

Abstract
Purpose:
Although remarkably effective in some patients, precision medicine typically induces only transient responses despite initial absence of resistance-conferring mutations. Using BRAF-mutated myeloma as a model for resistance to precision medicine we investigated if BRAF-mutated cancer cells have the ability to ensure their survival by rapidly adapting to BRAF inhibitor treatment.
Experimental Design:
Full-length single cell (sc)RNA-seq was conducted on three patients with BRAF-mutated myeloma and one healthy donor. We sequenced 1495 cells before, after one week and at clinical relapse to BRAF/ MEK inhibitor treatment. We developed an in vitro model of dabrafenib-resistance using genetically homogeneous single-cell clones from two cell lines with established BRAF mutations (U266, DP6). Transcriptional and epigenetic adaptation in resistant cells were defined by RNA-seq and H3K23ac ChIP-seq. Mitochondrial metabolism was characterized by metabolic flux analysis.
Results:
Profiling by scRNA-seq revealed rapid cellular state changes in response to BRAF/MEK inhibition in myeloma patients and cell lines. Transcriptional adaptation preceded detectable outgrowth of genetically discernible drug-resistant clones and was associated with widespread enhancer remodeling. As a dominant vulnerability, dependency on oxidative phosphorylation (OxPhos) was induced. In treated individuals, OxPhos was activated at the time of relapse and showed inverse correlation to MAPK activation. Metabolic flux analysis confirmed OxPhos as a preferential energetic resource of drug-persistent myeloma cells.
Conclusions:
This study demonstrates that cancer cells have the ability to rapidly adapt to precision treatments through transcriptional state changes, epigenetic adaptation and metabolic rewiring, thus facilitating the development of refractory disease while simultaneously exposing novel vulnerabilities.
Keywords: multiple myeloma, BRAF mutations, precision medicine, single-cell sequencing, metabolomics, epigenetics
Introduction
Precision medicine treatments rarely achieve cures in patients with most types of cancers. Patients with malignant melanoma, for instance, may experience an initial reduction in tumor load but experience disease recurrence later on (1,2). In multiple myeloma, a hematologic malignancy with oncogenic BRAF mutations in about ~5–12% of patients, recurrent mutations in the BRAF oncogene have been exploited with great therapeutic success (3,4). In the majority of these patients, however, responses have been transient and competitive outgrowth of clones harboring NRAS or KRAS mutations has been reported in relapsed disease (4,5). Many patients with BRAF-mutated myeloma who are treated with BRAF inhibitors only experience minimal responses or stable disease in the first place, despite initial absence of activating KRAS or NRAS mutations (6–8). Based on these observations we hypothesized that non-genetic mechanisms of resistance to precision medicine may allow myeloma cells to rapidly adapt to therapeutic challenges.
Using BRAF-mutated myeloma as a model disease in this study, we aimed to investigate why “precision treatment” of BRAF-mutated neoplasms with BRAF and/or MEK inhibition often fails to effectively elicit substantial treatment responses. We focused on BRAF-mutated myeloma, as sensitive liquid biopsy technology allows us to follow the cancer cell fate over time by interrogating circulating tumor cells from patients (9).
Cellular plasticity has recently been identified as an alternative mechanism driving non-genetic resistance as cancer cells acquire transcriptional states which no longer depend on the drug target (10). A better understanding of how these non-genetic programs integrate with genetic resistance may help with designing more effective therapies to overcome resistance to targeted therapies. We hypothesized that adaptive resistance mechanisms allow myeloma cells to rapidly alter their state, thus compensating for the loss of oncogenic stimulation. Utilizing single-cell RNA sequencing of myeloma cells we explored how inhibition of the BRAF/MEK pathway produces transcriptional state changes and exposes putative therapeutic vulnerabilities. We defined differential enhancer usage in cells that have the ability to persist in the presence of BRAF inhibitor treatment. We explored how transcriptional state changes mediate metabolic reprogramming and selective activation of genes involved in metabolic regulation. We defined the kinetics of state changes and determined if susceptibility to pharmacologic inhibition of oxidative phosphorylation may represent an opportunity to overcome drug resistance. These insights into the mechanisms that allow malignant plasma cells to escape precision treatments will enable the design of more effective targeted treatments in myeloma and other BRAF-mutated neoplasms.
Materials and Methods
Human subjects
Samples were collected from patients with relapsed/ refractory multiple myeloma (RRMM) harboring activating BRAF mutations (Table 1) who provided informed written consent and were treated according to the IRB-approved DFCI protocol 16–352 in accordance with the Declaration of Helsinki. Patients were treated with the BRAF inhibitor dabrafenib and the MEK inhibitor trametinib. Samples were retrieved at screening, after one week of treatment and at the time of clinical progression. Clinical progression for patients MM1, MM2 and MM3 was measured at cycle 4 day 1 (MM1), week 1 (MM2) and cycle 7 day 1 (MM3). In brief, dabrafenib was administered orally BID and trametinib was administered orally QD on a 28-day cycle until progression, unacceptable toxicity, patient refusal or changes in the patientś condition that prohibited further treatment. Normal donor plasma cells were obtained commercially from Research Blood Components, LLC.
Table 1.
Primary patient characteristics
| MM1 | MM2 | MM3 | |
|---|---|---|---|
| Age (years) | 64 | 60 | 65 |
| Gender | female | female | female |
| Ig subtype | IgD | IgG | IgA |
| Light-chain subtype | lambda | lambda | kappa |
| Prior therapies | 6 | 7 | 4 |
| -lenalidomide | yes | yes | yes |
| -bortezomib | yes | yes | yes |
| -dexamethasone | yes | yes | yes |
| -carfilzomib | yes | yes | yes |
| -pomalidomide | yes | yes | yes |
| -ASCT | no | yes | yes |
| -daratumumab | yes | yes | no |
| -cytoxan | yes | yes | no |
| BM PC infiltration | 80% | 90% | 80% |
| Evidence of EM-MM | no | no | no |
| Cytogenetics | +1q22, +3, +7, +9, −13, +15, XX | −1p, +3, +7, −9, +11, −13, +14, −17, XX | +1, +3, +5, +7, −8, +9, +11, −13, +15,XX |
| Snapshot-NGS | 7.9% NRAS Q61R
7.1% BRAF V600E |
BRAF D594N TP53 R267W (variant allele frequency not reported) |
34.0% NRAS Q61H 20.3% BRAF D594N |
| Best IMWG response on treatment | stable disease (SD) | progressive disease (PD | very good partial response (VGPR) |
Sample preparation and single cell isolation
Plasma cells were separated using the EasySep Human CD138 Positive Selection Kit (StemCell Technologies). Samples were next stained with anti-CD38-FITC (multi-epitope), anti-CD138-PE (clone 44F9), anti-CD319-APC (clone 162.1), anti-CD269-PE/Cyanine7 (clone 19F2) and DAPI according to the manufacturerś recommendations (Table S1). Single cells were sorted into 96-well plates containing TCL buffer (Qiagen) on a Sony SH800 sorter. Data were analyzed using the FlowJo software version 10.0 (Becton Dickinson).
Single-cell RNA-seq
Full-length single-cell RNA-seq libraries were prepared using Smart-seq2 (11). cDNA was fragmented by Nextera XT and amplified with indexed Nextera PCR primers. Products were purified with Agencourt Ampure XP beads (Beckman Coulter) and quantified using a Bioanalyzer High Sensitivity DNA Kit. Pooled sequencing of Nextera libraries was carried out using a NextSeq 500 (Illumina, C>A). All sequenced reads were paired-end and 36-base pair (bp) read length. A sequencing depth of 1 × 10e6 reads was aimed for in each cell.
Tumor cell lines
U266 was obtained from DMSZ and DP6 was provided by Dr. D. Jelinek (Mayo Clinic, MN). DP6 cells were maintained in Advanced RPMI 1640 with 4% FBS (not heat-inactivated), 1x Glutamax and IL-6 supplementation (2ng/ml QOD). Effective inhibition of the MAPK pathway in vitro was established at an IC90 dose of 1μM dabrafenib for U266 and 10nM dabrafenib for DP6. To account for genetic heterogeneity at baseline, single-cell clones were generated by single cell sorting using a Sony SH800 sorter. All single-cell clones were tested and found to be negative for mycoplasma contamination. Authentication was performed using DNA fingerprinting with small tandem repeat (STR) profiling.
Serial profiling in vitro
Single-cell clones were cultured with dabrafenib-containing media replaced twice per week. Aliquots of 5 × 10e5 cells were retrieved at defined time points throughout drug exposure for immunoblotting, low-pass whole genome sequencing (LPWGS) and bulk RNA-seq. For Western Blot analysis, antibodies were used as indicated in Table S2. For bulk RNA-seq, cells were resuspended in trizol and RNA was extracted using the Qiagen Rneasy FFPE kit. Libraries were prepared with the TruSeq RNA Access Library Prep Kit (Illumina). Paired-end sequencing, using 75 bp reads was performed on a NextSeq 500 (Illumina). For LPWGS, genomic DNA was extracted using the DNeasy Blood and Tissue Kit (Qiagen) and genomic DNA was quantified using the Qubit HS DNA kit (Invitrogen). An input of 5–20 ng DNA was used for library construction with custom adapters (IDT and Broad Institute, Cambridge, MA). Libraries were pooled and sequenced to an average sequencing depth of 0.2x.
Cytotoxicity analysis
DMSO-diluted dabrafenib (ThermoFisher), rotenone (Sigma Aldrich) and IACS-010759 (BioVision) were used. Cell viability was estimated using the CellTiter-Glo assay (Promega) on a SpectraMax M5 reader (Molecular Devices). If not stated otherwise, control cells were incubated for the same duration with equal amounts of DMSO and luminescence was normalized to the respective controls.
H3K27ac chromatin immunoprecipitation sequencing (ChIP-seq)
For ChIP-seq, 2 × 10e7 cells were cross-linked using formaldehyde and sonicated on a Covaris E220 focused ultrasonicator (~200–500 bp fragment size). Immunoprecipitation (IP) was performed with anti-H3K27ac antibody (Active Motif) and parts of the lysate were saved as whole cell extract (WCE). Drosophila chromatin and antibody spike-ins were done for normalization. IP pulldown was performed using Protein A/G dynabeads (Invitrogen), followed by washing and elution off the beads. Cross-linking was next reversed by proteinase K. Libraries were constructed with individual barcodes (IP and WCE).
Metabolic flux and functional assays
Cells were seeded overnight in 24-well XF microplates (Seahorse Bioscience) at a density of 15 × 10e4 cells per well (200 μL volume). The following day, media was replaced by XF Cell Mito Stress Test Assay Medium (Seahorse Bioscience) and adjusted to a pH of 7.4. Cells were next incubated for one hour at 37°C without CO2. Injection ports were loaded with oligomycin (1μM), carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP, 1μM+1μM) and rotenone/antimycin A (0.5μM each). Each drug was added sequentially using three basal rate measurements for OCR and ECAR prior to the first injection followed by three measurements after each injection. Intervals between these measurements were 3/ 2/ 3 minutes.
Computational Analysis
Processing of single-cell RNA-seq data
Processing and quality filtering of scRNA-seq data was carried out using trimmomatic, STAR, HTSeq and RSEM. Four different parameters were used to filter out low quality cells: i) library size, ii) number of genes detected, iii) percentage of reads mapping to mitochondrial genes, and iv) percentage of reads mapping to house-keeping genes (Figure S1–S2). Cells that fell below three median absolute deviations were excluded from downstream analyses. As an additional approach to remove bad-quality cells without any predefined cut-offs we used the R package mvoutlier. This resulted in the retention of 1153 cells with an average of 4836 genes detected per cell.
Clustering of single cells and identification of cell types
Clustering and marker gene analyses were performed using PAGODA2. In order to rule out the possibility of clusters being purely driven by cell cycle, each individual cell was additionally analyzed for expression of G1, G2M and S phase markers using SEURAT2. The marker genes for each cluster were determined using the findMarker function in the scran package. Copy number analysis was carried out with InferCNV (https://github.com/broadinstitute/inferCNV). Untreated normal donor plasma cells served as control to identify malignant plasma cells based on copy number alterations and transcriptional profile and were used for quality control in our analytical pipeline.
Processing of bulk RNA-seq data
Output for bulk RNA-seq was generated in FASTQ format (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) mapped to the human genome GRCh37 (hg19). Gene expression at transcript-level resolution was calculated using RSEM (v1.2.31) and Picard (https://broadinstitute.github.io/picard/). Annotations were derived from the Ensembl database (October 2018).
Differential gene expression in bulk RNA-seq data
Differential gene expression analysis and enrichment were performed using DESeq2 (v1.22.2) and GSEA (v4.0.3). Detection of copy number alterations was carried out using HMMcopy (v1.28.0).
Annotation of peaks in H3K27ac ChIP data
H3K27ac ChIP data was analyzed using the nf-core/chipseq pipeline as previously described (12). In brief, quality filtering was done by FastQC and TrimGalore. Reads were aligned to UCSC_hg19 using BWA and SAMtools. IP efficiency was calculated by normalizing peak counts to their WCE using deepTools. Narrow consensus peaks were called using MACS, HOMER, BEDTools and featureCounts. Enhancer characterization was done by ChIPseeker (v1.20.0) (13). Gene-enhancer relationships were annotated with GREAT. Gene set enrichment analysis was performed using GSEA (v4.0.3) and was visualized in IGV (v2.5.0).
Data sharing statement
Sequencing data have been deposited at GEO database (reference number GSE168951). Codes used for all analyses will be made available upon request.
Results
Transcriptional adaptation in myeloma with effective BRAF/MEK inhibition
We hypothesized that cancer cells are capable of rapidly adjusting to therapeutic pressure when challenged with targeted inhibitors. We previously described a methodology to isolate myeloma cells from blood or bone marrow at multiple time points during the course of the disease with high sensitivity (9). We used this approach to define transcriptional state changes in BRAF-mutated myeloma patients treated with the BRAF inhibitor dabrafenib and the MEK inhibitor trametinib. A total of 1495 single CD138+/CD38+ myeloma and normal plasma cells from three patients and one normal donor were examined by full-length single-cell RNA sequencing (Table 1). All three myeloma patients had received at least two prior therapeutic regimens. One patient harbored a BRAF V600E and two patients had D594N mutations (Table 1). Two of these patients also harbored additional NRAS Q61 mutations, confirmed by next generation sequencing. All patients showed varying degrees of clinical response to dabrafenib / trametinib treatment (Table 1). We isolated myeloma cells from the bone marrow or peripheral blood of these patients before and at various time points after the treatment was started (Figure S3). After employing quality control filtering steps, 1153 single cells remained for downstream analyses. We performed PAGODA2 and t-Stochastic Neighbor Embedding (t-SNE) visualization at screening, for the earliest available time point on treatment (one week) and the time of clinical relapse. Our analysis revealed eight distinct clusters (Figure 1A). One cluster comprised plasma cells from the normal donor, whereas the remaining clusters reflected the heterogeneity of myeloma and segregated according to the individual patients MM1, MM2 and MM3 (Figure 1A, left panel). Single myeloma cells that were obtained from patients at the time of clinical relapse clustered distinctly from myeloma cells that were obtained prior to initiation of treatment. Notably, transcriptional state changes were already evident as early as 7 days after patients had been started on treatment with dabrafenib/ trametinib (Figure 1A, right panel).
Figure 1. Effective BRAF/MEK inhibition in single myeloma cells is associated with transcriptional adaptation and novel putative therapeutic vulnerabilities.
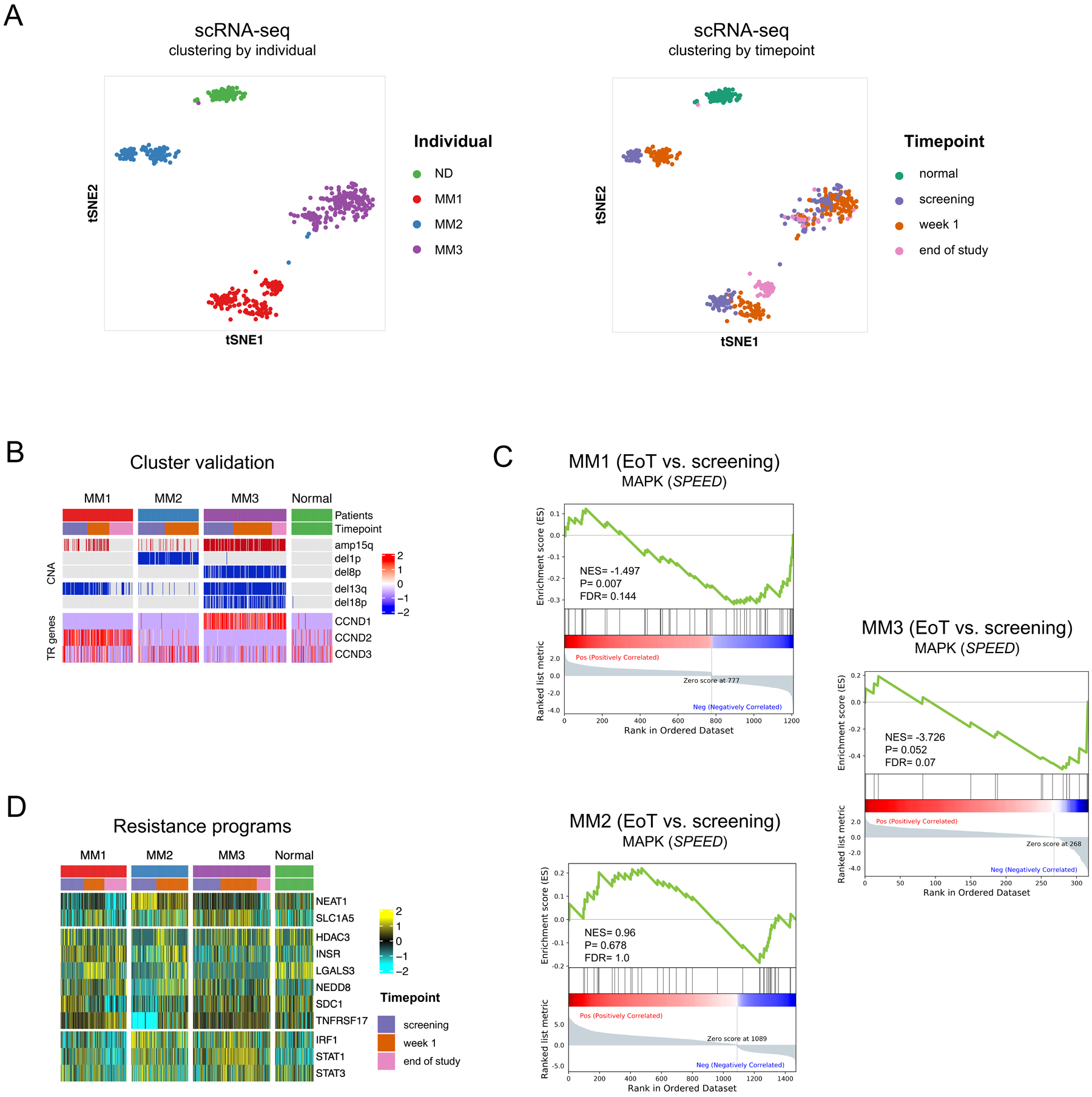
A. t-stochastic network embedding (t-SNE) plot of single cells colored by individual (left) and treatment time point (right) demonstrating distinct clustering of myeloma cells from different patients (MM1,2,3) and from normal donor plasma cells (ND) and distinct clustering by time point after treatment with dabrafenib/ trametinib. B. Malignant clusters were identified by determining pathogenic copy number alterations (CNA) in single cells and by determining overexpression of translocation-driven marker genes (TR genes). C. GSEA enrichment plots showing various degrees of signaling pathway deactivation of the SPEED database perturbation gene set for MAPK (https://speed.sys-bio.net) in single cells at end of treatment versus screening. Of note, treatment was stopped in MM2 after one week of treatment and the patient deceased shortly afterwards. D. Heatmap showing expression of genes representing putative therapeutic targets before and after treatment.
We postulated that the rapid transcriptional adaptation that we observed allows myeloma cells to compensate for the loss of oncogenic stimulation and to persist in the presence of BRAF/MEK inhibitor treatment. First, we ensured correct classification of myeloma cells and normal non-malignant hematopoietic cells, by determining large-segment copy number alterations (CNA) and expression of translocation target genes in all single cells (Figure 1B). Pathogenic CNA in single cells were consistent with aberrations as detected by clinical routine cytogenetics, including del13q in MM1 and MM3, del1p in MM2 and del8p in MM3 (Table 1). As expected, differential gene expression analysis of MAPK pathway genes showed various degrees of signaling pathway deactivation in MM1, MM2 and MM3 at the time of relapse to BRAF/MEK inhibitor treatment (Figure 1C).
While MAPK-related genes were down-regulated with varying kinetics in the individual patients, heterogeneous programs that have previously been linked to myeloma drug resistance were up-regulated at the same time (Figure 1D). In MM1, persisting cells showed an transient increase in cellular stress response (NEAT1) (14,15) and glutamine dependency (SLC1A5) (14,15), which was no longer detectable at the time of clinical relapse. At relapse, this patient showed significant downregulation of BCL2L11 coding for the tumor suppressor BIM (Figure S4), a BCL-2 family member which has previously been defined as a central mediator for BRAF and MEK inhibitor efficacy in myeloma (16). Persistent cells in MM2 showed up-regulation of genes involved in epigenetic modulation (HDAC3), tyrosine kinase signaling (INSR), regulation of cullin-ring ligases (NEDD8) and triggering of apoptosis (LGALS3) (17–20). In addition, persistent cells in MM2 showed increased expression of potential immunotherapeutic targets such as SDC1 and TNFRSF17 coding for the myeloma surface markers CD138 and BCMA, respectively (21,22).
The transcriptional state changes in persistent myeloma cells of MM3 differed markedly from MM1 and MM2 and included upregulation of the transcription factors IRF1, STAT1 and STAT3 (23–25). These data demonstrate that myeloma cells show rapid adaptation to therapeutic challenges in vivo with detectable transcriptional state changes as early as one week after BRAF/MEK inhibition, suggesting that these state changes may occur before genomic evolution. These observations also raised the question if transcriptional state changes generate potential therapeutically exploitable vulnerabilities.
Kinetics of transcriptional state changes and clonal selection in BRAF inhibitor-resistant myeloma in vitro
We next asked how transcriptional state changes relate to clonal selection as BRAF inhibitor resistance develops in BRAF-mutated myeloma. Since the characterization of clonal evolution in patients is confounded by extensive spatial heterogeneity and sampling bias, we established an in vitro model of BRAF inhibitor resistance. To this end we generated three single-cell clones from U266, a BRAFK601N-mutated myeloma cell line to optimize genetic homogeneity in the input population of myeloma cells (Figure 2A). This approach provided a genetically homogeneous input population and allowed for testing of multiple replicates, given that currently only the two myeloma cell lines U266 and DP6 have been described to harbor BRAF mutations (23,26). Effective inhibition of the MAPK pathway was established at an averaged IC90 dose of 1μM dabrafenib (Figure 2B–C).
Figure 2. Distinct phases of resistance with MAPK inhibition in BRAF-mutated myeloma cells.
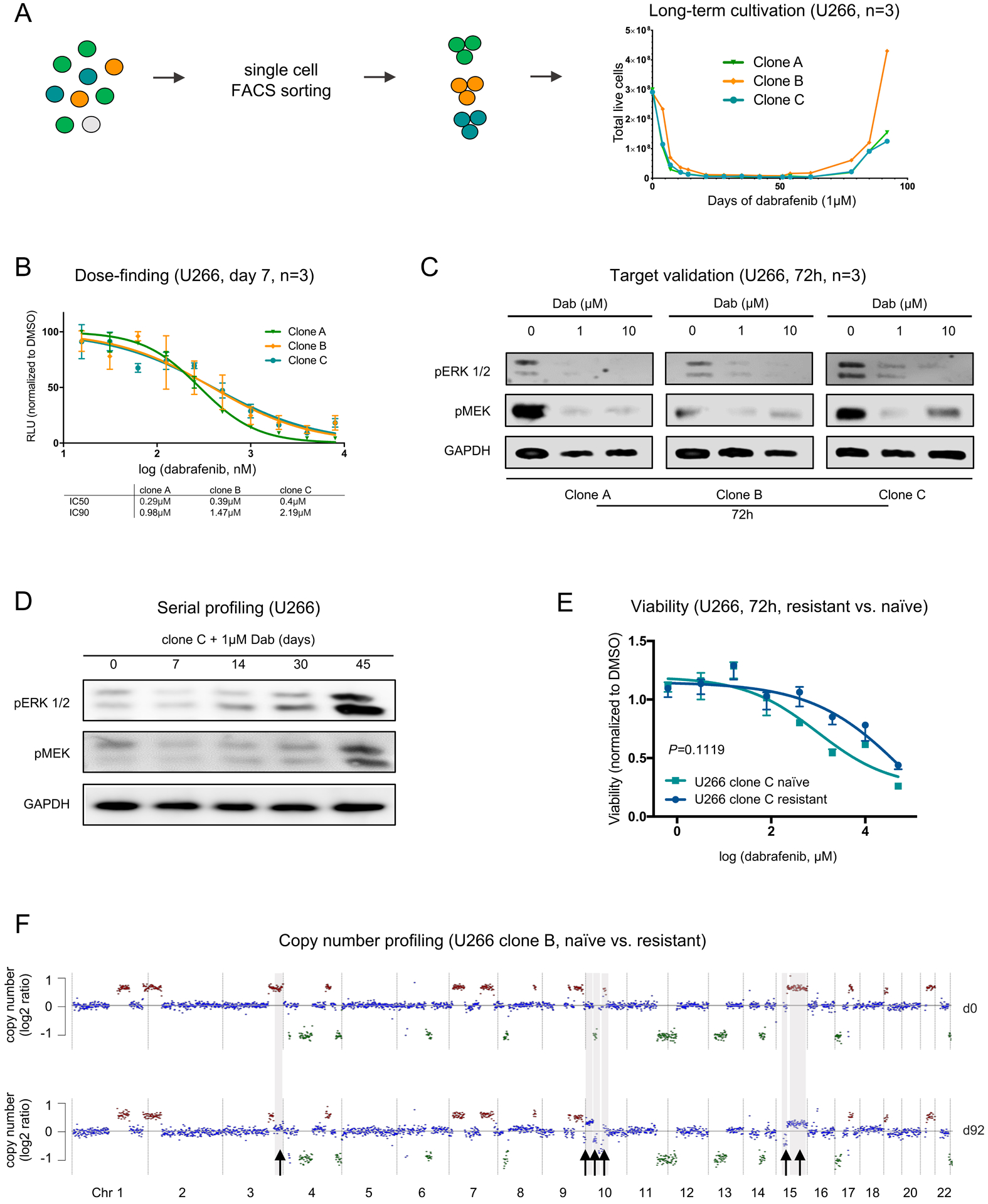
A. Schematic depicting the sorting and long-term cultivation of U266 single-cell clones treated with dabrafenib (1μM). Long-term dabrafenib exposure is characterized by a sharp decline in cell numbers, a plateau phase of persistence and outgrowth of resistant cells after day 60. B. Serial titrations for 7-day cultures to determine the IC50 and IC90 doses of dabrafenib in U266 (BRAFK601N) single-cell clones (RLU: relative light units normalized to DMSO control). C. Effective inhibition of the MAPK pathway members pERK1/2 and pMEK1/2 in U266 myeloma cells after 72h of treatment with dabrafenib at IC90 dose. D. Serial profiling of U266 cells by immunoblotting over time with transient downregulation of pERK1/2 and pMEK1/2 demonstrates initial decline and late recovery of MAPK pathway activity (data shown for clone C). E. Dabrafenib sensitivity in U266 naïve and resistant cells. Viability was measured by CellTiter-Glo assay after 72h (data shown for clone C, n=4). F. LPWGS was performed to determine large segmental copy number alterations (CNA) in U266 clone B. Arrows indicate amplifications (+1) or deletions (-1) that were newly acquired on day 92 after treatment compared to baseline (d0).
All three clones were next subjected to long-term dabrafenib treatment. The enumeration of viable cells followed a similar pattern in all U266 clones with a sharp decrease in viability, a plateau phase of persistence beginning at ~day 7 and outgrowth of resistant myeloma cells at ~day 60 of treatment (Figure 2A). Serial profiling by immunoblotting over time demonstrated late recovery of MAPK pathway activity (Figure 2D, Figure S5). As expected, dabrafenib sensitivity was lower in resistant clones as compared to their respective untreated controls after 72 hours of treatment (Figure 2E, Figure S6). In all clones, resistance was associated with the occurrence of heterogeneous CNA at these late time points as detectable by low-pass whole genome sequencing (LPWGS, Figure 2F, Figure S7), indicating evolving competitive outgrowth of genetically distinct myeloma cells at subclonal level. Notably, although all three replicate U266 clones had identical copy number profiles initially, the clones that were competitively selected after two months as BRAF inhibitor resistance had developed all had distinct CNA. These data demonstrate that the late phase of dabrafenib resistance is characterized by outgrowth of multiple drug-resistant clones, all of which are genetically distinct.
To determine if similar genetic heterogeneity was already present during the early phase of drug resistance to BRAF inhibition, we performed LPWGS of BRAF-mutated U266 myeloma cells after 7 days of BRAF inhibition. In contrast to the late phase of drug resistance we found identical CNA for all three U266 single-cell clones at baseline and persistent stage, indicating that this early persistence is not driven by clonal selection but by transcriptional plasticity of myeloma cells (Figure 3A, Figure S8).
Figure 3. Transcriptional reprogramming in persistent cells during early stages of BRAF inhibitor resistance precedes genomic evolution.
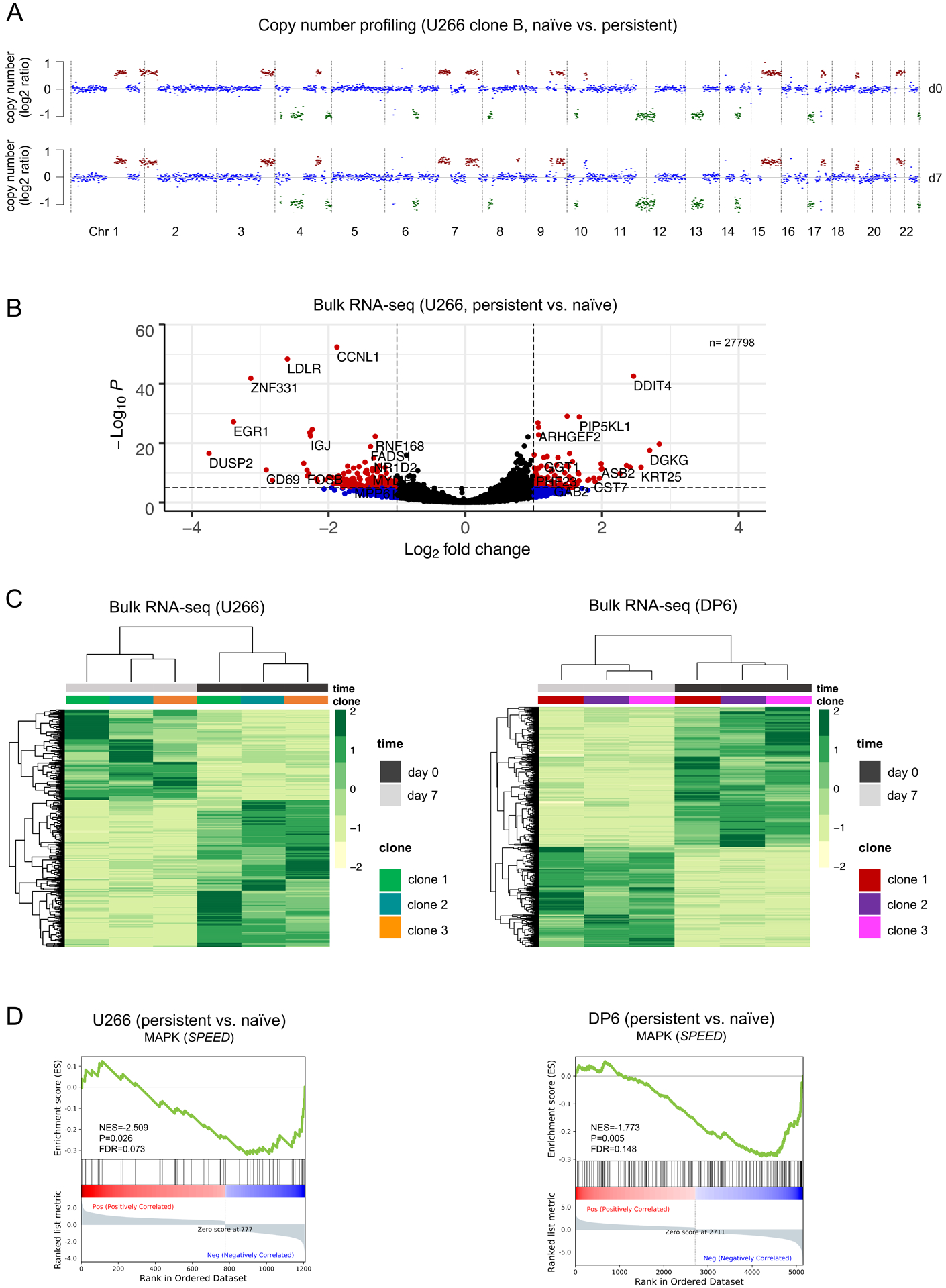
A. LPWGS was performed and copy number alterations were determined in persistent cells on day 7 of dabrafenib (d7) versus untreated BRAF-mutated cells from U266 clone B (d0). No copy number differences were detected between untreated (baseline) and persistent cells at this early stage of persistence. B. Volcano plot visualizing significantly upregulated and downregulated genes in dabrafenib-persistent (day 7) versus untreated U266 clones. C. Heatmaps of significant differentially regulated genes (P<0.05) shown as log2 gene expression indicate similar transcriptional changes in all single-cell clone-derived U266 (BRAFK601N) or DP6 (BRAFV600E) myeloma cells after 7 days of dabrafenib as compared to baseline (day 0). D. GSEA enrichment plots showing deactivation of the SPEED database perturbation gene set for MAPK (https://speed.sys-bio.net) in persistent (day 7 of dabrafenib) versus untreated U266 and DP6 cells.
To further characterize early persistent cells, we performed bulk RNA-seq and examined how transcriptional states in U266 and DP6, a second myeloma cell line with known BRAFV600E mutation (27), at baseline diverge from persistent cells after 7 days of dabrafenib exposure, respectively (Figure 3B, Figure S9–10). Interestingly, even though the genomics of resistant U266 clones ultimately differed substantially at the time of competitive outgrowth, the transcriptional changes after 7 days were homogeneous (Figure 3C). This was similar for DP6-derived single-cell clones, although differentially regulated genes were distinct between U266 and DP6. To explore if any differentially regulated transcriptional signatures were shared across both cell lines and all single-cell clones, we performed gene set enrichment analysis. As expected, BRAF inhibition led to downregulation of MAPK pathway perturbation genes in U266 and DP6 (Figure 3D), whereas oxidative phosphorylation (OxPhos) emerged as the most consistently enriched signaling pathway in persistent cells as compared to baseline (P=0.0001, FDR=0.0001, Figure 4A–B, Figure S11–12). These data suggest that BRAF inhibition in BRAF-mutated myeloma cells leads to transcriptional reprogramming with induction of OxPhos-related genes within a brief period of time.
Figure 4. Enhancer rewiring and adaptive induction of oxidative phosphorylation in dabrafenib-resistant persister cells.
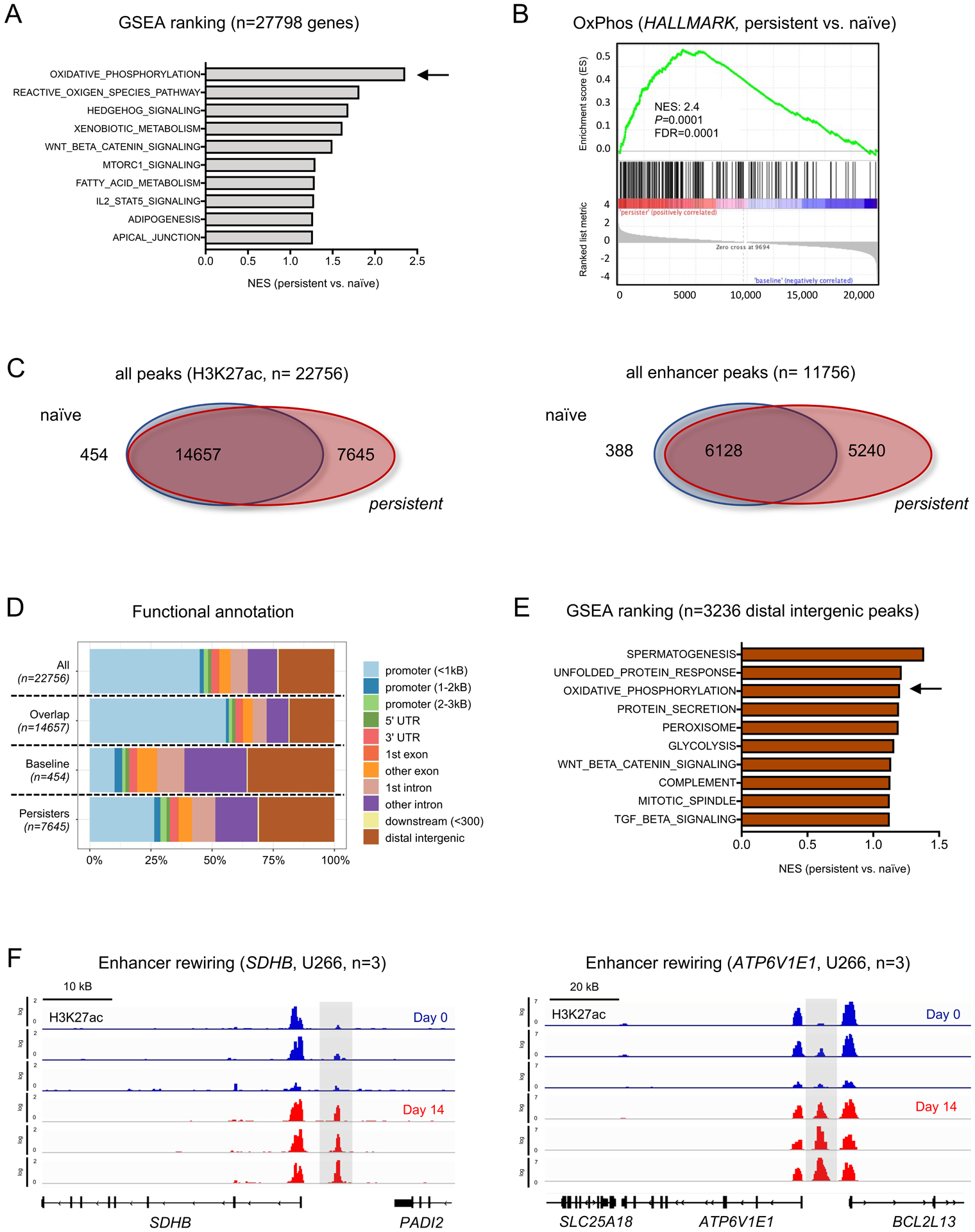
A. Most significantly enriched pathways in persistent (day 7 of dabrafenib) versus untreated U266 cells based on gene expression by GSEA HALLMARK pathway enrichment. Pathways are ranked by their normalized enrichment score (NES). B. GSEA plot showing enrichment of the HALLMARK oxidative phosphorylation gene set in persistent vs. untreated U266. C. Venn diagrams illustrating overlapping and distinct H3K27ac peaks (n= 22756) and distal (excluding promoter) peaks (n=11756) in persistent and untreated U266 cells. D. Peak annotation by genomic regions for peaks that are overlapping or distinct between untreated (baseline) and persistent U266 cells. E. Most significantly enriched pathways in distal intergenic H3K27ac peaks (n=3236) by GSEA for HALLMARK pathways. Listed pathways are ranked by their NES. F. H3K27ac signal intensities shown for two exemplary genes involved in the oxidative phosphorylation pathway (SDHB, ATP6V1E1). Grey boxes mark upregulated H3K27ac peaks in persistent vs. untreated U266 clones.
Altered transcriptional states are associated with differential enhancer usage in drug-resistant myeloma
We next examined the mechanisms of transcriptional adaptation in persistent cells and asked if transcriptional state changes can be attributed to alterations in enhancer activity. To this end, we performed ChIP-seq for H3K27ac as a key enhancer mark in three U266 single-cell clones at baseline versus persistent stage and asked to what extent H3K27ac peaks were distinct between naïve and persistent cells, and which cellular functions might be affected by differential epigenetic regulation.
We observed substantial remodeling illustrated by a greater number of unique H3K27ac peaks in dabrafenib-persistent cells as compared to dabrafenib-naïve controls. Out of a total of 22756 merged peaks, which were called in at least three samples, 7645 (34%) were unique to persistent cells compared to 454 (2%) in untreated cells, (Figure 4C, left panel), illustrating greater epigenetic activity being associated with transcriptional adaptation and persistence to BRAF inhibition. While the majority of overlapping peaks was found in promoter regions, there was a significant change to distal regulatory elements, which were exclusively detectable in clones at the persistent state (Figure 4C–D). As H3K27 acetylation of histones in these regions is typically associated with enhancer activity, we determined the number of those putative enhancer peaks. Notably, 5240/11756 (45%) of these peaks were specific for persistent cells, with the majority of those peaks mapping to intronic and distal intergenic regions, while only 388/11756 (3%) of the enhancer peaks were specific for cells at baseline (Figure 4C, right panel). We performed a gene set analysis for peak intensities of putative enhancers focusing on intergenic peaks and their corresponding genes as predicted by GREAT (28) (Figure 4E). This analysis demonstrated that the OxPhos pathway signature was enriched in persistent cells as third most enriched from a total of 186 gene sets for intergenic enhancers (n=3236 peaks). For example, H3K27ac signal intensities were increased at the enhancer sites of the OxPhos-related gene SDHB, which has been described to stimulate OxPhos via HIF-1α degradation and stabilization of mitochondrial cytochrome oxidase IV (COX4–1) (29,30), and at enhancers near ATP6V1E1, coding for a V-ATPase V1 subunit which has been shown to regulate senescence by promoting lysosomal acidification, metabolic reprogramming and mitochondrial recovery in yeast (31) (Figure 4F). These data therefore indicate that differential enhancer usage is associated with transcriptional adaptation to selective inhibition of the BRAF oncogenic pathway with induction of genes related to OxPhos.
Transcriptional reprogramming is associated with selective metabolic dependency and susceptibility to pharmacologic inhibition of oxidative phosphorylation
We next asked if the observed transcriptional shifts toward OxPhos translate into functional dependencies. To assess mitochondrial metabolism, we performed metabolic flux analysis in dabrafenib-persistent U266 and DP6 single-cell clones after 14 days of drug exposure. Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured as indicators of OxPhos and glycolysis, respectively. Sequential addition of the ATP synthase inhibitor oligomycin, the mitochondrial uncoupler FCCP and the complex III and complex I inhibitors antimycin A and rotenone enabled measurement of the largest possible capacity for OxPhos under maximal stress conditions (Figure 5A). The baseline ratio of OCR:ECAR indicating a preference for OxPhos over glycolysis was significantly elevated in dabrafenib-persistent cells versus DMSO controls (Figure 5B–C, Figure S13A+C). Correspondingly, the maximal OCR:ECAR ratio under FCCP stimulation was significantly elevated in dabrafenib-persistent cells versus controls (Figure S13B+D), demonstrating that while responding to cellular stress, dabrafenib-persistent myeloma cells rather maintain OxPhos than glycolysis as a preferential resource to recruit energetic metabolites.
Figure 5. Selective metabolic dependencies induce susceptibility to pharmacologic inhibitors of OxPhos in re-programmed myeloma cells.
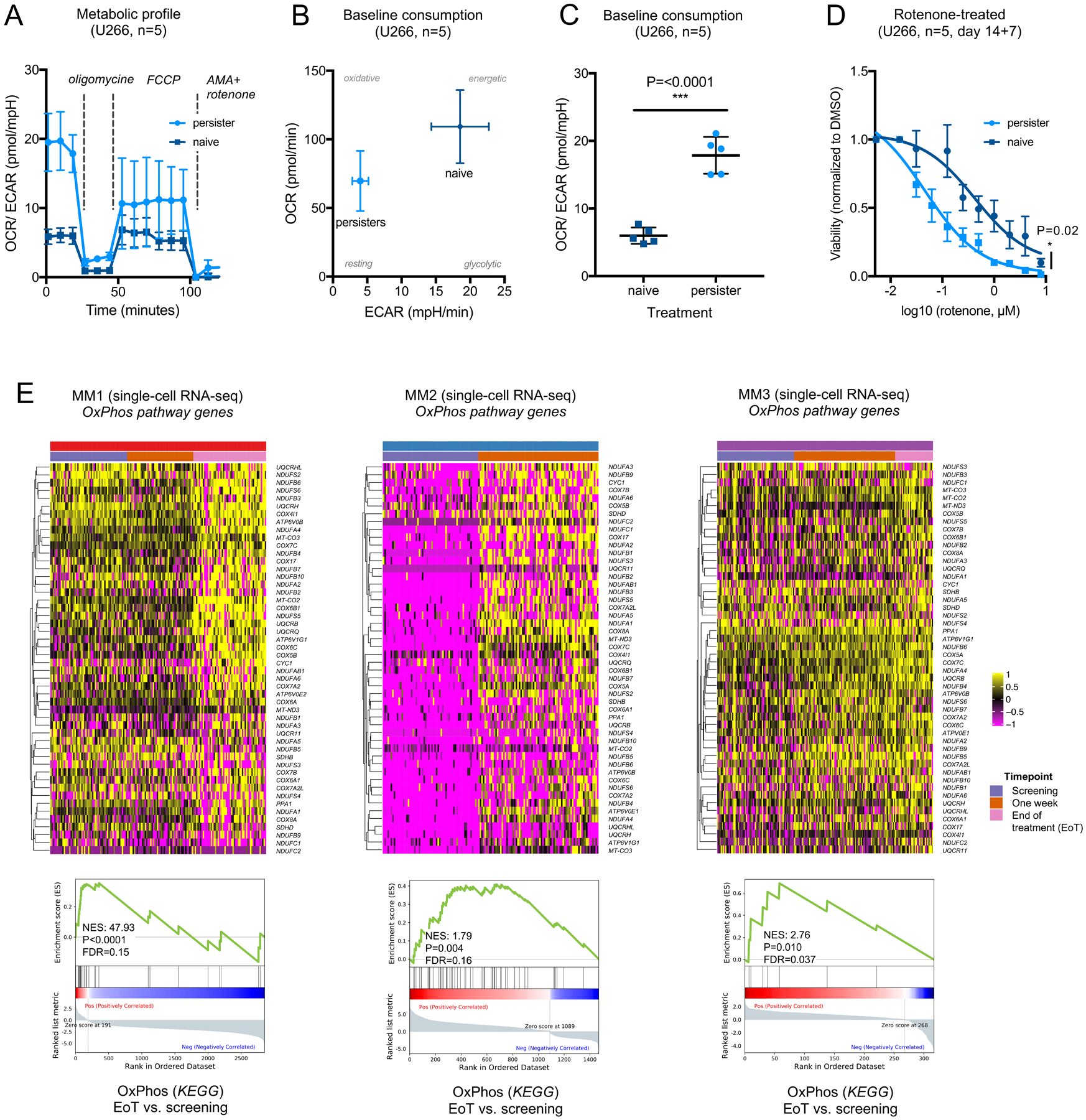
A. Kinetics of metabolic flux in untreated (naiïve) and persistent U266 myeloma cells (single-cell clone C). OCR/ECAR ratio is significantly higher in persistent cells at baseline and after adding FCCP to stimulate maximal mitochondrial capacity. B. Oxygen consumption rate (OCR, pmol/min) over extracellular acidification rate (ECAR, mpH/min) in persistent versus naïve U266 cells shown at baseline for U266 clone C. Disproportionally greater decline of ECAR compared to OCR levels in persistent cells. C. OCR/ECAR ratio in naïve and persistent U266 cells (clone C, baseline consumption, n=5, P<0.0001) D. Viability of U266 cells after treatment with dabrafenib (day 1–14) and subsequent rotenone titration (day 15–21). Pretreatment with dabrafenib significantly increases sensitivity to rotenone in U266 (P=0.02, clone C) as compared to DMSO control. E. In vivo assessment of metabolic reprogramming by examination of OxPhos-related genes in normal and malignant single plasma cells from patients with BRAF-mutated myeloma by single-cell RNA-seq. Heatmaps and GSEA plots show differentially expressed genes derived from the KEGG oxidative phosphorylation gene set (hsa00190) in treated vs. untreated primary cells (P<0.05 in ≥1 patient).
Next, we asked if this functionally validated OxPhos dependency could be therapeutically exploited to target dabrafenib persistence in vitro. To this end we generated dabrafenib-persistent cells by exposing U266 and DP6 single-cell clones to 1μM and 10nm dabrafenib as previously established for both cell lines. At day 14, persistent cells and corresponding DMSO controls were washed, purified by density gradient centrifugation and sequentially treated for seven days with increasing doses of rotenone or IACS-010759, a clinical-grade small-molecule inhibitor of complex I of the mitochondrial electron transport chain. All dabrafenib-persistent U266 and DP6 single-cell clones demonstrated greater sensitivity to rotenone or IACS-010759 than their respective controls (Figure 5D, Figure S14–15)
These data demonstrate that the transcriptional state changes and enhancer rewiring that occur with BRAF inhibitor resistance may create metabolic dependencies that can be targeted subsequently as therapeutic vulnerabilities.
To determine if the induction of OxPhos-related genes can also be found after inhibition of BRAF and MEK in vivo, we defined the transcriptional regulation of OxPhos-related genes in the single-cell dataset from patients with BRAF-mutated myeloma (Figure 1). Equivalent to the transcriptional regulation in myeloma cell lines, OxPhos-related genes were up-regulated in response to inhibition of the BRAF/MEK pathway in myeloma patients (Figure 5E), which was inversely correlated with MAPK-associated transcriptional profiles (Figure 1D). While gene set analysis demonstrated significant activation of OxPhos programs in all patients at single-cell level, the kinetics of this activation differed between individuals (Figure 5E). Moreover, the magnitude of OxPhos induction varied, suggesting that partial OxPhos induction may also be a consequence of resistance to other myeloma treatments, given that the patients received a number of different medications prior to BRAF/MEK inhibition (Table 1). Taken together these data demonstrate that successful inhibition of the BRAF/MEK pathway in myeloma patients is accompanied with a state of metabolic resistance characterized by preferential transcriptional activation of OxPhos.
Discussion
Precision medicine typically refers to targeted pharmacologic inhibition of oncogenes with well-defined mutations. While such approaches have proven to be a successful therapeutic concept in various neoplasms, they are rarely curative (32–34). BRAF mutations occur in 5–12% of myeloma patients and have attracted considerable attention because of their therapeutic potential for targeted inhibition (3,4,35). Although remarkably effective in some patients, BRAF inhibitors typically induce only transient responses despite initial absence of established resistance-conferring mutations (6–8,36). Using BRAF-mutated myeloma as a model for resistance to precision medicine we investigated if BRAF-mutated myeloma cells have the ability to ensure their survival by rapidly adapting to BRAF inhibitor treatment.
Cellular plasticity and dedifferentiation have recently emerged as a principle of non-genetic resistance in cancer, as cells acquire transcriptional states which no longer depend on the drug target (10). Here, we demonstrate that myeloma cells undergo transcriptional state changes when exposed to BRAF-inhibitor treatment in vitro and in patients. While clonal evolution and outgrowth of genetically distinct clones become apparent at late time points, transcriptional adaptation occurs very rapidly, and can be observed within one week of treatment when clonal selection has not yet commenced. Wide-spread enhancer recruitment appears to facilitate dynamic switching between transcriptional cell states, as emergence of transcriptionally altered myeloma cells coincides with significant changes in chromatin regulation. Epigenetic remodeling has previously been shown to be provoked by a diverse array of stimuli including macrophage activation, inflammation, atherogenesis and kinase inhibition (37–39). In this study, we demonstrate that BRAF inhibition is associated with differential enhancer usage and greater transcriptional promiscuity, likely in order to permit wider sampling of the transcriptome. These findings are in line with a previous report which indicates that transcriptional plasticity in myeloma is driven by trans-regulated epigenetic modulation and develops largely independent of predisposing genetics (40). The mechanisms that control permissive chromatin states and transcriptional adaptation remain interesting subjects of future investigations. In this study, pharmacologic inhibition of the oncogenic BRAF/MEK pathway led to induction of alternative survival signals, which may have been conveyed either by other oncogenes, by environmental regulatory pathways through cytokines and surface receptors, or by hardwired survival programs established for normal plasma cells.
Mitochondrial reprogramming has previously been described to promote metabolic recovery and attenuate senescence in fruit flies and yeast (31). OxPhos is normally utilized as the main pathway for energy breakdown in aerobic organisms and has raised interest as an emerging targetable pathway in several types of cancer: studies in melanoma have demonstrated that BRAF-mutant cells, which develop resistance to BRAF inhibitors, display increased activity of oxidative metabolism, increased dependency on mitochondria and higher levels of reactive oxygen species (41–43). OxPhos activation has also been linked to resistance to the Bruton’s tyrosine kinase inhibitor ibrutinib in mantle cell lymphoma (44) and has been observed in therapy-resistant myeloid leukemia stem cells (45,46).
In myeloma cell lines targeting of glycolysis and OxPhos by repurposing the FDA-approved drugs ritonavir and metformin has cytotoxic activity (47), and OxPhos overexpression has previously been associated with resistance to the proteasome inhibitor bortezomib (48). Pharmacologic inhibition of the transcriptional coactivator PGC-1α in myeloma cell lines has been reported to result in reduced proliferation, G2/M arrest and impaired OxPhos (49). Taken together, these data indicate that the OxPhos pathway may represent a putative target in myeloma patients but its regulation and the patient population that might benefit from OxPhos inhibition have been unclear thus far.
Our data provide understanding why OxPhos inhibitors may not be effective as single agents but require combinatory drugs to induce metabolic vulnerability. By performing metabolic flux analysis we demonstrate that functional dependency on mitochondrial ATP generation arises as myeloma cells become resistant to the BRAF inhibitor dabrafenib. Although OxPhos may be difficult to be targeted therapeutically, agents that exploit metabolic dependencies in cancer have been introduced. IACS-010759, a mitochondrial ETC complex I inhibitor, showed promising in vitro efficacy in this study and is currently being investigated as single agent in phase I clinical trials for advanced tumors, including triple-negative breast cancer, pancreatic adenocarcinoma, recurrent lymphoma and acute myeloid leukemia (NCT03291938, NCT02882321).
Upfront combination treatments rather than sequential administration may be a more effective path toward early killing of myeloma cells, which would also slow down clonal evolution, simply by reducing the number of remaining cells that can continue to divide and acquire new genomic aberrations. More studies will be needed to determine the best possible sequence or combinations of individual lines of therapy, including novel immunotherapeutic approaches and stem cell transplantation, which almost all myeloma patients require. Interestingly, in one patient (MM2) BRAF treatment induced up-regulation of TNFRSF17 (BCMA), which is a clinically actionable immunotherapeutic target, suggesting that precision-medicine treatment may unexpectedly sustain immunotherapeutic target expression in some patients. More studies are needed to identify which patients under which circumstances might benefit from this approach.
Although we observed that OxPhos activation is shared as a metabolic resistance program across myeloma patients and cell lines, the majority of activated pathways appeared to be proprietary to the individual patient or cell line. Indeed, our findings indicate that dependency on the BRAF/MEK pathway may be greater in some patients than in others. A deeper understanding on the relative contribution of different oncogenic pathways that drive myeloma growth in individual patients would aid to identify those pathways that should be prioritized for maximum therapeutic benefit.
Overcoming the apparent heterogeneity of alternate cell states and obvious diversity of regulatory programs between patients represents a therapeutic challenge and would argue for repeated molecular characterization of individual patients to identify resistance pathways, for example by liquid biopsy (9,50). Epigenetic drugs could potentially be used to inhibit epigenetic remodeling and subsequent metabolic reprogramming, especially since clinical efficacy of HDAC inhibitors has already been established in myeloma with panobinostat being FDA-approved for the treatment of double-refractory disease.
In summary, we demonstrate that differential enhancer usage is associated with transcriptional plasticity in BRAF-mutated myeloma cells that are exposed to BRAF/MEK inhibition. This transcriptional adaptation is rapid, precedes detectable outgrowth of genetically discernible resistance variants, and promotes metabolic reprogramming before genetic alterations provide additional survival advantages. While the majority of transcriptional state changes that occur with BRAF/MEK inhibition appear to be patient-specific, preferential utilization of oxidative phosphorylation appears to represent an inducible vulnerability across drug-resistant myeloma patients and cell lines. We propose that our findings inform successful implementation of precision medicine in BRAF-mutated neoplasms beyond myeloma. Similar mechanisms may also extend to patients with malignancies that harbor other targetable oncogenes and to a broader population of myeloma patients irrespective of their BRAF/MEK mutation status.
Therapeutic efficacy will require rational combination therapy as well as frequent molecular characterization, for example by liquid biopsy, to accurately identify resistance pathways and therapeutic escape (9,50). Targeting the interplay of epigenetic and metabolic adaptation as a basis of non-genetic plasticity may help to overcome refractory disease in myeloma and other BRAF-mutated cancers (6,51–54).
Supplementary Material
Statement of translational relevance.
Clinical responses to precision treatments with small molecules that target mutated oncogenes are short-lived in almost all types of cancer. Using BRAF-mutated multiple myeloma as a model disease, we examined how cellular state changes allow cancer cells to survive BRAF/MEK inhibition and how the kinetics of cellular adaptation relate to genomic evolution. We demonstrate that BRAF inhibition results in differential enhancer usage and occurs as early as one week after BRAF inhibition, whereas outgrowth of distinct clones with newly acquired copy number alterations is detected late. This epigenetic adaptation promotes transcriptional activation of oxidative phosphorylation pathway genes and induces a metabolic dependency to selective inhibition of oxidative phosphorylation. Our study characterizes the interplay of epigenetic and metabolic adaptation as a basis of non-genetic plasticity that precedes genomic evolution. Targeting cellular adaptation may help to avert refractory disease in BRAF-mutated cancers.
Acknowledgments
J.M.W. is supported by a postdoctoral fellowship of Deutsche Forschungsgemeinschaft (German Research Foundation, 391926441) and the International Myeloma Society Young Investigator Award (IMW, Boston, 2019). J.G.L. is supported by the NCI (K08CA191026), the V Foundation for Cancer Research and the Anna Fuller Fund. B.K. is supported by the NCI (K08CA191091).
We are grateful to D. Jelinek for generously providing the DP6 multiple myeloma cell line. I. C. Newbert and M. Young provided clinical information for correlative studies. J. Eismann and O. Sonzogni provided expertise for metabolic assays. W. Oldham and F. P. Bowman provided experimental expertise and kindly assisted with all Seahorse experiments. M. Wal, H. Yun, A. J. Rogers and J. Poller discussed data and provided methodological resources throughout the study.
Financial support:
No disclosures related to this publication
COI disclosure statement:
J.M.W.: Advisory boards of Janssen and Sanofi. J.G.L.: Research funding from Bristol Myers Squibb. N.S.R.: Consultant to Amgen, Bristol-Myers Squibb, Janssen, Sanofi, Takeda, AstraZeneca and C4 Therapeutics. Advisory boards of Caribou and Immuneel. Research funding from BluebirdBio. K.C.A.: Consultant to Bristol-Myers Squibb, Millennium, Janssen, Sanofi, Amgen, Gilead, Precision Biosciences. Scientific founder of Oncopep and C4 Therapeutics. P.G.R.: Research grants from Bristol-Myers Squibb, Oncopeptides, Celgene, Takeda, and Karyopharm. Advisory boards of Oncopeptides, Janssen, Sanofi, and Secura Bio. A.J.Y.: Consultant to Amgen, Adaptive Biotechnologies, Bristol-Myers Squibb, GSK, Jansen, Karyopharm, Oncopeptides, Sanofi, Takeda. All other authors declare no conflicts of interest.
References
- 1.Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol 2011;29:3085–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andrulis M, Lehners N, Capper D, Penzel R, Heining C, Huellein J, et al. Targeting the BRAF V600E mutation in multiple myeloma. Cancer Discov. 2013;3:862–9. [DOI] [PubMed] [Google Scholar]
- 4.Raab MS, Lehners N, Xu J, Ho AD, Schirmacher P, Goldschmidt H, et al. Spatially divergent clonal evolution in multiple myeloma: overcoming resistance to BRAF inhibition. Blood. 2016;127:2155–7. [DOI] [PubMed] [Google Scholar]
- 5.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay J-Y, et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N Engl J Med. 2015;373:726–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Subbiah V, Puzanov I, Blay J-Y, Chau I, Lockhart AC, Raje NS, et al. Pan-Cancer Efficacy of Vemurafenib in BRAFV600-Mutant Non-Melanoma Cancers. Cancer Discov. 2020;10:657–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raje N, Chau I, Hyman DM, Ribrag V, Blay J-Y, Tabernero J, et al. Vemurafenib in Patients With Relapsed Refractory Multiple Myeloma Harboring BRAFV600 Mutations: A Cohort of the Histology-Independent VE-BASKET Study. JCO Precis Oncol. Available from: https://ascopubs.org/doi/pdf/10.1200/PO.18.00070 [DOI] [PMC free article] [PubMed]
- 9.Lohr JG, Kim S, Gould J, Knoechel B, Drier Y, Cotton MJ, et al. Genetic interrogation of circulating multiple myeloma cells at single-cell resolution. Sci Transl Med. 2016;8:363ra147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shaffer SM, Dunagin MC, Torborg SR, Torre EA, Emert B, Krepler C, et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature. 2017;546:431–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Picelli S, Faridani OR, Björklund AK, Winberg G, Sagasser S, Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc. 2014;9:171–81. [DOI] [PubMed] [Google Scholar]
- 12.Ewels PA, Peltzer A, Fillinger S, Patel H, Alneberg J, Wilm A, et al. The nf-core framework for community-curated bioinformatics pipelines. Nat Biotechnol. 2020;38:276–8. [DOI] [PubMed] [Google Scholar]
- 13.Yu G, Wang L-G, He Q-Y. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinforma Oxf Engl. 2015;31:2382–3. [DOI] [PubMed] [Google Scholar]
- 14.Taiana E, Ronchetti D, Favasuli V, Todoerti K, Manzoni M, Amodio N, et al. Long non-coding RNA NEAT1 shows high expression unrelated to molecular features and clinical outcome in multiple myeloma. Haematologica. 2019;104:e72–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bolzoni M, Chiu M, Accardi F, Vescovini R, Airoldi I, Storti P, et al. Dependence on glutamine uptake and glutamine addiction characterize myeloma cells: a new attractive target. Blood. 2016;128:667–79. [DOI] [PubMed] [Google Scholar]
- 16.Ramakrishnan VG, Miller KC, Macon EP, Kimlinger TK, Haug J, Kumar S, et al. Histone deacetylase inhibition in combination with MEK or BCL-2 inhibition in multiple myeloma. Haematologica. 2019;104:2061–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mondello P, Tadros S, Teater M, Fontan L, Chang AY, Jain N, et al. Selective Inhibition of HDAC3 Targets Synthetic Vulnerabilities and Activates Immune Surveillance in Lymphoma. Cancer Discov. 2020;10:440–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang J, Zhou Y, Thomas GS, Gu Z, Yang Y, Xu H, et al. NEDD8 Inhibition Overcomes CKS1B-Induced Drug Resistance by Upregulation of p21 in Multiple Myeloma. Clin Cancer Res. 2015;21:5532–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hucthagowder V, Meyer R, Mullins C, Nagarajan R, DiPersio JF, Vij R, et al. Resequencing analysis of the candidate tyrosine kinase and RAS pathway gene families in multiple myeloma. Cancer Genet. 2012;205:474–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Streetly MJ, Maharaj L, Joel S, Schey SA, Gribben JG, Cotter FE. GCS-100, a novel galectin-3 antagonist, modulates MCL-1, NOXA, and cell cycle to induce myeloma cell death. Blood. 2010;115:3939–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berdeja JG, Hernandez-Ilizaliturri F, Chanan-Khan A, Patel M, Kelly KR, Running KL, et al. Phase I Study of Lorvotuzumab Mertansine (LM, IMGN901) in Combination with Lenalidomide (Len) and Dexamethasone (Dex) in Patients with CD56-Positive Relapsed or Relapsed/Refractory Multiple Myeloma (MM). Blood. 2012;120:728–728.22563087 [Google Scholar]
- 22.Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N Engl J Med. 2019;380:1726–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jelinek DF, Aagaard-Tillery KM, Arendt BK, Arora T, Tschumper RC, Westendorf JJ. Differential human multiple myeloma cell line responsiveness to interferon-alpha. Analysis of transcription factor activation and interleukin 6 receptor expression. J Clin Invest. 1997;99:447–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu YX, Shi C-X, Bruins LA, Wang X, Riggs DL, Porter B, et al. Identification of lenalidomide resistance pathways in myeloma and targeted resensitization using cereblon replacement, inhibition of STAT3 or targeting of IRF4. Blood Cancer J. 2019;9:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ujvari D, Nagy N, Madapura HS, Kallas T, Kröhnke MCL, Stenke L, et al. Interferon γ is a strong, STAT1-dependent direct inducer of BCL6 expression in multiple myeloma cells. Biochem Biophys Res Commun. 2018;498:502–8. [DOI] [PubMed] [Google Scholar]
- 26.Lodé L, Moreau P, Ménard A, Godon C, Touzeau C, Amiot M, et al. Lack of BRAF V600E mutation in human myeloma cell lines established from myeloma patients with extramedullary disease. Blood Cancer J. 2013;3:e163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Westendorf JJ, Ahmann GJ, Greipp PR, Witzig TE, Lust JA, Jelinek DF. Establishment and characterization of three myeloma cell lines that demonstrate variable cytokine responses and abilities to produce autocrine interleukin-6. Leukemia. 1996;10:866–76. [PubMed] [Google Scholar]
- 28.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7:77–85. [DOI] [PubMed] [Google Scholar]
- 30.Fukuda R, Zhang H, Kim J, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–22. [DOI] [PubMed] [Google Scholar]
- 31.Hughes AL, Gottschling DE. An early age increase in vacuolar pH limits mitochondrial function and lifespan in yeast. Nature. 2012;492:261–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kantarjian H, Giles F, Wunderle L, Bhalla K, O’Brien S, Wassmann B, et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med. 2006;354:2542–51. [DOI] [PubMed] [Google Scholar]
- 33.Trunzer K, Pavlick AC, Schuchter L, Gonzalez R, McArthur GA, Hutson TE, et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J Clin Oncol. 2013;31:1767–74. [DOI] [PubMed] [Google Scholar]
- 34.Choi YL, Soda M, Yamashita Y, Ueno T, Takashima J, Nakajima T, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363:1734–9. [DOI] [PubMed] [Google Scholar]
- 35.Rustad EH, Dai HY, Hov H, Coward E, Beisvag V, Myklebost O, et al. BRAF V600E mutation in early-stage multiple myeloma: good response to broad acting drugs and no relation to prognosis. Blood Cancer J. 2015;5:e299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Donnell E, Raje NS. Targeting BRAF in multiple myeloma. Cancer Discov. 2013;3:840–2. [DOI] [PubMed] [Google Scholar]
- 37.Zawistowski JS, Bevill SM, Goulet DR, Stuhlmiller TJ, Beltran AS, Olivares-Quintero JF, et al. Enhancer Remodeling during Adaptive Bypass to MEK Inhibition Is Attenuated by Pharmacologic Targeting of the P-TEFb Complex. Cancer Discov. 2017;7:302–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaikkonen MU, Spann NJ, Heinz S, Romanoski CE, Allison KA, Stender JD, et al. Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol Cell. 2013;51:310–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brown JD, Lin CY, Duan Q, Griffin G, Federation A, Paranal RM, et al. NF-κB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol Cell. 2014;56:219–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ledergor G, Weiner A, Zada M, Wang S-Y, Cohen YC, Gatt ME, et al. Single cell dissection of plasma cell heterogeneity in symptomatic and asymptomatic myeloma. Nat Med. 2018;24:1867–76. [DOI] [PubMed] [Google Scholar]
- 41.Vashisht Gopal YN, Gammon S, Prasad R, Knighton B, Pisaneschi F, Roszik J, et al. A Novel Mitochondrial Inhibitor Blocks MAPK Pathway and Overcomes MAPK Inhibitor Resistance in Melanoma. Clin Cancer Res. 2019;25:6429–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vazquez F, Lim J-H, Chim H, Bhalla K, Girnun G, Pierce K, et al. PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell. 2013;23:287–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schöckel L, Glasauer A, Basit F, Bitschar K, Truong H, Erdmann G, et al. Targeting mitochondrial complex I using BAY 87–2243 reduces melanoma tumor growth. Cancer Metab. 2015;3:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang L, Yao Y, Zhang S, Liu Y, Guo H, Ahmed M, et al. Metabolic reprogramming toward oxidative phosphorylation identifies a therapeutic target for mantle cell lymphoma. Sci Transl Med. 2019;11. [DOI] [PubMed] [Google Scholar]
- 45.Kuntz EM, Baquero P, Michie AM, Dunn K, Tardito S, Holyoake TL, et al. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat Med. 2017;23:1234–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013;12:329–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dalva-Aydemir S, Bajpai R, Martinez M, Adekola KUA, Kandela I, Wei C, et al. Targeting the metabolic plasticity of multiple myeloma with FDA-approved ritonavir and metformin. Clin Cancer Res. 2015;21:1161–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Song IS, Kim HK, Lee SR, Jeong SH, Kim N, Ko KS, et al. Mitochondrial modulation decreases the bortezomib-resistance in multiple myeloma cells. Int J Cancer. 2013;133:1357–67. [DOI] [PubMed] [Google Scholar]
- 49.Xiang Y, Fang B, Liu Y, Yan S, Cao D, Mei H, et al. SR18292 exerts potent antitumor effects in multiple myeloma via inhibition of oxidative phosphorylation. Life Sci. 2020;256:117971. [DOI] [PubMed] [Google Scholar]
- 50.Mithraprabhu S, Morley R, Khong T, Kalff A, Bergin K, Hocking J, et al. Monitoring tumour burden and therapeutic response through analysis of circulating tumour DNA and extracellular RNA in multiple myeloma patients. Leukemia. 2019;33:2022–33. [DOI] [PubMed] [Google Scholar]
- 51.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dietrich S, Glimm H, Andrulis M, von Kalle C, Ho AD, Zenz T. BRAF inhibition in refractory hairy-cell leukemia. N Engl J Med. 2012;366:2038–40. [DOI] [PubMed] [Google Scholar]
- 53.Haroche J, Cohen-Aubart F, Emile J-F, Arnaud L, Maksud P, Charlotte F, et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood. 2013;121:1495–500. [DOI] [PubMed] [Google Scholar]
- 54.Hong DS, Morris VK, El Osta B, Sorokin AV, Janku F, Fu S, et al. Phase IB Study of Vemurafenib in Combination with Irinotecan and Cetuximab in Patients with Metastatic Colorectal Cancer with BRAFV600E Mutation. Cancer Discov. 2016;6:1352–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.