

Abstract
This chapter will describe how mathematical modeling allows the RAS pathway to be studied with computational experiments. The mathematical model utilized simulates the biochemical reactions that regulate RAS signaling. This type of model incorporates knowledge of reaction mechanisms, including measured quantitative parameters that characterize these reactions for both wild-type and mutant RAS proteins. For an illustrative example, this chapter focuses on how modeling provided new insights that helped solve a problem that challenged the RAS community for nearly a decade: why do colorectal cancers with the KRAS G13D mutation, but not the other common KRAS mutations, benefit from EGFR inhibition. The methods described include computational dose response experiments and the use of “computational chimeric” RAS mutants.
Keywords: Computational Biology, Systems Biology, Systems Pharmacology, Targeted Therapy
1. Introduction
The RAS pathway has been exceptionally well-studied and is well-understood. However, unexpected pathway behaviors are still encountered. One such example involves how KRAS mutations influence the response to EGFR inhibitors in colorectal cancer. Activation of EGFR by its ligands can initiate multiple intracellular signaling events, including RAS activation, which in turn can activate the RAF/MEK/ERK MAPK cascade that can drive cellular proliferation (Figure 1). As cancer is a disease of excessive and uncontrolled cellular proliferation within the malignant cells, agents that target this pathway to impair proliferation have been developed and have demonstrated benefit in a variety of cancers, including colorectal cancer.
Figure. 1. KRAS G13D colorectal cancer and its unexplained response to EGFR inhibition.
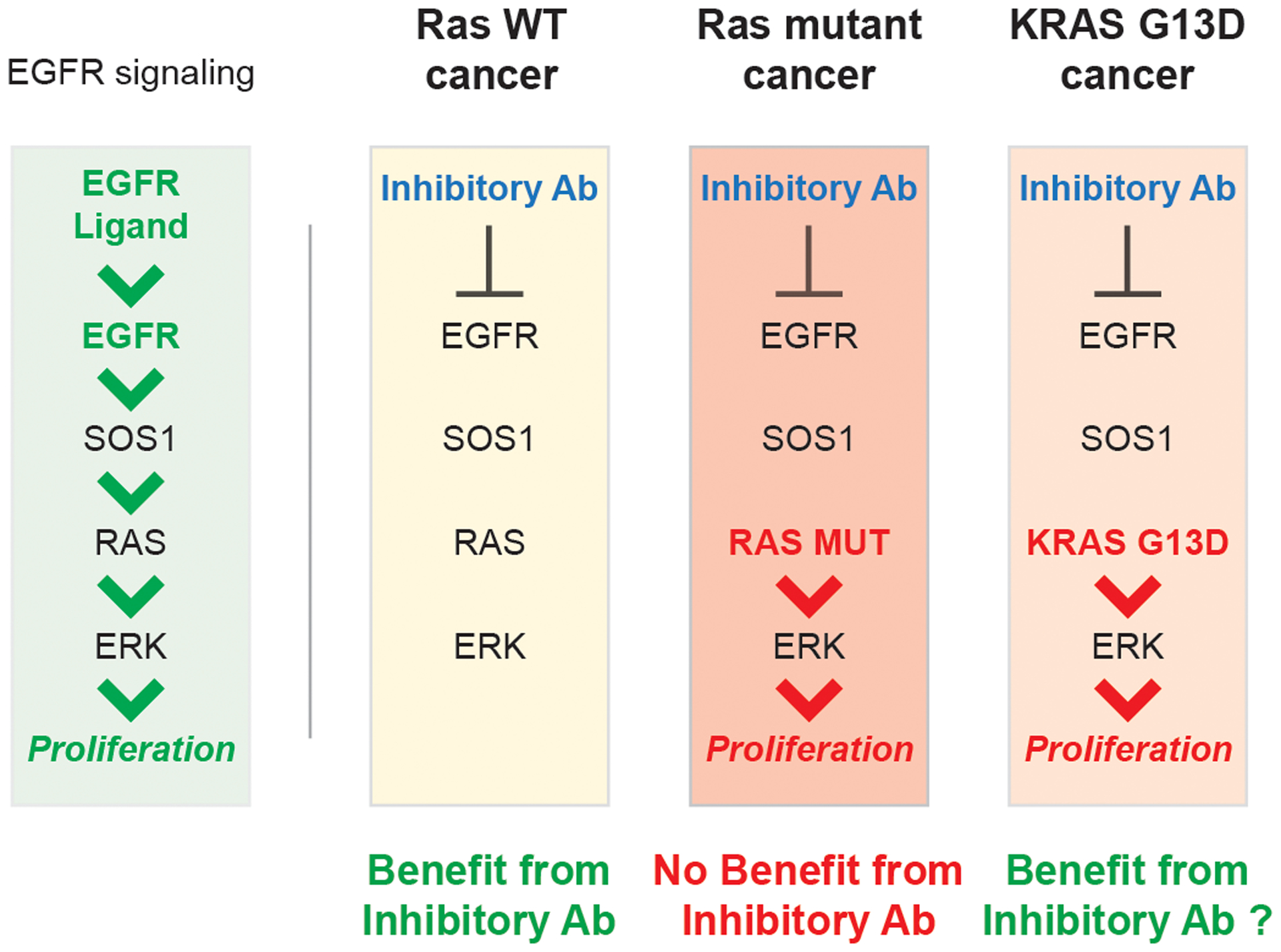
Activation of EGFR by its ligands leads to activation of the RAS/ERK pathway that drives cellular proliferation. This pathway is believed to be overactivated in colorectal cancer. Patients with KRAS WT colon cancers benefit from anti-EGFR agents that prevent the activation of EGFR. Patients with constitutively active KRAS mutants do not benefit from anti-EGFR agents, as the constitutively active RAS mutants can still drive proliferation. The constitutively active KRAS G13D mutation is an exception; patients with this mutation have been shown to benefit from anti-EGFR agents, although a mechanism to explain why this constitutively active mutation behaved differently had been unknown.
Approximately 40%-50% of colorectal cancer patients harbor oncogenic KRAS mutations [1] (see accompanying chapter by Prior, I.). As oncogenic KRAS mutant proteins are constitutively active and can initiate downstream signaling in the absence of EGFR stimulation, it was logical to hypothesize that EGFR inhibitors would offer less benefit to KRAS mutant colorectal cancer patients. Indeed, Phase 3 clinical trials revealed that the patients with a KRAS mutation, as a group, did not receive any benefit from treatment with EGFR inhibitors [2].
Many different KRAS mutations have been observed in colorectal cancer, with KRAS G12D, KRAS G12V, and KRAS G13D being the three most common [1]. An analysis of the Phase 3 clinical trial data that investigated whether any specific KRAS mutants behaved differently made the surprising observation that patients with the KRAS G13D tended to benefit from EGFR inhibitors, in contrast to the set of all other KRAS mutant colorectal cancers [3]. This was reproduced in cellular and in mouse xenograft experiments, and other, subsequent, studies also observed that KRAS G13D cancers appeared to be more sensitive to EGFR inhibitors [4,5]. However, it has also been controversial as it appears to contradict the well-accepted dogma on RAS biology, and studies have also been presented that claim KRAS G13D colorectal cancers treated with EGFR inhibitors responded the same as other KRAS mutant colorectal cancers [6,7].
The application of a computational model to a problem like this allows the problem to be studied from a new perspective. Additionally, the utilization of a mathematical model that simulates the biochemical reactions that regulate RAS signaling allows for one to investigate whether it is even consistent with the known principles of RAS biology for cancers with different constitutively active KRAS mutants to be more (or less) sensitive than one another when treated with an inhibitor. The incorporation of the fundamental biochemical properties, like reaction rate constants, that are specific to the different mutants allows one to investigate whether the known biochemical differences between these mutants are sufficient to cause the diverging responses to treatment. For example, KRAS G13D has been most notable for having fast, spontaneous, nucleotide dissociation relative to wild-type (WT) KRAS, which allows for increased GTP loading and autoactivation that is not GEF-dependent [8–11]. Mutations like this have been referred to as ‘fast-cycling’. Whether fast-cycling, the most well-known biochemical distinction of KRAS G13D, could mechanistically cause increased sensitivity to EGFR inhibition was unknown, but such a question can be investigated with a mechanism-based mathematical model. The first approach described here is the simulation of drug dose responses for modeled cellular conditions with different KRAS mutations present. The second approach discussed here is the development and utilization of chimeric RAS mutants to isolate specific biochemical parameters and determine which are responsible for specific phenotypes.
2. Materials
2.1. RAS model
If one wanted to study the effects of a drug on RAS mutant cancers in an animal model, that scientist might choose an established genetically engineered mouse model [12]. If one wanted to study the effects of a therapeutic on RAS mutant cancers with a cellular model, they might choose from existing cancer cell lines that are known to harbor an oncogenic RAS mutant. Similarly, to study RAS computationally, it may be possible to use an existing mathematical model. One advantage of using an existing model is that it may have already demonstrated an ability to make novel insights. If the available models could not be adapted to a specific problem, however, it may be necessary to develop a new model. The process of building a mass-action model of RAS has been described previously [13].
To study KRAS G13D colorectal cancer and its response to EGFR inhibition, a previously developed model that has been used to study KRAS G12V, KRAS G12D, and KRAS G12C [14,15] was utilized and extended to include KRAS G13D. The model focuses on the reactions that directly interact with RAS to influence the RAS nucleotide state (Figure 2). It includes RAS GTPases, RAS GEFs, RAS GAPs, RAS Effectors, and guanine nucleotides GTP and GDP. For RAS, intrinsic GTPase activities and spontaneous (non-GEF mediated) nucleotide dissociation and association are included and described with mass-action kinetics. Mass-action kinetics are also used to describe interactions between RAS and its effectors. GAP-facilitated GTP hydrolysis is modeled with Michaelis-Menten kinetics. GEF-facilitated nucleotide exchange is modeled with reversible Michaelis-Menten kinetics. For these enzymatic processes, competitive forms of the corresponding Michaelis-Menten equations are used to account for wild-type and mutant RAS proteins that may compete to bind to the same GEFs and GAPs. The parameters of these reactions (reaction rate constants, and Michaelis-Menten kcat and Km terms) are obtained from the experimental literature for wild-type RAS, and changes in these terms that follow from mutations are also obtained from the experimental literature. Computer code for this model can be downloaded as part of the supplementary material from one publication that utilized the model [15], and thorough descriptions of model development are also available [14,13,16]. The model allows for investigations of how the various reactions and their properties contribute to RAS activation and effector binding, such as how changes in the abundances and/or activity of different proteins in the network influences signaling, as well as how different RAS mutations respond to a specific cellular state.
Figure 2. The biochemical reactions of the RAS model.
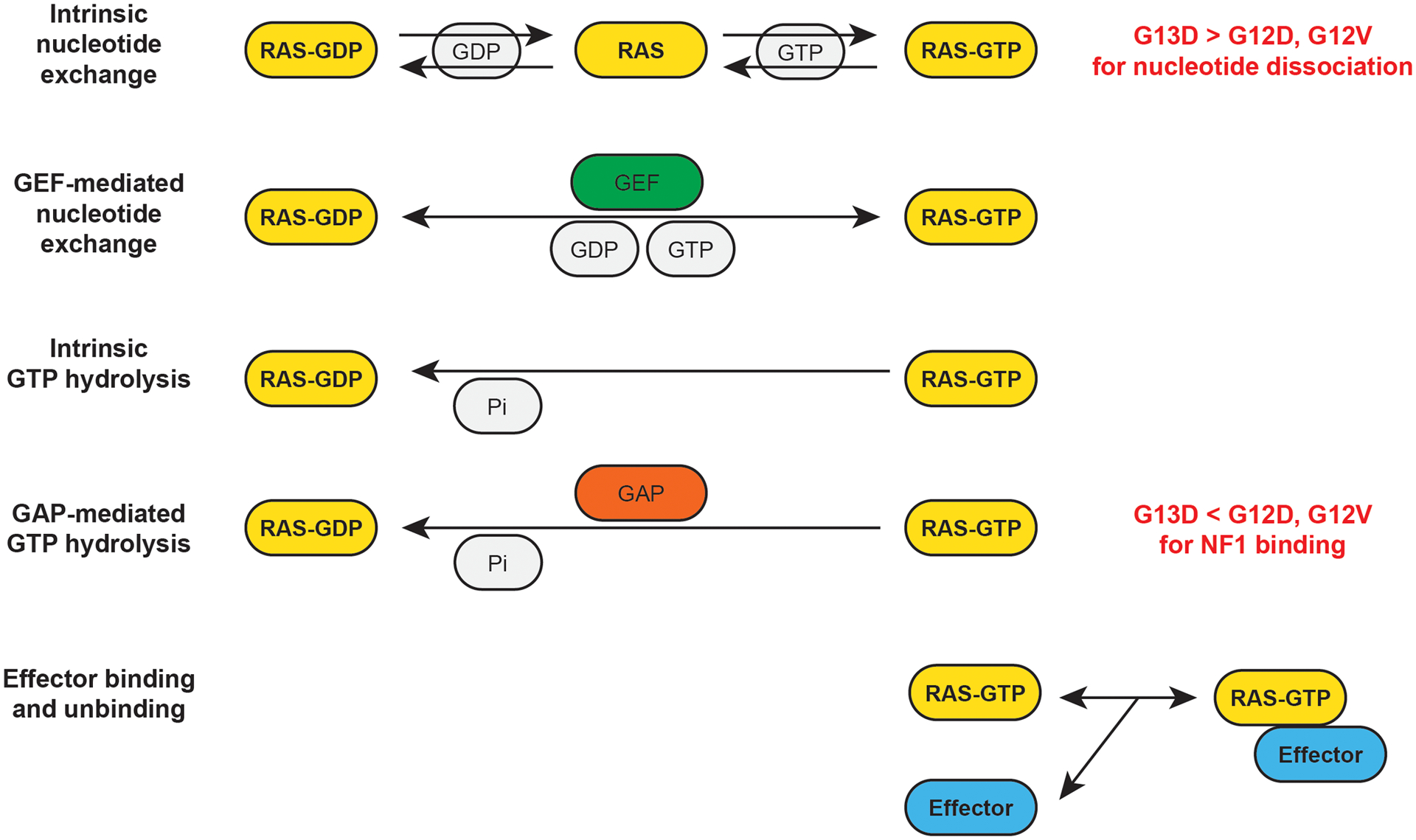
The model focuses on the reactions the directly influence RAS-GTP and RAS-GTP-Effector binding reactions. Each reaction (arrow) is modeled with mass-action or enzymatic kinetics. The same equations apply to wild-type and mutant proteins, but the specific parameter values (e.g. reaction rate constants) will vary based on whether the equation is describing the activity of a WT or mutant RAS protein. For example, KRAS G12V, G12D, and G13D have slower intrinsic GTPase reaction rate constants than WT RAS. When comparing KRAS G13D to KRAS G12V and KRAS G12D, the most notable differences are KRAS G13D has a more rapid rate of intrinsic nucleotide dissociation, and KRAS G13D binds less well to the RAS GAP NF1.
2.2. Data on the relevant mutants
Our goal is to use our computational model to investigate differences between KRAS G13D, KRAS G12D, and KRAS G12V. The original publication that described the RAS model and its development includes rate constant for G12D and G12V mutants, but not for G13D [14]. Although new data options continue to become available [8], we have chosen to utilize the original parameters to communicate the general value of the model – it does not need to be continually retuned, but can provide numerous insights in its current form. However, one could choose to update the parameters to newer data, if they choose, and we needed to specify parameters for KRAS G13D in order to model that mutant. To find these parameters, a search of manuscripts that characterized the biophysical properties of KRAS G13D uncovered data that report nucleotide dissociation rates 3.6625 times faster than for WT KRAS [11] (See Note 1) and that report poor binding of KRAS G13D to the RAS GAP NF1, leading us to estimate that the Km for binding to NF1 is approximately one hundred times weaker than for WT KRAS [17]. Additionally, it has also been long -appreciated that KRAS hotspot mutations at codon 12, 13, and 61 are partially impaired at nucleotide hydrolysis and severely impaired at GAP-mediated nucleotide hydrolysis. At the time we began our study, we could not find a value for the KRAS G13D GTPase reaction, so we utilized the impaired value of KRAS G12D as an estimate. For impaired GAP-mediated GTP hydrolysis, we assume that the GAP cannot increase GTP hydrolysis, as we do for other mutants (like G12D and G12V). Without clear data to define other parameter values, we began our modeling with a goal of determining whether these parameters were sufficient to explain the observed behavior of KRAS G13D. Alternatively, one may experimentally measure the other parameters or computationally explore the possible effects of the other parameters. Whatever is chosen for parameters, these are inserted into the model to specify the KRAS mutant, such as KRAS G13D.
2.3. Software to implement the model and hardware for simulations
The RAS model utilized here was simulated in MATLAB, and a version of the model that was written using MATLAB can be downloaded as part of a related publication [15]. The model can be implemented in other languages, such as Python or Mathematica. The RAS model is fairly limited in scope and does not present a computational challenge – it can quickly be simulated and studied on a common laptop computer. It is notable that even though the scope of the system may appear limited and less overwhelming than large pathway models, the model has revealed non-obvious behaviors within this pathway that have also been experimentally confirmed.
3. Methods
3.1. Simulate the effects of an EGFR inhibitor dose response.
Specify the basal conditions of the model. The original RAS model was parameterized for basal RAS signaling in the absence of induced activation. As RAS WT cancers are sensitive to EGFR inhibition, we assume colorectal cancers commonly have elevated EGFR activation that in turn activates RAS. The model thus needs to be adjusted to model the conditions of elevated EGFR driven RAS activation. When we originally developed our RAS model, we found a 10x increase in our basal level of GEF activity mimicked conditions of experimental receptor tyrosine kinase-mediated RAS activation [14]. Thus, a value of basal GEF activation that is 10x the value used in the original model appears to be a good approximation. (See Note 2)
Specify the conditions that may approximate an EGFR inhibitor dose response. The activation of EGFR is believed to lead to RAS activation through RAS GEFs like SOS1. Treatment with an EGFR inhibitor is assumed to result in reduced EGFR activation and reduced RAS GEF recruitment and activation. Thus, to simulate an EGFR inhibitor dose response with our model, one can consider different levels of GEF activity, ranging from the value used to mimic conditions of EGFR activation down through lower levels, such as the unstimulated level used in the original RAS model.
Specify a simulation protocol. The goal is to find the steady-state level of RAS signal, i.e. the level of RAS-GTP observed under typical conditions. This can be done by simulating the model for a period of time sufficiently long enough that RAS-GTP levels are no longer changing. Levels of total RAS-GTP, WT RAS-GTP, and Mutant RAS-GTP should be saved as outputs for each level of simulated GEF activity.
Simulate a dose response for conditions with no RAS mutation. To model a cell with all wild-type RAS, 100% of the total RAS abundance is specified with WT RAS biochemical parameters. The simulation protocol can then be followed for each level of GEF activity.
Simulate dose responses for conditions with a KRAS G12D, KRAS G12V, or KRAS G13D mutation. To model a cell with a KRAS mutation, 25% of total RAS is set to the mutant (which is specified with the mutant parameters) and 75% of total RAS is set to WT RAS (which is specified with the WT RAS parameters). The simulation protocol can then be followed for each level of GEF activity for each modeled RAS mutant.
Plot the resultant outputs. (See Note 3.)
Plot the simulation dose responses for the WT and KRAS Mutant subsets of total RAS. (See Note 4.)
3.2. Simulate KRAS mutant chimeras to evaluate the contribution of nucleotide cycling rate.
Generate chimeric mutants between KRAS G13D with KRAS G12V and between KRAS G13D and KRAS G12D that alternate the nucleotide dissociation rate constants. For the chimeric G13D with G12V cycling, the GDP and GTP dissociation rate constants from KRAS G13D are replaced with the (slower) rate constants for the same reactions from G12V. For the chimeric G12V with G13D cycling, the dissociation rate constants for GDP and GTP from KRAS G12V are replaced with the (faster) rate constants for the same reactions from G13D. The process for chimeras between KRAS G13D and KRAS G12D is similar.
Simulate EGFR dose responses, as in 3.1, but utilizing the chimeric mutants (Figure 3).
Compare to the simulated dose responses for the KRAS G12D, KRAS G12V, and KRAS G13D mutants to determine whether the change in nucleotide cycling rate made it more or less sensitive to EGFR inhibitors. (See Note 5.)
Figure 3. Fast-cycling (increased nucleotide exchange) does not explain why KRAS G13D responds to EGFR inhibition.
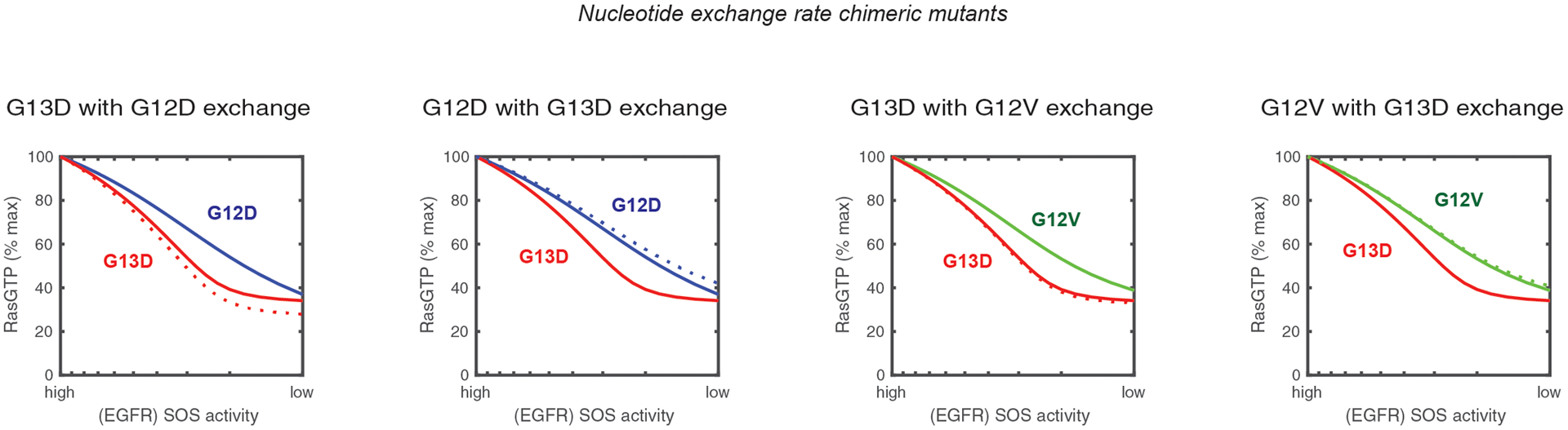
Simulated anti-EGFR dose responses for the Ras model with chimeric mutants involving the substitution of nucleotide properties between G13D, G12D, and G12V. Solid lines indicate the simulated dose responses for KRAS G12D (blue), KRAS G12V (green), and KRAS G13D (red). Dashed lines indicate the simulated dose responses for chimeric mutants. The color of the line indicates the mutant with which the chimera featured in the panel is most similar. The y-axis indicates the percentage of all RAS in the model (both WT and mutant) that is bound to GTP.
3.3. Simulate KRAS mutant chimeras to evaluate the contribution of impaired NF1 binding.
Generate chimeric mutants between KRAS G13D with KRAS G12V and between KRAS G13D and KRAS G12D that alternate the property that specifies the strength of binding to the RAS GAP NF1. For the chimeric G13D that has a G12V-like affinity for NF1, the Km for the NF1-G13D interaction is replaced with the value for the Km between NF1 and KRAS G12V. For the chimeric G12V that has a G13D-like affinity for NF1, the Km for the NF1-G12V interaction is replaced with the value for the Km between NF1 and KRAS G13D. The process for chimeras between KRAS G13D and KRAS G12D is similar.
Simulate EGFR dose responses, as in 3.1, but utilizing the chimeric mutants. (Figure 4).
Compare to the simulated dose responses for the KRAS G12D, KRAS G12V, and KRAS G13D mutants to determine whether the change in nucleotide cycling rate made it more or less sensitive to EGFR inhibitors. (See Note 6.)
Figure 4. Impaired binding to NF1 explains why KRAS G13D responds to EGFR inhibition.
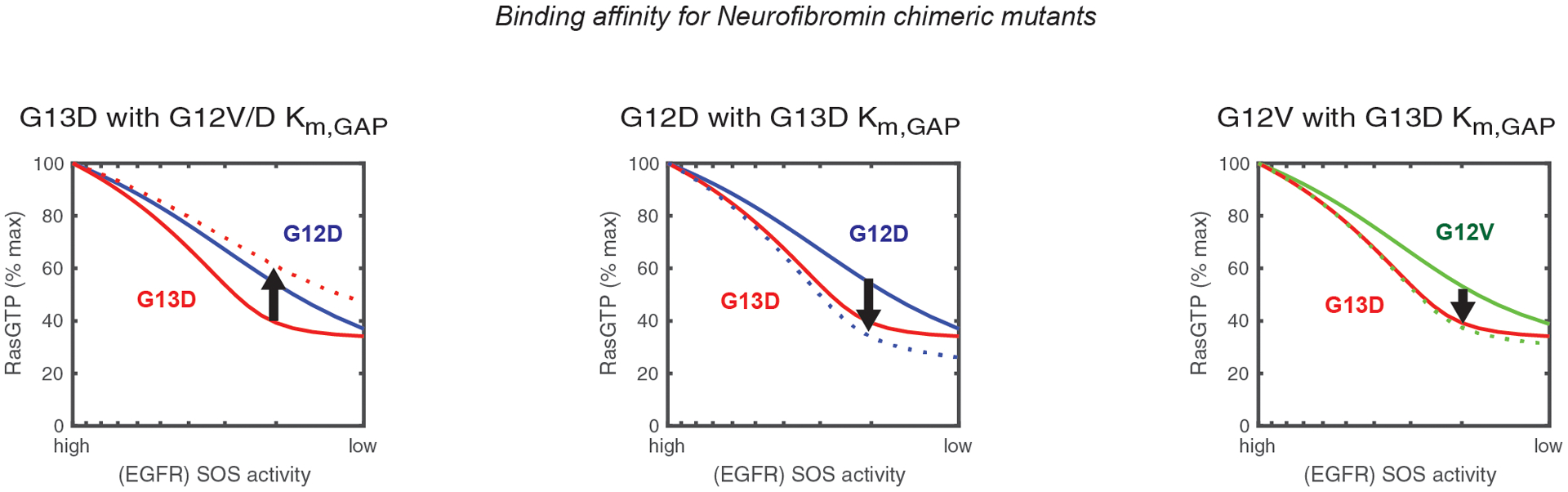
Simulated anti-EGFR dose responses for the Ras model with chimeric mutants involving the substitution of the Km for RAS-NF1 binding between G13D, G12D, and G12V. Solid lines indicate the simulated dose responses for KRAS G12D (blue), KRAS G12V (green), and KRAS G13D (red). Dashed lines indicate the simulated dose response for chimeric mutants. The color of the line indicates the mutant with which the chimera featured in the panel is most similar. The y-axis indicates the percentage of all RAS in the model (both WT and mutant) that is bound to GTP.
4. Notes
More recent measurements of parameters for KRAS G13D were published [8–10] after we began our work [18]. The manuscripts report KRAS G13D to have nucleotides dissociate at a rate that is 10 to 15 times larger than WT, in contrast to the approximately 4 times larger change we used based on the earlier publication with exchange rate data [11]. It would be possible to simulate with these alternative values. Of note, we do not believe values from the RAS GAP RASA1 can be used in place of NF1 as some studies have observed the biochemical consequences of RAS mutations can vary between these GAPS [19], and NF1 appears to be an essential GAP in colorectal cancer, as suggested by its frequent mutation rate within patient tumors.
The activation of wild-type RAS in colorectal cancer is undoubtedly more complex than RAS mutants and EGFR. Other receptor tyrosine kinases and other mutations have been shown to play a role, as well. The present study however investigates how KRAS G13D may be sensitive to EGFR inhibition, so we focused on KRAS mutations and EGFR activity level as the known variables to determine whether they alone may be sufficient to explain the observed and confusing clinical responses to EGFR inhibitors. Similarly, processes such as negative feedback, which are known to play an important role in the biology of RAS pathway mutations in colorectal cancer, are not included in the model [20,21]. That the insights generated by the model were then experimentally observable and that these data confirmed the model-based predictions suggests that that the computational model can generate new, non-obvious, and experimentally verifiable insights even when it necessarily limits itself to a subset of the RAS signaling network.
This revealed that the total RAS output decreased more rapidly for KRAS G13D than for KRAS G12D and KRAS G12V. This suggests first that it is perfectly consistent with the known principles of RAS signal regulation for different RAS mutants to respond with different intensities to EGFR inhibitors. This is notable, as disbelief of original clinical trial only makes sense if one assumes all mutants must behave essentially equivalently. Second, it is notable because the available data for KRAS G13D was sufficient to suggest it is more sensitive. There may be additional biochemical parameters that differ that have yet to be measured, but of the limited number that were measured, they were sufficient to cause an increased sensitivity. This is also notable, as once the G13D exceptional response was observed, the field wondered how this could be possible. That the data needed to explain increased sensitivity was readily available yet the mechanism was not elucidated until computational modeling was applied to the problem highlights both the complexity of the system and the value of computational modeling.
The further subdivision of total RAS GTP into WT RAS GTP and mutant RAS GTP shows that the differences in EGFR/GEF inhibition fall within the WT RAS GTP pool of RAS within a KRAS mutant cancer and shows little to no change in mutant RAS GTP for any of the three KRAS mutants [16].
These chimeras reveal that the higher nucleotide dissociation rate of KRAS G13D would actually make it less sensitive to EGFR inhibitors – the opposite of the observed behavior – thus suggesting that the known fast-cycling activity of KRAS-G13D does not explain why it is sensitive to EGFR inhibitors.
These chimeras reveal that impaired binding to NF1 makes a RAS mutant more sensitive to EGFR inhibition.
Acknowledgement
This work was supported by NIH grant K22CA216318 and P30CA014195.
References
- 1.Cancer Genome Atlas N (2012) Comprehensive molecular characterization of human colon and rectal cancer. Nature 487 (7407):330–337. doi: 10.1038/nature11252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, Price TJ, Shepherd L, Au HJ, Langer C, Moore MJ, Zalcberg JR (2008) K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 359 (17):1757–1765. doi: 10.1056/NEJMoa0804385 [DOI] [PubMed] [Google Scholar]
- 3.De Roock W, Jonker DJ, Di Nicolantonio F, Sartore-Bianchi A, Tu D, Siena S, Lamba S, Arena S, Frattini M, Piessevaux H, Van Cutsem E, O’Callaghan CJ, Khambata-Ford S, Zalcberg JR, Simes J, Karapetis CS, Bardelli A, Tejpar S (2010) Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. Jama 304 (16):1812–1820. doi: 10.1001/jama.2010.1535 [DOI] [PubMed] [Google Scholar]
- 4.Tejpar S, Celik I, Schlichting M, Sartorius U, Bokemeyer C, Van Cutsem E (2012) Association of KRAS G13D tumor mutations with outcome in patients with metastatic colorectal cancer treated with first-line chemotherapy with or without cetuximab. J Clin Oncol 30 (29):3570–3577. doi: 10.1200/JCO.2012.42.2592 [DOI] [PubMed] [Google Scholar]
- 5.Nakamura M, Aoyama T, Ishibashi K, Tsuji A, Takinishi Y, Shindo Y, Sakamoto J, Oba K, Mishima H (2017) Randomized phase II study of cetuximab versus irinotecan and cetuximab in patients with chemo-refractory KRAS codon G13D metastatic colorectal cancer (G13D-study). Cancer Chemother Pharmacol 79 (1):29–36. doi: 10.1007/s00280-016-3203-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peeters M, Douillard JY, Van Cutsem E, Siena S, Zhang K, Williams R, Wiezorek J (2013) Mutant KRAS codon 12 and 13 alleles in patients with metastatic colorectal cancer: assessment as prognostic and predictive biomarkers of response to panitumumab. J Clin Oncol 31 (6):759–765. doi: 10.1200/JCO.2012.45.1492 [DOI] [PubMed] [Google Scholar]
- 7.Segelov E, Thavaneswaran S, Waring PM, Desai J, Robledo KP, Gebski VJ, Elez E, Nott LM, Karapetis CS, Lunke S, Chantrill LA, Pavlakis N, Khasraw M, Underhill C, Ciardiello F, Jefford M, Wasan H, Haydon A, Price TJ, van Hazel G, Wilson K, Simes J, Shapiro JD (2016) Response to Cetuximab With or Without Irinotecan in Patients With Refractory Metastatic Colorectal Cancer Harboring the KRAS G13D Mutation: Australasian Gastro-Intestinal Trials Group ICECREAM Study. J Clin Oncol 34 (19):2258–2264. doi: 10.1200/JCO.2015.65.6843 [DOI] [PubMed] [Google Scholar]
- 8.Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, Westover KD (2015) Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol Cancer Res 13 (9):1325–1335. doi: 10.1158/1541-7786.MCR-15-0203 [DOI] [PubMed] [Google Scholar]
- 9.Smith MJ, Neel BG, Ikura M (2013) NMR-based functional profiling of RASopathies and oncogenic RAS mutations. Proc Natl Acad Sci U S A 110 (12):4574–4579. doi: 10.1073/pnas.1218173110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson CW, Lin YJ, Reid D, Parker J, Pavlopoulos S, Dischinger P, Graveel C, Aguirre AJ, Steensma M, Haigis KM, Mattos C (2019) Isoform-Specific Destabilization of the Active Site Reveals a Molecular Mechanism of Intrinsic Activation of KRas G13 D. Cell Rep 28 (6):1538–1550 e1537. doi: 10.1016/j.celrep.2019.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Palmioli A, Sacco E, Airoldi C, Di Nicolantonio F, D’Urzo A, Shirasawa S, Sasazuki T, Di Domizio A, De Gioia L, Martegani E, Bardelli A, Peri F, Vanoni M (2009) Selective cytotoxicity of a bicyclic Ras inhibitor in cancer cells expressing K-Ras(G13D). Biochem Biophys Res Commun 386 (4):593–597. doi: 10.1016/j.bbrc.2009.06.069 [DOI] [PubMed] [Google Scholar]
- 12.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA (2001) Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 15 (24):3243–3248. doi: 10.1101/gad.943001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stites EC, Ravichandran KS (2012) Mathematical investigation of how oncogenic ras mutants promote ras signaling. Methods Mol Biol 880:69–85. doi: 10.1007/978-1-61779-833-7_5 [DOI] [PubMed] [Google Scholar]
- 14.Stites EC, Trampont PC, Ma Z, Ravichandran KS (2007) Network analysis of oncogenic Ras activation in cancer. Science 318 (5849):463–467. doi: 10.1126/science.1144642 [DOI] [PubMed] [Google Scholar]
- 15.Stites EC, Shaw AS (2018) Quantitative Systems Pharmacology Analysis of KRAS G12C Covalent Inhibitors. CPT Pharmacometrics Syst Pharmacol 7 (5):342–351. doi: 10.1002/psp4.12291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McFall T, Diedrich JK, Mengistu M, Littlechild SL, Paskvan KV, Sisk-Hackworth L, Moresco JJ, Shaw AS, Stites EC (2019) A systems mechanism for KRAS mutant allele-specific responses to targeted therapy. Sci Signal 12 (600). doi: 10.1126/scisignal.aaw8288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gremer L, Gilsbach B, Ahmadian MR, Wittinghofer A (2008) Fluoride complexes of oncogenic Ras mutants to study the Ras-RasGap interaction. Biol Chem 389 (9):1163–1171. doi: 10.1515/BC.2008.132 [DOI] [PubMed] [Google Scholar]
- 18.Stites EC (2014) Differences in sensitivity to EGFR inhibitors could be explained by described biochemical differences between oncogenic Ras mutants. bioRxiv 10.1101/005397 [DOI] [Google Scholar]
- 19.Donovan S, Shannon KM, Bollag G (2002) GTPase activating proteins: critical regulators of intracellular signaling. Biochim Biophys Acta 1602 (1):23–45 [DOI] [PubMed] [Google Scholar]
- 20.Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, Bernards R (2012) Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 483 (7387):100–103. doi: 10.1038/nature10868 [DOI] [PubMed] [Google Scholar]
- 21.Stites EC (2012) The response of cancers to BRAF inhibition underscores the importance of cancer systems biology. Sci Signal 5 (246):pe46. doi: 10.1126/scisignal.2003354 [DOI] [PubMed] [Google Scholar]