

Abstract
The growth of most bony tuberosities, like the deltoid tuberosity (DT), rely on the transmission of muscle forces at the tendon-bone attachment during skeletal growth. Tuberosities distribute muscle forces and provide mechanical leverage at attachment sites for joint stability and mobility. The genetic factors that regulate tuberosity growth remain largely unknown. In mouse embryos with global deletion of fibroblast growth factor 9 (Fgf9), the DT size is notably enlarged. In this study, we explored the tissue-specific regulation of DT size using both global and targeted deletion of Fgf9. We showed that cell hypertrophy and mineralization dynamics of the DT, as well as transcriptional signatures from skeletal muscle but not bone, were influenced by the global loss of Fgf9. Loss of Fgf9 during embryonic growth led to increased chondrocyte hypertrophy and reduced cell proliferation at the DT attachment site. This endured hypertrophy and limited proliferation may explain the abnormal mineralization patterns and locally dysregulated expression of markers of endochondral development in Fgf9null attachments. We then showed that targeted deletion of Fgf9 in skeletal muscle leads to postnatal enlargement of the DT. Taken together, we discovered that Fgf9 may play an influential role in muscle-bone crosstalk during embryonic and postnatal development.
Keywords: Skeleton, Enthesis, Bone shape, Muscle, Mechanics
Introduction
The transmission of muscle forces to bone via tendons is required for the functional growth of the vertebrate skeleton (Arvind and Huang, 2017; Biewener et al., 1996; Blitz et al., 2009; Kahn et al., 2009; Rot-Nikcevic et al., 2006). Bone ridges and tuberosities are skeletal “superstructures” that transmit muscle forces to the periosteal surface of long bones and increase leverage at sites of tendon attachments (Benjamin et al., 2006; Blitz et al., 2009; Blitz et al., 2013; Zelzer et al., 2014). The presence of superstructures requires both initiation of a modular Sox9+/Scx+ progenitor cell pool (Blitz et al., 2013) and maintenance of skeletal muscle contraction (Blitz et al., 2009; Hamburger and Waugh, 1940; Pai, 1965; Rot-Nikcevic et al., 2006; Tremblay et al., 1998). One of the most prominent superstructures in the mouse skeleton is the deltoid tuberosity (DT) of the humerus, which is a migratory tendon attachment that projects anterolaterally from the shoulder with a fin-like shape (Felsenthal et al., 2018). Migratory tendon attachments are first established by a population of Sox9+ progenitor cells during embryonic development. Subsequently, these Sox9+ cells are then replaced by Gli1+ progenitor cells during postnatal growth (Felsenthal et al., 2018). As such, the growth of tendon attachments is likened to that of an arrested growth plate (Benjamin et al., 2006; Schwartz et al., 2015), with the attachment includes a layered arrangement of hypertrophic and pre-hypertrophic Gli1+ cells, similar to the growth plate during endochondral ossification, as well as a mineralized gradient at the enthesis between tendon and bone (Benjamin et al., 2006; Schwartz et al., 2015; Zelzer et al., 2014).
Because it is a site of tendon attachments, the DT is also a phenotypic readout for estimating muscle loads during embryonic development in mice. For example, in muscular-compromised mouse models (Myf5null: MyoDnull, Mdg, and Spd mutants), the DT fails to maintain its size and shape during the mid-to-late stages of embryonic growth (Blitz et al., 2009; Hamburger and Waugh, 1940; Pai, 1965; Rot-Nikcevic et al., 2006; Tremblay et al., 1998). Conversely, mice with genetic mutations that lead to increased muscle mass (e.g., myostatin−/− mice) exhibit an autonomous increase in the size and shape of the DT (Hamrick et al., 2002). Though many mutations related to skeletal muscle unloading result in smaller bone ridges (Blitz et al., 2009; Hamburger and Waugh, 1940; Pai, 1965; Rot-Nikcevic et al., 2006; Tremblay et al., 1998), embryos that lack the protein-encoding gene for Fibroblast growth factor 9 (Fgf9) have enlarged DTs (Hung et al., 2007). Surprisingly, even though the DTs are enlarged, the stylopods (i.e., humeri and femurs) of Fgf9 mutant (Fgf9null) embryos are shorter and experience delayed growth plate hypertrophy and proliferation, as well as reduced angiogenesis, ultimately restricting osteogenesis during long bone development (Hung et al., 2007; Hung et al., 2016). These phenotypic differences between DT growth and long bone development in Fgf9null embryos suggest a unique role of Fgf9 as a negative regulator of growth in attachment superstructures.
FGF9 is one of three ligands in the fibroblast growth factor (FGF) family known to be involved in bone development (Berendsen and Olsen, 2015; Coutu et al., 2011; Deng et al., 1996; Jacob et al., 2006; Karuppaiah et al., 2016; Ohbayashi et al., 2002; Ornitz and Itoh, 2015; Ornitz and Marie, 2002; Ornitz and Marie, 2015; Sahni et al., 1999; Su et al., 2014; Vaahtokari et al., 1996). In the developing limb, Fgf9 is expressed as early as embryonic day 10.5 (E10.5) within the apical ectodermal ridge (Colvin et al., 1999; Garofalo et al., 1999; Hung et al., 2007; Yang and Kozin, 2009). As limb development progresses, expression of Fgf9 coincides with expression of other chondrogenic markers, such as Fgfr2 and Fgf18 (Govindarajan and Overbeek, 2006; Hung et al., 2007; Lu et al., 2015). In periosteal and trabecular osteoblasts, overexpression of Fgf9 leads to smaller mice with shorter growth plates (Karuppaiah et al., 2016). Although Fgf9 expression is required for long bone growth, its expression is predominantly found in the surrounding muscle and perichondral cells and not in the bone anlagen, suggesting that FGF9 may also originate from surrounding soft tissues (Colvin et al., 1999; Garofalo et al., 1999; Hung et al., 2007). However, the role of tissue specific Fgf9 on the growth of bony superstructures like the DT has not been explored.
In this study, we hypothesized that global knockout of Fgf9 leads to increased expansion and growth of the arrested growth plate at sites of migratory tendon attachments. We quantified the size of the DT superstructure using whole mount staining and characterized cell shape and size using histomorphometry. We then used multiplexed fluorescence in situ hybridization (FISH) at key timepoints during embryonic growth to visualized expression of growth plate markers at the tendon attachment of the DT in wildtype and Fgf9null limbs. We used RNA sequencing (RNAseq) to identify differentially expressed genes between WT and Fgf9null bone anlagen and skeletal muscle during late embryonic development. Surprisingly, we found significant differential expression in muscle, but not bone. Therefore, we developed conditional muscle-specific Fgf9 knockout mice (Fgf9cKO) to determine the effect of muscle-specific deletion of Fgf9 on postnatal DT size and skeletal muscle innervation. Our findings suggest a muscle-specific role of FGF9 in the size of stylopod superstructures, further expanding the role of FGF signaling in musculoskeletal development.
Results
Global loss of Fgf9 results in an enlarged DT, even with a shortened humerus
To confirm the larger size of the DT in Fgf9null neonates suggested previously by Hung et al., we used whole-mount staining to visualize cartilage with Alcian Blue and mineralized bone with Alizarin Red (Hung et al., 2007). We found that Fgf9null neonates had a 68% larger ratio of the DT area to total humerus area compared to WT neonates at postnatal day 0 (P0) (Figure 1A,B). The DT was wider and shorter in Fgf9null neonates compared to WT littermates. Fgf9null neonates also had a similar total humerus area compared to WT neonates (Figure 2B). We did not find a sex-dependent effect of global Fgf9 deletion on DT size (Figure 1, blue and pink dots).
Figure 1: The deltoid tuberosities (DTs) of Fgf9 global knockout (Fgf9null) neonates were larger compared to WT neonates.
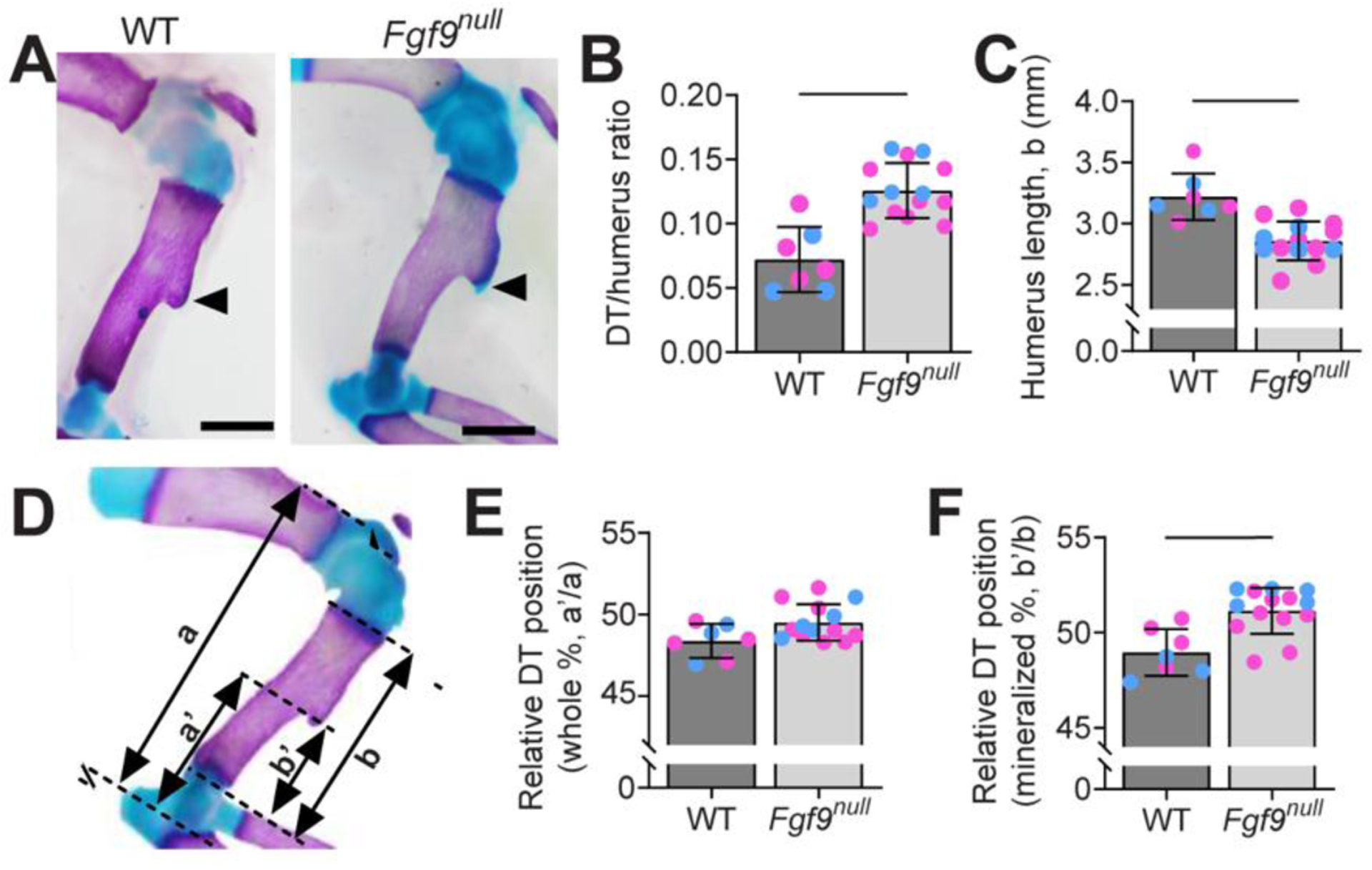
(A) Whole mount-stained forearms of a (left) WT mouse and (right) Fgf9null neonate at postnatal day 0 (P0) (Scale bar = 1mm). Black arrows point to the DT, which was noticeably larger on Fgf9null neonate humeri. Alcian Blue stain was present on the DT of Fgf9null neonates but absent in WT neonates. (B) Normalized area of the DT was significantly larger in Fgf9null neonates compared to WT neonates at P0. Whether the mice were male (blue) or female (pink) had no effect. (C) Contrastingly, the enlarged DT was a part of a shorter mineralized humerus. (D) The positioning of the DT on the humerus with respect to the total length (a, a’) and the mineralized length (b, b’). (E) Relative to the whole humeri, the DT of Fgf9null neonates was in a similar relative location compared to WT neonates. (F) Relative to the mineralized region, the DT of Fgf9null neonates was more proximally placed than WT neonates. (Data are biological replicates, mean ± SD. Lines in B, C, and F indicate significance (p < 0.05).
Figure 2: Fgf9null have a larger DT area with smaller primary humerus area than WT neonates.
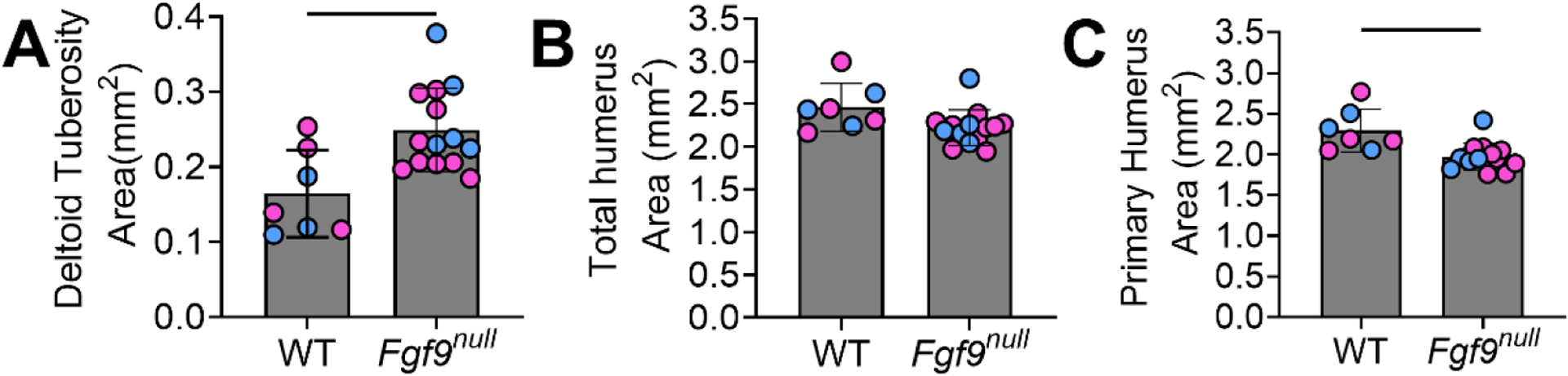
(A) Area of the DT was significantly larger in Fgf9null neonates compared to WT neonates at P0. Whether the mice were male (blue) or female (pink) had no effect. (B) There was no difference in the combined area of the DT and primary humerus between Fgf9null and WT neonates. (C) The area of just the primary humerus was significantly smaller in Fgf9null neonates compared to WT neonates. Data are biological replicates, mean ± SD; blue = male and magenta = female mice. Lines in A and C indicate significance (p < 0.05).
We observed increased Alcian Blue-stained cartilage on the crest of the DT in Fgf9null neonates, but this cartilage was absent on the crest of stage-matched WT DTs (Figure 1A, arrows). In line with previously published work, Fgf9null neonates had shorter humeri (Figure 1C) than WT neonates at P0 (Fgf9null = 3.2 ± 0.2mm, WT = 2.9 ± 0.2 mm) (Hung et al., 2007). DT migration was measured relative to the length of the humerus (Figure 1D). In Fgf9null neonates, the DT was located in a relatively similar position on the humerus compared to WT littermates (Figure 1E,F). Using dynamic bone histomorphometry, Fgf9null DTs had an increased and more variable mineral growth rate compared to WT embryos (mineral apposition rate between E16.5 and E18.5: Fgf9null = 133.1 ± 117.7%, WT = 32.9 ± 10.7%).
Global loss of Fgf9 led to larger attachment-site chondrocyte size but reduced cell proliferation at the attachment
Next, we compared the cellular and matrix morphology of the DT attachment between WT and Fgf9null embryos using Hematoxylin and Eosin Y (H&E) staining of transverse histological sections. At E15.5, the size and cell morphology of the DT was similar between WT and Fgf9null embryos (Figure 3A,E). At E16.5, the DT of Fgf9null embryos appeared larger and less mineralized compared to the DT of WT embryos (Figure 3B,F). At E18.5 and P0, we observed more hypertrophic chondrocytes in the DT of Fgf9null mice (Figure 3G,H) compared to WT mice (Figure 3C,D). These hypertrophic chondrocytes stained positive for type X collagen at E16.5 samples (Figure 4). At E16.5, we measured a similar number of hypertrophic chondrocytes at the DT for Fgf9null and WT littermates (Figure 3I). However, the average size of DT-localized hypertrophic chondrocytes in Fgf9null embryos at E16.5 was 72% larger compared to WT embryos (Figure 3L). Additionally, cell proliferation at E16.5, visualized using 5-ethynyl-2’-deoxyuridine (EdU), was 51% lower in attachments but not tendons or perichondrium, of Fgf9null embryos compared to similar regions of WT embryos (Figure 3N). We did not find measurable differences in cell density (Hoechst-stained; nuclei/100μm2) between E16.5 WT and Fgf9null embryos (Figure 3K).
Figure 3: The DTs of Fgf9null embryos undergo differences in cellular and morphological development when compared to WT embryos.
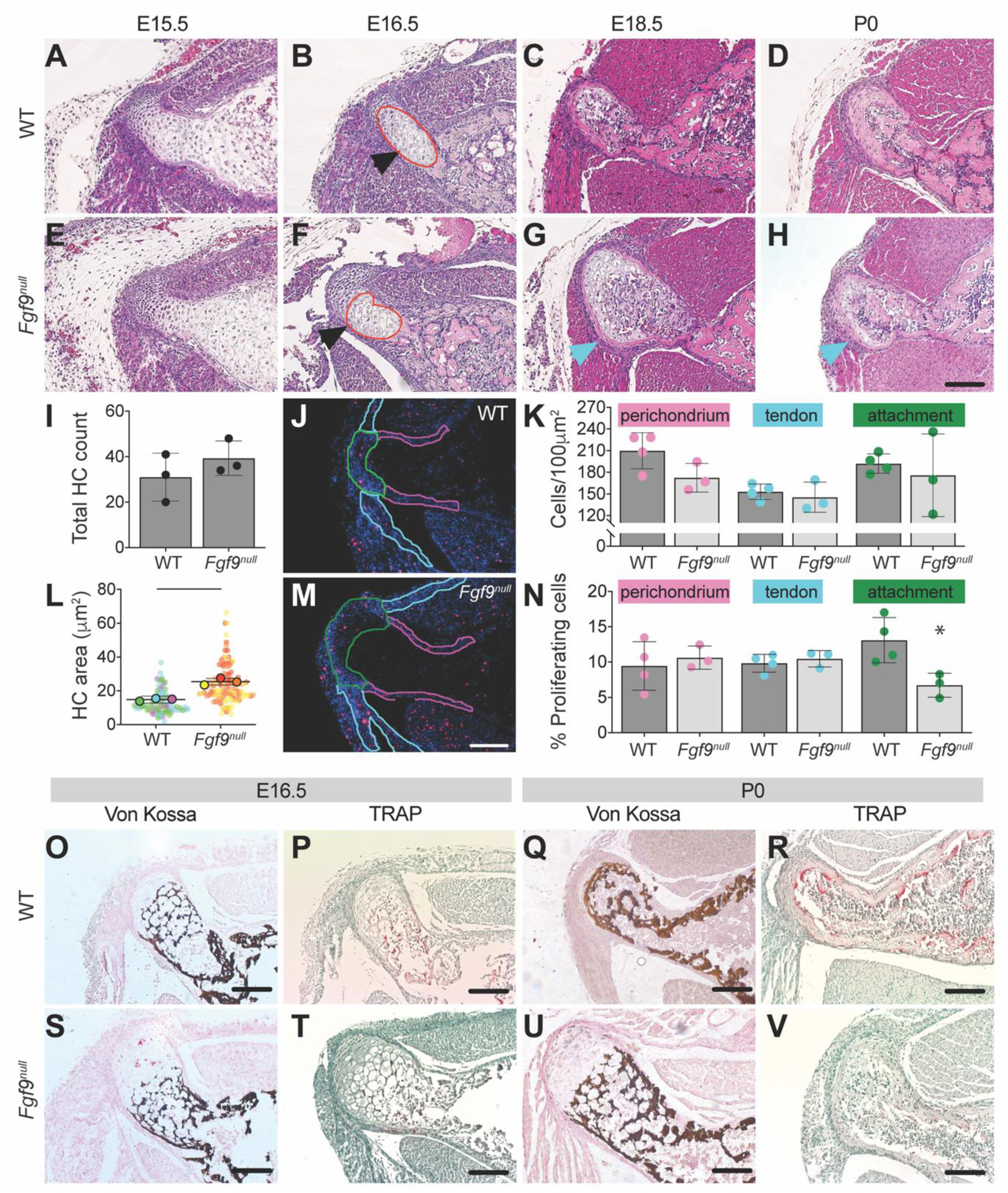
The DT of Fgf9null embryos was larger than the DT of WT embryos as early as E16.5 (black arrows; scale bar = 100μm). During later stages of development (E18.5 (C, G) and P0 (D, H)), hypertrophic chondrocytes (blue arrows) remained in the DT of Fgf9null embryonic limb and were absent in WT embryonic limb. H&E-stained transverse sections of the DT in (B) WT embryos and (F) Fgf9null embryos at E16.5. Red lines represent the region of hypertrophic chondrocytes. Fgf9null embryos had a (I) similar number of hypertrophic chondrocytes and (L) larger average area of hypertrophic chondrocytes compared to WT embryos. (J,M) Proliferating cells were labeled at E16.5 using EdU, in red. Outlined regions of interest include the perichondrium (pink), tendon (teal), and attachment (green). Fgf9null embryos had (K) similar cell density and (N) reduced proliferation rates at the DT attachment compared to WT embryos at E16.5. (U) At P0 Fgf9null neonates had abnormal mineral patterning (von Kossa) and (V) fewer osteoclasts (TRAP) than (Q,R) WT neonates (scale = 100μm). Data are biological replicates (faded circles are technical replicates), mean ± SD. Lines in L and * in N indicates significant difference between groups (p < 0.05).
Figure 4: Type X collagen immunostaining of the DT in WT and Fgf9null embryos at E16.5.
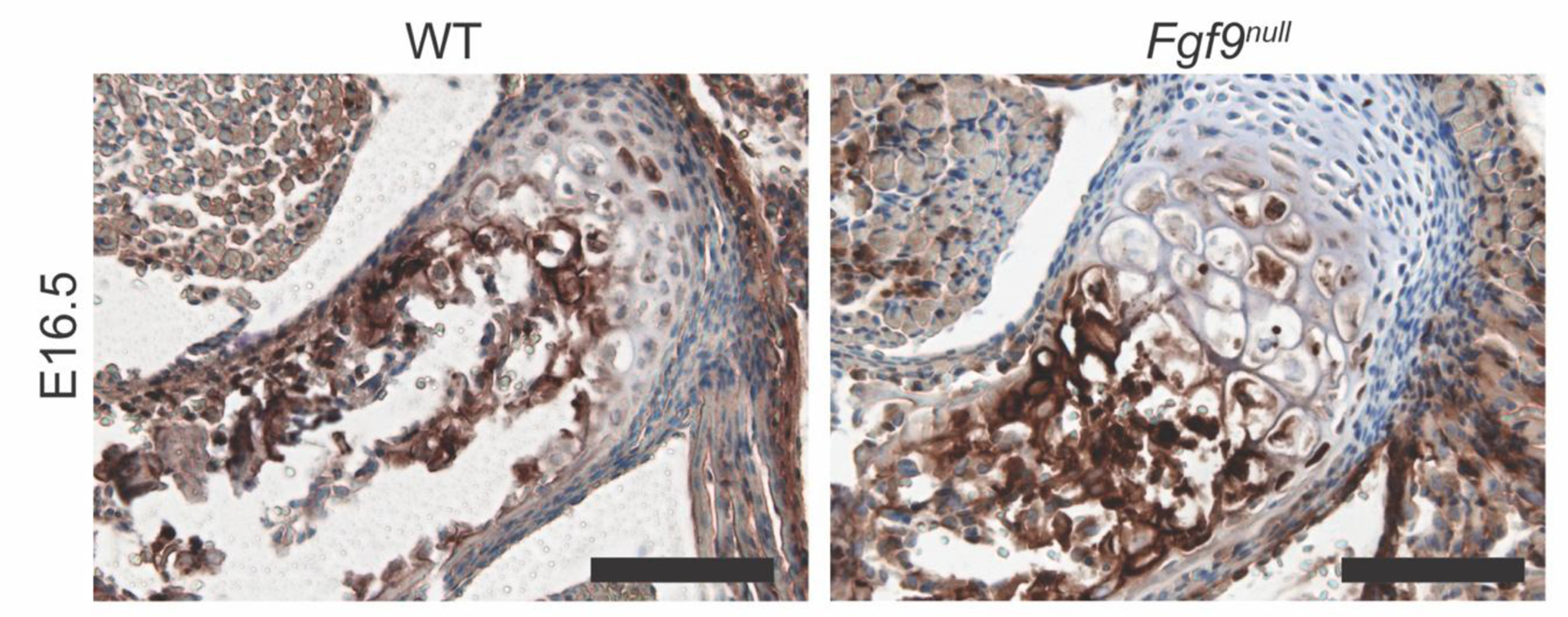
The tendon attachment has less ColX staining but more ColX+ hypertrophic cells in Fgf9null attachments compared to WT attachments (scale bar = 100μm).
We visualized mineralization of the attachment using von Kossa staining and found that at E16.5, WT and Fgf9null embryos both had a woven mineral pattern within the hypertrophic region. At P0, the remodeled mineral in WT DTs formed a layer of cortical bone (Figure 3Q), whereas in Fgf9null neonates, the woven mineral pattern remained (Figure 3U). In addition, fewer osteoclasts were present at both E16.5 and P0 (Figure 3R,V; Figure 5).
Figure 5: Fewer osteoclasts present in Fgf9null DTs.
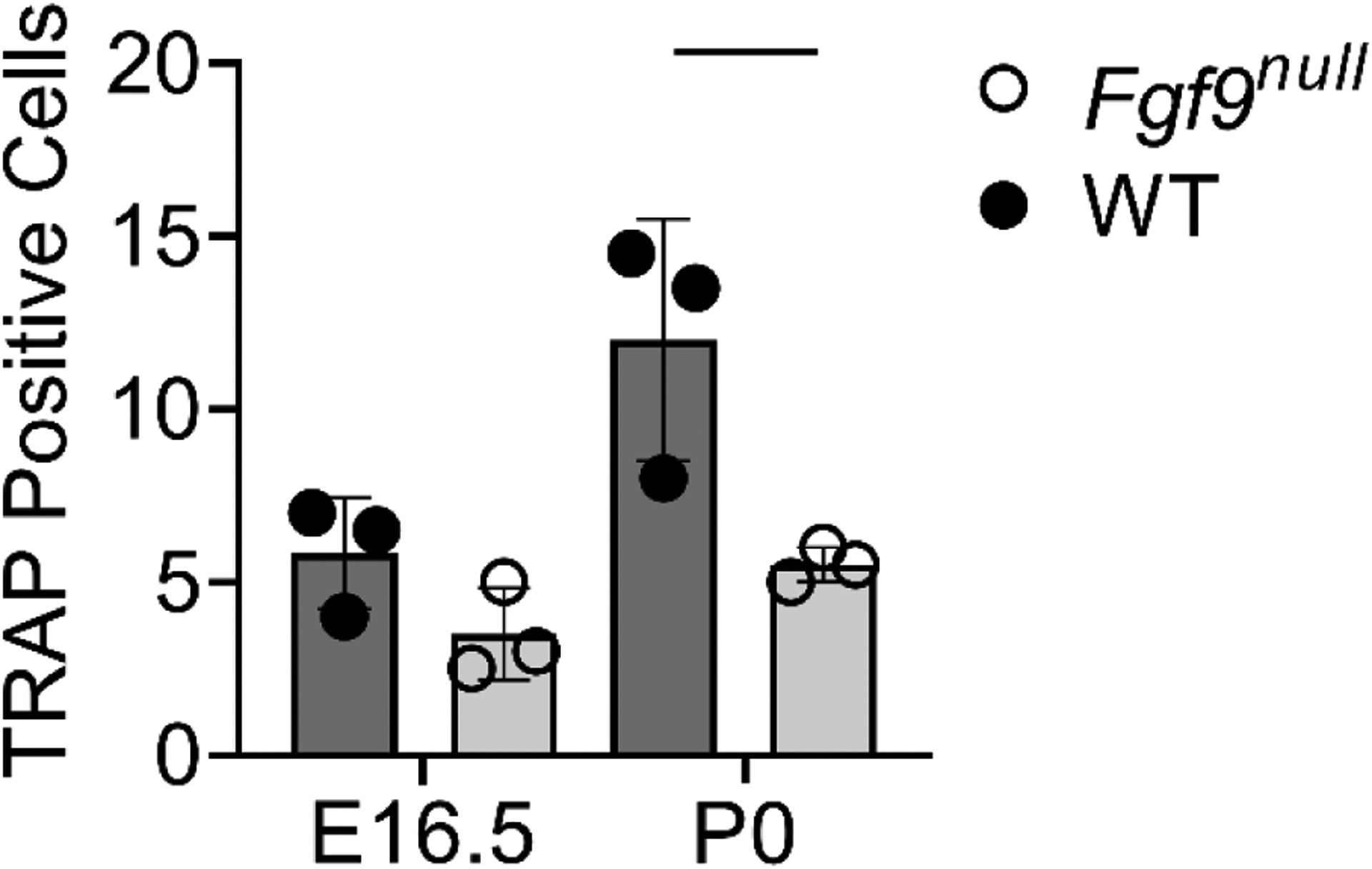
. Fgf9null mice have fewer osteoclasts at E16.5 and significantly fewer osteoclasts at p0. Data are biological replicates (1–2 technical replicates averaged per sample) mean ± SD. Line indicate significance (p < 0.05).
Global loss of Fgf9 leads to gene expression differences in muscle, but not bone
Fgf9 is expressed as early as E10.5 in the developing limb and later in the perichondrium and muscle (Colvin et al., 1999; Garofalo et al., 1999; Hung et al., 2007; Yang and Kozin, 2009). At E16.5, Fgf9null embryos had increased localized expression of Gli, Sox9, and Fgf18 (markers of bone and cartilage development) within and around the DT compared to WT embryos at the same developmental stage (Figure 6B,F,N). At P0, Fgf9null neonates had reduced localized Sox9 and Sost expression compared to WT neonates (Figure 6H,L).
Figure 6: Fluorescent in situ hybridization (FISH) of (A-D) Gli1, (E-H) Sox9, (I-L) Sost, and (M-P) Fgf18 at E16.5 and P0 of Fgf9null and WT mice.
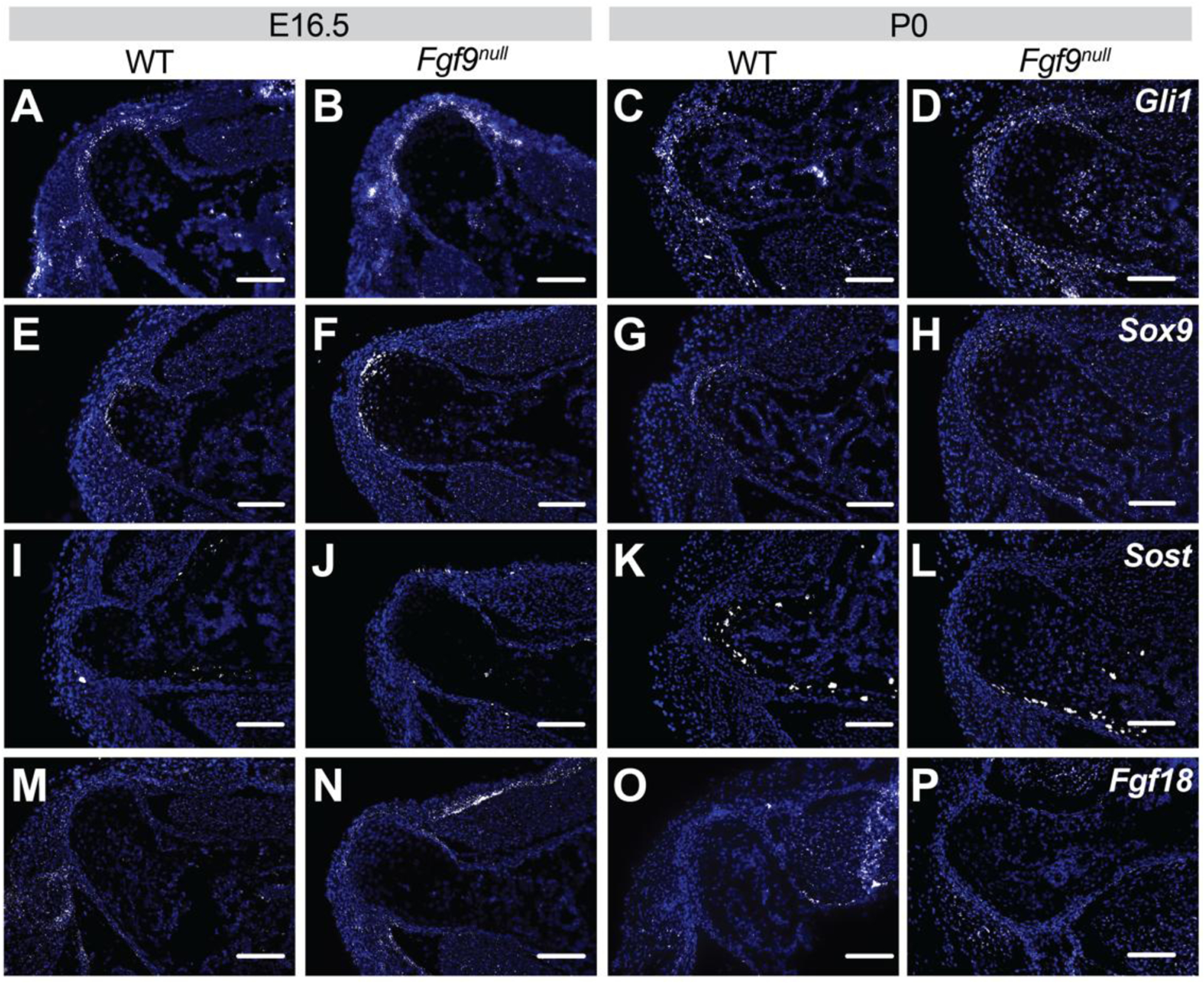
At E16.5, Fgf9null embryos had increased (B) Gli, (F) Sox9, and (N) Fgf18 expression compared to (A,E,I,M) WT embryos. At P0, Fgf9null neonates had less (H) Sox9 and (L) Sost, and similar (D) Gli1 and (P) Fgf18 expression compared to (C,G,L,P) WT neonates (scale bar = 100μm; n = 3 per genotype for each time point).
For an unbiased comparison of differential gene expression from bone and muscle of WT and Fgf9null embryonic limbs, we performed bulk RNAseq from micro-dissected tissue of stylopods at E18.5 (n=3/genotype). We used a cut off false discovery rate of 0.05 and discovered 805 differentially expressed (DE) genes between WT and Fgf9null muscle with only a single DE gene (Xist) in bone. A secondary analysis was performed to exclude DE genes with a fold change greater than two standard deviations between biological replicates. In our secondary analysis, we identified 135 unique genes (Figure 7). The primary and secondary DE gene lists were then analyzed using Database for Annotation, Visualization and Integrated Discovery (DAVID) (Huang et al., 2009a; Huang et al., 2009b) and the GOTERM cellular components direct, biological processes direct, and molecular functions direct libraries. DE genes in the primary pool of 805 genes were found in a broad set of terms. DE genes in the secondary pool of 135 genes were associated with decreased expression of mitochondria/energy and lipid associated genes in Fgf9null muscle compared to WT muscle (Figure 8). Additionally, of the genes investigated using FISH, we found that Gli1 was significantly upregulated in Fgf9null muscle compared to WT muscle (fold change = 2.8338; q-value false detection rate = 0.0042). However, Sox9, Sost, and Fgf18 were not statistically differentially expressed in muscle. Additionally, none of these genes were significantly differentially expressed between Fgf9null and WT bone.
Figure 7: Heat map of top 30 upregulated and downregulated genes from RNASeq secondary analysis of embryonic WT and Fgf9null muscle.
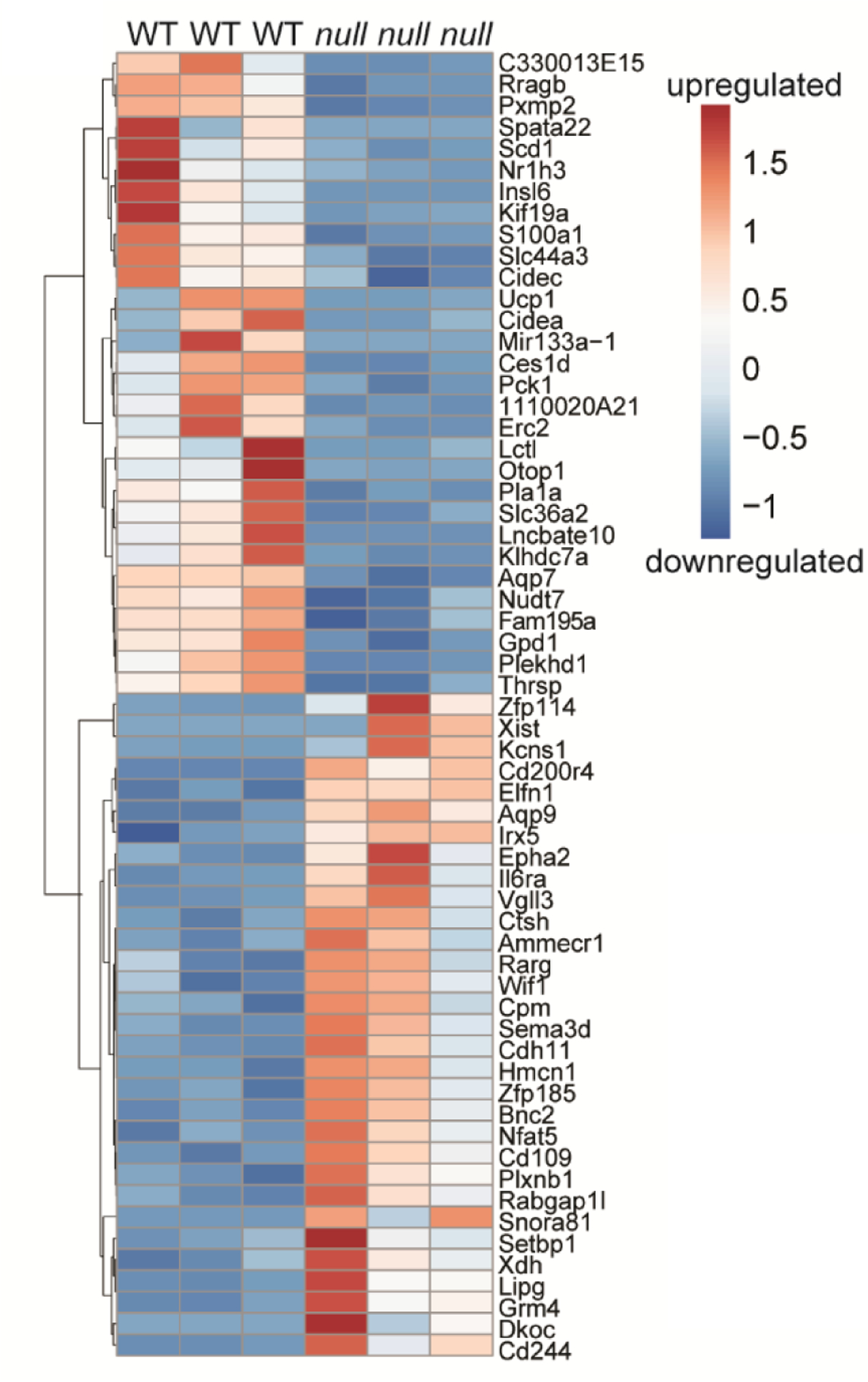
135 differentially expressed (DE) with a false discovery rate value < 0.05 and genes omitted with fold change outside 2 standard deviations of genotype average log2 fold change (FC). Color indicates z-score value as calculated and presented using Morpheus software. Scale bar = log2 FC.
Figure 8: Fgf9null muscle had predominantly downregulated mitochondrial and energy-related gene expression compared to WT muscle.
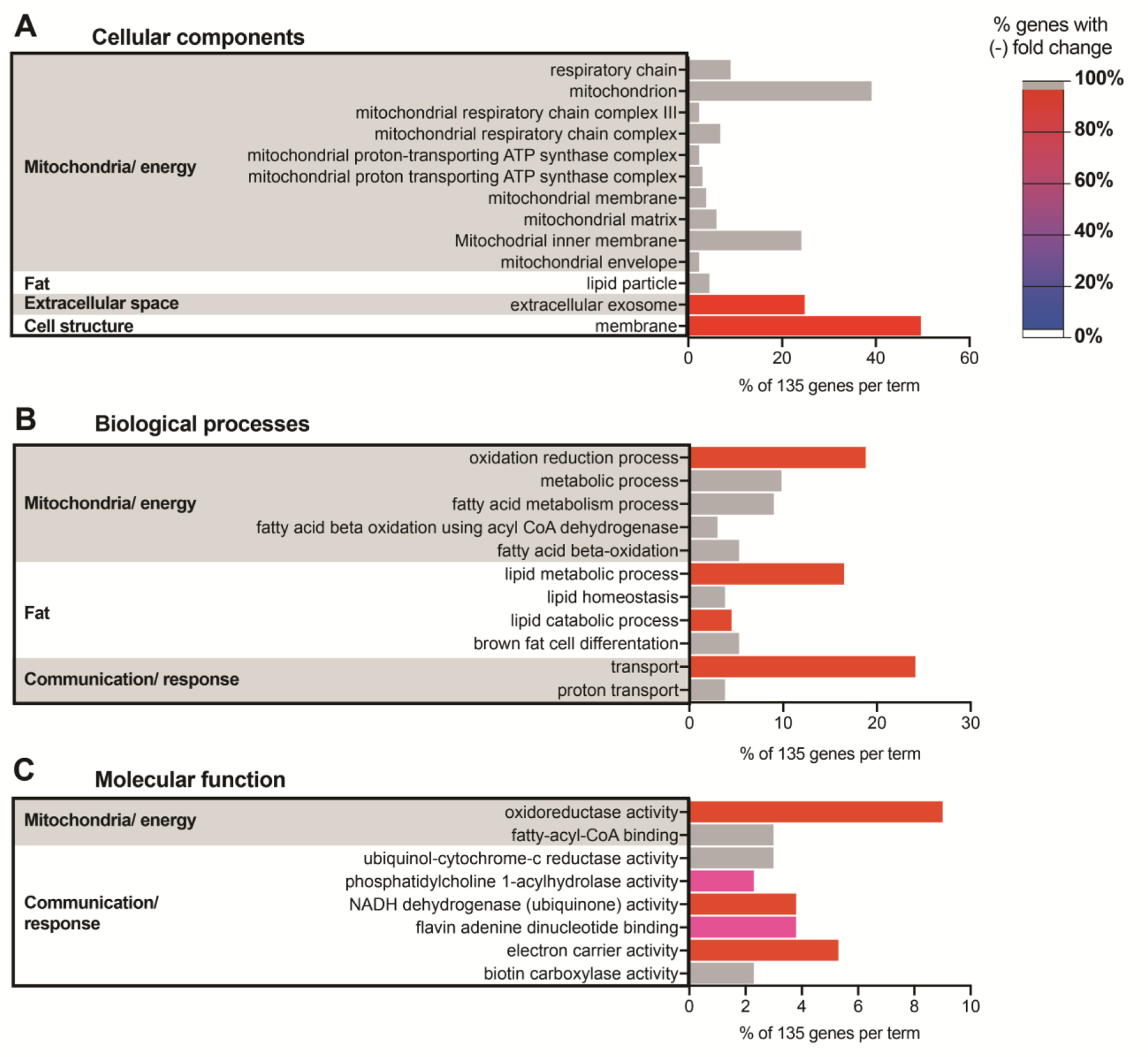
135 DE genes from secondary analyses were categorized using DAVID against a Mus musculus background. Terms with a Benjamini value < 0.05 were selected from the (A) GOTERM cellular components DIRECT, (B) GOTERM biological processes DIRECT, (C) and GOTERM molecular function DIRECT. Similar terms were then clustered into categories (bar chart = % gene in term out of 135; color = % genes in term with negative fold change). Terms with 100% negative FC (WT vs. Fgf9null) were assigned gray bars.
Skeletal-muscle specific deletion of Fgf9 results in enlarged tuberosities and decreased neural-related gene expression
Based on our RNAseq findings, we developed a doxycycline-inducible and skeletal-muscle specific Fgf9cKO mouse line, which, unlike Fgf9null embryos, were postnatal viable. Muscle-specific targeting was confirmed through Ai14 reporter (Acta1-Cre) (Figure 9G,H). At 8 weeks of age, Fgf9cKO mice were similar in size and had comparable humerus lengths (Figure 10A) compared to WT littermates. However, Fgf9cKO mice had ~35% larger DTs compared to WT littermates by 8-weeks of age (Figure 9C). However, the size of the DT was not measurably different (using microCT) between genotypes at 3-weeks of age, suggesting that the tuberosity is still developing until adulthood (~6–8 weeks of age). Bone quality of DTs between WT and Fgf9cKO mice was similar at 8-weeks of age (Figure 10B). In addition, another bony superstructure on the humerus, the lateral supracondylar ridge (LSR) near the elbow where extensor muscles originate, was ~30% larger in Fgf9cKO mice than WT mice (Figure 9D). Gene expression of Agrn (Figure 9I) and Ikbkb (Figure 9J, not statistically significant) in deltoid muscle from Fgf9cKOmice was increased compared to WT mice at 8 weeks but not 3 weeks of age. Acetylcholine receptor (AChR) density (Figure 9N) was 30% significantly higher in the deltoid muscles of Fgf9cKO mice when compared to that of WT mice at 3 weeks of age (Figure 9K,L). However, AChR density normalized between genotypes by 8 weeks. There was not a significant difference in total AChR count in the deltoid for either time point (Figure 9M).
Figure 9: Muscle-specific knockout of Fgf9 in mice (Fgf9cKO) resulted in larger superstructures and increased number of acetylcholine receptor (AChR) clusters in the deltoid muscle.
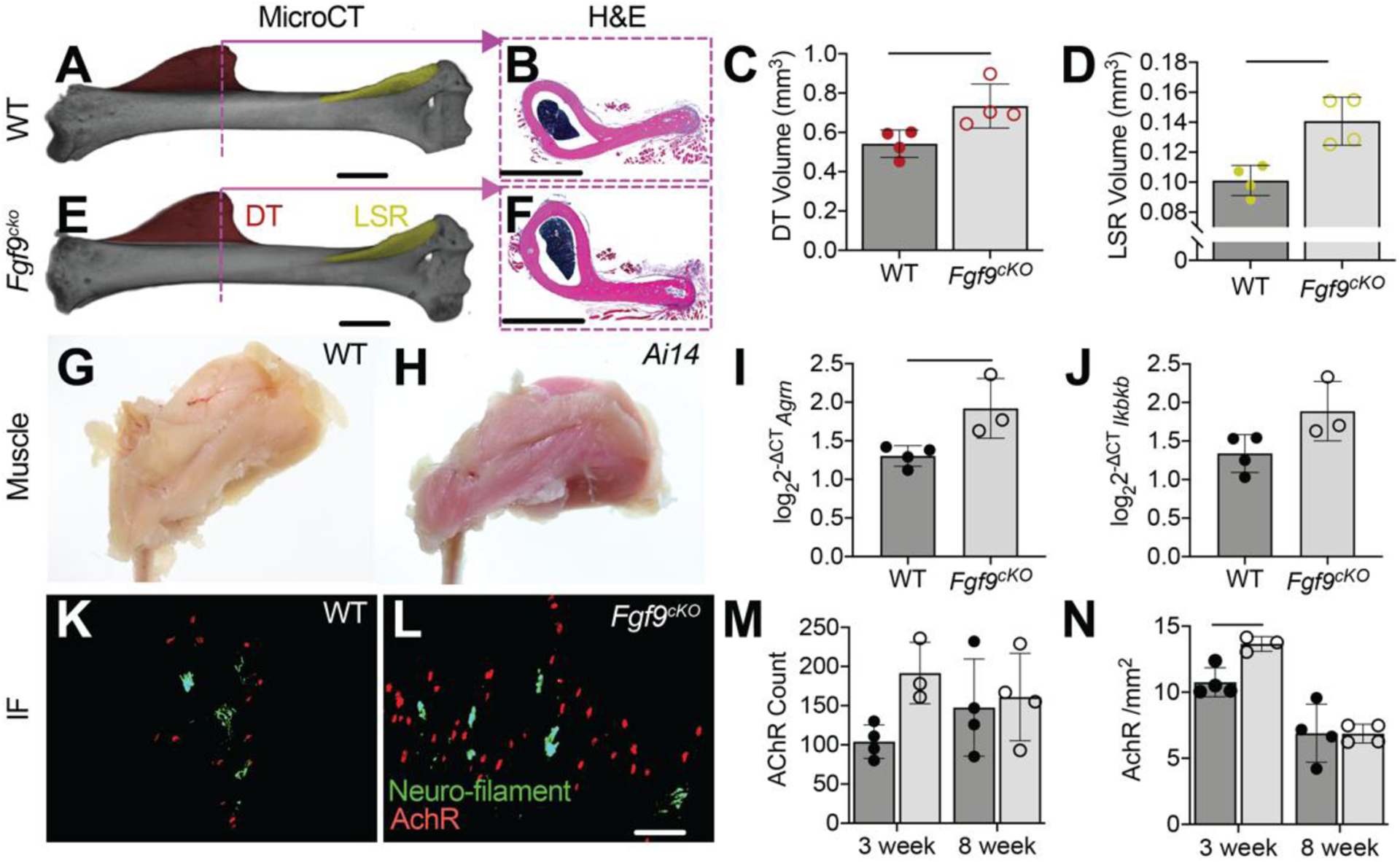
MicroCT scans of 8 week (A) WT and (E) Fgf9cKO mice show a significantly larger DT (DT;red) and lateral supracondylar ridge (LSR; yellow) in Fgf9cKO mice. Histology of the DT support this phenotype (B, F). Skeletal muscle specificity of Acta1-Cre was confirmed using Ai14 reporters (H,G). Gene expression of (I) Agrn (p = 0.0287) and (J) Ikbkb (p = 0.0675) were increased in Fgf9cKO deltoid muscle compared to WT muscle at 8 weeks of age (ΔCT calculated using average reference gene CT of ipo8 and Rn18s). Immunofluorescence (IF) was used to visualize nerves (neurofilament, green) and AChR clusters at neuromuscular junctions (α-bungarotoxin, red) in (K) WT and (L) Fgf9cKO mice. At 3 weeks postnatally, Fgf9cKO mice had significantly higher (N) AchR density than WT littermates and (M) more AChR. Data shown are biological replicates, mean ± SD. Lines in C, D, I, and N indicate significance (p < 0.05).
Figure 10: No difference in humerus length and DT bone volume/total volume (BV/TV) between Fgf9cKO and WT mice.
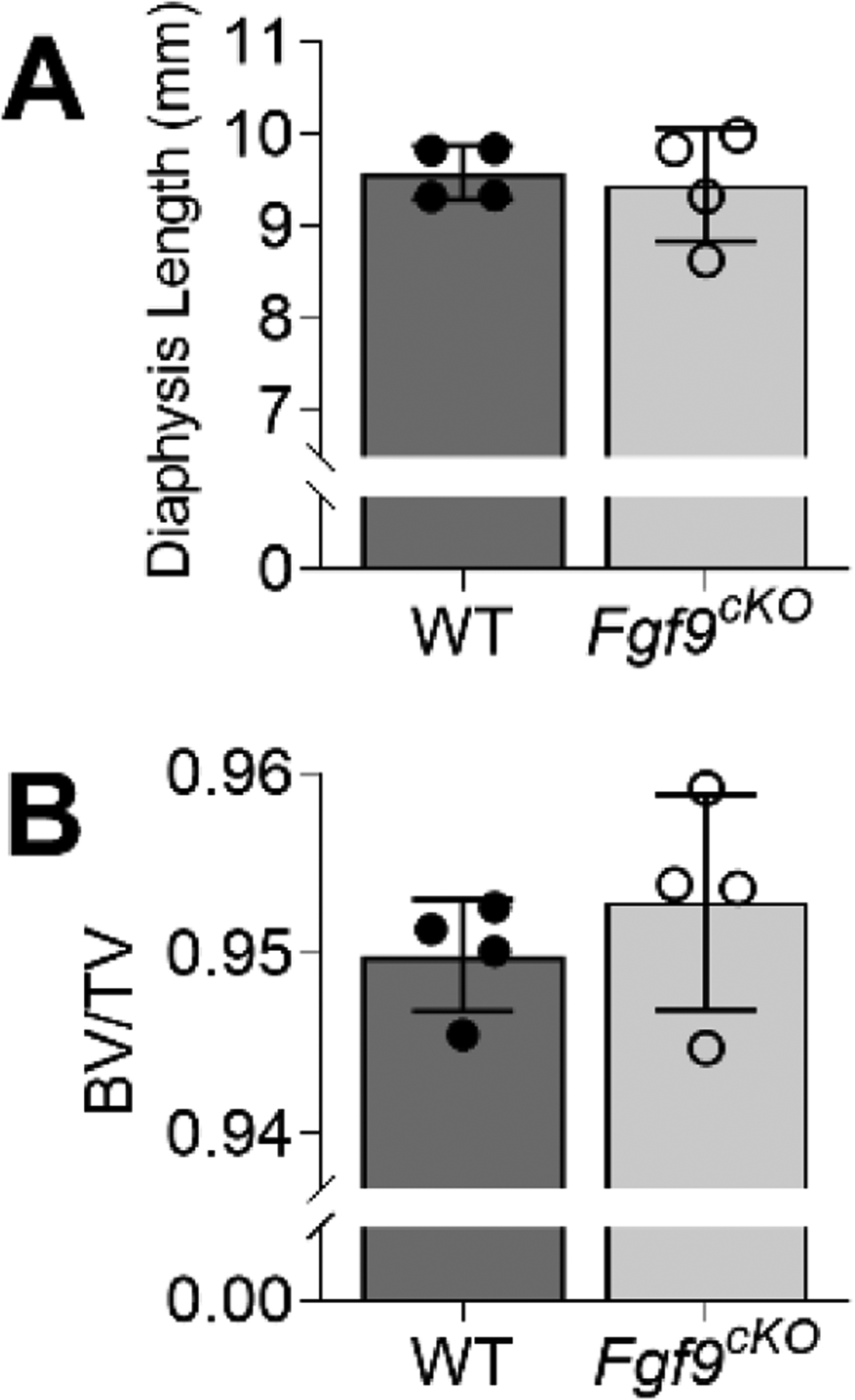
MicroCT scans of 8-week samples showed that (A) Fgf9cKO mice had a humerus of similar length when compared to WT littermates. (B) Though the DT was larger, BV/TV in Fgf9cKO and WT littermates. Data are biological replicates, mean ± SD.
Discussion
Intrinsic vs. extrinsic roles of FGF9 in DT development
In this study, we showed that Fgf9 may act as a negative regulator in growth of attachment superstructures such as the DT. Global deletion of Fgf9 led to enlarged superstructure size, and these superstructures had altered endochondral ossification of the arrested growth plate. This phenotype of enlarged DTs was modestly matched in conditional knockout of Fgf9 in skeletal muscle, suggesting a potential nonautonomous role of FGF9 during DT growth. The decreased proliferation we have shown in DTs of Fgf9null embryos, as well as enlarged hypertrophic chondrocytes, is similar to findings from previous work studying long bone development of Fgf9null embryos (Hung et al., 2007; Hung et al., 2016). The prevalence of hypertrophic chondrocytes at the tendon attachment in Fgf9null embryos may be suggestive of a prolonged progenitor cell maintenance along these superstructures (Blitz et al., 2013). Sox9 and hedgehog signaling (e.g., Gli1) are sequentially required for cellular expansion of growth plates during endochondral bone formation (Akiyama, 2002; Long, 2004; Mau et al., 2007; Pan et al., 2006; St-Jacques et al., 1999; Tan et al., 2018); however, migratory attachments (like the DT) form via replacement of Sox9 expressing cells by Gli1 expressing cells (Felsenthal et al., 2018). At E16.5, as DT enlargement begins in Fgf9null embryos, we observed increased Gli1 and Sox9 expression around the attachment. Indian hedgehog signaling also represses transcription of hypertrophy-related genes (Leung et al., 2011), and inhibits parathyroid hormone-like hormone (Pthlh) activity to maintain chondrocytes in a proliferative stage (Lanske et al., 1996). Though we saw similar levels of Pthlh expression in both WT and Fgf9null attachments, we did measure a decrease in cell proliferation in Fgf9null attachments, suggesting an accelerated chondrocyte maturation. This was supported by increased Gli1 expression in Fgf9null attachments compared to WT attachments. This decrease in proliferation was in parallel with increased hypertrophy of chondrocytes adjacent to the attachment cells within the Fgf9null DT, which may have led to a rapid expansion of matrix in the DT. Even though the DT was enlarged in Fgf9null mutants, we found fewer Sost+ cell clusters in their DTs compared to WT mice. Mature osteocytes express Sost (Winkler et al., 2003), and fewer Sost+ cells may indicate an impaired ability of Fgf9null osteoblasts to embed and mature into osteocytes. Overexpression of FGF9 in the perichondrium has been previously shown to suppress chondrocyte proliferation and limit bone growth in the limb (Karuppaiah et al., 2016); in our study, we found that loss of Fgf9 globally leads to an accelerated enlargement of chondrocytes in the tuberosity. This accelerated enlargement may limit the ability of these cells to deposit matrix and mineral and therefore limit osteocyte differentiation. We also found fewer osteoclasts in the Fgf9null DT which mirrors previous reports using the same mutation to study the length and vascularity of developing limb (Hung et al., 2007). Because the DT is enlarged and resides on the surface of a shortened bone, this phenotype may elucidate a divergent role of FGF9 in patterning of an arrested (e.g., attachment) growth plate compared to a regular (e.g., long bone) growth plate. This includes unexplored roles of FGF9 in vascularity of the tendon attachment and formation of bone ridges that overlap with or deviate from its role in growth plate development that are beyond the scope of the current study.
FGF ligands have varying affinities for receptors, and receptor expression changes throughout cell development (Su et al., 2014). The primary receptors of FGF9 during musculoskeletal development are Fgfr1, Fgfr2, and Fgfr3. Fgfr1 is highly expressed in hypertrophic chondrocytes (Peters et al., 1992), Fgfr1 and Fgfr2 are co-expressed in osteogenic cells (Ornitz and Marie, 2002; Peters et al., 1992), and Fgfr3 is predominately expressed in proliferating chondrocytes (Henderson et al., 2000). Given the striking role of Fgf9 deletion on tuberosity development shown in our study, FGF signaling may play a unique and cell-specific role during establishment and growth of the Sox9/Scx progenitor pool found within developing superstructures (Blitz et al., 2013). Previous work in craniofacial development has shown that Fgfr2 expression is necessary for maintaining Sox9/Scx progenitor pool cell fate (Roberts et al., 2019); however, spatial differences and regulatory roles of Fgf signaling between craniofacial and limb development are important to consider. For example, within the developing humerus, progenitor cell pools are temporally and transcriptionally different depending on location (e.g., proximal DT vs. distal olecranon superstructures) (Eyal et al., 2019). Additionally, Fgf9 predominantly effects proximal (stylopod) skeletal elements rather than distal (zeugopod, autopod) elements (Hung et al., 2007). Thus, the timing, spatial contribution, and expression patterns of Fgf9 on musculoskeletal development may be isolated to certain cells in proximal elements (Hung et al., 2007; Su et al., 2014).
The enlargement of the DT in Fgf9null embryos may also suggest differential rapid growth, as seen in the growth plate of some long bones in jerboa and bat (Cooper et al., 2013; Sears et al., 2006). For example, Cooper et al. showed that average chondrocyte volume was larger in the growth plate of the jerboa metatarsal compared to the mouse metatarsal, and the jerboa foot elongates 2.5 times more proportionally compared to the mouse foot (Cooper, 2011; Cooper et al., 2013). Additionally, increased proliferation and greater elongation of forelimb digits in bats compared to mice are likely driven by increased size of hypertrophic chondrocytes, which is predominantly mediated by BMP signaling (Sears et al., 2006). It is possible that the enlarged chondrocyte phenotype we observe histologically in the DT is an extended and accelerated growth of cell volume (Cooper et al., 2013). Notably, in our study, we did not see an increase in cell proliferation, which is attributed in part to rapid elongation of bat metatarsals (Galea et al., 2021; Sears et al., 2006). This divergent developmental pattern could differentiate the growth of arrested growth plates in superstructures like the DT from long bone growth plates.
Role of FGF9 in myogenesis
Fgf9 is expressed in muscle during embryonic stages, which we and others have observed using ISH (Colvin et al., 1999; Garofalo et al., 1999; Hung et al., 2007; Yang and Kozin, 2009). Previous work has established a connection between Fgf9 and muscle, as treatment of muscle and muscle progenitor cells with FGF9 slows maturation, enhances proliferation, and decreases expression of various myogenic genes (Huang et al., 2019). This study found supporting evidence that Fgf9 expression in muscle may be a limiting factor in tuberosity growth. However, it remains unknown how other FGFs and their receptors, FGFRs, regulate superstructure and attachment formation. In this study, we identified potential mediators of skeletal muscle metabolism in Fgf9null muscle, including downregulated mitochondrial-related genes associated with oxidative respiration and proton transport (i.e., Slc36a2 and Ucp1, amongst others). In cultured myoblasts, FGF9 can inhibit myogenic differentiation potentially via increased production of Myostatin (Huang et al., 2019), a well-established mediator of fast glycolytic muscle fibers (Girgenrath et al., 2005; Hennebry et al., 2009). While the role of FGF9 in myoblast fusion has been investigated in vitro, its effect on muscle fiber type and fiber metabolism (i.e., oxidative vs. glycolytic) has not yet been explored. Our findings from bulk RNA-seq of Fgf9null muscle point to potential mechanisms in muscle metabolism that may contribute to the enlarged phenotype that is mimetic of that found in Myostatin deficient mice and other animals (Elkasrawy and Hamrick, 2010; Hamrick et al., 2002). Conversely, following muscle paralysis in the developing duck jaw, the secondary cartilage at tendon entheses is lost (Solem et al., 2011). Additionally, this study found that expression of Fgfr2 in the enthesis of the duck jaw was regulated by activation of stretch-sensitive ion channels (Solem et al., 2011). Further investigations are needed to investigate the potential role of Fgf9 in mitochondrial function, ion transport, and lipid metabolism. Recent work by Huang et al. also identified FGF9 as a potent regulator of calcium signaling and homeostasis in myoblast culture in vitro, and calcium release from the sarcoplasmic reticulum in muscle plays a critical role during embryonic skeletal myogenesis via ryanodine receptor 1 (RYR1) (Huang et al., 2019). Although Ryr1 was not significantly different in between Fgf9null and WT muscle in the present study, we did find that calmodulin-associated genes (e.g., Calm4, Calml3, Camsap3, Calm5) were all significantly upregulated in Fgf9null muscle compared to WT muscle. Calmodulin interacts with RYR1 and its activation is required for intracellular binding of calcium (Newman et al., 2014, 1). Calmodulin is a crucial component of the calcium signal transduction pathway and also plays an important role in lipid and glucose metabolism (Nishizawa et al., 1988). Taken together, our findings along with recent work by Huang et al. support more mechanistic studies to investigate the metabolic effects of loss and gain of function of Fgf9 on skeletal muscle as well as the muscle secretome.
In conclusion, this work established a new role of skeletal muscle derived Fgf9 during skeletal development and tuberosity growth. Additionally, our unbiased transcriptomic approaches and rigorous analyses identified new potential mechanisms associated with muscle development, mitochondrial bioenergetics, and muscle metabolism that warrant further investigation into the role of FGF9 in muscle-bone crosstalk.
Material and Methods
Mice
Prrx1-Cre, Ai14(RCL-tdT)-D (Ai14), and Acta1-rtTA-tetO-Cre (Acta1-Cre) transgenic mice were obtained from Jackson Laboratory (The Jackson Laboratory, Bar Harbor, ME, USA). Fgf9-floxed mice were generated as previously described (Colvin et al., 1999; Colvin et al., 2001). Mice carrying the Fgf9 excised allele were maintained on a C57BL6J background. All experiments were performed with approval from the University of Delaware and University of Michigan Institutional Animal Care and Use Committees.
Breeding and genotyping
For global mutants, Fgf9ex/WT; Prrx1-Cre female mice were bred with Fgf9flx/flx male (mixed background) mice to generate pups with homozygous excised Fgf9 alleles (Fgf9null) and Fgf9WT/Ex (WT) littermates. The exploitation of the Prrx1-Cre female donors resulted in global excision of floxed alleles in utero, thereby generating global knockouts (Colvin et al., 2001). Embryos and neonates were used for all experiments. For timed matings, we considered noon on the day following mating as 0.5 days post coitum (0.5 dpc). For muscle-specific knockouts, Fgf9flx/WT; Acta1-Cre mice were crossed with Fgf9flx/flx mice and dams were given doxycycline chow ad libitum throughout gestation and weaning. To confirm that muscle was being targeted, Acta1-Cre mice were crossed with Ai14 reporter mice and dams were also given doxycycline chow ad libitum throughout gestation and weaning. Muscle targeting was confirmed via red fluorescent protein expression in Ai14; Acta1-Cre mouse muscle (Figure 6H). Genotypes were determined by PCR using commercial vendors (Transnetyx, Cordova, TN, USA). The efficiency of Cre-recombination and confirmation of Fgf9null gene knockout was determined using quantitative reverse transcription-polymerase chain reaction (qRT-PCR) (WT n=3; Fgf9null n=3). RNA was isolated from whole limbs using phenol/chloroform and on-column commercially available kits (Invitrogen, ThermoFisher). RNA was converted to cDNA at 5ng/ul concentrations for all samples using Superscript (VILO, ThermoFisher) and cDNA samples were amplified as technical replicates (in triplicate) using SYBR green (PerfeCTa, QuantBio) and primers (IDT). ΔCT values were calculated using Rn18s as the reference gene. Relative expression was then calculated using 2−ΔCT. Gene expression of Fgf9 was not detected and Scx expression was reduced in Fgf9null limbs (Figure 11).
Figure 11: Gene expression of Scx, Bmp4, and Fgf9 from WT and Fgf9null embryonic limbs at E18.5.
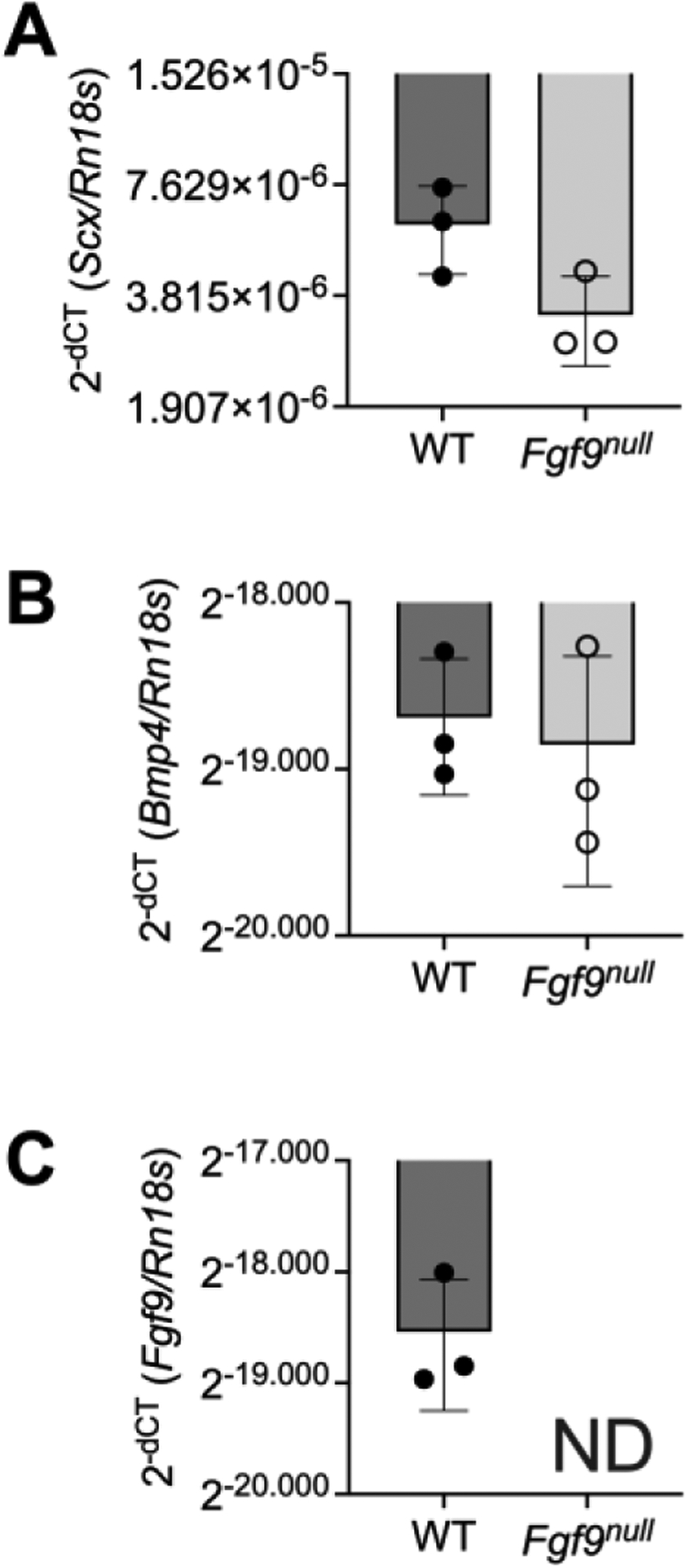
Relative expression are biological replicates (2−ΔCT), mean ± SD. ND = not detected or CT value greater than or equal to 35.
Measurement of DT size
For global mutants, whole-mount staining was used following a standard protocol (Rigueur and Lyons, 2014). Arms of P0 neonates (WT n=7; Fgf9null n=14) were removed from whole-mount skeletons and placed in a Petri dish filled with glycerol with the anterior side facing up. In-plane with the arm, we used a ruler at the bottom of the petri dish for scaling. Samples were imaged using a Stemi dissection microscope (Carl Zeiss, Gottingen, Germany) and imaged using (EOS REBEL T5, Canon, Melville, New York, USA). The size of the DT and humerus were measured using ImageJ (Schneider et al., 2012). When we observed no significant difference in mineralized humerus area and DT area combined (Figure 2B), we based DT size on the ratio of DT area to mineralized humerus area. Red mineral staining determined the mineralized zone, and DT measurement included any cartilage observed at the crest of the DT. We determined the relative placement of the DT based on a ratio of length distal to DT over the length of the humerus. Both mineralized humerus and full humerus were measured. The distal length was based on the distance between the most distal part of the DT and the most distal part of the mineralized/full humerus. Humerus length was based on the distance between the most proximal part of the mineralized/full humerus and the most distal part of the mineralized/full humerus. Once we determined no difference between left and right arms, values were averaged together.
Histology and in situ hybridization
Embryonic and neonatal mice were eviscerated and fixed in 4% paraformaldehyde (PFA) for 24 hours. Forelimbs for histology of a subset of embryonic age 18.5 (E18.5) embryos and P0 neonates were decalcified using formic acid (Formical 2000, Statlab, McKinney, TX, USA) for 24 hours. Histomorphometry was carried out on 7 μm paraffin sections of samples from E15.5 to P0 (n= at least 3 per genotype/time point). Samples were stained using hematoxylin and eosin (H&E), coverslipped with acrymount (Statlab, McKinney, TX, USA), and imaged with an Upright microscope (Carl Zeiss, Gottingen, Germany). The area and number of hypertrophic chondrocytes were measured in the DT of E16.5 embryos using the freehand and multipoint tools in ImageJ (Schneider et al., 2012). Non-decalcified E16.5 and P0 samples had mineralized bone visualized by von Kossa staining (n=3 per age and genotype) with Nuclear Fast Red counterstaining as part of an established protocol (Abcam, Cambridge, UK). For osteoclast staining, P0 neonates were decalcified using 14% ethylene-diamine-tetraacetic acid (EDTA), paraffin sectioned, and stained for TRAP (n=3 per age and genotype). We performed in situ hybridization using the RNAscope Multiplex Fluorescent Reagent Kit v2 (Advanced Cell Diagnostics, Hayward, CA, USA). Non decalcified E16.5 and P0 samples (n=3 per age and genotype) were treated with Fgf9, Fgf18, Gli1, Pthlh, Scx, Sost, Sox9, and Wnt7b probes (Advanced Cell Diagnostics, Hayward, CA, USA) for 2 hours at 40C. A positive control slide used a mixture of probes targeting housekeeping genes Polr2A, Ppib, and Ubc. A negative control slide used dapB (a bacteria gene). Nuclei were counterstained with DAPI and mounted with Citifluor Antifade mounting medium (Electron Microscopy Science, Hatfield, PA, USA). Slides were imaged using Zeiss Axio-observer Z1 microscope (Axio.Observer Z1, Carl Zeiss, Gottingen, Germany).
Immunohistochemistry
Embryonic and neonatal mice were eviscerated and fixed in 4% PFA for 24 hours (n=3 age and genotype). Forelimbs for histology of P0 neonates were decalcified using 14% EDTA for 24 hours. Histomorphometry was carried out on 7 μm paraffin sections of samples from E15.5 and P0 (n= at least 3 per genotype/time point). Slides were deparaffinized and hydrated to 70% ETOH, followed by heat mediated antigen retrieval (Sodium Citrate Buffer, pH 6.0) at 65°C for 2 hours. Slides were kept in the solution until equilibrated to room temperature, quenched in 0.3% hydrogen peroxide (Santa Cruz, Dallas, TX) for 30 minutes and blocked with 5% goat serum in 1% phosphate buffer saline in room temperature for 1 hour. For primary antibody, rabbit monoclonal anti-collagen X antibody (ab260040; 1:1000 dilution) (ABCAM, Cambridge, MA, USA) was used for overnight incubation at 4°C. Following incubation, HRP/DAB system (Millipore Sigma) was used for detection. Samples were then counterstained with hematoxylin and mounted with a xylene-based mounting medium (Acrymount, Stat Lab, McKinney, TX, USA) and imaged using a Lionheart FX (BioTek, Winooski, Vermont, USA). 2 P0 (n=1 per genotype) samples were omitted from analysis due to surplus nonspecific staining.
EdU proliferation
A dam at E16.5 in gestation was given an intraperitoneal (IP) injection with EdU (5-ethynyl- 2’-deoxyuridine) from the Click-iT™ EdU Alexa FluorTM 647 Imaging Kit (Invitrogen, Carlsbad, CA, USA) at a dosage of 0.25mg /10g body weight. The dam was euthanized two hours later through CO2 asphyxiation. Embryos were eviscerated and fixed in 4% PFA for 24 hours. Forelimbs were processed for paraffin histology, and humeri were cut in the transverse plane at 7 μm. Proliferating cells were stained with Alexa Fluor 647 and Hoechst 33342 as a counterstain. Slices were mounted with Citifluor Antifade Mountant. Two histological sections per sample treated with EdU (WT n=4; Fgf9null n=3) were imaged using a Zeiss Axio-observer Z1 microscope (Axio.Observer. Z1, Carl Zeiss, Gottingen, Germany). Differential interference contrast (DIC) images and fluorescence images were collected from the same observed tissue to visualize tissue morphology. DIC allowed us to observe the highly organized structure of the tendon and perichondrium. Regions were separated into perichondrium, tendon, and attachment. Tissue area, nuclei number, and the number of EdU positive proliferating cells were measured and counted within ImageJ using the freehand and multipoint tools. Two slides per sample were averaged together. For the tendon and perichondrium, tissues on the medial and lateral side of the DT were measured separately. Once no phenotypic difference based on the side of tissue was observed, medial and lateral values were summed together.
Evaluation of bone deposition and resorption
For dynamic bone histomorphometry, Calcein (Sigma, C0875; 15 mg/kg body weight; 12 pm) was injected into pregnant females intraperitoneally (IP) at E16.5 and Alizarin Complexone (Sigma, a3882; 45 mg/kg IP; 2 days later at 6 pm) at E18.5 with a set time in between of 42 hours. Neonates were collected at birth, eviscerated, and fixed for 24 hours in 4% PFA. After paraffin embedding, samples were sectioned at 7um. Slides were deparaffinized, dehydrated, and mounted with Acrymount. Slides were imaged using Zeiss Axio-observer Z1 microscope (Axio.Observer. Z1, Carl Zeiss, Gottingen, Germany). Calcein Green and Alizarin Red were imaged using fluorescence microscopy and quantified using ImageJ by using the freehand tool to measure the area of green and red stained mineral. The mineralization area (MA) of the DT was measured, and the mineral growth rate was calculated using:
| Eqn 1. |
Bulk RNA-Sequencing
E18.5 embryos were collected, toed, and eviscerated under sterile conditions. RNA was extracted from each sample (n=3 WT and n = 3 Fgf9null) using Trizol/chloroform and spin-column assembly with on-column DNase treatment (RNeasy mini kits; Qiagen Germantown, MD). We assessed the quality of RNA using fragment analysis to obtain RNA integrity number (RIN) before proceeding with Library preparation (NEBNext® Ultra™ RNA Library Prep Kit, Illumina, San Diego, CA, USA). Each sample was sequenced in Illumina HiSeq 2000 (Single read, 50 base pair). Sequence reads from each sample were quality assessed (FastQC) & all the reads were quality trimmed and filtered (threshold length of the sequence was 30 & Phred score of each base was 28). Reads were mapped to the Mus musculus reference genome (mm10) using TopHat2 with default settings (ver 2.1.0)(Kim et al., 2013) and mapping quality control metrics were obtained fro RSeQC (ver 2.6.1)(Wang et al., 2012). Per gene counts were determined using HTseq (ver 0.6.1p1)(Anders et al., 2015) and used for pairwise determination of differential expression analyses using EdgeR (ver 3.28.1) (Robinson et al., 2010) with a significance cutoff of FDR-corrected p<0.05. To control for consistency within biological replicates, a secondary analysis was performed to exclude DE within a group if the mean count per million for a gene feature in a given sample was >log2FC different than its biological replicates (yielding 135 genes). These 135 genes were then uploaded to Morpheus, which assigned a z score to determine the top 30 upregulated and downregulated genes and generate a heatmap (Morpheus, https://software.broadinstitute.org/morpheus). Both sample pools (primary analysis of 805 and secondary analysis of 135) were then uploaded to DAVID Bioinformatics Resources 6.8 (Huang et al., 2009a; Huang et al., 2009b) and run against a mm10 background. GOTERM cellular components direct, GOTERM biological processes direct, and GOTERM molecular functions direct libraries were examined, and terms were ranked based on Benjamini number (DAVID adjusted p value) (Benjamini and Hochberg, 1995). Terms with a Benjamini number < 0.05 were percentiled and categorized based on similarity.
Skeletal Muscle Immunofluorescence
Muscle-specific Fgf9 knockouts and WT littermates were euthanized at 3 weeks of age (n=4 WT, n=3 Fgf9cKO) and 8 weeks of age (n=4 WT, n=4 Fgf9cKO) for histology, structural analysis, and RNA isolation. Immediately following euthanasia, the right deltoid muscles of each mouse were dissected in an RNase-free environment, flash-frozen in liquid nitrogen, and stored at −80°C until RNA isolation. The left deltoid muscles were then dissected, fixed in 4% PFA for 20 minutes, cryo-protected, and then embedded in optimal cutting temperature (OCT) media for cryosectioning (longitudinal plane). The left humerus from each mouse was also dissected, fixed in 4% PFA, and stored in 70% ethanol.
The immunofluorescence staining protocol used in this study was based on previously established protocol (Mead et al., 2011). For 3-week and 8-week old deltoids, 35 μm thick sections were used immunohistochemistry (IHC) of acetylcholine receptors (α-Bungarotoxin extracted from Bungarus muticinctus, conjugated to Alexa Fluor 647, Thermofisher #B35450) and nerve fibers (chicken anti-neurofilament-H antibody, Thermofisher #PA1–10002; Alexa Fluor 488 donkey anti-chicken, Jackson Immunofluorescence #703-545-155). Samples were then imaged using a Zeiss Axio-observer Z1 microscope (Axio.Observer Z1, Carl Zeiss, Gottingen, Germany) at 20X magnification subfields which were then stitched in order to obtain a complete image of the ventral, middle, and dorsal heads of the deltoid. Images were visualized using fluorescent microscopy (neurofilament conjugated to 488 nm and neuromuscular junctions conjugated to 647 nm). AChR were counted in ImageJ (Schneider et al., 2012) using the multipoint tool. Muscle area was collected by opening the file in Dragonfly software (Object Research Systems, Montreal, Quebec, Canada) and separated by color channel. Muscle regions of interest were selected and area measured based on autofluorescence in GFP channel.
Micro-computed Tomography
Micro-computed tomography (microCT) was used to obtain 3-dimensional renderings of the left humerus of 8-week old mice (n=4 WT, n=4 Fgf9cKO) to visualize topographical data on DT volume, LSR volume, DT bone volume/ total volume (BV/TV), humerus diaphysis length. MicroCT scans were performed using a Scanco μCT 35 (X-ray energy: 60kV; X-ray intensity: 95ma; integration time: 1100msec; voxel size: 10.6μm; rotation step: 0.4°)(Bouxsein et al., 2010). All raw scans were reconstructed and analyzed using Dragonfly software (Object Research Systems, Montreal, Quebec, Canada). Scans were aligned down the humeral axis and a box form was created to extract the bone ridges of interest. Using the spit at OTSU function, bone volume was selected based on voxel value. The bone analysis function was then used to fill in the bone volume and give the total volume. Diaphysis length was measured from the proximal growth plate to the Coronoid Fossa. After scans were obtained, humeri were decalcified in 14% EDTA, processed for paraffin embedding, sectioned at 7um thick, and stained with H&E. One Fgf9cKO sample and one WT sample were omitted due to processing errors.
Neural/Muscle-related Gene Expression
Fgf9cKO and WT littermates were aged to 3 weeks (n=3 WT, n=3 Fgf9cKO) and 8 weeks of age (n=4 WT, n=3 Fgf9cKO). qRT-PCR was used to compare differential gene expression of skeletal muscle using muscle contractility genes (Dmpk, Mef2c, and Ikbkb) and neural genes (Nes, Agrn, Gap43, Ache, Rapsn, Chrne, and Musk). Neural genes were selected for their role in the development and stabilization of neurons and neuromuscular junctions as well as for their role in the signal propagation and firing pathway. Contractility genes were chosen for their role in encoding genes necessary for cell-cell communication and differentiation, skeletal muscle contraction and relaxation, myogenesis, and homeostasis (Bakkar et al., 2008). qRT-PCR, samples were amplified in triplicate and averaged together to calculate and ΔCT values with the average ΔCT of ipo8 and Rn18s serving as the reference gene value. ΔCT values were converted to log-scale (log2ΔCT). Log scale was then presented as mean ± SD.
Statistical Analysis
Statistical analyses were performed in Prism (Prism GraphPad 6.0, LaJolla, CA, USA). For whole-mount area measurements, DT area was divided by the humerus area. For relative DT placement, distal length was divided by the whole length across the mineralized and whole humerus. The resulting ratios were compared through separate two-sample t-tests. Hypertrophic cell area measurements were averaged together per mouse and a two-sample t-test compared the number of hypertrophic chondrocytes and the average hypertrophic chondrocyte area between Fgf9null embryos and WT embryos. A two-way ANOVA was run to compare cell density, proliferating cell density, and percent of proliferating cells for the EdU proliferation assay. The two-way ANOVA looked for differences between WT and Fgf9null embryos in the perichondrium, tendon, and deltoid-tendon attachment. Whole-mount, hypertrophic cell, and EdU quantifications were repeated by a separate observer through coded files to minimize bias. Two-way ANOVA was used to compare the osteoclast number between WT and Fgf9null samples at E16.5 and P0. For dynamic bone histomorphometry, 3 slides per sample were measured, and mineral growth rate was calculated. Technical replicates were averaged together per sample, and a two-sample t-test was performed. A two-sample t-test was used to compare Fgf9cKO gene expression. A two-sample t-test was used to analyze LSR volume, DT volume, DT BV/TV, and humeri length between Fgf9cKO and WT mice at 8 weeks. Two-way ANOVA was used to compare gene expression and AchR clustering between Fgf9cKO and WT mice at 3 week and 8-week time points.
Acknowledgments:
We thank Brewster Kingham, Summer Thompson, and Mark Shaw in the University of Delaware DNA Sequencing & Genotyping Center for assistance with for assistance with RNA sequencing; Shaneaka Anderson, Kierstyn Hendricks, and Kayleigh Denner for help with histology; Crystal Idleburg (Washington University) for TRAP staining; Karol Miaskiewicz for high performance computing support.
Funding:
National Institutes of Health (NIGMS P30GM103333 (IDeA); NICHD R03HD094594; NICHD K12HD073945; NIGMS P20GM103446 (IDeA); NIAMS P30AR074992; R01HD049808); NSF (CAREER 1944448); University of Delaware Research Foundation; University of Delaware Doctoral Fellowship.
Works Cited
- Akiyama H (2002). The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes & Development 16, 2813–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvind V and Huang AH (2017). Mechanobiology of limb musculoskeletal development. Ann. N. Y. Acad. Sci 1409, 18–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkar N, Wang J, Ladner KJ, Wang H, Dahlman JM, Carathers M, Acharyya S, Rudnicki MA, Hollenbach AD and Guttridge DC (2008). IKK/NF-κB regulates skeletal myogenesis via a signaling switch to inhibit differentiation and promote mitochondrial biogenesis. Journal of Cell Biology 180, 787–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin M, Toumi H, Ralphs JR, Bydder G, Best TM and Milz S (2006). Where tendons and ligaments meet bone: attachment sites (‘entheses’) in relation to exercise and/or mechanical load. Journal of Anatomy 208, 471–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y and Hochberg Y (1995). Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society: Series B (Methodological) 57, 289–300. [Google Scholar]
- Berendsen AD and Olsen BR (2015). Bone development. Bone 80, 14–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biewener AA, Fazzalari NL, Konieczynski DD and Baudinette RV (1996). Adaptive changes in trabecular architecture in relation to functional strain patterns and disuse. Bone 19, 1–8. [DOI] [PubMed] [Google Scholar]
- Blitz E, Viukov S, Sharir A, Shwartz Y, Galloway JL, Price BA, Johnson RL, Tabin CJ, Schweitzer R and Zelzer E (2009). Bone ridge patterning during musculoskeletal assembly is mediated through SCX regulation of Bmp4 at the tendon-skeleton junction. Dev Cell 17, 861–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitz E, Sharir A, Akiyama H and Zelzer E (2013). Tendon-bone attachment unit is formed modularly by a distinct pool of Scx- and Sox9-positive progenitors. Development 140, 2680–2690. [DOI] [PubMed] [Google Scholar]
- Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ and Müller R (2010). Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J Bone Miner Res 25, 1468–1486. [DOI] [PubMed] [Google Scholar]
- Colvin JS, Feldman B, Nadeau JH, Goldfarb M and Ornitz DM (1999). Genomic organization and embryonic expression of the mouse fibroblast growth factor 9 gene. Dev. Dyn 216, 72–88. [DOI] [PubMed] [Google Scholar]
- Colvin JS, White AC, Pratt SJ and Ornitz DM (2001). Lung hypoplasia and neonatal death in Fgf9-null mice identify this gene as an essential regulator of lung mesenchyme. Development 128, 2095–2106. [DOI] [PubMed] [Google Scholar]
- Cooper KL (2011). The lesser Egyptian jerboa, Jaculus jaculus: a unique rodent model for evolution and development. Cold Spring Harb Protoc 2011, 1451–1456. [DOI] [PubMed] [Google Scholar]
- Cooper KL, Oh S, Sung Y, Dasari RR, Kirschner MW and Tabin CJ (2013). Multiple phases of chondrocyte enlargement underlie differences in skeletal proportions. Nature 495, 375–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutu DL, François M and Galipeau J (2011). Inhibition of cellular senescence by developmentally regulated FGF receptors in mesenchymal stem cells. Blood 117, 6801–6812. [DOI] [PubMed] [Google Scholar]
- Deng C, Wynshaw-Boris A, Zhou F, Kuo A and Leder P (1996). Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell 84, 911–921. [DOI] [PubMed] [Google Scholar]
- Elkasrawy MN and Hamrick MW (2010). Myostatin (GDF-8) as a key factor linking muscle mass and bone structure. Journal of Musculoskeletal Neuronal Interactions 10, 56–63. [PMC free article] [PubMed] [Google Scholar]
- Eyal S, Kult S, Rubin S, Krief S, Felsenthal N, Pineault KM, Leshkowitz D, Salame T-M, Addadi Y, Wellik DM, et al. (2019). Bone morphology is regulated modularly by global and regional genetic programs. Development 146, dev167882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenthal N, Rubin S, Stern T, Krief S, Pal D, Pryce BA, Schweitzer R and Zelzer E (2018). Development of migrating tendon-bone attachments involves replacement of progenitor populations. Development 145,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galea GL, Zein MR, Allen S and Francis-West P (2021). Making and shaping endochondral and intramembranous bones. Dev Dyn 250, 414–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garofalo S, Kliger-Spatz M, Cooke JL, Wolstin O, Lunstrum GP, Moshkovitz SM, Horton WA and Yayon A (1999). Skeletal dysplasia and defective chondrocyte differentiation by targeted overexpression of fibroblast growth factor 9 in transgenic mice. J. Bone Miner. Res 14, 1909–1915. [DOI] [PubMed] [Google Scholar]
- Girgenrath S, Song K and Whittemore L-A (2005). Loss of myostatin expression alters fiber-type distribution and expression of myosin heavy chain isoforms in slow- and fast-type skeletal muscle. Muscle Nerve 31, 34–40. [DOI] [PubMed] [Google Scholar]
- Govindarajan V and Overbeek PA (2006). FGF9 can induce endochondral ossification in cranial mesenchyme. BMC Dev. Biol 6, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamburger V and Waugh M (1940). The Primary Development of the Skeleton in Nerveless and Poorly Innervated Limb Transplants of Chick Embryos. Physiological Zoology 13, 367–382. [Google Scholar]
- Hamrick MW, McPherron AC and Lovejoy CO (2002). Bone Mineral Content and Density in the Humerus of Adult Myostatin-Deficient Mice. Calcif Tissue Int 71, 63–68. [DOI] [PubMed] [Google Scholar]
- Henderson JE, Naski MC, Aarts MM, Wang D, Cheng L, Goltzman D and Ornitz DM (2000). Expression of FGFR3 with the G380R Achondroplasia Mutation Inhibits Proliferation and Maturation of CFK2 Chondrocytic Cells. J Bone Miner Res 15, 155–165. [DOI] [PubMed] [Google Scholar]
- Hennebry A, Berry C, Siriett V, O’Callaghan P, Chau L, Watson T, Sharma M and Kambadur R (2009). Myostatin regulates fiber-type composition of skeletal muscle by regulating MEF2 and MyoD gene expression. Am J Physiol Cell Physiol 296, C525–534. [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT and Lempicki RA (2009a). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Research 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT and Lempicki RA (2009b). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Huang J, Wang K, Shiflett LA, Brotto L, Bonewald LF, Wacker MJ, Dallas SL and Brotto M (2019). Fibroblast growth factor 9 (FGF9) inhibits myogenic differentiation of C2C12 and human muscle cells. Cell Cycle 18, 3562–3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung IH, Yu H, Lavine KJ and Ornitz DM (2007). FGF9 regulates early hypertrophic chondrocyte differentiation and skeletal vascularization in the developing stylopod. Dev Biol 307, 300–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung IH, Schoenwolf GC, Lewandoski M and Ornitz DM (2016). A combined series of Fgf9 and Fgf18 mutant alleles identifies unique and redundant roles in skeletal development. Dev. Biol 411, 72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob AL, Smith C, Partanen J and Ornitz DM (2006). Fibroblast growth factor receptor 1 signaling in the osteo-chondrogenic cell lineage regulates sequential steps of osteoblast maturation. Dev. Biol 296, 315–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn J, Shwartz Y, Blitz E, Krief S, Sharir A, Breitel DA, Rattenbach R, Relaix F, Maire P, Rountree RB, et al. (2009). Muscle contraction is necessary to maintain joint progenitor cell fate. Dev. Cell 16, 734–743. [DOI] [PubMed] [Google Scholar]
- Karuppaiah K, Yu K, Lim J, Chen J, Smith C, Long F and Ornitz DM (2016). FGF signaling in the osteoprogenitor lineage non-autonomously regulates postnatal chondrocyte proliferation and skeletal growth. Development 143, 1811–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanske B, Karaplis AC, Lee K, Luz A, Vortkamp A, Pirro A, Karperien M, Defize LHK, Ho C, Mulligan RC, et al. (1996). PTH/PTHrP Receptor in Early Development and Indian Hedgehog--Regulated Bone Growth. Science 273, 663–666. [DOI] [PubMed] [Google Scholar]
- Leung VYL, Gao B, Leung KKH, Melhado IG, Wynn SL, Au TYK, Dung NWF, Lau JYB, Mak ACY, Chan D, et al. (2011). SOX9 Governs Differentiation Stage-Specific Gene Expression in Growth Plate Chondrocytes via Direct Concomitant Transactivation and Repression. PLoS Genet 7, e1002356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long F (2004). Ihh signaling is directly required for the osteoblast lineage in the endochondral skeleton. Development 131, 1309–1318. [DOI] [PubMed] [Google Scholar]
- Lu J, Dai J, Wang X, Zhang M, Zhang P, Sun H, Zhang X, Yu H, Zhang W, Zhang L, et al. (2015). Effect of fibroblast growth factor 9 on the osteogenic differentiation of bone marrow stromal stem cells and dental pulp stem cells. Molecular Medicine Reports 11, 1661–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mau E, Whetstone H, Yu C, Hopyan S, Wunder JS and Alman BA (2007). PTHrP regulates growth plate chondrocyte differentiation and proliferation in a Gli3 dependent manner utilizing hedgehog ligand dependent and independent mechanisms. Developmental Biology 305, 28–39. [DOI] [PubMed] [Google Scholar]
- Mead RJ, Bennett EJ, Kennerley AJ, Sharp P, Sunyach C, Kasher P, Berwick J, Pettmann B, Battaglia G, Azzouz M, et al. (2011). Optimised and Rapid Pre-clinical Screening in the SOD1G93A Transgenic Mouse Model of Amyotrophic Lateral Sclerosis (ALS). PLoS ONE 6, e23244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman RA, Sorensen BR, Kilpatrick AM and Shea MA (2014). Calcium-Dependent Energetics of Calmodulin Domain Interactions with Regulatory Regions of the Ryanodine Receptor Type 1 (RyR1). Biophys Chem 0, 35–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizawa Y, Okui Y, Inaba M, Okuno S, Yukioka K, Miki T, Watanabe Y and Morii H (1988). Calcium/calmodulin-mediated action of calcitonin on lipid metabolism in rats. J Clin Invest 82, 1165–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohbayashi N, Shibayama M, Kurotaki Y, Imanishi M, Fujimori T, Itoh N and Takada S (2002). FGF18 is required for normal cell proliferation and differentiation during osteogenesis and chondrogenesis. Genes Dev. 16, 870–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz DM and Itoh N (2015). The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip Rev Dev Biol 4, 215–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz DM and Marie PJ (2002). FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 16, 1446–1465. [DOI] [PubMed] [Google Scholar]
- Ornitz DM and Marie PJ (2015). Fibroblast growth factor signaling in skeletal development and disease. Genes Dev. 29, 1463–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai AC (1965). Developmental genetics of a lethal mutation, muscular dysgenesis (mdg), in the mouse. Developmental Biology 11, 82–92. [DOI] [PubMed] [Google Scholar]
- Pan Y, Bai CB, Joyner AL and Wang B (2006). Sonic hedgehog Signaling Regulates Gli2 Transcriptional Activity by Suppressing Its Processing and Degradation. Molecular and Cellular Biology 26, 3365–3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters KG, Werner S, Chen G and Williams LT (1992). Two FGF receptor genes are differentially expressed in epithelial and mesenchymal tissues during limb formation and organogenesis in the mouse. Development 114, 233–243. [DOI] [PubMed] [Google Scholar]
- Rigueur D and Lyons KM (2014). Whole-mount skeletal staining. Methods Mol. Biol 1130, 113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RR, Bobzin L, Teng CS, Pal D, Tuzon CT, Schweitzer R and Merrill AE (2019). FGF signaling patterns cell fate at the interface between tendon and bone. Development 146, dev170241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rot-Nikcevic I, Reddy T, Downing KJ, Belliveau AC, Hallgrímsson B, Hall BK and Kablar B (2006). Myf5−/− :MyoD−/− amyogenic fetuses reveal the importance of early contraction and static loading by striated muscle in mouse skeletogenesis. Dev. Genes Evol 216, 1–9. [DOI] [PubMed] [Google Scholar]
- Sahni M, Ambrosetti DC, Mansukhani A, Gertner R, Levy D and Basilico C (1999). FGF signaling inhibits chondrocyte proliferation and regulates bone development through the STAT-1 pathway. Genes Dev. 13, 1361–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz AG, Long F, Thomopoulos S, Karp S, Gaffield W, McMahon AP and Vortkamp A (2015). Enthesis fibrocartilage cells originate from a population of Hedgehog-responsive cells modulated by the loading environment. Development (Cambridge, England) 142, 196–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears KE, Behringer RR, Rasweiler JJ and Niswander LA (2006). Development of bat flight: morphologic and molecular evolution of bat wing digits. Proc Natl Acad Sci U S A 103, 6581–6586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solem RC, Eames BF, Tokita M and Schneider RA (2011). Mesenchymal and mechanical mechanisms of secondary cartilage induction. Developmental Biology 356, 28–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Jacques B, Hammerschmidt M and McMahon AP (1999). Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 13, 2072–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su N, Jin M and Chen L (2014). Role of FGF/FGFR signaling in skeletal development and homeostasis: learning from mouse models. Bone Res 2, 14003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Z, Niu B, Tsang KY, Melhado IG, Ohba S, He X, Huang Y, Wang C, McMahon AP, Jauch R, et al. (2018). Synergistic co-regulation and competition by a SOX9-GLI-FOXA phasic transcriptional network coordinate chondrocyte differentiation transitions. PLoS Genet 14, e1007346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay P, Dietrich S, Mericskay M, Schubert FR, Li Z and Paulin D (1998). A crucial role for Pax3 in the development of the hypaxial musculature and the long-range migration of muscle precursors. Dev. Biol 203, 49–61. [DOI] [PubMed] [Google Scholar]
- Vaahtokari A, Aberg T and Thesleff I (1996). Apoptosis in the developing tooth: association with an embryonic signaling center and suppression by EGF and FGF-4. Development 122, 121–129. [DOI] [PubMed] [Google Scholar]
- Wang L, Wang S and Li W (2012). RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185. [DOI] [PubMed] [Google Scholar]
- Winkler DG, Sutherland MK, Geoghegan JC, Yu C, Hayes T, Skonier JE, Shpektor D, Jonas M, Kovacevich BR, Staehling-Hampton K, et al. (2003). Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J 22, 6267–6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y and Kozin SH (2009). Cell signaling regulation of vertebrate limb growth and patterning. J Bone Joint Surg Am 91 Suppl 4, 76–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelzer E, Blitz E, Killian ML and Thomopoulos S (2014). Tendon-to-bone attachment: from development to maturity. Birth Defects Res. C Embryo Today 102, 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]