Endothelial sICAM Protein Expression
At baseline, sICAM was significantly greater in ZDF (10.6 ± 0.9 ng/mL) when compared with ZLC (8.1 ± 0.4 ng/mL) (p ≤ 0.05). Higher levels of soluble sICAM in ZDF vs. ZLC persisted after MCAO-R (ZDF = 11.3 ± 0.1; ZLC = 9.3 ± 0.2 ng/mL, p < 0.05) (Figure 6). Consistent with this finding, immunohistochemical staining for sICAM protein in cerebral microvessels was also increased approximately twofold at baseline in the ZDF when compared with ZLC animals, as the mean area of sICAM staining in the ZDF was 16.8 ± 4.4 mm2 compared with 7.8 ± 0.8 mm2 in the ZLC.
Figure 6.

Serum levels of sICAM in ZLC and ZDF at baseline and at 2.5 hours of reperfusion following MCAO. *p ≤ 0.05 compared with ZLC. #p ≤ 0.05 compared with pre-ischemia. n = 5 animals/group.
Cerebral Inflammatory Gene Expression
After only 2.5 hours of reperfusion in animals subjected to MCAO-R, cerebral gene expression for the proinflammatory cytokines, TNF-α and IL-1β; neutrophil chemokine, GRO/KC; endothelial adhesion molecules, E-selectin and sICAM, and endothelial and inducible nitric oxide synthases, eNOS and iNOS, were significantly increased in ZDF rats over baseline levels of expression (p < 0.05) (Table 3). In contrast, at this time point in non-diabetic ZLC animals, only TNF-α gene expression was statistically increased over baseline values (p < 0.05). However, in comparing gene expression between ZDF and ZLC rats subject to MCAO-R at this single early time point after reperfusion, only iNOS gene expression was significantly increased in ZDF when compared with ZLC animals (p < 0.05). Of note, expression of these same genes was not statistically different between ZDF vs. ZLC animals at baseline prior to stroke (data not shown).
Table 3.
Cerebral inflammatory gene expression after MCAO-R (mRNA levels, fold-increase relative to baseline expression in each strain)
| Gene | ZLC | ZDF |
|---|---|---|
| TNF-α | 11.2* | 12 1** |
| IL-1 β | 9.9 | 8.4** |
| GRO/KC | 58.2 | 168.2** |
| E-selectin | 3.2 | 3.9** |
| sICAM | 3.5 | 4.5** |
| eNOS | 1.9 | 2.6** |
| iNOS | 5.9 | 22.4**,*** |
Measure of variance: SEM.
p < 0.05 vs. ZLC baseline.
p < 0.05 vs. ZDF baseline.
p < 0.05 vs. ZLC after MCAO-R (n = 3 animals/group).
DISCUSSION
Mortality rates after ischemic stroke have been reported to be up to five times greater in people with untreated diabetes compared with those without diabetes [1,2,9,19]. The precise reasons for the high mortality in this population remain unclear, but may extend beyond the contribution of conventional risk factors such as hyperglycemia and hypertension [9]. It is now clear that inflammatory pathways are persistently activated in type 2 diabetes [5,7,26]. It is also well documented that activation of inflammatory pathways, including those that result in neutrophil–endothelial adhesion in the microcirculation, contributes to ischemia–reperfusion injury in the brain. Thus, we hypothesized that the combined chronic inflammatory profile of type 2 diabetes plus the acute activation of inflammatory pathways during middle cerebral artery occlusion (ischemia) and reperfusion would result in exaggerated neutrophil-mediated reperfusion injury after ischemic stroke, and thus worsen stroke outcome in the ZDF model.
The results of the present study confirm this hypothesis. Specifically, as early as four hours after reperfusion, significantly worse infarction size, edema and neurologic function was evident in the ZDF vs. ZLC. Preceding this by a matter of hours, neutrophil adhesion in the cerebral microcirculation was significantly increased, with notable aggregation of cells, and was associated with a further decrease in shear rate when compared with that occurring in ZLC. Cerebral expression of eNOS increased approximately twofold in both ZDF and ZLC after I–R, although this increase only achieved statistical significance in ZDF. Based on the observation that vessel diameters were similar and shear rates were lower in the ZDF vs. ZLC after I–R, it is unlikely that an increase in eNOS offered any vasodilatory protection [30] in this diabetic group. Of note, neutrophil adhesion and shear rate values in the non-diabetic ZLC are similar to those previously reported in Sprague–Dawley rats after cerebral I–R, where, for example, shear rate is decreased to 33% of baseline following I–R [34]. Thus, in toto, our findings suggest that excessive neutrophil aggregation following I–R injury in T2DM may be associated with a decrease in shear rate upon reperfusion of up to 75% relative to flow in the non-diabetic brain.
To our knowledge, these are the first studies examining effects of T2DM on neutrophil-mediated inflammation and brain injury using an experimental model of focal cerebral I–R, an experimental model that emulates the majority of human stroke events [39]. Although, to our knowledge, isolated effects of hyperglycemia (without insulin resistance) on neutrophil adhesion in focal cerebral I–R have not been examined, increased neutrophil adhesion and worse neurologic outcome has been reported in STZ-induced hyperglycemic rats following global cerebral ischemia [25,41,49]. In addition, using a murine model of obesity and isolated insulin resistance (young ob/ob mice prior to onset of hyperglycemia), increased neutrophil–platelet adhesion to the microcirculation following focal cerebral I–R has been described [47]. Also of note is that exaggerated neutrophil adhesion in the coronary microcirculation after I–R in a STZ-rat model has been described [16]. Taken together, evidence suggests that the exaggerated neutrophil-mediated reperfusion injury observed in the brain in our studies in ZDF rats may be secondary to hyperglycemia and/or insulin resistance. Undoubtedly, each of these pathogenic factors may lead to unique or shared responses that contribute to the increases in cardiovascular disease risk in T2DM [27].
To determine the mechanisms that contribute to the findings in the current study, we examined both sides of neutrophil–endothelial interface, both pre- and post-stroke. The number and activation state of neutrophils (CD11b) in diabetic animals prior to stroke were both increased two-fold. Thus, more neutrophils are primed, or chronically activated [8] prior to I–R injury in T2DM. With I–R injury, activated CD11b positive neutrophils remained almost twofold higher in diabetic animals than in their non-diabetic counterparts. Although the role of lymphocytes was not evaluated in the current study, previous studies in non-diabetic animal models indicate that T lymphocytes (but not B lymphocytes) contribute to cerebral I–R injury [17,50]. The role of T lymphocytes in I–R injury in the T2DM brain remains essentially unexplored.
On the other side of the neutrophil–endothelial interface, endothelial sICAM serves as the ligand for firm neutrophil adhesion [51]. No difference in cerebral sICAM gene expression could be detected in ZDF vs. ZLC rats at baseline prior to I–R injury. However, circulating levels of sICAM protein at baseline in ZDF rats were increased twofold, a finding consistent with a state of chronic endothelial activation in clinical T2DM [26]. There appears to be an increase in protein levels of sICAM at baseline in cerebral vessels of diabetic animals, although further characterization of these and other inflammatory protein levels at baseline and after I–R injury are clearly needed. After I–R injury, sICAM levels remained higher in ZDF than in ZLC. Cerebral ICAM gene expression was also already significantly increased (4.5-fold) at this early time point post-I–R (2.5 hours), whereas gene expression in ZLC was not yet statistically changed. These findings are consistent with reports of increased sICAM protein levels in other vascular bed after ischemia–reperfusion in the ZDF [21].
Additional new findings from this study indicate an exaggerated inflammatory profile in the post-ischemic T2DM brain, evidenced by increased gene expression of systemic cytokines TNF-α and IL-1β, neutrophil chemokine GRO/KC, endothelial cell E-selectin and cytotoxic iNOS in the ZDF after cerebral I–R. Thus, a greater potential to attract [29], activate and capture [40] damaging neutrophils [18] in the cerebral microcirculation may exist in the injured, post-ischemic T2DM brain.
In summary, the findings from this study indicate that the ZDF model of T2DM replicates worse stroke outcome observed in human T2DM ischemic stroke. The findings also support the postulate that both sides of the neutrophil–endothelial interface appear to be primed prior to, and remain significantly more activated during I–R in T2DM. Consequently, reperfusion injury plays a significant role in poor stroke outcome in T2DM. Clearly, additional studies to define the effects of treatments aimed at these chronic and acute pathophysiologic processes, including the effects of conventional monotherapies (for example, glucose lowering) and novel multi-target therapies, may be of particular interest to minimize stroke burden in this increasingly prevalent population.
PERSPECTIVE
Diabetic complications are traditionally separated into those occurring secondary to microvascular disease vs. macrovascular disease of the arterioles and arteries on the “red side” of the circulation. As increased stroke risk in diabetes has been largely attributed to disease in the “red side,” the finding in this study of a strong association between increased microvascular injury in venules and worse stroke outcome in an experimental model of Type 2 diabetes calls into question this existing paradigm. Reformulation of our view of the pathogenesis of diabetic stroke to include an important role for microvascular injury on the “blue side” of the circulation provides novel areas of study and new targets for therapeutic interventions.
Abbreviations used:
- T2DM
type 2 diabetes mellitus
- ZDF
Zucker Diabetic Fatty rat
- ZLC
Zucker Lean Control rat
- I-R
ischemia-reperfusion
- MCAO-R
middle cerebral artery occlusion-reperfusion
- sICAM
soluble intercellular adhesion molecule
- IL-1B
interleukin-1 beta
- GRO/KC
growth-related oncogene/keratinocyte chemoattractant
- TNF-alpha
tumor necrosis-alpha
- eNOS
endothelial nitric oxide synthase
- iNOS
inducible nitric oxide synthase
- WBC
white blood cell
- TTC
triphenyl tetrazolium chloride
- FITC
fluorescein isothiocyanate
- TFI
total fluorescence intensity
- aCSF
artificial cerebral spinal fluid
- MAP
mean arterial pressure
- CD11b
cluster of differentiation molecule 11B
- STZ
streptozotocin
REFERENCES
- 1.Almdal T, Scharling H, Jensen JS, Vestergaard H. The independent effect of type 2 diabetes mellitus on ischemic heart disease, stroke, and death: a population-based study of 13,000 men and women with 20 years of follow-up. Arch Intern Med 164: 1422–1426, 2004. [DOI] [PubMed] [Google Scholar]
- 2.Andersen KK, Olsen TS. One-month to 10-year survival in the copenhagen stroke study: interactions between stroke severity and other prognostic indicators. J Stroke Cerebrovasc Dis 20: 117–123, 2011. [DOI] [PubMed] [Google Scholar]
- 3.Boyle JP, Honeycutt AA, Narayan KM, Hoerger TJ, Geiss LS, Chen H, Thompson TJ. Projection of diabetes burden through 2050: impact of changing demography and disease prevalence in the U.S. Diabetes Care 24: 1936–1940, 2001. [DOI] [PubMed] [Google Scholar]
- 4.Cohen Z, Gonzales RF, Davis-Gorman GF, Copeland JG, McDonagh PF. Thrombin activity and platelet microparticle formation are increased in type 2 diabetic platelets: a potential correlation with caspase activation. Thromb Res 107: 217–221, 2002. [DOI] [PubMed] [Google Scholar]
- 5.Dandona P, Aljada A, Bandyopadhyay A.Inflammation: the link between insulin resistance, obesity and diabetes. Trends Immunol 25: 4–7, 2004. [DOI] [PubMed] [Google Scholar]
- 6.Dandona P, Aljada A, Chaudhuri A, Mohanty P, Garg R. Metabolic syndrome: a comprehensive perspective based on interactions between obesity, diabetes, and inflammation. Circulation 111: 1448–1454, 2005. [DOI] [PubMed] [Google Scholar]
- 7.Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol 11: 98–107, 2011. [DOI] [PubMed] [Google Scholar]
- 8.El-Benna J, Dang PM, Gougerot-Pocidalo MA. Priming of the neutrophil NADPH oxidase activation: role of p47phox phosphorylation and NOX2 mobilization to the plasma membrane. Semin Immunopathol 30: 279–289, 2008. [DOI] [PubMed] [Google Scholar]
- 9.Ergul A, Li W, Elgebaly MM, Bruno A, Fagan SC. Hyperglycemia, diabetes and stroke: focus on the cerebrovasculature. Vascul Pharmacol 51: 44–49, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferreira L, Teixeira-de-Lemos E, Pinto F, Parada B, Mega C, Vala H, Pinto R, Garrido P, Sereno J, Fernandes R, Santos P, Velada I, Melo A, Nunes S, Teixeira F, Reis F. Effects of sitagliptin treatment on dysmetabolism, inflammation, and oxidative stress in an animal model of type 2 diabetes (ZDF rat). Mediators Inflamm 2010: 592760, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Funk JL, Frye JB, Oyarzo JN, Chen G, Lantz RC, Jolad SD, Solyom AM, Timmermann BN. Efficacy and mechanism of action of turmeric supplements in the treatment of experimental arthritis. Arthritis Rheum 54: 3452–3464, 2006. [DOI] [PubMed] [Google Scholar]
- 12.Funk JL, Migliati E, Chen G, Wei H, Wilson J, Downey KJ, Mullarky PJ, Coull BM, McDonagh PF, Ritter LS. Parathyroid hormone-related protein induction in focal stroke: a neuroprotective vascular peptide. Am J Physiol Regul Integr Comp Physiol 284: R1021–R1030, 2003. [DOI] [PubMed] [Google Scholar]
- 13.Gao X, Picchi A, Zhang C. Upregulationof TNF-alpha and receptors contribute to endothelial dysfunction in Zucker Diabetic Rats. Am J Biomed Sci 2: 1–12, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Griffen SC, Wang J, German MS. A genetic defect in beta-cell gene expression segregates independently from the fa locus in the ZDF rat. Diabetes 50: 63–68, 2001. [DOI] [PubMed] [Google Scholar]
- 15.Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol 9: 367–377, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hokama JY, Ritter LS, Davis-Gorman G, Cimetta AD, Copeland JG, McDonagh PF. Diabetes enhances leukocyte accumulation in the coronary microcirculation early in reperfusion following ischemia. J Diabetes Complications 14: 96–107, 2000. [DOI] [PubMed] [Google Scholar]
- 17.Hurn PD, Subramanian S, Parker SM, Afentoulis ME, Kaler LJ, Vandenbark AA, Offner H. T- and B-cell-deficient mice with experimental stroke have reduced lesion size and inflammation. J Cereb Blood Flow Metab 27: 1798–1805, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iadecola C, Zhang F, Xu S, Casey R, Ross ME. Inducible nitric oxide synthase gene expression in brain following cerebral ischemia. J Cereb Blood Flow Metab 15: 378–384, 1995. [DOI] [PubMed] [Google Scholar]
- 19.Janghorbani M, Hu FB, Willett WC, Li TY, Manson JE, Logroscino G, Rexrode KM. Prospective study of type 1 and type 2 diabetes and risk of stroke subtypes: the Nurses’ Health Study. Diabetes Care 30: 1730–1735, 2007. [DOI] [PubMed] [Google Scholar]
- 20.Johns DG, Ao Z, Eybye M, Olzinski A, Costell M, Gruver S, Smith SA, Douglas SA, Macphee CH. Rosiglitazone protects against ischemia/reperfusion-induced leukocyte adhesion in the zucker diabetic fatty rat. J Pharmacol Exp Ther 315: 1020–1027, 2005. [DOI] [PubMed] [Google Scholar]
- 21.La Bonte LR, Davis-Gorman G, Stahl GL, McDonagh PF. Complement inhibition reduces injury in the type 2 diabetic heart following ischemia and reperfusion. Am J Physiol Heart Circ Physiol 294: H1282–H1290, 2008. [DOI] [PubMed] [Google Scholar]
- 22.La Bonte LR, Dokken B, Davis-Gorman G, Stahl GL, McDonagh PF. The mannose-binding lectin pathway is a significant contributor to reperfusion injury in the type 2 diabetic heart. Diab Vasc Dis Res 6: 172–180, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leonard BL, Watson RN, Loomes KM, Phillips AR, Cooper GJ. Insulin resistance in the Zucker diabetic fatty rat: a metabolic characterisation of obese and lean phenotypes. Acta Diabetol 42: 162–170, 2005. [DOI] [PubMed] [Google Scholar]
- 24.Lesniewski LA, Donato AJ, Behnke BJ, Woodman CR, Laughlin MH, Ray CA, Delp MD. Decreased NO signaling leads to enhanced vasoconstrictor responsiveness in skeletal muscle arterioles of the ZDF rat prior to overt diabetes and hypertension. Am J Physiol Heart Circ Physiol 294: H1840–H1850, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin B, Ginsberg MD, Busto R, Li L. Hyperglycemia triggers massive neutrophil deposition in brain following transient ischemia in rats. Neurosci Lett 278: 1–4, 2000. [DOI] [PubMed] [Google Scholar]
- 26.Mastej K, Adamiec R. Neutrophil surface expression of CD11b and CD62L in diabetic microangiopathy. Acta Diabetol 45: 183–190, 2008. [DOI] [PubMed] [Google Scholar]
- 27.Mathis D, Shoelson SE. Immunometabolism: an emerging frontier. Nat Rev Immunol 11: 81, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsuo Y, Kihara T, Ikeda M, Ninomiya M, Onodera H, Kogure K. Role of neutrophils in radical production during ischemia and reperfusion of the rat brain: effect of neutrophil depletion on extracellular ascorbyl radical formation. J Cereb Blood Flow Metab 15: 941–947, 1995. [DOI] [PubMed] [Google Scholar]
- 29.McColl BW, Rothwell NJ, Allan SM. Systemic inflammatory stimulus potentiates the acute phase and CXC chemokine responses to experimental stroke and exacerbates brain damage via interleukin-1- and neutrophil-dependent mechanisms. J Neurosci 27: 4403–4412, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moro MA, Cardenas A, Hurtado O, Leza JC, Lizasoain I. Role of nitric oxide after brain ischaemia. Cell Calcium 36: 265–275, 2004. [DOI] [PubMed] [Google Scholar]
- 31.Morrison H, McKee D, Ritter L. Systemicneutrophil activation in a mouse model of ischemic stroke and reperfusion. Biol Res Nurs 13: 154–163, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oltman CL, Richou LL, Davidson EP, Coppey LJ, Lund DD, Yorek MA. Progression of coronary and mesenteric vascular dysfunction in Zucker obese and Zucker diabetic fatty rats. Am J Physiol Heart Circ Physiol 291: H1780–H1787, 2006. [DOI] [PubMed] [Google Scholar]
- 33.van Poelje PD, Potter SC, Chandramouli VC, Landau BR, Dang Q, Erion MD. Inhibition of fructose 1,6-bisphosphatase reduces excessive endogenous glucose production and attenuates hyperglycemia in Zucker diabetic fatty rats. Diabetes 55: 1747–1754, 2006. [DOI] [PubMed] [Google Scholar]
- 34.Ritter LS, Orozco JA, Coull BM, McDonagh PF, Rosenblum WI. Leukocyte accumulation and hemodynamic changes in the cerebral microcirculation during early reperfusion after stroke. Stroke 31: 1153–1161, 2000. [DOI] [PubMed] [Google Scholar]
- 35.Ritter LS, Stempel KM, Coull BM, McDonagh PF. Leukocyte-platelet aggregates in rat peripheral blood after ischemic stroke and reperfusion. Biol Res Nurs 6: 281–288, 2005. [DOI] [PubMed] [Google Scholar]
- 36.Ritter LS, Wilson DS, Williams SK, Copeland JG, McDonagh PF. Early in reperfusion following myocardial ischemia, leukocyte activation is necessary for venular adhesion but not capillary retention. Microcirculation 2: 315–327, 1995. [DOI] [PubMed] [Google Scholar]
- 37.Rodrigues SF, Granger DN. Role of blood cells in ischaemia-reperfusion induced endothelial barrier failure. Cardiovasc Res 87: 291–299, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, Carnethon MR, Dai S, de Simone G, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Greenlund KJ, Hailpern SM, Heit JA, Ho PM, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, McDermott MM, Meigs JB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Rosamond WD, Sorlie PD, Stafford RS, Turan TN, Turner MB, Wong ND, Wylie-Rosett J, American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics – 2011 update: a report from the American Heart Association. Circulation 123: e18–e209, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roger VL. Executive summary: heart disease and stroke statistics – 2011 update: a report from the American Heart Association. Circulation 123: 459–463, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ruehl ML, Orozco JA, Stoker MB, McDonagh PF, Coull BM, Ritter LS. Protective effects of inhibiting both blood and vascular selectins after stroke and reperfusion. Neurol Res 24: 226–232, 2002. [DOI] [PubMed] [Google Scholar]
- 41.Santizo RA, Xu HL, Ye S, Baughman VL, Pelligrino DA. Loss of benefit from estrogen replacement therapy in diabetic ovariectomized female rats subjected to transient forebrain ischemia. Brain Res 956: 86–95, 2002. [DOI] [PubMed] [Google Scholar]
- 42.Schmid-Schonbein GW. The damaging potential of leukocyte activation in the microcirculation. Angiology 44: 45–56, 1993. [DOI] [PubMed] [Google Scholar]
- 43.Suematsu M, Schmid-Schonbein GW, Chavez-Chavez RH, Yee TT, Tamatani T, Miyasaka M, Delano FA, Zweifach BW. In vivo visualization of oxidative changes in microvessels during neutrophil activation. Am J Physiol 264(3 Pt 2): H881–H891, 1993. [DOI] [PubMed] [Google Scholar]
- 44.Suzaki Y, Ozawa Y, Kobori H. Intrarenal oxidative stress and augmented angioten-sinogen are precedent to renal injury in Zucker diabetic fatty rats. Int J Biol Sci 3: 40–46, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Szocs Z, Brunmair B, Stadlbauer K, Nowotny P, Bauer L, Luger A, Furnsinn C. Age-dependent development of metabolic derangement and effects of intervention with pioglitazone in Zucker diabetic fatty rats. J Pharmacol Exp Ther 326: 323–329, 2008. [DOI] [PubMed] [Google Scholar]
- 46.Teixeira de Lemos E, Reis F, Baptista S, Pinto R, Sepodes B, Vala H, Rocha-Pereira P, Correia da Silva G, Teixeira N, Silva AS, Carvalho L, Teixeira F, Das UN. Exercise training decreases proinflammatory profile in Zucker diabetic (type 2) fatty rats. Nutrition 25: 330–339, 2009. [DOI] [PubMed] [Google Scholar]
- 47.Terao S, Yilmaz G, Stokes KY, Ishikawa M, Kawase T, Granger DN. Inflammatory and injury responses to ischemic stroke in obese mice. Stroke 39: 943–950, 2008. [DOI] [PubMed] [Google Scholar]
- 48.Tipton A, Downey K, Frank OC, Migliati E, Wilson J, McDonagh P, Funk J, Ritter L. Neutrophil function in aged versus young rats subjected to global cerebral ischemia and reperfusion. FASEB J 17: A1075–A1076, 2003. [Google Scholar]
- 49.Xu HL, Vetri F, Lee HK, Ye S, Paisansathan C, Mao L, Tan F, Pelligrino DA. Estrogen replacement therapy in diabetic ovariectomized female rats potentiates postischemic leukocyte adhesion in cerebral venules via a RAGE-related process. Am J Physiol Heart Circ Physiol 297: H2059–H2067, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation 113: 2105–2112, 2006. [DOI] [PubMed] [Google Scholar]
- 51.Zhang RL, Chopp M, Zaloga C, Zhang ZG, Jiang N, Gautam SC, Tang WX, Tsang W, Anderson DC, Manning AM. The temporal profiles of ICAM-1 protein and mRNA expression after transient MCA occlusion in the rat. Brain Res 682: 182–188, 1995. [DOI] [PubMed] [Google Scholar]