

Abstract
Multiple small molecule immune modulators have been identified as synergistic with immune checkpoint blockade (ICB) in their effects on T lymphocytes, but are limited in their successful application to combination cancer immunotherapy due to their short in vivo retention and lack of affinity for T cells. We engineered an antibody-nanoparticle conjugate (ANC) platform consisting of 30 nm polymer nanoparticles that, due to their size and formulation, efficiently distribute after administration to lymph nodes, tissues highly enriched in lymphocytes that contribute to tumor control mediated by ICB. Displaying monoclonal antibodies against surface-expressed T cell markers, NP delivery in vivo to circulating and lymph node-resident lymphocytes was substantially enhanced, as was delivery of small molecules formulated into the NP by passive encapsulation. Using ICB monoclonal antibodies as both targeting moiety and signal-blocking therapeutic, ANCs improved the local and systemic anti-tumor effects of small molecule TGFβ receptor 1 inhibitor and an adenosine 2A antagonist when administered either locoregionally or systemically into the circulation in two syngeneic, aggressive tumor models, slowing tumor growth and prolonging animal survival. As these benefits were lost in the absence of ANC targeting, co-formulation strategies enabling the targeted co-delivery of multiple immunotherapeutics to T lymphocytes have high potential to improve ICB cancer immunotherapy by concurrent inhibition of non-redundant suppressive pathways.
Keywords: Controlled release, Targeted drug delivery, Lymph node, Tumor immunotherapy
1. Introduction
Cancer immunotherapy utilizes the immune system rather than cytotoxic agents, such as chemotherapeutics, to recognize and eradicate cancers.1 One way this is achieved is through activation and invigoration of tumor-specific T cells via immune checkpoint blockade (ICB) using monoclonal antibody (mAb) immunotherapies that include anti-programmed cell death 1 (aPD1) and anti-cytotoxic T lymphocyte associated protein 4 (aCTLA4).2,3 While ICB therapies have generated tremendous improvements in the survival of responsive patients, response rates to ICB therapies remain low and thus combinations with other small molecule immune modulators that block non-redundant suppressive pathways are being actively explored to overcome ICB resistance.4,5 However, the ability to deliver small molecule therapeutics to cells of interest at bioactive doses to completely harness the potential of ICB remains challenging as these drugs distribute in a non-affinity based manner and have short circulation times.6 Furthermore, lymph nodes (LNs) that are tissue niches where a substantial fraction of the total lymphocytes in the body reside and that have been shown to mediate the effects of ICB,7 are difficult to address with small molecules or using conventional drug formulations.8 A crucial hurdle to unleashing the potential of combination immunotherapies for cancer therapy is thus a lack of appropriate drug delivery vehicles.
While many nanoparticle (NP) platforms for cancer therapy have been described, most primarily rely on their effects being mediated through passive NP accumulation within the tumor microenvironment.9 However, tumor accumulation can vary dramatically and primarily relies on the enhanced permeability and retention effect, which has not improved anti-tumor efficacy as initially hoped.10 Furthermore, the development of targeted platforms including ANCs and antibody drug conjugates, so-called ADCs, have mainly focused on tumor targeting strategies to deliver chemotherapeutic agents to directly eliminate targeted cancer cells.11,12 While promising, these require large doses to fully saturate target cells and may require stimuli for payload release, including tumor-associated changes in pH or enzymatic microenvironments.13 As many immune checkpoints, including but not limited to PD1 and CTLA4, are T cell-intrinsic, lymphocytes rather than cancer cells are the target cells of ICB. As such, approaches that target these cells directly after injection into the systemic circulation have demonstrated high promise. For example, anti-Thy1.1 F(ab’)2 fragment-decorated liposomes enable targeting of circulating adoptively transferred T cells14 and CD8-targeting NPs have been shown to label T cells in circulation and tumors after intravenous (i.v.) injection.15 T cells implicated in tumor control reside at high levels within LNs,16 however, and we and others have shown that directed delivery of ICB mAb delivery to LNs potentiates the effects of ICB to afford dose sparing.7,17,18 Drug delivery innovations that potentiate effects on T lymphocytes resident within LNs and like circulating cells can traffic to the tumor to exert their anti-tumor effects16,19 have the potential to realize the benefits of combination immunotherapy.
To this end, we engineered an antibody-nanoparticle conjugate (ANC) platform consisting of NP that, due to their size and composition, both distribute to LNs after administration and are capable of encapsulating small molecule drugs as well as NP surface-conjugated antibodies. We hypothesized that sustained delivery of small molecule immune modulators using ANCs directed to immune checkpoint-expressing lymphocytes would improve anti-tumor responses compared to non-targeted delivery. ANC targeting to immune checkpoints improved both NP retention at the site of injection and accumulation within draining LNs after intratumoral injection, and NP association with and small molecule cargo delivery to circulating and LN-resident lymphocytes after locoregional or systemic administration. ANC targeting also improved NP association with and NP-encapsulated cargo delivery to target lymphocytes, benefits that in the context of ICB augmented the therapeutic effects of ICB with small molecular inhibitors of non-redundant immune pathways, including TGFβ signaling and the adenosine pathway. These so-called drug-eluting ICB-ANCs thus demonstrate the benefits of co-formulation leveraging a unique LN-targeting NP technology for improving the effects of combination therapy of ICB mAb with small molecule immunomodulators.
2. Materials and Methods
2.1. Synthesis of PDS-NPs.
Poly(propylene sulfide) NPs were synthesized according to previous literature. Briefly, 500 mg of carboxyl Pluronic F127 was dissolved in 10 mL of DI water for 30 min under argon, followed by addition of 400 µL of propylene sulfide for 15 min. Initiator20 was activated using sodium methoxide for 15 min after which time it was added to the reaction for another 15 min. 1,8-Diazabicyclo[5.4.0]undec-7-ene was added and the reaction was stirred overnight followed by exposure to oxygen (uncapping) for 2 h to mediate crosslinking of the NP core. NPs were dialyzed against DI water using a 100,000 Da MW cutoff cellulose membrane dialysis tubing (SpectrumLabs). Following dialysis, core thiols were capped using either N-ethylmaleimide or AlexaFluor488 or 647-maleimide, followed by N-ethylmaleimide in 1X PBS overnight. NPs were functionalized with pyridyl disulfide (PDS) on the carboxy Pluronic using EDC/NHS crosslinking for 24 h followed by dialysis against DI water for 3 days, yielding PDS-NPs.
2.2. Antibody thiolation.
mAb were thiolated by reacting with 2-iminothiolane (Traut’s reagent) in 1X PBS at room temperature for 60 min with stirring. Thiolated mAb was subsequently purified using a 7 kDa MW cutoff zeba filter according to manufacturer’s instructions. Alternatively, fluorescently labeled mAbs were generated by reacting with AlexaFluor700 NHS ester (ThermoFisher) for 2.5 h with stirring before reacting with Traut’s reagent as described above. The number of thiols per IgG molecule was determined by Ellman’s assay using a thiomalic acid standard curve. Thiolated mAb was immediately used for NP conjugation and characterization.
2.3. ANC synthesis and purification.
Thiolated mAb was mixed with PDS-NPs and reacted overnight at RT while stirring, resulting in mAb conjugation to the NP surface. Following reaction, ANCs were purified from unreacted mAb by size exclusion chromatography using a Sepharose 4B resin. ANC-containing fractions were identified using fluorescamine and concentrated using a 4 mL 30 kDa MW cutoff spin filter, then stored until use at 4° C. Final mAb concentrations in ANC suspensions were determined using the bicinchoninic acid protein assay kit (ThermoFisher). Following initial synthesis parameter optimization, subsequent ANCs were synthesized using a 40-fold molar excess of Traut’s reagent for antibody thiolation, which resulted in 6–8 thiols per antibody. 0.2–0.6 mg of thiolated antibody were reacted per mg of NP pending the application, which resulted in 4–6 antibodies conjugating to each NP.
2.4. ANC binding to target ligand.
Rhodamine-labeled aCD3 (clone:KT3, BioXcell) was conjugated to Alexa Fluor 647 labeled PDS-NPs. To test ANC T cell binding, murine splenocytes were incubated with saline, control PDS-NPs, free rhodamine-labeled aCD3 mAb, or aCD3 ANCs for 15 min on ice, then stained for surface markers and analyzed by flow cytometry as described below. To test aPD1-ANC binding to EL4 cells, unlabeled aPD1 (clone 29F.1A12, BioXCell) or isotype were conjugated to Alexa Fluor 647-labeled PDS-NPs. EL4 cells were incubated with either isotype ANCs or aPD1 ANCs for 15 min on ice, then stained for surface markers and analyzed by flow cytometry. For aPD1 mAb blocking experiments, EL4 cells were incubated with saline or non-fluorescent aPD1-ANCs for 15 min on ice, then stained with Brilliant Violet 785 aPD1 and other cell surface markers for analysis by flow cytometry.
2.5. Tissue and single cell preparations.
Following tissue collection, tissues were processed by cutting rather than enzymatic digestion to prevent cell surface receptor degradation. Single cell suspensions were generated by disrupting organs through a 70 μm cell strainer followed by washing in PBS. Red blood cells were lysed with lysing buffer hybri-max (Sigma) for 7 min at room temperature followed by quenching in PBS. Cells were plated in 96-well U bottom plates for staining in PBS.
2.6. Flow cytometry and antibodies.
Single cell suspensions from tumors, LNs, and spleens were prepared as described above. After washing, live/dead staining was performed using Zombie Aqua fixable viability kit (Biolegend), followed by wash steps and surface staining using mAb (all from Biolegend): CD45 (clone: 30-F11), CD3 (clone: 17A2), CD4 (GK1.5.), CD8 (clone: 53–6.7), NK1.1 (PK136), B220 (RA3–6B2), CD19 (6D5), IFN-γ (clone: XMG1.2), PD1 (clone: 29F.1A12 or RMP1–14). Surface staining was carried out on ice for 30 min. All flow cytometric analyses were performed using a Fortessa flow cytometer (BD Biosciences) and analyzed using FlowJo software (Tree Star, Inc.).
2.7. Small molecule encapsulation and drug release profiles.
To encapsulate small molecule drugs in the ANC core, lyophilized drug was reconstituted at 10 mg/ml in DMSO. Stock drug was subsequently dispersed into 30 uL ANC suspension or PBS by simple pipette mixing. Upon simple mixing, hydrophobic drugs partitioned into the hydrophobic NP core with >95% encapsulation efficiency without the need for chemical conjugation or other modification of the small molecule drug.21 Drug-loaded ANC suspensions or drug solutions were then loaded into dialysis cups (100 kDa MWCO, ThermoFisher) and dialyzed against PBS with constant stirring. The retentate was periodically sampled over 26–96 h and drug absorbance measured by plate reader (Synergy H4 BioTek). In these and all other experiments using drug-loaded ANCs, drug was loaded immediately before use.
2.8. Animals and cell lines.
Cell lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin/amphotericin B, referred to as complete media, and periodically checked for mycoplasma contamination. C57Bl/6 and BalbC mice were purchased from Jackson Laboratories. All protocols were approved by the Institutional Animal Case and Use Committee. Tumors were implanted intradermally (C57Bl/6) or in the 4th mammary fatpad (BalbC) in 6–12 wk old mice and were monitored in anesthetized mice by caliper measurements of width, length, and depth. Mice were sacrificed when tumors led to ulceration or maximum tumor size of 1.5 cm in any dimension.
2.9. In vitro cytotoxic effects of PXL.
EL4 cells were cultured and plated in 96-well plates at a density of 105 cells per well in 198 µL of complete media. 2 µL of sample [vehicle, paclitaxel (PXL), ANC formulation] was added to each well and allowed to incubate for 4 days. Cells were then stained for flow cytometry with LiveDead and viability assessed by flow cytometric analysis of staining. To investigate ANC benefits on cell targeting, EL4 cells were plated and incubated for 15 min with isotype-ANCs or aPD1-ANCs loaded with 185, 275, or 550 mol PXL per NP. After incubation with cells on ice in 1% BSA in PBS, cells were then pelleted at 300 g, washed twice with complete media, and incubated for 2 days in complete media free of PXL. Control cells were incubated with PXL-loaded aPD1-ANCs without washing, or without NPs/PXL. Cells were then stained with Live/Dead and treatment toxicity evaluated by flow cytometry.
2.10. SCH-58261 effects on CD8+ T cell activation.
CD8+ T cells were isolated from a mouse spleen using a MojoSort CD8+ isolation kit (Biolegend) according to manufacturer instructions. Cells were resuspended at 1 million cells/mL in RPMI medium and incubated for 24 h with aCD3/aCD28 Dynabeads at a 1:1 ratio alongside treatment with either 1 mM adenosine, 0.01 mM adenosine receptor antagonist SCH-58261, or vehicle control. After 24 h cells were stained for cell surface markers and Ki67, and were analyzed by flow cytometry.
2.11. In vivo mAb biodistribution.
Fluorescently (Alexa Fluor 700) labeled aPD1 (clone 29F.1A12), aCTLA4 (clone UC10–4F10–11), or isotype control (rat IgG2a) was conjugated to Alexa Fluor 647-labeled PDS-NPs as described above. Free mAb and ANCs were dose-matched to 10ug of mAb/30uL saline and injected intratumorally into B16F10 tumor-bearing mice (day 5 post-implantation). For retention studies, fluorescent imaging was performed with an IVIS Spectrum instrument (Perkin Elmer) at the injection site over 3 days and of tissues following sacrifice. 24, 48, and 72 h after injection, mice were euthanized and tissues collected for imaging and homogenization. mAb concentration in tissues was determined following homogenization using the injected mAb or ANC solutions to generate a standard curve in a background of naïve tissue.
Fluorescent labeling of isotype or aCD3 (KT3, BioXCell) mAb was done using Alexa Fluor 700 NHS-Ester for 1–2.5 h followed by purification using a Sepharose CL-6B column with fluorescent mAb fractions pooled and concentrated using a 10 kDa (Millipore) spin filter. mAb concentrations were determined using a BCA assay. Fluorescent NPs (Alexa Fluor 647) were synthesized as previously mentioned. For aCD3 labeling of T cells within LNs following i.d. administration, the lateral dorsal skin of naïve C57Bl6 mice was shaved and aCD3 free mAb, control NPs, or aCD3-ANCs (6.25 μg of aCD3) were administered i.d. Mice were euthanized 24 h after injection. LNs were either collagenase treated for 30 min or immediately digested with scissors to prevent ex vivo T cell labeling followed by processing into single cell suspensions and flow cytometry (described above). For biodistribution of Cy5.5-loaded ANC, aCD3- or isotype-ANC synthesis was done as previously described using unlabeled aCD3 (clone: KT3, BioXCell) or isotype (rat anti-mouse IgG2a, clone: 2A3, BioXCell) and fluorescent NPs (Alexa Fluor 488). 10% v/v of 5mg/mL Cyanine5.5 carboxylic acid/DMSO (Cy5.5, Lumiprobe) was added to aCD3-or isotype-ANC stock solutions and mixed by inversion for 5 MIN to allow encapsulation into the NP core. To clean ANCs of free, un-encapsulated Cy5.5, ANC solutions were run through two successive 7kDa zeba spin desalting columns (Thermo Fisher) in saline and diluted to the appropriate concentration (40 or 6.25 μg mAb dose; 6.25 or 1 μg Cy5.5 dose, respectively). 4T1 tumor-bearing mice 6 days post-implant were shaved and injected intradermally into the flank ipsilateral to the tumor with the above formulations, and mice were euthanized 24 h post-injection. Alternatively, 4T1 tumor-bearing mice 6 days post-implant were injected i.v. with aCD3- or isotype-ANCs (40 μg mAb; AlexaFluor647-labeled NP) and euthanized 1 h post i.v. administration. Immediately prior to sacrifice, blood was collected from mice and LNs, spleens, and tumors were immediately digested with scissors to prevent ex vivo T cell labeling followed by processing into single cell suspensions as previously described.
2.12. B16F10 melanoma bearing mice treatments.
The dorsal skin of C57Bl6 mice was shaved and B16F10 cells (105) implanted in the right lateral dorsal skin on day 0. In aPD1 monotherapy experiments, animals were treated on days 5, 7, and 9 with either saline i.t., 150 μg aPD1 mAb as monotherapy or 150 μg each of aPD1+aCTLA4 combination therapy mAb i.t., or 150 μg aPD1 or 150 μg each of aPD1+aCTLA4 as aPD1- or combination ICB-ANCs (respectively) i.t.. In this and all other ANC experiments, mAb delivery was dose-matched between free and ANC-conjugated treatment groups. In SB-431542 treatment experiments, after 5 (when all tumors were visible), 7, 9, 11, and 13 days, mice were injected i.d. with 30 μl containing either control treatment (vehicle); a mixture of unloaded, targeted ANCs [6 µg each of anti-mouse CTLA4- (clone:UC10–4F10–11, BioXCell) and rat anti-mouse PD1-ANCs (clone: 29F.1A12, BioXCell)] and SB431542-loaded isotype ANCs [Armenian hamster IgG- (BioXCell) and rat isotype control-ANCs (clone: 2A3, BioXCell)]; or SB431542-loaded targeted ANCs [6 µg of CTLA4-ANCs and PD1-ANCs]. 5 µg of SB-431542 (Sigma) was encapsulated in drug-loaded ANCs immediately prior to injection. Mice were monitored every other day for survival studies. For immunofluorescence studies, mice were euthanized at endpoint and tumors were harvested.
2.13. Investigation of adenosine and PD1 expression in 4T1 model.
3.5 × 105 4T1 cells re-suspended in 30 uL of PBS were implanted i.d. in the left mammary fatpad (4th) in BalbC mice. On day 15, mice were euthanized for serum collection or LNs were collected and mechanically disrupted in a 96 well round bottom plate for ELISA assays. Following disruption, debris was pelleted and supernatants collected and adenosine levels measured by ELISA (Abcam) according to the manufacturer’s protocol. To determine PD1 expression, on day 15 post tumor implantation, tissues were collected and processed for flow cytometry.
2.14. 4T1 breast tumor bearing mouse treatments.
3.5 × 105 4T1 cells re-suspended in 30 uL of PBS were implanted i.d. in the left mammary fatpad (4th) in BalbC mice. Mice were treated with either vehicle, SCH58261-loaded isotype-ANCs + free aPD1 mAb, or SCH58261-loaded aPD1-ANCs; treatments were given on days 6, 9, 12, and 15, and were administered i.p. or i.d. in the ipsilateral flank in separate experiments. All ANCs contained 2 μg of SCH58261, and all aPD1 treatments contained 60 μg total of aPD1. Tumors were monitored every 2–3 days until endpoint for survival analysis. For analysis of metastatic disease, mice were sacrificed on day 35 and the spleen and LNs collected. TdLNs and NdLNs were harvested and prepared for flow cytometry analysis as described above and the spleen areas were analyzed using ImageJ.
2.15. Statistical analysis.
Statistical significance of differences between experimental groups was calculated with Prism software (GraphPad). All data is expressed as mean ± standard deviation, except for tumor growth which is presented as mean ± standard error of the mean. ****p<0.0001, ***p<0.001, **p<0.01, and *p<0.05 by unpaired two-tailed t-tests or one- or two-way ANOVA followed by Tukey post-hoc test for multiple comparisons. For survival curves, log-rank (Mantel-Cox) test was performed.
3. Results
3.1. ANCs retain mAb binding and immunotherapeutic effects
As mAbs rely on their tertiary structure for their epitope recognition functions, we utilized a previously described NP platform functionalization and bioconjugation scheme based on reacting thiol-containing species with pyridyl disulfide (PDS) groups on the corona of poly(propylene sulfide) NP20 (referred to as PDS-NPs) synthesized by emulsion polymerization to form ANCs with surface-conjugated antibodies (Fig. 1A). PDS-NPs are advantageous for this application, as they can be synthesized to ~30 nm sizes ideal for accumulation in LNs after administration8, and their amphiphilic structure provides both a semi-solid22 hydrophobic core for small molecule encapsulation and a modifiable corona with surface reactive groups for surface conjugation.22,23 PDS-NP additionally allow surface conjugation using gentle, aqueous solvents and rapid reaction times to minimize loss of mAb functions. Thiols were amended onto commercially purchased mAbs by treatment with 2-iminothiolane (Traut’s reagent). The resulting extent of thiolation was proportional to Traut’s molar excess used (Fig. 1B). After removal of excess Traut’s reagent via spin desalting columns, thiolated mAb were incubated overnight with PDS-NPs. Through a disulfide displacement reaction monitored by absorbance at 340 nm of the displaced pyridyl group, the generation of ANCs could be monitored, which occurred in a manner proportional to both the mAb thiolation (Fig. 1C) and ratio of thiolated mAb to PDS-NP (Fig. 1D). While other conjugation schema, like 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide or maleimide coupling, may also be feasible, conjugation to PDS-NP using antibodies thiolated by Traut’s treatment is a simple process that can be done in aqueous solvents and at neutral pH, a significant benefit for maintaining antibody activity. ANCs were purified from unreacted mAb by size exclusion chromatography (Fig. 1E–F) and ANC-containing fractions were collected and concentrated, prior to further investigations. As expected, ANCs formed only after reaction of PDS-NP with thiolated mAb (Fig. 1F). Resulting ANCs were ~30 nm in diameter as measured by dynamic light scattering (Fig. 1G).
Figure 1: Method of antibody-nanoparticle conjugate (ANC) formation.
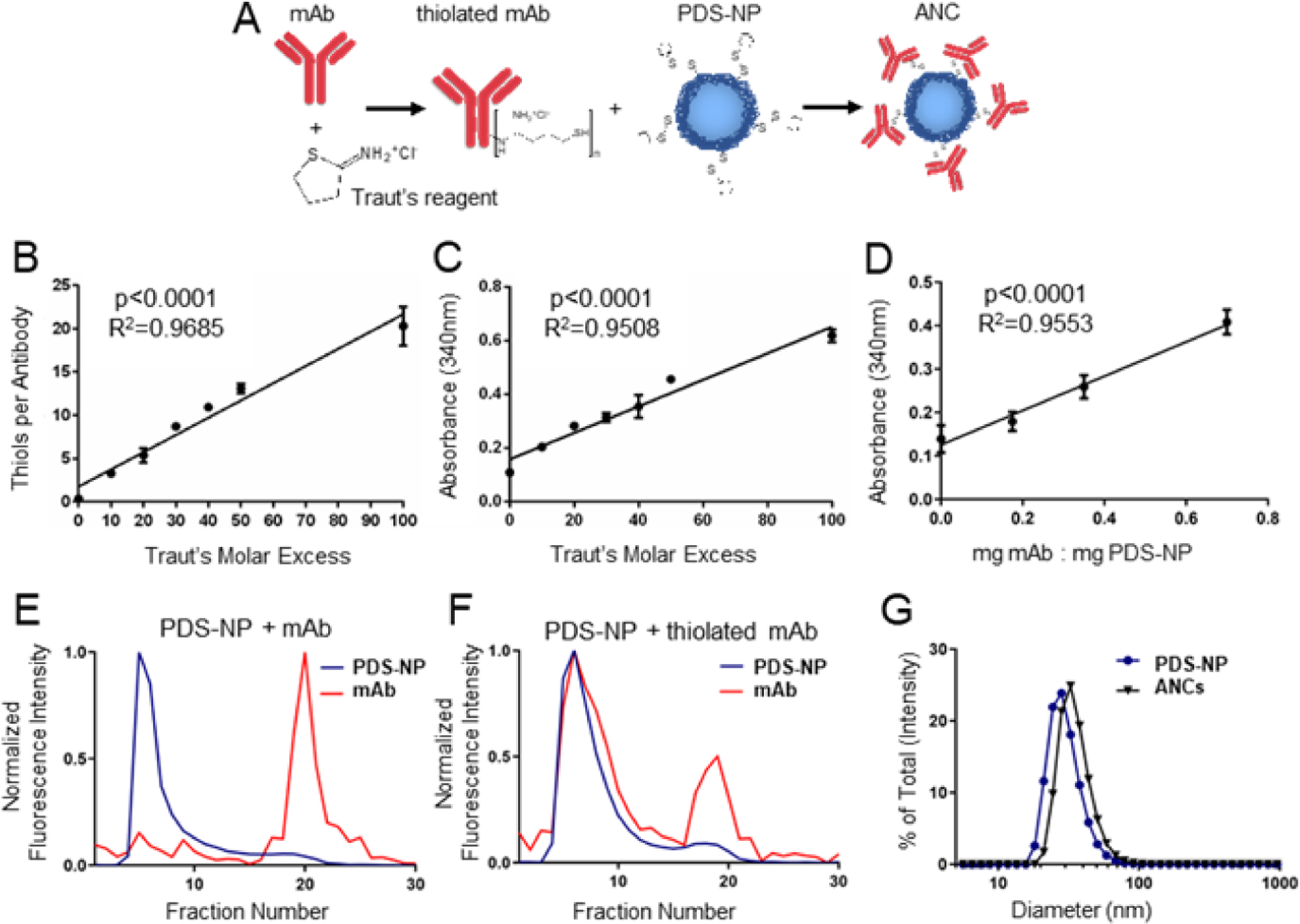
A) Schema of ANC formation wherein monoclonal IgG antibody (mAb) that is thiolated using Traut’s reagent is mixed with pyridyl disulfide (PDS)-derivatized PPS-NP (PDS-NP) to result in conjugation of mAb to PPS-NP via a disfulide linkage. The molar excess of Traut’s reagent controls number of thiols per mAb, as measured by Ellman’s assay (B), and extent of mAb conjugation to PDS-NP, as measured by 340 nm absorbance of the released pyridyl at 340 nm (C). D) Loading ratio of thiolated mAb to PDS-NP additionally controls the extent of mAb conjugation to PDS-NP, as measured by 340 nm absorbance of released pyridyl. Intensity of fluorescently labeled mAb or PDS-NP in eluted fractions from size exclusion chromatography when mAb was left unmodified (E) or thiolated using Traut’s reagent (F). G) Dynamic light scattering plots of plain NPs and ANCs post size exclusion purification and concentration. Statistical analyses were done using linear regression.
Rhodamine labeled anti-CD3 (aCD3) was conjugated to AlexaFluor647 labeled PDS-NPs yielding aCD3-ANCs that when incubated with murine splenocyte suspensions engaged with T cells (CD3+) to similar extents (as quantified in the double positive gate) compared to free (non-NP conjugated, rhodamine labelled) aCD3 mAb (single positive for mAb) as control (Fig. 2A–B), suggesting successful in vitro interaction of ANCs with T cells. To evaluate whether mAb binding was preserved in vivo, AlexaFluor700-labelled aCD3 in its free and ANC (AlexaFluor647-labeled NP) forms was administered to naïve mice via injection in the skin (6.25 μg aCD3 mAb dose) and labeling of CD4+ and CD8+ leukocytes within dLN assessed 24 h post injection (Fig. 2C–E). Strikingly, approximately 40% of dLN T cells were labeled with aCD3-ANCs, levels comparable to labeling achieved using free aCD3 (Fig. 2D). Correspondingly, ~40% of T cells were also NP+ (AlexaFluor647+), in sharp contrast to the <1% of T cells were labeled with control, non-targeted NPs (Fig. 2D), results in line with previous literature.24 Very little association with off-target cells was noted, including B (CD19+CD45+) cells (Fig. 2E). Thus, the ability of mAbs to specifically bind target cells is preserved following conjugation to PDS-NP, yielding ANC formulations, in both in vitro and in vivo settings.
Figure 2: ANCs retain mAb binding in vitro and in vivo.
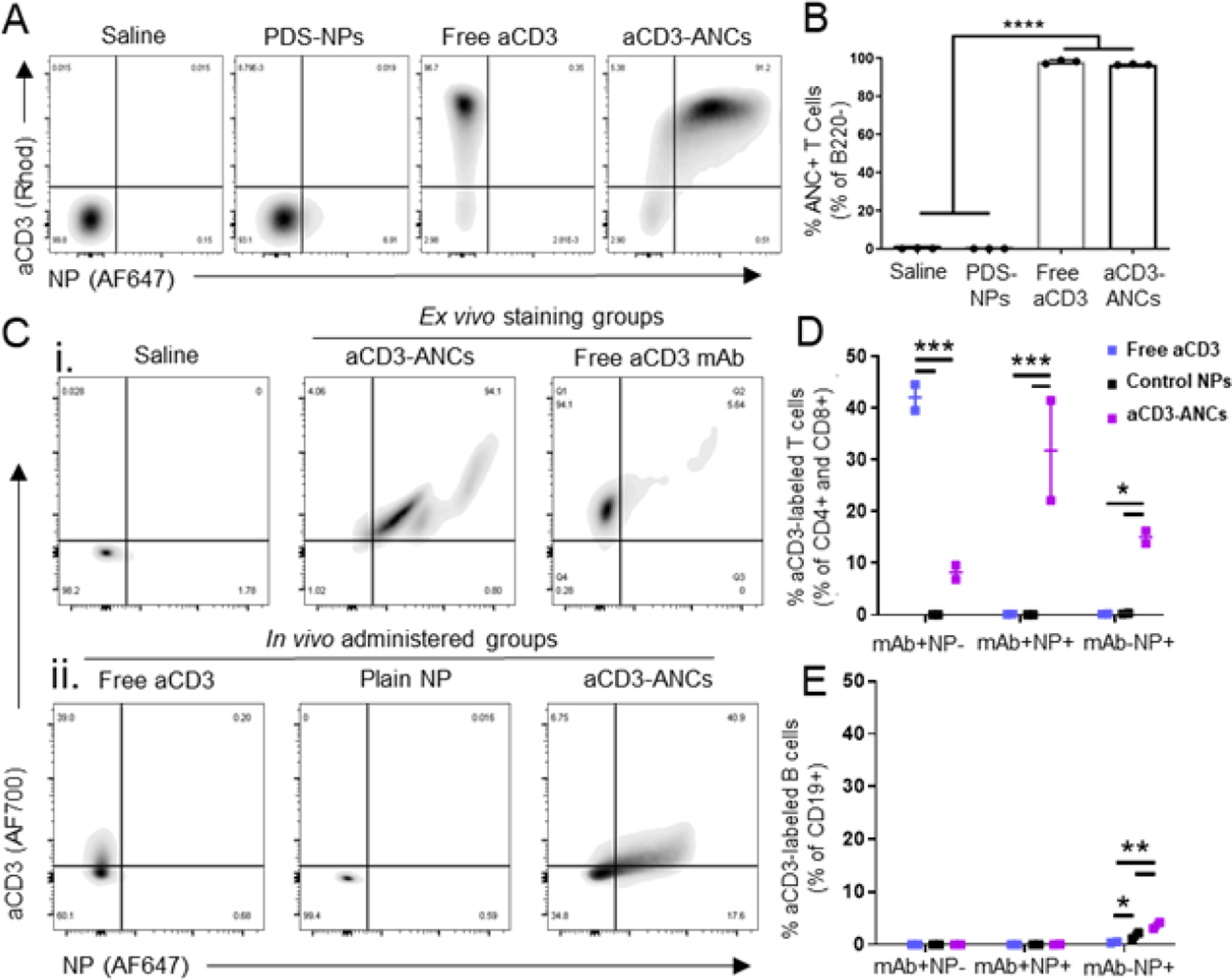
A) Representative flow cytometry plots of binding by various aCD3 mAb (AlexaFluor700-labeled) and NP (AlexaFluor647-labeled) formulations to B220− murine splenocytes after ex vivo co-incubation for 15 min. B) Quantification of NP+ (AlexaFluor647+) B220− murine splenocytes by various aCD3 mAb (AlexaFluor700-labeled) and NP (AlexaFluor647-labeled) formulations after ex vivo co-incubation for 15 min, as shown in A. C) Flow cytometry plots of aCD3 mAb (AlexaFluor700-labeled) and NP (AlexaFluor647-labeled) association with LN-resident CD4+ & CD8+ T cells after ex vivo incubation with free or ANC-formulated aCD3 (i) or in vivo 24 h post i.d. injection of free aCD3, unconjugated PDS-NPs, or aCD3-ANCs in naïve mice (6.25 μg total mAb dose) (ii). Quantified in vivo flow cytometry data from Cii. Frequencies represent the proportion of total CD4+ and CD8+ (D) or CD19+ (E) cells in each subgate: mAb+NP− (AF700+AF647−), mAb+NP+ (AF700+AF647+), and mAb−NP+ (AF700−AF647+). Statistical analyses were done using one- (B) or two-way (D-E) ANOVA with Tukey’s test. *p<0.05, **p<0.01, ***p<0.001, ****p< 0.0001.
As immune checkpoint molecules like PD1 and CTLA4 are expressed by T cells within B16F10 melanomas (Fig. 3A), the therapeutic potential of ANCs as combination ICB therapy targeting both PD1 and CTLA4 was explored in the B16F10 model in vivo. aPD1-ANCs were verified to specifically stain PD1, as demonstrated by impeded staining PD1-expressing EL4 cells with aPD1 after pre-incubation with aPD1-ANCs (Fig. 3B). When administered i.t., aPD1-ANCs were found to be as efficacious as free aPD1 in eliciting anti-tumor therapeutic effects, including slowed tumor growth (Fig. 3C), demonstrating that mAb functionality is sustained upon ANC conjugation and that ANCs maintain the beneficial therapeutic effects of ICB. Similarly, i.t. administered aPD1- and aCTLA4-ANCs in combination were found to provide comparable anti-tumor therapeutic effects as combination therapy with the free mAb (Fig. 3D). Interpreted another way, however, non-drug loaded ANCs did not improve the effects of i.t. combination ICB.
Figure 3: ANCs maintain in vitro binding and in vivo therapeutic efficacy of immune checkpoint blockade (ICB) mAbs.
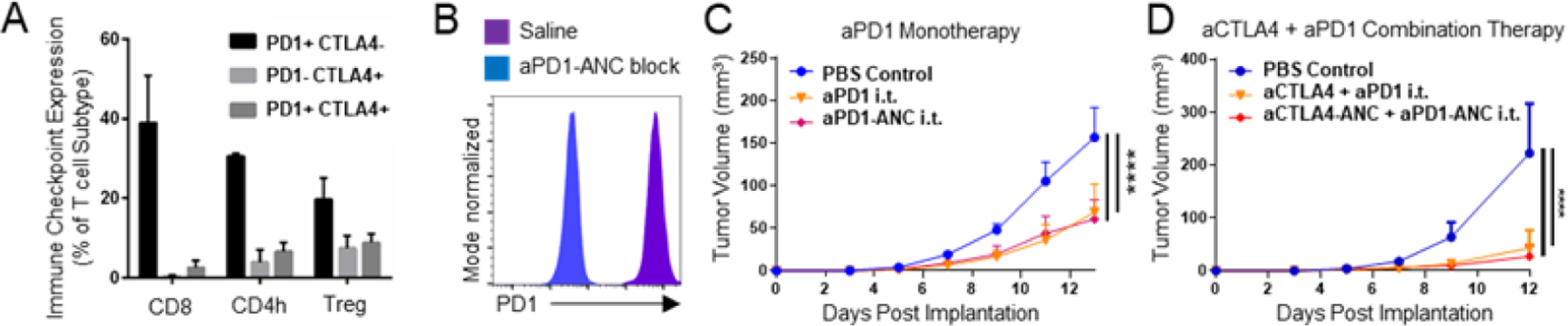
A) Expression of immune checkpoint molecules PD1 and CTLA4 by CD8 (CD8+), CD4h (CD4+FoxP3−) and Treg (CD4+FoxP3+) T lymphocytes (CD3+CD45+). B) Flow cytometry histograms of fluorescent labeling of PD1-expressing EL4 murine lymphoma cells with aPD1 with or without 15 min pre-incubation with aPD1-ANCs. Tumor control by aPD1 (150 ug) as monotherapy (C) or in combination with aCTLA4 (150 ug of each) (D) administered i.t. on days 5, 7, and 9 post-B16F10 implant is equivalent between ICB in its free or ANC form. Statistical analyses were done using two-way ANOVA with Tukey’s test (C-D). ****p < 0.0001.
To better understand the biodistribution of ANCs in vivo, the influence of NP conjugation on mAb biodistribution and vice versa after i.t. administration into B16F10 tumor-bearing mice was assessed. Retention at the site of injection and accumulation within the LN draining the tumor injection site were evaluated for AlexaFluor700-labeled isotype mAb, aPD1, or aCTLA4 in its free form or conjugated to AlexaFluor647-labeled NPs for 72 h post administration (Fig. S1A–F). By IVIS imaging of AlexaFluor700 signal, NP conjugation was found to have no influence on the extent of retention in the tumor by any tested mAb type (Fig. S1A, C, E). No difference in the extent of mAb accumulation in TdLN between ANCs or free mAb control was seen at any analysis time point (Fig. S1B, D, F). However, when NP signal (measured via AlexaFluor647) was assessed, conjugation to aCTLA4 modestly improved NP retention in the tumor injection site (Fig. 4A–B). Conjugation to either ICB mAb also improved NP levels in TdLNs post injection over 72 h post injection (Fig. 4C–D). Results suggest whereas NP conjugation does not appear to substantially alter mAb biodistribution after i.t. injection, ANC targeting to immune checkpoints alters NP biodistribution profiles to favor both tumor and LN retention.
Figure 4: ANCs targeted to immune checkpoints increases retention in tumor injection site and accumulation in TdLNs.
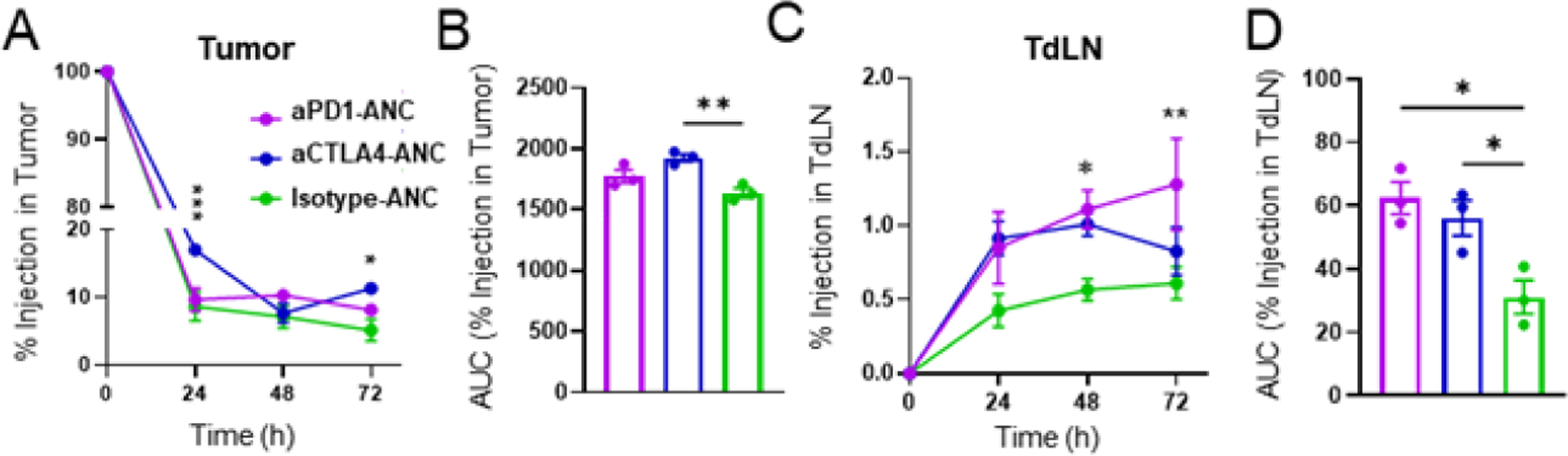
ANC-formulated aPD1, aCTLA4, or isotype mAb (10μg) were injected i.t. into mice bearing d5 B16F10 tumors. NPs were labeled with AlexaFluor647, mAb with AlexaFluor700. A) AlexaFluor647 levels in tumors from aPD1-, aCTLA4-, or isotype-ANCs over 72 h. B) Cumulative area under the curve (AUC) of AlexaFluor647 signal over 72 h post injection, quantified from A. C) AlexaFluor647 levels in TdLNs from aPD1-, aCTLA4-, or isotype-ANCs over 72 h. D) Cumulative AUC of AlexaFluor647 signal over 72 h post injection, quantified from C. Statistical analyses were done using two- (A, C) or one-way (B, D) ANOVA with Tukey’s test. * p<0.05, ** p<0.01, *** p<0.001.
3.2. Targeting of drug-eluting ANCs to immune checkpoints improves melanoma control from i.t. administered combination immunotherapy
ANCs offer multiple unique advantages to combination immunotherapies, including mAb multivalency, tailorable biodistribution profiles, and the capacity to co-formulate agents with synergistic or orthogonal bioactivities to potentiate drug effects. Given favorable retention of NP in the tumor and TdLN, tissues relevant in the context of ICB immunotherapy, with immune checkpoint-targeting ANC, the ability of these ANC features to potentiate the therapeutic effects of ICB and co-delivered immunomodulatory drugs was thus explored. First, the potential for ANC formed from therapeutic mAb with amphiphilic NP to encapsulate drug to facilitate its release and effects on T cells in a targeted manner was first investigated in vitro using a PD1-expressing EL4 cells (Fig. 5A). ANC association with PD1-expressing EL4 cells was found to be enhanced by functionalization with aPD1 compared to isotype mAb (Fig. 5B–C). As proof-of-concept, hydrophobic chemotherapeutic PXL, well established for its cytostatic activities and poor solubility in aqueous solutions, was encapsulated in the NP,23 which enabled sustained PXL release over days (Fig. 5D). Its dose-dependent effects on EL4 cell viability with 15 min of co-incubation in vitro when incorporated into the NP were only modestly reduced (Fig. 5E). Targeting to PD1 resulted in concentration-dependent EL4 cell death by PXL-loaded ANCs (Fig. 5F). These results highlight the ability of ANCs to deliver small molecule cargo in vitro in a targeted manner.
Figure 5: ICB-ANCs target NP-encapsulated small molecules to immune checkpoint-expressing cells in vitro.
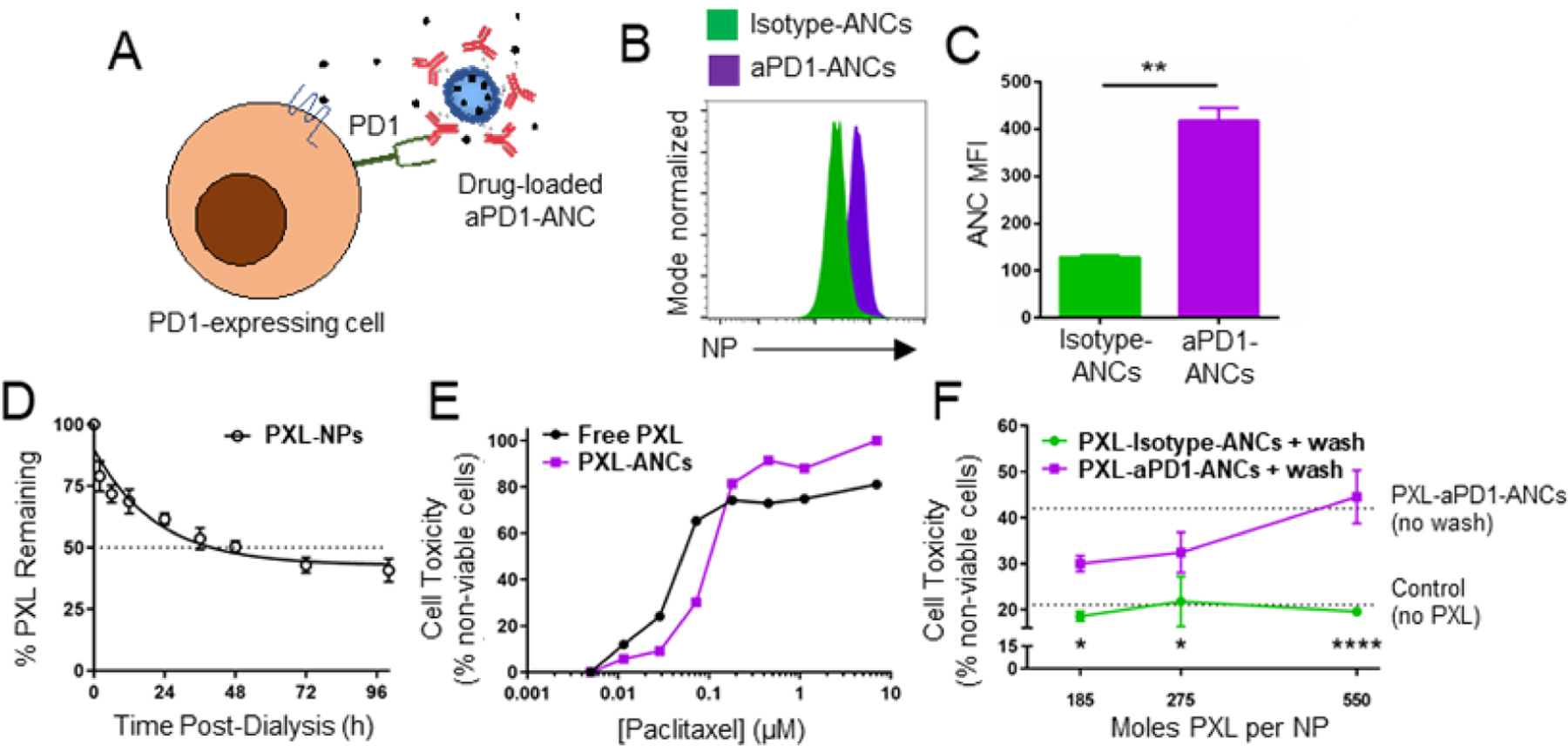
A) Schematic of ICB-ANC-mediated co-delivery of small molecule drugs to immune checkpoint-expressing T cells. Histogram (B) and MFI (C) of flow cytometrically measured NP signal of fluorescently labeled (AlexaFluor647) NPs from isotype- or aPD1-ANCs after 15 min co-incubation in vitro with PD1-expressing EL4 cells and serial washing. D) Fraction of initial loaded paclitaxel (PXL) amount remaining in NP during in vitro release with dialysis. E) Fraction of plated EL4 cells non-viable after 72 h treatment with free or NP-encapsulated PXL. F) aPD1 targeting improves cytotoxic effects of PXL-loaded ANCs on PD1-expressing EL4 cells. Cells were co-incubated in vitro with PXL encapsulated within isotype- or aPD1-ANCs for 15 min followed by two washes and subsequently cultured in drug- and NP-free media for 72 h. Controls include untreated cells or cells treated with ANCs without washing (dotted lines). Statistical analyses were done using two-tailed unpaired t-test (C) or two-way ANOVA with Sidak’s test (F). *p<0.05, **p < 0.01, ****p < 0.0001.
Transforming growth factor beta (TGFβ) has garnered attention recently as it is commonly upregulated in tumors and lymphoid tissues where it can promote immune suppressive milieus, in part by suppression of T cell cytotoxic function.25,26 Furthermore, heightened TGFβ signaling can reduce T cell infiltration into the TME leading to immune excluded tumors of which have been shown to respond poorly to ICB therapies.27,28 We hypothesized that co-delivery of SB-431542, a TGFβ receptor 1 inhibitor, and ICB to immune checkpoint-expressing T cells using ANCs could combat immune-excluded tumors and thereby improve anti-tumor efficacy, given that ANC targeting improves drug retention at the tumor/injection site and in TdLNs. To assess the potential of drug eluting ANCs to potentiate combination cancer immunotherapy in vivo, delivery of TGFβ receptor 1 inhibitor SB-431542 to immune checkpoint expressing T cells was investigated. Encapsulated SB-431542 was released from NP over hours (half-life ~12 h, Fig. 6A). When administered i.t., targeting of SB-431542-encapsulating NP via aPD1 and aCTLA4 (6 µg each) dramatically improved the therapeutic effects of combination therapy with respect to reduced tumor growth compared to non-targeted SB-431542-encapsulating NP functionalized with isotype mAb co-administered alongside aPD1- and aCTLA4-ANCs (Fig. 6B–C), resulting in prolonged animal survival (Fig. 6D). Targeting of NP-encapsulated SB-431542 via ICB thus improves the effects of this combination immunotherapy.
Figure 6: TGFβ inhibitor-loaded ICB-ANCs improve anti-tumor effects of i.t. combination immunotherapy in B16F10 melanoma model.
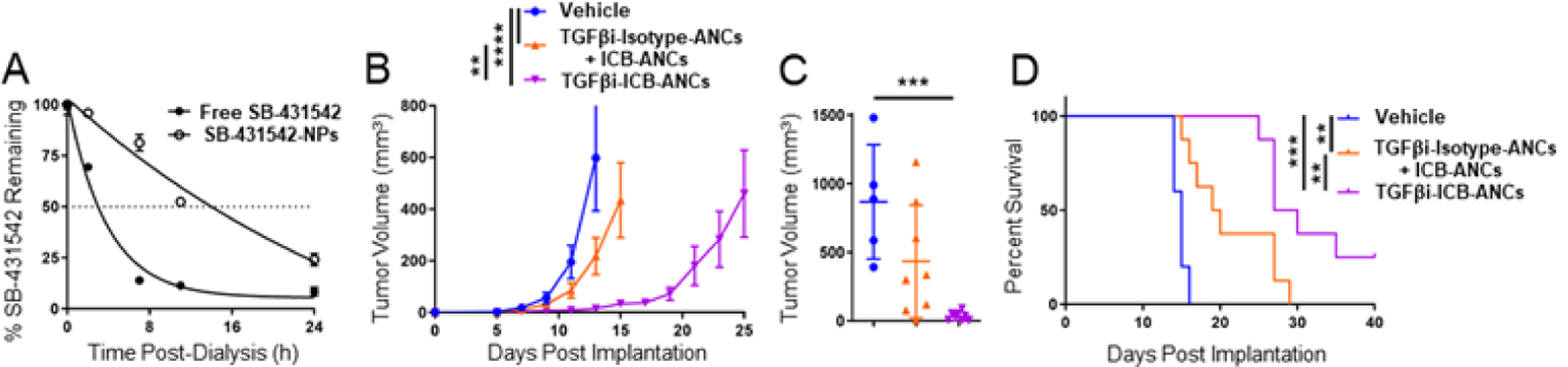
A) Fraction of initial loaded SB-431542 (TGFβi, starting concentration 4 mM) amount remaining in NP during in vitro release with dialysis. B) B16F10 tumor growth in response to i.t. treatment on day 5, 7, 9, 11, and 13 post tumor implantation with ICB- [aPD1 (6 μg total mAb dose) and aCTLA4 (6 μg total mAb dose)] ANCs co-encapsulating TGFβi or co-injected with isotype-ANCs encapsulating TGFβi (5 μg total dose). ICB-ANCs consisted of an equal mixture of ANCs functionalized with either aPD1 or aCTLA-4. Each tumor growth curve ends on the day of the first animal death. C) Tumor volume on day 15 post-implant. D) Survival curves of animals whose tumor volumes are shown in (B). Statistical analyses were done using two- (B) or one-way (C) ANOVA with Tukey’s test. Log-rank (Mantel-Cox) test for survival curve (D). **p < 0.01, ***p < 0.001.
3.3. Targeting improves NP and encapsulated cargo delivery to target cells in vivo
Given the benefit of NP retention in the tumor and TdLNs after i.t. administration was observed to confer benefits on combination immunotherapy when SB-431542 was co-delivered via the targeted ANCs, the potential for this delivery system to improve immunotherapeutic delivery after injection in locoregional tissues that results in NP accumulation in TdLNs was next explored. Previously, favorable effects of CD3 targeting on NP delivery to lymphocytes was demonstrated after locoregional administration in vivo (Fig. 2C–E), thus the potential for this ANC platform to improve targeting of small molecular cargo passively encapsulated within the ANC NP to target-expressing cells, previously shown in vitro, to confer these delivery benefits to TdLNs in vivo was assessed. For this application, the 4T1 orthotopic model of murine triple negative breast cancer (TNBC) was utilized because of the well-established involvement of the TdLN in tumor metastasis and progression,29 presence of active immune checkpoints and other suppressive mechanisms in TdLNs of 4T1-bearing mice,18,30 and current lack of therapies that leverage the benefits of targeted delivery due to the absence of molecular targets.31
To first characterize the biodistribution of ANCs and delivery of co-encapsulated small molecule drug to TdLNs and other relevant tissues, model aCD3- or control isotype-ANCs (AlexaFluor488-labeled NP) were used to encapsulate fluorescent cyanine5.5 (Cy5.5) as model cargo, which exhibits a release half-life of ~11h from NP.22 Cy5.5-aCD3 or isotype-ANCs (6.25 μg total aCD3 or isotype mAb dose; 1 μg Cy5.5 dose) were then administered locoregionally into the flank skin ipsilateral to the tumor (thus resulting in drainage to LNs co-draining the 4T1 tumor) and extent of ANC or Cy5.5 association with leukocytes harvested from various tissues were assessed by flow cytometry (Fig. 7A). After 24 h post-i.d. administration, NP (n.s., p=0.12) and Cy5.5 were found to accumulate to greater extents with T lymphocytes (B220−CD45+) in dLNs but not the circulation (blood) or tumor, as a result of ANC functionalization with aCD3 (Fig. 7B–C). Notably, CD3-targeting ANC also enhanced Cy5.5 (n.s., p=0.10), but not NP, association with B220+ leukocytes in TdLNs from i.d. administration (Fig. 7D–E), presumably due to the benefit afforded by multistage delivery into lymph on delivery to LN resident lymphocytes. Perhaps unsurprisingly, the benefit of ANC targeting was more pronounced for Cy5.5 delivery to T lymphocytes (~10x), which tend to be more restricted from lymph access,8,32 compared to B cells (~2x) that reside in the afferent lymph-proximal LN paracortex (Fig. 7C, E). Additionally, i.d. administration of a higher mAb and Cy5.5 dose (40 μg total aCD3 or isotype mAb dose; 6.25 μg Cy5.5 dose) revealed that while the benefit of ANC targeting on NP association with T lymphocytes in the TdLN is maintained, there was no targeting benefit on Cy5.5 association with T cells (Fig. S2A–B), while B cells (B220+CD45+) were efficiently labeled with Cy5.5 but not NP regardless of formulation (Fig. S2C–D). This demonstrates the ability to control potential off-target accumulation by using a smaller dose of mAb or co-encapsulated small molecule, another potentially advantageous feature of ANC targeting. NP targeting by mAbs directed to T lymphocytes thus improves NP-encapsulated cargo delivery to T lymphocytes resident within TdLNs.
Fig. 7. ANCs targeted to T cell-expressed surface receptor increases delivery of NP and NP-encapsulated small molecule cargo to T cells in LNs and blood after locoregional administration.
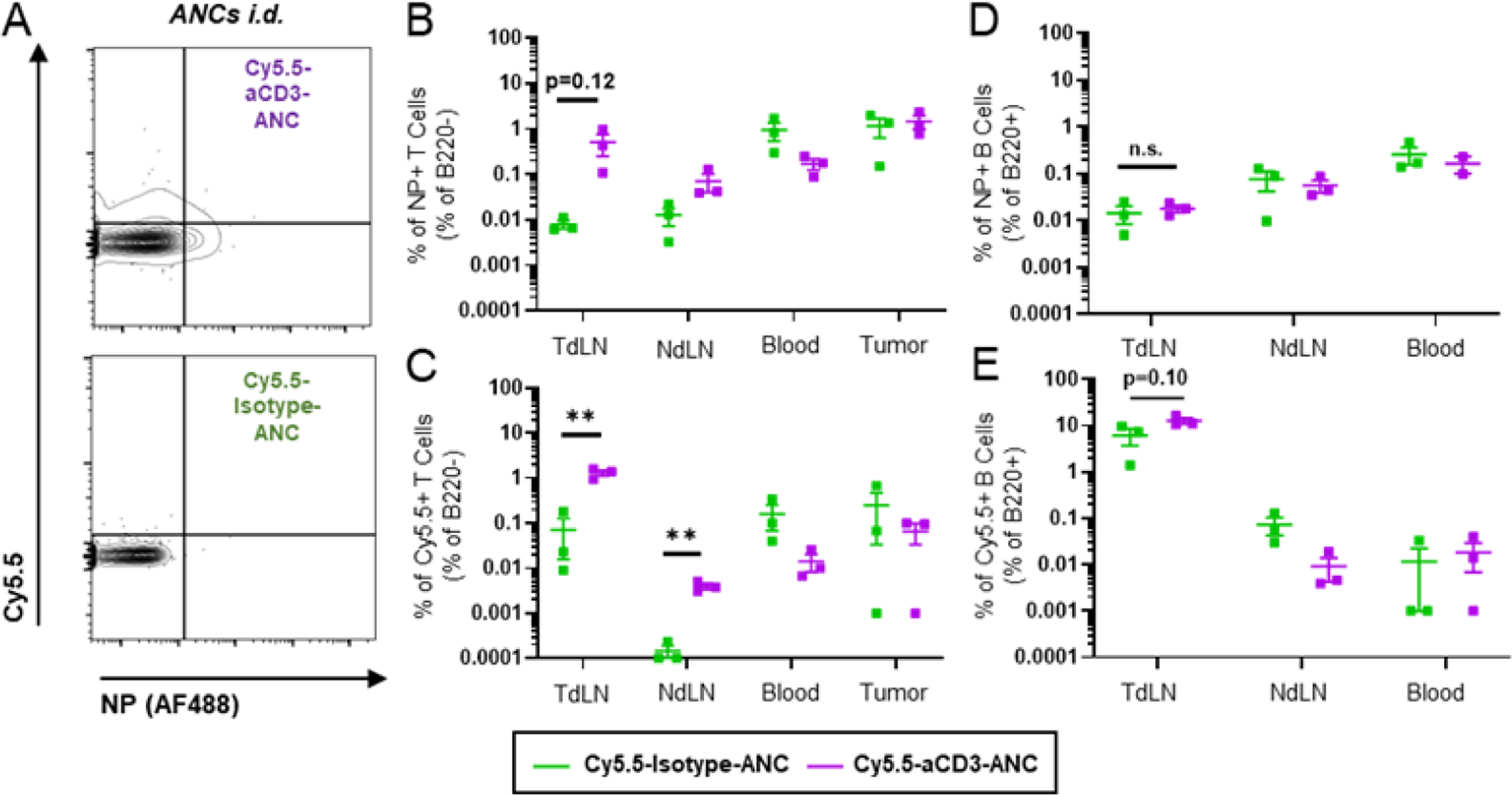
Representative flow cytometry plots (A) and quantification (B-C) of B220- leukocyte association with aCD3- or isotype-ANCs (AlexaFluor488-labeled NP) co-encapsulating Cy5.5 24 h post i.d. injection into flanks of day 6 4T1 tumor-bearing mice (6.25µg total mAb dose, 1µg Cy5.5 dose). B) Total frequency of ANC+ B220- leukocytes [sum of ANC+Cy5.5- and ANC+Cy5.5+ gates in (A)]. C) Total frequency of Cy5.5+ B220- leukocytes [sum of ANC-Cy5.5+ and ANC+Cy5.5+ gates in (A)]. D-E) Quantification of B220+ leukocyte association with aCD3- or isotype-ANCs (AlexaFluor488-labeled NP) co-encapsulating Cy5.5 24 h post i.d. injection into flanks of day 6 4T1 tumor-bearing mice (6.25 µg total mAb dose, 1 µg Cy5.5 dose). Total frequency of total ANC+ (D) or Cy5.5+ (E) B220+ leukocytes quantified as above. Gating controls included LN cells from saline-treated mice. Statistical analyses were done using two-tailed unpaired t-test. **p < 0.01. n.s., not significant.
3.4. Targeted antagonism of adenosine signaling with ICB via ICB-ANCs improves both systemic and locoregional combination immunotherapy for TNBC
Adenosine signaling pathways have recently emerged as new targets for combination immunotherapy approaches, as adenosine concentrations are greatly increased within tumors since cancer cells rapidly turnover and release intracellular adenosine triphosphate which is broken down to free adenosine by CD39 and CD73, two surface ectonucleotidases.33,34 Free adenosine acts to suppress effective CD8+ T cell priming within LNs and helps protect tumor cells via suppressed CD8+ T cell cytotoxic function.35,36 This has spurred the development of various adenosine modulation strategies including several small molecule adenosine receptor antagonists37 including those for the adenosine receptor 2A which has a high adenosine affinity.38 We hypothesized that co-delivery of adenosine receptor 2A antagonist SCH-58261 to PD1 expressing T cells in LNs would improve anti-tumor responses as these pathways, while non-redundant, have been shown to upregulate the other’s expression upon modulation of either pathway.39,40
Adenosine is commonly upregulated in cancers and contributes to immune suppression, including in TNBC. We thus explored the levels of adenosine and PD1 in the 4T1 tumor model of TNBC. Adenosine concentrations in the serum and TdLNs of tumor bearing mice were found to be significantly increased (Fig. 8A), which is presumably due to the high metastatic propensity of this model to infiltrate systemic tissues and TdLNs.29 Within these same tissues, frequencies of PD1+ cells were also found to be high within CD8+ and CD4+ T cells and NK cells (Fig. 8B). In line with this, CD8+ T cells incubated with aCD3/aCD28 Dynabeads in the presence of adenosine showed suppressed IFNγ production and Ki-67 expression, effects reversed by co-treatment with adenosine receptor 2A antagonist SCH-58261 (Fig. 8C–E). These results highlight the adenosine and PD1 mediated suppression in the 4T1 TNBC model and the ability for adenosine receptor antagonism to rescue immune function. Thus, the potential for aPD1 targeting of ANCs co-delivering SCH-58261 to PD1-expressing cells in TdLNs to augment anti-tumor immune response compared to non-targeted therapy was next explored. NP encapsulated SCH-58261 was released with a half-life of ~9 h (Fig. 8F); like all other small molecule drugs explored in this work, SCH-58261 was released from the NPs without the need for external stimuli, likely by simple diffusion driven by a concentration sink. On days 6, 9, 12, and 15 following 4T1 tumor implantation, 60 µg of aPD1 mAb, either in the free or ANC formulation along with 2 µg of encapsulated SCH-58261 in isotype- or aPD1-ANCs were administered i.d. into the flank ipsilateral to the tumor (thus resulting in drainage to LNs co-draining the tumor). SCH-58261 loaded aPD1-ANCs greatly reduced tumor growth and prolonged mouse survival compared to non-targeted SCH-58261 loaded isotype-ANCs, which provided no benefit compared to vehicle controls (Fig. 8G–H). These results highlight the ability of drug-eluting, targeted ANC to improve combination immunotherapy for TNBC in the context of TdLN-directed locoregional administration.
Figure 8: Targeting of adenosine antagonist-loaded ANCs to immune checkpoint PD1 improves anti-tumor effects of locoregional combination immunotherapy in 4T1 TNBC tumor model.
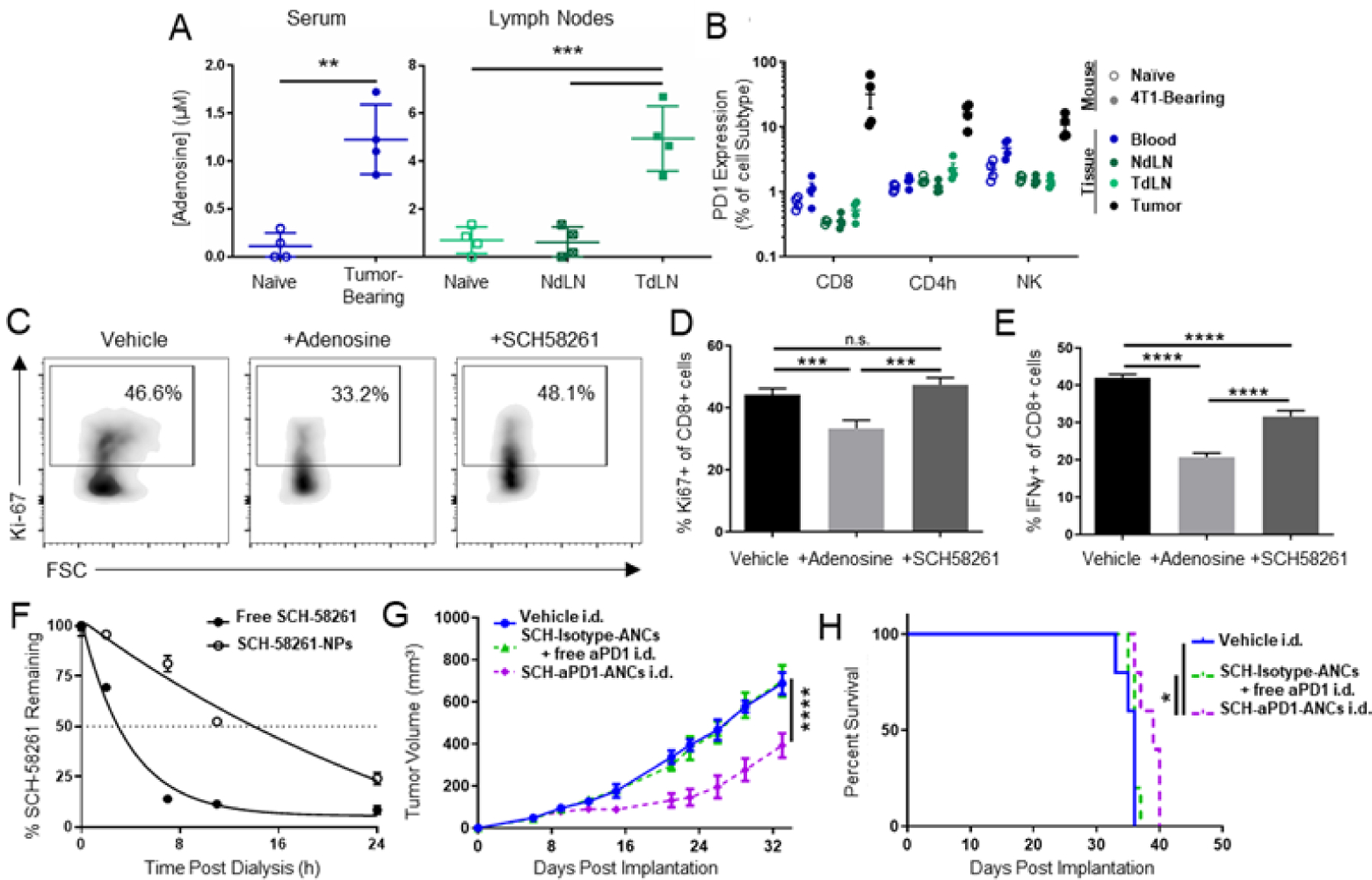
A) Adenosine concentrations in serum and LN homogenates from mice bearing d15 4T1 tumors. B) PD1 expression by CD8, CD4, and NK leukocytes in various tissues from mice bearing d15 4T1 tumors. C) Flow cytometry plots of Ki-67 staining in CD8 T cells after 24 h ex vivo activation with Dynabeads and co-incubation with adenosine (1mM) and adenosine antagonist SCH-58261 (0.01 mM). Quantified flow cytometry data of Ki-67 (D) and IFNγ (E) expression by CD8 T cells after 24 h ex vivo activation with Dynabeads and co-incubation with adenosine (1mM) and adenosine antagonist SCH-58261 (0.01 mM). F) Fraction of initial loaded SCH-58261 (adenosine antagonist) amount remaining in NP during in vitro release with dialysis. Tumor growth curves (G) and animal survival (H) in response to combination therapy administered i.d. in the flank ipsilateral to the 4T1 tumor on d 6, 9, 12, and 15 post implantation. Treatment groups include aPD1-ANCs (60 µg mAb) encapsulating SCH-58261 (2 µg) or isotype ANCs encapsulating SCH-58261 co-injected with free aPD1 mAb. Statistical analyses were done using one- (A, D-E) or two-way (G) ANOVA with Tukey’s test. Log-rank (Mantel-Cox) test for survival curves (H). *p < 0.05, **p < 0.01, ***p<0.001, ****p < 0.0001. n.s. not significant.
In addition to a LN-mediated approach, we next sought to explore the ability of T lymphocyte-targeted ANCs to enhance NP delivery to target cells in systemic tissues using systemic routes of administration most commonly used for clinical cancer immunotherapy. Given the presence of elevated adenosine concentration in the serum and PD1 expression on CD8+ and CD4 T+ cells in the blood and tumors of 4T1-bearing mice, the potential for the ANC system to potentiate co-delivery of SCH-58261 and aPD1 combination immunotherapy delivered systemically was next explored in the 4T1 model.
To assess the biodistribution of systemically administered ANCs, AlexaFluor647-labeled NP functionalized with isotype or aCD3 (40 μg aCD3 or isotype mAb dose) were administered i.v. into 4T1 tumor-bearing mice. 1 h post i.v. injection, NPs of CD3-targeted ANCs were found to distribute to a substantially higher frequencies of T lymphocytes (B220−CD45+ cells) in the blood, spleen, and TdLNs compared to isotype-ANCs (Fig. 9A–B). Interestingly, no benefit on targeting to the tumor directly was observed (Fig. 9B). CD3 targeting ANCs also increased NP delivery to B lymphocytes in the blood, spleen, and TdLN, though to a less dramatic extent compared to T lymphocytes (10x vs. 5x, 100x vs. 20x, and 10x vs. 5x, respectively) (Fig. 9C). Thus, targeting of T lymphocyte-expressed surface receptors appears to uniquely improve delivery of ANCs administered systemically to both circulating and LN-resident T lymphocytes.
Figure 9: Targeting of adenosine antagonist-loaded ANCs to immune checkpoint PD1 improves NP delivery to circulating and LN-resident T cells and anti-tumor effects of systemic combination immunotherapy in 4T1 TNBC tumor model.

Representative flow cytometry plots (A) and quantification (B) of NP+ frequency of B220− leukocytes 1 h post i.v. administration of aCD3- or isotype-ANC (AlexaFluor647-labeled NP; 40μg mAb dose) in TdLN, spleen, blood, and tumor tissues of day 6 4T1-bearing mice. C) Quantification of aCD3- or isotype-ANCs binding to B220+ leukocytes 1 h post i.v. administration. D) Tumor growth curves after combination immunotherapy administered i.p. on days 6, 9, 12, and 15 post-implantation of 4T1 tumors. Treatments included aPD1-ANCs (60 µg mAb) encapsulating SCH-58261 (2 µg) or isotype ANCs encapsulating SCH-58261 co-injected with free aPD1 mAb. E) Representative gating of TdLN lymphocytes on day 35 after 4T1 tumor implantation. LN metastases quantified as the fraction of analyzed total events within the shown lymphocyte gate. Gating controls include cells harvested from 4T1 tumors (Tumor) and cells from LNs of tumor-free mice (Tumor-free LN). F) Quantified levels of LN lymphocytes on day 35 from (E). Dotted line represents % lymphocytes in LN of non-tumor bearing mice. G) Spleen area. Dotted line represents spleen area in non-tumor bearing mice, statistics relative to non-tumor bearing mouse spleen size. H) Animal survival. Statistical analyses were done using two-tailed unpaired t-test (B-C), two- (D) or one-way (F-G) ANOVA with Tukey’s test. Log-rank (Mantel-Cox) test for survival curves (H). *p<0.05, **p<0.01, ****p<0.0001. n.s., not significant.
To extend these results in a therapeutic setting, 60 µg of aPD1 mAb, either in the free or ANC formulation along with 2 µg of encapsulated SCH-58261 in isotype- or aPD1-ANCs were administered systemically (i.p.) on days 6, 9, 12, and 15 following 4T1 tumor implant. The combination of free aPD1 mAb and SCH-58261-loaded isotype-ANCs i.p. resulted in modestly reduced 4T1 tumor growth, which was further improved by targeting of SCH-58261-loaded aPD1-ANCs (Fig. 9D). Metastatic tumor burden in LNs was also reduced, as indicated by normalized immune cell frequencies relative to non-TdLNs and LNs from non-tumor bearing mice (Fig. 9E–F). Furthermore, spleen size, which has previously been correlated with metastatic disease burden,29 was found to be reduced in animals treated with aPD1-ANC encapsulating SCH-58261 and comparable to the sizes of non-tumor bearing mice (Fig. 9G). Survival of a separate mouse cohort treated using the same schedule demonstrated improved animal survival by aPD1 targeting of NP-encapsulated SCH-58261 (Fig. 9H). These results highlight the ability of drug eluting targeted ANC to improve combination immunotherapy for TNBC in the context of clinically relevant systemic administration routes, which enables targeted ANC accumulation within circulating and LN-resident lymphocytes.
4. Discussion
The ability of cancers to develop and metastasize relies on numerous factors including the upregulation of multiple suppressive pathways, for example exclusion of T cells within the tumor and expression of numerous checkpoint pathways.41 As a result, effective immunotherapy must modulate multiple pathways simultaneously, which has led to the development of combination therapies in order to generate a robust anti-tumor immune response. Conventional combination therapies consist of co-administered agents but may not to lead to concurrent delivery to and thus modulation to the same cell target, thereby diminishing potential synergistic effects.
Herein, we explored the ability to co-deliver multiple immunotherapies to immune checkpoint (PD1 and CTLA4)-expressing lymphocytes in vivo using a biomaterial NP platform. We hypothesized that sustained co-delivery would improve anti-tumor responses compared to conventional formulations that do not necessarily ensure co-delivery and thus co-modulation of pathways within immune cells of interest. Using this system, we show delivery of three unique small molecule compounds to T cells: a model chemotherapeutic drug PXL, a TGFβ receptor 1 inhibitor, and an adenosine 2A receptor antagonist. Each drug works in a different cellular location, the nucleus, cytoplasm, and extracellular surface, respectively, highlighting the ability of NP-released small molecule to drug pathways at a variety of cellular locations. Of interesting note, while all three drugs tested released from ANCs with no need for external stimulus, presumably driven by diffusion down a concentration gradient, the rate of release differed between drugs: 50% release occurred in as little as 9 h for SCH-58261, and up to 2 days for PXL. While the reason for these differences can be further investigated, trends in these and other drugs suggest that drug size plays an important role, with larger molecules taking longer to release. Additionally, we demonstrate that drug activity is retained after release, a benefit enabled by this carrier and encapsulation strategy which requires no chemical modifications of NP-formulated cargo, a major barrier that can affect drug activity in other platforms. Furthermore, the mAb conjugation synthesis can be applied to multiple mAb while retaining binding activity as the reaction conditions are mild (require no heating, organic solvents, etc.). These data highlight the versatility of ANCs, which can be applied to deliver numerous small, hydrophobic molecules to T cells by targeting surface receptors with the cognate mAb.
Due to their low levels of phagocytosis and pinocytosis, T cells represent a challenging target to drug via passive targeting, yet are immensely important for generating robust anti-cancer immune responses.24,42 Our results demonstrate the ability to target T cells both in vitro and in vivo using targeting mAbs that is not impaired by conjugation to NPs and thus dramatically improves co-association of drug-encapsulating ANCs with T cells relative to non-targeted, isotype-ANCs. Furthermore, ANC formulations did not diminish effects of ICB when used as monotherapies. Emerging literature suggests that immune suppression and exclusion is a multi-pathway process, thus requiring multiple modes of modulation to fully restore cytotoxic function. Therefore, the potential of enabling co-delivery of multiple immunotherapies and immune synergies is vast.43 When leveraging the NP as a drug reservoir, anti-tumor responses induced by the co-delivery of ICB with immune modulators using targeted ANCs were improved relative to that of conventional combination therapies, or formulations lacking co-delivery. We highlight the importance of co-delivery as we explored this approach using three different administration schemes: intratumoral, locoregional skin, and systemic. In a melanoma model, i.t. administration of targeted ANC improved NP delivery to tumor and TdLN tissues, and in a TNBC model both locoregional i.d. and systemic administration of targeted ANCs resulted in enhanced NP association with LN lymphocytes, with the additional benefit of NP association with circulating lymphocytes from systemic administration. In each therapeutic model and administration scheme, drug-eluting, targeted ANCs manifested improved anti-tumor responses to formulations lacking co-delivery, though we did not directly demonstrate the relative importance of delivery to circulating, tumor, or LN-resident immune cells in each of the different administration route applications. Because ANCs improved the efficacy of these combination therapies in the tested models, this platform may allow for dose reduction compared to free ICB and small molecule drug formulations, with the potential to reduce side effects that may be associated with high doses of free formulations. Additionally, given our recent demonstration of the advantages of delivery of ICB to LNs in potentiating tumor control and affording doses-sparing,16,18 this platform in combination with locoregional administration routes may allow for potent and facile co-delivery of ICB and small molecule drugs to LN-resident immune cells or metastases, an effect which is novel to this ANC platform. Considering the emerging interest in combining ICB therapies with other small molecule modulators including those targeting the TGFβ and adenosine pathways, these results highlight the potential for co-delivery approaches, such as the drug eluting immune checkpoint inhibitor ANC approach described herein, to substantially improve the efficacy of systemic and lymphatic-directed immunotherapies.
5. Conclusions
We engineered a versatile ANC platform capable of delivering small molecule therapeutics in a targeted fashion to immune checkpoint expressing cells in the circulation and LNs to augment targeted therapy and sustain drug synergies to improve combination cancer immunotherapies. Use of our ANC platform allows for encapsulation of a variety of small hydrophobic molecules within the NP core while retaining surface-conjugated mAb binding affinity to enable targeted delivery of encapsulated drug to cells of interest. The use of this ANC platform enabled co-delivery of ICB and TGFβ or adenosine antagonists to immune cells of interest which reduced tumor growth and extended survival after both systemic and locoregional administration, highlighting the potential for engineered drug delivery system to unleash the therapeutic potential of modulating multiple suppressive pathways within cells of interest for cancer immunotherapy.
Supplementary Material
Acknowledgements
This work was supported by National Institutes of Health (NIH) grants R01CA207619 (S.N.T.), R01CA247484 (S.N.T.), T32EB021962 (D.M.R.), T32EB006343 (L.F.S.), and S10OD016264, CCR15330478 grant from Susan G. Komen® (S.N.T.), and Department of Defense Grant CA150523 (S.N.T.). M.P.M. was supported by a National Science Foundation Graduate Research Fellowship. L.F.S. and A.S. were American Heart Association Pre-doctoral Fellows.
Footnotes
Credit Author Statement
D.M.F.: Conceptualization, Formal analysis, Investigation, Writing – Original Draft, Writing- Review & Editing, Visualization. M.P.M.: Validation, Formal analysis, Investigation, Writing- Review & Editing, Visualization. P.A.A.: Validation, Formal analysis, Investigation, Writing- Review & Editing, Visualization. L.F.S.: Validation, Formal analysis, Investigation, Writing- Review & Editing, Visualization. A.J.H.: Investigation, Writing- Review & Editing. A.S.: Investigation. S.N.T.: Conceptualization, Writing – Original Draft, Writing- Review & Editing, Supervision, Project Administration, Funding Acquisition.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
The authors declare no conflicts of interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- (1).Chen DS; Mellman I Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39 (1), 1–10. 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- (2).Pardoll DM The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat. Rev. Cancer 2012, 12 (4), 252–264. 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Topalian SL; Drake CG; Pardoll DM Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell 2015, 27 (4), 450–461. 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Fares CM; Van Allen EM; Drake CG; Allison JP; Hu-Lieskovan S Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. B 2019, No. 39, 147–164. 10.1200/edbk_240837. [DOI] [PubMed] [Google Scholar]
- (5).Larkin J; Chiarion-Sileni V; Gonzalez R; Grob JJ; Cowey CL; Lao CD; Schadendorf D; Dummer R; Smylie M; Rutkowski P; Ferrucci PF; Hill A; Wagstaff J; Carlino MS; Haanen JB; Maio M; Marquez-Rodas I; McArthur GA; Ascierto PA; Long GV; Callahan MK; Postow MA; Grossmann K; Sznol M; Dreno B; Bastholt L; Yang A; Rollin LM; Horak C; Hodi FS; Wolchok JD Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med 2015, 373 (1), 23–34. 10.1056/nejmoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Chen ZG Small-Molecule Delivery by Nanoparticles for Anticancer Therapy. Trends Mol. Med 2010, 16 (12), 594–602. 10.1016/j.molmed.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Fransen MF; Schoonderwoerd M; Knopf P; Camps MG; Hawinkels LJ; Kneilling M; van Hall T; Ossendorp F Tumor-Draining Lymph Nodes Are Pivotal in PD-1/PD-L1 Checkpoint Therapy. JCI insight 2018, 3 (23). 10.1172/jci.insight.124507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Schudel A; Francis DM; Thomas SN Material Design for Lymph Node Drug Delivery. Nature Reviews Materials Nature Publishing Group June 1, 2019, pp 415–428. 10.1038/s41578-019-0110-7. [DOI] [PMC free article] [PubMed]
- (9).Blanco E; Shen H; Ferrari M Principles of Nanoparticle Design for Overcoming Biological Barriers to Drug Delivery. Nat. Biotechnol 2015, 33 (9), 941–951. 10.1038/nbt.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Jain RK; Stylianopoulos T Delivering Nanomedicine to Solid Tumors. Nat. Rev. Clin. Oncol 2010, 7 (11), 653–664. 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ducry L; Stump B Antibody-Drug Conjugates: Linking Cytotoxic Payloads to Monoclonal Antibodies. Bioconjug. Chem 2010, 21 (1), 5–13. 10.1021/bc9002019. [DOI] [PubMed] [Google Scholar]
- (12).Carter T; Mulholland P; Chester K Antibody-Targeted Nanoparticles for Cancer Treatment. Immunotherapy 2016, 8 (8), 941–958. 10.2217/imt.16.11. [DOI] [PubMed] [Google Scholar]
- (13).Zhu L; Torchilin VP Stimulus-Responsive Nanopreparations for Tumor Targeting. Integr. Biol. (United Kingdom) 2013, 5 (1), 96–107. 10.1039/c2ib20135f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zheng Y; Stephan MT; Gai SA; Abraham W; Shearer A; Irvine DJ In Vivo Targeting of Adoptively Transferred T-Cells with Antibody- and Cytokine-Conjugated Liposomes. J. Control. Release 2013, 172 (2), 426–435. 10.1016/j.jconrel.2013.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Schmid D; Park CG; Hartl CA; Subedi N; Cartwright AN; Puerto RB; Zheng Y; Maiarana J; Freeman GJ; Wucherpfennig KW; Irvine DJ; Goldberg MS T Cell-Targeting Nanoparticles Focus Delivery of Immunotherapy to Improve Antitumor Immunity 10.1038/s41467-017-01830-8. [DOI] [PMC free article] [PubMed]
- (16).O’Melia MJ; Rohner NA; Manspeaker MP; Francis DM; Kissick HT; Thomas SN Quality of CD8+ T Cell Immunity Evoked in Lymph Nodes Is Compartmentalized by Route of Antigen Transport and Functional in Tumor Context. Sci. Adv 2020, 6 (50), 7134–7145. 10.1126/sciadv.abd7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Fransen MF; Van Der Sluis TC; Ossendorp F; Arens R; Melief CJM Controlled Local Delivery of CTLA-4 Blocking Antibody Induces CD8 + T-Cell-Dependent Tumor Eradication and Decreases Risk of Toxic Side Effects. Clin. Cancer Res 2013, 19 (19), 5381–5389. 10.1158/1078-0432.CCR-12-0781. [DOI] [PubMed] [Google Scholar]
- (18).Francis DM; Manspeaker MP; Schudel A; Sestito LF; O’Melia M; Kissick H; Pollack BP; Waller EK; Thomas SN Blockade of Immune Checkpoints in Lymph Nodes through Locoregional Delivery Augments Cancer Immunotherapy. Sci. Transl. Med 2020. [DOI] [PMC free article] [PubMed]
- (19).Iwai T; Sugimoto M; Patil NS; Bower D; Suzuki M; Kato C; Yorozu K; Kurasawa M; Shames DS; Kondoh O Both T Cell Priming in Lymph Node and CXCR3-Dependent Migration Are the Key Events for Predicting the Response of Atezolizumab. Sci. Rep 2021, 11 (1), 1–15. 10.1038/s41598-021-93113-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).van der Vlies AJ; O’Neil Conlin P.; Hasegawa Urara; Hammond N; Hubbell JA Synthesis of Pyridyl Disulfide-Functionalized Nanoparticles for Conjugating Thiol - Containing Small Molecules, Peptides and Proteins.Pdf. Bioconjug. Chem 2010, 21, 653–662. [DOI] [PubMed] [Google Scholar]
- (21).Thomas Susan N. a, b, Efthymia Vokali a, Lund Amanda W. a, b, Hubbell Jeffrey A. a, c, *; Swartz MA. Biomaterials Targeting the Tumor-Draining Lymph Node with Adjuvanted Nanoparticles Reshapes the Anti-Tumor Immune Response. Biomaterials 2014, 35 (2), 814–824. 10.1016/j.biomaterials.2013.10.003. [DOI] [PubMed] [Google Scholar]
- (22).Schudel A; Chapman AP; Yau M-K; Higginson CJ; Francis DM; Manspeaker MP; Avecilla ARC; Rohner NA; Finn MG; Thomas SN Programmable Multistage Drug Delivery to Lymph Nodes. Nat. Nanotechnol 2020, 15 (6), 491–499. 10.1038/s41565-020-0679-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Thomas SN; Vokali E; Lund AW; Hubbell JA; Swartz MA Targeting the Tumor-Draining Lymph Node with Adjuvanted Nanoparticles Reshapes the Anti-Tumor Immune Response. Biomaterials 2014, 35 (2), 814–824. 10.1016/j.biomaterials.2013.10.003. [DOI] [PubMed] [Google Scholar]
- (24).Kourtis IC; Hirosue S; de Titta A; Kontos S; Stegmann T; Hubbell JA; Swartz MA Peripherally Administered Nanoparticles Target Monocytic Myeloid Cells, Secondary Lymphoid Organs and Tumors in Mice. PLoS One 2013, 8 (4). 10.1371/journal.pone.0061646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Massagué J TGFβ in Cancer. Cell 2008, 134 (2), 215–230. 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Connolly EC; Freimuth J; Akhurst RJ Complexities of TGF-β Targeted Cancer Therapy. Int. J. Biol. Sci 2012, 8 (7). 10.7150/ijbs.4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Tauriello DVF; Palomo-Ponce S; Stork D; Berenguer-Llergo A; Badia-Ramentol J; Iglesias M; Sevillano M; Ibiza S; Cañellas A; Hernando-Momblona X; Byrom D; Matarin JA; Calon A; Rivas EI; Nebreda AR; Riera A; Attolini CSO; Batlle E TGFβ Drives Immune Evasion in Genetically Reconstituted Colon Cancer Metastasis. Nature 2018, 554 (7693), 538–543. 10.1038/nature25492. [DOI] [PubMed] [Google Scholar]
- (28).Mariathasan S; Turley SJ; Nickles D; Castiglioni A; Yuen K; Wang Y; Kadel EE; Koeppen H; Astarita JL; Cubas R; Jhunjhunwala S; Banchereau R; Yang Y; Guan Y; Chalouni C; Ziai J; Şenbabaoǧlu Y; Santoro S; Sheinson D; Hung J; Giltnane JM; Pierce AA; Mesh K; Lianoglou S; Riegler J; Carano RAD; Eriksson P; Höglund M; Somarriba L; Halligan DL; Van Der Heijden MS; Loriot Y; Rosenberg JE; Fong L; Mellman I; Chen DS; Green M; Derleth C; Fine GD; Hegde PS; Bourgon R; Powles T TGFβ Attenuates Tumour Response to PD-L1 Blockade by Contributing to Exclusion of T Cells. Nature 2018, 554 (7693), 544–548. 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Pulaski BA; Ostrand‐ Rosenberg S Mouse 4T1 Breast Tumor Model. Curr. Protoc. Immunol 2000, 39 (1), 20.2.1–20.2.16. 10.1002/0471142735.im2002s39. [DOI] [PubMed] [Google Scholar]
- (30).Jana S; Muscarella RA; Jones D The Multifaceted Effects of Breast Cancer on Tumor-Draining Lymph Nodes. Am. J. Pathol 2021, 191 (8), 1353–1363. 10.1016/J.AJPATH.2021.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Loi S; Pommey S; Haibe-Kains B; Beavis PA; Darcy PK; Smyth MJ; Stagg J CD73 Promotes Anthracycline Resistance and Poor Prognosis in Triple Negative Breast Cancer. Proc. Natl. Acad. Sci. U. S. A 2013, 110 (27), 11091–11096. 10.1073/pnas.1222251110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Sixt M; Kanazawa N; Selg M; Samson T; Roos G; Reinhardt DP; Pabst R; Lutz MB; Sorokin L The Conduit System Transports Soluble Antigens from the Afferent Lymph to Resident Dendritic Cells in the T Cell Area of the Lymph Node. Immunity 2005, 22 (1), 19–29. 10.1016/J.IMMUNI.2004.11.013. [DOI] [PubMed] [Google Scholar]
- (33).Vijayan D; Young A; Teng MWL; Smyth MJ Targeting Immunosuppressive Adenosine in Cancer. Nat. Rev. Cancer 2017, 17 (12), 709–724. 10.1038/nrc.2017.86. [DOI] [PubMed] [Google Scholar]
- (34).Ohta A A Metabolic Immune Checkpoint: Adenosine in Tumor Microenvironment. Front. Immunol 2016, 7 (MAR), 109. 10.3389/fimmu.2016.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Ohta A; Gorelik E; Prasad SJ; Ronchese F; Lukashev D; Wong MKK; Huang X; Caldwell S; Liu K; Smith P; Chen JF; Jackson EK; Apasov S; Abrams S; Sitkovsky M A2A Adenosine Receptor Protects Tumors from Antitumor T Cells. Proc. Natl. Acad. Sci. U. S. A 2006, 103 (35), 13132–13137. 10.1073/pnas.0605251103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Ohta A; Ohta A; Madasu M; Kini R; Subramanian M; Goel N; Sitkovsky M A2A Adenosine Receptor May Allow Expansion of T Cells Lacking Effector Functions in Extracellular Adenosine-Rich Microenvironments. J. Immunol 2009, 183 (9), 5487–5493. 10.4049/jimmunol.0901247. [DOI] [PubMed] [Google Scholar]
- (37).Leone RD; Lo YC; Powell JD A2aR Antagonists: Next Generation Checkpoint Blockade for Cancer Immunotherapy. Comput. Struct. Biotechnol. J 2015, 13, 265–272. 10.1016/j.csbj.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Leone RD; Emens LA Targeting Adenosine for Cancer Immunotherapy. J. Immunother. Cancer 2018, 6 (1), 57. 10.1186/s40425-018-0360-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Allard B; Pommey S; Smyth MJ; Stagg J Targeting CD73 Enhances the Antitumor Activity of Anti-PD-1 and Anti-CTLA-4 MAbs. Clin. Cancer Res 2013, 19 (20), 5626–5635. 10.1158/1078-0432.CCR-13-0545. [DOI] [PubMed] [Google Scholar]
- (40).Beavis PA; Milenkovski N; Henderson MA; John LB; Allard B; Loi S; Kershaw MH; Stagg J; Darcy PK Adenosine Receptor 2A Blockade Increases the Efficacy of Anti-PD-1 through Enhanced Antitumor T-Cell Responses. Cancer Immunol. Res 2015, 3 (5), 506–517. 10.1158/2326-6066.CIR-14-0211. [DOI] [PubMed] [Google Scholar]
- (41).Kalbasi A; Ribas A Tumour-Intrinsic Resistance to Immune Checkpoint Blockade. Nat. Rev. Immunol 2020, 20 (1), 25–39. 10.1038/s41577-019-0218-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Xia A; Zhang Y; Xu J; Yin T; Lu XJ T Cell Dysfunction in Cancer Immunity and Immunotherapy. Front. Immunol 2019, 10, 1719. 10.3389/fimmu.2019.01719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Guerrouahen BS; Maccalli C; Cugno C; Rutella S; Akporiaye ET Reverting Immune Suppression to Enhance Cancer Immunotherapy. Front. Oncol 2020, 9 (January). 10.3389/fonc.2019.01554. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.