

Abstract
Objective:
An attractive target to interfere with epileptic brain hyperexcitability is the enhancement of GABAergic inhibition by inactivation of the GABA metabolizing enzyme GABA aminotransferase (GABA-AT). GABA-AT inactivators were designed to control seizures by raising brain GABA levels. OV329, a novel drug candidate for the treatment of epilepsy and addiction, has been shown in vitro to be substantially more potent as a GABA-AT inactivator than vigabatrin, an antiseizure drug approved as an add-on therapy for adult patients with refractory complex partial seizures and monotherapy for pediatric patients with infantile spasms. Thus, we hypothesized that OV329 should produce pronounced anticonvulsant effects in two different rat seizure models.
Methods:
We therefore examined the effects of OV329 (5, 20, and 40 mg/kg, i.p.) on the seizure threshold of female Wistar Unilever rats, using the timed intravenous pentylenetetrazole seizure threshold (ivPTZ-ST) model as a seizure test particularly sensitive to GABA-potentiating manipulations, and amygdala-kindled rats as a model of difficult-to-treat temporal lobe epilepsy.
Results:
GABA-AT inactivation by OV329 clearly increased the threshold of both ivPTZ-induced and amygdala-kindled seizures. OV329 further showed a 30-fold greater anticonvulsant potency on ivPTZ-induced myoclonic jerks and clonic seizures compared to vigabatrin investigated previously (see1). Notably, all rats were responsive to OV329 in both seizure models.
Significance:
These results reveal an anticonvulsant profile of OV329 that appears to be superior in both potency and efficacy to vigabatrin and highlight OV329 as a highly promising candidate for the treatment of seizures and pharmacoresistant epilepsies.
Keywords: amygdala kindling, GABA-AT, epilepsy, metrazol, PTZ, vigabatrin
1. Introduction
Epilepsy is one of the most common neurological diseases globally, affecting more than 70 million people worldwide. In light of the fact that ~30% of epilepsy patients have persistent seizures despite use of different currently approved drugs and combinations thereof, there is a critical need for the development of new antiseizure drugs (ASDs), particularly for drug-resistant seizures2, 3. Among various approaches to interfere with epileptic brain hyperexcitability, an attractive target is the enhancement of GABAergic inhibition by inactivation of GABA aminotransferase (GABA-AT), the enzyme responsible for GABA catabolism4, 5. Therefore, GABA-AT inactivators were designed to control seizures by raising brain GABA levels6.
To date, the only approved drug that acts as an inactivator of GABA-AT is the rationally designed ASD vigabatrin. Vigabatrin is approved as add-on therapy for adult patients with refractory complex partial seizures and monotherapy for pediatric patients with infantile spasms, a severe form of epilepsy. However, the antiseizure effect of the irreversible GABA-AT inhibitor vigabatrin has been associated with severe adverse effects during long-term use7, 8, which led to its limited approval. Due to the relatively poor GABA-AT inhibition and ability to cross the blood–brain barrier (CSF/plasma ratio is only about 0.1)5, 9, vigabatrin is generally taken in large doses (1–3 g/day) that may cause visual field defects resulting from retinal damage in 25–50% of patients and limit its clinical utility10–12.
To increase therapeutic potency and to eliminate retinal toxicity, the vigabatrin analogue CPP-115 was designed. CPP-115 was described to be 187 times more potent in inactivating GABA-AT than vigabatrin in vitro and suppressed spasms at 1/100th the mass dose of vigabatrin in a rat model of infantile spasms13. Meanwhile, CPP-115 has completed a Phase I safety clinical trial without revealing adverse effects, and markedly reduced seizures with no evidence of retinal dysfunction in a patient with refractory infantile spasms14, 15. Recently, the structure of CPP-115 has been further refined, leading to the development of OV329, a novel drug candidate for the treatment of pharmacoresistant epilepsies that was shown in vitro to be even 10 times more potent as an inactivator of GABA-AT than CPP-11515. Moreover, it has recently been demonstrated that pretreatment with OV329 reduces the severity of N‐methyl‐d‐aspartate (NMDA)‐induced seizures in mice16 in: 17.
We previously demonstrated that systemic administration of high doses of vigabatrin increase seizure thresholds in the timed intravenous pentylenetetrazole seizure threshold (ivPTZ-ST) test in rats1. Vigabatrin has also been shown to increase seizure thresholds and to reduce seizure severity and duration in amygdala-kindled rats18–20. Yet systemic treatments of high doses of vigabatrin were associated with marked adverse effects, such as sedation, ataxia, and weight loss1, 18. Since OV329 appears to be substantially more potent as a GABA-AT inactivator than vigabatrin, we hypothesized that OV329 produces pronounced anticonvulsant effects in the two different rat models mentioned above, i.e., the ivPTZ-ST model and the amygdala-kindling model. We therefore examined the effects of OV329 on the seizure thresholds of rats, using the ivPTZ-ST model as a seizure test particularly sensitive to GABA-potentiating drugs21, and amygdala-kindled rats as a highly predictive model of drug efficacy against difficult-to-treat types of seizures as in temporal lobe epilepsies22. Based on previous studies showing pharmacoresistance to vigabatrin in a subset of patients and animal models19, 23–26, we additionally determined the OV329 responder rate in both rat models. Finally, drug-induced behavioral and physiological side effects were assessed. Our study helps characterize the antiseizure potential and tolerability of the novel GABA-AT inactivator OV329 and whether it shows improved efficacy compared to previous findings with vigabatrin1.
2. Material and methods
2.1. Animals
As in our previous studies with vigabatrin1, 23, adult female Wistar Unilever rats (~220–360 g) were used. The animals were obtained from Envigo (Venray, The Netherlands) and housed in groups of up to four. To consider estrous cycle-linked effects on brain physiology and behavior27, 28, females were housed without males in order to keep them acyclic or asynchronous with respect to their estrous cycle29. We have previously shown that the PTZ seizure threshold is not affected by the estrous cycle in rats1. Rats were kept under controlled ambient conditions (23–24 °C, 45–50% relative humidity, 12 h/12 h light/dark cycle, lights on at 7 a.m.). Standard laboratory chow (Altromin 1324 standard diet) and water were available ad libitum. Nesting material and tubes served as environmental enrichment. All procedures complied with the European Union directive 2010/63/EU and were formally approved by the animal subjects review board of our institution “Lower Saxony State Office for Consumer Protection and Food Safety” (LAVES, Oldenburg, Germany; File number 14/1727).
2.2. Drugs
OV329 was donated by Ovid Therapeutics (New York, NY, USA) as a hydrochloride salt. All OV329 doses refer to the free base. OV329 and PTZ (Sigma Aldrich, Taufkirchen, Germany) were freshly prepared on each test day and dissolved in sterile 0.9% saline.
2.3. The timed intravenous PTZ infusion seizure threshold test
The ivPTZ-ST model is generally considered as an acute screening model, which provides a sensitive parametric method for evaluating seizure thresholds in individual animals21, 30, 31. It allows determining drug effects on different components of seizure behavior32, such as myoclonic twitches and clonic seizures. For a fast screening of the anticonvulsant potential of OV329, rats received iv infusion of PTZ31, 33, a CNS stimulant that induces convulsions by GABAA receptor antagonism34. Thus, the ivPTZ-ST test is particularly sensitive to drugs that increase GABAergic inhibition21, such as GABA-AT inactivators, and can be used to determine the ability of drugs to alter seizure thresholds by applying the drug at appropriate time before onset of PTZ infusion. To avoid a kindling effect, the number of ivPTZ-ST tests was limited to a maximum of five in the same individual, which has been proven not to change seizure thresholds35, 36.
To quantify the threshold doses to the different components of ivPTZ-induced seizures, a 0.8% PTZ solution was continuously infused (Pump 11 Elite, Harvard Apparatus, Holliston, MA, USA; constant flow rate of 1 ml/min) via flexible polyethylene tubing (PE20 tubing, non-sterile; Instech Laboratories, Plymouth Meeting, PA, USA) connected to a 24 G needle inserted into the lateral tail vein of rats that were allowed to move freely inside a Makrolon® cage type III. We measured two endpoints: the occurrence of the first myoclonic twitch and the subsequent occurrence of the first clonic seizure, the latter of which was set as end of infusion. The seizure thresholds were calculated in mg/kg PTZ based on the body weight of the animal (measured immediately before seizure threshold testing), the rate of infusion, the PTZ concentration, and the latency to induce the seizure.
We first determined the basal seizure threshold once for each rat (pre-drug control). Two days later, the rats (n = 8) entered the drug testing phase where they received intraperitoneal (i.p.) injections of three OV329 doses (5, 20, and 40 mg/kg) and saline as vehicle control at a volume of 3 ml/kg body weight in a randomized crossover design (Fig. 1A). Since full GABA-AT recovery after irreversible inhibition by OV329 is expected to require re-synthesis of the enzyme and full enzyme activity does not return within 48 h in vitro15, ivPTZ-ST tests were done 7 days apart. We have previously shown that the maximum effect of vigabatrin on the PTZ-ST occurs 6 h post injection1. Therefore, we decided to conduct the ivPTZ-ST test 6 h after OV329 or saline injections in the present study. OV329 doses were based on previous studies15, 37 and our own preliminary investigation (unpublished results).
Figure 1. Outline of the experimental design using two different seizure threshold models.
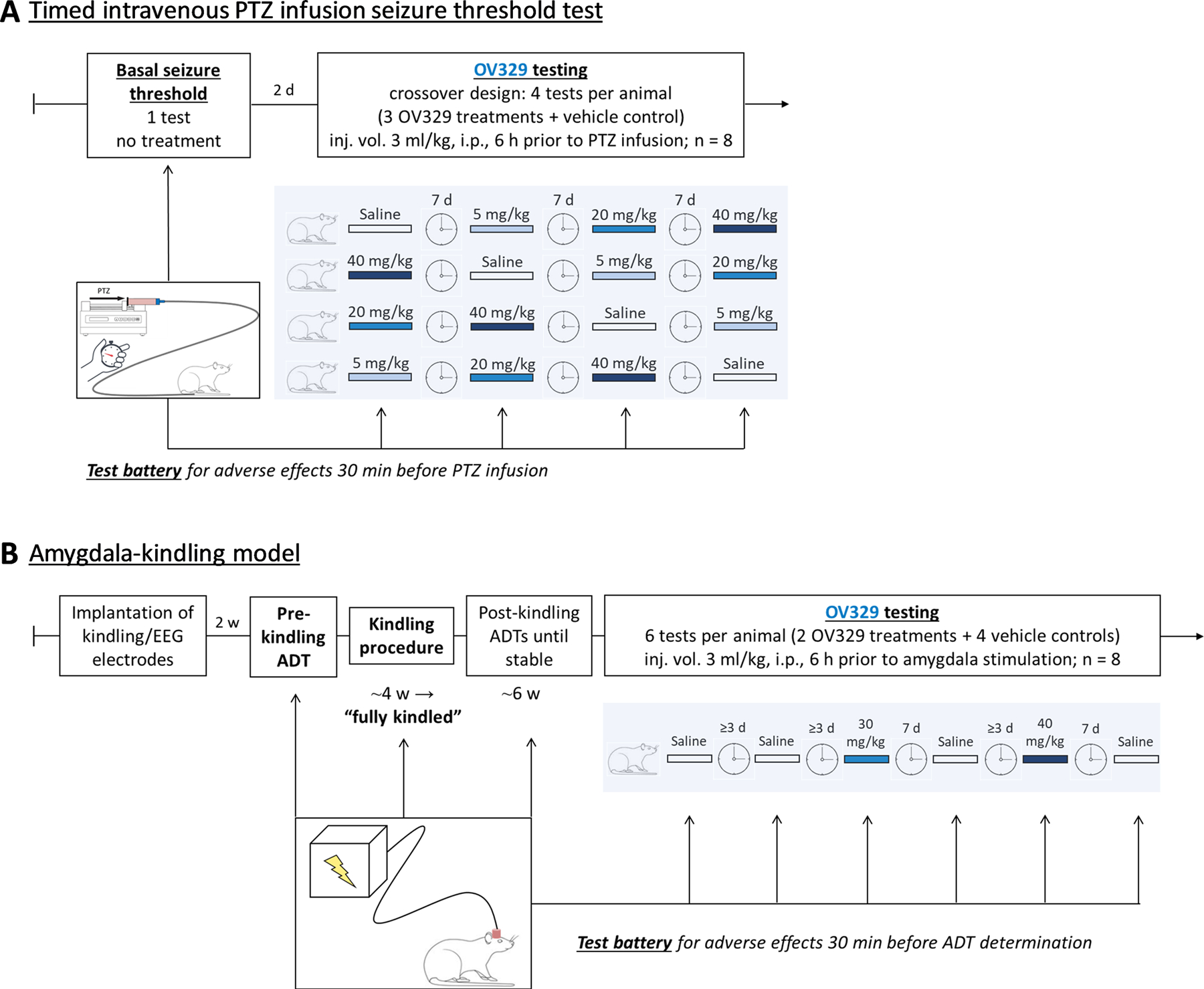
A) Seizure thresholds were determined by timed intravenous pentylenetetrazole (PTZ) infusion. Following determination of basal seizure thresholds, rats (n = 8) received intraperitoneal (i.p.) injections of OV329 (5, 20, and 40 mg/kg) and saline as vehicle control (3 ml/kg) in a crossover design 5½ h before test battery for adverse effects and 6 h before PTZ infusion. For more details, refer to text. B) Seizure thresholds were determined in fully amygdala-kindled rats. Rats were kindled via a chronically implanted electrode in the right amygdala until fully kindled. Determination of seizure thresholds (afterdischarge thresholds, ADT) was performed until reproducible before onset of drug and vehicle trials. Rats (n = 8) received intraperitoneal (i.p.) injections of OV329 (30 and 40 mg/kg) and saline as vehicle control (3 ml/kg) in an alternating design starting with vehicle as indicated in the figure. The post-control for the previous dose served as pre-control for the subsequent dose. Thus, each rat served as its own control. Pretreatment time was 6 h. Adverse effects were assessed 30 min before ADT determination. In addition to the ADT, further seizure parameters (generalized seizure threshold, seizure severity, seizure duration, afterdischarge duration) were determined.
Based on previous experiments with vigabatrin23, we determined the OV329 antiseizure response/nonresponse rate. In our present study, responders were defined as showing at least 25% increase in seizure threshold (for the myoclonic jerk and clonic seizure separately) after OV329 treatment compared to saline control.
2.4. Amygdala-kindling model
In addition to the acute seizure model described above, we used a chronic seizure/epilepsy model, i.e., fully amygdala-kindled rats, to verify and extend our findings. Fully amygdala-kindled rats are characterized by permanent brain alterations reflecting epileptic brain networks38. The amygdala-kindling model is highly predictive for detecting clinically effective drugs for the treatment of focal onset seizures as in temporal lobe epilepsies22 and is thus considered a model for difficult-to-treat types of seizures. We previously published kindling protocols comparable to the one used here23, 36, 39, 40, which is described in detail in the Supplement. Briefly, rats were electrically kindled via a chronically-implanted electrode in the right amygdala. After establishing reproducible stimulation thresholds for eliciting afterdischarges (ADT, afterdischarge threshold) in fully-kindled rats, the rats were alternately treated with vehicle (pre-control and post-control) and OV329 (refer to Fig. 1B). The post-control for the previous dose served as pre-control for the subsequent dose. Thus, each rat served as its own control. During this drug-testing phase, the ADT determination performed by an ascending staircase procedure (refer to Supplement) always started three steps below the previous vehicle ADT. Because OV329 is expected to induce an irreversible inactivation of GABA-AT requiring re-synthesis of the enzyme41, the vehicle post-control ADT determination was determined not earlier than one week following the previous drug trial (equally to the ivPTZ-ST protocol). This was not necessary after vehicle injections, which is why the testing interval could be shorter after vehicle injections. As in the ivPTZ-ST test, saline at a volume of 3 ml/kg body weight served as vehicle. Because the lower OV329 doses of 5 and 20 mg/kg i.p. induced only subtle effects in the ivPTZ-ST test, while the higher dose of 40 mg/kg i.p. was clearly anticonvulsant (refer to results), we used doses of 30 and 40 mg/kg i.p. in the amygdala-kindling model. As in the ivPTZ-ST test, the pretreatment time was 6 h and responders were defined as showing at least 25% increase in seizure threshold (here, ADT and GST, respectively) following OV329 compared to saline.
Usually, fully-kindled rats show generalized motor seizures (stage 4 or 5 seizures according to Racine, 1972) at the ADT. Otherwise, the electrical current was further elevated up to the generalized seizure threshold (GST). Apart from the ADT, we recorded the seizure severity (SS), seizure duration (SD), and afterdischarge duration (ADD) at ADT and GST, respectively.
2.5. Assessment of adverse effects
The assessment of drug-induced behavioral alterations was conducted 30 min before each seizure threshold determination and is detailed in the Supplement.
2.6. Histological verification
Histological verification of the kindling site is detailed in the Supplement.
2.7. Statistical analysis
Normally distributed data were compared using one-way mixed-effects model analysis or one-way repeated measures (RM) analysis of variance (ANOVA). Nonparametric statistics were used in case data did not follow a normal distribution (for details, see Supplement).
3. Results
OV329 causes anticonvulsant effects in the ivPTZ-ST rat model
The basal (pre-drug) PTZ seizure thresholds (mean±SEM) were 15.8±0.97 mg/kg for the myoclonic twitch and 19.5±1.34 mg/kg for the clonic seizure. As depicted in Fig. 2, 40 mg/kg OV329 i.p. significantly increased the threshold for the myoclonic twitch (33.1±2.54 mg/kg, P=0.0026, Fig. 2A) and for the clonic seizure (40.98±3.04 mg/kg, P=0.0027, Fig. 2B) compared to the basal threshold. In contrast, vehicle injections did not alter myoclonic and clonic seizure thresholds (P>0.05, Figs. 2A, 2B). Considering inter-individual differences in basal seizure thresholds, we further calculated the effects of vehicle control and OV329 as percent change to the basal seizure threshold for a more reliable quantification and better comparison between vehicle- and drug-treated rats. We found that OV329 (40 mg/kg i.p.) significantly increased the ivPTZ-induced seizure threshold for the myoclonic twitch (102.74±14.07%, P=0.0006, Fig. 2C) and the clonic seizure (105.18±14.66%, P=0.0012, Fig. 2D) compared to vehicle-injected animals (myoclonic twitch: −1.63±5.91%; clonic seizure: 4.58±6.66%). The anticonvulsant effects of OV329 were dose-dependent, since we did not observe significant effects on seizure thresholds at lower doses, i.e., 0.1 mg/kg (data not shown), 1 mg/kg (data not shown), 5 mg/kg (Fig. 2), and 20 mg/kg OV329 (Fig. 2). Higher OV329 doses, i.e., 50 mg/kg, did not result in a more pronounced antiseizure effect, but were associated with severe adverse effects, e.g., catalepsy, and therefore excluded from further investigation. Dose-response analyses of OV329 on ivPTZ-induced seizure thresholds revealed ED50 = 25.83 mg/kg for the myoclonic twitch and ED50 = 20.8 mg/kg for the clonus.
Figure 2. OV329 dose-dependently increases the thresholds of intravenous pentylenetetrazole (ivPTZ)-induced seizures.
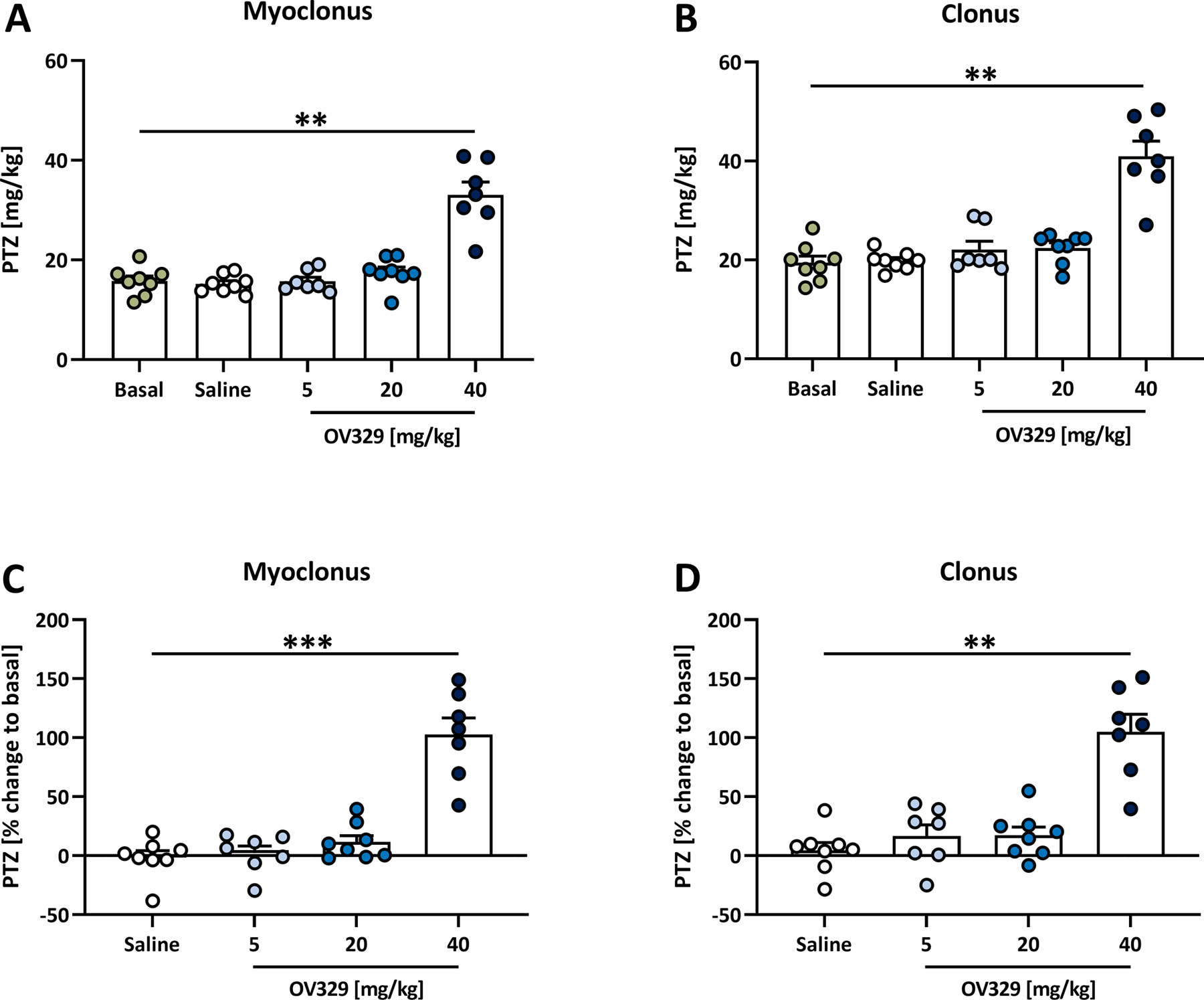
Effects of 5, 20, and 40 mg/kg OV329 injected intraperitoneally on the thresholds of the ivPTZ-induced (A) first myoclonic twitch and (B) first clonic seizure in rats (n = 8). Treatment effects were further calculated as percent change to the basal seizure threshold for the myoclonic twitch (C) and the clonic seizure (D). Data are expressed as mean+SEM. Statistically significant differences between OV329 and the basal seizure threshold (A, B), and between OV329 and saline as vehicle control (C, D) are indicated by asterisks (one-way mixed-effects model analysis, post-hoc Sidak correction, **P<0.01, ***P<0.001).
All rats are responsive to OV329
Since previous experiments have shown that not all rats respond to the treatment with vigabatrin in a high dose with tolerable adverse effects23, we conducted an intra-individual response analysis between OV329 and saline as vehicle control. As shown in Fig. 3, we found a dose-dependent increase in the responder rate. While only one out of eight (12.5%) rats showed at least 25% increase in ivPTZ-induced clonic seizure threshold after 5 mg/kg OV329 compared to saline, the responder rate increased to three out of eight (37.5%) for the myoclonic jerk and the clonic seizure at a dose of 20 mg/kg OV329. Importantly, all eight rats were responsive to 40 mg/kg OV329 with regard to the myoclonic twitch as well as the clonic seizure (100% responder rate).
Figure 3. OV329 produces a dose-dependent increase in the responder rate.
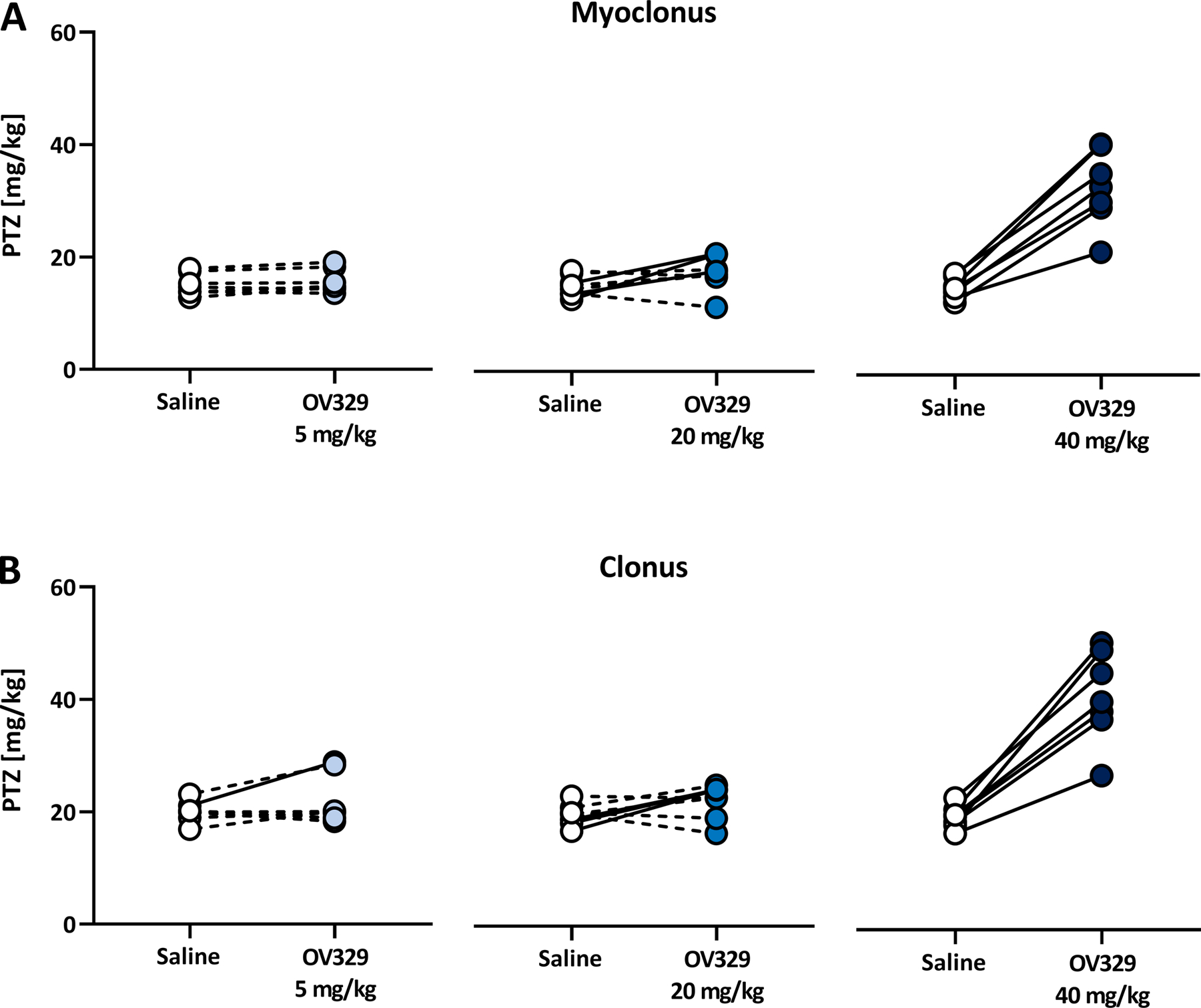
Responders (marked by a continuous line) were defined as showing at least 25% increase in seizure threshold, for either the myoclonic twitch (A) or the clonic seizure (B), after OV329 treatment compared to saline control.
OV329 causes anticonvulsant effects in amygdala-kindled rats
As for PTZ seizure thresholds, kindled seizure parameters were determined 6 h after drug or vehicle injection. For evaluation of ADTs and GSTs and seizure parameters at both thresholds (seizure severity, seizure duration, afterdischarge duration), mean drug values were compared with mean pre-drug and post-drug control values. During pre-drug control testing, the animals showed stable ADTs after saline (S) injection [mean ADT±SEM: 143.3±18.2 μA (range 110–200 μA) at S1 vs. 130±16.9 μA (range 90–200 μA) at S2; Fig. 4A].
Figure 4. OV329 increases the afterdischarge threshold (ADT), suppresses generalized seizures and reduces seizure severity in amygdala-kindled rats.
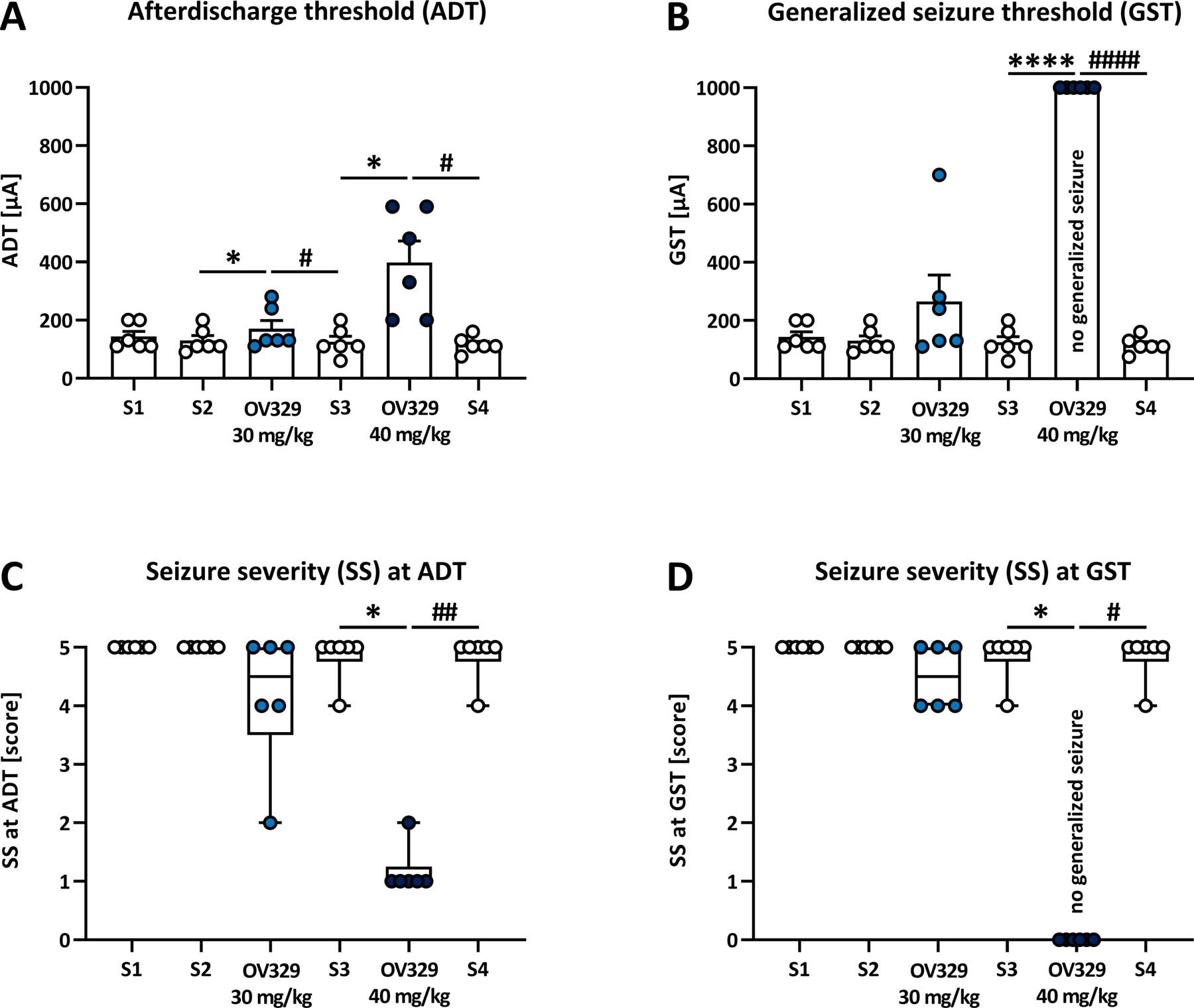
Effects of 30 and 40 mg/kg OV329 injected intraperitoneally on the ADT (A), the generalized seizure threshold (GST, B), and the seizure severity (SS) at ADT (C) and GST (D) in rats (n = 6). Saline (S) injections served as pre- and post-drug controls. Data are expressed as mean+SEM (A, B) and as box plots with whiskers (min to max; C, D). At 40 mg/kg OV329, none of the animals expressed generalized motor seizures, and the GST was set at 1000 μA (B) and the SS at GST was set at score 0 (D) for data evaluation. Statistically significant differences between OV329 and saline as pre-drug control are indicated by asterisks, and differences between OV329 and saline as post-drug control are indicated by hashtags (A, B: one-way mixed-effects model analysis, post-hoc Sidak correction; C, D: Friedman test for one-way repeated measures ANOVA by ranks, with Dunn’s correction for post hoc multiple comparisons; *, #P<0.05, ##P<0.01, ****, ####P<0.0001).
OV329 significantly increased the ADT (Fig. 4A) compared to pre-drug saline control at doses of 30 mg/kg (mean±SEM: 170±29.1 μA; range: 110–280 μA; P=0.049) and 40 mg/kg (398.3±73.8 μA; range: 200–590 μA; P=0.027). The saline post-drug control ADTs (measured one week after OV329 treatment) were as low as pre-drug control values and thus also differed significantly from the ADTs after OV329 treatment (125±19.8 μA; range: 60–200 μA; P=0.027 for 30 mg/kg, and 115.8±11.4 μA; range: 75–160 μA; P=0.023 for 40 mg/kg). Remarkably, OV329 (40 mg/kg) suppressed generalized seizures in all rats (Fig. 4B). Following injection of vehicle or 30 mg/kg OV329, a secondarily generalized seizure [stage 4/5;42] was always inducible in kindled rats. After injection of 40 mg/kg OV329, none of the animals expressed generalized motor seizures up to the highest current intensity tested (840 μA). In these cases, the GST was set at 1000 μA (the next theoretical 20% step in seizure threshold testing) for data evaluation (Fig. 4B). Because 40 mg/kg OV329 prevented the animals from generalized seizure expression, further kindling parameters at GST (seizure severity, seizure durations) were set at zero. Seizure severity at ADT was significantly reduced after 40 mg/kg OV329 compared to pre-drug control (Fig. 4C, P=0.011) and post-drug control (Fig. 4C; P=0.0086).
OV329 (40 mg/kg) further significantly reduced the duration of motor seizures (SD1) and the duration of motor seizures plus the adjacent time of immobility (SD2) at the ADT (Fig. 5; SD1: 24.3±4.6 s, range 10–42 s, P=0.0022; SD2: 24.3±4.1 s, range 12–40 s, P<0.0001) compared to pre-drug control (SD1: 65.5±5.5 s, range 46–81 s; SD2: 550±36.1 s, range 420–665 s). In OV329-treated rats, ADD1 was always equal to ADD2, i.e., no distinguishable afterdischarge patterns occurred. At 40 mg/kg OV329, the ADD at ADT was significantly reduced compared to pre-drug control (Fig. 5; ADD1 at ADT: 24.3±4.1 s, range 12–40 s, P=0.0232) and post-drug control (P=0.0021), the latter showing that we did not observe any carry-over effects on kindled seizure parameters. Indeed, multiple comparisons between S1 and subsequent saline controls revealed no differences regarding kindled seizure parameters. OV329 (40 mg/kg i.p.) also significantly decreased the SD and ADD at GST (Fig. 5), since generalized seizures were completely suppressed. The fact that vehicle control injections given 7 days following an OV329 treatment did not alter seizure thresholds in the ivPTZ-ST model and in amygdala-kindled rats indicates the return of GABA-AT activity within 7 days in our rats.
Figure 5. OV329 reduces seizure and afterdischarge durations in amygdala-kindled rats.
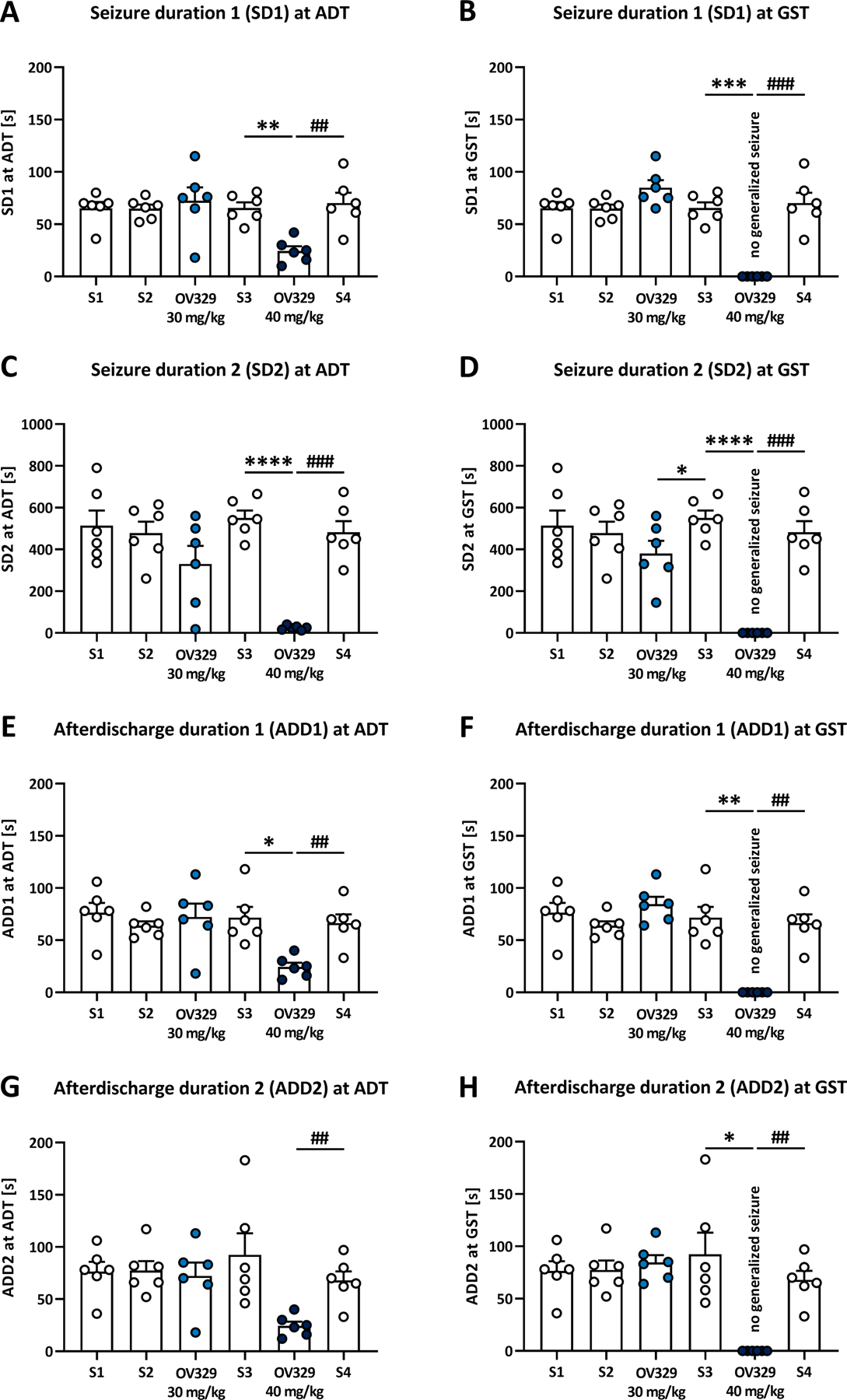
Effects of 30 and 40 mg/kg OV329 injected intraperitoneally on the duration of motor seizures (seizure duration 1, SD1; A, B) and the time of motor seizures plus the adjacent time of immobility (seizure duration 2, SD2; C, D) at the afterdischarge threshold (ADT) and the generalized seizure threshold (GST), and on the afterdischarge durations ADD1 (E, F) and ADD2 (G, H) in rats (n = 6). Saline (S) injections served as pre- and post-drug controls. At 40 mg/kg OV329, none of the animals expressed generalized motor seizures and the SD and ADD at GST were set at 0 s (B, D, F, G) for data evaluation. Data are expressed as mean+SEM. Statistically significant differences between OV329 and saline as pre-drug control are indicated by asterisks, and differences between OV329 and saline as post-drug control are indicated by hashtags (one-way mixed-effects model analysis, post-hoc Sidak correction, *P<0.05, **, ##P<0.01, ***, ###P<0.001, ****P<0.0001).
All amygdala-kindled rats respond to OV329
As in the ivPTZ-ST model, we also evaluated the efficacy of OV329 in individual amygdala-kindled animals and therefore again conducted an intra-individual analysis between drug and control treatment. We found a responder rate of 2/6 (33.3%) for the ADT and 3/6 (50%) for the GST at 30 mg/kg OV329. Strikingly, all rats were responsive to 40 mg/kg OV329 concerning the ADT and GST (100% responder rate; Fig. 6).
Figure 6. OV329 produces a dose-dependent increase in the responder rate concerning the afterdischarge threshold (ADT) and the generalized seizure threshold (GST).
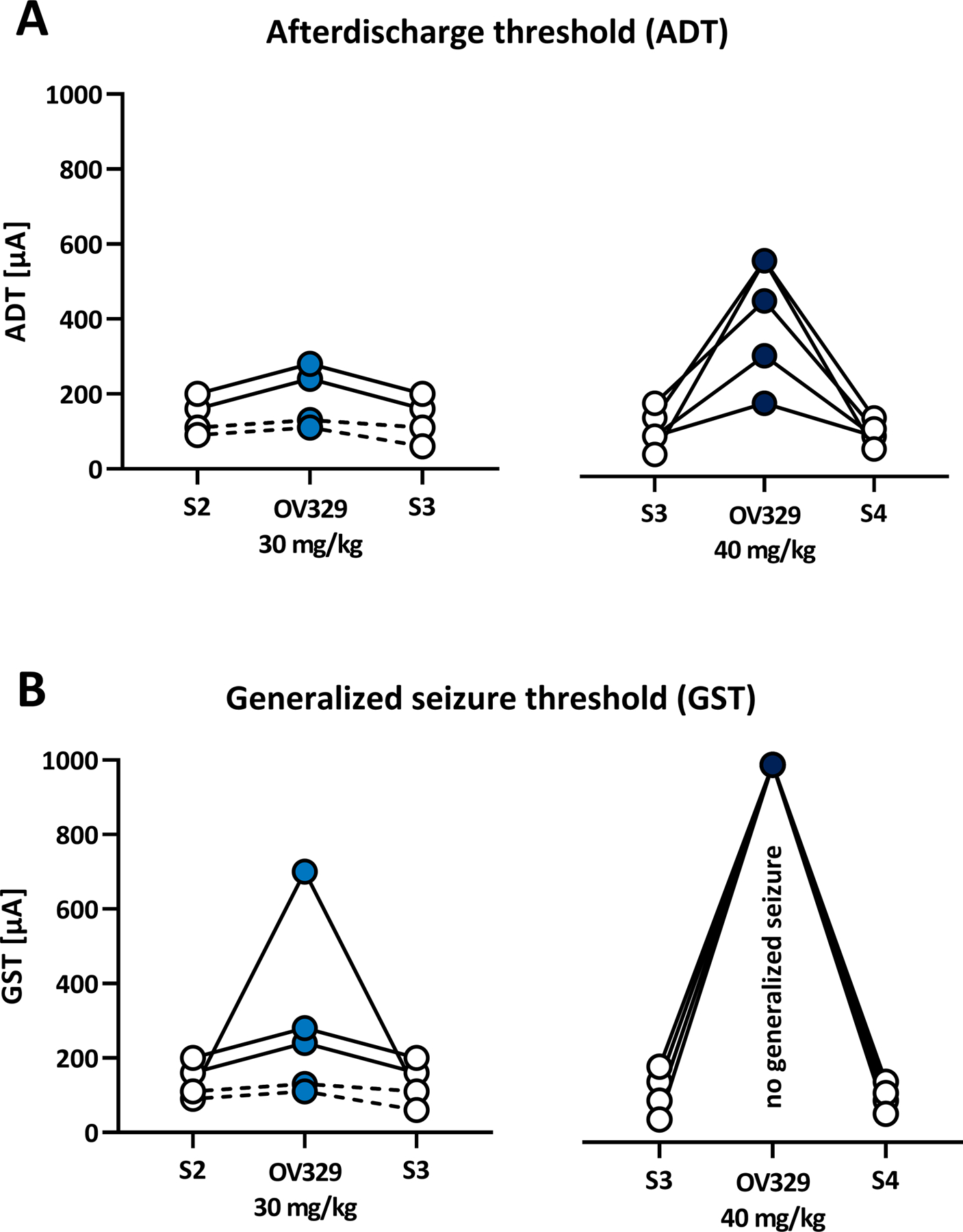
Responders (marked by a continuous line) were defined as showing at least 25% increase in ADT (A) or GST (B) after OV329 treatment compared to saline (S) control. At 40 mg/kg OV329, none of the animals expressed generalized motor seizures, and the GST was set at 1000 μA (B) for data evaluation.
OV329 causes mild adverse effects
Low doses of OV329 were well tolerated, while higher doses were associated with decreased body temperature, moderate loss of body weight (Fig. S1), and sedation in about one third of rats. Half of the animals (3/8 iv-PTZ-tested rats, 4/6 amygdala-kindled rats) did not pass the rotarod test, indicating moderate but not significant deficits in motor coordination compared to saline. The evaluation of toxic effects of OV329 on the rotarod performance yielded TD50 = 46.16 mg/kg (for details, see Supplement).
4. Discussion
This study sought to determine the anticonvulsant potential of the GABA-AT inactivator OV329 in two well-established seizure/epilepsy models. We discovered three main novel findings. First, OV329 increased the threshold of ivPTZ-induced acute seizures. Second, OV329 increased the threshold of amygdala-kindled chronic, difficult-to-treat seizures (focal and generalized). Third, all rats were responsive to OV329 in both seizure models. In addition, OV329 showed a 30-fold greater anticonvulsant potency on ivPTZ-induced myoclonic jerks and clonic seizures compared to previous findings on vigabatrin (1; see below). These results reveal an anticonvulsant profile of OV329 that appears to be superior in potency and efficacy to vigabatrin.
As one mechanism, seizure control can be achieved by increasing brain GABA concentrations4, 6. Initial attempts to increase GABA levels in the brain directly by iv injection of the neurotransmitter were unsuccessful due to impermeability of the blood-brain barrier to GABA43, 44. Therefore, mechanism-based, selective inactivators of GABA-AT, like vigabatrin and OV329, were rationally designed to enhance GABAergic transmission in the CNS by suppressing GABA degradation5, 45. Vigabatrin demonstrated inhibition of GABA catabolism to be effective in decreasing seizures in both animal models and humans45. In previous animal studies, systemic vigabatrin blocked generalized convulsions induced by subcutaneous PTZ with an ED50 of 940 mg/kg and increased ivPTZ-ST at 490 mg/kg 6 h after i.p. administration in mice46, 47. More recently, increased ivPTZ-ST 6 h after systemic vigabatrin (600 and 1200 mg/kg i.p.) was confirmed in rats1. Our results reaffirm increased ivPTZ-ST by GABA-AT inactivation and further strengthen the concept of mechanism-based GABA-AT inactivators acting as effective anticonvulsant agents. Vigabatrin has long been the most effective of these mechanism-based GABA-AT inactivators as an anticonvulsant agent48 until its analogues CPP-115 and OV329 showed higher in vitro efficacy13, 15. CPP-115 decreased infantile spasms in a rat model at considerably lower and better tolerated doses than vigabatrin49. OV329 revealed pharmacokinetic properties superior to CPP-115 and reduced NMDA-induced seizure severity in juvenile mice15, 16. Here, we demonstrated that systemic OV329 injections caused clear antiseizure effects at a dose of 40 mg/kg (the mean raise in seizure threshold was 103% above the basal threshold for the myoclonic twitch and 105% above the basal threshold for the clonic seizure) in rats. In the same rat seizure model, a high dose of 1200 mg/kg vigabatrin was necessary to induce comparable seizure threshold elevations (59% and 87% above the basal threshold for the myoclonic twitch and for the clonic seizure, respectively), revealing OV329 is equieffective at 30-fold lower mass doses than vigabatrin (40 mg/kg OV329 vs. 1200 mg/kg vigabatrin) on ivPTZ-induced myoclonic jerks and clonic seizures.
In addition to the primary screening in our acute seizure model, we verified and further investigated the anticonvulsant activity of OV329 in the only clinically validated chronic seizure model, the kindled rodent model of mesial temporal lobe epilepsy50. Indeed, the amygdala kindling model is highly predictive of drug efficacy against focal limbic seizures in humans22. Furthermore, especially focal, and to a lesser extent also focal to bilateral tonic-clonic seizures in amygdala-kindled rats are considered difficult to suppress, so that amygdala-kindling was the first proposed animal model of drug-resistant focal epilepsy in humans51. Using this model of difficult-to-treat types of seizures, we found that six hours after its administration, OV329 (40 mg/kg) increased the threshold for electrographic seizures recorded from the stimulated amygdala. Furthermore, although the suppression of generalized motor seizures was more pronounced, we found that OV329 decreased the incidence not only of the generalized seizures, but also of the difficult-to-treat focal seizures. Although less predictive with regard to drug activity against epilepsies in humans, we additionally observed that OV329 decreased the duration of electrographic and motor seizures, and reduced seizure severity in fully-kindled rats. A similar dose of vigabatrin (50 mg/kg) failed to exert anticonvulsant effects in kindled rats 4 h after i.p. administration, but rather increased the duration of motor seizures20. Most studies on vigabatrin observed antiseizure effects only at much higher doses (800–1500 mg/kg) in amygdala-kindled rats18, 52, 53, again indicating the much greater potency of OV329 relative to vigabatrin. In comparison to the only study on vigabatrin, in which a lower dose (200 mg/kg) displayed anticonvulsant activity after acute i.p. injection in kindled rats20, OV329 resulted in a more than 6-fold increase in ADT and more pronounced reductions in seizure severity and durations. The decreased seizure severity at increased ADT in the present study suggests that OV329 not only elevates seizure threshold but also reduces seizure propagation from the focus.
Despite the fact that in the last 30 years more than 20 new ASDs have been introduced, there is continuing clinical demand for more efficacious and better-tolerated ASDs, especially with regard to the large portion of about 30% drug-resistant epilepsy patients. Since vigabatrin is typically effective only in high doses that are often associated with serious side effects, more potent and less toxic GABA-AT inhibitors are desired. The favorable pharmacokinetic profile of OV329 including a higher binding affinity and a larger inactivation rate constant for GABA-AT compared to previous GABA-AT inhibitors15, 37 becomes manifest in vivo in the present study. Here, we prove for the first time the potent anticonvulsant properties of the new GABA-AT inactivator, OV329, in two different rat models. Several vigabatrin studies revealed pharmacoresistance in a subset of patients and rodents in PTZ tests and kindling models24, 26, 52–54. Therefore, despite of its tremendous efficacy as an inactivator of GABA-AT, we presumed not all rats to respond sensitively to OV329. Remarkably, however, all rats were responsive to the highest (and tolerable) dose of OV329 in both seizure models. OV329 (40 mg/kg) not only increased the thresholds for ivPTZ-induced myoclonic twitch and clonic seizure but also elevated the ADT and completely suppressed generalized seizures throughout all kindled rats. Together with the above-described characteristics of amygdala-kindled rats being considered as a model for difficult-to-treat types of seizures, our data indicate that OV329 may be a promising candidate for the treatment of drug-resistant epilepsies.
Systemic administration of OV329 was associated with tolerable adverse effects including decreased body temperature, reduced body weight, and impaired motor coordination in a few animals, most probably due to sedation. The highest dose decreased body temperature and weight by about 0.7–0.9 °C and 6%, respectively, within 6 h after administration. This is in line with previous studies where GABA-AT inhibition by vigabatrin resulted in comparable or greater reductions in body temperature and weight1, 53. Comparison of median effective doses of OV329 on ivPTZ-induced seizure thresholds (ED50 = 25.83 mg/kg and 20.8 mg/kg for the myoclonic twitch and the clonus, respectively) and median toxic doses on rotarod performance indicate a narrow therapeutic index for OV329 regarding PTZ-induced myoclonic twitches and clonic seizures. However, previous studies demonstrated that vigabatrin doses (1000–1200 mg/kg) required for an anticonvulsant effect equivalent to what we observed here with OV329 cause significant motor deficits in rats1, 55, whereas OV329 did not significantly impair rotarod performance across the animals. Moreover, we previously observed the anticonvulsant effects of vigabatrin (1200 mg/kg i.p.) being accompanied with ataxia and sedation in each rat1, while in the present study, only one rat displayed ataxia along with increased seizure thresholds. These findings suggest that the improved (30-fold) in vivo potency of OV329 is also coupled to an improved tolerability compared to vigabatrin, even though future studies are needed to comparably analyze the therapeutic indices of vigabatrin and OV329 more specifically. Since this study focused on the acute effects of OV329 and its antiseizure potential, we did not determine the retinal toxicity of OV329. However, previous studies with CPP-115 indicate that the retinotoxicity of vigabatrin appears uncoupled to the GABA-AT activity14, 15.
In summary, we collected important data that could help to define the clinical potential of OV329. We provide proof-of-concept evidence that OV329, a novel GABA-AT inactivator, is highly active against ivPTZ-induced myoclonic twitches and clonic seizures as well as difficult-to-treat amygdala-kindled focal and focal-to-bilateral tonic-clonic seizures. OV329 showed robust anticonvulsant efficacy with only mild adverse effects in the two well-established animal seizure and epilepsy models. Together, the present findings highlight OV329 as a highly promising anticonvulsant agent and a promising candidate for the treatment of drug-resistant epilepsies.
Supplementary Material
Key points.
The GABA aminotransferase inhibitor OV329 increased the threshold of intravenous pentylenetetrazole (ivPTZ)-induced acute seizures
OV329 also increased the threshold of amygdala-kindled chronic, difficult-to-treat seizures (focal and generalized)
All rats were responsive to OV329 in both seizure models (100% responder rate)
OV329 showed a 30-fold greater anticonvulsant potency on ivPTZ-induced seizures compared to previous findings on vigabatrin
OV329 is a highly promising candidate for the treatment of seizures and pharmacoresistant epilepsies
Acknowledgments
We thank Doris Möller, Martina Gramer, and Ivo Denden for technical assistance. This work was supported by a grant (GE 1103/9-1) from the Deutsche Forschungsgemeinschaft (Bonn, Germany) to M.G., and a National Institutes of Health - National Institute on Drug Abuse Grant to R.B.S. (Grant No. R01 DA030604). S.M. received a scholarship from the Prof. Dr. Peter and Jytte Wolf Foundation for Epilepsy (Bielefeld, Germany).
Footnotes
Ethical publication statement
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Disclosure of conflicts of interest
R.B.S. is an inventor on the patent that covers OV329, which has been licensed to the biotechnology company Ovid Therapeutics by Northwestern University. M.J.D. is a consultant to Ovid Therapeutics.
Supporting information
Additional supporting information may be found online in the Supporting Information section.
References
- 1.Bröer S, Backofen-Wehrhahn B, Bankstahl M, Gey L, Gernert M, Löscher W. Vigabatrin for focal drug delivery in epilepsy: bilateral microinfusion into the subthalamic nucleus is more effective than intranigral or systemic administration in a rat seizure model Neurobiol Dis. 2012. May;46:362–376. [DOI] [PubMed] [Google Scholar]
- 2.Janmohamed M, Brodie MJ, Kwan P. Pharmacoresistance - Epidemiology, mechanisms, and impact on epilepsy treatment Neuropharmacology. 2020. May 15;168:107790. [DOI] [PubMed] [Google Scholar]
- 3.Kalilani L, Sun X, Pelgrims B, Noack-Rink M, Villanueva V. The epidemiology of drug-resistant epilepsy: A systematic review and meta-analysis Epilepsia. 2018. December;59:2179–2193. [DOI] [PubMed] [Google Scholar]
- 4.Sills GJ, Rogawski MA. Mechanisms of action of currently used antiseizure drugs Neuropharmacology. 2020. January 14;168:107966. [DOI] [PubMed] [Google Scholar]
- 5.Silverman RB. Design and Mechanism of GABA Aminotransferase Inactivators. Treatments for Epilepsies and Addictions Chem Rev. 2018. April 11;118:4037–4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petroff OA, Rothman DL, Behar KL, Mattson RH. Low brain GABA level is associated with poor seizure control Ann Neurol. 1996. December;40:908–911. [DOI] [PubMed] [Google Scholar]
- 7.Hardus P, Verduin WM, Postma G, Stilma JS, Berendschot TT, van Veelen CW. Long term changes in the visual fields of patients with temporal lobe epilepsy using vigabatrin Br J Ophthalmol. 2000. July;84:788–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ponjavic V, Andreasson S. Multifocal ERG and full-field ERG in patients on long-term vigabatrin medication Doc Ophthalmol. 2001. January;102:63–72. [DOI] [PubMed] [Google Scholar]
- 9.Menachem EB, Persson LI, Schechter PJ, Haegele KD, Huebert N, Hardenberg J, et al. Effects of single doses of vigabatrin on CSF concentrations of GABA, homocarnosine, homovanillic acid and 5-hydroxyindoleacetic acid in patients with complex partial epilepsy Epilepsy Res. 1988. Mar-Apr;2:96–101. [DOI] [PubMed] [Google Scholar]
- 10.Sabers A, Gram L. Pharmacology of vigabatrin Pharmacol Toxicol. 1992. April;70:237–243. [DOI] [PubMed] [Google Scholar]
- 11.Wild JM, Chiron C, Ahn H, Baulac M, Bursztyn J, Gandolfo E, et al. Visual field loss in patients with refractory partial epilepsy treated with vigabatrin: final results from an open-label, observational, multicentre study CNS Drugs. 2009. November;23:965–982. [DOI] [PubMed] [Google Scholar]
- 12.Willmore LJ, Abelson MB, Ben-Menachem E, Pellock JM, Shields WD. Vigabatrin: 2008 update Epilepsia. 2009. February;50:163–173. [DOI] [PubMed] [Google Scholar]
- 13.Silverman RB. The 2011 E. B. Hershberg award for important discoveries in medicinally active substances: (1S,3S)-3-amino-4-difluoromethylenyl-1-cyclopentanoic acid (CPP-115), a GABA aminotransferase inactivator and new treatment for drug addiction and infantile spasms J Med Chem. 2012. January 26;55:567–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doumlele K, Conway E, Hedlund J, Tolete P, Devinsky O. A case report on the efficacy of vigabatrin analogue (1S, 3S)-3-amino-4-difluoromethylenyl-1-cyclopentanoic acid (CPP-115) in a patient with infantile spasms Epilepsy Behav Case Rep. 2016;6:67–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Juncosa JI, Takaya K, Le HV, Moschitto MJ, Weerawarna PM, Mascarenhas R, et al. Design and Mechanism of (S)-3-Amino-4-(difluoromethylenyl)cyclopent-1-ene-1-carboxylic Acid, a Highly Potent gamma-Aminobutyric Acid Aminotransferase Inactivator for the Treatment of Addiction J Am Chem Soc. 2018. February 14;140:2151–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abrahams BS, Crean CS, Chadchankar H, Silverman RB, Davies PA, During MJ, et al. Progress report on new antiepileptic drugs: A summary of the Fourteenth Eilat Conference on New Antiepileptic Drugs and Devices (EILAT XIV). I. Drugs in preclinical and early clinical development Epilepsia. 2018. October;59:1811–1841. [DOI] [PubMed] [Google Scholar]
- 17.Bialer M, Johannessen SI, Koepp MJ, Levy RH, Perucca E, Tomson T, et al. Progress report on new antiepileptic drugs: A summary of the Fourteenth Eilat Conference on New Antiepileptic Drugs and Devices (EILAT XIV). I. Drugs in preclinical and early clinical development Epilepsia. 2018. October;59:1811–1841. [DOI] [PubMed] [Google Scholar]
- 18.Löscher W, Jäckel R, Müller F. Anticonvulsant and proconvulsant effects of inhibitors of GABA degradation in the amygdala-kindling model Eur J Pharmacol. 1989. April 12;163:1–14. [DOI] [PubMed] [Google Scholar]
- 19.Löscher W, Rundfeldt C, Hönack D. Pharmacological characterization of phenytoin-resistant amygdala-kindled rats, a new model of drug-resistant partial epilepsy Epilepsy Res. 1993. July;15:207–219. [DOI] [PubMed] [Google Scholar]
- 20.Rundfeldt C, Löscher W. Development of tolerance to the anticonvulsant effect of vigabatrin in amygdala-kindled rats Eur J Pharmacol. 1992. March 31;213:351–366. [DOI] [PubMed] [Google Scholar]
- 21.Green AR, Murray TK. A simple intravenous infusion method in rodents for determining the potency of anticonvulsants acting through GABAergic mechanisms J Pharm Pharmacol. 1989. December;41:879–880. [DOI] [PubMed] [Google Scholar]
- 22.Löscher W Critical review of current animal models of seizures and epilepsy used in the discovery and development of new antiepileptic drugs Seizure. 2011. June;20:359–368. [DOI] [PubMed] [Google Scholar]
- 23.Gey L, Gernert M, Löscher W. Continuous bilateral infusion of vigabatrin into the subthalamic nucleus: Effects on seizure threshold and GABA metabolism in two rat models Neurobiol Dis. 2016. July;91:194–208. [DOI] [PubMed] [Google Scholar]
- 24.Hahn J, Park G, Kang HC, Lee JS, Kim HD, Kim SH, et al. Optimized Treatment for Infantile Spasms: Vigabatrin versus Prednisolone versus Combination Therapy J Clin Med. 2019. October 2;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mueller SG, Weber OM, Duc CO, Weber B, Meier D, Russ W, et al. Effects of vigabatrin on brain GABA+/CR signals in patients with epilepsy monitored by 1H-NMR-spectroscopy: responder characteristics Epilepsia. 2001. January;42:29–40. [DOI] [PubMed] [Google Scholar]
- 26.O’Callaghan FJK, Edwards SW, Alber FD, Cortina Borja M, Hancock E, Johnson AL, et al. Vigabatrin with hormonal treatment versus hormonal treatment alone (ICISS) for infantile spasms: 18-month outcomes of an open-label, randomised controlled trial Lancet Child Adolesc Health. 2018. October;2:715–725. [DOI] [PubMed] [Google Scholar]
- 27.Lovick TA, Zangrossi H Jr. Effect of Estrous Cycle on Behavior of Females in Rodent Tests of Anxiety Front Psychiatry. 2021;12:711065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scharfman HE, Mercurio TC, Goodman JH, Wilson MA, MacLusky NJ. Hippocampal excitability increases during the estrous cycle in the rat: a potential role for brain-derived neurotrophic factor J Neurosci. 2003. December 17;23:11641–11652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kücker S, Töllner K, Piechotta M, Gernert M. Kindling as a model of temporal lobe epilepsy induces bilateral changes in spontaneous striatal activity Neurobiol Dis. 2010. March;37:661–672. [DOI] [PubMed] [Google Scholar]
- 30.Löscher W Preclinical assessment of proconvulsant drug activity and its relevance for predicting adverse events in humans Eur J Pharmacol. 2009. May 21;610:1–11. [DOI] [PubMed] [Google Scholar]
- 31.Pollack GM, Shen DD. A timed intravenous pentylenetetrazol infusion seizure model for quantitating the anticonvulsant effect of valproic acid in the rat J Pharmacol Methods. 1985. April;13:135–146. [DOI] [PubMed] [Google Scholar]
- 32.Mandhane SN, Aavula K, Rajamannar T. Timed pentylenetetrazol infusion test: a comparative analysis with s.c.PTZ and MES models of anticonvulsant screening in mice Seizure. 2007. October;16:636–644. [DOI] [PubMed] [Google Scholar]
- 33.Orloff MJ, Williams HL, Pfeiffer CC. Timed intravenous infusion of metrazol and strychnine for testing anticonvulsant drugs Proc Soc Exp Biol Med. 1949. February;70:254–257. [DOI] [PubMed] [Google Scholar]
- 34.Ramanjaneyulu R, Ticku MK. Interactions of pentamethylenetetrazole and tetrazole analogues with the picrotoxinin site of the benzodiazepine-GABA receptor-ionophore complex Eur J Pharmacol. 1984. March 2;98:337–345. [DOI] [PubMed] [Google Scholar]
- 35.Bankstahl M, Bankstahl JP, Bloms-Funke P, Löscher W. Striking differences in proconvulsant-induced alterations of seizure threshold in two rat models Neurotoxicology. 2012. January;33:127–137. [DOI] [PubMed] [Google Scholar]
- 36.Handreck A, Mall EM, Elger DA, Gey L, Gernert M. Different preparations, doses, and treatment regimens of cyclosporine A cause adverse effects but no robust changes in seizure thresholds in rats Epilepsy Res. 2015. May;112:1–17. [DOI] [PubMed] [Google Scholar]
- 37.Pan Y, Gerasimov MR, Kvist T, Wellendorph P, Madsen KK, Pera E, et al. (1S, 3S)-3-amino-4-difluoromethylenyl-1-cyclopentanoic acid (CPP-115), a potent gamma-aminobutyric acid aminotransferase inactivator for the treatment of cocaine addiction J Med Chem. 2012. January 12;55:357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goddard GV, McIntyre DC, Leech CK. A permanent change in brain function resulting from daily electrical stimulation Exp Neurol. 1969;25:295–330. [DOI] [PubMed] [Google Scholar]
- 39.Gernert M, Fedrowitz M, Wlaz P, Löscher W. Subregional changes in discharge rate, pattern, and drug sensitivity of putative GABAergic nigral neurons in the kindling model of epilepsy Eur J Neurosci. 2004. November;20:2377–2386. [DOI] [PubMed] [Google Scholar]
- 40.Töllner K, Wolf S, Löscher W, Gernert M. The anticonvulsant response to valproate in kindled rats is correlated with its effect on neuronal firing in the substantia nigra pars reticulata: a new mechanism of pharmacoresistance J Neurosci. 2011. November 9;31:16423–16434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weerawarna PM, Moschitto MJ, Silverman RB. Theoretical and Mechanistic Validation of Global Kinetic Parameters of the Inactivation of GABA Aminotransferase by OV329 and CPP-115 ACS Chem Biol. 2021. April 16;16:615–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Racine RJ. Modification of seizure activity by electrical stimulation: II. Motor seizure Electroencephalogr Clin Neurophysiol. 1972;32:281–294. [DOI] [PubMed] [Google Scholar]
- 43.Gulati OD, Stanton HC. Some effects on the central nervous system of gamma-amino-n-butyric acid (GABA) and certain related amino acids administered systemically and intracerebrally to mice J Pharmacol Exp Ther. 1960. June;129:178–185. [PubMed] [Google Scholar]
- 44.Knudsen GM, Schmidt J, Almdal T, Paulson OB, Vilstrup H. Passage of amino acids and glucose across the blood-brain barrier in patients with hepatic encephalopathy Hepatology. 1993. June;17:987–992. [PubMed] [Google Scholar]
- 45.Ben-Menachem E Mechanism of action of vigabatrin: correcting misperceptions Acta Neurol Scand Suppl. 2011:5–15. [DOI] [PubMed] [Google Scholar]
- 46.Löscher W Comparative assay of anticonvulsant and toxic potencies of sixteen GABAmimetic drugs Neuropharmacology. 1982. August;21:803–810. [DOI] [PubMed] [Google Scholar]
- 47.Löscher W A comparative study of the pharmacology of inhibitors of GABA-metabolism Naunyn Schmiedebergs Arch Pharmacol. 1980;315:119–128. [DOI] [PubMed] [Google Scholar]
- 48.Storici P, De Biase D, Bossa F, Bruno S, Mozzarelli A, Peneff C, et al. Structures of gamma-aminobutyric acid (GABA) aminotransferase, a pyridoxal 5’-phosphate, and [2Fe-2S] cluster-containing enzyme, complexed with gamma-ethynyl-GABA and with the antiepilepsy drug vigabatrin J Biol Chem. 2004. January 2;279:363–373. [DOI] [PubMed] [Google Scholar]
- 49.Briggs SW, Mowrey W, Hall CB, Galanopoulou AS. CPP-115, a vigabatrin analogue, decreases spasms in the multiple-hit rat model of infantile spasms Epilepsia. 2014. January;55:94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barker-Haliski M, Steve White H. Validated animal models for antiseizure drug (ASD) discovery: Advantages and potential pitfalls in ASD screening Neuropharmacology. 2020. May 1;167:107750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Albright PS, Burnham WM. Development of a new pharmacological seizure model: effects of anticonvulsants on cortical- and amygdala-kindled seizures in the rat Epilepsia. 1980. December;21:681–689. [DOI] [PubMed] [Google Scholar]
- 52.Kalichman MW, Burnham WM, Livingston KE. Pharmacological investigation of gamma-aminobutyric acid (GABA) and fully-developed generalized seizures in the amygdala-kindled rat Neuropharmacology. 1982. February;21:127–131. [DOI] [PubMed] [Google Scholar]
- 53.Shin C, Rigsbee LC, McNamara JO. Anti-seizure and anti-epileptogenic effect of gamma-vinyl gamma-aminobutyric acid in amygdaloid kindling Brain Res. 1986. November 29;398:370–374. [DOI] [PubMed] [Google Scholar]
- 54.Holland KD, McKeon AC, Canney DJ, Covey DF, Ferrendelli JA. Relative anticonvulsant effects of GABAmimetic and GABA modulatory agents Epilepsia. 1992. Nov-Dec;33:981–986. [DOI] [PubMed] [Google Scholar]
- 55.Sayin U, Purali N, Ozkan T, Altug T, Buyukdevrim S. Vigabatrin has an anxiolytic effect in the elevated plus-maze test of anxiety Pharmacol Biochem Behav. 1992. October;43:529–535. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.