

Abstract
Aging is a complex physiological process that leads to the progressive decline of metabolic and immune function, among other biological mechanisms. As global life expectancy increases, it is important to understand determinants of healthy aging—including environmental and genetic factors—and thus slow the onset or progression of age-related disease. Environmental enrichment (EE) is a housing environment wherein laboratory animals engage with complex physical and social stimulation. EE is a prime model to understand environmental influences on aging dynamics, as it confers an anti-obesity and anti-cancer phenotype that has been implicated in healthy aging and health span extension. While EE is frequently used to study malignancies in young mice, fewer studies characterize EE-cancer outcomes in older mice. Here, we used young (3-month-old) and aged (14-month-old) female C57BL/6 mice to determine whether EE would be able to mitigate age-related deficiencies in metabolic function and thus alter Lewis lung carcinoma (LLC) growth. Overall, EE improved metabolic function, resulting in reduced fat mass, increased lean mass, and improved glycemic processing; many of these effects were stronger in the aged cohort than in the young cohort, indicating an age-driven effect on metabolic responses. In the aged-EE cohort, subcutaneously-implanted LLC tumor growth was inhibited and tumors exhibited alterations in various markers of apoptosis, proliferation, angiogenesis, inflammation, and malignancy. These results validate EE as an anti-cancer model in aged mice and underscore the importance of understanding environmental influences on cancer malignancy in aged populations.
Prevention Relevance:
Environmental enrichment serves as a model of complex physical and social stimulation. This study validates environmental enrichment as an anti-cancer intervention paradigm in aged mice and underscores the importance of understanding environmental influences on cancer malignancy in aged populations.
Keywords: environmental enrichment, aging, cancer, metabolism, Lewis lung carcinoma
INTRODUCTION
Aging is accompanied by various biological changes which influence the balance between health and disease states [1]. As such, aging can be considered a risk factor for cancer, ischemic stroke, neurodegeneration, cardiovascular disease, diabetes, and obesity, among others [2]. Recent improvements in healthcare, water and food access, immunization, hygiene, and housing have increased life expectancy, consequently resulting in an increasingly aged population [3]. While researchers and clinicians previously focused on improving life span, much of the current work focuses on improving health span. The former is defined by life expectancy, while health span is defined by the length of time one is able to live a healthy life [4]. Increases in life span are worrisome if not accompanied by increases in health span; in absence of good health, global life span increases will result in a large burden for healthcare providers. As such, there is a pressing need to understand the processes through which health span can be increased and a compression of morbidity might occur [3, 5].
Aging is influenced by genetic, behavioral, and environmental factors. Accordingly, researchers have utilized a variety of laboratory modeling systems to understand how such influences can induce biological processes implicit in healthy aging. Environmental enrichment (EE) is one such model that can be used to study the interaction between lifestyle factors and aging. EE combines complex stimuli to provide enhanced social, somatosensory, cognitive, and motor stimulation to laboratory animals [6, 7]. In EE, laboratory animals encounter varied stimuli, including toys, running wheels, increased bedding, and social encounters. Our work with EE has revealed a novel anti-cancer and anti-obesity phenotype that is mediated by a brain-fat axis, dubbed the hypothalamic-sympathoneural-adipocyte (HSA) axis [8, 9]. EE induces upregulation of hypothalamic brain-derived neurotrophic factor (BDNF), resulting in elevated sympathetic tone to adipose tissue, wherein tissue remodeling—characterized by adipose browning/beiging and suppression of leptin—occurs [8]. The drop of circulating level of leptin plays a critical role in the anticancer effects of EE in melanoma and breast cancer models [9, 10]. Physical activity, although an important component of EE, does not account for the entirety of EE-associated benefits on metabolism, cancer, and aging [8, 9, 11, 12]. Various researchers have applied EE to multiple tumor models, including melanoma, glioma, intestinal, breast, and pancreatic cancer models, revealing additional anti-cancer mechanisms beyond the HSA axis [9, 10, 13-16].
Recent work by our lab suggests that EE promotes healthy aging and extension of health span. EE-induced healthy aging is characterized by reduced adiposity, improved glycemic control, improved motor abilities, reduced anxiety-like behavior, and reduced hepatosteatosis [12]. The anti-aging effects of EE were largely recapitulated following hypothalamic administration of an autoregulatory AAV-BDNF vector, implicating BDNF as one mechanistic mediator of the EE-aging phenotype [17]. Further work shows that EE alters microglial morphology in aged animals and reduces age-associated neuroinflammation [12, 18-20], the latter of which has been shown to be positively related to systemic metabolic function [21-23].
Given the interplay between aging, metabolism, and cancer, we began to consider how EE might influence cancer development in aged models. While the metabolic and immune benefits of EE have been well characterized in young mice, there exists a paucity of data investigating its use to mitigate cancer in aged models. Previous work has displayed the importance of host age on Lewis lung carcinoma (LLC) tumor progression, suggesting a link between cancer progression and age-related dysregulation in angiogenesis, apoptosis, and metabolism [24]. Since age has been described as an “organizing axis” for the biological mediators of LLC progression [24], we considered it to be a cancer model worthy of investigation in the aging-EE space. Here, we investigated LLC progression in an aging-EE model, hypothesizing that EE would mitigate age-related declines in metabolism, thus altering LLC tumor growth.
MATERIALS AND METHODS
To investigate whether EE could mitigate age-related declines in metabolism and thus alter LLC tumor growth, young adult (3-month-old) and middle-aged (14-month-old) female C57BL/6 mice were randomized to live in standard environment (SE) or EE (Fig. 1A). Body weight and food intake were monitored weekly. Beginning at 4 weeks post housing, mice were subject to metabolic assessments to assess metabolic function prior to tumor inoculation. At 8 weeks post housing, mice were subcutaneously injected with LLC cells and tumor progression was monitored (Fig. 1B).
Figure 1. EE ameliorates aged-related metabolic deficits.

(A) SE and EE housing examples. (B) Experimental timeline. (C) Body weight (D) Daily food intake, normalized to body weight. (E) Percent fat mass, as measured echoMRI body composition assessment at 4 weeks post housing. (F) Percent lean mass, as measured echoMRI body composition assessment at 4 weeks post housing. (G) Glucose tolerance test (GTT), performed 5 weeks post housing. (H) Area under the curve of the GTT. For all panels: Young SE n=10, Young EE n=9, Aged SE n=10, Aged EE n=9. Data are means ± SEM. SE vs. EE: * P < 0.05, *** P < 0.001. Aged (EE-SE) vs Young (EE –SE): # P < 0.05, ## P < 0.01.
EE housing protocol [7]
3-month-old (Charles River Laboratories) and 14-month-old (National Institute on Aging, Aged Rodent Colonies) female C57BL/6 mice were randomized to live in SE or EE for 11 weeks. SE mice were group housed (5 mice) in standard laboratory environment cages (30.5 cm x 17 cm x 15 cm). EE mice were group housed (10 mice) in large cages (120 cm x 90 cm x 76 cm) supplemented with running wheels, huts, shelters, toys, tunnels, a maze, and nesting material. All mice had ad libitium access to food (normal chow diet, 11% fat, caloric density 3.4 kcal/g, Teklad) and water. Mice were housed in temperature (22-23°C) and humidity (30-70%) controlled rooms under a 12:12 light:dark cycle. All animal experiments were in accordance with the regulations of The Ohio State University’s Institutional Animal Care and Use Committee (IACUC) and were performed at The Ohio State University.
Body composition by echoMRI
At 4 weeks post housing, echoMRI was utilized to measure body composition of fat, lean, free water, and total water masses in live mice without anesthesia. Body composition analysis was performed with an echoMRI 3-in-1 Analyzer at the Small Animal Imaging Core of The Dorothy M. Davis Heart & Lung Research Institute, The Ohio State University.
Glucose tolerance test (GTT)
At 5 weeks post housing, mice were injected intraperitoneally with glucose solution (2.0 g glucose per kg body weight) after a 16 h overnight fast. Blood was obtained from the tail at 0, 15, 30, 60, 90, and 120 min after glucose injection. Blood glucose concentrations were measured with a portable glucometer (Bayer Contour Next).
LLC culture, inoculation, and measurement
The LLC line was obtained from ATCC (#CRL-1642) and was additionally verified by IDEXX BioAnalytics using short tandem repeat profiling. LLC cells were cultured with RPMI, 10% fetal bovine serum, and 1% glutamine, and 1% penicillin streptomycin. No mycoplasma testing was performed. After four passages, cells were isolated and counted during the logarithmic growth phase. To allow for measurement by calipers, 2.5 x 105 LLC cells were implanted in mouse subcutaneous tissue with 100 μL of serum-free, antibiotic-free RPMI. Tumors were measured every 2-3 days to obtain length and width measurements. Estimates of volume were calculated using the formula V = L * W2 * (π/6). When the largest tumors approached the IACUC-determined size removal criteria, all mice were euthanized to provide direct comparisons of biomarkers, unaffected by asynchronous endpoints.
Tissue collection
Following 11 weeks of housing, mice were sacrificed. Tissues to be used for mRNA and protein analysis were flash frozen on dry ice and stored at −80°C until further analysis. Tissues to be used for histology were fixed in 4% paraformaldehyde for 48h, then dehydrated in 70% ethanol prior to paraffin sectioning and staining.
Serum harvest and analysis
Trunk blood was collected at euthanasia and allowed to clot on ice for at least 30 min before centrifugation at 10,000 rpm for 10 min at 4°C. The serum component was collected and stored at −20°C until further analysis. DuoSet ELISA kits were used to assay serum leptin (R&D Systems #DY498), adiponectin (R&D Systems #DY1119), and insulin-like growth factor (IGF-1) (R&D Systems #DY791). Triglyceride (Cayman Chemical #10010303) and glucose levels (Cayman Chemical #10009582) were measured using colorimetric assay kits.
Histology
Standard procedures were used for immunohistochemistry staining. Paraformaldehyde-fixed, paraffin-embedded tumor samples was sectioned at 3μm and deparaffinized followed by blocking of endogenous peroxidase activity with H2O2 before epitope retrieval. Anti–Ki-67 (clone ab 16667, abcam, 1:200) was used as a primary antibody. Dako REAL EnVision Detection System kits (K5007, DaKo) were used as secondary antibodies. Additional slides underwent hematoxylin and eosin (H&E) staining.
Quantitative analysis of Ki-67
Immunocytochemistry was used to detect Ki-67 expression in tumor cells to represent the cell proliferation index. The slides were scanned by 3D-HISTECH (Pannoramic MIDI, 3DHISTECH Ltd., Hungary) and the positive cells were evaluated by Cognition Master Professional Suite (VMscope GmbH Berlin, Germany). The percentage of Ki-67 expression = Positive tumor cells / Total tumor cells.
TUNEL assay
A terminal deoxynucleotidyl transferase dUTP Nick-End Labeling (TUNEL) assay was performed to detect cell apoptosis. Tissue sections were stained with the In Situ Cell Death Detection Kit (Roche, Basel, Switzerland) according to the manufacturer’s instructions.
Quantitative RT-PCR
Hypothalamus was collected under dissection scope at sacrifice. Tissues were flash frozen on dry ice and stored at −80°C until further analysis. Following sonication, RNA was isolated using the QIAGEN RNeasy Mini kit with RNase-free DNase treatment. cDNA was reverse transcribed using Taqman Reverse Transcription Reagents (Applied Biosystems). qRT-PCR was completed on StepOnePlus Real-Time PCR System using Power SYBR Green (Applied Biosystems) PCR Master Mix. Primers are available upon request. We calibrated data to endogenous controls— Actinb for adipose tissue and hypothalamus, Gapdh for tumor, and quantified the relative gene expression using the 2 −ΔΔCT method.
Western blotting
Tissue samples were homogenized in ice-cold RIPA buffer (Pierce #89901) containing 1x Phosstop (Roche #4906845001) and protease inhibitor cocktail III (Calbiochem #539134). Tissue lysates were separated by gradient gel (4-20%, Mini-PROTEAN TGX, Bio-Rad #4561096), transferred to a nitrocellulose membrane (Bio-Rad #1620115). Blots were incubated overnight at 4 °C with the following primary antibodies: Vinculin (Cell Signaling Technology #13901, 1:1000), COX2 (Cell Signaling Technology #D5H5, 1:1000), CD31 (Cell Signaling Technology #D8V9E, 1:1000), CXCR4 (Novus Biologicals #NB100-56437, 1:500). Blots were rinsed and incubated with HRP-conjugated secondary antibodies (Bio-Rad). Chemiluminescence signal was detected and visualized by LI-COR Odyssey Fc imaging system (LI-COR Biotechnology, Lincoln, NE). Quantification analysis was carried out with Image Studio software version 5.2 (LI-COR Biotechnology).
Statistical analysis
Data analyses were performed by using statistics software SAS 9.4 (SAS, Inc; Cary, NC). Longitudinal measures—including body weights and tumor volumes—were analyzed by using mixed effect models, incorporating repeated measures for each mouse/tumor. Other data with single time measurements were analyzed by using analysis of variance (ANOVA), with the exception of the Ki-67 proliferative index, which was analyzed using the Student’s t-test. Multiplicities were adjusted using Holm’s procedure. Differences with adjusted p-values of less than 0.05 were defined as significant. All data were analyzed untransformed since they were normally distributed based on residual plots of statistical models, with the exception of qPCR data, which were log2 transformed to ensure normality. Age-related EE effects were tested across 4 groups (EE-aged, SE-aged, EE-young, SE-young) in the model. Two mice were erroneously injected intraperitoneally and were excluded from analyses.
RESULTS
EE ameliorates age-related metabolic deficits.
To investigate whether EE would be able to mitigate age-related declines in metabolism and thus alter LLC tumor growth, young (3-month old) and aged (14-month old) female mice were housed in either standard environment (SE) or EE housing (Fig. 1A) for 11 weeks. Throughout the housing period, mice were subject to various metabolic measures (Fig. 1B). Consistent with previous data [12], aged EE mice showed a marked reduction in weight when compared to aged SE controls (P < 0.001, slope comparison) (Fig. 1C). In contrast, young mice showed no significant differences in weight change (Fig. 1C) when compared to young SE controls (P = 0.348). EE mice consumed more food than SE controls in both the young (P < 0.001) and aged (P < 0.001) groups (Fig. 1D), suggesting EE-induced weight loss due to elevated energy expenditure rather than food intake suppression. The effect on increase in food intake (difference EE - SE) was comparable in young and aged mice (P = 0.162). At 4 weeks post housing, an in vivo echoMRI revealed EE reduced relative body fat in both young (P < 0.001) and aged mice (P < 0.001) (Fig. 1E); the effect on the reduction in body fat (difference EE - SE) was more dramatic in aged mice (Aged (EE - SE) vs Young (EE - SE), P = 0.048). Results were similar for relative lean mass percentage; EE increased relative lean mass in both young (P = 0.022) and aged mice (P < 0.001) (Fig. 1F), with a more pronounced effect in aged mice (P = 0.002). At 5 weeks post housing, mice were subject to an intraperitoneal glucose tolerance test (GTT) to assess systemic glycemic processing (Fig. 1G and 1H). Young EE mice displayed no improvement in glycemic processing over SE controls. In contrast, EE induced a marked improvement in glycemic processing in aged mice (P < 0.001). In summary, EE had a significant effect in aged mice for fat mass percentage, lean mass percentage, and glycemic processing, consistent with our previous report [12]; many of these effects were observed to be stronger in the aged cohort than in the young cohort, indicating an age-driven effect on EE metabolic response.
EE slows LLC tumor growth and reduces tumor mass in aged mice.
At 8 weeks post housing, mice were subcutaneously injected with 2.5 x 105 LLC cells and tumor progression was monitored with caliper measurements. We observed no difference in tumor volume progression in young mice (Fig. 2A) (P = 0.377, slope comparison). In contrast, EE slowed tumor progression in aged mice, as compared to aged SE controls (P = 0.0006, slope comparison) (Fig 2A). At sacrifice, EE did not alter final tumor volume (P = 0.966) and weight (P = 0.738) in young mice (Fig. 2B and 2C). In contrast, EE reduced final tumor volume (P < 0.001) and weight (P < 0.001) in aged mice (Fig. 2B and 2C). At the gross level, aged SE tumors appeared larger (Fig. 2D), while the aged EE tumors appeared more like the young mouse SE and EE tumors. Together, these data suggested that EE induced an anti-tumor response in aged mice.
Figure 2. EE slows LLC tumor growth and reduces tumor mass in aged mice.

(A) LLC tumor growth curve, as measured by calipers. (B) LLC tumor volume at sacrifice. (C) LLC tumor weight at sacrifice. (D) Excised LLC tumors, scale equalized. (E) Gross tissue weight. For all panels: Young SE n=10, Young EE n=9, Aged SE n=10, Aged EE n=9. Data are means ± SEM. SE vs. EE: * P < 0.05, *** P < P<0.001. Aged (EE-SE) vs Young (EE –SE): # P < 0.05.
At sacrifice, we collected tumor-naïve inguinal white adipose tissue (iWAT), gonadal white adipose tissue (gWAT), and liver (Fig. 2E). EE reduced tumor-naïve iWAT (n-iWAT) in aged mice (P < 0.001), while no significant differences were observed in young mice (P = 0.106). EE reduced gWAT weight in both young (P = 0.016) and aged (P < 0.001) groups; the effect was more pronounced in aged mice (Aged (EE - SE) vs Young (EE - SE), P = 0.040). No significant differences in liver weight were observed.
Leptin is an adipokine that serves as a central-peripheral messenger to maintain energy homeostasis. Leptin production is positively correlated with adipose tissue mass and additionally has been described as an proinflammatory link between immune and neuroendocrine systems [25]. Consistent with previous reports [12] and present measures of adiposity (Fig. 1E and 2E), EE reduced serum leptin in young (P = 0.018) and aged (P < 0.001) mice (Fig 3A); the EE effect was more distinct in aged mice (Aged (EE - SE) vs Young (EE - SE), P = 0.024). Adiponectin is an adipokine that has been shown to influence insulin sensitivity, glucose homeostasis, and systemic metabolism [26]. Inconsistent with previous reports in juvenile male mice [9], EE reduced serum adiponectin in young mice (P = 0.028), whereas no change was observed in the aged cohort (P = 0.322) (Fig 3B). In young and aged mice, EE increased serum IGF-1 (P = 0.044 for both) (Fig. 3C). Furthermore, EE induced a mild decrease in serum triglycerides in aged (P = 0.052), but not young mice (Fig. 3D). EE had no effect on non-fasting serum glucose levels in young and aged cohorts (Fig. 3E).
Figure 3. Serum profile at sacrifice.
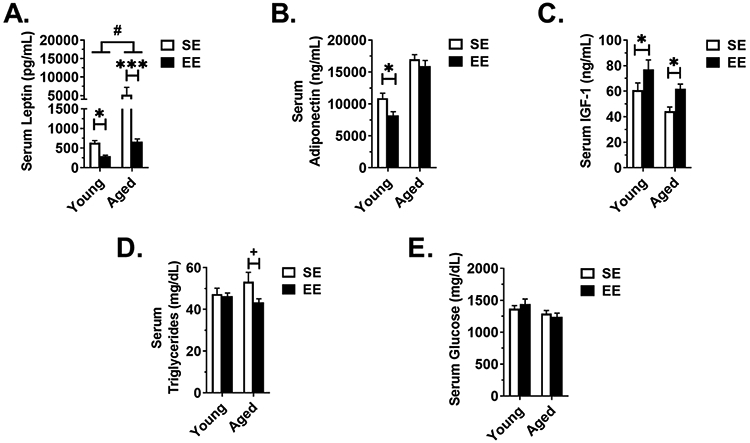
(A) Leptin (Young SE n=9, Young EE n=9, Aged SE n=10, Aged EE n=9). (B) Adiponectin. (C) IGF-1. (D) Triglycerides. (E) Glucose. For all panels except (A): Young SE n=10, Young EE n=9, Aged SE n=10, Aged EE n=9. Data are means ± SEM. SE vs. EE: + P < 0.06, * P < 0.05, *** P < 0.001. Aged (EE-SE) vs Young (EE –SE): # P < 0.05.
EE alters metabolic and inflammatory biomarkers in central and peripheral tissues.
As in previous EE studies, we sought to determine whether the HSA axis could be implicated in these metabolic phenotypes [8, 9]. Increased age has been associated with reduced central and peripheral BDNF levels [27-29]; such changes are thought to be tied with various aging-related maladies including hippocampal dysfunction [30], memory impairment [30] and metabolic dysfunction [17]. EE induced an upregulation of hypothalamic Bdnf mRNA in aged (P < 0.001) but not young mice (Fig. 4A) (P = 0.145). Similar results were observed for expression of Mc4r (encoding melanocortin 4 receptor), which is located upstream of BDNF in the leptin-proopiomelanocortin pathway [31]. EE induced an upregulation of Mc4r in aged mice (P = 0.008), while no change was observed in young mice (P = 0.194) (Fig. 4A).
Figure 4. EE alters metabolic and inflammatory biomarkers in central and peripheral tissues.

(A) Gene expression profile of hypothalamic tissue (n=6 per group). (B) Gene expression profile of iWAT (n=6 per group). (C) Gene expression profile of LLC tumors (Young SE n=9, Young EE n=8, Aged SE n=9, Aged EE n=9). (D) C-X-C motif chemokine ligand and receptor gene expression in LLC tumors (Young SE n=9, Young EE n=8, Aged SE n=9, Aged EE n=9). (E) Inflammation gene expression profile of LLC tumors (Young SE n=9, Young EE n=8, Aged SE n=9, Aged EE n=9). Data are means ± SEM. SE vs. EE: + P < 0.06, * P < 0.05, ** P < 0.01, *** P<0.001.
Upregulation of hypothalamic Bdnf leads to increased sympathetic tone to adipose tissue, thereby inducing adipose remodeling [8, 9, 11]. As such, we profiled mRNA expression in the iWAT for markers indicative of this process. Consistent with both adipose weight and serum measurements, Lep (encoding leptin) was downregulated in iWAT of both aged (P = 0.006) and young mice (P = 0.008) (Fig. 4B). Pten (encoding phosphatase and tensin homolog) has been implicated in anti-obesity and anti-cancer states [32-34] and has been described as one downstream mediator of EE-induced adipose remodeling [35]. In the iWAT, Pten was upregulated in both young (P = 0.002) and aged (P < 0.001) mice (Fig. 4B). Furthermore, we profiled Adrb3 (encoding β3 adrenergic receptor), which is involved in adipose browning and lipolysis. Similar to previous reports [8, 12], both aged (P = 0.06) and young mice (P = 0.06) exhibited a trending increase of Adrb3 in EE (Fig. 4B). In summary, these data indicate EE upregulated hypothalamic Bdnf, thus activating the HSA axis to induce adipose remodeling. The extent of EE-induced fat remodeling was not observed to be affected by age (Aged (EE-SE) vs Young (EE –SE), P > 0.05 for Lep, Pten, and Adrb3).
In the LLC tumors, we profiled mRNA expression of several markers of apoptosis, angiogenesis, inflammation, and immune response. Cox2 (encoding cyclooxygenase-2) is expressed in many cancer types and promotes apoptotic resistance, proliferation, angiogenesis, inflammation, and invasion of cancer cells [36]. Here, we observed that EE reduced Cox2 in tumors of aged mice (P = 0.014) (Fig. 4C), consistent with the observed reduction in tumor growth rate and size (Fig. 2A-D). No EE-induced changes in Cox2 expression were observed in the young cohort (P = 0.934). Vegf (encoding vascular endothelial growth factor) is implicated in tumor angiogenesis [37, 38]. Aged EE tumor samples displayed a trend toward decreased Vegf (P = 0.056) when compared to aged SE controls (Fig. 4C). No significant difference in Vegf expression between housing conditions was observed in young tumor samples (P = 0.885) (Fig 4C). The expression of vascular marker Cd31 (encoding cluster of differentiation 31) appeared lower in EE for both young (P = 0.090) and aged (p = 0.090) cohorts, but did not reach statistical significance (Fig. 4C).
C-X-C motif chemokine ligands and their receptors are implicated in various cancer-relevant processes, including chemotaxis, apoptosis, hematopoiesis, angiogenesis, and inflammatory responses [39, 40]; accordingly, some are described as prognostic cancer biomarkers [41]. Thus, we profiled several of these ligand-receptor pairs, including Cxcl12 and Cxcr4, Cxcl10 and Cxcr3, and Cxcl1 and Cxcr2 (Fig. 4D). In aged mice, EE reduced Cxcr4 expression (P < 0.001) with no concurrent change in ligand Cxcl12. In contrast, EE had no effect on Cxcr4 or Cxcl12 expression in young mice. No EE-induced changes in Cxcl10 or Cxcr3 expression were observed in young or aged mice. Interestingly, we observed an EE-induced reduction in ligand Cxcl1 (P = 0.028) but not receptor Cxcr2 in aged mice. No EE-induced changes in Cxcl1 or Cxcr2 were observed in young mice. Cxcr1 expression remained unchanged following EE in both young and aged cohorts.
Various additional markers of inflammation, macrophages, apoptosis, and vascularity were profiled (Fig. 4E). We observed no EE- or age-driven differences in Bcl2, F4/80, Ikbkb (encoding inhibitor of nuclear factor kappa-B kinase subunit beta), Notch1 (encoding notch receptor 1), or Tgfb1 (encoding transforming growth factor beta 1). We additionally profiled protein expression in LLC tumor samples by immunoblotting (Fig. 5A). Consistent with qPCR data, we observed EE induced a downregulation of COX2 protein expression (Fig. 5B) in aged tumor samples (P = 0.036), but not in young mice (P = 0.3473). No significant EE- or age-driven differences were observed in CD31 (Fig. 5C) or CXCR4 (Fig. 5D) protein expression.
Figure 5. LLC tumor immunoblotting.
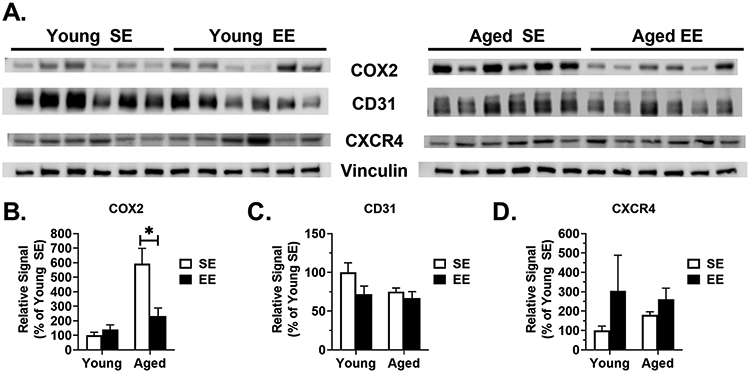
(A) LLC tumor immunoblots (n=6 per group). (B) COX2 protein expression in LLC tumors (n=6 per group). (C) CD31 protein expression in LLC tumors (n=6 per group). (D) CXCR4 protein expression in LLC tumors (n=6 per group). Data are means ± SEM. SE vs. EE: * P < 0.05.
EE alters tumor pathology and reduces proliferation in aged mice.
A pathologist examined tumor histology from the aged cohort and provided a qualitative report (Fig. 6). Architecturally, all tumors were characterized by a predominantly solid or syncytial infiltrative pattern with thin-walled vascular stroma. Nuclei presented as pleomorphic, with multiple nucleoli. In aged SE tumors, more cells were in mitosis (Fig 6A, yellow arrows) and some abnormal mitotic figures were present. Tumor necrosis (Fig. 6A, white arrows; Fig. 6B, black arrows) and apoptosis measured by TUNEL staining (Fig. C) was less obvious in the aged EE group. Multinucleated or mononuclear giant cells were common in SE tumor samples. Overall, the tumor cells appeared polygonal and moderate in size, with the cytoplasm being moderately more abundant in the EE group. More dilated vascular cavities were observed in the EE group. Altogether, these observations indicate that EE altered tumor pathology in aged mice. Aged SE tumors showed signs of fast growth and higher malignancy than aged EE tumors. Furthermore, aged EE tumors exhibited a lower proliferation index than aged SE tumors, as measured by quantitative measurement of Ki-67 staining (Fig. 6D and 6E, P = 0.039); these observations were consistent with tumor weight and volume observations.
Figure 6. Enriched environment alters tumor pathology and reduces proliferation in aged mice.

(A) Representative H&E staining of LLC tumors (400X, 50 μm scale bar, yellow arrows indicate abnormal mitotic figures, white arrows indicate necrosis and apoptosis). (B) Additional representative H&E staining of LLC tumors (200X, 100 μm scale bar, black arrows indicate necrotic foci). (C) Representative TUNEL staining for apoptosis in LLC tumors (400X, 50 μm scale bar). (D) Representative Ki-67 staining for proliferation in LLC tumors (400X, 50 μm scale bar). (E) Percentage of tumor cells positive for Ki-67 proliferation stain (n=7 animals per group, 3 images per animal). Data are means ± SEM. SE vs. EE: * P < 0.05.
DISCUSSION
Aging, metabolic function, and cancer risk are inextricably connected. With age, many individuals lose lean mass and experience increases in total adiposity. Fat becomes dysfunctional and is redistributed from subcutaneous and visceral adipose depots to ectopic sites, including liver, skeletal muscle, heart, and the pancreas [42]. Accordingly, age is considered a risk factor for the development of metabolic syndrome, obesity, insulin resistance, and related inflammation of adipose tissue [43]. Obesity-driven inflammatory states, dyslipidemia, insulin resistance, hyperglycemia, and adipokine aberrations can promote cancer initiation, proliferation, and favorable microenvironments [44, 45], so it is of upmost importance that we characterize multi-faceted therapeutics and interventions that can target all three of these actors. Perhaps unsurprisingly, researchers and clinicians have turned their focus toward understanding and implementing lifestyle interventions—like EE—that improve metabolic function and slow the development and progression of cancer across the life span.
In young mice, EE is capable of modifying tumor growth—and in some cases, lengthen survival [9, 10, 13-16]. The EE-induced slowing of cancer growth is thought to occur due in part to induction of hypothalamic BDNF, which improves metabolic health and systemic immune function [9, 15, 16]. Previous work by our lab described the ability of EE and hypothalamic BDNF to promote healthy aging in mice [12, 17]. Both short- and long-term exposure to EE in aged mice induced marked health benefits, including: reduced adiposity, improved glucose tolerance, decreased serum leptin, enhanced motor abilities, reduced anxiety-like behavior, reduced hepatic steatosis and hepatic glucose production, and increased glucose uptake by the liver—underscoring improvements in glycemic control [12]. Moreover, EE reduced inflammatory genes in the brain, adipose, and liver [12]. Interestingly, EE mice displayed a trend toward increased mean life span [12] and many of these observations were replicated following hypothalamic gene transfer of BDNF [17]. Together, these studies implicate a molecular hub—BDNF—which connects EE stimuli and downstream metabolic and immune phenotypes implicit in healthy aging, increased health span, and reduced cancer risk.
Here, we expanded upon previous work to determine whether EE would be able to mitigate age-related declines in metabolic function and thus alter LLC growth. To our knowledge, this is the first report to characterize an EE-related anti-cancer effect within aged mice. Consistent with our previous results [12], we observed an increase in hypothalamic Bdnf in aged EE mice and an overall favorable metabolic profile. Notably, aged mice responded more readily to EE than young counterparts across various metabolic metrics, perhaps indicative of aging-related baseline deficits in metabolic function. This notion is consistent with previous reports which indicate aging negatively influences insulin resistance [46, 47], glucose tolerance [48], body composition [49, 50], and adipose function and inflammation [51]. Tumor phenotypes were consistent with metabolic observations, given that age-related increases in adiposity serve as a risk factor for cancer. In aged EE mice, implanted LLC tumors grew slower and weighed less at sacrifice when compared to aged SE controls. Furthermore, changes in growth were associated with favorable pathological observations and molecular changes—indicative of alterations in apoptosis, proliferation, angiogenesis, inflammation, and malignancy. Overall, these data suggest that EE can attenuate age-related declines in metabolism, thus slowing LLC growth. In sum, these outcomes validate EE as an anti-cancer model in aged mice and underscore the importance of understanding environmental influences on cancer malignancy in aged populations.
Beheshti and colleagues report that host age has a considerable impact on the growth of implanted LLC, describing age as an “organizing axis” for the biological determinants of LLC progression. They employed male C57BL/6 mice across the life span (68-, 143-, 551-, and 736-days) and found that the growth rate of LLC was higher in young adult mice (143-days, 4.8-months) as compared to older mice (551-days, 18.4-months) [24]. In contrast, we observed a faster growth rate and larger tumor mass at sacrifice in older female mice (16-months at tumor implantation, aged-SE) as compared to observations in young mice (5-months old at tumor implantation, young-SE) (Fig. 2). Age discrepancies between the two studies might contribute to the inconsistency in findings, however, sex may be a more critical biological variable. It is thought that menopause occurs between 12 and 14 months of age in mice [52]. Menopause is associated with the risk of weight gain and developing central obesity, as well as metabolic dysfunctions including insulin resistance, glucose intolerance, and dyslipidemia [53]. The propensity to postmenopausal obesity and associated metabolic syndromes is caused clearly—if not entirely—by the natural decrease in estrogen levels [54]. Here, our data suggest postmenopausal female mice exhibit metabolic declines that are associated with accelerated LLC tumor growth, as compared to young adults. Implementing EE during menopause alleviated this metabolic decline and inhibited LLC progression. As postmenopausal obesity is associated with a ~50% higher risk of breast cancer [55], it will be particularly interesting to investigate whether EE can mitigate the increased risks of postmenopausal metabolic disturbance and breast cancer.
Our results here did not characterize an EE anti-tumor effect in young mice, contrary to previous results in various tumor models [9, 10, 15]. It is feasible that these results are inherent to the LLC model, as—to our knowledge—this is the first described combination of the LLC model and EE. Alternatively, these observations could be explained by the use of females in this experiment, as we have observed sexual dimorphism in response to EE interventions. In general, EE results in more pronounced metabolic effects in male mice than females—we have observed this across multiple strains (C57BL/6 and BTBR T+ Itpr3tf/J) and ages (adult and younger) [56]. The underlying mechanisms remain unclear. Of consideration, estrogens act on estrogen receptor α (ERα) to regulate the gene and protein expression of BDNF in the brain [57]. Hypothalamic BDNF has been identified as the upstream brain mediator driving EE-induced adipose remodeling and anti-cancer effects. Therefore, the female brain may be more sensitive to the signaling molecules critical to energy homeostasis—such as leptin and BDNF. As a result, females have a fitter baseline, at least before menopause, which might partially explain the more modest metabolic response to EE when compared to male mice. We did not use male mice in this experiment, as males are prone to fighting in EE, especially when introduced to novel social conspecifics later in life. Special labor- and cost-intensive study design considerations—such as long-term housing of EE males from a juvenile state—may limit fighting and associated confounding inflammatory states resulting from wounds and aggression. Further work will be needed to optimize male EE-aging housing protocols and understand whether sex-related differences in EE-induced tumor inhibition exist.
Interestingly, we observed an EE-associated increase in IGF-1 in both young and aged mice. This result was unexpected, as IGF-1 increases are implicated in tumorigenesis and are considered a risk factor for various cancers [58]. IGF-1 has pleotropic effects—including modulation of inflammation [59] and body composition [60]—and is known to decrease over the lifespan [58, 61]. Furthermore, IGF-1 is positively associated with lean mass [60]. The mechanisms underlying our IGF-1 observations remain unclear. Cancer-driven cachexia depends on IGF-1 axis impairment [62, 63] and IGF-1 administration was found to be protective from cachexia [63], so it is feasible that we observed a compensatory increase in IGF-1 before mice reached this state in our study. Alternatively, EE-associated increases in IGF-1 could be attributed to observed increases in lean mass. Interpretations within the context of three overlapping systems, aging, EE, and cancer—all known to influence IGF-1 [9, 59-63]—remain difficult. Accordingly, we remain unable to delineate whether circulating IGF-1 alters tumor progression in this study.
The current study has several limitations. In some cases, protein levels were found to be incongruent with relative mRNA expression. Secondly, this study lacks examination of immune responses. Previously, we and others demonstrated EE enhances cytotoxicity and mobilization of natural killer (NK) cells and CD8+ cytotoxic T-cells (CTLs) in young mice [9, 15, 16]. Recent reports describe the ability of EE to enhance NK cell maturation [64, 65] through hypothalamic BDNF [65]. Future studies should investigate whether EE can modify the body’s immune response, thus boosting antitumor immunity in advanced age. Furthermore, we did not investigate the glycolytic pathways which have been shown to regulate insulin sensitivity, glycemic processing, and cancer growth. Cancer cells reprogram their metabolism—catabolizing large amounts of glucose through glycolysis—to facilitate growth [66]. Multiple oncogenes—including RAS, MYC, and PI3K, favor glycolysis over oxidative phosphorylation, while many tumor suppressors—including PTEN—negate this effect [34]. Notably, EE induces an upregulation in PTEN within white adipose tissue [35]. Of relevance to the previous comments on estrogen, glycolysis has been shown to be altered by estrogen and its receptors [67, 68]. Thus, it will be important to delineate whether/how EE alters the glycolytic capacity of cancers and other metabolic-relevant tissues in future studies.
On another note, our experiment lacks comparisons of pre- and post-tumor metabolic and inflammatory biomarkers in serum. While we acknowledge this limitation, we performed many measures of systemic metabolic function prior to tumor inoculation. In tandem, we observed several EE metabolic signatures (e.g. Lep, Pten, Adrb3, Bdnf, and Mc4r mRNA, serum leptin) were maintained even after tumor inoculation. Together, these observations lend credence to the notion that the EE metabolic phenotype persisted through tumor treatment.
Finally, we must consider the experimental endpoint choice. Near the study sacrifice, the largest LLC tumor had a length which approached the maximum tumor diameter allowed by our IACUC’s policy. We decided to terminate the study to preserve power and minimize the number of mice lost to the early removal criteria. In doing so, we were able to analyze all tumors at the same experimental time point rather than one dictated by animal welfare. This allowed for (1) direct comparisons of biomarkers, unaffected by asynchronous endpoints and (2) observation of tumors in a midpoint state, rather than at a terminal state when they would be excessively necrotic and display high levels of intratumor heterogeneity, thus complicating IHC analysis. Other study designs (e.g. survival measurements without interference) may provide additional biological context and aid in interpretation.
It remains to be seen whether similar EE-aging results hold in other tumor models. Research in young mice has characterized anti-cancer EE phenotypes across melanoma, glioma, intestinal, breast, and pancreatic cancer models [9, 10, 13-16], providing several experimental targets for future EE-aging investigations. Additional work should be done to further characterize the EE anti-cancer phenotype in aged animals and describe mechanisms implicit in cancer immunity beyond the induction of hypothalamic BDNF.
It will be interesting to see whether similar results can be obtained with spontaneous tumors and/or hematopoietic malignancies in aged EE mice. Here, we did not perform a spontaneous tumor study in aged animals, as it would be cost- and labor- prohibitive, given the need to maintain EE cages and closely monitor growth over a period longer than the 19 days presented here. We previously investigated EE’s ability to extend life span in C57BL/6 mice – housing 80 female mice (40 SE and 40 EE) until natural death [12]. Necropsies revealed very few spontaneous tumors during this experiment and none of the observed tumors occurred in the lungs. While carcinogen administration would be a potential approach to avoid long-term housing of wild type or transgenic animals, we worry about variance in tumor latency and growth, which might necessitate large sample sizes for sufficient statistical power. Well-designed studies may overcome this limitation and have the potential to better characterize an aging-EE cancer prevention phenotype. In conclusion, this is not a strict cancer prevention study per se, as all mice developed tumors; spontaneous tumor models would address this concern. Nonetheless, the concept of cancer prevention is inextricably intertwined with maintenance of a healthy lifestyle and avoidance of negative environmental exposures—EE is a valuable paradigm to model these contributing factors.
While the observations reported here are promising, the question still remains: how do we translate these findings to humans? Obvious improvements in physical activity and diet can lead to lean states and extended health span, but our work with EE has led us to consider less-intuitive environmental factors and their contribution toward healthy aging. Although it may seem difficult to connect the running wheels, bedding, and toys of EE to human life, we should consider complex stimulation in our own lives. Individuals who seek meaning and purpose, have stimulating jobs or hobbies, exercise frequently, are engaged in meaningful social circles, etc. may engage similar biologically-rooted eustress pathways as those observed in EE. Such engagement may prove more important with advanced age. We encourage readers to reference our recent reviews on the promise of the EE model and lifestyle interventions for health span extension [5, 69] and cancer prevention [70], which discuss these topics at a greater depth. By engaging in a complex lifestyle, perhaps we can prevent or delay biological states that lead to cancer.
ACKNOWLEDGEMENTS
This work was supported by NIH grants AG041250, CA166590, and CA163640, as well as internal funding from The Ohio State University Comprehensive Cancer Center.
Footnotes
Conflict of Interest Disclosure: All authors declare no conflicts of interest.
REFERENCES
- 1.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, and Kroemer G, The hallmarks of aging. Cell, 2013. 153(6): p. 1194–217. PMC3836174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Niccoli T and Partridge L, Ageing as a risk factor for disease. Curr Biol, 2012. 22(17): p. R741–52. [DOI] [PubMed] [Google Scholar]
- 3.Partridge L, Deelen J, and Slagboom PE, Facing up to the global challenges of ageing. Nature, 2018. 561(7721): p. 45–56. [DOI] [PubMed] [Google Scholar]
- 4.Crimmins EM, Lifespan and Healthspan: Past, Present, and Promise. The Gerontologist, 2015. 55(6): p. 901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Queen NJ, Hassan QN, and Cao L, Improvements to Healthspan Through Environmental Enrichment and Lifestyle Interventions: Where Are We Now? Frontiers in Neuroscience, 2020. 14: p. 605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nithianantharajah J and Hannan AJ, Enriched environments, experience-dependent plasticity and disorders of the nervous system. Nat Rev Neurosci, 2006. 7(9): p. 697–709. [DOI] [PubMed] [Google Scholar]
- 7.Slater AM and Cao L, A Protocol for Housing Mice in an Enriched Environment. J Vis Exp, 2015(100): p. e52874. PMC4544994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao L, Choi EY, Liu X, Martin A, Wang C, Xu X, et al. , White to brown fat phenotypic switch induced by genetic and environmental activation of a hypothalamic-adipocyte axis. Cell Metab, 2011. 14(3): p. 324–38. PMC3172615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cao L, Liu X, Lin EJ, Wang C, Choi EY, Riban V, et al. , Environmental and genetic activation of a brain-adipocyte BDNF/leptin axis causes cancer remission and inhibition. Cell, 2010. 142(1): p. 52–64. PMC3784009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foglesong GD, Queen NJ, Huang W, Widstrom KJ, and Cao L, Enriched environment inhibits breast cancer progression in obese models with intact leptin signaling. Endocr Relat Cancer, 2019. 26(5): p. 483–495. PMC6717689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.During MJ, Liu X, Huang W, Magee D, Slater A, McMurphy T, et al. , Adipose VEGF Links the White-to-Brown Fat Switch With Environmental, Genetic, and Pharmacological Stimuli in Male Mice. Endocrinology, 2015. 156(6): p. 2059–73. PMC4430610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McMurphy T, Huang W, Queen NJ, Ali S, Widstrom KJ, Liu X, et al. , Implementation of environmental enrichment after middle age promotes healthy aging. Aging (Albany NY), 2018. 10(7): p. 1698–1721. PMC6075449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li G, Gan Y, Fan Y, Wu Y, Lin H, Song Y, et al. , Enriched environment inhibits mouse pancreatic cancer growth and down-regulates the expression of mitochondria-related genes in cancer cells. Sci Rep, 2015. 5: p. 7856. PMC4297951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bice BD, Stephens MR, Georges SJ, Venancio AR, Bermant PC, Warncke AV, et al. , Environmental Enrichment Induces Pericyte and IgA-Dependent Wound Repair and Lifespan Extension in a Colon Tumor Model. Cell Rep, 2017. 19(4): p. 760–773. PMC5474344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiao R, Bergin SM, Huang W, Slater AM, Liu X, Judd RT, et al. , Environmental and Genetic Activation of Hypothalamic BDNF Modulates T-cell Immunity to Exert an Anticancer Phenotype. Cancer Immunol Res, 2016. 4(6): p. 488–497. PMC4891265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garofalo S, D'Alessandro G, Chece G, Brau F, Maggi L, Rosa A, et al. , Enriched environment reduces glioma growth through immune and non-immune mechanisms in mice. Nat Commun, 2015. 6(1): p. 6623. PMC4389244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McMurphy T, Huang W, Liu X, Siu JJ, Queen NJ, Xiao R, et al. , Hypothalamic gene transfer of BDNF promotes healthy aging in mice. Aging Cell, 2019. 18(2): p. e12846. PMC6413658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ali S, Liu X, Queen NJ, Patel RS, Wilkins RK, Mo X, et al. , Long-term environmental enrichment affects microglial morphology in middle age mice. Aging (Albany NY), 2019. 11(8): p. 2388–2402. PMC6519992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu H, Gelyana E, Rajsombath M, Yang T, Li S, and Selkoe D, Environmental Enrichment Potently Prevents Microglia-Mediated Neuroinflammation by Human Amyloid beta-Protein Oligomers. J Neurosci, 2016. 36(35): p. 9041–56. PMC5005718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Williamson LL, Chao A, and Bilbo SD, Environmental enrichment alters glial antigen expression and neuroimmune function in the adult rat hippocampus. Brain Behav Immun, 2012. 26(3): p. 500–10. PMC3294275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cai D and Khor S, "Hypothalamic Microinflammation" Paradigm in Aging and Metabolic Diseases. Cell Metab, 2019. 30(1): p. 19–35. [DOI] [PubMed] [Google Scholar]
- 22.Tang Y, Purkayastha S, and Cai D, Hypothalamic microinflammation: a common basis of metabolic syndrome and aging. Trends Neurosci, 2015. 38(1): p. 36–44. PMC4282817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang G, Li J, Purkayastha S, Tang Y, Zhang H, Yin Y, et al. , Hypothalamic programming of systemic ageing involving IKK-β, NF-κB and GnRH. Nature, 2013. 497(7448): p. 211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beheshti A, Benzekry S, McDonald JT, Ma L, Peluso M, Hahnfeldt P, et al. , Host age is a systemic regulator of gene expression impacting cancer progression. Cancer Res, 2015. 75(6): p. 1134–43. PMC4397972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abella V, Scotece M, Conde J, Pino J, Gonzalez-Gay MA, Gomez-Reino JJ, et al. , Leptin in the interplay of inflammation, metabolism and immune system disorders. Nat Rev Rheumatol, 2017. 13(2): p. 100–109. [DOI] [PubMed] [Google Scholar]
- 26.Lihn AS, Pedersen SB, and Richelsen B, Adiponectin: action, regulation and association to insulin sensitivity. Obes Rev, 2005. 6(1): p. 13–21. [DOI] [PubMed] [Google Scholar]
- 27.Lommatzsch M, Zingler D, Schuhbaeck K, Schloetcke K, Zingler C, Schuff-Werner P, et al. , The impact of age, weight and gender on BDNF levels in human platelets and plasma. Neurobiol Aging, 2005. 26(1): p. 115–23. [DOI] [PubMed] [Google Scholar]
- 28.Croll SD, Ip NY, Lindsay RM, and Wiegand SJ, Expression of BDNF and trkB as a function of age and cognitive performance. Brain Research, 1998. 812(1-2): p. 200–208. [DOI] [PubMed] [Google Scholar]
- 29.Hayashi M, Mistunaga F, Ohira K, and Shimizu K, Changes in BDNF-immunoreactive structures in the hippocampal formation of the aged macaque monkey. Brain Res, 2001. 918(1-2): p. 191–6. [DOI] [PubMed] [Google Scholar]
- 30.Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, et al. , The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell, 2003. 112(2): p. 257–269. [DOI] [PubMed] [Google Scholar]
- 31.Xu B, Goulding EH, Zang K, Cepoi D, Cone RD, Jones KR, et al. , Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat Neurosci, 2003. 6(7): p. 736–42. PMC2710100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ortega-Molina A, Efeyan A, Lopez-Guadamillas E, Munoz-Martin M, Gomez-Lopez G, Canamero M, et al. , Pten positively regulates brown adipose function, energy expenditure, and longevity. Cell Metab, 2012. 15(3): p. 382–94. [DOI] [PubMed] [Google Scholar]
- 33.Garcia-Cao I, Song MS, Hobbs RM, Laurent G, Giorgi C, de Boer VC, et al. , Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell, 2012. 149(1): p. 49–62. PMC3319228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ortega-Molina A and Serrano M, PTEN in cancer, metabolism, and aging. Trends Endocrinol Metab, 2013. 24(4): p. 184–9. PMC3836169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang W, Queen NJ, McMurphy TB, Ali S, Wilkins RK, Appana B, et al. , Adipose PTEN acts as a downstream mediator of a brain-fat axis in environmental enrichment. Comprehensive Psychoneuroendocrinology, 2020. 4: p. 100013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hashemi Goradel N, Najafi M, Salehi E, Farhood B, and Mortezaee K, Cyclooxygenase-2 in cancer: A review. J Cell Physiol, 2019. 234(5): p. 5683–5699. [DOI] [PubMed] [Google Scholar]
- 37.Hoeben A, Landuyt B, Highley MS, Wildiers H, Van Oosterom AT, and De Bruijn EA, Vascular endothelial growth factor and angiogenesis. Pharmacol Rev, 2004. 56(4): p. 549–80. [DOI] [PubMed] [Google Scholar]
- 38.Jain RK, Tumor angiogenesis and accessibility: role of vascular endothelial growth factor. Semin Oncol, 2002. 29(6 Suppl 16): p. 3–9. [DOI] [PubMed] [Google Scholar]
- 39.Karin N and Razon H, Chemokines beyond chemo-attraction: CXCL10 and its significant role in cancer and autoimmunity. Cytokine, 2018. 109: p. 24–28. [DOI] [PubMed] [Google Scholar]
- 40.Meng W, Xue S, and Chen Y, The role of CXCL12 in tumor microenvironment. Gene, 2018. 641: p. 105–110. [DOI] [PubMed] [Google Scholar]
- 41.Samarendra H, Jones K, Petrinic T, Silva MA, Reddy S, Soonawalla Z, et al. , A meta-analysis of CXCL12 expression for cancer prognosis. Br J Cancer, 2017. 117(1): p. 124–135. PMC5520200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sepe A, Tchkonia T, Thomou T, Zamboni M, and Kirkland JL, Aging and regional differences in fat cell progenitors - a mini-review. Gerontology, 2011. 57(1): p. 66–75. PMC3031153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.De Pergola G and Silvestris F, Obesity as a major risk factor for cancer. Journal of Obesity, 2013. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Avgerinos KI, Spyrou N, Mantzoros CS, and Dalamaga M, Obesity and cancer risk: Emerging biological mechanisms and perspectives. Metabolism, 2019. 92: p. 121–135. [DOI] [PubMed] [Google Scholar]
- 45.Deng T, Lyon CJ, Bergin S, Caligiuri MA, and Hsueh WA, Obesity, Inflammation, and Cancer. Annu Rev Pathol, 2016. 11: p. 421–49. [DOI] [PubMed] [Google Scholar]
- 46.Fink RI, Kolterman OG, Griffin J, and Olefsky JM, Mechanisms of insulin resistance in aging. J Clin Invest, 1983. 71(6): p. 1523–35. PMC370358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rowe JW, Minaker KL, Pallotta JA, and Flier JS, Characterization of the insulin resistance of aging. J Clin Invest, 1983. 71(6): p. 1581–7. PMC370364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shimokata H, Muller DC, Fleg JL, Sorkin J, Ziemba AW, and Andres R, Age as Independent Determinant of Glucose-Tolerance. Diabetes, 1991. 40(1): p. 44–51. [DOI] [PubMed] [Google Scholar]
- 49.Guo SS, Zeller C, Chumlea WC, and Siervogel RM, Aging, body composition, and lifestyle: the Fels Longitudinal Study. Am J Clin Nutr, 1999. 70(3): p. 405–11. [DOI] [PubMed] [Google Scholar]
- 50.Baumgartner RN, Body composition in healthy aging. Ann N Y Acad Sci, 2000. 904(1): p. 437–48. [DOI] [PubMed] [Google Scholar]
- 51.Palmer AK and Kirkland JL, Aging and adipose tissue: potential interventions for diabetes and regenerative medicine. Exp Gerontol, 2016. 86: p. 97–105. PMC5001933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Siler LM, Mouse Genetics: concepts and applications. 1995, New York: Oxford University Press. [Google Scholar]
- 53.Kwasniewska M, Pikala M, Kaczmarczyk-Chalas K, Piwonnska A, Tykarski A, Kozakiewicz K, et al. , Smoking status, the menopausal transition, and metabolic syndrome in women. Menopause, 2012. 19(2): p. 194–201. [DOI] [PubMed] [Google Scholar]
- 54.Palmer BF and Clegg DJ, The sexual dimorphism of obesity. Mol Cell Endocrinol, 2015. 402: p. 113–9. PMC4326001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trentham-Dietz A, Newcomb PA, Egan KM, Titus-Ernstoff L, Baron JA, Storer BE, et al. , Weight change and risk of postmenopausal breast cancer (United States). Cancer Causes Control, 2000. 11(6): p. 533–42. [DOI] [PubMed] [Google Scholar]
- 56.Queen NJ, Boardman AA, Patel RS, Siu JJ, Mo X, and Cao L, Environmental enrichment improves metabolic and behavioral health in the BTBR mouse model of autism. Psychoneuroendocrinology, 2020. 111: p. 104476. PMC6914218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Solum DT and Handa RJ, Estrogen regulates the development of brain-derived neurotrophic factor mRNA and protein in the rat hippocampus. J Neurosci, 2002. 22(7): p. 2650–9. PMC6758321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sonntag WE, Lynch CD, Cefalu WT, Ingram RL, Bennett SA, Thornton PL, et al. , Pleiotropic effects of growth hormone and insulin-like growth factor (IGF)-1 on biological aging: inferences from moderate caloric-restricted animals. J Gerontol A Biol Sci Med Sci, 1999. 54(12): p. B521–38. [DOI] [PubMed] [Google Scholar]
- 59.Higashi Y, Sukhanov S, Anwar A, Shai SY, and Delafontaine P, IGF-1, oxidative stress and atheroprotection. Trends Endocrinol Metab, 2010. 21(4): p. 245–54. PMC2848911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu JM, Zhao HY, Ning G, Chen Y, Zhang LZ, Sun LH, et al. , IGF-1 as an early marker for low bone mass or osteoporosis in premenopausal and postmenopausal women. J Bone Miner Metab, 2008. 26(2): p. 159–64. [DOI] [PubMed] [Google Scholar]
- 61.Ashpole NM, Logan S, Yabluchanskiy A, Mitschelen MC, Yan H, Farley JA, et al. , IGF-1 has sexually dimorphic, pleiotropic, and time-dependent effects on healthspan, pathology, and lifespan. Geroscience, 2017. 39(2): p. 129–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Costelli P, Muscaritoli M, Bossola M, Penna F, Reffo P, Bonetto A, et al. , IGF-1 is downregulated in experimental cancer cachexia. Am J Physiol Regul Integr Comp Physiol, 2006. 291(3): p. R674–83. [DOI] [PubMed] [Google Scholar]
- 63.Schmidt K, von Haehling S, Doehner W, Palus S, Anker SD, and Springer J, IGF-1 treatment reduces weight loss and improves outcome in a rat model of cancer cachexia. J Cachexia Sarcopenia Muscle, 2011. 2(2): p. 105–109. PMC3117996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meng Z, Liu T, Song Y, Wang Q, Xu D, Jiang J, et al. , Exposure to an enriched environment promotes the terminal maturation and proliferation of natural killer cells in mice. Brain Behav Immun, 2019. 77: p. 150–160. [DOI] [PubMed] [Google Scholar]
- 65.Mansour AG, Xiao R, Bergin SM, Huang W, Chrislip LA, Zhang J, et al. , Enriched environment enhances NK cell maturation through hypothalamic BDNF in male mice. Eur J Immunol, 2021. 51(3): p. 557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hanahan D and Weinberg RA, Hallmarks of cancer: the next generation. Cell, 2011. 144(5): p. 646–74. [DOI] [PubMed] [Google Scholar]
- 67.O'Mahony F, Razandi M, Pedram A, Harvey BJ, and Levin ER, Estrogen modulates metabolic pathway adaptation to available glucose in breast cancer cells. Mol Endocrinol, 2012. 26(12): p. 2058–70. PMC3517720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cai Q, Lin T, Kamarajugadda S, and Lu J, Regulation of glycolysis and the Warburg effect by estrogen-related receptors. Oncogene, 2013. 32(16): p. 2079–86. PMC3435484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cao L, Ali S, and Queen NJ, Hypothalamic gene transfer of BDNF promotes healthy aging. Vitamins and Hormones, 2021. 115: p. 39–66. [DOI] [PubMed] [Google Scholar]
- 70.Hassan QN II, Queen NJ, and Cao L, Regulation of aging and cancer by enhanced environmental activation of a hypothalamic-sympathoneural-adipocyte axis. Translational Cancer Research, 2020. 9(9): p. 5687–5699. [DOI] [PMC free article] [PubMed] [Google Scholar]