

Abstract
Sex differences in opioid analgesia occur in rodents and humans, and could be due to differences in drug and metabolite levels. Thus, we investigated the sex and cycle differences in analgesia (nociception) from oxycodone in rats and related these to sex and cycle differences in brain and plasma oxycodone and metabolite levels. Since numerous opioids are CYP2D enzyme substrates and variation in CYP2D alters opioid drug levels and response, we also initiated studies to see if the sex and cycle differences observed might be due to differences in brain CYP2D activity. Across oxycodone doses, females in diestrus had higher analgesia (using tail flick latency) compared to males and females in estrus; we also demonstrated a direct effect of estrous cycle on analgesia within females. Consistent with the analgesia, females in diestrus had highest brain oxycodone levels (assessed using microdialysis) compared to males and females in estrus. Analgesia correlated with brain oxycodone, but not brain oxymorphone or noroxycodone levels, or plasma drug or metabolite levels. Propranolol (a CYP2D mechanism-based inhibitor), versus vehicle pretreatments, increased brain oxycodone, and decreased brain oxymorphone/oxycodone drug level ratios (an in vivo CYP2D activity phenotype in the brain) in males and females in estrus, but not in females in diestrus. Brain oxymorphone/oxycodone inversely correlated with analgesia. Together, both sex and estrous cycle impact oxycodone analgesia and brain oxycodone levels, likely through regulation of brain CYP2D oxycodone metabolism. As CYP2D6 is expressed in human brain, perhaps similar sex and cycle influences also occur in humans.
Keywords: sex differences, estrous cycle differences, analgesia, brain drug levels, microdialysis, oxycodone
Introduction
Opioids are effective pain relievers used for moderate-to-severe pain. There has been an increased prevalence of non-medical use of prescription opioids leading to opioid dependence. In 2017, the prevalence of opioid dependence was estimated to be 40.5 million people globally, with the highest estimated prevalence in the United States of 4.8 million people [1]. Efforts have been made to reduce opioid use; however, emergency room visits for overdose and intoxication have continued to increase. In the United States, opioid death overdoses reached nearly 47,000 in 2018 with 32% of those deaths involving prescription opioids [2]. In Canada, the opioid-related hospitalization rate per 100,000 population increased from 15.5% in 2019 to 17.1% in 2020, and the opioid-related death rate increased from 10.2% in 2019 to 16.0% in 2020 [3].
Following similar doses, there are inter-individual differences in pain relief and in the risk for dependence creating therapeutic challenges. One potentially important source of variation is sex differences in opioid analgesia [4–6]. Sex differences in analgesia may involve 1) opioid pharmacokinetic factors such as opioid absorbance, altered permeability (both distribution and blood brain barrier), central and peripheral metabolism, excretion, and/or 2) opioid pharmacodynamic factors such as differences in μ-opioid receptors (μ-ORs) density or function [7–9]. However, there are many inconsistencies in the literature on sex differences in opioid analgesia. Of the clinical studies that have examined differences between men and women, the majority of the studies have used I.V. morphine and have found either lower [10], equal [11], or higher [12] analgesic response in women compared to men. Preclinical studies on sex differences in nociception (referred to as analgesia herein) also conflict; females have either lower [13] or equal [14] analgesic response compared to males following subcutaneous administration of morphine. Of the 53 studies included in a review of sex differences in μ-opioid analgesia in rats, 66% found lower, 30% found equal, and 4% found higher analgesia in females compared to males [6]. When examining oxycodone specifically, tail-flick analgesia after I.P. administration was higher in female rats compared to males [15]. There were no sex differences found in a separate study looking at oxycodone analgesic potency (ED50), where males and females required similar subcutaneous doses of 1.53 ± 0.29 mg/kg and 1.71 ± 0.30 mg/kg respectively to attain a 50% analgesic response using a warm-water tail-withdrawal assay [16]. At doses of 1, 3.2 and 5.6 mg/kg oxycodone I.P. in mice, there were no sex differences in analgesia tested using the hot plate, while at a higher dose of 10 mg/kg, females had lower analgesic response compared to males [17]. Differences between the specific opioid tested, the route of administration, the duration and the modality of pain, may contribute to the inconsistences. In addition, the impact of sex differences could be masked or modified if the estrous cycle also alters opioid analgesia among the females [4].
Oxycodone is metabolized by CYP enzymes to three main metabolites – oxymorphone, noroxycodone, and noroxymorphone. In humans, the CYP2D6-mediated metabolite oxymorphone constitutes 11% of urinary recovery, the CYP3A4-mediated metabolite noroxycodone is 45% of urinary recovery, and the CYP3A4- and CYP2D6-mediated metabolite noroxymorphone is 14% of urinary recovery [18]. Similar enzymatic pathways are observed in rats (in vitro using liver S9 fraction), where the CYP2D-mediated oxymorphone formation has a Vmax of 0.273 ± 0.033 nmol/min/mg and a km of 33.5 ± 12.2 uM, while the CYP3A-mediated noroxycodone formation has a Vmax of 1.13 ± 0.17 nmol/min/mg and a km of 47.6 ± 19.4 uM [19]. Activation of the μ-ORs contributes to the analgesic properties of oxycodone; oxycodone and its metabolites have different affinities for μ-ORs. Relative to the affinity of morphine for the μ-ORs, the affinity of oxycodone is 5-times lower, oxymorphone is 9-times higher, and noroxycodone is 18-times lower [18]. While the contributions of the parent drug oxycodone and its metabolites to analgesia are related to their μ-OR affinities, other aspects may also contribute to their relative analgesic properties for example, their relative metabolism/concentrations in the systemic and central systems, their ability to cross the blood brain barrier, and their rate of efflux from the brain [20,21,18]. Here we explored sex and estrous cycle differences in oxycodone-induced analgesia, complementing this with the assessment of central and systemic drug and metabolite levels to determine if the differences in analgesia are related to differences in pharmacokinetics. We tested sex and estrous cycle effects using oxycodone-elicited analgesic response (measured as tail-flick latency) between and within female rats. We also measured brain drug and metabolite levels (by microdialysis), and plasma drug and metabolite levels, and then related these to analgesia. Lastly, we piloted studies to investigate if differences observed in brain oxycodone levels were mechanistically related to differences in activity of CYP2D in the brain.
Methods
Animals
Adult male and female Wistar rats (Charles River Laboratories, Saint-Constant, Canada) were individually housed and were food restricted to maintain a weight range of 250-350 g for females and 350-450 g for males. Rats were maintained on a 12-hour light/dark cycle with experimentation occurring during the light cycle. Stress-related confounders were reduced through acclimation of animals to handling, equipment, and room prior to each experiment. All procedures were approved by the Animal Care Committee at the University of Toronto and conform to the guidelines of the Canadian Council on Animal Care.
Visual Inspection and Lavage
Female rats were tested during two main phases of the estrous cycle: the estrus (proestrus plus estrus) phase and the diestrus (metestrus plus diestrus) phase. The estrous cycle was checked visually and confirmed through vaginal lavage as previously described with minor modifications [22]. In brief, a transfer pipette (1.2 ml Narrow-Stem, UltiDent Scientific, Quebec, Canada) with approximately 0.5 ml filtered distilled water (dH2O) was inserted into the vaginal orifice. Filtered dH2O was flushed 2 or 3 times until the fluid turned cloudy. After the lavage, a drop of the fluid was placed on the microscope slide and allowed to air-dry. Both the unstained slides (observed before experiment) and stained slides (confirming cycle after the experiment) were used to differentiate estrous phases. Crystal violet was used for staining, and the cells were observed through a light microscope using 10x and 20x objective lenses. Three types of cells were identified to differentiate phases of the estrous cycle; nucleated epithelial cells and anucleated cornified cells were predominant in the estrus phase, while leukocyte cells were predominant in the diestrus phase [22].
Treatment
Oxycodone hydrochloride (Professional Compounding Centers of America, London, Canada) was dissolved in filtered dH2O and administered by oral gavage. Oxycodone dose ranging was initially tested at 12.5 mg/kg, a dose previously used in male rats [20]. The dose was subsequently decreased to 10 mg/kg and then to 7.5 mg/kg to achieve a submaximal peak in analgesia for all groups (males, females in estrus, and females in diestrus) tested. Oxycodone treatment had no effect on estrous cycle, which remained regular throughout the experiment.
Nociceptive Testing
Antinociception (describe here as analgesia) was measured as the tail-flick latency (TFL) using a tail-flick meter (Columbus Instruments, Columbus, OH, USA). The TFL was recorded as the time from the onset of a thermal stimulus to the withdrawal of the tail, as previously described [23,24]. The thermal strength of the apparatus was adjusted prior to experimentation to obtain baseline TFLs of approximately 3-4 s. A cut-off of 12 s was used to minimize tissue damage and was therefore considered the maximal TFL. Baseline TFLs were reported as the average of two measurements prior to drug administration. As before [24], analgesia in the TFL test was expressed as a percentage of the maximal possible effect (%MPE):
Plasma drug levels
A saphenous blood draw was taken at 30 min post-oxycodone injection. The blood sample (200 μl) was collected using lithium heparin coated microvettes (CB 300 LH, SARSTEDT AG & Co. KG, Numbretcht, Germany) and was centrifuge at 5,000 rpm to isolate plasma. Samples were analyzed as previously described [20]. In brief, internal standards (oxymorphone-D3 and noroxycodone-D3) were added to the sample, and the sample was centrifuged at 12,000 rpm for 10 min. The supernatant was evaporated under nitrogen at 38°C, and reconstituted in the mobile phase. Drug and metabolite concentrations were measured using an Agilent 6430 LC-ESI-MS/MS system (Agilent Technologies, Santa Clara, CA), using a gradient elution of 5-20% in 15 min of acetonitrile in 10 mM ammonium acetate buffer through an Agilent SB C18 column (5 μm, 15 cm x 2.1 mm). The mass spectrometer was operated with multiple-ion monitoring under positive-ion mode for oxycodone (316.2→298.1), oxymorphone (302.1→284.1) and noroxycodone (302.1→284.1).
Surgery
Rats were anesthetized with isoflurane and injected with the analgesic meloxicam (5 mg/kg, subcutaneous). After induction, rats were surgically implanted with microdialysis guide-cannulae (MD-2257, Bioanalytical System, Inc. (BASi), West Lafayette, IN, USA) into the right lateral ventricles (Bregma coordinates: anteroposterior: −0.9 mm; lateral: −1.4 mm; dorsoventral, −3.6). Four screws (BASi) were implanted into the skull to serve as an anchor, and DuraLay inlay pattern resin (Reliance Dental Mfg. Co., Worth, IL, USA) was used as an adhesive to build a head cap around the microdialysis guide-cannula. Rats were given one week for recovery, and meloxicam was given daily (5 mg/kg, subcutaneous) for up to 72 hours post-surgery.
Microdialysis
Microdialysis probes (2 mm, MD-2201, BASi) were introduced into the microdialysis guide-cannulae. Animals were allowed to habituate for at least 10 minutes while perfusion medium (Ringer’s solution; 147 mM Na+, 2 mM Ca2+, 4 mM K+, 155 mM Cl−, pH 6.0) was circulated at a flow rate of 2 μl/min. Three baseline dialysate samples were collected prior to drug administration. After 7.5 mg/kg oral oxycodone, dialysate samples were collected for 15 min time bins over 120 min as previously described [20]. In brief, drug and metabolite concentrations for each microdialysis collection period were measured as follow: the entire 30 μl dialysate was mixed with 10 μl of the internal standards (oxymorphone-D3 and noroxycodone-D3) in water and assessed by LCMS, as described for plasma drug levels above. The microdialysis tubing dead volume was equivalent to one-time bin and thus the first-time bin following drug treatment was removed in the data analyses to adjust for this.
Pre-treatment
Propranolol hydrochloride (Sigma-Aldrich, Oakville, Canada) was dissolved in a 20% (w/v) solution of 2-hydroxypropyl-β-cyclodextrin (cyclodextrin; Sigma-Aldrich) in water. Propranolol is a CYP2D mechanism-based inhibitor in humans and rats; it undergoes 4-hydroxylation by CYP2D to a reactive metabolite that is capable of covalently binding to, and thereby inactivating, the CYP2D enzyme [25,26]. Propranolol was formulated to deliver 20 μg base in a 4 μl volume by intracerebroventricular (i.c.v) injection, 24-hour prior to oral oxycodone administration. To deliver propranolol or vehicle i.c.v., a microdialysis infusion probe (2 mm, MD-2258, BASi) was used. Propranolol was injected over 4 min, with the injector left in place for an additional minute. This inhibitor dose and 24-hour timing has been used in previous studies to selectively inhibit CYP2D in the brain, but not in the liver, as assessed both in vivo and in vitro [20,27]. The effects of pre-treatments on in vivo CYP2D activity were measured using brain microdialysis and plasma drug monitoring. Neither propranolol nor vehicle pre-treatments had any effect on estrous cycle, which remained regular throughout the experiment.
Protocol
In each experiment, baseline analgesia (immediately before opioid treatment) and 30-min plasma drug levels were assessed. In the first experiment, a between animal study design, analgesia was tested over 120 min after three different doses of oral oxycodone (12.5, 10, and 7.5 mg/kg) in males (M), females in estrus (FE), and females in diestrus (FDE). Experiment 2 was a within-animal study design with a two-week wash out between the oral oxycodone treatments. Analgesia was tested during the estrus and diestrus phases of the estrous cycle within female rat, at two different doses of oral oxycodone (10 and 7.5 mg/kg). In experiments 3, males and female rats were surgically implanted with microdialysis cannulae. Females were divided into two groups for testing in estrus phase or diestrus phase, and they were only tested in this specific phase, along with males. A full pre-treatment cross-over study design was performed with a two-week washout between the 7.5 mg/kg oral oxycodone treatments. Each animal in the three groups (M, FE, FDE) went through pre-treatment crosses (propranolol and vehicle) with microdialysis after each pre-treatment. During microdialysis, 30-min analgesia and 30-min saphenous blood draws were taken. A second pre-treatment cross (propranolol and vehicle) was performed for analgesia, resulting in a total of 4 sessions per animal (2 microdialysis followed by 2 analgesia). However, propranolol pre-treated animals in M, FE, FDE reached maximum analgesia and thus this session (of the four) was not included in the results.
Statistical Analyses
Data were analyzed with GraphPad Prism v.6.01 (GraphPad Software Inc., La Jolla, CA, USA) by one-way, two-way, and mixed ANOVA, where appropriate. Data from animals treated with oral oxycodone alone (Experiment 1 and 2) were analyzed using two-way ANOVA with Bonferroni post-hoc test for analgesic-time curve, followed by a one-way ANOVA with Bonferroni post-hoc test for the total area under the curves (AUCs). AUCs were determined by the linear trapezoid rule. Paired t-tests (within animal) were used for the total AUCs in Experiment 2. Data from animals with propranolol and/or vehicle pre-treatments followed by oral oxycodone in Experiment 3 were analyzed using two-way ANOVA (for analgesia) or mixed ANOVA (for drug levels with propranolol (vs vehicle) pre-treatment) with Bonferroni post-hoc tests for full-time curves of analgesia, brain drug levels, and brain OM/OC ratios, followed by a two-way ANOVA with Bonferroni post-hoc tests for total AUCs. Repeated measures were used in within-animal experiments. Relationships between data sets were assessed using Pearson correlation coefficients (normally distributed) or Spearman correlation coefficients.
Results
Analgesia varies with sex and estrous cycle over a range of oral oxycodone doses
Females in diestrus (FDE) had higher analgesia compared to males (M) and females in estrus (FE) following 12.5 mg/kg oral oxycodone, demonstrated in the analgesic-time curve (FSex/Cycle (2,6) = 123.70, p<0.0001, Fig. 1a) and when examining the total analgesic area under the curves (AUC0-120; (F (2,6) = 123.70, p<0.0001, Fig. 1b). Specifically, M and FE had similar analgesic responses whereas FDE reached maximum analgesia at 100% MPE from 30 min to 40 min (Fig. 1a). At lower doses of 10 mg/kg (FSex/Cycle (2,15) = 18.84, p<0.0001, Fig. 1c) and 7.5 mg/kg (FSex/Cycle (2,15) = 27.17, p<0.0001, Fig. 1e) oral oxycodone, FDE also had higher analgesia compared to M and FE; analgesia in the three groups peaked at 30 minutes and was submaximal. At 10 mg/kg, FDE had a 3.4-times and 2.3-times higher analgesic AUC0-120 (F (2,15) = 14.94, p=0.0003, Fig. 1d) compared to M and FE respectively, while at 7.5 mg/kg, FDE had 2.8-times and 1.9-times higher analgesic AUC0-120 (F (2,15) = 23.39, p<0.0001), Fig. 1f) compared to M and FE respectively. Within each group, there were significant dose-response relationships (Supplementary Fig. 1 and 2).
Figure 1:
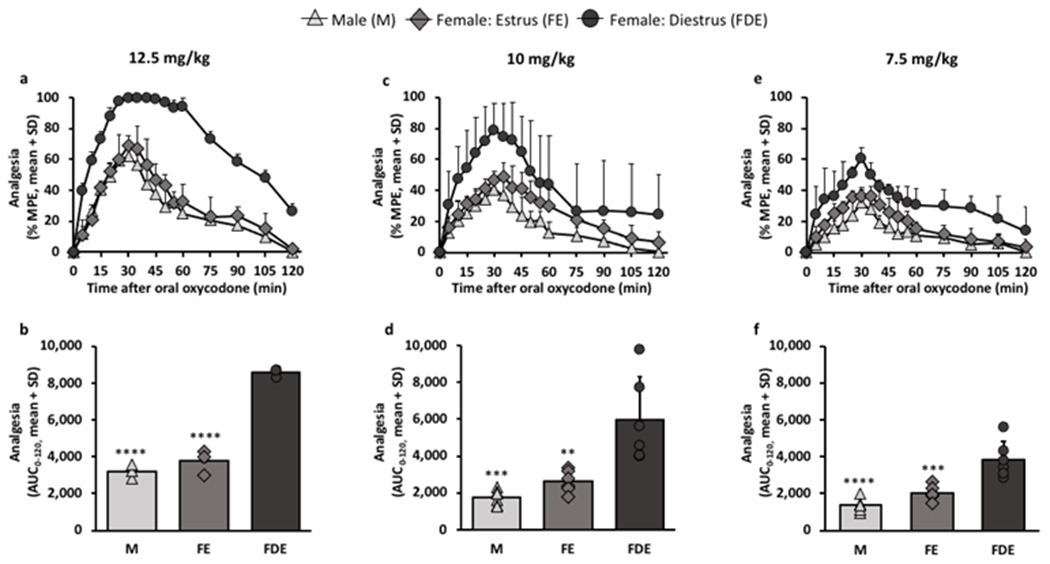
Females in diestrus had higher analgesia compared to males and females in estrus at varying oral oxycodone doses.
Analgesia was assessed after the following oral oxycodone doses- a, b, 12.5 mg/kg (n=3/group), c, d, 10 mg/kg (n=6/group), and e, f, 7.5 mg/kg (n=6/group). a, analgesic-time curve at 12.5 mg/kg (FSex/Cycle(2,6) = 123.70, p<0.0001; FTime(16,96) = 59.20, p<0.0001; FInteraction(32, 96) = 3.47, p<0.0001). b, analgesic AUC0-120 at 12.5 mg/kg (F(2,6) = 123.70, p<0.0001). c, analgesic-time curve at 10 mg/kg (FSex/Cycle(2,15) = 18.84, p<0.0001; FTime(16,240) = 80.67, p<0.0001; FInteraction(32, 240) = 4.65, p<0.0001). d, analgesic AUC0-120 at 10 mg/kg (F(2,15) = 14.94, p=0.0003). e, analgesic-time curve at 7.5 mg/kg (FSex/Cycle(2,15) = 27.17, p<0.0001; FTime(16,240) = 52.61, p<0.0001; FInteraction(32, 240) = 1.83, p=0.0059). f, analgesic AUC0-120 at 7.5 mg/kg (F(2,15) = 23.39, p<0.0001). Analgesic-time curves were assessed using two-way ANOVA with repeated measures by time. Analgesic AUCs0-120 were assessed using one-way ANOVA; **p<0.01, ***p<0.001, ****p<0.0001 relative to females in diestrus phase using Bonferroni post-hoc analysis.
Baseline analgesia was also tested before any given dose of oral oxycodone to determine if there were any sex and estrous cycle differences in analgesia in response to pain attributed to these physiological differences in males and females. There were no group differences in baseline analgesia before any oxycodone doses (12.5 mg/kg: F (2,6) = 0.94, p=0.4414; 10 mg/kg: F (2,15) = 1.65, p=0.2251; 7.5 mg/kg: F (2,15) = 1.38, p=0.2816, Supplementary Fig. 3), suggesting that the sex and cycle differences in analgesia were related to differences in oxycodone response.
Analgesia varies with estrous cycle within females
To confirm the association of estrous cycle with oxycodone-induced analgesia observed between animals and across doses (Fig. 1), a second cohort of female rats was tested during both estrus and diestrus phases within animal at both lower doses. During diestrus, female rats had higher analgesia, demonstrated in the analgesic-time curve (10 mg/kg: Fcycle (1,10) = 13.21, p=0.0046, Fig. 2a; 7.5 mg/kg: Fcycle (1, 10) = 18.71, p=00015, Fig. 2d) and when examining the AUC0-120 (10 mg/kg: t (10) = 2.91, p=0.0154, Fig. 2b; 7.5 mg/kg: t (10) = 4.20, p=0.0018, Fig. 2e) than when tested during estrus. The impact of estrous cycle was consistent for all animals, at both doses, as indicated by the lines denoting changes in the analgesic AUC0-120 in Fig. 2b and 2e. Overall, the analgesic AUC0-120 in diestrus was 1.7- and 1.9-times higher than in estrus at 10 (Fig. 2b) and 7.5 mg/kg (Fig. 2e) oral oxycodone, respectively. There were no opioid/cycle order effects on analgesia (Supplementary Fig. 4), indicating sufficient washout between analgesic tests, and no within animal cycle effects on 30-min plasma oxycodone levels at 10 (t (10) = 0.16, p=0.8793, Fig. 2c) or 7.5 mg/kg (t (10) = 0.55, p=0.5932, Fig. 2f) oral oxycodone.
Figure 2:
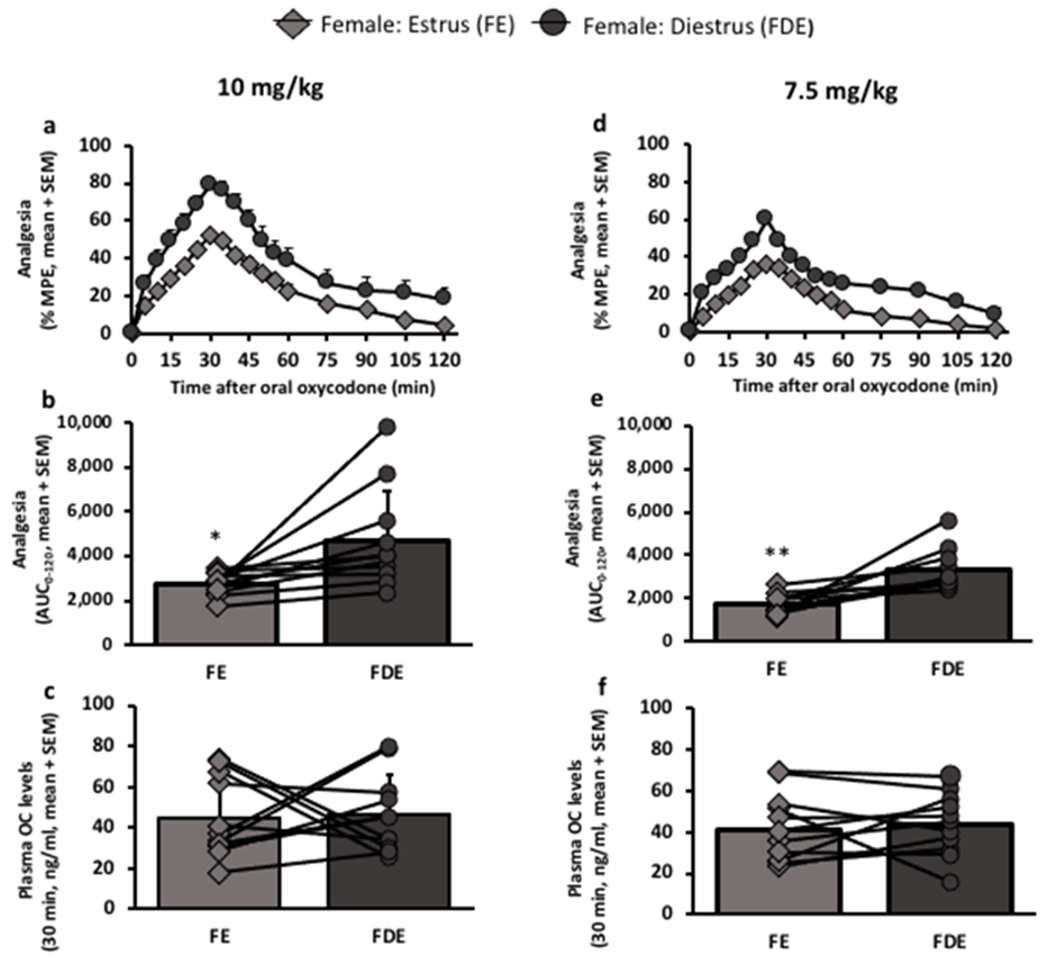
Analgesia was higher in females during diestrus than during estrus; a within animal study design.
Females were each tested during their estrus phase and during their diestrus phase; analgesia and 30-min oxycodone plasma levels were assessed, both at a, b, c, 10 mg/kg (n=11 female rats) and d, e, f, 7.5 mg/kg (n= 11 female rats) oral oxycodone. Lines represent data within animal tested during their estrus phase and during their diestrus phase. a, analgesic-time curve at 10 mg/kg (FCycle(1,10) = 13.21, p=0.0046; Ftime(16,160) = 89.52, p<0.0001; FInteraction(16, 160) = 4.12, p<0.0001). b, analgesic AUC0-120 at 10 mg/kg, *p=0.0154. c, 30-min plasma oxycodone levels at 10 mg/kg, p=0.8793. d, analgesic-time curve at 7.5 mg/kg (FCycle(1,10) = 18.71, p=0.0015; Ftime(16,160) = 107.60, p<0.0001; FInteraction(16, 160) = 3.21, p<0.0001). e, analgesic AUC0-120 at 7.5 mg/kg, **p=0.0018. f, 30-min plasma oxycodone levels at 7.5 mg/kg, p=0.5932. Analgesic-time curves were assessed using two-way ANOVA with repeated measures by both factors. AUCs0-120 and 30-min plasma oxycodone levels were assessed using paired t-test.
Analgesia correlates with brain oxycodone but not the brain metabolites, and not with plasma levels
Using microdialysis, we examined the impact of sex and cycle on in vivo brain drug levels and related these drug levels to analgesia at 7.5 mg/kg oxycodone. As observed with analgesia (Fig. 1 and 2), there were significant group differences in brain oxycodone levels (Fig. 3a). Brain oxycodone levels were higher in FDE compared to M and FE at 7.5 mg/kg oral oxycodone (FSex/Cycle (2,17) = 30.42, p<0.0001, Fig. 3a); analgesic AUC0-120 correlated with brain oxycodone AUC0-105 (r=0.8355, p=0.0001, Fig. 3b). In contrast, there were no group differences in the CYP2D metabolite, oxymorphone (FSex/Cycle (2,17) = 1.33, p=0.2904, Fig. 3d), or the CYP3A metabolite, noroxycodone (FSex/Cycle (2,17) = 2.15, p=0.1471, Fig. 3g). Analgesic AUC0-120 did not correlate with brain oxymorphone AUC0-105 (r=−0.1053, p=0.6587, Fig. 3e) or brain noroxycodone AUC0-105 (r=−0.3617, p=0.1171, Fig. 3h). In addition, there was no relationship between analgesia and plasma oxycodone (r=−0.0015, p=0.9950, Fig. 3c), plasma oxymorphone (r=0.1789, p=0.4504, Fig. 3f), or plasma noroxycodone (r=−0.4066, p=0.0753, Fig. 3i) levels.
Figure 3:
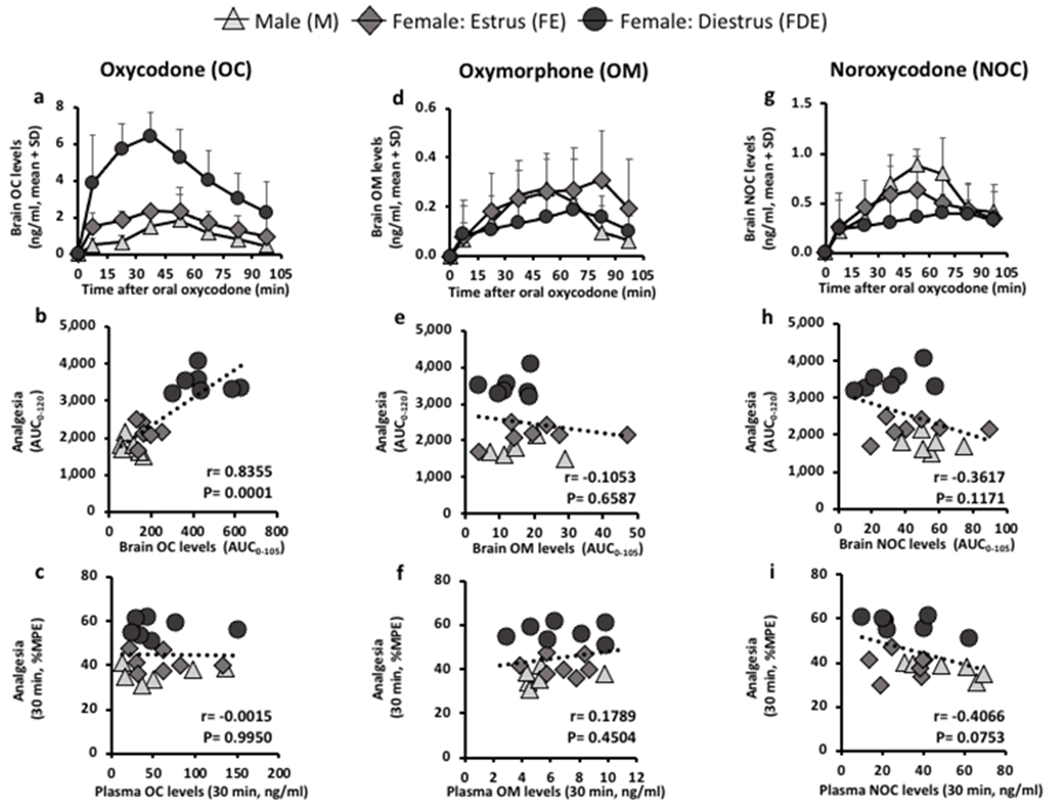
Analgesia correlated with brain oxycodone levels, but not brain oxymorphone, plasma oxycodone, or plasma oxymorphone levels.
A cross-over design was used between analgesia and microdialysis at 7.5 mg/kg oral oxycodone; males (n=6), females in estrus phase (n=7), and females in diestrus phase (n=7). a, b, c, oxycodone (OC), d, e, f, oxymorphone (OM), and g, h, i, noroxycodone (NOC) levels were measured in the brain through microdialysis and in the plasma through saphenous blood draw. a, brain OC-time curve (FSex/Cycle(2,17) = 30.42, p<0.0001; FTime(7,119) = 30.42, p<0.0001; FInteraction(14, 119) = 6.52, p<0.0001). Analgesia correlated with b, brain OC AUC0-105 (r=0.8355, p=0.0001) but not c, 30-min plasma OC levels (r=−0.0015, p=0.9950). d, brain OM-time curve (FSex/Cycle(2,17) = 1.33, p=0.2904; FTime(7,119) = 13.91, p<0.0001; FInteraction(14, 119) = 1.62, p=0.0834). Analgesia did not correlate with e, brain OM AUC0-105 (r=−0.1053, p=0.6587) and not with f, 30-min plasma OM levels (r=0.1789, p=0.4504). g, brain NOC-time curve (FSex/Cycle(2,17) = 2.15, p=0.1471; FTime(7,119) = 25.13, p<0.0001; FInteraction(14, 119) = 2.69, p=0.0018). Analgesia did not correlate with h, brain NOC AUC0-105 (r=−0.3617, p=0.1171) and not with i, 30-min plasma NOC levels (r=−0.4066, p=0.0753). Brain drug level-time curves were assessed using two-way ANOVA with repeated measures by time. Correlations were assessed using Pearson Correlation (normally distributed) and Spearman Correlation.
Selective inhibition of CYP2D in the brain, but not liver, increases brain oxycodone levels in males and females in estrus but not females in diestrus
The selective irreversible inhibition of CYP2D in the brain of male rats, by 24-hour propranolol pre-treatment, increased brain, not plasma, oxycodone levels and analgesia relative to the vehicle pre-treatment group [20]; this provided evidence that in male rats CYP2D activity within the brain contributed to the brain oxycodone levels and resulting analgesia. Herein we piloted studies to determine if sex and estrous cycle effects on analgesia and oxycodone brain levels observed here were, at least in part, due to sex and cycle differences in CYP2D activity in brain. In M and FE, propranolol versus vehicle pre-treatment elevated brain oxycodone AUC0-105 (Fpretreatment (1,24) = 34.36, p<0.0001, Bonferroni post-hoc analysis (M and FE: p<0.001), Fig. 4a). In contrast in FDE, there was no change in brain oxycodone levels following propranolol versus vehicle pre-treatment. Specifically, propranolol versus vehicle pre-treatment increased the brain oxycodone AUC0-105 in M by 1.9-times, in FE by 1.4-times, but not in FDE (1.1-times) (Fig. 4a). After vehicle pre-treatment alone, FDE had higher brain oxycodone AUC0-105 compared to M and FE (Fsex/cycle (2,24) = 10.94, p=0.0004, Fig. 4a). Together this suggests that M and FE may have more CYP2D in the brain available for propranolol inhibition than FDE, and that the higher brain oxycodone in FDE may be due to lower CYP2D activity in the brain.
Figure 4:
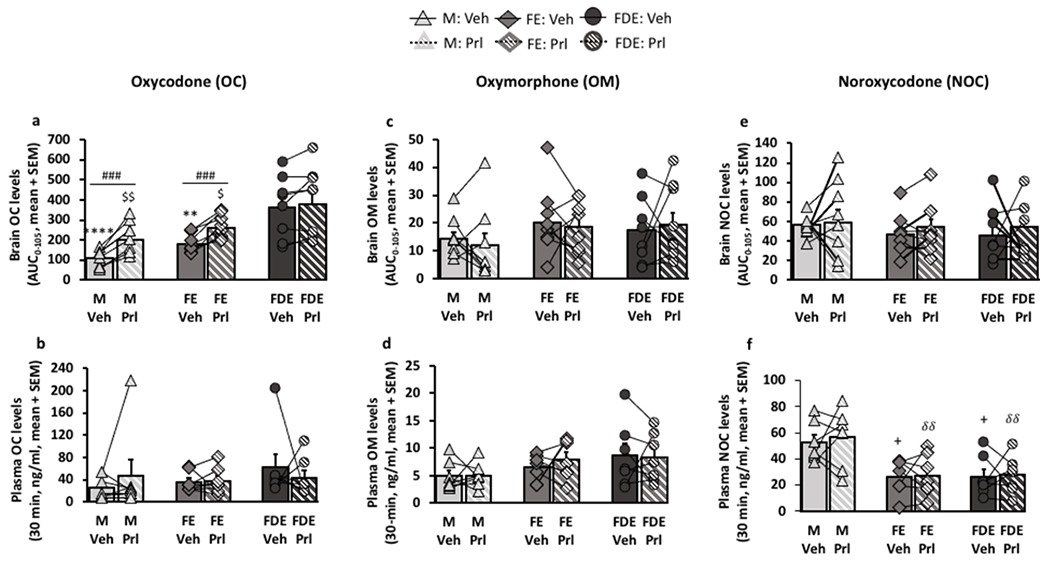
Selective inhibition of CYP2D in the brain, but not liver, with propranolol pre-treatment; main impact was on brain oxycodone levels in males and females in estrus but not females in diestrus.
A cross-over design was used between pre-treatments 24-hour prior to 7.5 mg/kg oral oxycodone; lines represent data within animal tested with vehicle pre-treatment and propranolol pre-treatment. a, b, oxycodone (OC), c, d, oxymorphone (OM), and e, f, noroxycodone (NOC) levels were measured in the brain through microdialysis (n=9/group) and in the plasma through saphenous blood draw (n=7/group). a, brain OC AUC0-105 (FSex/Cycle(2,24) = 10.94, p=0.0004; FPre-treatment(1,24) = 34.36, p<0.0001; FInteraction(2,24) = 4.18, p=0.0257). b, 30-min plasma OC levels (FSex/Cycle(2,18) = 0.60, p=0.5620; FPre-treatment(1,18) = 0.01, p=0.9215; FInteraction(2,18) = 0.66, p=0.5270). c, brain OM AUC0-105 (FSex/Cycle(2,24) = 1.28, p=0.2967; FPre-treatment(1,24) = 0.08, p=0.7794; FInteraction(2,24) = 0.28, p=0.7582). d, 30-min plasma OM levels (FSex/Cycle(2,18) = 2.46, p=0.1139; FPre-treatment(1,18) = 0.16, p=0.6918; FInteraction(2,18) = 0.27, p=0.7666). e, brain NOC AUC0-105 (FSex/Cycle(2,24) = 0.43, p=0.6524; FPre-treatment(1,24) = 0.48, p=0.4973; FInteraction(2,24) = 0.08, p=0.9211). f, 30-min plasma NOC levels (FSex/Cycle(2,18) = 10.10, p=0.0011; FPre-treatment(1,18) = 0.24, p=0.6313; FInteraction(2,18) = 0.06, p=0.9404). Brain drug AUCs0-105 were assessed using two-way ANOVA; **p<0.01, ****p<0.0001 relative to vehicle pre-treated FDE, $p<0.05, $$p<0.01 relative to propranolol pre-treated FDE, and ###p<0.001, relative to its respective vehicle using Bonferroni post-hoc analysis. 30 min plasma levels were assessed using two-way ANOVA; +p<0.05 relative to vehicle pre-treated males, δδp<0.01, relative to propranolol pre-treated males using Bonferroni post-hoc analysis.
There were no group differences and no propranolol versus vehicle pre-treatment effects in brain oxymorphone AUC0-105 (Fsex/cycle (2,24) = 1.28, p=0.2967; Fpretreatment (1,24) =0.08, p=0.7794, Fig. 4c), and brain noroxycodone AUC0-105 (Fsex/cycle (2,24) = 0.43, p=0.6524; Fpretreatment (1,24) =0.48, p=0.4973, fig. 4e). Further, there were no group differences in the 30-min plasma oxycodone levels (Fsex/cycle (2,18) =0.60, p=0.5620, Fig. 4b), and 30-min plasma oxymorphone levels (Fsex/cycle (2,18) =2.46, p=0.1139, Fig. 4d). Males had higher plasma noroxycodone levels compared to females, both FE and FDE, (Fsex/cycle (2,18) =10.10, p=0.0011, Bonferroni post-hoc analysis (p<0.05 relative to M-vehicle and p<0.01 relative to M-propranolol, Fig. 4f), consistent with the literature on sex differences in hepatic CYP3A [28,29]. There were no effects of propranolol versus vehicle pre-treatment on plasma levels of oxycodone (Fpretreatment (1,18) =0.01, p=0.9215, Fig. 4b), oxymorphone (Fpretreatment (1,18) =0.16, p=0.6918, Fig. 4d), or noroxycodone (Fpretreatment (1,18) =0.24, p=0.6313, Fig. 4f), indicating no impact of the propranolol pre-treatment on CYP2D or CYP3A in the liver.
Brain OM/OC, a phenotypic measure of CYP2D in the brain, is higher and has greater impact of propranolol in males > females in estrus > females in diestrus
To further examine if CYP2D in the brain was responsible for the differences among the three groups, the brain oxymorphone/oxycodone (OM/OC) drug level ratios were used to phenotype in vivo CYP2D activity in the brain. In M, propranolol versus vehicle pre-treatment demonstrated decreases in both the brain OM/OC-time curve (FPretreatment (1,127) = 19.02, p<0.0001, Fig. 5a) and the brain OM/OC ratio AUC0-105 (FPretreatment (1,24) = 10.74, p=0.0032, Bonferroni post-hoc analysis (M: p<0.001), Fig. 5b). Although the differences in propranolol versus vehicle pre-treatment in FE was not significant, the rank order impact of propranolol versus vehicle pre-treatment was higher in males by 3.0-times, followed by FE by 1.4-times, while having no effect in FDE (0.92-times) (Fig. 5b). Analgesia at 30-min negatively correlated with brain OM/OC ratio AUC0-105 (r=−0.3748, p=0.0057, Fig. 5c). After removing the animals that reached 100% MPE (15 in total), the correlation between 30-min analgesia and brain OM/OC ratio AUC0-105 was still significant (r=−0.3722, p=0.0214). Together these data provide initial evidence that CYP2D activity in the brain is reduced during FDE, resulting in elevated analgesia and brain oxycodone levels, which is unaffected by propranolol pre-treatment.
Figure 5:
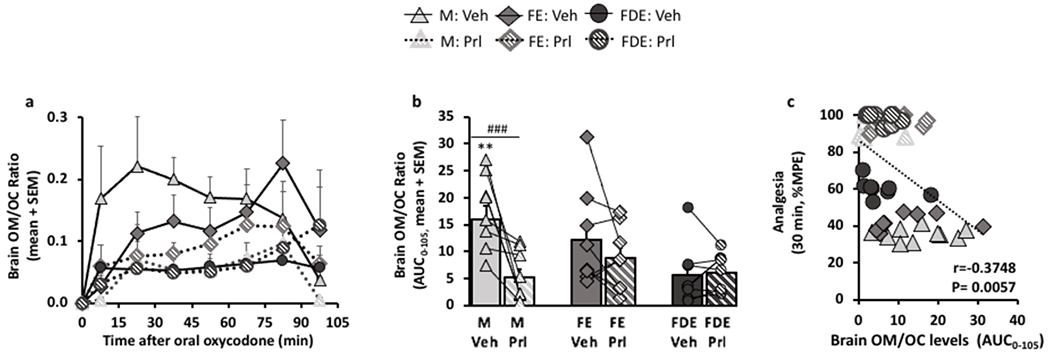
Selective inhibition of CYP2D in the brain with propranolol decreased brain oxymorphone/oxycodone ratio in males > females in estrus > females in diestrus.
Brain oxycodone (OC) and brain oxymorphone (OM) assessed by microdialysis after 7.5 mg/kg oral oxycodone (N=9/group) were used for the metabolite/parent ratio (brain OM/OC ratio), a phenotype measure for in vivo CYP2D in the brain; a, brain OM/OC-time curve in M (FPre-treatment(1,127) = 19.02, p<0.0001; FTime(7,127) = 3.05, p=0.0053; FInteraction(7,127) = 1.06, p=0.3962), in FE (FPre-treatment(1,128) = 2.67, p=0.1047; FTime(7,128) = 3.69, p=0.0012; FInteraction(7,128) = 0.52, p=0.8202), and in FDE (FPre-treatment(1,126) = 0.20, p=0.6565; FTime(7,126) = 1.70, p=0.1155; FInteraction(7,126) = 0.49, p=0.8432). b, brain OM/OC ratio AUC0-105 (FSex/Cycle(2,24) = 2.77, p=0.0828; FPre-treatment(1,24) = 10.74, p=0.0032; FInteraction(2,24) = 5.47, p=0.0110). c, 30-min analgesia correlated with brain OM/OC ratio AUC0-105 (Spearman r=−0.3748, p=0.0057). Brain OM/OC-time curve was assessed using mixed ANOVA. Brain OM/OC ratio AUC0-105 was assessed using two-way ANOVA repeated by pre-treatment; **p<0.01, relative to vehicle pre-treated FDE, and ###p<0.001 relative to its respective vehicle using Bonferroni post-hoc analysis. Correlations were assessed using Spearman Correlation.
Discussion
Sex and cycle differences were observed in analgesia which were concordant with differences in brain oxycodone levels. Some studies have found sex differences in analgesia only at specific oxycodone doses [17]; however, in this study, at all oral oxycodone doses tested, females in diestrus had higher analgesia compared to males and females in estrus (FDE>M=FE). This was consistent with a study done by Holtman et. al, where tail-flick analgesia was higher in female rats compared to males after I.P. administration of oxycodone [15]; however, this was not consistent with Peckham et. al, which found no sex differences in analgesic potency after subcutaneous administration of oxycodone [16]. When tested within females, analgesia was higher during diestrus than during estrus, demonstrating an estrous cycle effect (FDE>FE). Analgesia correlated with the brain levels of oxycodone, but not oxymorphone and noroxycodone. Sex and cycle differences in analgesia were not due to differences in baseline analgesia or plasma drug levels. Moreover, in our pilot study to examine if the differences in brain oxycodone were mediated by CYP2D in the brain, we found there were sex and cycle differences in the degree of propranolol inhibition in brain oxycodone levels, and brain OM/OC ratio (M>FE>FDE) which were related to differences in analgesia. Propranolol (vs vehicle) pre-treatment increased brain oxycodone in males and females in estrus but had little to no impact in females in diestrus. Although there were no significant differences in brain OM/OC between propranolol vs vehicle pre-treatment in females in estrus, the impact of propranolol followed the same rank order as in brain oxycodone levels (i.e. M>FE>FDE). Together, these data suggest that the sex and cycle differences in analgesia were due to differences in the brain oxycodone, that may be mediated by the local brain metabolism of the drug by CYP2D.
Sex differences in opioid analgesia in the literature are inconsistent, and may be due to the specific opioid tested, opioid dose, and the lack of consideration of estrous cycle in the females [4]. The majority of the data on sex differences in opioid analgesia was generated using morphine [6]; however the pharmacokinetics, and specific metabolic enzymes involved, differ between morphine [30] and oxycodone [18,20,19]. The lower [13] and/or equal [14] analgesia in females compared to males after morphine administration may not be comparable to our results that showed that the sex and cycle differences in analgesia were due to brain drug levels, specifically brain oxycodone levels, potentially mediated by oxycodone metabolism by CYP2D in the brain. Moreover, the lack of sex differences in analgesia in other papers may be confounded by the female cycle differences. Here, we looked at females in estrus phase and diestrus phase allowing our experiment to be more comprehensive. For example, as illustrated here, if the majority of the females were previously tested in estrus phase, the differences between males and females may not be seen.
Oxycodone is metabolized by CYP2D to an active metabolite, oxymorphone, and by CYP3A to a non-active metabolite noroxycodone [18]. It was originally thought that similar to the bioactivation of codeine to morphine, CYP2D metabolism of oxycodone to oxymorphone is the principal activating pathway where a more potent metabolite, oxymorphone, contributes significantly to the oxycodone-induced analgesia [31]. However, further evidence has shown, from both pre-clinical and clinical studies, that hepatic CYP2D metabolism has little contribution to oxycodone-induced analgesia. For example, inhibition of rat CYP2D (in vivo) with quinine did not alter oxycodone-induced analgesia [32]. Likewise, in humans, inhibition CYP2D6 (in vivo) with either quinidine or paroxetine, reduced plasma oxymorphone but did not alter plasma oxycodone levels or analgesia [33]. The lack of contribution of hepatic CYP2D and peripheral drug levels suggest that other factors may contribute to the inter-individual variation in drug response. In the current study, we found that brain oxycodone, despite being a less potent μ-opioid receptor activator than oxymorphone, is responsible for analgesia in females in estrus, and females in diestrus, demonstrated in the correlation of analgesia with brain oxycodone, but not brain oxymorphone, as well as in males [20]. We also emphasized the lack of contribution of noroxycodone on analgesia, demonstrated in the lack of correlation of analgesia with brain noroxycodone levels. Moreover, oxycodone, despite lower potency than oxymorphone, readily crosses the blood-brain barrier in the central nervous system, whereas oxymorphone has low systemic levels and blood-brain barrier permeability [21,34,35,18,36]. The differences in brain-to-plasma ratio, relative amounts, and clearance in the brain between oxycodone and oxymorphone may be responsible, at least in part, for the lack of contribution of oxymorphone in oxycodone-induced analgesia despite having higher μ-opioid binding affinity. Together this suggests that brain oxycodone drug levels contribute substantially to analgesia, and specifically to the sex and cycle differences observed here. Together, this suggests that local brain (vs systemic) metabolism of oxycodone may contribute to the sex and cycle differences in oxycodone drug levels and resulting response.
Sex and cycle differences in analgesia may be due to either pharmacokinetics or pharmacodynamics. Here we provide strong evidence that there are sex and cycle pharmacokinetic impacts on analgesia mediated by differences in brain oxycodone levels. Although estrous cycle phases were verified through both visual inspection and cytological lavage, plasma hormone levels were not assessed. We also explored the potential contribution of local metabolism in the brain, where we saw that brain oxymorphone/oxycodone (OM/OC) ratio, an in vivo phenotype of CYP2D enzyme activity, followed the same rank order as the degree of propranolol inhibition in brain oxycodone levels (M>FE>FDE), consistent with highest brain oxycodone levels and analgesia in females in diestrus. Further experiments at lower dose of oral oxycodone with selective inhibition of CYP2D in the brain, as well as characterized sex- and estrous cycle-dependent regulation of CYP2D within the brain (and not liver) are warranted. A lower dose of oral oxycodone could avoid observation of maximal analgesia following propranolol pre-treatment, allowing better quantification of change in analgesia effected by inhibition of CYP2D in the brain by propranolol. Moreover, pharmacodynamic differences between sexes and estrous cycle phases could also contribute to differences in analgesia observed here. Although analgesia correlated with brain oxycodone levels, it is also important to understand if there are additional impacts of downstream differences in μ-opioid receptors and effectors between sexes and estrous cycle phases. Another limitation was that the tail-flick latency was the only behavioral assay used in this study, chosen because of its reliability and reproducibility [37]; however, previous studies in male rats using a hot plate have shown a similar role of CYP2D in the brain for codeine-induced analgesia [24]. Lastly, these studies were conducted in adult animals. It remains to be seen what the impact of adolescence and menopausal/estropause stages will have on brain drug levels and resulting analgesia.
Understanding the sex and cycle differences in opioid analgesia preclinically paves the way to exploring variation in oxycodone response between men and women and among women, which may assist in choice and timing of treatment, and may lead to optimization of treatment. Cyclical regulation of oxycodone levels within the female brain may result in variation in drug response within women over their menstrual cycle.
Supplementary Material
Acknowledgements
We would like to thank Dr. Bin Zhao for his invaluable technical assistance with the LC-MS analyses, Dr. Ahmed El-Sherbeni for assistance with MD calculations, and Fariba Baghai Wadji for her expert support with stereotaxic surgeries and microdialysis animal procedures.
Funding
This work was supported by the R01 grant from National Institutes of Health (DA043526-01A1); Canada Research Chairs program (Dr. Tyndale, the Canada Research Chair in Pharmacogenomics) a Canadian Institutes of Health Research (Foundation grant FDN-154294); the Centre for Addiction and Mental Health and the CAMH Foundation.
Footnotes
Conflicts of interest/Competing interests
Dr. Rachel F Tyndale has consulted for Quinn Emanuel and Ethismos on topics unrelated to this current work. Nicole Arguelles and Dr. Sharon Miksys have no conflicts to declare.
Availability of data and material
Supporting data and material can be found in the additional files and can be requested from the corresponding author.
Code availability
Not applicable
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. Experiments were performed in accordance with the NIH guidelines for the care and use of laboratory animals, and with approval of the University of Toronto Animal Care Committee.
Consent to participate
Not applicable
Consent to publish
Not applicable
References
- 1.Degenhardt L, Grebely J, Stone J, Hickman M, Vickerman P, Marshall BDL, Bruneau J, Altice FL, Henderson G, Rahimi-Movaghar A, Larney S (2019) Global patterns of opioid use and dependence: harms to populations, interventions, and future action. Lancet 394 (10208):1560–1579. doi: 10.1016/S0140-6736(19)32229-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilson N, Kariisa M, Seth P, Smith H, Davis NL (2020) Drug and Opioid-Involved Overdose Deaths - United States, 2017-2018. MMWR Morb Mortal Wkly Rep 69 (11):290–297. doi: 10.15585/mmwr.mm6911a4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Canada PHAo (March 2021) Opioids and Stimulant-related Harms in Canada. Ottawa [Google Scholar]
- 4.Averitt DL, Eidson LN, Doyle HH, Murphy AZ (2019) Neuronal and glial factors contributing to sex differences in opioid modulation of pain. Neuropsychopharmacology 44 (1):155–165. doi: 10.1038/s41386-018-0127-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bodnar RJ, Kest B (2010) Sex differences in opioid analgesia, hyperalgesia, tolerance and withdrawal: central mechanisms of action and roles of gonadal hormones. Horm Behav 58 (1):72–81. doi: 10.1016/j.yhbeh.2009.09.012 [DOI] [PubMed] [Google Scholar]
- 6.Craft RM (2003) Sex differences in opioid analgesia: “from mouse to man”. Clin J Pain 19 (3):175–186 [DOI] [PubMed] [Google Scholar]
- 7.Chan S, Edwards SR, Wyse BD, Smith MT (2008) Sex differences in the pharmacokinetics, oxidative metabolism and oral bioavailability of oxycodone in the Sprague-Dawley rat. Clin Exp Pharmacol Physiol 35 (3):295–302 [DOI] [PubMed] [Google Scholar]
- 8.Lee CW, Ho IK (2013) Sex differences in opioid analgesia and addiction: interactions among opioid receptors and estrogen receptors. Mol Pain 9:45. doi: 10.1186/1744-8069-9-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li J, Xie M, Wang X, Ouyang X, Wan Y, Dong G, Yang Z, Yang J, Yue J (2015) Sex hormones regulate cerebral drug metabolism via brain miRNAs: down-regulation of brain CYP2D by androgens reduces the analgesic effects of tramadol. Br J Pharmacol 172 (19):4639–4654. doi: 10.1111/bph.13206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cepeda MS, Carr DB (2003) Women experience more pain and require more morphine than men to achieve a similar degree of analgesia. Anesth Analg 97 (5):1464–1468. doi: 10.1213/01.ane.0000080153.36643.83 [DOI] [PubMed] [Google Scholar]
- 11.Fillingim RB, Ness TJ, Glover TL, Campbell CM, Hastie BA, Price DD, Staud R (2005) Morphine responses and experimental pain: sex differences in side effects and cardiovascular responses but not analgesia. J Pain 6 (2):116–124. doi: 10.1016/j.jpain.2004.11.005 [DOI] [PubMed] [Google Scholar]
- 12.Sarton E, Olofsen E, Romberg R, den Hartigh J, Kest B, Nieuwenhuijs D, Burm A, Teppema L, Dahan A (2000) Sex differences in morphine analgesia: an experimental study in healthy volunteers. Anesthesiology 93 (5):1245–1254; discussion 1246A. doi: 10.1097/00000542-200011000-00018 [DOI] [PubMed] [Google Scholar]
- 13.Cicero TJ, Nock B, Meyer ER (1996) Gender-related differences in the antinociceptive properties of morphine. J Pharmacol Exp Ther 279 (2):767–773 [PubMed] [Google Scholar]
- 14.Kanarek RB, Homoleski B (2000) Modulation of morphine-induced antinociception by palatable solutions in male and female rats. Pharmacol Biochem Behav 66 (3):653–659. doi: 10.1016/s0091-3057(00)00251-3 [DOI] [PubMed] [Google Scholar]
- 15.Holtman JR, Wala EP (2006) Characterization of the antinociceptive effect of oxycodone in male and female rats. Pharmacol Biochem Behav 83 (1):100–108. doi: 10.1016/j.pbb.2005.12.013 [DOI] [PubMed] [Google Scholar]
- 16.Peckham EM, Traynor JR (2006) Comparison of the antinociceptive response to morphine and morphine-like compounds in male and female Sprague-Dawley rats. J Pharmacol Exp Ther 316 (3):1195–1201. doi: 10.1124/jpet.105.094276 [DOI] [PubMed] [Google Scholar]
- 17.Collins D, Reed B, Zhang Y, Kreek MJ (2016) Sex differences in responsiveness to the prescription opioid oxycodone in mice. Pharmacol Biochem Behav 148:99–105. doi: 10.1016/j.pbb.2016.06.006 [DOI] [PubMed] [Google Scholar]
- 18.Lalovic B, Kharasch E, Hoffer C, Risler L, Liu-Chen LY, Shen DD (2006) Pharmacokinetics and pharmacodynamics of oral oxycodone in healthy human subjects: role of circulating active metabolites. Clin Pharmacol Ther 79 (5):461–479. doi: 10.1016/j.clpt.2006.01.009 [DOI] [PubMed] [Google Scholar]
- 19.Salmin SF, Giroux MC, Vachon P, Beaudry F (2017) In vitro metabolism of specific CYP2D and CYP3A opioid substrates using rat liver S9 fractions and mass spectrometry reveal a severe metabolic impairment with increasing age. Biomed Chromatogr 31 (2). doi: 10.1002/bmc.3786 [DOI] [PubMed] [Google Scholar]
- 20.McMillan DM, Miksys S, Tyndale RF (2019) Rat brain CYP2D activity alters in vivo central oxycodone metabolism, levels and resulting analgesia. Addict Biol 24 (2):228–238. doi: 10.1111/adb.12590 [DOI] [PubMed] [Google Scholar]
- 21.Bostrom E, Simonsson US, Hammarlund-Udenaes M (2006) In vivo blood-brain barrier transport of oxycodone in the rat: indications for active influx and implications for pharmacokinetics/pharmacodynamics. Drug Metab Dispos 34 (9):1624–1631. doi: 10.1124/dmd.106.009746 [DOI] [PubMed] [Google Scholar]
- 22.Cora MC, Kooistra L, Travlos G (2015) Vaginal Cytology of the Laboratory Rat and Mouse: Review and Criteria for the Staging of the Estrous Cycle Using Stained Vaginal Smears. Toxicol Pathol 43 (6):776–793. doi: 10.1177/0192623315570339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Le Bars D, Gozariu M, Cadden SW (2001) Animal models of nociception. Pharmacol Rev 53 (4):597–652 [PubMed] [Google Scholar]
- 24.McMillan DM, Tyndale RF (2015) Nicotine Increases Codeine Analgesia Through the Induction of Brain CYP2D and Central Activation of Codeine to Morphine. Neuropsychopharmacology 40 (7):1804–1812. doi: 10.1038/npp.2015.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rowland K, Yeo WW, Ellis SW, Chadwick IG, Haq I, Lennard MS, Jackson PR, Ramsay LE, Tucker GT (1994) Inhibition of CYP2D6 activity by treatment with propranolol and the role of 4-hydroxy propranolol. Br J Clin Pharmacol 38 (1):9–14. doi: 10.1111/j.1365-2125.1994.tb04315.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Masubuchi Y, Narimatsu S, Hosokawa S, Suzuki T (1994) Role of the CYP2D subfamily in metabolism-dependent covalent binding of propranolol to liver microsomal protein in rats. Biochem Pharmacol 48 (10):1891–1898. doi: 10.1016/0006-2952(94)90587-8 [DOI] [PubMed] [Google Scholar]
- 27.Miksys S, Wadji FB, Tolledo EC, Remington G, Nobrega JN, Tyndale RF (2017) Rat brain CYP2D enzymatic metabolism alters acute and chronic haloperidol side-effects by different mechanisms. Prog Neuropsychopharmacol Biol Psychiatry 78:140–148. doi: 10.1016/j.pnpbp.2017.04.030 [DOI] [PubMed] [Google Scholar]
- 28.Li J, Wan Y, Na S, Liu X, Dong G, Yang Z, Yang J, Yue J (2015) Sex-dependent regulation of hepatic CYP3A by growth hormone: Roles of HNF6, C/EBPα, and RXRα. Biochem Pharmacol 93 (1):92–103. doi: 10.1016/j.bcp.2014.10.010 [DOI] [PubMed] [Google Scholar]
- 29.Waxman DJ, Ram PA, Pampori NA, Shapiro BH (1995) Growth hormone regulation of male-specific rat liver P450s 2A2 and 3A2: induction by intermittent growth hormone pulses in male but not female rats rendered growth hormone deficient by neonatal monosodium glutamate. Mol Pharmacol 48 (5):790–797 [PubMed] [Google Scholar]
- 30.De Gregori S, De Gregori M, Ranzani GN, Allegri M, Minella C, Regazzi M (2012) Morphine metabolism, transport and brain disposition. Metab Brain Dis 27 (1):1–5. doi: 10.1007/s11011-011-9274-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaiko RF, Benziger DP, Fitzmartin RD, Burke BE, Reder RF, Goldenheim PD (1996) Pharmacokinetic-pharmacodynamic relationships of controlled-release oxycodone. Clin Pharmacol Ther 59 (1):52–61. doi: 10.1016/S0009-9236(96)90024-7 [DOI] [PubMed] [Google Scholar]
- 32.Cleary J, Mikus G, Somogyi A, Bochner F (1994) The influence of pharmacogenetics on opioid analgesia: studies with codeine and oxycodone in the Sprague-Dawley/Dark Agouti rat model. J Pharmacol Exp Ther 271 (3):1528–1534 [PubMed] [Google Scholar]
- 33.Gronlund J, Saari TI, Hagelberg NM, Neuvonen PJ, Olkkola KT, Laine K (2010) Exposure to oral oxycodone is increased by concomitant inhibition of CYP2D6 and 3A4 pathways, but not by inhibition of CYP2D6 alone. Br J Clin Pharmacol 70 (1):78–87. doi: 10.1111/j.1365-2125.2010.03653.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klimas R, Witticke D, El Fallah S, Mikus G (2013) Contribution of oxycodone and its metabolites to the overall analgesic effect after oxycodone administration. Expert Opin Drug Metab Toxicol 9 (5):517–528. doi: 10.1517/17425255.2013.779669 [DOI] [PubMed] [Google Scholar]
- 35.Lalovic B, Phillips B, Risler LL, Howald W, Shen DD (2004) Quantitative contribution of CYP2D6 and CYP3A to oxycodone metabolism in human liver and intestinal microsomes. Drug Metab Dispos 32 (4):447–454. doi: 10.1124/dmd.32.4.447 [DOI] [PubMed] [Google Scholar]
- 36.Sadiq MW, Boström E, Keizer R, Björkman S, Hammarlund-Udenaes M (2013) Oxymorphone active uptake at the blood-brain barrier and population modeling of its pharmacokinetic-pharmacodynamic relationship. J Pharm Sci 102 (9):3320–3331. doi: 10.1002/jps.23492 [DOI] [PubMed] [Google Scholar]
- 37.Grossman ML, Basbaum AI, Fields HL (1982) Afferent and efferent connections of the rat tail flick reflex (a model used to analyze pain control mechanisms). J Comp Neurol 206 (1):9–16. doi: 10.1002/cne.902060103 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.