

Abstract
The hepatic MAPK cascade leading to JNK activation has been implicated in the pathogenesis of nonalcoholic fatty liver /non-alcoholic steatohepatitis (NAFL/NASH). In acute hepatotoxicity we previously identified a pivotal role for mitochondrial SH3BP5 (SAB) as a target of JNK which sustains its activation through promotion of reactive oxygen species (ROS) production.
Aim:
Assess the role of hepatic SAB in experimental NASH and metabolic syndrome.
Results:
In mice fed high-fat, high-calorie, high-fructose (HFHC) diet, SAB expression progressively increased through a sustained JNK/ATF2 activation loop. Inducible deletion of hepatic SAB markedly decreased sustained JNK activation and improved systemic energy expenditure at 8 weeks followed by decreased body fat at 16 weeks of HFHC diet. After 30 weeks mice treated with control-ASO developed steatohepatitis and fibrosis which was prevented by Sab-ASO treatment. P-JNK and P-ATF2 were markedly attenuated by Sab-ASO treatment. After 52 weeks of HFHC feeding control N-acetylgalactosamine antisense oligonucleotide (GalNAc-Ctl-ASO) treated mice fed the HFHC diet exhibited progression of steatohepatitis and fibrosis but GalNAc-Sab-ASO treatment from weeks 40 to 52 reversed these findings while decreasing hepatic SAB, P-ATF2, and P-JNK to chow fed levels.
Conclusions:
Hepatic SAB expression increases in HFHC diet fed mice. Deletion or knockdown of SAB inhibited sustained JNK activation and steatohepatitis, fibrosis, and systemic metabolic effects, suggesting that induction of hepatocyte Sab is an important driver of the interplay between the liver and the systemic metabolic consequences of overfeeding. In established NASH, hepatocyte targeted GalNAc-Sab-ASO treatment reversed steatohepatitis and fibrosis.
Keywords: c-Jun-N-terminal kinase (JNK), antisense oligonucleotide (ASO), insulin resistance, fibrosis, NASH
Introduction
Obesity, diabetes, and the metabolic syndrome have merged as major causes of cirrhosis and its complications, including HCC.(1–3) This disease begins as non-alcoholic fatty liver (NAFL) which progresses in some patients to steatohepatitis (NASH) and fibrosis. The pathogenesis of NAFLD (NAFL/NASH) is complex and incompletely understood. Critical in the development of this disease is the interplay of the systemic effects of the metabolic syndrome on the liver as well as the intrinsic effects in the liver and the crosstalk between the liver and the peripheral manifestations of the metabolic syndrome. (4–6)
Considerable evidence has emerged supporting the contribution of the MAPK signaling cascade in the response of hepatocytes to overfeeding induced metabolic dysregulations and the influence of these intrinsic effects in the liver on the systemic metabolic syndrome.(7–9) Critical in this context is sustained activation of JNK in hepatocytes which affects a broad range of targets in metabolic dysregulation, lipotoxicity, inflammation, and fibrosis.(10–15) In mouse and human NAFL/NASH, activated JNK (P-JNK) but not total JNK is increased, and deletion of hepatic JNK 1 and 2 prevents diet induced hepatic steatosis and systemic metabolic effects.(8)
Our previous work in models of acute liver injury identified an interaction of activated JNK with mitochondria which is mediated by a protein in the mitochondrial outer membrane referred to a SAB (SH3BP5).(16–19) SAB is a P-JNK docking protein and substrate of JNK which promotes an intramitochondrial signaling pathway impairing mitochondrial function and promoting ROS production, which feedback to activate upstream MAP3Kinases and sustain JNK activation.(10,12,17–20) Based on the known importance of the hepatic MAPK pathway in fatty liver disease and the pivotal role of the P-JNK-mitoSAB-ROS activation loop in mediating sustained JNK activation in acute liver injury, the current work has addressed the expression and importance of hepatocyte SAB in sustaining JNK activation and its hepatic and systemic consequences in diet-induced metabolic syndrome and the progression of NAFL/NASH in mice. Furthermore, we provide evidence using inducible hepatocyte specific deletion or antisense oligonucleotide (ASO) knockdown to support the potential of SAB as a therapeutic target.
Materials and Methods
Mice and diet
All animal procedures were conducted utilizing protocols and methods approved by the Institutional Animal Care and Use Committee and were in compliance with Animal Welfare Act and NIH Guide for the Care and Use of Laboratory Animals. All animal procedures and diets are described in supporting materials and methods.
Statistical analysis
Statistical tests included two-way analysis of variance (ANOVA) and co-variance (ANCOVA), Student t-test, Welch’s t-test, linear regression, and Spearman correlation analysis. Data were expressed as means ± SEM from a minimum of three independent experiments and p < 0.05 or FDR < 0.05 was considered statistically significant. Statistical tests for RNA-seq and IPA analysis followed according to the program.
Results
Increased hepatic SAB expression and P-JNK activation in diet-induced NAFLD
We first examined the expression of SAB and P-JNK in an animal model of diet induced fatty liver disease. Two month old male wild type littermates were fed chow or high-fat, high-calorie, high-fructose (HFHC) diet which provides 58% of calories from saturated fat and 6.5% from fructose.(6) To assess the effects of HFHC in the early stage of liver disease, RNA-seq analysis after 8 weeks of HFHC versus chow feeding confirmed that hepatic genes of lipid/cholesterol metabolism were significantly upregulated in HFHC fed mice and de novo lipogenesis/cholesterol synthesis and lipid-oxidation were major pathways affected in the HFHC diet fed mice (GSE154426) (Fig.S1A,B). Expression of hepatic SAB increased over the course of prolonged HFHC diet feeding, increasing at 8 weeks and reached a plateau from 16 weeks (> 6 fold) to 30 weeks of feeding (Fig.1A,B). Sab mRNA expression progressively increased over the course of HFHC diet feeding (Fig.1C). Of note, the magnitude of the increased SAB protein levels was greater than the mRNA. Although both SAB and P-JNK were significantly increased by 8 weeks, the time to peak levels of P-JNK lagged behind that of SAB (Fig.1A,B, S1C).
Fig.1. Expression of hepatic SAB, P-JNK and P-ATF2.
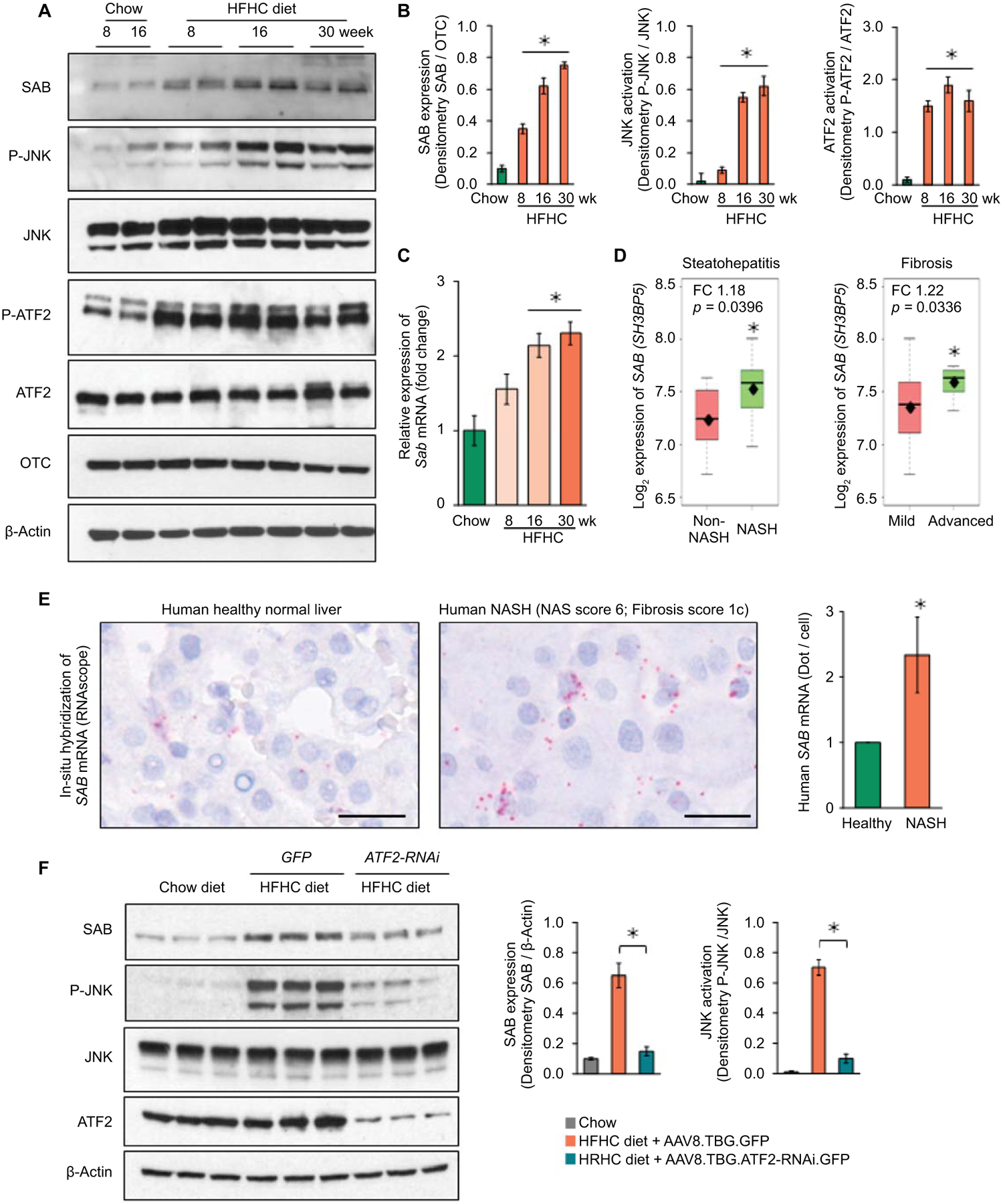
Male wild type C57BL/6N mice were fed with chow or HFHC diet. (A) SAB, P-JNK/JNK and P-ATF2/ATF2 examined by immunoblot of liver extracts. OTC and β-Actin are loading controls. (B) Densitometric analyses. (C) qPCR of Sab mRNA. n=5 mice/group, ✱=p<0.05 versus chow diet group by unpaired, 2-tailed Student’s t-test. Data represents mean ± SEM. (D) SAB (SH3BP5) expression in non-NASH (n=7) vs NASH (n=16) patients, ✱=p<0.05, by Student’s t-test; mild-fibrosis (n=15) vs advanced-fibrosis (n=8), ✱= p<0.05, by Welch’s t-test. The middle line of the box is the median, the top of the box is the third quartile and the bottom is the first quartile. The diamond (⧫) is the mean value of Log2 expression of SH3BP5 gene. The dashed lines represent the range of the data. FC = fold change. (E) In-situ hybridization (ISH) of SAB transcript in human liver: representative of 3 normal (average score1) and 3 NASH livers (average score 2.33), summarized in bar graph. Scale bar = 200 μm (digital high magnification) ✱=p<0.05, by unpaired, two-tailed Student’s t-test. (F) AAV8.TBG.ATF2-RNAi or GFP were given to mice after 12 weeks on HFHC diet and the diet continued to week 16. Immunoblots of SAB, P-JNK, JNK and ATF2. β-Actin, loading control. Representative immunoblots from 3 separate experiments. Each lane represents a single mouse liver. Densitometry of 5 mice/group, ✱=p<0.05, unpaired, 2-tailed Student’s t-test.
Heretofore, there has been no recognition of the potential role of increased SAB expression in NAFL/NASH. Using Base Space Correlation Engine and IPA, we identified statistically significant increased SAB expression in human fatty liver diseases – NAFL and NASH. In independent clinical studies, SAB expression was significantly increased in the liver from obese and NASH patients compared to normal healthy controls (Table S1). In addition, we analyzed the SAB expression in microarray data of a clinical study of NAFL/NASH designed to identify genes related to high- and low-risk.(21) SAB was significantly upregulated in NASH (hepatocyte ballooning ≥ 1 or lobular inflammation ≥ 2) versus non-NASH, and in advanced fibrosis (stageF3) versus mild fibrosis (stageF0-F1) (Fig.1D). The NASH activity scores (NAS), especially steatosis and lobular inflammation scores, were significantly correlated with SAB expression in 23 male patients diagnosed with NAFL/NASH (Table S2). Furthermore, in situ hybridization of human liver samples confirmed increased Sab mRNA in NASH livers (Fig.1E, S1D).
Studies in H9c2 cells previously found that SAB was induced by chronic rotenone or hypoxic stress in an AP-1 dependent fashion.(22) We identified AP-1 response elements in the mouse and human SAB promoters. Hepatic P-ATF2, a subunit of AP-1 transcription factors, increased in the liver of HFHC fed mice in parallel with increased SAB (Fig.1A). ATF2 is known to be activated by MAPkinases.(23) To verify that ATF2 was directly involved in Sab expression we exposed HEK293 cells to palmitic acid which activates JNK. This lead to increased SAB levels which was inhibited by treating cells with siATF2 (Fig.S2A,B). In addition, in AML12 hepatocytes, tunicamycin increased SAB expression which was prevented by deletion of ATF2 by CRISPR/Cas9 (Fig.S2C). Furthermore, in mice fed HFHC diet for 16 weeks, the administration of AAV8.TBG.ATF2-RNAi at week 12 lead to a marked decrease in SAB and P-JNK at week 16, along with decreased fatty liver and serum ALT (Fig.1F,S2D,E).
Deletion of hepatic Sab prevents liver steatosis and insulin resistance
To determine the role of Sab expression in diet induced hepatic steatosis, we fed chow or HFHC diet to Sabf/f and SabiΔHep mice (inducible hepatocyte specific Sab deletion) for up to 16 weeks. Weight gain in chow fed mice was not different between the two groups. However, SabiΔHep mice gained less body weight at 16 weeks but not at 8 weeks compared to Sabf/f mice receiving HFHC diet (Fig.2A, 3F). The difference in weight gain at 16 weeks was due to increased whole body fat tissue in the Sabf/f group (Fig.2A). Calorie (food and drink) intake, locomotor activity, respiratory exchange ratio (RER) and body temperature were not different over the course of 16 weeks HFHC diet feeding in Sab deleted versus floxed controls (Fig.S3A–D). RER results indicated that both Sabf/f and SabiΔHep mice were utilizing HFHC diet (rich in fat; giving RER = around 0.8) without variation in intestinal absorption and nutritional metabolism (Fig.S3C). Though RER was not different, hepatic steatosis (liver triglyceride and Oil-Red-O staining) was markedly decreased in HFHC 16 weeks fed SabiΔHep mice compared to Sabf/f littermates. (Fig.2B,C). In addition, deletion of Sab prevented hepatic steatosis and P-JNK activation caused by another type of high fat diet (Western diet containing 1% cholesterol and lard) (Fig.S3E,F). Serum ALT was significantly increased (3-fold) by 16 weeks of HFHC diet in floxed controls but deletion of Sab almost completely prevented the rise of ALT (Fig.2D). Peri-sinusoidal or peri-hepatocytic fibrosis identified by Sirius red staining of collagen fibers exhibited minimal increase with a very sparse patchy distribution after 16 weeks in HFHC fed Sabf/f mice (Fig.2E). However, collagen staining was barely detectable in HFHC fed SabiΔHep mice. At this early stage, immunoblotting showed no difference in Col1α in HFHC fed Sabf/f liver compared to SabiΔHep littermates (Fig.S3G). P-JNK was progressively increased at 8, 12 and 16 weeks of hepatic steatosis in Sabf/f mice, but was completely prevented in SabiΔHep littermates (Fig.2F). P-p38, P-AMPK, CPT1 and HADHA were not different between HFHC fed Sabf/f and SabiΔHep mice (Fig.S3H). The increase of hepatic SAB with HFHC feeding in Sabf/f mice was prevented in SabiΔHep mice (Fig.2F), indicating that HFHC-diet induces Sab expression primarily in hepatocytes.
Fig.2. Hepatic Sab deletion prevents diet-induced obesity, fatty liver and insulin resistance.
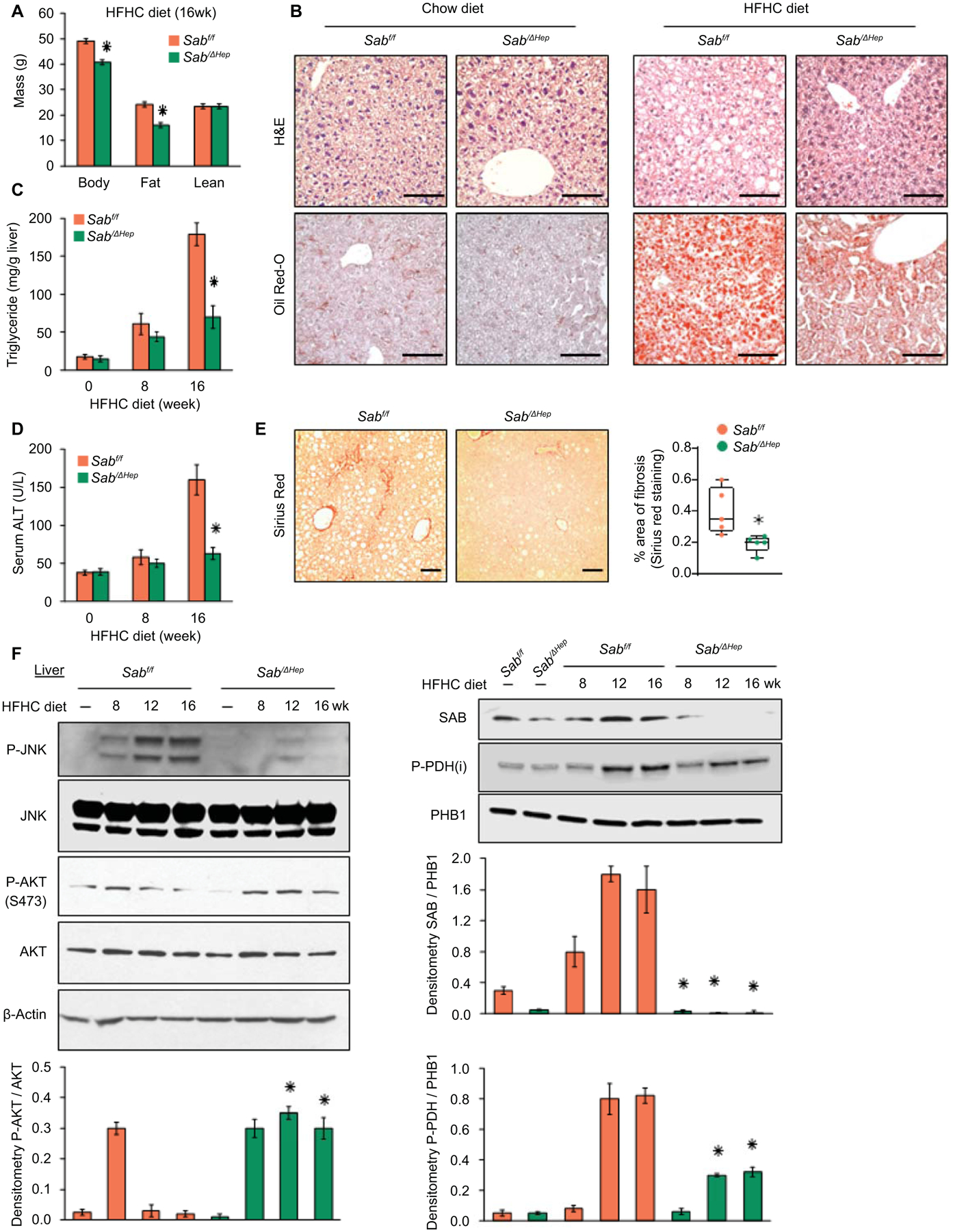
Male Sabf/f and SabiΔHep mice were fed chow or HFHC diet for up to 16 weeks. (A) Body, fat and lean mass, n=6 mice/group, ✱=p<0.05 SabiΔHep versus Sabf/f by unpaired, 2-tailed Student’s t-test. (B) Representative liver histology of 16 weeks of chow or HFHC fed Sabf/f and SabiΔHep mice. n=5 mice/group. Scale bar = 100 μm. (C, D) Liver triglyceride and serum ALT in 5 mice per group. ✱=p<0.05 SabiΔHep versus Sabf/f by unpaired, 2-tailed Student’s t-test. (E) Sirius red staining and comparison of area of fibrosis in 16 weeks HFHC fed Sabf/f and SabiΔHep mice. Scale bar = 100 μm. (F) Immunoblots of SAB, JNK, P-JNK (active), AKT, P-AKT (S473) (active), P-PDH (inactive). PHB1 and β-Actin are loading controls. Representative of 3 separate blots with bar graphs depicting densitometric summary. n=3–5/group, ✱=p<0.05 SabiΔHep versus corresponding Sabf/f by unpaired, 2-tailed Student’s t-test.
Fig.3. Sab deletion prevents diet-induced metabolic intolerance and inflammatory response.
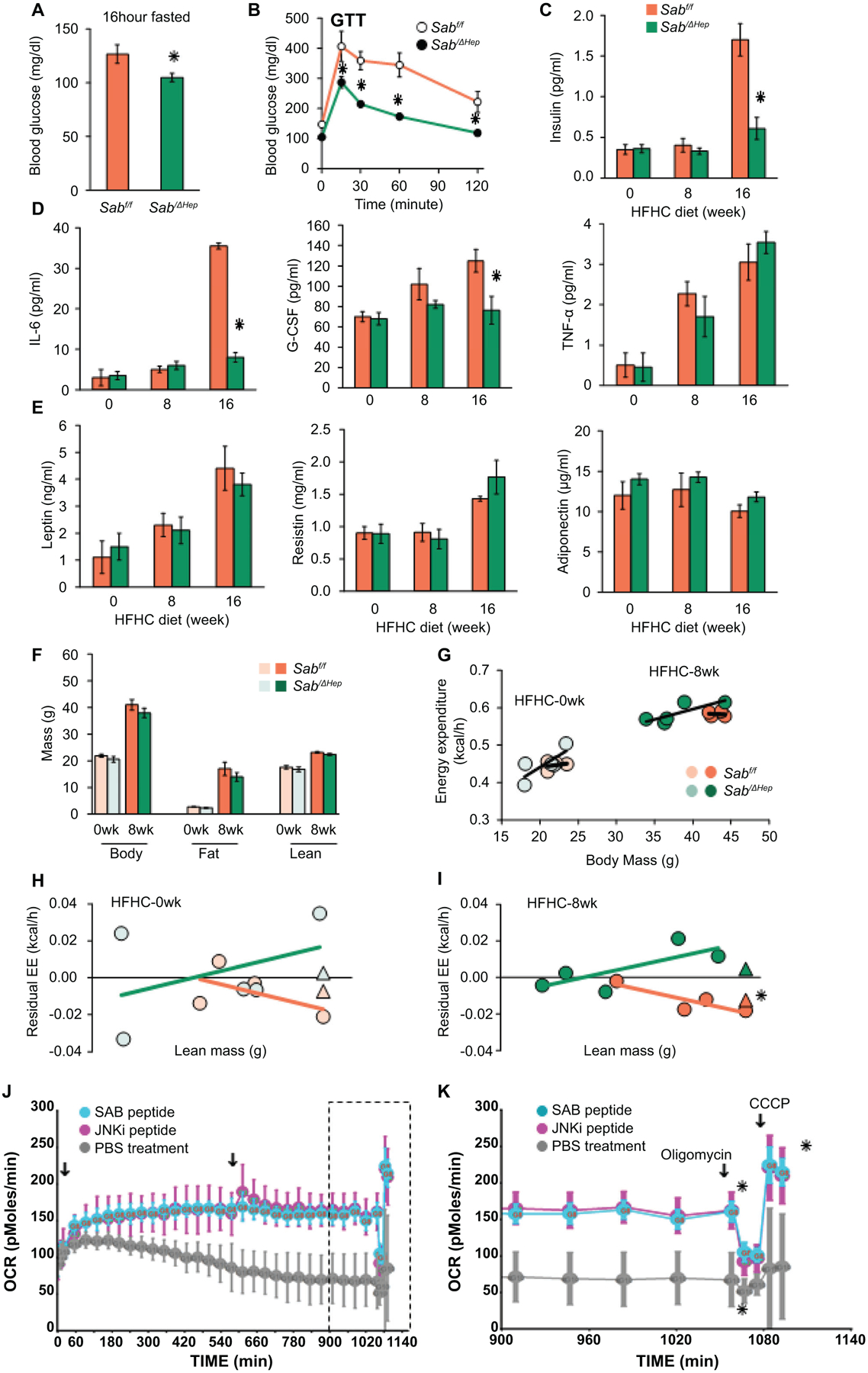
Male Sabf/f and SabiΔHep mice were fed the HFHC diet up to 16 weeks. (A) 16 hours fasting blood glucose, (B) glucose tolerance test (GTT) (C) serum insulin after 4 hours fast. (D) Serum inflammatory cytokines: IL-6, G-CSF, TNF, and (E) serum leptin, resistin, and adiponectin in 4 hours fasted mice. All data are presented as mean ± SEM. n=5 mice/group, ✱=p<0.05 SabiΔHep versus Sabf/f by unpaired, 2-tailed Student’s t-test. (F) Body, fat and lean mass, n=5 mice per group. (G) Energy expenditure (EE) measured by PhenoMaster/LabMaster home cage System. (H,I) Residual EE obtained from regression plot was graphed against lean mass. Mean value of residuals of EE are shown as (Δ). Statistical significant difference (✱=p<0.05) of EE in SabiΔHep versus Sabf/f treatment by ANCOVA. n=4–5 mice/group. (J) Lipid metabolism of steatotic hepatocytes examined by Seahorse XF analyzer. PMH from wild type C57BL/6N mice fed HFHC diet for 12 weeks were seeded on collagen coated Seahorse cell culture plate. Cells were washed in serum, glucose and pyruvate-free-XF medium. After measurement of basal level, SAB-peptide or JNKi-peptide (5 μM) was injected. OCR was measured over the time course (18 hours) with second injection (arrow) 9 hours after first injection. (K) Final 3 hours measurements (boxed in J) showing the oligomycin inhibitable (ATP coupled) respiration and CCCP induced maximal (uncoupled) respiration. n=3 separate experiments, ✱=P<0.05 peptide versus vehicle treatment by two-way ANOVA. Data represents mean ± SEM.
Hepatic Sab mediates metabolic intolerance and inflammatory response
At 16 weeks of HFHC feeding, fasting blood glucose was significantly higher in Sabf/f mice and serum insulin was increased 3-fold compared to SabiΔHep mice, indicating global insulin resistance. However, these changes were prevented in SabiΔHep mice (Fig.3A–C). Glucose tolerance test (GTT) confirmed decreased insulin sensitivity in the HFHC fed Sabf/f mice and this was prevented in the SabiΔHep mice (Fig.3B). The, lower tolerance to high glucose in Sabf/f mice was not due to low blood insulin level but associated with high HOMA-IR in Sabf/f mice. The HOMA-IR was normal in SabiΔHep mice (Fig.S4A). Therefore, 16 weeks HFHC diet feeding caused fatty liver with insulin resistance in Sabf/f but not in SabiΔHep mice. It is known that P-JNK inhibits insulin receptor activation of AKT in the liver.(24) P-AKT (S473) mediates insulin signaling. Decreased hepatic P-AKT (S473) in steatotic Sabf/f liver in response to HFHC diet was consistent with insulin resistance (Fig.2F). The decreased hepatic P-AKT contributes to gluconeogenesis and hyperglycemia. The decreased P-AKT, increased P-PDH and insulin resistance in HFHC fed Sabf/f mice were prevented in SabiΔHep littermates (Fig.2F,S4A). Serum IL-6, a marker of adipose tissue lipolytic metabolic stress, and G-CSF, an inflammatory marker, were increased in HFHC fed floxed controls but were significantly decreased in SabiΔHep mice to the level of chow fed mice (Fig.3D). Of note, serum ALT, insulin and IL-6 in HFHC fed mice at 8 weeks were not different than chow fed mice (Fig.2D,3C,D), suggesting that these changes were associated with increased mitochondrial SAB and JNK activation which were evident at 16 weeks. Serum TNF-α, leptin, resistin and adiponectin were not different (Fig.3D,E). Therefore, deletion of hepatic Sab prevents high-fat diet induced metabolic intolerance and metabolic stress induced inflammatory/cytokine response.
Since deletion of Sab in hepatocytes prevented diet-induced weight gain, fat mass, hepatic steatosis, insulin resistance and glucose intolerance at 16 weeks of HFHC feeding, we examined the effect of deletion of hepatic Sab on whole body energy expenditure (EE) (Fig.3F–I) Since body weight, fat and lean mass were not different at 8 weeks in HFHC diet fed Sabf/f and SabiΔHep mice (Fig.3F), we focused on this time point to dissociate from weight gain and attempt to better understand the reason for the subsequent prevention of weight gain in hepatic Sab deleted mice. Thus, we measured the EE at 8 weeks of HFHC feeding (Fig.3G,S4B) and determined the difference of residual-EE between Sabf/f and SabiΔHep mice (Fig.3H,I). At this stage, P-JNK and SAB had already increased in Sabf/f but not in SabiΔHep mice (Fig.2F). Indeed, deletion of hepatic Sab significantly prevented the decrease of EE observed in the HFHC diet controls (Fig.3I). Calorie intake, locomotor activity and RER were not different (Fig.S4C–E). Thus, HFHC feeding decreased systemic energy expenditure and the deletion of Sab prevented this in advance of preventing weight gain.
To directly assess the role of hepatic SAB on mitochondria mediated lipid metabolism, we examined the respiratory function of intact steatotic hepatocytes isolated from wild type mice fed HFHC diet for 12 weeks when P-JNK and SAB had increased (Fig.3J,K). Steatotic-PMH cultured in glucose-free, pyruvate-free, serum-free, low-amino acid XF-medium utilized intracellular triglyceride for mitochondrial β-oxidation (inhibitable by etomoxir) and mitochondrial respiration (oxidative-phosphorylation) (inhibitable by rotenone) (Fig.S4F). In steatotic hepatocytes, fatty acid oxidation dependent mitochondrial respiration declined over time and was restored either by membrane permeant 20 amino acid SAB-peptide corresponding to the JNK binding site of SAB or by membrane permeant peptide JNK inhibitor, D-JNKi (Fig.3J,K). The SAB-peptide has previously been shown to inhibit binding of P-JNK to mitochondria and protect against the direct effects of P-JNK on mitochondrial function.(12,13,17,20) The findings underscore the key role of the JNK-SAB interaction in mitochondrial dysfunction of steatotic hepatocytes. Since HFHC diet upregulates SREBP1 and 2 and de novo lipogenesis genes (Fig.S1A–C), hepatic SREBP activity of 16 weeks HFHC fed Sabf/f and SabiΔHep mice was determined. Indeed, diet induced SREBP target gene expression was prevented in SabiΔHep mice (Fig.S4G). Of note, SREBP-1 is activated by JNK.(7,15) Therefore, hepatic SAB controls whole body EE and liver triglyceride metabolism in HFHC diet induced obesity and fatty liver.
Effect of Sab-ASO treatment on the effects of HFHC feeding
First, we verified if Sab-ASO treatment phenocopies the SabiΔHep mice fed HFHC diet for 16 weeks. Indeed, wild type mice which were treated with Sab-ASO 50mg/kg, three times per week exhibited reduced hepatic steatosis, body fat mass and improved glucose tolerance (Fig. S5A–H), as seen in SabiΔHep mice fed HFHC diet for 16 weeks. This high dose of Sab-ASO almost completely depletes SAB in the liver as we previously described.(16) Next, wild type mice were fed chow or HFHC diet for 30 weeks to address progression to NASH and weight gain (Fig.4A). At the same time, a lower dose of Ctl-ASO or Sab-ASO (25 mg/kg, three times per week) was given to dampen Sab induction in HFHC fed mice in contrast to complete depletion of SAB in SabiΔHep mice or high dose Sab-ASO treated mice (Fig.4B,2F,S5A). The Sab-ASO or control–ASO was given for 12 weeks beginning after 17 weeks and ending at 30 weeks of HFHC feeding. Body composition of mice was measured before and at the end of the course of Ctl-ASO or Sab-ASO injection (Fig.4C). HFHC feeding increased body fat mass and caused hepatic lipid accumulation and high liver triglyceride levels in Ctl-ASO treated mice, whereas Sab-ASO treatment reduced the accumulation of body fat mass and lipid in the liver (Fig.4C–E). By 30 weeks of HFHC diet feeding, increased liver fibrosis (Sirius red staining Fig.4F, and Col1α levels Fig.5A) developed in Ctl-ASO treated mice but was prevented in Sab-ASO treated mice. Furthermore, Sab-ASO markedly decreased the serum ALT, fasting glucose levels (Fig.4G), and the upregulated expression of SREBP1 and 2 target genes (Fig.S5I). The Sab-ASO treatment decreased SAB to the levels of chow fed mice, suggesting that the higher levels of SAB induced by HFHC diet feeding is a key determinant of the severity of hepatic steatosis and fibrosis, and that prevention of SAB induction abrogates progression of NASH. Sab-ASO treatment also prevented the gain of body mass and fat mass as was seen in the hepatocyte specific deletion of Sab. Therefore, the complex interplay of the liver and systemic consequences of HFHC feeding was abrogated by targeting Sab in the liver.
Fig.4. ASO treatment suppresses Sab expression and progression of steatohepatitis and fibrosis.
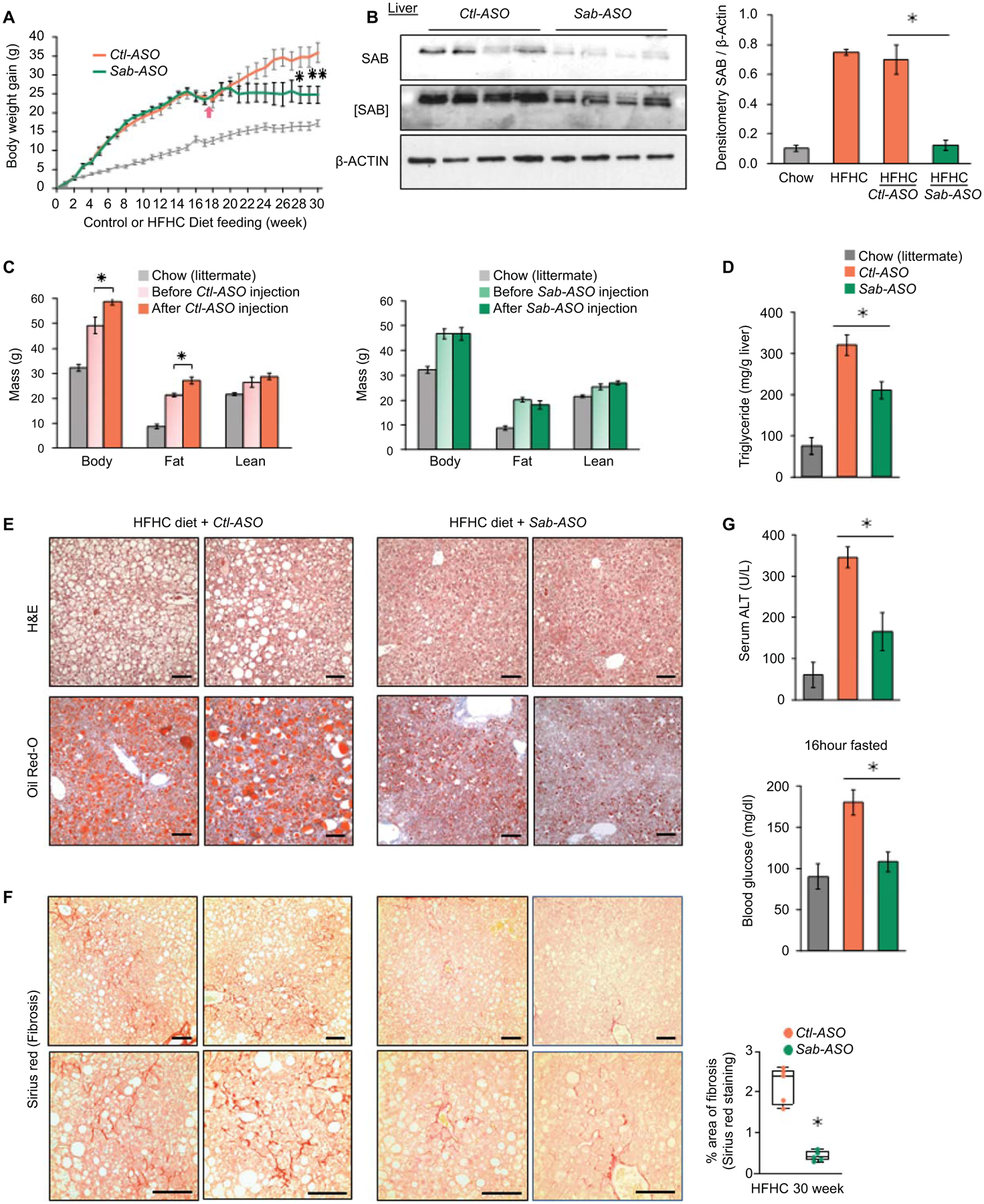
Male wild type littermates were fed HFHC diet for 30 weeks. Mice were treated with Control-ASO (Ctl-ASO) or Sab-ASO 25mg/kg ip three times per week starting after 17 weeks of HFHC diet. (A) Weight gain in mice fed HFHC diet, n=6 mice/group; chow diet fed mice shown as gray line. (B) Level of SAB after Sab-ASO injection in HFHC fed mice at week 30. Each lane represents a single mouse liver; n=4 mice per group. Representative image of 3 separate blots. Densitometry results shown as bar graph. All data shown as mean ± SEM. ✱=p<0.05 Sab-ASO versus Control-ASO by unpaired, 2-tailed Student’s t-test. (C) Body composition of Ctl-ASO or Sab-ASO treated groups before versus after 12 weeks course of ASO injections. n=5–6 mice/group, ✱=p<0.05 after- versus before-ASO by paired, 2-tailed Student’s t-test. (D) Liver triglyceride, (E) Representative histology images of H&E, Oil-Red-O and (F) Sirius red staining (scale bar = 100μm) and quantitation of area of fibrosis. (G) Serum ALT and fasting blood glucose examined at 30 weeks of HFHC feeding with ASO injections for the last 12 weeks. All data shown as mean ± SEM. n=5–6 mice/group. ✱=p<0.05 Sab-ASO versus Control-ASO by unpaired 2-tailed Student’s t-test.
Fig.5. Sab knockdown prevents decreased energy expenditure, hepatic inflammation and fibrosis.
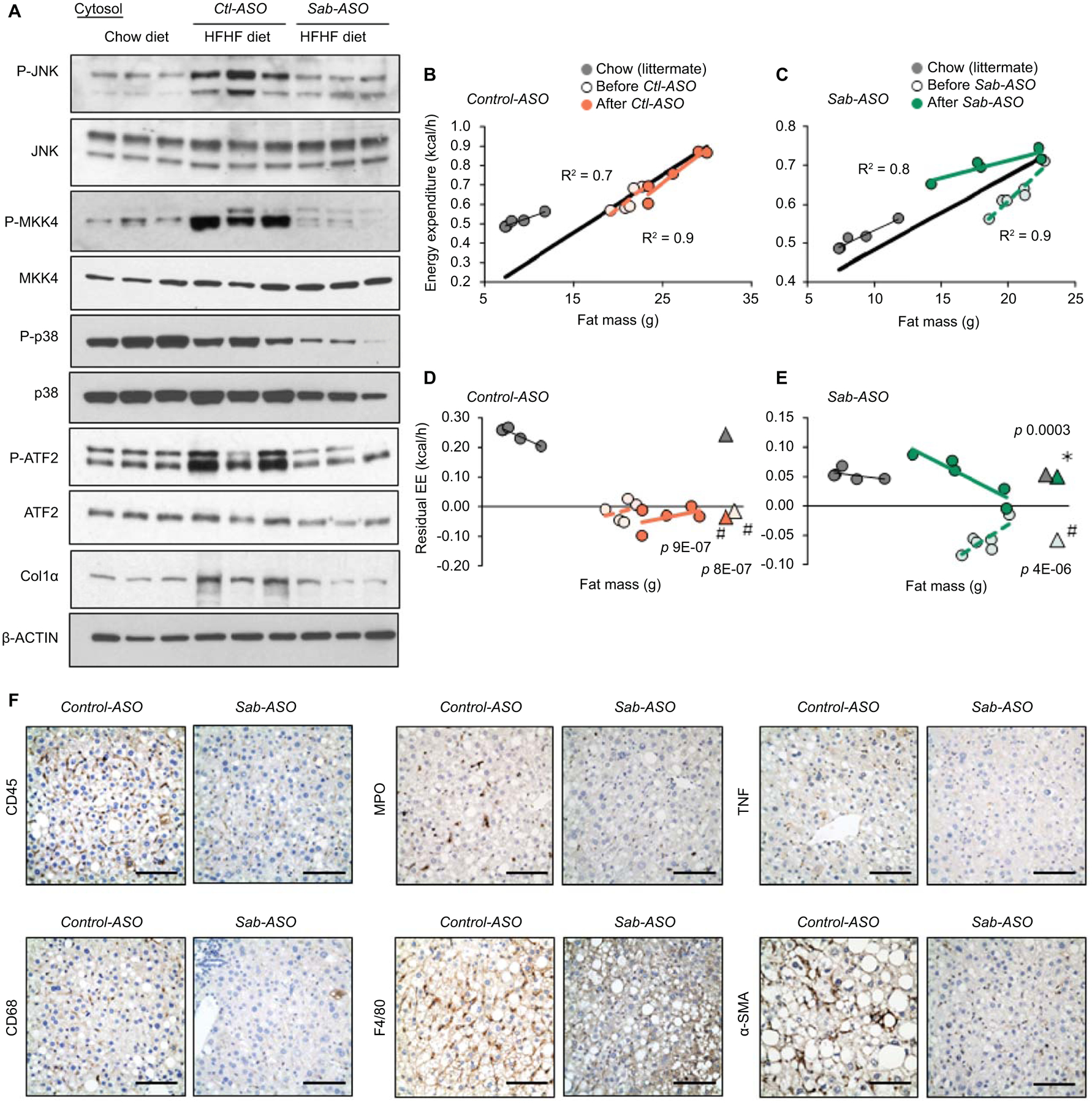
Feeding and ASO as in Fig.4. (A) Immunoblot of JNK, p38, ATF2 and MKK4 and phospho-forms, and Col1α. β-Actin is the loading control. Each lane represents a single mouse liver; n=4 mice/group. Representative images of 3 separate blots. (B,C) Regression plot of EE and (D,E) residual EE before and after ASO treatment of the same mice. Regression plot of residual EE against fat mass. Mean values of residual EE shown as (Δ). Statistical significance (p<0.05) of EE in HFHC vs chow fed groups shown as (#), before vs after ASO treatment shown as (✱)determined by ANCOVA. n=5–6 mice/group. (F) Representative IHC of liver sections from Control-ASO (Ctl-ASO) or Sab-ASO treated mice fed the HFHC diet for 30 weeks. CD45, CD68, MPO, F4/80, TNF and α–SMA stained in formalin fixed paraffin embedded tissue sections and color developed by DAB substrate. n=5–6 mice/group. Scale bar = 100μm.
Sab-ASO treatment reduces MAPK pathway activation and hepatic inflammation, and improves energy expenditure
Since activation of JNK and upstream MAPKs are regulated by the level of mitochondrial SAB(16–18,25), we examined P-JNK and upstream P-MKK4 in Ctl-ASO or Sab-ASO treated mice fed HFHC diet for 30 weeks compared to chow fed littermates. Hepatic P-JNK and P-MKK4 were increased in Ctl-ASO treated mice, whereas Sab-ASO treatment decreased P-JNK and P-MKK4 to levels comparable to chow fed littermates (Fig.5A,S6A). Furthermore, Sab-ASO treatment decreased P-ATF2 to the levels of chow fed mice. Since Sab-ASO treatment prevented diet-induced weight gain, EE of HFHC fed mice was examined. EE of each mouse before (15 weeks HFHC) vs after ASO injection (30 weeks HFHC) was measured and residual-EE was plotted as a function of fat mass, lean mass, or body mass (Fig.5B–E,S5B,C). HFHC feeding decreased EE compared to chow fed littermates (Fig. 5D,E). Treatment with Sab-ASO but not Ctl-ASO markedly improved the EE to the level of chow fed mice (Fig.5E). The regression plot using the lean mass or body mass also confirmed that Sab-ASO treatment significantly improved the EE (Fig.S6B,C) whereas respiratory exchange ratio, locomotor activity, calorie intake and body heat production were not different (Fig.S6D–G).
Metabolic stress induced P-JNK activation in hepatocytes in vivo causes lipotoxicity and an inflammatory response.(1,5–9) Therefore, hepatic inflammation and the inflammatory response were assessed in Ctl-ASO vs Sab-ASO treated mice. CD45, CD68, F4/80, MPO, and TNF were examined by immunohistochemical staining. Infiltrating lymphocytes, tissue macrophages, neutrophils and TNF staining were markedly decreased by Sab-ASO treatment compared to Ctl-ASO treatment (Fig.5F). Steatohepatitis due to HFHC diet feeding for 30 weeks was accompanied by increased α–smooth muscle actin, indicating stellate cell activation, which is associated with hepatic fibrosis. In addition, to examine the effect of Sab-ASO treatment on genetic obesity and steatosis, ob/ob mice were treated with ASO. Sab-ASO treatment decreased P-JNK levels and hepatic lipid accumulation (Fig.S6H–K). These data indicate that knockdown of Sab with Sab-ASO treatment prevents the progression of hepatic steatosis, steatohepatitis, fibrosis, metabolic stress and obesity which correlates with inhibiting sustained P-JNK activation and improving energy expenditure.
Reversal of steatohepatitis and fibrosis by targeting hepatocyte Sab
To exclude the possible effect of weight loss on steatohepatitis and fibrosis in Sab-ASO treatment, mice were fed the HFHC diet for 52 weeks to reach a plateau of weight-gain which occurred after 40 weeks (Fig.6A). Beginning at 42 weeks of HFHC diet, mice were treated with GalNAc-Control-ASO (GalNAc-Ctl-ASO) or GalNAc-Sab-ASO, 2.5mg/kg intraperitoneally three times per week for 10 weeks. GalNAc-ASO, which selectively targets hepatocytes, (18) became available after Sab-ASO work had been completed. In HFHC fed mice, GalNAc-Sab-ASO dampened the increased expression of SAB and the sustained activation of JNK and ATF2 (Fig.6B). HFHC feeding for 52 weeks in GalNAc-ctrl-ASO treated mice featured a histological pattern similar to human NASH. Massive lobular hepatic steatosis with widespread macro- and micro-vesicular fat (H&E, Oil-Red-O staining), prominent, diffuse peri-sinusoidal/peri-hepatocyte fibrosis (chicken wire appearance) determined by Sirius red staining, and scattered ballooned hepatocytes were found in HFHC fed mice at 52 weeks in GalNAc-Ctl-ASO treated mice (Fig.6C, D, S6A, B). In contrast, 10 weeks of GalNAc-Sab-ASO treatment markedly reduced hepatic steatosis as well as liver triglyceride, serum ALT, blood glucose and HOMA-IR (Fig.6C, E). NAS score 5.4±0.6 in the GalNAc-Ctl-ASO treatment group was improved to 4.2±0.4 in the GalNAc-Sab-ASO treatment group (n=5/group; p=0.005) (Fig.6F). Fibrosis became barely visible with very rare areas of chicken wire appearance by Sirius staining and ballooned cells were markedly decreased in GalNAc-Sab-ASO group (Fig.6D,F,S7B). The area of fibrosis decreased from 4.1±2.7% in GalNAc-Ctl-ASO to 0.9±0.3% in Sab-ASO (n=5/group, p=0.033) (Fig.6D,F). Furthermore, IPA pathway analysis and hepatic RNAseq data confirmed that de novo lipogenesis, including cholesterol synthesis, was markedly decreased by GalNAc-Sab-ASO compared to GalNAc-Ctl-ASO treatment (Fig.S7C). GalNAc-Sab-ASO treatment markedly reduced SREBP target gene expression (Fig.S7D) and decreased chemokines, and inflammatory gene expression, (Fig.S6E) and macrophage infiltration (Fig.S7F). In addition, GalNAc-Sab-ASO treatment decreased α-SMA (Fig.S7F) and Col1α levels in comparison to GalNAc-Ctl-ASO treatment by immunoblot analyses (Fig.6G,S7F). Moreover, expression of various fibrogenic genes in 52 weeks HFHC diet fed mice were reduced by GalNAc-Sab-ASO treatment, including Wnt5b, Ptch2, Notch1, Notch2, Notch4, Spp2 and Vwf (Fig.S7G), while markedly increasing the expression of the hepatic fibrinolytic gene Mmp12 (Fig.S7H). Mmp12 plays pivotal role in extracellular collagen degradation.(26) Interestingly, improvement of steatohepatitis and fibrosis by GalNAc-Sab-ASO treatment after 52 weeks of HFHC feeding was associated with marked adaptive up-regulation of genes involved in mitochondrial oxidative-phosphorylation (Cox6b2, Sdhaf3) and mitochondrial ADP/ATP nucleotide translocase (ANT; Slc25a4) (Fig.S7I). The role of these gene changes in GalNAc-Sab-ASO treatment will require further detailed time course experiments. Finally, to support the role of the JNK-SAB-ROS activation loop, we examined the levels of liver protein carbonyls, and found that the Sab deletion or knockdown inhibited the HFHC-induced oxidatively modified protein levels in all the experimental cohorts (Fig.S8).
Fig.6. Hepatic specific GalNAc-Sab-ASO treatment.
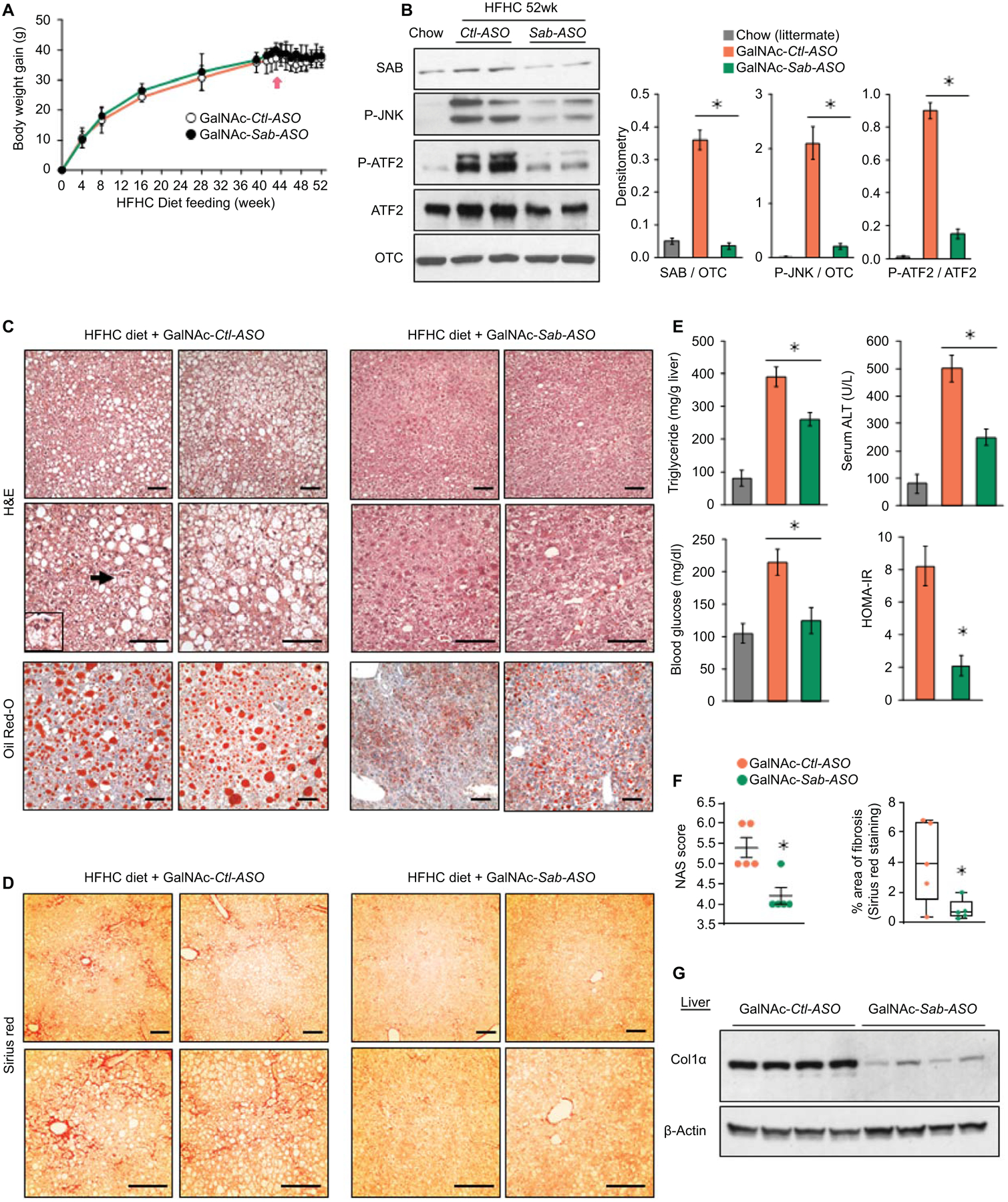
Male wild type littermates were fed HFHC diet for 52 weeks. Mice were given GalNAc-Control-ASO (GalNAc-Ctl-ASO) or GalNAc-Sab-ASO 2.5mg/kg ip three times per week starting after 42 weeks of HFHC diet. n=6 mice/group. (A) Time course of body weight gain (B) Immunoblots of SAB, P-JNK, JNK, P-ATF2 and ATF2; OTC, loading control. Representative of 3 separate experiments. Each lane is a single mouse liver. Bar graph of densitometry of 5 mice/group, ✱=p<0.05, unpaired, 2-tailed Student’s t-test. (C) Representative images of H&E and Oil-Red-O staining of liver sections of HFHC diet fed mice after 10 weeks course of ASO injections. Ballooned hepatocyte (arrow and insert). Scale bar = 100μm. (D) Sirius red staining. (E,F) Bar graphs of liver triglyceride, serum ALT, fasting blood glucose, HOMA-IR, NAS score, and area of fibrosis. Area of fibrosis of Sirius red stain quantitated by Image-J. Each dot is an average of 5–10 fields per mouse (10x magnification). n=5–6 mice/group. ✱=p < 0.05 GalNAc-Sab-ASO versus GalNAc-Ctl-ASO by unpaired, 2-tailed Student’s t-test. (G) Immunoblot of sonicated tissue lysate using anti-Col1α. β-Actin, loading control. Representative immunoblots from 3 separate experiments. Each lane is a single mouse liver.
Discussion
Studies from our laboratory in acute liver injury models identified a novel target of JNK, referred to as SAB, which is a mitochondrial outer transmembrane protein with a P-JNK docking site facing the cytoplasm.(16–18) P-JNK phosphorylates SAB leading to an intramitochondrial signal transduction pathway causing decreased respiration and increased ROS production.(12,17) ROS released from mitochondria then continue to activate the MAPK cascade, sustaining JNK activation.(12,27) Therefore, we aimed to explore the potential role of hepatic SAB in the pathogenesis of NAFL/NASH and to explore the possibility that SAB could serve as a therapeutic target.
Mice fed the HFHC diet exhibited a gradual increase in hepatic SAB mRNA and protein as well as P-JNK/JNK, which reached maximum at 16 weeks and was sustained thereafter. Human transcriptomic data in NAFL and NASH cohorts demonstrated a correlation between NASH activity score, steatohepatitis and fibrosis with SAB expression. In addition, we observed increased Sab mRNA in human NASH livers by in-situ hybridization. Our results implicate AP-1 transcription factor dimer subunit ATF2 in inducing Sab. Sustained activation of ATF2 was associated with sustained JNK activation and increased SAB expression. Importantly, knockdown of ATF2 reversed the diet induced induction of SAB and the sustained JNK activation. Since AP-1 dimer subunits such as ATF2 are phosphorylated by JNK, we propose the notion that a self-amplifying mechanism of increased AP-1 activation, SAB expression, and increased JNK activation leads to higher steady state levels of expression of SAB and P-JNK (Fig.7).
Fig.7. Working model depicting the mechanism of SAB induction and its pivotal role in fatty liver, steatohepatitis, and fibrosis.
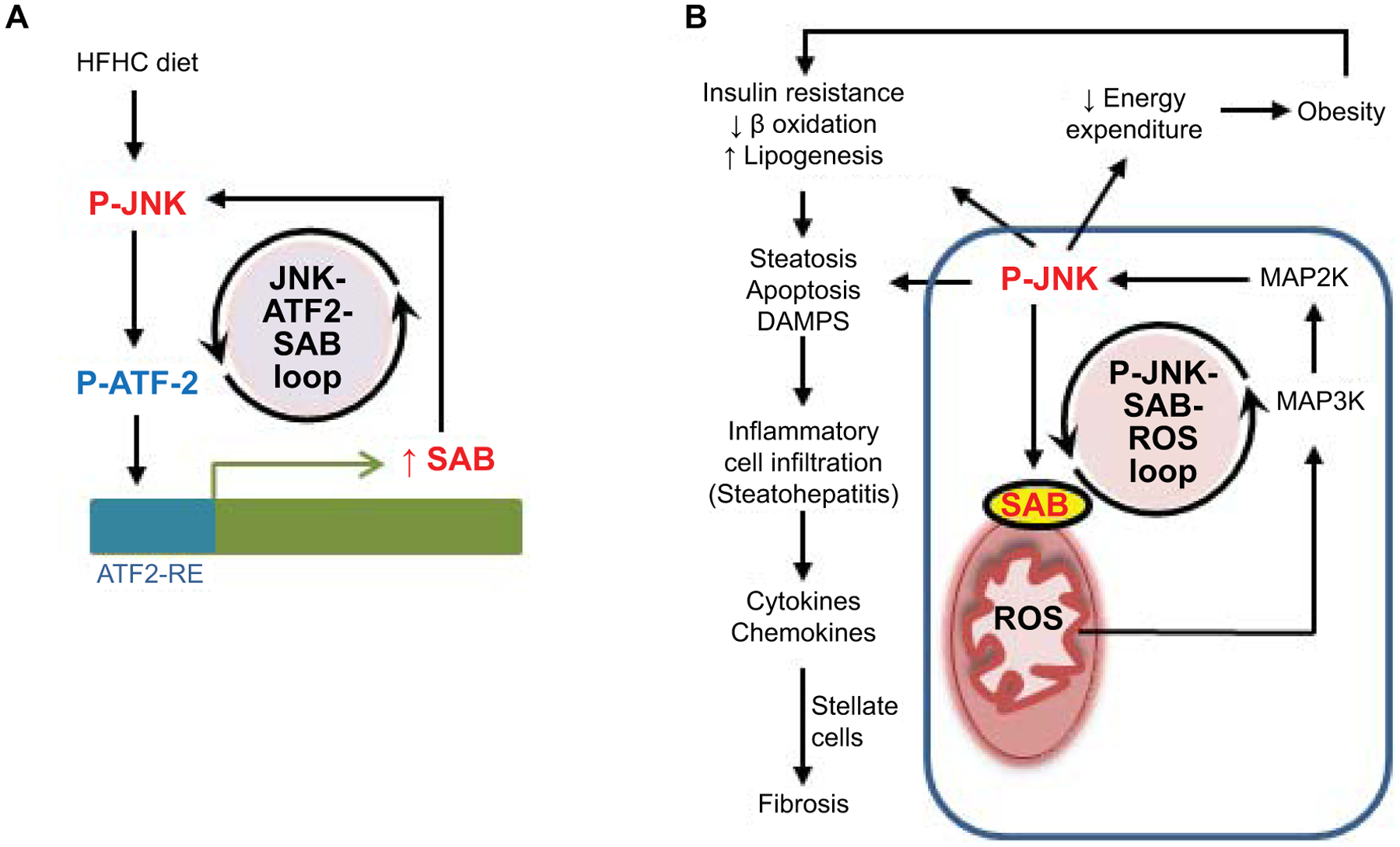
(A) A regulatory loop activated by metabolic stress leads to feedforward activation of JNK and ATF2 which then induces Sab expression. (B) High steady state levels of SAB promote high levels of sustained JNK activation through mitochondrial SAB-ROS self-amplification during ongoing metabolic stress. The enhanced sustained JNK activation, through a JNK-SAB-ROS activation loop, leads to downstream effects in hepatocytes on mitochondrial function, insulin resistance, and lipogenesis as well as systemic effects leading to decreased energy expenditure and obesity.
Hepatocyte deletion of Sab markedly decreased sustained JNK activation in HFHC fed mice indicating that SAB is required to sustain JNK activation, which heretofore has not been appreciated in NAFLD. Although deletion of hepatic Sab decreased weight gain at 16 weeks of HFHC feeding. At 8 weeks when the body weight increase was the same in Sabf/f and SabiΔHep mice, Sab deletion improved energy expenditure which had been decreased by feeding the HFHC diet, likely accounting for the subsequent resistance to weight gain. Inhibition of obesity and metabolic syndrome by Sab deletion or knockdown therefore is an important aspect of the beneficial effect of targeting Sab in the liver. The interplay between the effects of SAB and JNK in the liver and cross talk with systemic metabolic effects is complex and not fully understood, but together likely contribute to the phenotype of NAFL/NASH, insulin resistance and obesity (Fig.7).
Although inducible hepatic Sab deletion or global embryonic knockout (unpublished data, Win et al) do not exhibit any phenotypic differences compared to Sabf/f, an important aspect of our findings was that knockdown of elevated Sab to the levels of chow fed controls exerted marked protection against NASH and obesity. This underscores the importance of hepatic Sab induction as a driver of the disease progression. We have previously shown that overexpression of Sab drives high P-JNK levels and liver injury in acute toxicity of acetaminophen.(18) Such a scenario also is likely to be the case in chronic diet-induced liver disease. Targeted knockdown of hepatic Sab as a therapeutic approach offers advantages over targeting MAPK cascade enzymes which have important physiological functions as well as pathological effects. Decreasing Sab expression to normal levels would not be expected to interfere with the physiologic functions of MAPK enzymes. Finally, although we show that increased hepatic Sab leads to systemic impairment in EE, likely a major factor in development of obesity, the identification of the signal(s) emanating from the liver that control EE and body fat have not been identified and this is an important question to pursue.
There are some limitations in our studies. We did not perform a direct comparison of Sab versus JNK deletion or knockdown. However, the work of the Davis’ lab using hepatic JNK1/2 deletion generated a very similar protective effect on hepatic and systemic effects of diet induced NAFL and weight gain which supports our conclusion that JNK mediates the effects of SAB. Also, we did not perform a side-by-side comparison of Sab-ASO and GalNAc-Sab-ASO. Therefore, we do not know if Sab-ASO and more targeted GalNAc-Sab-ASO would provide the same or different results at the early and late stages of disease development.
In conclusion, we have found that overfeeding increases SAB expression in the liver by sustaining a JNK-ATF2 mediated induction of SAB. Increased SAB expression during metabolic stress drives sustained high levels of P-JNK which leads to NAFL/NASH and metabolic syndrome. Deletion or targeted knockdown of Sab abrogates the adverse effects of the JNK-SAB-ROS activation loop suggesting that SAB is a potential therapeutic target.
Supplementary Material
Acknowledgments
We thank Dr. Anna Mae Diehl, Dr. Manal F. Abdelmalek and Duke NAFLD Clinical-Translational Research Program for facilitating access to the Duke microarray data set of human NASH severity, Dr. Eddie Loh, informatics specialist for Bioinformatics, Health Sciences Libraries, USC for input in data analysis of RNA-seq. We gratefully acknowledged the services of the USC Research Center for Liver Disease’s Cell Separation and Culture, Cell and Tissue Imaging, Histology, and Metabolic, Analytical, and Instrumentation Cores, and the Rodent Metabolic Core at the Saban Research Institute of Children’s Hospital Los Angeles.
Financial support statement:
This research was supported by NIH grants R01DK067215 (NK), the Veronica Gerrie Budnick Chair in Liver Disease (NK), the Donald E. and Delia Baxter Foundation Faculty Fellows award (SW), a pilot project award (SW) by USC Research Center for Liver Diseases (P30DK048522), and a pilot project grant funding (SW) by the Rodent Metabolic Core of the Saban Research Institute of Children’s Hospital Los Angeles. Histology service and tissue imaging were supported by the USC Research Center for Liver Disease’s Core (P30DK048522). Steatotic primary mouse hepatocytes were isolated by the NIAAA-funded Integrative Liver Cell Core (R24AA012885). The bioinformatics software and computing resources used in the RNA-seq analysis and data mining were funded by the USC Office of Research and the Norris Medical Library. Ionis Pharmaceuticals supported the in-situ hybridization in human liver samples.
Competing interests
The authors have no relevant conflicts of interest to disclose except S.F.M & M.A, who are employees of Ionis Pharmaceuticals. Ionis prepared oligonucleotides and supported commercially available in-situ hybridization services to assess SAB expression in human livers but provided no direct research funding.
Abbreviations
- ADP
adenosine diphosphate
- AKT
Protein kinase B
- ALT
alanine aminotransferase
- AMPK
AMP-activated protein kinase
- ASO
antisense oligonucleotide
- ATF2
Activating transcription factor 2
- ATP
adenosine triphosphate
- AP-1
Activator protein 1
- AUC
area under the curve
- Col1
Collagen1
- CPT1
Carnitine palmitoyltransferase-1
- GalNAc
N-acetylgalactosamine
- G-CSF
granulocyte-colony stimulating factor
- c-Src
SRC proto-oncogene (nonreceptor tyrosine kinase)
- Ctl
control
- Cox6b2
Cytochrome c oxidase subunit 6b2
- EE
energy expenditure
- Eto
Etomoxir
- GTT
glucose tolerance test
- HADHA
Hydroxyacyl-CoA Dehydrogenase
- HFHC
High fat high calorie fructose
- HOMA
homeostatic model assessment
- IHC
immunohistochemistry
- ip
intraperitoneal
- ISH
in situ hybridization
- JNK
c-Jun N-terminal kinase (JNK1, MAPK8; JNK2, MAPK9)
- KIM1
kinase interacting motif 1
- IL6
Interleukin 6
- KO
knockout
- LDH
Lactate dehydrogenase
- MKK4
mitogen-activated protein kinase kinase 4
- NASH
Nonalcoholic Steatohepatitis
- Ob/Ob
B6.Cg-Lepob/J
- OCR
oxygen consumption rate
- OTC
Ornithine transcarbamylase
- P38
p38 MAP Kinase
- PA
palmitic acid
- P-AKT
phosphorylated Protein kinase B
- p-JNK
phosphorylated JNK
- P-AMPK
Phospho-AMPK
- PDH
Pyruvate Dehydrogenase
- PHB1
Prohibitin 1
- P-MKK4
phosphorylated mitogen-activated protein kinase kinase 4
- P-PDH(i)
Phosphorylated Pyruvate Dehydrogenase (inactive)
- P-p38
Phosphorylated p38 MAP Kinase
- P-Src
Phosphorylated proto-oncogene tyrosine-protein kinase
- RER
Respiratory exchange ratio
- RNA
ribonucleic acid
- ROS
reactive oxygen species
- SAB (SH3BP5)
SH3 homology associated BTK binding protein
- SabiΔHep
inducible hepatocyte-specific Sab knockout
- Src
Proto-oncogene tyrosine-protein kinase
- SREBF1
Sterol Regulatory Element Binding Transcription Factor 1
- Tbp
TATA box binding protein
- TG
Triglyceride
- TNF
tumor necrosis factor
Data and materials availability
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Accession numbers for data relating to the paper were mentioned in the paper and deposited in a public database.
References
- 1.Vallerie SN, Hotamisligil GS. The role of JNK proteins in metabolism. Sci Transl Med 2010;2:60rv5. [DOI] [PubMed] [Google Scholar]
- 2.Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol 2013;10:656–665. [DOI] [PubMed] [Google Scholar]
- 3.Woo Baidal JA, Lavine JE. The intersection of nonalcoholic fatty liver disease and obesity. Sci Transl Med 2016;8:323rv1. [DOI] [PubMed] [Google Scholar]
- 4.Priest C, Tontonoz P. Inter-organ cross-talk in metabolic syndrome. Nat Metab 2019;1:1177–1188. [DOI] [PubMed] [Google Scholar]
- 5.Czaja MJ. JNK regulation of hepatic manifestations of the metabolic syndrome. Trends Endocrinol Metab 2010;21:707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kohli R, Kirby M, Xanthakos SA, Softic S, Feldstein AE, Saxena V, et al. High-fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology 2010;52:934–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Knebel B, Lehr S, Hartwig S, Haas J, Kaber G, Dicken HD, et al. Phosphorylation of sterol regulatory element-binding protein (SREBP)-1c by p38 kinases, ERK and JNK influences lipid metabolism and the secretome of human liver cell line HepG2. Arch Physiol Biochem 2014;120:216–227. [DOI] [PubMed] [Google Scholar]
- 8.Vernia S, Cavanagh-Kyros J, Garcia-Haro L, Sabio G, Barrett T, Jung DY, et al. The PPARα-FGF21 hormone axis contributes to metabolic regulation by the hepatic JNK signaling pathway. Cell Metab 2014;20:512–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang PX, Ji YX, Zhang XJ, Zhao LP, Yan ZZ, Zhang P, et al. Targeting CASP8 and FADD-like apoptosis regulator ameliorates nonalcoholic steatohepatitis in mice and nonhuman primates. Nat Med 2017;23:439–449. [DOI] [PubMed] [Google Scholar]
- 10.Win S, Than TA, Zhang J, Oo C, Min RWM, Kaplowitz N. New insights into the role and mechanism of c-Jun-N-terminal kinase signaling in the pathobiology of liver diseases. Hepatology 2018;67:2013–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seki E, Brenner DA, Karin M. A liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology 2012;143:307–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Win S, Than TA, Fernandez-Checa JC, Kaplowitz N. JNK interaction with Sab mediates ER stress induced inhibition of mitochondrial respiration and cell death. Cell Death Dis 2014;5:e989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Win S, Than TA, Le BH, García-Ruiz C, Fernandez-Checa JC, Kaplowitz N. Sab (Sh3bp5) dependence of JNK mediated inhibition of mitochondrial respiration in palmitic acid induced hepatocyte lipotoxicity. J Hepatol 2015;62:1367–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kluwe J, Pradere JP, Gwak GY, Mencin A, De Minicis S, Osterreicher CH, et al. Modulation of hepatic fibrosis by c-Jun-N-terminal kinase inhibition. Gastroenterology 2010;138:347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kotzka J, Knebel B, Haas J, Kremer L, Jacob S, Hartwig S, et al. Preventing phosphorylation of sterol regulatory element-binding protein 1a by MAP-kinases protects mice from fatty liver and visceral obesity. PLoS One 2012;7:e32609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Win S, Than TA, Han D, Petrovic LM, Kaplowitz N. c-Jun N-terminal kinase (JNK)-dependent acute liver injury from acetaminophen or tumor necrosis factor (TNF) requires mitochondrial Sab protein expression in mice. J Biol Chem 2011;286:35071–35078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Win S, Than TA, Min RWM, Aghajan M, Kaplowitz N. c-Jun N-terminal kinase mediates mouse liver injury through a novel Sab (SH3BP5)-dependent pathway leading to inactivation of intramitochondrial Src. Hepatology 2016;63:1987–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Win S, Min RW, Chen CQ, Zhang J, Chen Y, Li M, et al. Expression of mitochondrial membrane-linked SAB determines severity of sex-dependent acute liver injury. J Clin Invest 2019;129:5278–5293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Win S, Than TA, Kaplowitz N. The regulation of JNK signaling pathways in cell death through the interplay with mitochondrial SAB and upstream post-translational effects. Int J Mol Sci 2018;19:3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chambers JW, LoGrasso PV. Mitochondrial c-Jun N-terminal kinase (JNK) signaling initiates physiological changes resulting in amplification of reactive oxygen species generation. J Biol Chem 2011;286:16052–16062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moylan CA, Pang H, Dellinger A, Suzuki A, Garrett ME, Guy CD, et al. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology 2014;59:471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chambers TP, Santiesteban L, Gomez D, Chambers JW. Sab mediates mitochondrial dysfunction involved in imatinib mesylate-induced cardiotoxicity. Toxicology 2017;382:24–35. [DOI] [PubMed] [Google Scholar]
- 23.Kirsch K, Zeke A, Tőke O, Sok P, Sethi A, Sebő A, et al. Co-regulation of the transcription controlling ATF2 phosphoswitch by JNK and p38. Nat Commun. 2020;11:5769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aguirre V, Uchida T, Yenush L, Davis R, White MF. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J Biol Chem 2000;275:9047–9054. [DOI] [PubMed] [Google Scholar]
- 25.Zhang J, Min RWM, Le K, Zhou S, Aghajan M, Than TA, et al. The role of MAP2 kinases and p38 kinase in acute murine liver injury models. Cell Death Dis 2017;8:e2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duarte S, Baber J, Fujii T, Coito AJ. Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix Biol 2015;44–46:147–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 2005;120:649–661. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Accession numbers for data relating to the paper were mentioned in the paper and deposited in a public database.