

Abstract
Purpose:
Glioblastoma (GBM) is an incurable primary brain tumor that has not benefited from immunotherapy to date. More than 90% of GBM expresses the tryptophan (Trp) metabolic enzyme, indoleamine 2,3-dioxygenase 1 (IDO). This observation supported the historical hypothesis that IDO suppresses the antitumor immune response solely through a mechanism that requires intratumoral Trp depletion. However, recent findings led us to investigate the alternative hypothesis that IDO suppresses the anti-GBM immune response independent of its association with Trp metabolism.
Experimental Design:
IDO-deficient GBM cell lines reconstituted with IDO wild-type or IDO enzyme–null cDNA were created and validated in vitro and in vivo. Microarray analysis was conducted to search for genes that IDO regulates, followed by the analysis of human GBM cell lines, patient GBM and plasma, and The Cancer Genome Atlas (TCGA) database. Ex vivo cell coculture assays, syngeneic and humanized mouse GBM models, were used to test the alternative hypothesis.
Results:
Nonenzymic tumor cell IDO activity decreased the survival of experimental animals and increased the expression of complement factor H (CFH) and its isoform, factor H like protein 1 (FHL-1) in human GBM. Tumor cell IDO increased CFH and FHL-1 expression independent of Trp metabolism. Increased intratumoral CFH and FHL-1 levels were associated with poorer survival among patients with glioma. Similar to IDO effects, GBM cell FHL-1 expression increased intratumoral regulatory T cells (Treg) and myeloid-derived suppressor cells while it decreased overall survival in mice with GBM.
Conclusions:
Our study reveals a nonmetabolic IDO-mediated enhancement of CFH expression and provides a new therapeutic target for patients with GBM.
Translational Relevance.
Since the ECHO-301 phase III clinical trial results were reported, questions have been raised as to why indoleamine 2,3-dioxygenase 1 (IDO) enzyme inhibition fails to improve the survival of patients with cancer. In this study, we addressed this question and confirmed that tumor cell IDO possesses activities that extend beyond tryptophan metabolism and suppress the anticancer immune response. This study discovered that nonmetabolic IDO increased the expression of immunosuppressive complement factor H (CFH) expression and in turn, suppressed the antitumor immune response and decreased the survival of experimental animals with brain tumors. High intratumoral CFH levels were associated with a substantial decrease in the survival of patients with glioma. These findings help elucidate our understanding of clinical trial results that have targeted IDO enzyme activity to-date and provide a new target for improving immunotherapeutic efficacy in patients with malignant glioma.
Introduction
Glioblastoma (GBM) is the most common malignant primary central nervous system (CNS) cancer in adults (1). Despite an aggressive standard-of-care treatment that includes maximal surgical resection when possible, followed by tumor-targeted radiation and chemotherapy with temozolomide, the prognosis remains dismal. Median survival for GBM is 14.6 months (2) with a five-year overall survival (OS) of approximately 4.7% in the United States (3). These grim statistics provide compelling rationale to develop more effective treatments for patients with GBM.
Immune checkpoint blockade and chimeric antigen receptor (CAR) T-cell treatment have improved the lifespan of patients diagnosed with select advanced cancers (4). Patients with GBM are among the malignancies that are uniquely unresponsive to cancer immunotherapy and have yet to benefit from this approach in accordance with all phase III clinical trials to date (5–7). A contributing factor to the immune resistance of GBM cells is indoleamine 2,3-dioxygenase 1 (IDO) that is frequently expressed in wild-type (WT) isocitrate dehydrogenase (IDH) GBM (8). IDO is canonically characterized as a rate-limiting immunosuppressive enzyme that converts the essential amino acid, tryptophan (Trp), into downstream metabolites that are collectively referred to as kynurenines (Kyn; ref.9). Tumor-cell expression of IDO increases the intratumoral accumulation of immunosuppressive regulatory T cells (Treg; CD4+CD25+FoxP3+) and decreases OS in experimental mice with brain tumors (10). Although GBM cells do not normally express IDO, its expression is induced by tumor-infiltrating T cells (11). Higher levels of GBM-infiltrating T cells are therefore associated with higher intratumoral IDO expression levels and an associated decreased OS of patients with GBM (11, 12). Since IDO is expressed among a wide variety of adult cancers (8), pharmacologic enzyme-inhibitor treatment approaches have been evaluated for their potential to improve survival outcomes of patients with cancer (13, 14). There have been no objective survival benefits noted among randomized clinical trials evaluating this approach in patients with aggressive cancer to date (15). This may be due to a combination of factors including: (i) a requirement to inhibit IDO and other immunosuppressive pathways simultaneously (16, 17), (ii) age-dependent increases of IDO that are unresponsive to pharmacologic enzyme inhibition (18), and/or (iii) immunosuppressive IDO effects that are independent of its association with acting as a Trp metabolic enzyme (19).
Previous work in mice demonstrated that Tregs accumulate in IDO-expressing brain tumors despite the treatment with a potent blood–brain barrier–penetrating pharmacologic IDO enzyme inhibitor (17). Also unexpectedly, IDO-mediated Trp metabolism was predominantly mediated by non-GBM cells rather than by the tumor cells in the brain (20). These findings collectively challenge the historical hypothesis that tumor cell IDO increases Tregs and decreases survival through a mechanism that solely depends on Trp metabolism and motivated us to explore the alternative hypothesis that GBM cell IDO suppresses antitumor immunity independent of its enzyme function(s).
Materials and Methods
Patient samples
Peripheral blood from patients with GBM and aneurysm were collected from the Northwestern Central Nervous System Tissue Bank (NSTB). Plasma samples were stored at −80°C until batch analysis. Peripheral blood mononuclear cells (PBMC) were isolated using Ficoll-Paque (GE Healthcare) density gradient separation and stored in liquid nitrogen prior to initiating coculture experiments. Snap-frozen tissue from surgically-resected GBMs were collected from the NSTB. All tumors were diagnosed according to WHO diagnostic criteria by C. Horbinski,. Detailed information for patient tissue samples used in this study is provided in Supplementary Table S1.
The Cancer Genome Atlas sample description
The Cancer Genome Atlas (TCGA) data for all cancer types analyzed in the current study were accessed from the UCSC Xena browser (http://xena.ucsc.edu/). RNA expression data assayed by RNA sequencing (RNA-seq; Illumina Hi-seq platform) includes RSEM normalized level-3 data that is present in the TCGA as of April 13, 2017. DNA methylation data were extracted from the same TCGA dataset. TCGA GBM gene-expression data by AffyU133a array analysis were acquired from the UCSC Xena browser.
Glioblastoma cell lines and patient-derived GBM xenografts
The human malignant glioma cell line U87 stably expressing luciferase (U87), the human IDO-overexpressing U87 cells (IDO-OE U87), and the mouse IDO−/−tGBM cell line were created and maintained as previously described (11, 20, 21). To generate the mIDO-overexpressing tGBM cell lines, a lentiviral vector that expresses mIDO-mGFP fusion protein was purchased from Origene (catalog # CW303099). To obtain lentiviral vector encoding enzyme–null mIDO, mutagenesis of His350 into Ala was performed on the WT mIDO-mGFP lentiviral vector using QuickChange II Site-directed Mutagenesis kit (Agilent Technologies, catalog # 200523) following the product protocol. Mutagenic primers were designed by the online QuickChange Primer Design Program (www.agilent.com/genomics/qcpd) and sequence information was provided in Supplementary Table S2. A GFP lentiviral vector, pCDH-CMV-MCS-EF1-copGFP-T2A-Puro (System Biosciences, catalog # CD513B-1) was provided by the Northwestern University SBDRC Gene Editing, Transduction and Nanotechnology (GET iN) Core as a control vector. The lentiviral vector that expresses FHL-1–mGFP fusion protein was purchased from Origene (catalog # CW304772). All vectors were sequenced before applied for viral packaging and cell transduction. Lentiviral particles were generated by transfecting 293FT cells with the lentiviral-expression vector and packaging vectors following routine protocol at the SBDRC GET iN Core which is supported by NIH award P30AR075049. IDO−/−tGBM cells were transduced with the lentiviral particles at a ratio of 5 infectious units of virus per cell in the presence of 8 μg/mL polybrene for 6 hours. The transduced cells were further selected by FACS based on the GFP intensity. Cells within the top 1% GFP intensity were enriched for subsequent experiments.
IDO-deficient U87 cells (IDO-KO U87) were generated by the Applied StemCell Inc. using CRISPR-CAS9 technique. Briefly, human IDO guide RNAs targeting the exon 8 of human IDO gene were designed at CRISPR design web tool (Deskgene and CRISPOR) with at least three mismatches for NGG PAM sites. The crRNA-tracrRNA duplex were prepared by mixing equimolar concentration of Alt-R crRNA, Alt-R tracrRNA, and ATTO 550 (catalog # 1075298; Integrated DNA Technologies) followed by heating at 95°C for 5 minutes and slowly cooled to room temperature. To prepare the Cas9/RNP complex, the crRNA-tracrRNA duplex and Alt-R S.p Cas9 nuclease V3 (catalog # 1081059; Integrated DNA Technologies) were gently mixed and incubated at room temperature for 20 minutes. U87 cells were resuspended in SE nucleofection buffer (SE cell Line 4D-Nucleofector X kit L; V4XC-1024, Lonza) and incubated with Cas9/RNP complex at room temperature for 2 minutes and electroporated using a 4D nucleofector (4D-Nucleofector Core Unit: AAF-1002B; Lonza, 4D-Nucleofector X Unit: AAF-1002X, Lonza). Forty-eight hours after transfection, cells were trypsinized and resuspended in PBS with 1% FBS and sorted by FACS based on ATTO signal intensity. After 7 to 14 days of culture or formation of visible cell clones, genomic DNA were extracted and subjected to PCR. The PCR products were then Sanger-sequenced to identify clones that would result in frameshift mutation. IDO knockout at both mRNA and protein levels were confirmed by analyzing parental and IDO-KO U87 cells using RT-PCR and Western blotting, respectively.
The glioma cells from patient-derived GBM xenografts were provided by the laboratory of Dr. C. David James at Northwestern University and prepared as previously reported (22, 23). Except for the PDX-derived human GBM cells, all the other cell lines used in this study were tested for Mycoplasma prior to analysis and cultured in DMEM/F12 medium (Thermo Fisher Scientific, catalog # 11320) supplemented with 10% FBS and 100 U/mL penicillin as well as 100 μg/mL streptomycin under 5% CO2 incubation condition unless described for specific experiments.
Animal and tissue preparation
Humanized mice reconstituted with human immune cells (NSG-SGM3-BLT), NOD.CB17-Prkdcscid/J (NOD/SCID) mice, CrTac:NCr-Foxn1nu mice were used as previously described (11). Cre−/−Ido1−/− tGBM mice were previously generated (20) by crossing transgenic mice that spontaneously develop glioblastoma after intraperitoneal injections of tamoxifen (24) with B6.129-Ido1tm1Alm/J (Jackson Laboratories). Mice were maintained under specific pathogen-free conditions in the Northwestern University Center for Comparative Medicine. For T-cell depletion experiments, 200 μg anti-mouse CD4 (clone YTS191; BioXCell), 200 μg anti-mouse CD8 (clone YTS169.4; BioXCell), and 200 μg anti-mouse NK1.1 (clone PK136; BioXCell) were administered by intraperitoneal injection 3 days prior to and every 3 days after tumor cell engraftment up to 30 days after intracranial injection or at the declared experimental endpoints. Rat IgG2b (clone LTF-2, BioXCell) and mouse IgG2a (clone C1.18.4, BioXCell) were administrated at the same concentration and dosing schedule as for the leukocyte-depleting antibodies. For orthotopic brain tumor mouse modeling, 3 × 105 tGBM or patient-derived xenograft (PDX) cells were intracranially engrafted similar to previous studies (10). PDX tumor tissue was kindly provided by Dr. C. David James at Northwestern University from continuously propagated patient-resected GBM that was subcutaneously engrafted into nude mice. Mice were euthanized at the indicated time point(s). Brain tumor and nontumor contralateral brain hemisphere tissue was collected, dissected, and washed in ice-cold PBS, frozen in liquid nitrogen, and stored at −80°C until analysis or processed for other techniques. Procedures for all mouse experiments were reviewed and approved by the Institutional Animal Care and Use Committee at Northwestern University and were in compliance with national and institutional guidelines.
Coculture assays
For the tGBM cell-splenic monocyte coculture, monocytes were isolated and enriched from mouse spleens using EasySep Mouse CD11b Positive Selection Kit (catalog # 18970, STEMCELL) according to the product protocol. Viability of the isolated cells was typically more than 90% as seen by trypan blue staining. CD11b+ cells were seeded onto a 12-well plate at a density of approximately 1.5×106 per well and cultured in RPMI1640 medium supplemented with 10% heat-inactivated FBS, 100 mg/mL streptomycin, 100 U/mL penicillin, and 10 ng/mL mouse recombinant IL2 (R&D Systems, catalog # 402-ML-020) overnight. The next day, nonattached cells were removed and adherent cells were washed once with PBS and incubated in fresh RPMI1640 medium as described above for another 5 days. After counting the macrophages, tGBM cells were seeded on a 0.4-μm Transwell insert at a 1:1 ratio and placed into the 12-well plate for 48 hours. At the end of coculture, tGBM cells and some macrophage cells were lysed using RNA Lysis Buffer from the PureLink RNA Mini Kit (Thermo Fisher Scientific, catalog # 12183020) and stored at −80°C for RT-PCR. The remaining macrophages were washed with PBS containing 5 mmol/L EDTA and gently deattached by cell scrappers followed by twice washing with PBS containing 2% FBS, then subject to flow cytometry analysis. Conditioned media from the coculture were collected and filtered through a 40- μm cell strainer and stored at −80°C for high-performance liquid chromatography (HPLC) analysis. The coculture of U87 cells with patient PBMC-derived T cells was performed as described in our previous study (11).
Hematoxylin and eosin staining and IHC
Brain tumors were dissected and fixed in 10% (w/v) neutral buffered formalin for 24 to 72 hours. Formalin-fixed tissues were processed into paraffin blocks and sectioned at a thickness of 4 μm. After deparaffinization, antigen retrieval was performed using sodium citrate pH6 buffer. The slides were incubated in a decloaking chamber (Biocare Medical) at 110°C for 5 minutes, rinsed in distilled water 2 times and in 1x PBS for 5 minutes, then incubated with anti-mGFP antibody (Origene, catalog # TA150122; 1:5000 dilution) in antibody diluent, overnight at 4°C. After rinsing with Tris-Buffered NaCl Solution (TBST) with 0.1% Tween 20, sections were further incubated with HRP-labelled antirabbit secondary antibody (BioCare Mach2 #RHRP520MM) for 1 hour. Slides were then washed for 3 minutes. IHC reactions were visualized using a DAB substrate (DAKO). Tissue sections were counterstained with hematoxylin Gill II (Surgipath), mounted in the xylene-based mounting medium, and visualized under a light microscope. Both hematoxylin and eosin (H&E) and IHC images were taken using a CRI Nuance camera on Zeiss Axioskop microscope at the Northwestern University Center for Advanced Microscopy Core. Histology services were provided by the Northwestern University Research Histology and Phenotyping Laboratory supported by NCI P30-CA060553.
Microarray analysis
The microarray analysis was carried out at the Northwestern University NUSeq Core Facility using the human transcriptome analysis system, Clariom D Assay (Thermo Fisher Scientific). Briefly, 1×105 cells per well of U87 cells or IDO-OE U87 cells were seeded on a 12-well plate. After attachment, cells were transfected with human IDO-specific siRNA at a final concentration of 20 nmol/L (GE Health Dharmacon) using either Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific, catalog # 13778030) or jetPRIME siRNA transfection reagent (Polyplus-transfection). The sequences of hIDO siRNA duplex are given in Supplementary Table S2. Sixteen to 18 hours after siRNA transfection, U87 cells were further incubated for another 48 hours with or without human recombinant IFNγ (Shenandoah Biotechnology, SKU# 100–77–100ug) at a concentration of 100 ng/mL. After the incubation, cells were lysed using RNA Lysis Buffer from the PureLink RNA Mini Kit (Thermo Fisher Scientific, catalog # 12183020) and stored at −80°C. The IDO-OE U87 cells were transfected with human IDO-specific siRNA for 24 hours then subjected to cell lysis as described above. The above experiment was repeated twice at different time points. Total 36 samples (6 groups × 2 duplicate × 3 repeats) were subject to the microarray analysis. Total RNA was quantified with a NanoDrop 3000 then further evaluated by a Bioanalzyer. Only samples with a RIN value ≥9.0 were used for downstream analysis. The RNA selected was amplified and hybridized using the GeneChip WT PLUS Reagent Kit (Thermo Fisher Scientific) and further analyzed by the GeneChip Scanner 3000 platform (Thermo Fisher Scientific).
The Affymetrix Transcriptome Analysis Console (TAC Version 4.0.2, Thermo Fisher Scientific) was used for normalization, summarization, and quality control of the resulting microarray data using the signal space transformation–robust multi-array average (SST-RMA) algorithm. ANOVA empirical Bayes (eBayes) method using adjusted statistical P values (P < 0.05; fold change ±2), was used for determination of the differentially-expressed genes within the TAC console. The eBayes method, which is suitable for small sample sizes, uses moderated t statistics, where instead of the global or single-gene estimated variances, a weighted average of the global and single-gene variances is used (25). Sixty-five genes were identified as the most differentially expressed genes by IDO siRNA treatment between U87 cells and IDO-OE U87 cells (Supplementary Table S3). Two genes without a curated gene symbol (Supplementary Table S3) associated with the Affymetrix probe set were excluded from downstream analyses. Gene-expression pattern and Kaplan–Meier analysis of these 63 genes were further compared with those of GBM IDO using the GlioVis online data portal (https://gliovis.shinyapps.io/GlioVis/). Pearson correlation was also performed between mRNA level of each of these 65 genes with that of IDO using the TCGA GBM RNA-seq data (as described in Methods). After the above screening, 4 genes showing highest correlation with IDO were identified, MYADM, GADD45A, TSPAN4, and CFH, of which only CFH showed the correlative expression with IDO as confirmed by the real-time RT-PCR analysis. The microarray data have been deposited in Gene Expression Omnibus (GEO) and the accession number is GSE175700 (https://urldefense.com/v3/__https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE175700__;!!Dq0×2DkFhyF93HkjWTBQKhk!Bep13fF052qW8RRWldy7Muamf9Ml8NMyGQJ8RTGR18jcBVqEAn7xY5YD8fm4gu4C6Qp1vwE$).
Western blotting
For cultured cell samples, media were removed, and cells were lysed in ice-cold RIPA buffer supplemented with 1x Halt protease/phosphatase-inhibitor cocktail (Thermo Fisher Scientific). For GBM tissue samples, approximately 50 mg tissue sample was homogenized in the above protein lysis buffer using the gentleMACS Dissociator (Miltenyi Biotec) following product protocol. The protein lysate was centrifuged at 12,600 × g for 15 minutes; the supernatant was stored at −80°C for further analysis. Protein concentration was measured by the bicinchoninic acid assay (Thermo Fisher Scientific). Equal amounts of protein were loaded in precast Mini-PROTEAN TGX Stain-Free gels (Bio-Rad). After electrophoresis, protein was transferred to the polyvinylidene difluoride (PVDF) membrane followed by blocking in 5% (w/v) nonfat milk in 1x TBST for 1 hour, then probed with primary antibodies: anti-mGFP antibody (Origene, catalog # TA150122; 1:1000 dilution), anti-hIDO (Cell Signaling Technology, clone: D5J4E; 1:1,000 dilution), anti-FH/FHL-1 antibody (Origene, clone: OTI5H5, catalog # TA804532; 1:1,000 dilution), and anti-GAPDH (Cell Signaling Technology, clone: 14C10l; 1:1,000 dilution) overnight at 4°C. After washing 5 times with 1x TBST, membrane was incubated with donkey anti-rabbit/goat IgG antibodies conjugated with horseradish peroxidase (Jackson ImmunoResearch Inc.). The blotting membrane was then incubated with SuperSignal West Pico/Femto ECL substrate (Thermo Fisher Scientific) and visualized on a ChemiDoc (Bio-Rad).
Flow cytometry
Flow cytometry was performed as previously described (10). All of the conjugated antibodies were purchased from eBioscience and detailed information is given in Supplementary Table S2. Fc Blocking Ab (catalog # 14–9161–73), anti-mouse CD16/CD32 (catalog # 14–0161–82), fixation/permeabilization concentrate (catalog # 00–5123–56), fixation/permeabilization diluent (catalog # 00–5223–56), and permeabilization buffer (catalog # 00–8333–56) were also purchased from eBioscience. Cytometry data were acquired on a BD LSRFortessa 6-Laser flow cytometer and analyzed on FlowJo 6 software. This work was supported by the Northwestern University – Flow Cytometry Core Facility supported by NCI CA060553.
RNA isolation and real-time RT-PCR
Total RNA was extracted from freshly dissected tissue samples and cultured cells using the TRIzol reagent and PureLink RNA Mini Kit (Thermo Fisher Scientific), respectively. One microgram of total RNA was reverse transcribed into mRNA using iScript cDNA Synthesis Kit (Bio-Rad). qRT-PCR was performed on a CFX96 Touch Real-Time PCR Detection System using the default program setting (Bio-Rad). The sequences for all PCR primers are listed in the Supplementary Table S2. The relative quantitation of gene expression was calculated using the 2−ΔΔCt method (26) with normalization of the target threshold cycle (Ct) values to the internal housekeeping gene (GAPDH).
Trp and Kyn analysis by HPLC
Procedures for HPLC sample processing and analysis have been previously described (27).
Mass spectrometry quantification of CFH and FHL-1
We previously described how to evaluate plasma levels for CFH and FHL-1 (28). Briefly, plasma samples were thawed and vortexed, and a 5 μL aliquot taken and diluted with 90 μL 50 mmol/L ammonium bicarbonate, 2 μL of 1% (w/v) ProteaseMax (Promega), and 1 μL 500 mmol/L dithiothreitol. This was incubated at 56°C for 25 minutes to reduce cysteine residues. Three microlitres of 500 mmol/L iodoacetamide was then added and sample incubated in the dark for a further 15 minutes. To digest the protein, a further 43 μL of 50 mmol/L ammonium bicarbonate and 1 μL ProteaseMAX solution was added alongside 5 μL of 1 μg/μL endoproteinase Glu-C (Roche). Sample was mixed and digested for 16 hours at 25°C. Digested samples were spiked with heavy labeled synthetic peptide standards (FH: VTYKCFE; FHL-1: NGWSPTPRCIRVSFTL, each containing S-carboxymethylated cysteine and heavy labeled amino acids at the underlined residues; Cambridge Research Biochemicals) to a final concentration of 47.6 pg/μL for the FH and 0.95 pg/μL for FHL-1. Peptides were dried in a centrifugal evaporator and resuspended in 50 μL 0.1% (v/v) trifluroacetic acid. Four microlitres of this peptide solution was analyzed, providing final on-column standard peptide loads of 200 fmol FH and 2 fmol FHL-1 peptides, respectively. Peptides were separated using an Agilent 1200 series liquid chromatography system with a C18 column (250 mm × 2.1 mm I.D., Thermo Scientific Acclaim 120, 3 μmol/L particle size) at 50°C. Peptides were eluted using a gradient elution increasing from 5% acetonitrile to 25%. The flow rate was maintained at 250 μL/minute with an initial composition of 5% Buffer B [acetonitrile with 0.1% (v/v) formic acid]. The following gradient elution profile was used to separate the peptides (time: % acetonitrile): 0 minutes: 5%; 2 minutes: 5%; 3 minutes: 12%; 12 minutes: 15%; 15 minutes: 20%; 30 minutes: 25%; 31 minutes: 90%; 39 minutes: 90%; 40 minutes: 5%; 49 minutes: 5%. Eluted peptides were detected using an Agilent 6595 triple quadrupole mass spectrometer in SRM mode monitoring three transitions per peptide as shown in Supplementary Table S4. Data were extracted using Skyline software (https://skyline.ms/) and protein concentration was calculated by comparison of peak areas between the heavy labeled standard peptides and its endogenous counterpart.
Cell-proliferation assay
Two thousand to 3,000 tGBM cells per well were seeded on a 96-well plate. Cell growth at different time points was measured using the Cell Counting Kit 8 (Abcam, catalog # ab228554) following the product protocol. Absorbance at 460 nm was measured using a Synergy 2 multi-mode microplate reader (BioTek).
Statistical analysis
The cut-off value for gene-expression levels were determined with Cutoff Finder software (http://molpath.charite.de/cutoff/) using significance as the cutoff optimization method (29). Kaplan–Meier survival analysis was performed to estimate the survival distribution, while the Bonferroni-corrected, Mantel–Cox, or Gehan–Breslow–Wilcoxon log-rank tests were used to assess the statistical significance of differences between the stratified survival groups using GraphPad Prism (version 9, GraphPad Software, Inc.). Renyi family of test statistics was computed via SAS software (version 9.4, SAS Institute Inc.) to determine the survival difference between two groups given the presence of crossing hazard rates. Pearson correlation was used to analyze the relationship between each two genes’ mRNA expression level. Canonical-correlation analysis (CCA) was performed using concor() function in R package CCR. F-approximations of Wilks Lambda was used to test the statistical significance of canonical correlation coefficients, using p.asym() function in R package CCP. Comparisons between multiple groups were analyzed by one-way ANOVA using GraphPad Prism software. Differences were considered to be statistically significant when P < 0.05. SEM is presented as the error bar in all bar graphs and mean ± SEM was utilized to describe the data throughout the text unless specifically noted.
Results
Tumor cell IDO increases immune suppression through nonenzyme activity
To determine if GBM cell IDO possesses nonenzyme activity, we bred B6.129-Ido1tm1Alm/J (Ido1−/−) mice with GFAP(ERT2)→Cre;p53fl/fl;Rbfl/fl;pTENfl/fl mice (24) to generate an IDO-deficient tamoxifen-inducible transgenic mouse model of GBM (IDO−/−tGBM; Fig. 1A; top left). Tumor tissue was isolated from the mouse brain and disseminated into single cells (IDO−/−tGBM cells) until growing at a rate of exponential proliferation, in vitro (Fig. 1A, top right). Because the amino-acid sequence for the mouse and human IDO enzyme active site is conserved (30), site-directed mutagenesis was performed on a WT mouse IDO-mGFP cDNA fusion construct such that the derivative protein would change from a histidine to an alanine at the 350th amino acid (H350A). IDO−/−tGBM cells were then transduced with either an empty plasmid vector (VectorEMPTY), a vector expressing WT murine IDO cDNA (IDOWT), or a vector expressing the IDO-enzyme null cDNA (IDOH350A; Fig. 1A, bottom left). Fluorescence microscopy (Fig. 1A, bottom right), real-time RT-PCR, Western blotting, and HPLC for Trp and Kyn confirmed that both IDOWT- and IDOH350A-modified IDO−/−tGBM cells express IDO mRNA and protein as compared with the VectorEMPTY-expressing IDO−/−tGBM cells that are absent for IDO expression (Fig. 1B). The modified IDOWT-expressing IDO−/−tGBM cells show a significant increase of Kyn accumulation as compared with both the VectorEMPTY- and IDOH350A-expressing IDO−/−tGBM cells. The expression of IDO has no effect on the proliferation of IDO−/−tGBM cells, in vitro (Fig. 1B, right most panel). These data validate the successful reconstitution of IDO-deficient tumor cells with constructs expressing enzymatically-active or enzymatically-null IDO protein.
Figure 1.
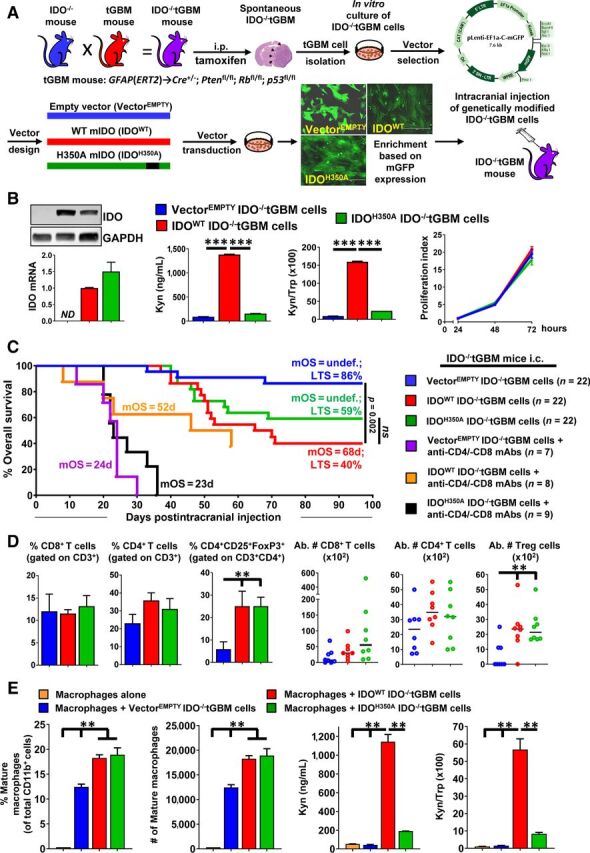
Tumor cell IDO mediates immune suppression in part through an IDO-dependent Trp metabolism-independent mechanism. A, Schematic representation of the protocol used to generate IDO−/−tGBM cells that express vectors with or without IDO cDNA. B, Left, Western blotting and qRT-PCR for the detection of IDO in IDO−/−tGBM cells expressing an empty vector (VectorEMPTY), WT IDO cDNA (IDOWT), or enzyme-null IDO cDNA (IDOH350A). Center, HPLC quantification of Kyn and Trp levels in cell-culture supernatants of genetically modified IDO−/−tGBM cell lines (n = 3 per cell line reflecting one representative experiment from more than 15 experimental repeats). Right, Cell-proliferation assay to compare the in vitro growth of genetically modified IDO−/−tGBM cells (n = 4 per cell line reflecting one representative experiment from 8 experimental repeats). C, IDO−/−tGBM mice were intracranially engrafted with genetically modified IDO−/−tGBM cells, with or without treatment of anti-CD4 and anti-CD8 mAbs beginning at day 3 prior to intracranial engraftment and twice/week for up to 30 days post intracranial engraftment followed by monitoring for OS (n = 7–22/group). The long-term survival rate (LTS) and mOS is labeled on the graph. Survival monitoring of mice depleted for leukocytes ended at 58 days post engraftment. D, Tumor-tissue samples were isolated at 4 weeks post tumor cell engraftment followed by the analysis of tumor-infiltrating leukocyte phenotypes. The percentage and absolute cell numbers of CD8+ T cells, CD4+ T cells, and CD4+CD25+FoxP3+ were quantified (n = 8 per group). E, Left, Flow cytometric analysis of genetically modified IDO−/−tGBM cells cocultured with splenic CD11b+ monocytes isolated from IDO−/−tGBM mice (n = 3 per group that reflect one representative experiment from 5 experimental repeats). Mature macrophage surface markers: CD11b+Ly6G−/lowLy6C+. Right, HPLC measurement of Trp and Kyn from the coculture cells experiment. **, P < 0.01; ***, P < 0.001. ND, not detectable; ns, not significant. All bar graphs represent mean ± SEM and dot plots show the median value as a horizontal line.
We next characterized the in vivo role of VectorEMPTY-, IDOWT-, and IDOH350A-expressing IDO−/−tGBM cells after their intracranial injection into syngeneic IDO−/−tGBM mice (Fig. 1A). Mice with intracranial IDOWT- and IDOH350A-expressing tumors have decreased OS with a median overall survival (mOS) of 67.5 days and undefined as compared with mice with VectorEMPTY-expressing tumors, respectively (Fig. 1C). The percentage of mortalities due to IDOWT and IDOH350A are not different from one another (P = 0.294). To further address whether the survival difference is caused by IDO-mediated immunosuppression, this experiment was repeated in IDO−/−tGBM mice treated with CD4+ T- and CD8+ T-cell depleting antibodies. T-cell depletion decreases survival as compared with mice that are not depleted for those leukocytes with the fastest mortality rates in mice with intracranial VectorEMPTY and IDOH350A-expressing tGBM cells (Fig. 1C). Similar survival effects are also found in IDO−/−tGBM mice with modified tumor cells and depleted for T cells and natural killer (NK) cells (Supplementary Fig. S1). The phenotype of tumor-infiltrating lymphocytes at 4 weeks post injection show a similar increase of Treg levels in IDOWT-expressing (25.06 ± 6.67%) and IDOH350A-expressing (25.05 ± 4.03%) brain tumors as compared to mice engrafted with VectorEMPTY-expressing tumors (5.89 ± 3.25%, P < 0.05; Fig. 1D). The expression of IDO is stable in both the IDOWT- and IDOH350A-expressing brain tumors as compared with the VectorEMPTY-expressing tumors that are absent for IDO expression in vivo (Supplementary Fig. S2). The in vitro coculture of both the IDOWT- and IDOH350A-expressing tumor cells with splenic CD11b+ monocytes induce a greater number of CD11b+Ly6c+Ly6g−/low mature macrophages at 18.25 ± 0.65% and 18.9 ± 1.4%, respectively (Fig. 1E) as compared with only 12.45 ± 0.55% among macrophages cocultured with VectorEMPTY-expressing tumor cells (P < 0.01; Fig. 1E, left). The effects on macrophage differentiation are tumor-cell IDO-dependent and Trp-metabolism independent (Fig. 1E, right).
IDO enhances complement factor H expression in human GBM cells
Since Fig. 1 collectively demonstrated that IDO enzyme activity does not fully account for its associated immunosuppressive and maladaptive effects in subjects with IDO-expressing glioma cells, we next questioned the mechanism by which IDO facilitates nonenzyme-mediated effects. Utilizing the Clariom D microarray platform, unmodified human U87 GBM cells were either left untreated or treated with human IFNγ and/or human IDO siRNA. A human U87 GBM cell line expressing WT human IDO cDNA that we previously described (11) was also treated with or without human IDO siRNA and all samples were analyzed collectively (Fig. 2A, top left). Principle component analysis (PCA) confirms the experimental reproducibility among the treatment conditions that were performed at different times and confirm the intragroup molecular similarity and intergroup molecular differences. Sixty-three gene candidates were identified based on their similar pattern of gene expression with IDO (Fig. 2A, Venn diagram). CFH has the closest correlation with IDO gene-expression changes in human GBM cells (Fig. 2A, bottom right). qRT-PCR confirms the IFNγ-dependent increase of CFH expression, and in contrast, the decreased expression of CFH when IDO expression is either absent or inhibited with IDO-specific siRNA (Fig. 2B). The IDO-mediated enhancement of CFH expression is independent of Trp metabolism since the treatment of U87 cells with the IDO enzyme inhibitor we previously characterized, BGB-5777 (17), has no effect on CFH expression levels in U87 (Fig. 2B) and PDX43 (Fig. 2C) GBM cells. In contrast, the inhibition of CFH expression has no effect on IDO expression levels (Fig. 2D). Since previous work showed that human GBM-infiltrating T cells induce IDO expression in vitro and in vivo (11, 31), CFH expression levels were analyzed in human immune system reconstituted humanized mice with intracranial human PDX43 and in patient-resected human GBM. Similar to intratumoral IDO and CD3ϵ levels, human CFH expression is absent in PDX43 when engrafted into humanized mice that are codepleted for human CD4+ and CD8+ T cells (Fig. 2E). Notably, the presence of IDO, CD3ϵ, and CFH are detectable in both newly-diagnosed and recurrent patient-resected GBM. These data collectively suggest that while tumor cell IDO and CFH are increased through a mechanism that depends on human tumor-infiltrating T cells, maximal CFH expression potential requires IDO-dependent Trp metabolism-independent effects.
Figure 2.
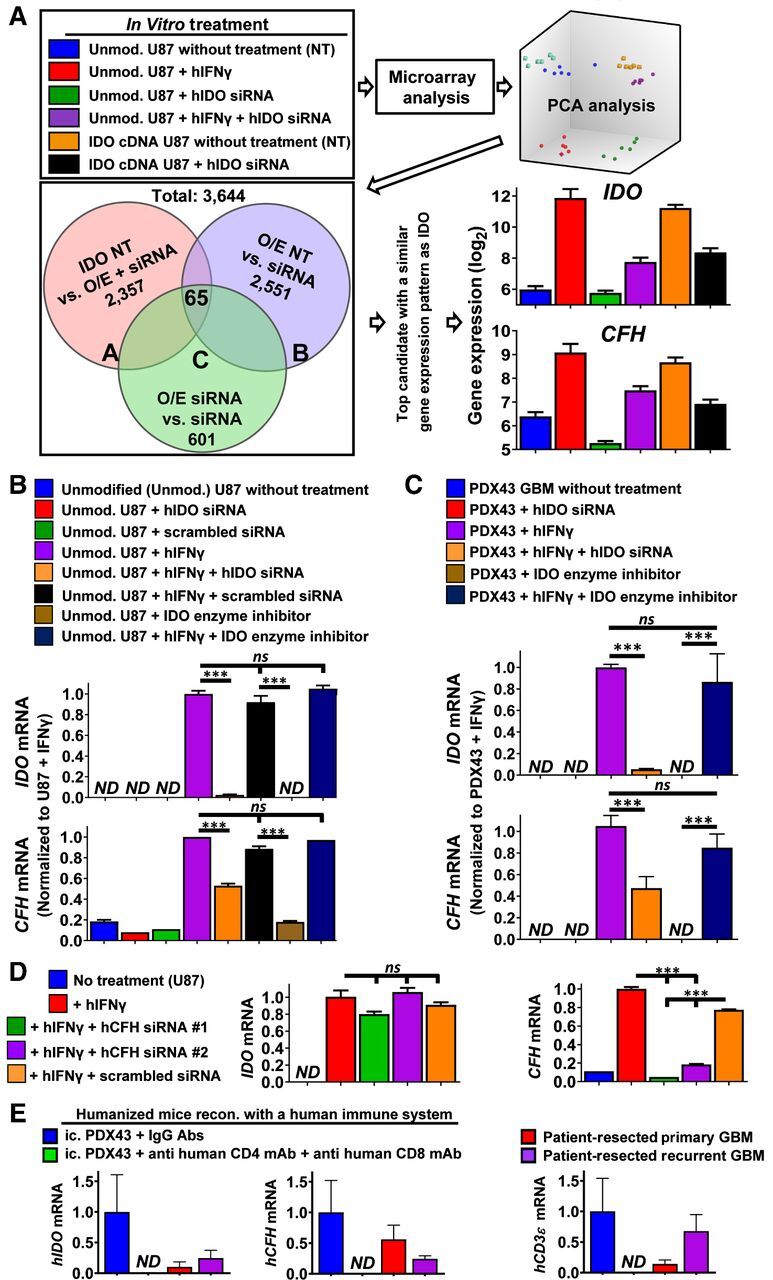
IDO nonmetabolically increases CFH levels in human GBM. A, Flow chart for the microarray experimental design, data analysis, as well as validation of results. B and C, Real-time RT-PCR confirmation of IDO regulatory effects on CFH expression in cultured U87 (B) or PDX43 GBM (C). Cells were exposed to a variety of conditions including transfection with IDO-specific siRNA (20 nmol/L) or scrambled control siRNA for 16 hours; treatment with or without 100 ng/mL human IFNγ; treatment with or without the IDO enzyme inhibitor, BGB-5777 (1 μmol/L); or IFNγ plus BGB-5777 for 24 hours. qRT-PCR was performed (n = 3 reflecting one representative experiment from four experimental repeats). D, qRT-PCR analysis of IDO and CFH mRNA expression on IFNγ-stimulated U87 cells treated with CFH siRNA or scrambled control siRNA. Same experimental conditions as in B (n = 3/group reflecting one representative experiment from three experimental repeats). E, mRNA expression levels for human IDO, CFH, and CD3ϵ were quantified and compared among intracranial PDX43 and patient-resected newly-diagnosed or recurrent intracranial human GBM (n = 4–9/group). Intracranially-engrafted (ic.) PDX43 was isolated from humanized mice with or without treatment of CD4+ and CD8+ T-cell depleting antibodies between 14 and 21 days post intracranial injection. ***, P < 0.001. All bar graphs represent mean ± SEM.
IDO and CFH demonstrate similar patterns of expression in patient-resected GBM and correlate with survival of patients with glioma
Based on the similar patterns of IDO and CFH expression in the established human U87 GBM cell line (Fig. 2B) and in human PDX43 GBM (Fig. 2C), we next explored whether such a relationship exists in patient-resected GBM. Figure 3A shows TCGA analysis of IDO and CFH mRNA levels, both of which progressively increase with glioma grade and are maximally expressed in GBM. Pearson correlation analysis indicates that CFH and IDO mRNA levels positively correlate in patient-resected GBM (r = 0.3962, P < 0.0001) as well as when all grades of glioma are analyzed simultaneously (r = 0.549, P < 0.0001; Fig. 3B). Kaplan–Meier analysis further demonstrates that higher CFH mRNA levels are inversely associated with survival of patients with glioma (Fig. 3C) and predictive of a faster rate to GBM recurrence (Fig. 3D). Intraglioma CFH expression is also affected by isocitrate dehydrogenase (IDH) status such that a higher level of CFH expression is observed among WT IDH (IDHwt)-expressing tumors (within grade II gliomas, P = 0.0037; within grade III gliomas, P < 0.0001; within GBM, P = 0.0056; Fig. 3E). CFH methylation for the cg23557926 locus is significantly different among grade II and grade III IDHwt and mutant IDH (IDHmut)-expressing glioma (P < 0.0001) but is not significantly different within GBM (Fig. 3F). Consistent with the analysis of humanized mice with intracranial PDX43 (Fig. 2D), GBM samples with a higher mRNA profile indicative of CD8+ T cells (CD3E, CD8A) possess significantly higher levels of CFH and IFNγ expression (P < 0.01; Figs. 2D and 3G). To further understand the relationship of human T cells with IDO and CFH in human GBM, the coculture of either naïve or activated T cells, conditioned media from activated T cells, and/or the treatment of anti-IFNγ was assessed in cultures of U87 GBM cells. Figure 3H demonstrates that activated human T cells and the associated conditioned media containing human IFNγ directly induce both IDO and CFH gene expression. This observation was further extended with the in vitro culturing of U87, PDX12, PDX39, and PDX43 treated with or without human IFNγ (Fig. 3I). Under all conditions, IFNγ increased CFH expression in human GBM. Collectively, the data indicate that IDO and CFH coordinately increase in patient-resected glioma, and higher CFH expression is inversely associated with survival of patients with glioma regardless of tumor grade, and that IFNγ-expressing GBM-infiltrating T cells enhance the expression of intratumoral CFH levels.
Figure 3.
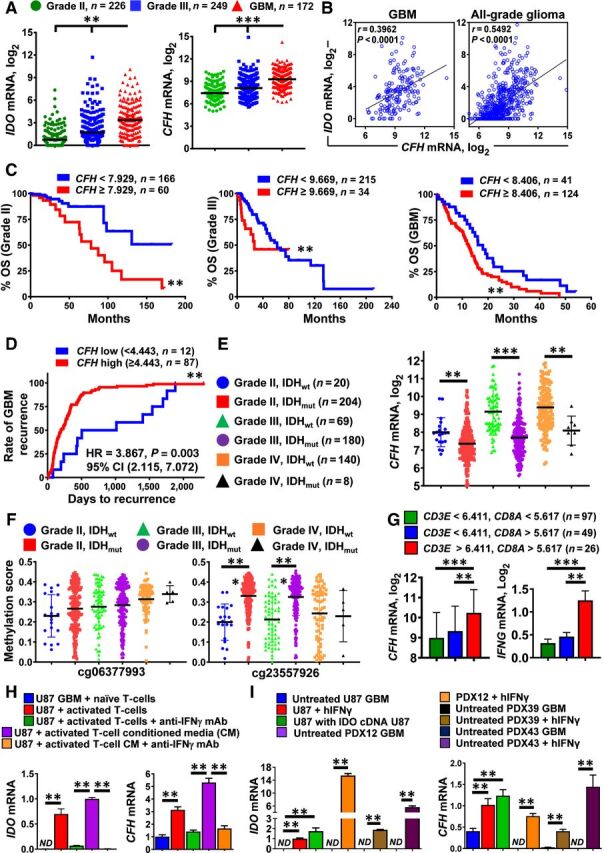
IDO and CFH mRNA levels positively correlate with T-cell infiltration in patient-resected GBM. A, mRNA expression levels for IDO and CFH in grade II (green; n = 226), grade III (blue; n = 249), and grade IV (GBM; red; n = 172) glioma of the RNA Hi-Seq. Illumina dataset as analyzed in TCGA. Horizontal lines in the scatter plots represent mean ± SEM. B, Pearson correlation analysis for IDO and CFH mRNA levels within GBM and all-grade glioma. C, Kaplan–Meier survival analysis of grade II (left), grade III (center), and grade IV (GBM, right) patients with glioma stratified by low CFH (blue) and high CFH (red) expression levels. D, Kaplan–Meier analysis of GBM recurrence. Recurrent GBM samples were identified from the ‘days to tumor recurrence’ section listed in the TCGA GBM clinical dataset. CFH mRNA expression levels were extracted from the Affymetrix U133a microarray dataset. E, CFH mRNA levels were compared among grade II (IDHwt; blue circle and IDHmut; red square), grade III (IDHwt; green triangle and IDHmut; purple circle), as well as grade IV (IDHwt; orange circle and IDHmut; black triangle) glioma. F, CFH DNA methylation analysis at two distinct genomic loci, cg06377993 and cg23557926 in grade II (IDHwt; blue circle and IDHmut; red square), grade III (IDHwt; green triangle and IDHmut; purple circle), and grade IV (IDHwt; orange circle and IDHmut; black triangle) glioma. G, Expression of CFH and IFNγ mRNA levels in patient-resected GBM tissue samples as categorized by CD3E and CD8A expression levels while accessing the TCGA GBM RNA-seq dataset. H, Detection of CFH mRNA in the human GBM cell T-cell coculture system in vitro. CD3+ human T cells were isolated under positive selection from PBMCs of patients with GBM. CFH mRNA levels were analyzed in U87 GBM cells cocultured with either naïve or activated T cells or conditioned medium from activated T cells in the presence or absence of IFNγ-neutralizing antibodies. Data were compiled from 3 independent experiments. I, In vitro expression analysis of human CFH mRNA in different GBM cells with or without the addition of human IFNγ. Data represent pooled data from 4 independent experiments. **, P < 0.01; ***, P < 0.001. All bar graphs represent mean ± SEM.
IDO enhances CFH isoform expression
The human CFH gene locus is on chromosome 1q32 in the regulators of complement activation (RCA) gene cluster. CFH encodes for an approximately 155 kDa secreted glycoprotein comprised of 20 contiguous complement control protein (CCP) modules (Fig. 4A, top) and a truncated splice variant referred to as FHL-1 that encodes a second isoform composed of CCPs 1 to 7 followed by a unique 4-amino acid sequence (Fig. 4A, bottom; ref. 32). Analysis of the TCGA indicates that both the full-length CFH and truncated CFH variant, FHL-1, positively correlate with IDO (Fig. 4B) and the T-cell surface marker, CD3ϵ (Fig. 4C), in patient-resected GBM. Both CFH isoforms are present in tumor lysate isolated from fresh patient-resected newly-diagnosed GBM or recurrent GBM (Fig. 4D, left). Protein expression for CFH and FHL-1 is also expressed in cell-culture supernatants but not in intracellular U87 GBM cell lysate and is higher after treatment with IFNγ (Fig. 4D, right). To further elucidate the dynamic mRNA expression of both CFH transcript variants as they relate to IDO levels over time, primers were designed for targeting the different CFH variants including the full-length variant by targeting CCPs 10 to 11 and the truncated variant by targeting the unique 4-amino acid sequence (Fig. 4A). IDO mRNA expression is detectable as early as 4 hours after treatment with IFNγ. In contrast, the earliest that the full-length and truncated CFH variants increased is at 12 hours post IFNγ treatment in U87 (Fig. 4E, top row) and PDX43 (Fig. 4E, bottom row) GBM cells. The protein expression kinetic profile (Fig. 4F) is similar to the mRNA profile (Fig. 4E). To confirm that IDO possesses a direct regulatory effect on CFH expression, we created a homologously IDO-deleted U87 (IDOKO U87) cell line using the CRISPR-Cas9 gene-editing approach (Fig. 4G). Whereas unmodified U87 treated with IFNγ expresses IDO protein and metabolizes Trp into Kyn, IDOKO U87 fails to express IDO protein and does not metabolize Trp. Strikingly, while IDO and both CFH splice variants are induced and upregulated after treatment with IFNγ in unmodified U87, respectively, no such increase takes place in IDOKO U87 cells (Fig. 4H). These data collectively confirm that upon stimulation with the T-cell effector cytokine, IFNγ, the increase of CFH splice variant expression levels is dependent on the coexpression of IDO in human GBM cells.
Figure 4.
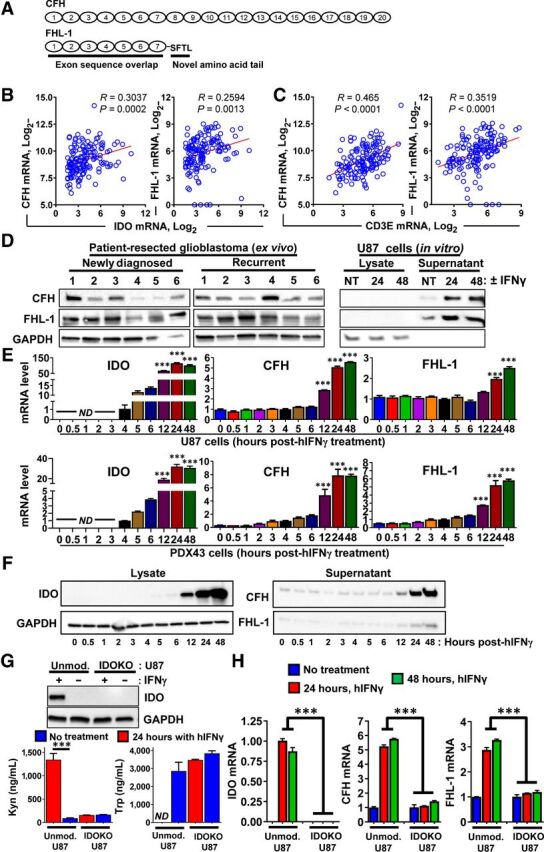
IDO enhances the expression of both CFH isoforms in human GBM. A, Schematic representation of CFH transcript variants reflecting the full-length (CFH) and truncated (FHL-1) sequences. B and C, Pearson correlation analysis of IDO (B) and CD3E (C) with CFH and FHL-1 using the TCGA GBM RNA-seq dataset. D, Left, Western blot analysis of surgically-resected tumor tissue samples from both patients with newly diagnosed GBM and patients with recurrent GBM. Right, Protein expression of full-length and truncated CFH variants in IFNγ-stimulated U87 cells. Cells were treated with IFNγ and cultured in serum-free medium for 24 to 48 hours. Both cell lysates and cell-culture supernatants were collected. The supernatants were concentrated with ultrafiltration and analyzed by Western blot along with cell lysate samples. One representative result is shown that reflects 4 experimental repeats. E, Time-course analysis of IDO, CFH, and FHL-1 mRNA expression after treatment with IFNγ in U87 (top row) and PDX43 (bottom row) GBM cells. The U87 cells were stimulated with 100 ng/mL human IFNγ and RNA lysates were extracted followed by qRT-PCR analysis (n = 4). F, Western blot of cell lysates and supernatants collected from the same experimental design as in E. Data from one representative experiment from two experiments are shown. G, Top, Western blot showing IDO protein levels in unmodified (Unmod.) as well as in CRISPR-Cas9 IDO-deleted (IDOKO) U87 cells. Unmod. U87 cells and IDOKO U87 cells were stimulated with 100 ng/mL human IFNγ for 24 hours. Protein lysates were collected from both untreated and IFNγ-treated cells followed by Western blotting analysis. Bottom, HPLC measurement of Kyn and Trp in Unmod. U87 and IDOKO U87 cells. H, Comparison of IDO, CFH, and FHL-1 mRNA expression induction in Unmod. and IDOKO U87 cells using qRT-PCR (n = 3 per group that reflects 1 representative experiment after two repeats). ***, P < 0.001. All bar graphs represent mean ± SEM.
Tumor cell CFH isoform expression enhances intratumoral immune suppression and decreases survival in a syngeneic brain tumor model
To better understand the downstream effects of IDO-enhanced tumor cell CFH expression, IDO−/−tGBM cells were engineered to express the truncated CFH splice variant, FHL-1 cDNA (Fig. 1A). The coculture of FHL-1–expressing tumor cells with splenic CD11b+ macrophages leads to a maximal expression of ARG1, CCL2, and IL6 in macrophages as compared with VectorEMPTY-expressing cells that are cocultured with macrophages or in macrophages cultured alone (P < 0.05; Fig. 5A). FHL-1 expression also directly increases ARG1 and CCL2 levels in tumor cells (Fig. 5B) suggesting that FHL-1 binds to at least one receptor on macrophages and on tumor cells for carrying out its immunosuppressive gene reprogramming effects. The intracranial injection of FHL-1 cDNA-expressing IDO−/−tGBM cells into syngeneic IDO−/−tGBM mice leads to 100% mortality and a mOS of 33 days that is significantly lower than mice with brain tumor cells expressing VectorEMPTY (P < 0.0001; Fig. 5C). The survival benefit of mice with tumor cells expressing VectorEMPTY and treated with a nonspecific IgG antibody is reduced to a mOS of 20.5 days when T cells and NK cells are codepleted (P < 0.0001; Fig. 5D). The decreased mOS of 35 days in mice with tumors expressing FHL-1 cDNA treated with nonspecific IgG antibodies is also reduced to a mOS of 17 days in mice codepleted for T and NK cells. Flow cytometric analysis of brain tumors isolated at 3 weeks post intracranial injection show a marked decrease of tumor-infiltrating CD8+ T cells (45.17% ± 3.78% vs. 17.52% ± 1.72%, P < 0.001), an increase of Tregs (12.23% ± 2.79% vs. 29.23% ± 3.535, P < 0.01), and an increase of total MDSCs (CD3−CD45+CD11b+Ly6C+; 6.98% ± 1.56% vs. 17.71% ± 2.80%, P < 0.01) that primarily reflected monocytic-type MDSCs (M-MDSCs: CD11b+Ly6G−Ly6Chi) in FHL-1–expressing tumors as compared with tumors expressing VectorEMPTY (Fig. 5E). Gene-expression analysis of FHL-1–expressing brain tumors isolated at 3 weeks post intracranial injection showed an immunosuppressive signature with increases of ARG1, CCL2, IL6, and Foxp3 expression as compared with VectorEMPTY brain tumors (P < 0.01), as well as to contralateral nontumor brain (P < 0.05; Fig. 5F). Collectively, the data show that when IDO-deficient brain tumor cells are genetically engineered to express a CFH variant, the resulting cells potently increase intratumoral immune suppression and decrease OS.
Figure 5.
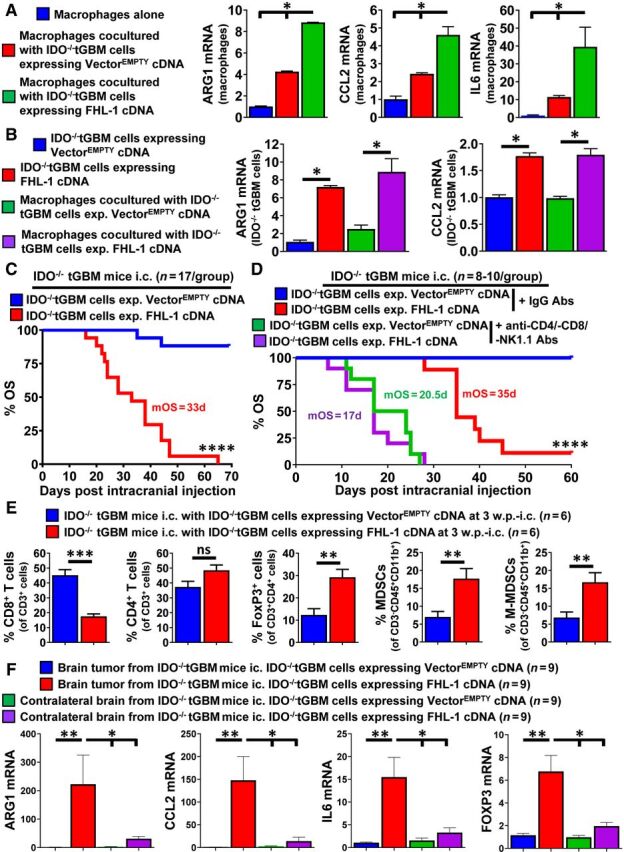
CFH increases immunosuppressive factor expression and decreases OS in a syngeneic mouse brain-tumor model. A, Splenic CD11b+ monocytes were isolated from IDO−/−tGBM mice and cocultured with either IDO−/−tGBM cells expressing VectorEMPTY or FHL-1 cDNA. RT-PCR quantification for ARG1, CCL2, and IL6 mRNA levels were determined in cultured macrophages (blue bar), macrophages cocultured with IDO−/−tGBMs expressing VectorEMPTY (red bar), or macrophages cocultured with FHL-1 cDNA (green bar; n = 3 per group reflecting data compiled from 2 independent experiments). B, RT-PCR quantification for ARG1 and CCL2 in cultured IDO−/−tGBMs alone (blue bar), IDO−/−tGBM cells expressing FHL-1 cDNA (red), IDO−/−tGBM cells expressing VectorEMPTY cocultured with macrophages (green bar) and IDO−/−tGBM cells expressing FHL-1 cDNA cocultured with macrophages (purple bar; n = 3 per group reflecting data compiled from 2 independent experiments). C, Kaplan–Meier survival analysis of IDO−/−tGBM mice intracranially engrafted with either IDO−/−tGBM cells expressing VectorEMPTY (blue) or IDO−/−tGBM cells expressing FHL-1 cDNA (red; n = 17/group). Colorful numbers represent median survival (MS). Plots without labeled numbers indicate undefined MS. D, Kaplan–Meier survival analysis of IDO−/−tGBM mice intracranially engrafted with either IDO−/−tGBM cells expressing VectorEMPTY or IDO−/−tGBM cells expressing FHL-1 cDNA in the presence or absence of antimouse-CD4 mAb, -CD8 mAb, and -NK1.1 mAb (n = 8–10/group). Colorful numbers represent MS. E, Flow-cytometry analysis of tumor-infiltrating lymphocytes and MDSCs from GBM tissue samples collected at 3-week post intracranial injection. F, Gene-expression analysis on mouse GBM tumor tissues and contralateral nontumor brain samples. Mice intracranially injected with modified IDO−/−tGBM cells were euthanized when displaying endpoint symptoms. Brain tumor tissues and contralateral brain tissues were collected and stored in Trizol reagent. At the end of survival analysis (65 days post injection; Fig. 5C), all samples were subjected to RNA extraction and real-time RT-PCR. Plots without labeled numbers indicate undefined MS. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. All bar graphs represent mean ± SEM.
Circulating and intratumoral CFH correlations in patients with GBM
Since the two CFH isoforms are normally found in human plasma (32), we next compared the protein levels for the full length and truncated CFH variants in nontumor aneurysm- and age-matched GBM-patient plasma. Figure 6A shows the systemic levels of CFH at 865 nmol/L ± 37.42 nmol/L and 788 nmol/L ± 43.85 nmol/L in plasma from patients with aneurysm and GBM, respectively. It further shows the systemic levels for FHL-1 at 20.67 nmol/L ± 1.34 nmol/L in patients with aneurysm that is decreased to 14.69 ± 1.39 nmol/L in patients with GBM (P < 0.05). The ratio of CFH:FHL-1 is also decreased in patients with GBM as compared with the aneurysm control group (P < 0.05; Fig. 6A). No difference was observed regarding systemic CFH and FHL-1 levels when comparing the plasma of patients with newly diagnosed and recurrent GBM (Fig. 6B). Intratumorally, CFH expression positively correlates with mRNA levels for many other immunosuppressive modulators including PD-L1, PD-L2, PD-1, CTLA-4, LAG3, BTLA, and FGL2 in GBM (Fig. 6C). Notably, CFH also broadly correlates with mRNA signatures for infiltrating leukocytes. The data collectively suggest that there is a unique profile of CFH variant expression in the circulation as compared with the intratumoral CFH expression. In addition, increases of intratumoral CFH expression positively correlate with increases for other molecular and cellular mediators, inflammation, and immune suppression (Fig. 6D). A hypothetical model based on the data here and previously published shows how the CFH contributes to immunosuppressive Treg accumulation in GBM (Fig. 6E).
Figure 6.
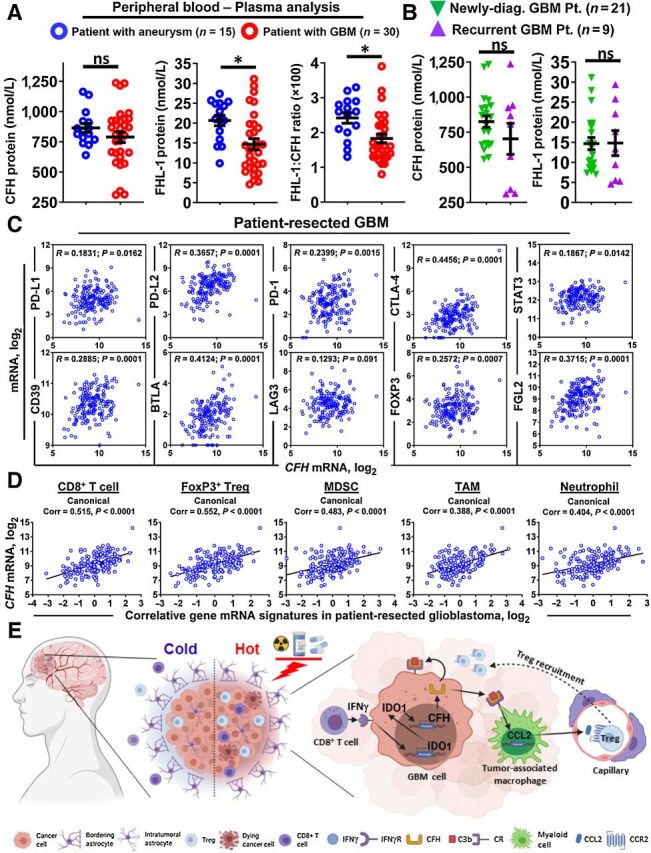
Systemic and local complement factor H levels and the relationship with other immunosuppressive factors in patient-resected GBM. A and B, Quantification of protein levels of full-length and truncated CFH in patient plasma samples by mass spectrometry. C, Pearson correlation analysis for CFH mRNA with PD-L1, PD-L2, PD-1, CTLA-4, STAT3, CD39, BTLA, LAG3, FOXP3, and FGL2 in GBM. Each small circle in the plot represents the expression in a single patient. D, Canonical correlation analysis of CFH with major tumor immune cell types. The signature genes of each type of immune cells are defined as: CD8+ T cell (CD3ϵ, CD8α), Treg (CD3ϵ, CD4, CD25, FoxP3), MDSC (CD14, CD11b, CD33, Arg1), TAM (CD14, HLA-DR, CD312, CD115, CD163, CD204, CD301, CD206), and neutrophil (CD11b, CD16, CD66b, ELANE). E, Schematic presentation of a hypothesis based on the findings reported here and elsewhere. Standard of care treatment radiation (RT) and TMZ enhances inflammatory mechanisms that alter the immune-tolerant (cold) GBM microenvironment into more inflamed (hot) conditions which is partially caused by tumor-infiltrating IFNγ+ CD8+ T cells. The IFNγ acts on human GBM cells to induce IDO expression, which in turn, enhances CFH expression levels through a nonenzymatic mechanism. CFH acts on complement receptors in an autocrine and paracrine manner; the latter of which elicits CCL2 expression in TAMs. The TAMs then facilitate Treg and additional monocyte recruitment into the GBM which reinforces the immunosuppressive microenvironment.
Discussion
The relationship between IDO and its regulatory effects on the complement cascade was initially described in the anatomical setting of placenta (33). At the time, there was no description of how treatment with the IDO pathway inhibitor, 1-methyl Trp (1-MT), affected intra-placental CFH expression levels or Trp and Kyn levels. In a subsequent study, Li and colleagues demonstrated that pharmacologic IDO pathway modulation with either 1-MT or NLG919 triggered chemo-radiation–dependent complement C3 deposition at sites of tumor growth in the GL261-based mouse orthotopic brain-tumor model (34). However, there was no description of how 1-MT or NLG919 affected intratumoral CFH expression levels or Trp and Kyn levels. This is of significant notability since D1-MT, which has the most potent anti–brain tumor effects (16) and is used as the exclusive stereoisomer of 1-MT in clinical trials (14), does not effectively inhibit Trp metabolism (35, 36).
Complement C3 functions as a pivotal inducer by activating the complement-mediated inflammatory pathways, while CFH/FHL-1 plays a critical inhibitory role that suppresses complement-mediated inflammatory responses. With respect to the observations of our study, it's possible that the mechanistic effects of IDO enzyme activity on C3 activation are independent from those underlying the IDO nonenzyme effects on CFH/FHL-1 regulation. Since IDO protein is expressed in a majority of human cancers including GBM (8), further investigation that focuses on the molecular mechanism(s) underlying IDO regulation of C3 activation and CFH/FHL-1 is warranted. In the present study, we showed for the first time that human tumor cells utilize IDO nonenzyme activity to enhance the expression level of immunosuppressive CFH and its truncated isoform, FHL-1. We further demonstrated that tumor cell FHL-1: (i) enhanced macrophage maturity; (ii) enhanced macrophage expression for ARG1, CCL2, and IL6; and (iii) decreased the survival of mice with brain tumors in part by suppressing the anti-GBM T- and NK-cell immune response. Translationally relevant, we also showed that IDO and CFH expression are positively correlated in patient-resected GBM and that increased intratumoral CFH/FHL-1 levels are associated with decreased survival of patients with GBM. This study therefore contributes a mechanistic understanding for why pharmacologic IDO enzyme inhibitor treatment fails to reverse the immunosuppressive effects of IDO when administered as a monotherapy (14).
Questions regarding the immunosuppressive role of CFH/FHL-1 remain to be addressed in the setting of GBM. First, does full-length CFH play an identically immune-tolerant role as the isoform, FHL-1? Previous work showed that CFH-treated monocyte-derived dendritic cells (MoDC) had a tolerogenic state, such as the production of immunomodulatory mediators including IL10 and TGFβ, a reduced expression for CCR7 and chemotactic migration, impaired CD4+ T-cell alloproliferation, and an induction of CD4+CD127low/−CD25highFoxp3+ Tregs (37). One future direction is to investigate how CFH/FHL-1 regulates complement pathway activation in GBM. However, as demonstrated in Fig. 6E and by Olivar and colleagues, CFH/FHL-1 may possess immunomodulatory activities that are independent of complement regulation, raising important considerations for further mechanistic study (37).
Our microarray analysis discovered a similar pattern of regulation between IDO and CFH in human GBM, in vitro, and this effect was confirmed in patient-resected tumors, in vivo. We further showed that both the full-length CFH and truncated variant, FHL-1, suppress the immune response in GBM. Although both CFH and FHL-1 have been previously associated with mechanisms of immune evasion (38, 39), no previous investigation focused on the role of FHL-1 in tumor-induced immunosuppression. Interestingly, FHL-1 is not expressed by mice (40) which allowed us to ignore the potential for multi-species gene expression competition of similar protein products during our investigation of IDO−/−tGBM cells expressing human FHL-1 cDNA. In summary, we have revealed a nonenzymic function of IDO in human tumor cells that nonmetabolically increases immunosuppression and contributes to poorer survival outcomes.
Authors' Disclosures
J. Griffiths reports grants from MCRC during the conduct of the study, as well as grants from MCRC outside the submitted work. R.D. Unwin reports other support from Complement Therapeutics outside the submitted work; in addition, R.D. Unwin has a patent for Complementome Assay (GB2014570.2) pending and licensed to Complement Therapeutics. C. Horbinski reports grants from NIH during the conduct of the study. R.V. Lukas reports other support from BMS, as well as personal fees from Novocure outside the submitted work. G.E. Schiltz reports grants from NIH during the conduct of the study. No disclosures were reported by the other authors.
Supplementary Material
Supplementary Table 1. Demographics of patient-isolated tissue samples.
Supplementary Table 2. Nucleic acid-based reagents utilized.
Supplementary Table 3. Differentially expressed genes that possess the strongest correlation with IDO in human GBM cells.
Supplementary Table 4. Mass spectrometry parameters for detecting CFH and FHL-1 in plasma.
Supplemental Figure 1. Overall survival of syngeneic mice with intracranial IDO-/-tGBM tumors and depleted for CD4+ T, CD8+ T, and NK1.1+ immune cells.
Supplemental Figure 2. Histological evaluation of tumor progression and confirmation of IDO-mGFP expression in brain tumors.
Acknowledgments
This work was supported in part by NIH grants R01NS102669 (C. Horbinski), P50CA221747 (R.V. Lukas, C. Horbinski, D.A. Wainwright), R01NS097851 (D.A. Wainwright), and K02AG068617 (D.A. Wainwright); UK Medical Research Council facilitated by the Manchester NIHR Biomedical Research Centre and the Greater Manchester Comprehensive Local Research Network MR/P025838/1 and MR/M008959/1 (J. Griffiths, R.D. Unwin, S.J. Clark); BrainUp grant 2136 (R.V. Lukas and D.A. Wainwright); and American Cancer Society RSG-21–058–01 - CCE (D.A. Wainwright).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Contributions
L. Zhai: Conceptualization, investigation, writing–original draft. A. Bell: Investigation. E. Ladomersky: Investigation. K.L. Lauing: Resources, investigation, project administration. L. Bollu: Investigation. B. Nguyen: Investigation. M. Genet: Investigation. M. Kim: Investigation. P. Chen: Conceptualization, investigation. X. Mi: Formal analysis. J.D. Wu: Supervision, investigation. M.J. Schipma: Formal analysis, supervision. B. Wray: Formal analysis. J. Griffiths: Resources, formal analysis. R.D. Unwin: Resources, formal analysis. S.J. Clark: Resources, formal analysis, supervision, investigation. R. Acharya: Formal analysis. R. Bao: Conceptualization, resources, data curation, formal analysis, supervision. C. Horbinski: Resources, investigation. R.V. Lukas: Investigation, writing–review and editing. G.E. Schiltz: Resources, validation, investigation, writing–review and editing. D.A. Wainwright: Conceptualization, resources, data curation, software, formal analysis, supervision, funding acquisition, validation, investigation, visualization, methodology, writing–original draft, project administration, writing–review and editing.
References
- 1. Adamson C, Kanu OO, Mehta AI, Di C, Lin N, Mattox AK, et al. Glioblastoma multiforme: a review of where we have been and where we are going. Expert Opin Investig Drugs 2009;18:1061–83. [DOI] [PubMed] [Google Scholar]
- 2. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352:987–96. [DOI] [PubMed] [Google Scholar]
- 3. Ries LAGSM, Gurney JG, Linet M, Tamra T, Young JL, Bunin GR. Cancer Incidence and Survival among Children and Adolescents: United States SEER Program 1975–1995. Bethesda (MD): National Cancer Institute, SEER Program; 1999. [Google Scholar]
- 4. Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science 2013;342:1432–3. [DOI] [PubMed] [Google Scholar]
- 5. Kong D-S, Nam D-H, Kang S-H, Lee JW, Chang J-H, Kim J-H, et al. Phase III randomized trial of autologous cytokine-induced killer cell immunotherapy for newly diagnosed glioblastoma in Korea. Oncotarget 2017;8:7003–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liau LM, Ashkan K, Tran DD, Campian JL, Trusheim JE, Cobbs CS, et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J Trans Med 2018;16:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A, et al. Effect of nivolumab vs. bevacizumab in patients with recurrent glioblastoma: the CheckMate 143 phase 3 randomized clinical trial. JAMA Oncol 2020;6:1003–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med 2003;9:1269–74. [DOI] [PubMed] [Google Scholar]
- 9. Shimizu T, Nomiyama S, Hirata F, Hayaishi O. Indoleamine 2,3-dioxygenase. Purification and some properties. J Bio Chem 1978;253:4700–6. [PubMed] [Google Scholar]
- 10. Wainwright DA, Balyasnikova IV, Chang AL, Ahmed AU, Moon KS, Auffinger B, et al. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin Cancer Res 2012;18:6110–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhai L, Ladomersky E, Lauing KL, Wu M, Genet M, Gritsina G, et al. Infiltrating T cells increase IDO1 expression in glioblastoma and contribute to decreased patient survival. Clin Cancer Res 2017;23:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhai L, Ladomersky E, Lenzen A, Nguyen B, Patel R, Lauing KL, et al. IDO1 in cancer: a Gemini of immune checkpoints. Cell Mol Immunol 2018;15:447–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhai L, Lauing KL, Chang AL, Dey M, Qian J, Cheng Y, et al. The role of IDO in brain tumor immunotherapy. J Neurooncol 2015;123:395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhai L, Spranger S, Binder DC, Gritsina G, Lauing KL, Giles FJ, et al. Molecular pathways: targeting IDO1 and other tryptophan dioxygenases for cancer immunotherapy. Clin Cancer Res 2015;21:5427–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Muller AJ, Manfredi MG, Zakharia Y, Prendergast GC. Inhibiting IDO pathways to treat cancer: lessons from the ECHO-301 trial and beyond. Semin Immunopath 2019;41:41–8. [DOI] [PubMed] [Google Scholar]
- 16. Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res 2014;20:5290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ladomersky E, Zhai L, Lenzen A, Lauing KL, Qian J, Scholtens DM, et al. IDO1 inhibition synergizes with radiation and PD-1 blockade to durably increase survival against advanced glioblastoma. Clin Cancer Res 2018;24:2559–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ladomersky E, Zhai L, Lauing KL, Bell A, Xu J, Kocherginsky M, et al. Advanced age increases immunosuppression in the brain and decreases immunotherapeutic efficacy in subjects with glioblastoma. Clin Cancer Res 2020;26:5232–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhai L, Bell A, Ladomersky E, Lauing KL, Bollu L, Sosman JA, et al. Immunosuppressive IDO in cancer: mechanisms of action, animal models, and targeting strategies. Front Immunol 2020;11:1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhai L, Ladomersky E, Dostal CR, Lauing KL, Swoap K, Billingham LK, et al. Non-tumor cell IDO1 predominantly contributes to enzyme activity and response to CTLA-4/PD-L1 inhibition in mouse glioblastoma. Brain, Behav Immun 2017;62:24–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hashizume R, Ozawa T, Dinca EB, Banerjee A, Prados MD, James CD, et al. A human brainstem glioma xenograft model enabled for bioluminescence imaging. J Neurooncol 2010;96:151–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Giannini C, Sarkaria JN, Saito A, Uhm JH, Galanis E, Carlson BL, et al. Patient tumor EGFR and PDGFRA gene amplifications retained in an invasive intracranial xenograft model of glioblastoma multiforme. Neuro-oncol 2005;7:164–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sarkaria JN, Carlson BL, Schroeder MA, Grogan P, Brown PD, Giannini C, et al. Use of an orthotopic xenograft model for assessing the effect of epidermal growth factor receptor amplification on glioblastoma radiation response. Clin Cancer Res 2006;12:2264–71. [DOI] [PubMed] [Google Scholar]
- 24. Chow LM, Endersby R, Zhu X, Rankin S, Qu C, Zhang J, et al. Cooperativity within and among Pten, p53, and Rb pathways induces high-grade astrocytoma in adult brain. Cancer Cell 2011;19:305–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Efron B, Tibshirani R. Empirical bayes methods and false discovery rates for microarrays. Genet Epidemiol 2002;23:70–86. [DOI] [PubMed] [Google Scholar]
- 26. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001;25:402–8. [DOI] [PubMed] [Google Scholar]
- 27. Zhai L, Dey M, Lauing KL, Gritsina G, Kaur R, Lukas RV, et al. The kynurenine to tryptophan ratio as a prognostic tool for glioblastoma patients enrolling in immunotherapy. J Clin Neurosci 2015;22:1964–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cipriani V, Tierney A, Griffiths JR, Zuber V, Sergouniotis PI, Yates JRW, et al. Beyond factor H: The impact of genetic-risk variants for age-related macular degeneration on circulating factor-H-like 1 and factor-H-related protein concentrations. Am J Hum Genet 2021;108:1385–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Budczies J, Klauschen F, Sinn BV, Gyorffy B, Schmitt WD, Darb-Esfahani S, et al. Cutoff Finder: a comprehensive and straightforward Web application enabling rapid biomarker cutoff optimization. PLoS One 2012;7:e51862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Littlejohn TK, Takikawa O, Truscott RJW, Walker MJ. Asp274 and His346 are essential for heme binding and catalytic function of human indoleamine 2,3-dioxygenase. J Biol Chem 2003;278:29525–31. [DOI] [PubMed] [Google Scholar]
- 31. O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med 2017;9:eaaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Parente R, Clark SJ, Inforzato A, Day AJ. Complement factor H in host defense and immune evasion. Cell Mol Life Sci 2017;74:1605–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science 1998;281:1191–3. [DOI] [PubMed] [Google Scholar]
- 34. Li M, Bolduc AR, Hoda MN, Gamble DN, Dolisca SB, Bolduc AK, et al. The indoleamine 2,3-dioxygenase pathway controls complement-dependent enhancement of chemo-radiation therapy against murine glioblastoma. J Immunother Cancer 2014;2:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lob S, Konigsrainer A, Zieker D, Brucher BL, Rammensee HG, Opelz G, et al. IDO1 and IDO2 are expressed in human tumors: levo- but not dextro-1-methyl tryptophan inhibits tryptophan catabolism. Cancer immunology, immunotherapy: CII 2009;58:153–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Qian F, Liao J, Villella J, Edwards R, Kalinski P, Lele S, et al. Effects of 1-methyltryptophan stereoisomers on IDO2 enzyme activity and IDO2-mediated arrest of human T cell proliferation. Cancer immunology, immunotherapy: CII 2012;61:2013–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Olivar R, Luque A, Cárdenas-Brito S, Naranjo-Gómez M, Blom AM, Borràs FE, et al. The complement inhibitor factor H generates an anti-inflammatory and tolerogenic state in monocyte-derived dendritic cells. J Immunol 2016;196:4274. [DOI] [PubMed] [Google Scholar]
- 38. Junnikkala S, Hakulinen J, Jarva H, Manuelian T, Bjørge L, Bützow R, et al. Secretion of soluble complement inhibitors factor H and factor H-like protein (FHL-1) by ovarian tumour cells. Br J Cancer 2002;87:1119–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Junnikkala S, Jokiranta TS, Friese MA, Jarva H, Zipfel PF, Meri S. Exceptional resistance of human H2 glioblastoma cells to complement-mediated killing by expression and utilization of factor H and factor H-like protein 1. J Immunol 2000;164:6075–81. [DOI] [PubMed] [Google Scholar]
- 40. Pouw RB, Vredevoogd DW, Kuijpers TW, Wouters D. Of mice and men: The factor H protein family and complement regulation. Mol Immunol 2015;67:12–20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Demographics of patient-isolated tissue samples.
Supplementary Table 2. Nucleic acid-based reagents utilized.
Supplementary Table 3. Differentially expressed genes that possess the strongest correlation with IDO in human GBM cells.
Supplementary Table 4. Mass spectrometry parameters for detecting CFH and FHL-1 in plasma.
Supplemental Figure 1. Overall survival of syngeneic mice with intracranial IDO-/-tGBM tumors and depleted for CD4+ T, CD8+ T, and NK1.1+ immune cells.
Supplemental Figure 2. Histological evaluation of tumor progression and confirmation of IDO-mGFP expression in brain tumors.