

Summary
The proto-oncogene Anaplastic Lymphoma Kinase (ALK) is a receptor tyrosine kinase (RTK) expressed primarily in the developing nervous system. After development, ALK activity is associated with learning and memory1. ALK also controls energy expenditure and ALK inhibition can prevent diet-induced obesity2. Aberrant ALK signaling causes numerous cancers3. In particular, full length ALK is an important driver in pediatric neuroblastoma4,5, where it is either mutated6 or activated by ligand7. Here we report crystal structures of ALK’s unusual extracellular glycine-rich domain (GRD), which regulates receptor activity by binding to activating peptides (ALKALs)8,9. Fusing ALK’s GRD together with its ligand allowed us to capture a dimeric receptor complex that reveals how ALK responds to its regulatory ligands. We show that repetitive glycines in the GRD unexpectedly form rigid helices that separate the major ligand-binding site from a distal poly-glycine extension loop (PXL) that mediates ALK dimerization. The PXL of one receptor acts as a complex ‘sensor’ by interacting with a ligand bound second receptor. ALK activation can be abolished through PXL mutation or with PXL targeting antibodies. Together, our results explain how ALK uses an unusual architecture for its regulation, and suggest new therapeutic opportunities for ALK-expressing cancers like pediatric neuroblastoma.
ALK has an atypical domain architecture for an RTK10, including an unusual membrane-proximal glycine-rich domain (GRD). The GRD is a loosely defined region that includes a juxtamembrane cysteine rich region thought to adopt an EGF-like fold. This unusual GRD is shared between ALK and the closely related receptor leukocyte tyrosine kinase (LTK). Despite high glycine content –typically associated with structural disorder – the GRD alone is sufficient for ligand regulated receptor activity11,12. Ligands for vertebrate ALKs are designated ALKALs (ALK and LTK Activating Ligands), also known as Fam150 and AUG8,9,12. ALKALs are peptides of ~100 amino acids, with a highly conserved domain referred to as the ALKAL domain (AD) that is sufficient to stimulate ALK13. We crystallized the ALK’s GRD, the AD, and their complex to understand the physical basis for ligand-regulated activation of this RTK family.
Structure of ALK bound to activating ligand
To visualize the ALK GRD-ALKAL AD complex, we employed a fusion construct that links the C-terminus of ALK’s ECR to the N-terminus of the ALKAL2 conserved domain (AD) through a 14-residue linker. Use of this fusion was necessary to keep the AD soluble at sufficiently high concentration to saturate the GRD. The fusion construct purified as a monomer (Extended Data Fig. 1a). We solved the crystal structure to 3.05 Å by molecular replacement using phases of an isolated receptor that we solved separately (Extended Data Table 1). In the crystal, the fusion protein produces a dimeric complex yielding a 2:2 (receptor:ligand) structure (Fig. 1a). Two separate AD molecules (green) stabilize the ALK dimer. The ALK GRD is an elongated domain (Fig. 1a) with three largely distinct structural regions: beta sheets (red), short alpha helices (orange) and glycine helices (yellow). In the dimer, two ALK-AD complexes are related by 2-fold rotational symmetry (Fig. 1a, b). The AD forms a disulfide ‘stapled’ helical hairpin (Fig. 1a, b) similar to other helix-loop-helix peptides14–16. Despite being included in the fusion construct, the membrane proximal cysteine rich region of ALK’s GRD could not be reliably modeled.
Fig. 1 |. Structure of the ALK-ALKAL complex and ALK’s unusual glycine rich domain.
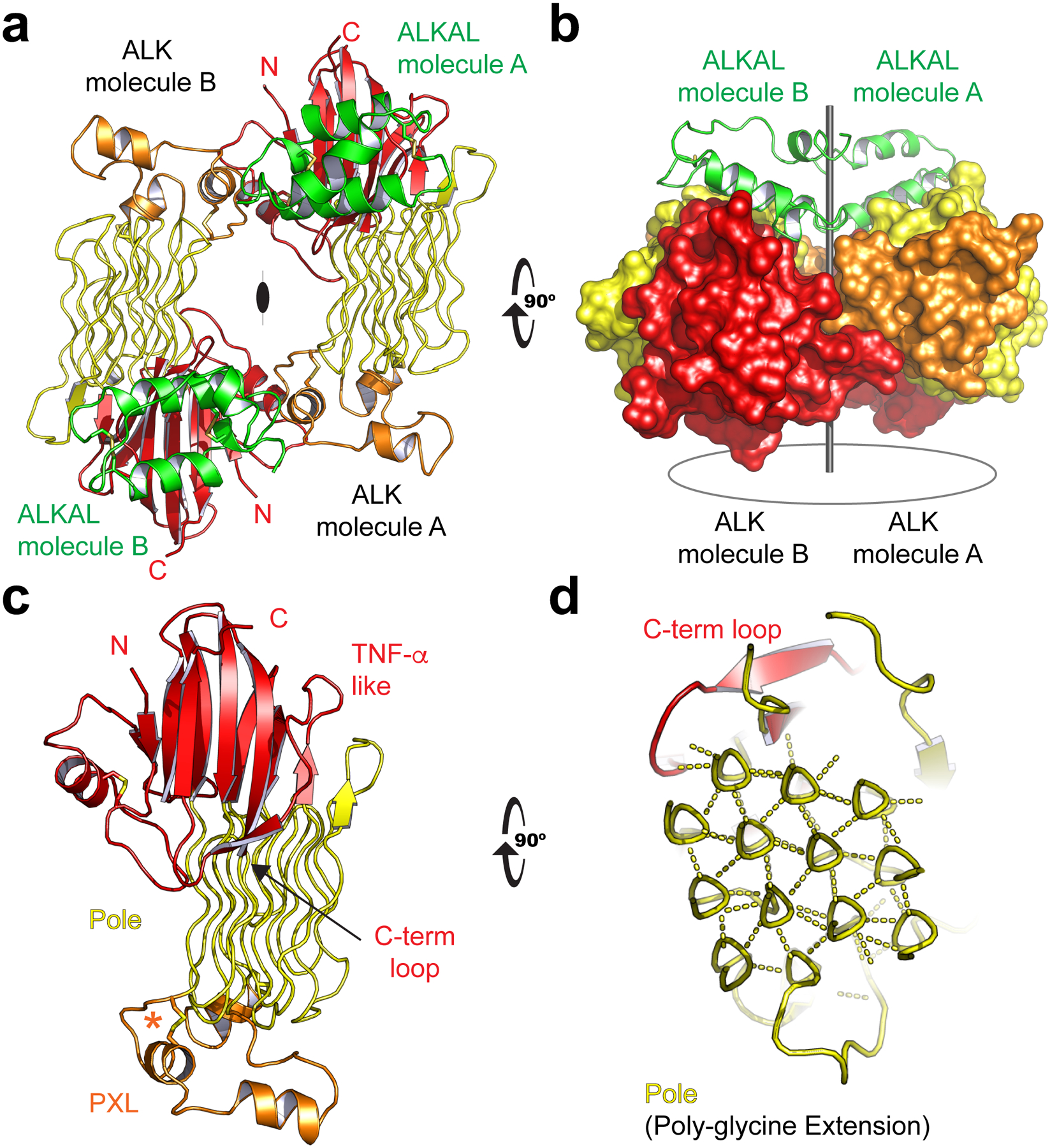
a, The (2:2, ALK:ALKAL) complex. One of two complexes in the crystal’s asymmetric unit (ASU) is shown. ALK’s GRD is colored by regions of predominant secondary structure (β-strands, red; α-helices, orange; strands, yellow). ALKAL is colored green. b, ALK’s GRD shown in surface representation. View orthogonal to (a). c, ALK’s GRD has a TNF-α like region (red) with an unusual Poly-Glycine Extension (Pole, yellow). Displayed on one end of the Pole are Poly-glycine eXtension Loops (PXL, orange). The primary loop helix is stabilized by a single disulfide, “disulfided helix” (asterisk). d, An enlarged orthogonal view details the Pole’s hexagonal lattice of rare glycine helices. Hydrogen bonds (dashed lines) link the polypeptide backbone to neighboring helices.
ALK’s unusual Glycine Rich Domain (GRD) structure
To better understand the unusual structure of the ALK ECR and how it responds to ligand, we also determined a high-resolution (1.91Å) crystal structure of an uncomplexed human ALK GRD, truncated prior to the cysteine rich region. In this additional structure, the N and C termini are adjacent to one another (Fig. 1c). The N- and C-terminal strands are part of a β-sandwich that resembles a tumor necrosis factor-α (TNF-α) domain (Fig. 1c)16, but has a distinct topology (Extended Data Fig. 1b)17. We therefore name this region “TNF-α like”. One key consequence of ALK’s unusual β-sandwich topology is that a prominent C-terminal loop (Fig. 1c, d) passes over two strands to complete the fold. This feature generates an important binding epitope that is discussed below.
Contrary to previous expectations, the multiple glycines characteristic of ALK’s GRD are not disordered. Rather, they extend from the TNF-α like region to form a highly rigid structure that weaves back-and-forth to the β-strands of the TNF-α like fold (Fig. 1c). Unexpectedly, the poly-glycine stretches adopt well-ordered poly-glycine II (PG-II) helices (Fig. 1d). We refer to this region as the Poly-Glycine Extension or ‘Pole’. The Pole forms a rarely observed hexagonal array of hydrogen-bonded glycine-helices (Fig. 1d)18. Such arrays were originally predicted from powder diffraction of glycine peptides19, and have recently been described in phage20. The Pole of human ALK has 14 helices that form 3 complete hexagons (Extended Data Fig. 1c–e). The fully saturated hydrogen-bonded main chain helices central to each hexagon contain only glycines and range from 6–8 residues in length. The Pole’s extensive hydrogen bonding and tight packing – permitted by the absence of side chains – creates a very compact and stable extension that projects linearly from the β-strands of the TNF-α like region and functions like a pole.
At the end of the Pole, opposite the TNF-α like fold, are loops that are not enriched in glycine (Fig. 1c), which we call the Poly-glycine eXtension Loops (PXL). The primary PXL loop links glycine helices 3 and 4 of the Pole (Extended Data Fig. 1b–e). Two α-helices are seen in this primary loop, one stabilized by a disulfide bond.
We also solved the structure of an invertebrate ALK GRD (C. elegans) to 2.60 Å, which reveals an overall similar architecture – although the PXL is structurally divergent and there are fewer glycine helices (Extended Data Fig. 1f–h). The invertebrate structure additionally includes the C-terminal cysteine-rich region that leads up to the TM helix. This region contains two EGF-like domains (Extended Data Fig. 2a, b). The EGF-like domains are packed against the GRD through multiple hydrophobic contacts (Extended Data Fig. 2c). Using the invertebrate structure as a template, human ALK is predicted to have a single EGF-like domain.
Ligand recognition by ALK
In the complex between ALK and ALKAL, the AD interacts primarily with the TNF-α like region in the GRD of the receptor (Fig. 2a). The highly positively-charged surface of the AD faces the TNF-α like region (Fig. 2b). A series of salt bridges form between the oppositely charged AD arginines and ALK GRD glutamate side-chains. The binding epitope on ALK centers around the C-terminal loop of the TNF-α like region and includes two conserved glutamates (E974, E978). A third conserved acidic residue sits next to this loop (E859) and interacts with R140 on the AD. In addition to these ionic interactions, the initial part of the C-terminal loop (Y966-L970) makes mostly non-polar interactions with the AD (Fig. 2c).
Fig. 2 |. Ligand binding is directed by ALK’s C-terminal loop.
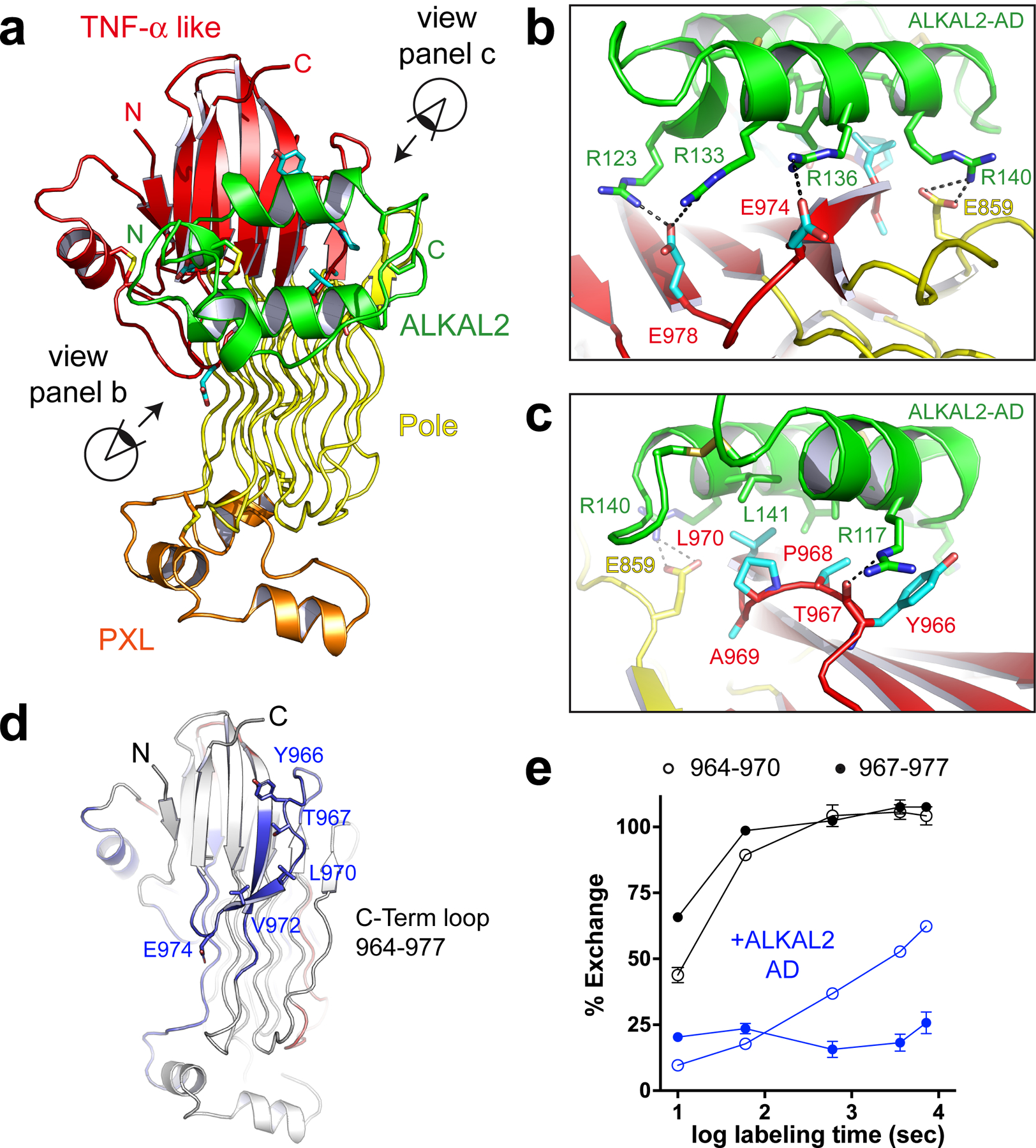
a, ALKAL binds to the TNF-α like region of ALK. b, The primary ligand-interacting residues on ALK involve the C-terminal loop of the TNF-α like region. The distal segment of the loop, including glutamates E974 and E978 is detailed. An additional glutamate, E859, outside the TNF-α like region interacts with the ALKAL2-AD. The positively charged arginine-containing surface of ALKAL faces the negatively charged glutamate cluster of ALK. c, Interactions involving the non-polar initial segment of ALK’s C-terminal loop (Y966-L970) are detailed. d, Statistically significant protection seen in HDX-MS studies mapped onto the crystal structure of human ALK GRD. Regions that show statistically significant protection when bound by ALKAL are colored blue. e, Percent exchange plots for the C-terminal loop peptides 964–970 and 967–977 with or without ALKAL2-AD as a function of exposure time. Data represent mean ± SD of two biologically independent measurements of which three technical repeats were carried out.
To validate the ALK-ALKAL interface observed in our crystal structure we used Hydrogen Deuterium Exchange Mass Spectrometry (HDX-MS) to map the binding epitope for the AD. HDX-MS reveals peptides protected from backbone amide proton exchange at complex interfaces21. Comparing deuterium exchange peptide profiles of ALK’s GRD alone with those seen in the presence of excess AD revealed two peptides that become significantly more protected upon ALKAL binding: 964–970 and 967–977 (Fig. 2d, e, Extended Data Table 2, Extended Data Fig. 3). These peptides correspond precisely to the C-terminal loop of the TNF-α like region that mediates AD binding in the crystal structure.
Using Bio-Layer Interferometry (BLI) we determined the impact of point mutations at the ligand binding interface on binding (Extended Data Table 3, Extended Data Fig. 4a). High-affinity ligand binding to ALK’s GRD requires the juxtamembrane EGF-like domain, deletion of which reduces affinity by ~100 fold. Mutating any of several interfacial glutamates on ALK to arginine (E859R, E974R, E978R) greatly impaired ALKAL binding. Swapping two of these glutamates to their corresponding residues in C. elegans (E974L/E978Y) completely abrogated detectable ALKAL binding and signaling (Extended Data Fig. 4a, b). Mutating the uncharged C-terminal loop residues identified by HDX to charged residues prevented AD binding. Furthermore, mutating arginines on ALKAL to glutamates (R133E, R140E) greatly reduced binding affinity. Together, these data support the importance of the receptor/ligand interface observed in our ALK-AD fusion crystal structure.
Nature of the ligand-induced ALK dimer
The dimer interface between two ligand-bound receptors results in two nearly equivalent interfaces and places the PXL of one receptor adjacent to the ligand-bound TNF-α like region on the other (Fig. 3a). The PXL of one ligand/receptor complex makes contacts with both the ligand and part of the TNF-α like region of the receptor in a second complex, but the interactions appear to be relatively weak. A surface exposed PXL side-chain (I795) makes hydrophobic contacts with the AD (Fig. 3b). Intriguingly, when compared to unliganded ALK, the GRD of ligand-bound dimeric ALK is rotated slightly clockwise (Extended Data Fig. 5a). This rotation appears to be coupled to ALK dimerization, which stabilizes the PXL’s disulfided helix (residues 783–797) (Extended Data Fig. 5b). The PXL’s disulfided helix also makes direct contact with the second receptor’s TNF-α like region involving N703 and A704 (Fig. 3c). Consistent with these regions playing an important role in dimerization, we observe protection of these dimerization interface peptides by HDX (Extended Data Fig. 5c, d). As expected, the rigid AD itself does not change significantly upon complex formation (Extended Data Fig. 5e).
Fig. 3 |. Dimerization of ALK is directed by the PXL.
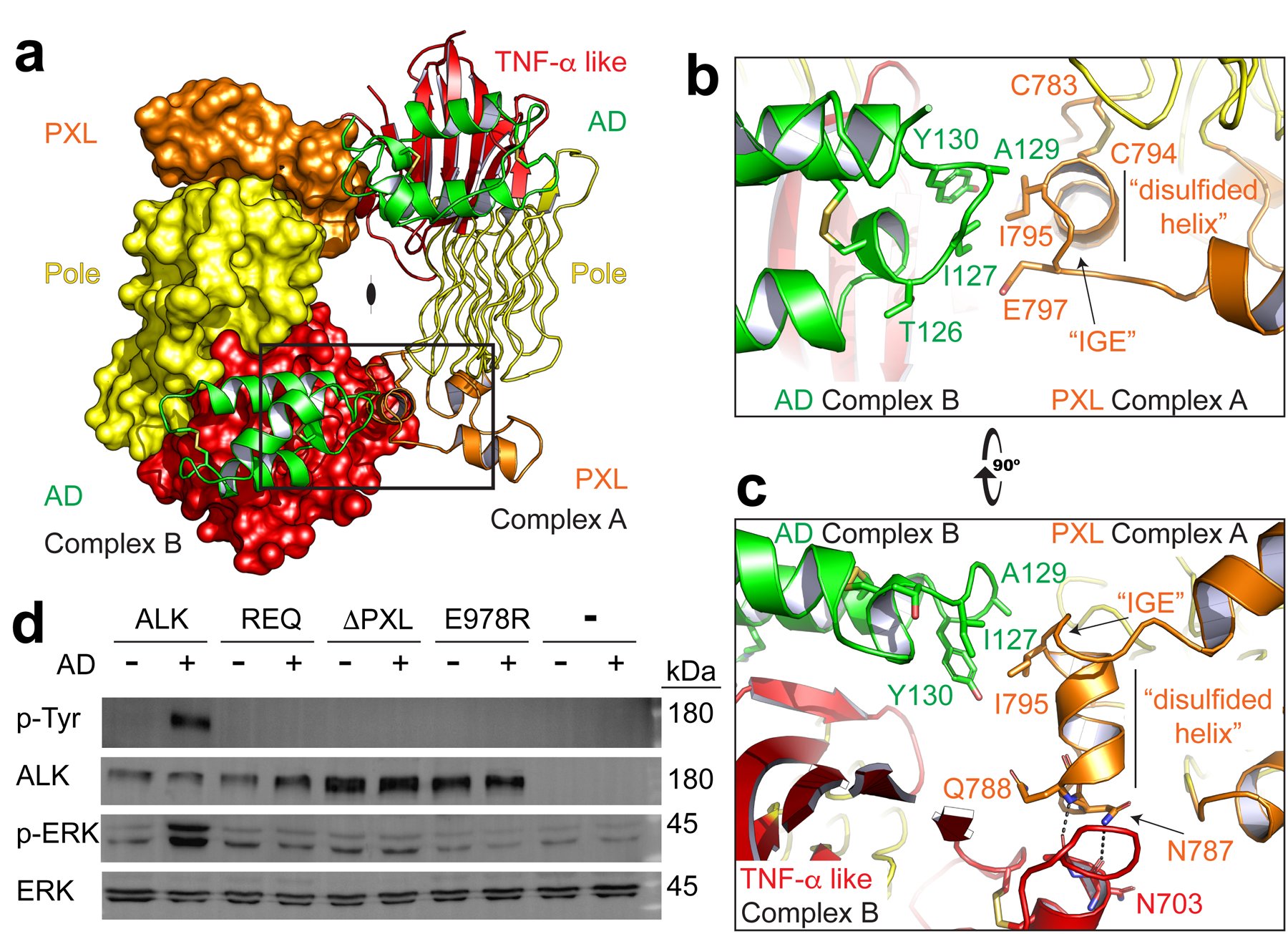
a, The ALK-ALKAL fusion complex forms a dimer (2:2, ALK:ALKAL). For clarity, the second receptor is shown in surface representation (left) with both bound ligands in green cartoon. b, An enlarged view of the boxed dimer interface from (a) to show details of the interaction. The PXL of one receptor (orange, Complex A) makes contacts with an epitope that combines ALKAL (green) and the TNF-α like region (red) of a second ligand-bound receptor (Complex B). c, An orthogonal view of the interface looking down the long axis of the Pole. d, NIH/3T3 cells stably expressing Halo-tagged ALK or ALK mutants were stimulated with a low concentration of purified ALKAL2-AD (0.5 nM for 10 min) to assess ligand responsiveness. REQ is a mutant that alters the three interface residues 795–797 (IGE, shown in b, c) to REQ. ΔPXL is a mutant that removes the entire “disulfided helix” 783–797 (shown in b,c). Data represent five independent measurements. For gel source data, see Supplementary Figure 1.
To determine whether the interface seen in the crystallographic ALK dimer is important for ALK activation in cellular contexts, we evaluated the impact of mutations in this interface on ALK activation. We designed two different mutation strategies in the PXL region. First, we mutated PXL residues 795–797 from (IGE) to REQ, “REQ”. This removes the hydrophobic side-chain of the isoleucine (I795) that interacts with the bound ligand on the adjacent complex (Fig. 3b). A second, more substantial modification (ΔPXL), removes the entire disulfided helix, including I795 and N787/Q788 that make dimer contacts with the ligand and receptor, respectively (Fig. 3c). Binding studies with the isolated GRD of these purified mutants demonstrated that each maintains high-affinity ligand binding (Extended Data Fig. 6a). We generated NIH3T3 stable lines expressing Halo-tagged ALK – beginning at the GRD and including the kinase domain (Halo-ALK). Using a cell-impermeable dye, we observed that each variant was properly expressed at the plasma membrane surface (Extended Data Fig. 6b). Upon adding a low dose of ALKAL2-AD (0.5 nM), both the REQ and ΔPXL variants remained unphosphorylated, whereas wild-type was robustly phosphorylated, leading to ERK activation (Fig. 3d). The ligand binding-defective C-terminal loop mutant E978R was also non-responsive to ALKAL2-AD. The lack of signaling from these PXL mutants is consistent with the crystallographic dimer model for ALK activation.
Antibody-based ALK inhibitors
We next studied two monoclonal ALK antibodies (mAbs) that have been developed as antibody-drug conjugates in preclinical studies of neuroblastoma: Celldex®-mAb123 (CDX123) and Celldex®-mAb125 (CDX125). CDX123 is an ‘activating’ antibody that likely functions by crosslinking receptor monomers22, whereas CDX125 is an inhibitory ALK antibody22. Using HDX-MS, we found that the activating mAb CDX123 protects a peptide in the PXL region that includes the dimer interface residue I795 (Fig. 4a). By contrast, the inhibitory mAb CDX125 protects a peptide in the C-terminal loop of the TNF-α like region of ALK (Y966-V972) – which overlaps with the ALKAL binding site (Fig. 4b). Consistent with these findings, BLI binding experiments demonstrated that mAb CDX125 efficiently prevents ALK from interacting with ALKAL whereas CDX123 binding does not affect ligand recognition by ALK (Fig. 4c). Indeed, the binding response in the presence of CDX123 is nearly 4 times higher due to the increased mass of the complex. Thus, CDX125 but not CDX123 inhibits ligand binding to ALK.
Fig. 4 |. Antibodies to the C-terminal loop or PXL prevent ALK activation.
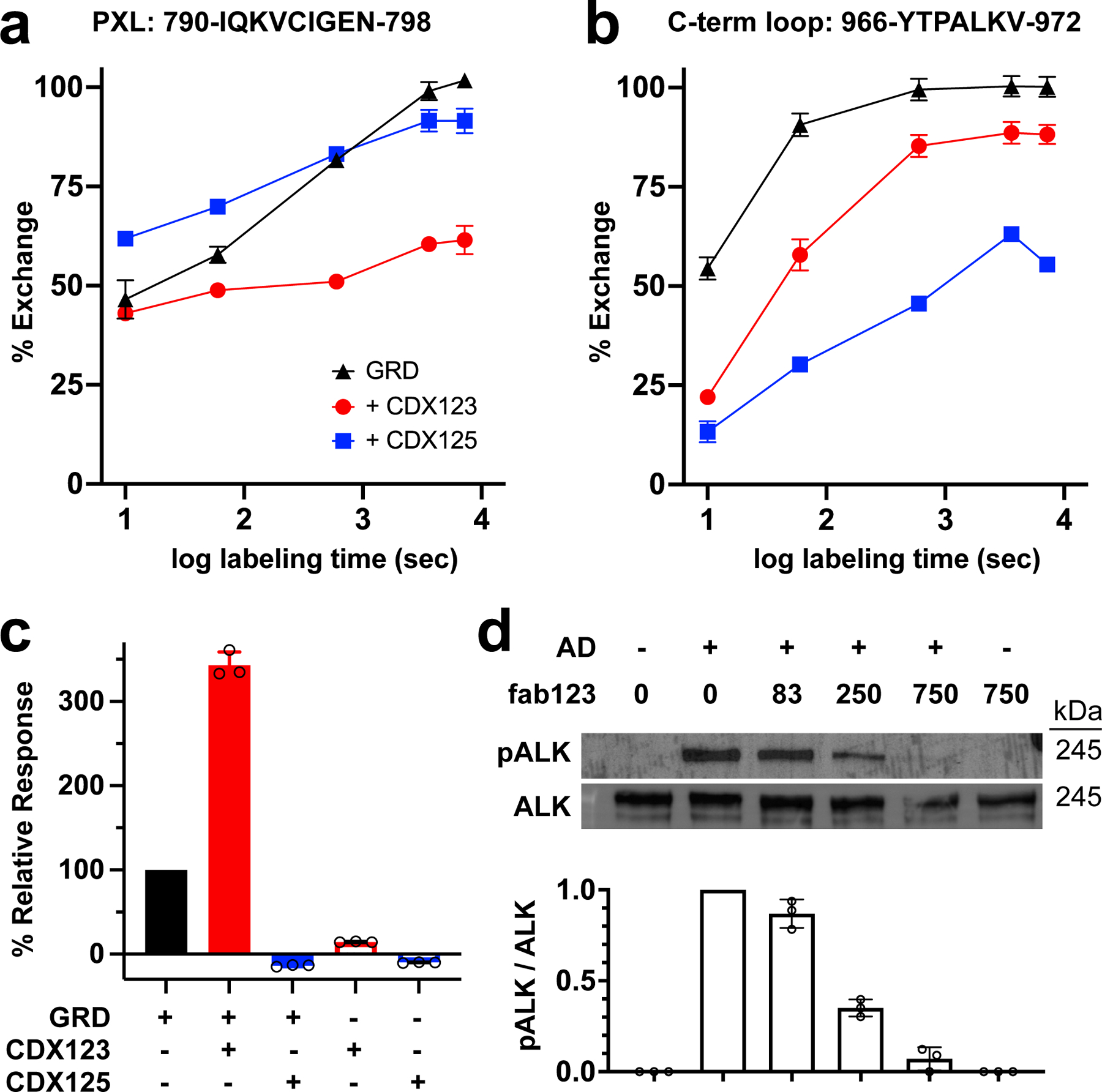
a, Percent exchange plots for residues 790–798 of the PXL as a function of exposure time. b, Percent exchange plots for the residues 966–972 of the C-term loop as a function of exposure time. c, Binding of GRD or a GRD-antibody complex to ALKAL2-AD detected by BLI. d, Neuroblastoma (IMR32) cells preincubated with the indicated concentrations of fab123 were stimulated with purified ALKAL2-AD. Phosphorylation of endogenous ALK at Y1604 (pALK) and total ALK were assessed by immunoblotting and quantified. Data represent mean ± SD of three independent measurements. For gel source data, see Supplementary Figure 1.
Having found that the CDX123-binding epitope includes the ALK dimer interface (Fig. 4a and Fig. 3b) but does not impact ligand recognition, we reasoned that it might block formation of the activated ALK dimer. We confirmed that the PXL region is the epitope for CDX123 by showing that the PXL deletion construct loses ability to bind CDX123 but retains CDX125 binding (Extended Data Fig. 6c). Since bivalent CDX123 can cross-link and dimerize ALK (leading to activation) we reasoned that a monovalent antigen-binding fragment (Fab) of CDX123 (fab123) should inhibit ligand-induced ALK activation on cells by blocking the dimer interface. Indeed, preincubating neuroblastoma IMR32 cells (which express wild-type ALK) with fab123 prevented AD induced ALK phosphorylation and ERK activation in a dose dependent manner (Fig. 4d). These data strongly support the importance of the ALK dimerization interface identified in our crystallographic and HDX studies for ALK activation in cells.
Discussion
Our structural studies of ALK reveal both a rare fold and an unexpected mode of regulation among receptor tyrosine kinases. Two GRD peptide loops drive ligand binding and receptor dimerization – the C-terminal loop and the PXL, respectively. The rigid Pole separates these two loops. However, the GRD’s unique architecture presents folding challenges that likely limits its evolutionary occurrence, possibly explaining the rarity of this fold. Indeed, substitution of any glycine in the Pole would disrupt proper folding. Notably, multiple glycine to acidic residues mutations have been described for Drosophila ALK9 and many similar acidic mutations have been catalogued in the COSMIC database23 (Extended Data Fig. 7a, b).
Although ALK’s GRD is conserved from invertebrates through vertebrates, its ligand is not. Jeb and Hen-1, reported to be ligands for Drosophila and C. elegans ALK, respectively, both share an LDL-A domain that is required for receptor stimulation24–26. No related ligand has been identified in vertebrates, however. We could not detect AD binding to C. elegans ALK (Extended Data Fig. 7c). Moreover, overlays of invertebrate and vertebrate GRDs reveals that the AD binding epitope is not preserved (Extended Data Fig. 7d) – consistent with the need for a structurally different ligand.
We note that our fusion construct likely enforced the observed near-symmetric complex. It remains possible, however, that a single AD could stabilize an ALK dimer. Furthermore, alternate modes of oligomerization or additional interactions may help regulate ALK’s activity27. We additionally note that findings from an independent study using complementary techniques strongly support the model we present here28.
Together, the regulation of ALK dimerization by ALKAL points to clear ways to inhibit ALK activity and may offer new therapeutic strategies in multiple disease settings. Moreover, LTK maintains the critical epitopes for ligand binding and dimerization described here – suggesting that the findings reported here for ALK should extend to LTK.
Methods
Recombinant Protein Expression and Purification
Codon-optimized cDNAs for the GRD of human ALK (678–1030), C. elegans SCD-2 (551–900), and the human ALK ligand ALKAL (ALKAL2-AD, 71–152) were synthesized (IDT). The sequences were incorporated into a pFastBac plasmid (Invitrogen) for baculovirus (Autographa californica multiple nucleopolyhedrovirus; AcMNPV) driven expression in Trichoplusia ni (Hi5) cells. The mature protein sequence followed a Wnt3 signal peptide (MEPHLLGLLLGLLLGGTRVLAG)29. An octa-histidine tag combined with a Factor Xa cut site was added to the amino terminus. A Spitz glycosylation peptide (GSSTSNGSSISSSMSGTALPPTQAPVTSSTTMRTTTTTTPRPNIT) was added between the His tag and the Factor Xa cut site for ALKAL2-AD expression30. The fusion protein linked the human ALK-GRD after the EGF domain and ALKAL2 with a linker (GGSGGSGGSGGSGG). Mutations of human ALK-GRD and ALKAL constructs were introduced by site directed mutagenesis using the QuickChange Kit (Agilent Technologies). The proteins were purified from baculovirus-infected Hi5 cells grown to a density of 1×106 cells/ml in Ex-Cell405 medium (Sigma-Aldrich). Media were harvested 2 days post infection and flowed directly over Ni-Penta™ Agarose-Base Resin (Marvelgent Biosciences) by gravity. The resins were washed three times with 50 ml of 20 mM Imidazole (pH 8.0), 100 mM NaCl, and bound proteins were eluted with 35 ml of 200 mM and 35 ml of 500 mM imidazole (pH 8.0), 100 mM NaCl. Eluted proteins were further purified by size exclusion chromatography with a HiLoad 26/600 superdex 200 pg or 75 pg column (GE Healthcare Life Sciences) equilibrated in 20 mM HEPES (pH 7.5) with 100 mM NaCl. Fractions were evaluated by SDS-PAGE, then concentrated in a Centricon 10 kDa spin concentrator (Millipore) and saved at 4°C. The His-tags of ALK-GRD and its mutants were cleaved by Factor Xa protease (New England Biolab) then flowed over Ni-Penta™ Agarose-Base Resin. The tag-free flow through was collected and further purified by Superdex 200 or 75 Increase 10/300 GL size exclusion column (GE Healthcare Life Sciences) equilibrated in 20 mM HEPES (pH 7.5) with 100 mM NaCl. For C. elegans SCD-2 (551–900) selenium-methionine substituted protein expression, Hi5 cells grown to a density of 1 × 106 cells/ml were first infected with baculovirus for 8 hours then centrifuged at 250 × g for 20 min at 4°C and re-suspended in methionine-free medium and starved of methionine for 8 h. Seleno-L-methionine (Sigma-Aldrich) was then added to the culture to a final concentration of 166.7 mg/l.
Crystallography
Crystals of C. elegans SCD-2-GRD were grown at 18°C by mixing equal volumes of protein (28 mg/ml) and reservoir solution (0.1 M Bicine (pH 9.0), 1.84 M (NH4)2SO4) using the hanging-drop method. Crystals were cryoprotected in 0.1 M Bicine (pH 9.0), 2.2 M (NH4)2SO4, 20% glycerol or 2.2 M melonate (pH 8.0) then shot at the Advanced Photon Source (APS) beamline NE-CAT. The structure was solved with Se-Met substituted protein by multi-wavelength anomalous diffraction (MAD). SHARP/autoSHARP was used for locating the anomalous atoms and generating phases31. The density was sufficient for manual building with Coot32. Crystals of human ALK-GRD without EGF domain were grown at 18°C in hanging drops containing equal volumes of protein (26 mg/ml) and reservoir solution (0.1 M imidazole (pH 8.0), 10% PEG8000, 0.2 M Ca(OAc)2). Crystals were frozen in reservoir solution with 9% glycerol. The structure was solved by Phaser molecular replacement and autobuild on ARP/wARP using trimmed a C. elegans SCD-2-GRD structure as a starting model33,34. Crystals of the ALK-ALKAL fusion protein were obtained after several months by adding equal volumes of protein (16 mg/ml) and reservoir solution (0.1 M citrate (pH 5.5), 20% PEG3000) and hanging at 16°C. Crystals were frozen in reservoir solution plus 9% glycerol. The structure was solved by molecular replacement using a human GRD model with Phaser33 in the Phenix version 1.19.1–412235 platform curated by SBGrid version 1.0.69536.
Hydrogen-deuterium exchange and mass spectrometry (HDX-MS)
The purified protein, with a concentration of 0.3 mg/ml, was diluted 20-fold with 20 mM HEPES, 100 mM NaCl (pH 7.4) D2O buffer for the deuterium labeling experiments. All deuterium labeling experiments were performed using the HDX-2 PAL platform (LEAP Technologies) interfaced with an Acquity UPLC-M class (Waters) and a Synapt G2-Si (Waters) mass spectrometer. After the deuterium labeling step for different time periods (10, 60, 600, 3600, 7200 seconds) at 25°C, the labeling reaction was quenched by adding 200 mM sodium phosphate, 2 M guanidine-HCl, 0.1 M TCEP (pH 2.5) to lower the sample pH to ~2.5 at 0°C. The labeled sample was injected onto an Enzymate BEH pepsin column (Waters) maintained at 15°C to digest the labeled protein for 3 min, and the peptic peptides were trapped using an Acquity UPLC BEH C18 pre-column (2.1 × 5 mm, 1.7 μm, Waters). Peptic peptides were separated using an Acquity UPLC BEH C18 column (1.0 × 100 mm, 1.7 μm, Waters) at 0°C, and were eluted using 5% to 40 % acetonitrile gradient over 7 min to assess the deuterium uptake of each peptic peptide by mass spectrometry analysis. The MSe data were collected using ramp collision energy of 15 V to 35 V with continuous lock mass (Leu-Enk) for mass accuracy correction. To prepare the fully-deuterated protein sample, the protein was unfolded in 20 mM HEPES, 6 M guanidine-HCl at pH 7.4 and deuterium-labeled for 1 min. Subsequent experimental steps for the fully-deuterated standard sample were the same as described above.
HDX-MS data analysis
Peptides were sequenced using ProteinLynx Global Server 3.0.3 (PLGS, Waters), and the deuterium uptake of each peptic peptide was determined using Waters DynamX 3.0. The percent exchange of each peptic peptide (%D) was determined by equation 1:
where mt = the centroid mass of a peptic peptide at time, t, m0 = the centroid mass of a peptic peptide without deuterium labeling, and mf = the centroid mass of a peptic peptide for the fully-deuterated standard sample. The difference in the deuterium uptake of each peptide between unliganded and the complex (Δ%Ex) was calculated using the percent deuterium uptake obtained by equation 1 as the following:
Bio-Layer Interferometry
Using an Octet RED96e instrument (ForteBio), HIS1K biosensors (ForteBio) were first loaded with ALKAL to a response of about 1 nm followed by quenching in 10 ng/ml biocytin (Sigma-Aldrich) for 120 s and equilibrated in the working buffer (20 mM HEPES, pH 7.5, containing 100 mM NaCl) for 15 s. The sensors were then dipped in indicated concentrations of GRD or GRD mutants for 60 s, then dissociated in the working buffer for 60 s. Equilibrium (plateau) responses were plotted against the concentrations (GraphPad Prism 8.1.1 and 9.1) and a one site specific binding model was used to fit the data. The Bmax was shared for the same batch of experiments. The responses divided by the Bmax were termed as relative binding. All the relative bindings were combined and plotted against the concentrations in GraphPad Prism. Each point represents mean ± SD (of 15 measurements for WT, 3 measurements for No EGF, E859R, E978R, E974L&E978Y, Y966R, T967R, L970R, V972R, 2 measurements for E974R, R123E, R133E and R140E). The fitted KD and the error of the KD were summarized in the Extended Data Table 3. The interactions between antibodies and GRD were analyzed in the same way, except the antibodies were loaded onto protein A biosensors. The antibody competition assay was done in the same way with ALKAL loaded on the HIS1K biosensors. The analytes were either 1 μM GRD or antibody or their combinations. Responses at the plateau were normalized to GRD. Data represent mean ± SD of three independent measurements.
Cell Stimulation Assays
IMR-32 cells (ATCC CCL-127, Homo sapiens brain neuroblast cell from 13 month old male neuroblastoma) were cultured in Opti-MEM I reduced serum medium (Gibco) with 10% HI FBS and 1% penicillin/streptomycin (Sigma Aldrich) in 10 cm plates until reaching 90% confluence, after which cells were serum starved overnight. The starved cells were stimulated with 0.5 nM ALKAL2-AD or ALKAL2-AD mutants at 37°C for 10 min. The effects of Fabs were evaluated by adding the indicated concentration of Fabs 30 min prior to ALKAL stimulation. Fab123 and Fab125 were generated by treating the CDX123 and CDX125 monoclonal antibodies (mAbs) with immobilized papain agarose (Goldbio) according to instructions with the buffer adjusted to contain 100 mM NaCl. The flow-throughs were then incubated with protein A agarose resin (Goldbio) to eliminate fragment constant (Fc) and uncut mAbs. The flow-throughs of protein A agarose resin were further purified using a Superdex 200 Increase 10/300 GL size exclusion column (GE Healthcare Life Sciences) equilibrated in 20 mM HEPES (pH 7.5) with 100 mM NaCl.
An NIH/3T3 (ATCC CRL-1658, Mus musculus embryo fibroblast cell) cell line stably expressing ALK with the domains before GRD substituted by HaloTag (NIH/3T3-HaloGRD) was established. The pcDNA3.1(−)-ALK plasmid was a gift from Lemmon Lab37. The HaloTag (HALO7 1–297, NCBI GenBank: KY511555) plus a GSG linker was incorporated between A18 and D678 of ALK using the NEBuilder HiFi DNA Assembly Cloning Kit (New England Biolabs). The constructed pcDNA3.1(−)-HaloGRD plasmid was transfected into NIH/3T3 using Lipofectamine 2000 (Thermo Fisher Scientific). The NIH/3T3-HaloGRD stable cell line was selected with Opti-MEM I reduced serum medium (Gibco) containing 10% HI FBS, 1% penicillin/streptomycin (Sigma Aldrich), and 400 μg/ml G-418 (Santa Cruz Biotechnology). The stimulations were done the same as with IMR-32 cells.
Western Blotting
After stimulation, cells were immediately placed on ice. One milliliter of ice-cold PBS was added to the dish, and cells were scraped and transferred into ice-cold EP tube. Cells were then centrifuged at 300 × g for 5 min at 4°C and washed with ice-cold PBS twice. The cell pellets were lysed on ice for 40 min in lysis buffer (20 mM HEPES (pH 7.5), 140 mM NaCl, 1% NP-40) with 1× protease inhibitor cocktail (Roche) and 1× phosphatase inhibitor cocktail (Santa Cruz Technology). After lysis, the solutions were centrifugated at 15,000 rpm for 15 min and the supernatants were collected. The concentrations of the total proteins were measured by Bradford assay (Thermo Fisher Scientific). For detecting ALK and phosphorylated ALK, five dishes of IMR-32 cells or two dishes of NIT/3T3-HaloGRD cells lysates were combined and incubated overnight at 4°C with protein A agarose resin coated with ALK monoclonal antibody C26G7 (Cell Signaling Technology). The resins were washed with cold PBS three times prior to boiling in 1× reducing protein loading buffer at 100°C for 7 min. The flow-throughs were collected and prepared for detecting ERK and phosphorylated ERK by adding reducing protein loading buffer and boiling at 100°C for 7 min. Samples were stored at −20°C. For detecting ERK1/2 and p-ERK1/2, 100 μg total proteins were loaded on a 4–20% gradient SDS-PAGE gel. For detecting ALK and p-ALK, one pull-down sample was loaded on two 4–12% gradient SDS-PAGE gels. BLUEstain 2 protein ladder (GoldBio) was used as a molecular weight ladder. After electrophoresis, proteins were transferred to PVDF membrane in transfer buffer (25 mM Tris, 192 mM glycine, 20% methanol (v/v)) using 90 V constant voltage for 47 min. The membranes were then blocked with 5% BSA in TBST buffer (20 mM Tris (pH 7.5), 150 mM NaCl, 0.1% Tween 20) for 1 h at room temperature followed by incubation with the primary antibodies overnight at 4°C. The dilutions for the primary antibodies were: p44/42 MAPK (Erk1/2) Antibody (Cell Signaling Technology), 1:1000; Phospho-p44/42 MAPK (p-Erk1/2) (Thr202/Tyr204) Antibody (Cell Signaling Technology), 1:1000; ALK (C26G7) Rabbit mAb (Cell Signaling Technology), 1:1000; Anti-ALK (phospho Y1604) antibody (Abcam), 1:500. The horseradish peroxidase (HRP) conjugated Goat Anti-Rabbit IgG - H&L Polyclonal antibody (Abcam) and Rabbit Anti-Mouse IgG H&L (Abcam) was then diluted 1:5000 and incubated with the membranes at room temperature for 1 h. The membranes were developed with Amersham ECL prime western blotting detection reagent (GE healthcare Life Sciences) and chemiluminescence scanned using ChemiDoc (Bio-Rad). The bands were quantified using ImageJ 1.51h.
Imaging
Cells were seeded in 2 cm dishes at 25% confluence the day before imaging. Membrane impermeable dye, JF-549i from Luke Lavis’ lab at Janelia38, was added to the cells to a final concentration of 400 nM and incubated at 37°C for 15 min followed by three times wash with DPBS. Cells were kept in live cell imaging solution (Gibco) during imaging. The fluorescence images were taken at 500 ms exposure and 200 ms brightfield exposure. The untransfected image was adjusted to reflect the same intensities as the GRD image.
Full length ALK stimulation assay
NIH/3T3 cell stably expressing WT ALK or its various mutants (described in Extended Data Fig. 6) were generated using the retroviral pBABE expression vector containing the puromycin resistant gene. The cells were maintained at 37°C, 5% CO2, in DMEM with 10% FBS, 1% penicilin-streptomycin and 1μg/ml puromycin. Cells were starved overnight in serum free medium before stimulation for 10 minutes with 10nM of ALKAL2-AD and cell lysates were incubated with anti ALK antibodies (homemade) and protein A Sepharose (Invitrogen) overnight at 4°C. The immunocomplexes were washed with lysis buffer, separated on SDS-PAGE and immunoblotted with anti-pTyr antibodies followed by immunoblotting with anti-Alk antibodies.
Purification and Crystallization of AD
AD-β (residues 60–129) with a N-terminal 6×His tag followed by a TEV cleavage site and a C-terminal crystallizable MBP-tag lacking N-terminal residues KIEEG was cloned into a pET42 vector. Protein expression and refolding were performed following a previously described protocol13. Protein expression was performed using BL21-CodonPlus (DE3)-RIPL cells (Agilent) in LB media for 3 h at 37°C. Cell pellets were resuspended in lysis buffer (2×PBS, 0.5% IGEPAL, 1 mg/ml DNAse I (Roche), 1 mg/ml lysozyme (AmercanBio)), and lysed by sonification. Inclusion bodies were isolated by centrifugation (5000 × g, 15 min), and pellets were washed three times with 2×PBS, 0.5% IGEPAL. Washed inclusion bodies were solubilized in solubilization buffer (6 M guanidine chloride, 50 mM Tris pH 8.8, and 20 mM DTT) at a concentration of 15 mg/ml, and incubated overnight. The suspension was centrifugation (5000 × g, 15 min), and the supernatant was diluted with a buffer composed of 2.6 M guanidine chloride, 500 mM arginine chloride, 50 mM Tris pH 8.8, 10 mM reduced glutathione, 1 mM oxidized glutathione, 0.001% IGEPAL-CA630 to final concentration of 50 μg/ml. Protein was refolded by dialysis against refolding buffer (10 mM HEPES pH 7.4, 10% glycerol, 150 mM NaCl, 5 mM maltose, 0.001% IGEPAL-CA630) with a Spectra/Por MWCO 6–8 kD dialysis membrane (overnight, 4°C). Refolded protein was purified in batch mode with Ni-NTA agarose, followed by size exclusion chromatography (HiLoad Superdex 200 16/60 column in buffer containing 15 mM HEPES pH 7.4, 500 mM NaCl, 0.5 M urea, and 0.1% CHAPS). After removal of the His-tag by TEV protease cleavage, protein was further purified by cation exchange chromatography (Source 15S resin in 15 mM HEPES pH 7.4, 150 mM NaCl, and 0.1% CHAPS; eluted with a linear gradient of 0.15–1 M NaCl). Buffer was exchanged to 15 mM HEPS pH 7.4, 150 mM NaCl, and 5 mM maltose using a Superdex 75 10/300 column. AD-β-MBP was crystallized by hanging drop vapor diffusion at room temperature, using a protein concentration of 14.5 mg/ml and a reservoir solution containing 0.1 M sodium citrate pH 4.9, and 16% PEG 4000. Crystals were cryo-protected with reservoir solution containing 20% glycerol, and flash frozen in liquid nitrogen.
Data were collected at the Advanced Photon Source (APS; Argonne, Illinois, USA) on beamline 24-ID-C (λ=0.979200 Å) on a Dectris Pilatus 6M pixel detector at 100 K. Diffraction data were indexed, integrated, and scaled using XDS39 curated by SBGrid36. Initial phases were calculated via molecular replacement using Phaser33 from CCP440, using the structure of MBP (PDB ID 1ANF) as search model. Refinement was performed with PHENIX version 1.11.1–257541. For cross validation of the model, 5% of randomly selected reflections were omitted from refinement and used for the calculation of Rfree. Initially, Cartesian simulated annealing was performed, followed by alternating cycles of manual model building and TLS refinement using Coot32 and PHENIX, respectively.
Extended Data
Extended Data Figure 1 |. Structural comparison of human and invertebrate ALK.
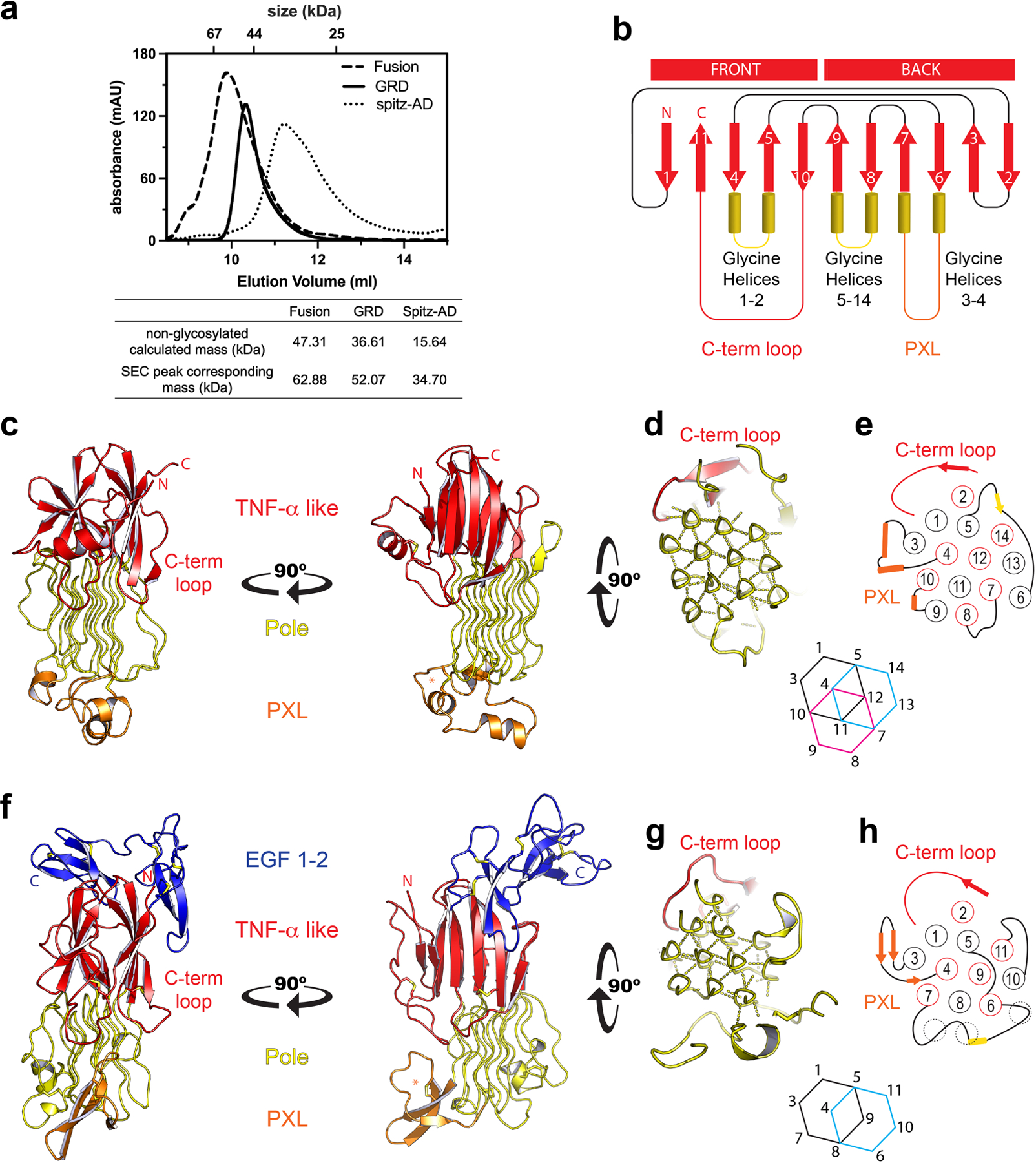
a, Size exclusion chromatography of each protein was carried out using a Superdex 75 Increase 10/300 GL column. The non-glycosylated mass was calculated from the protein sequence. The mass corresponding to each SEC peak was determined based on the log (MW) versus elution volume plot of standards for the column. The SEC peak mass of each protein is consistent with a glycosylated monomer. b, ALK’s glycine helices project downward from the TNF-α like region. Unique to ALK, strand 10 crosses over strands 4 and 5 to terminate the fold, producing a surface terminal loop, “C-term loop”. The GRD has a distinct topology, and does not form the jelly-roll characteristic of TNF-α domains c, The GRD of human ALK. d, The hexagonal array of the Pole, and e, the order and topology of the Pole (red circles into the page, black out of the page). f, C. elegans ALK adopts a similar overall architecture to the human GRD. However, the invertebrate PXL is structurally different, forming a β-hairpin rather than helices (orange). g, The Pole of invertebrate ALK is also smaller, with 11 glycine helices that form 2 complete hexagons. h, The helical strands are in the same order and topology as in human ALK. Interestingly, the missing glycine helices of the invertebrate Pole are partially matched by non PG-II loops that interact with the hexagonal array (dashed circles). The invertebrate GRD structure additionally includes a C-terminal cysteine-rich region that leads up to the transmembrane domain. This region has 10 cysteines and forms 2 EGF-like domains (f, blue).
Extended Data Figure 2 |. Structure of invertebrate ALK’s EGF-like domains.
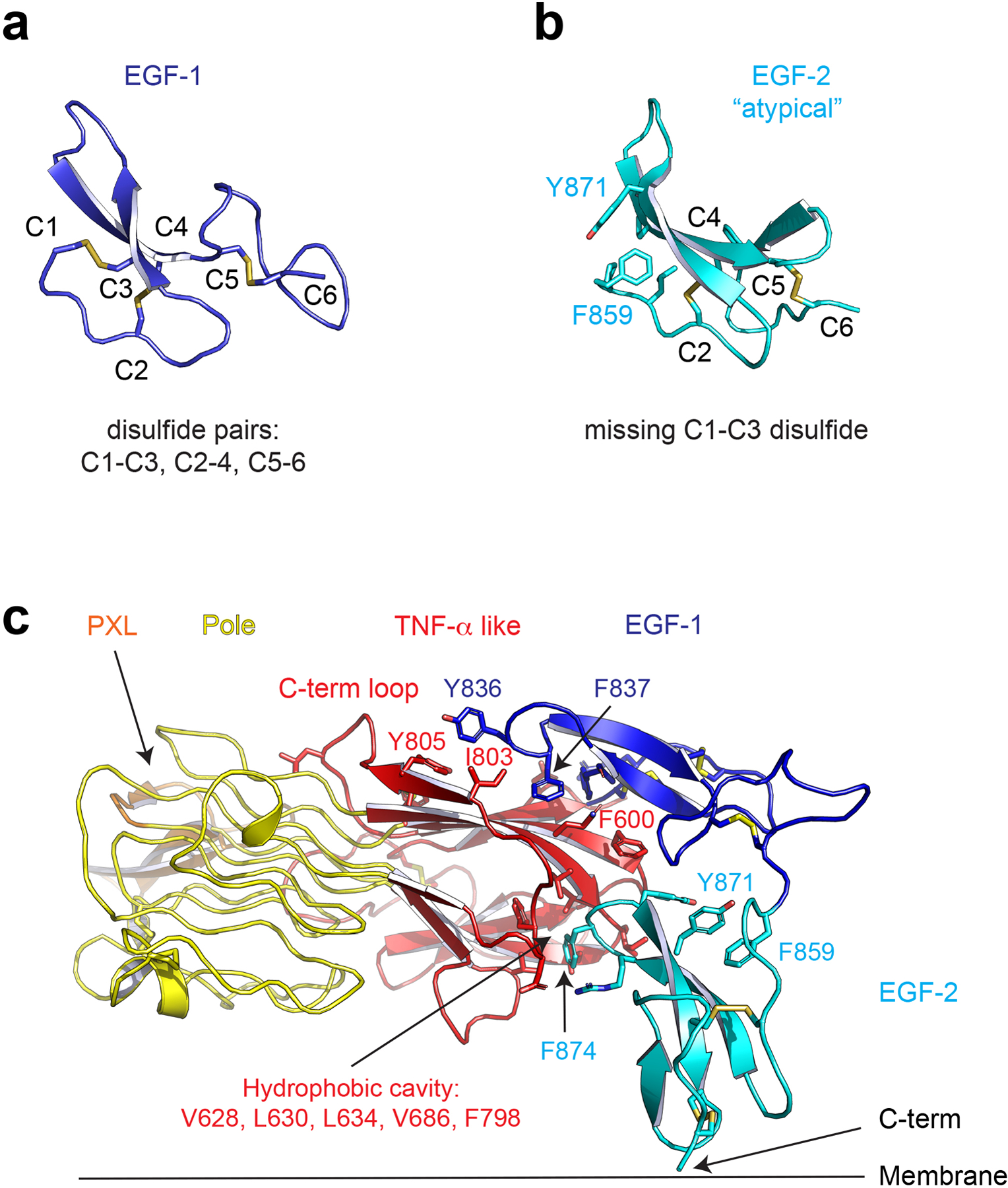
a, The first EGF-like domain has canonical disulfide pairing. b, The second EGF-like domain is atypical in that it lacks the first 1–3 disulfide bond, the stabilizing role of which is replaced by hydrophobic interactions involving Y871 and F859. c, The EGF-like domains pack tightly to the TNF-α like domain with hydrophobic interactions. Y836 and F837 of the first EGF domain bind to the proximal C-terminal loop. F874 of the second EGF like domain is buried in a hydrophobic cavity.
Extended Data Figure 3 |. HDX analysis of ALKAL binding to ALK.
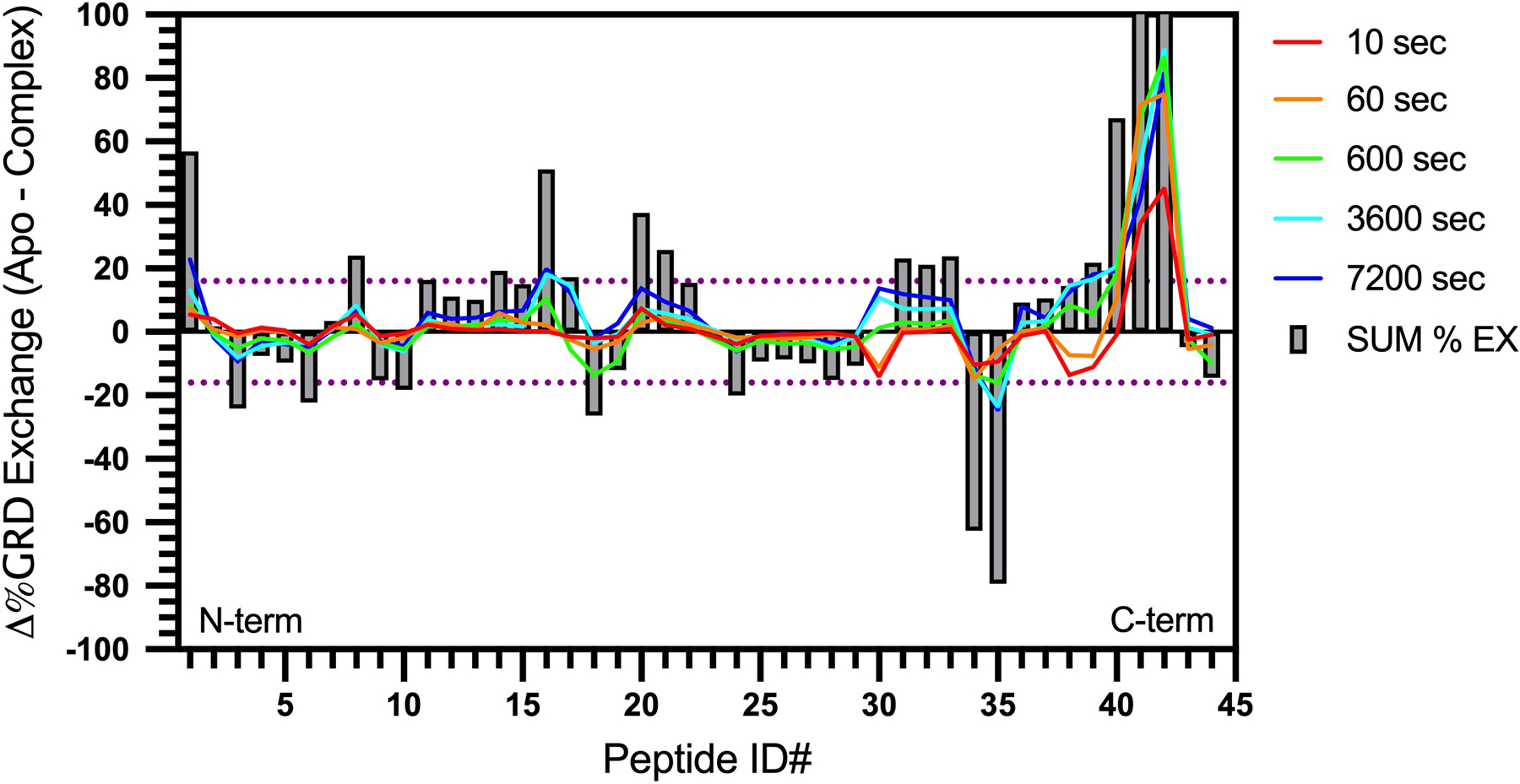
HDX-MS percent exchange butterfly plot for ALKAL2-AD binding to human ALK GRD. Each peptide is assigned a peptide ID number (Extended Data Table 2) from the N- (left) to the C-terminus (right). Each grey bar shows the sum of Δ%Exchange at all labeling timepoints (Δ%sum) for each peptide. The dotted purple line corresponds to statistically significant Δ%sum (16%, or ± 0.77 Da difference in deuterium uptake between unliganded and ALKAL-bound GRD) with 98 % confidence limit calculated based on the measured standard deviation of deuterium uptake for each peptide. Regions with positive or negative Δ%Exchange become more stable or flexible, respectively, upon ALKAL binding. Statistics were derived from two independent biological repeats, each with three technical repeats. Data represent mean ± SD.
Extended Data Figure 4 |. ALK binding and signaling by ALKAL2-AD.
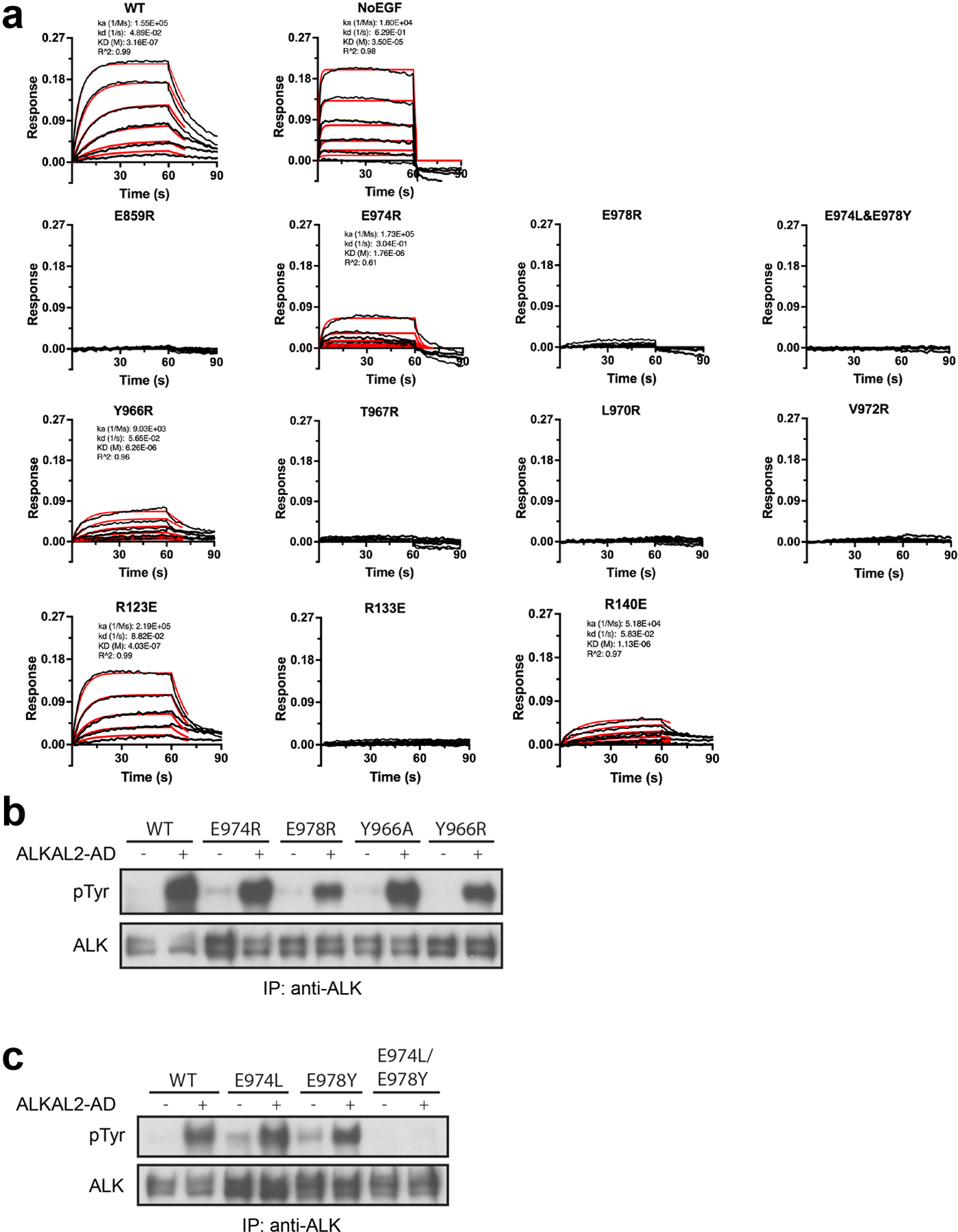
a, Representative BLI sensograms traces (black) and kinetic fittings (red). Fitting was carried out using ForteBio Data Analysis 10.0 software using 1:1 model with Rmax linked global fitting. Where appropriate, kinetic fit parameters are included. b-c, NIH 3T3 cells stably expressing wild type (WT) or mutated full length ALK, stimulated with high concentrations (10 nM) of ALKAL2 to assess residual signaling ability. b, Single point mutations of conserved binding-site residues. E978R has the greatest impact on ALKAL induced ALK signaling in agreement with binding data. c, C-terminal conserved glutamates mutated to the residues found at the same position in invertebrate ALK. The double (E974L/E978Y) mutant fails to signal, consistent with it being the only mutation shown here that completely abolished ligand binding in (a) (Extended Data Table 3). For gel source data, see Supplementary Figure 2. The stimulation experiment was repeated three times with similar results.
Extended Data Figure 5 |. Ligand binding and receptor dimerization are coupled to Pole rotation and PXL changes.
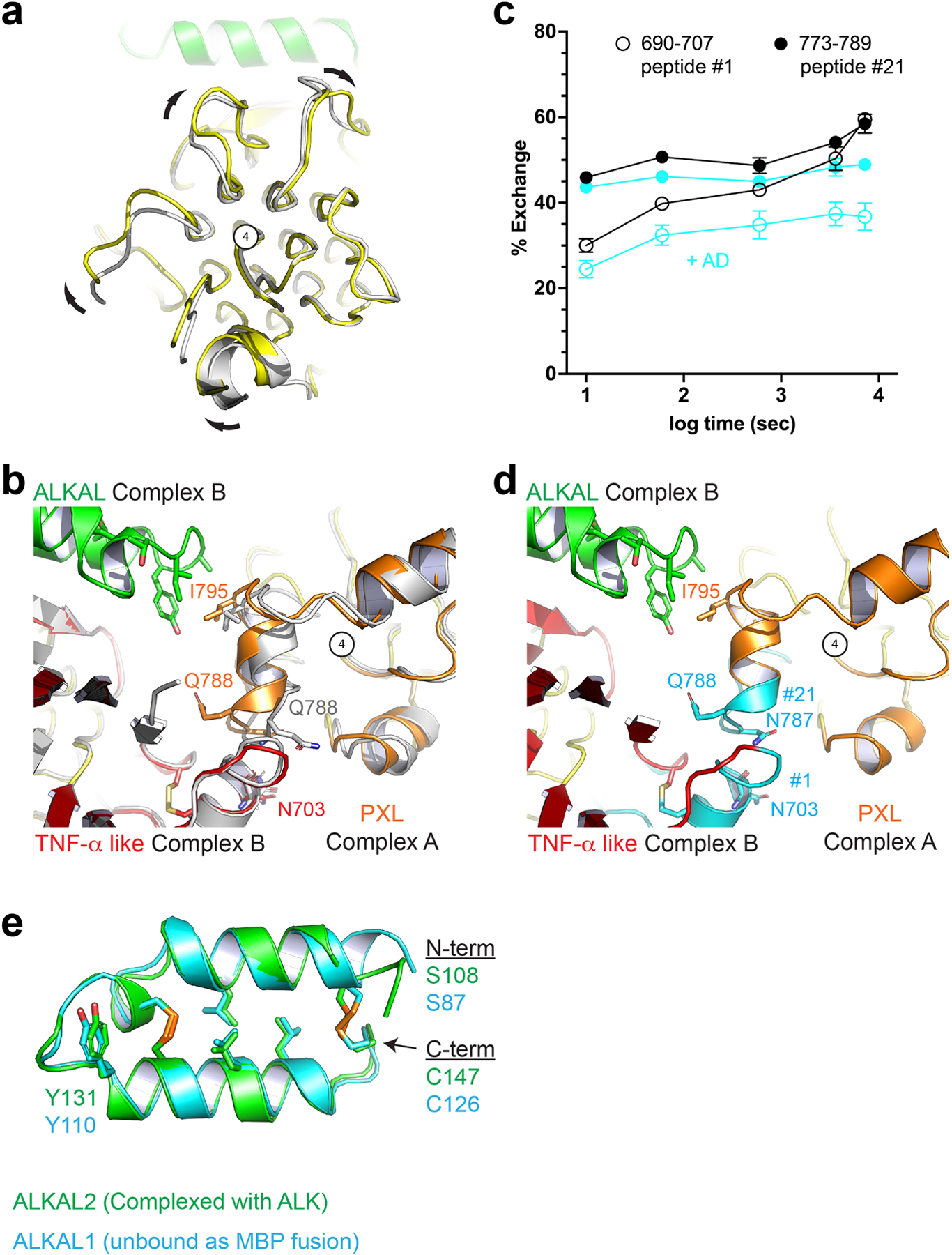
a, View looking down the long axis of the Pole with apo ALK (gray) aligned to the complex structure (color). Compared to unliganded ALK, the ligand-bound ALK dimer undergoes a clockwise rotation of the Pole about the central glycine helix (number 4). b, Unliganded ALK (gray) aligned to both protomers of the complex dimer (color). The PXL residues surrounding Q788 adopt a helical structure upon ligand binding and dimerization. c, Upon ligand binding, two additional peptides – not directly involved in ligand binding and discussed in Fig. 2 – are significantly protected. Statistics were derived from two independent biological repeats, each with three technical repeats. Data represent mean ± SD. d, these peptides (cyan) (peptide #1 and #21, Extended data Fig. 3) correspond to the dimer interface including the disulfide linked helix of the PXL (#21) and the helix on the TNF-α like domain it forms a bond with (#1). e, The core helix-turn-helix of the AD is largely unaltered upon binding. ALKAL2 from the ALK-ALKAL fusion complex structure (green) is aligned to an un-complexed ALKAL1-AD MBP (Maltose Binding Protein) fusion (cyan).
Extended Data Figure 6 |. The PXL region is necessary for ALK signaling.
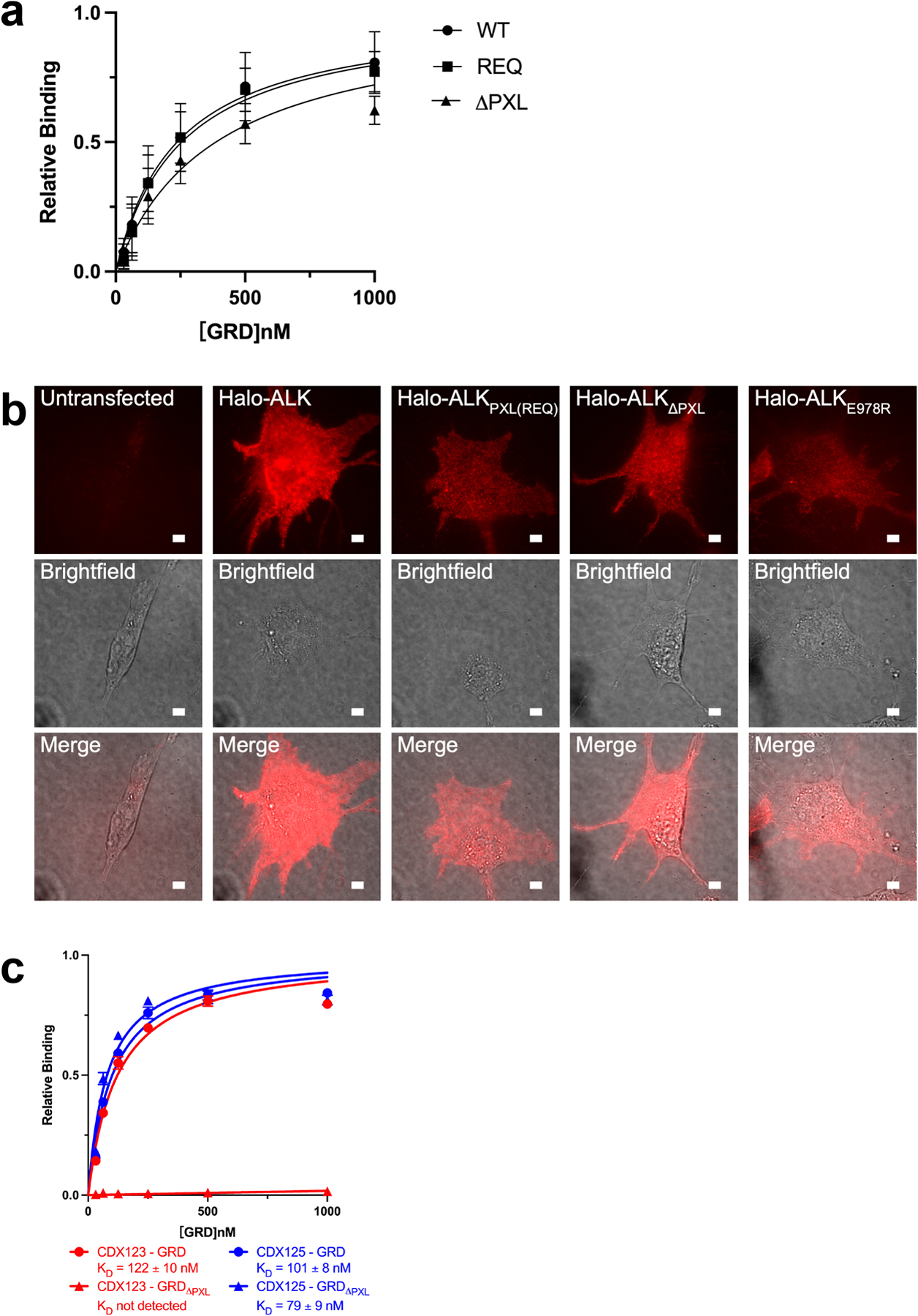
a, Binding of ALKAL2-AD to GRD PXL mutations detected by BLI. The sensors were loaded with ALKAL2-AD. REQ is a mutant that alters the interface residues 795-“IGE”−797 (shown in Fig. 3b, c) to REQ. ΔPXL is a mutant that removes the entire “disulfided helix” 783–797 (shown in Fig. 3b, c). The relative binding is the binding normalized to the maximum responses. Data represent mean ± SD of four measurements. b, Expression of Halo-ALK on NIH/3T3 cell membranes. NIH/3T3 cells were untransfected, transfected with wildtype Halo-ALK or ALK mutants. Cells were stained with membrane impermeable dye, JF549i, before imaging. For each construct fluorescence (top row), brightfield (middle row), and a merge (bottom row) were shown. Scale bar 5 μm. The experiment was repeated two times with similar results. c, Binding of CDX123 or CDX125 to GRD or GRDΔPXL detected by BLI. Data represent mean ± SD from three independent experiments.
Extended Data Figure 7 |.
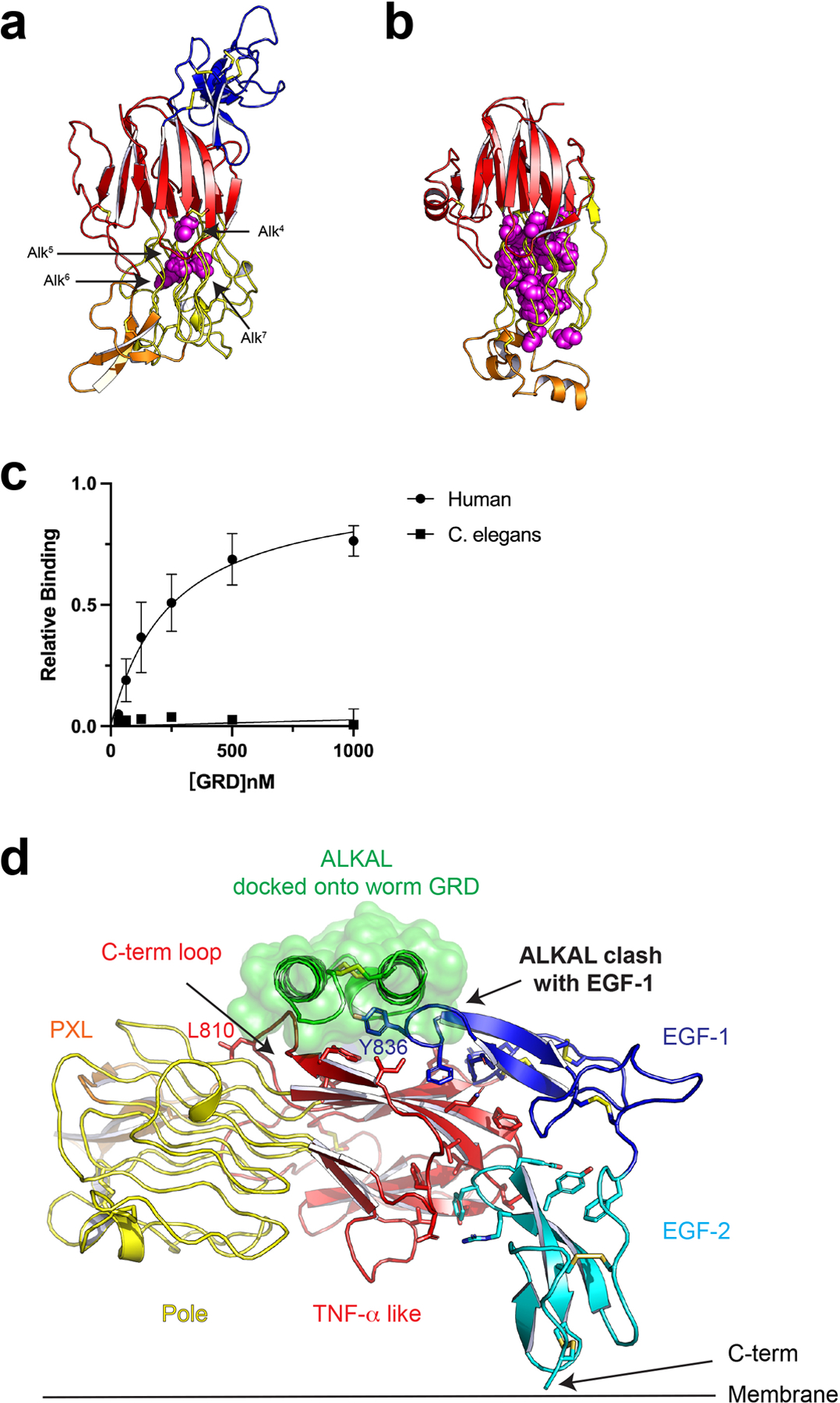
Implications for GRD mutation and evolution a, Invertebrate ALK GRD structure with known glycine-to-acidic residue (D/E) mutations highlighted (purple spheres)9. b, Human ALK GRD structure with all of glycine-to-acidic residue mutations found in the COSMIC database (https://cancer.sanger.ac.uk) highlighted (purple spheres). c, Invertebrate ALK GRD does not bind to human ALKAL2-AD. The relative binding is the binding normalized to the maximum responses. Data represent mean ± SD of three measurements. d, The EGF-like domain of invertebrate ALK occupies a region of the C-terminal loop required for ALKAL binding. This would prevent binding of any similar helix-loop-helix like ligand.
Extended Data Table 1 |. Data collection and refinement statistics.
Values in parentheses are for highest-resolution shell.
| ALK-ALKAL | Human ALK | Nematode ALK | AD-MBP | |
|---|---|---|---|---|
| Data collection | ||||
| Space group | P 1 21 1 | P 61 2 2 | I 41 2 2 | P1 |
| Cell dimensions | ||||
| a, b, c (Å) | 66.3, 177.8, 94.8 | 76.1, 76.1, 230.8 | 224.4, 224.4, 114.5 | 53.6, 56.8, 94.4 |
| α, β, γ (°) | 90.0, 104.8, 90.0 | 90.0, 90.0, 120.0 | 90.0, 90.0, 90.0 | 75.6, 77.4, 61.8 |
| Resolution (Å) | 49.8–3.05 (3.12–3.05) | 63.5–1.91 (1.96–1.91) | 50–2.60 (2.70–2.60) | 50.0–2.43 (2.57–2.43) |
| Rsym or Rmerge | 16.6 (78.3) | 14.3 (59.9) | 19.6 (808.6) | 12.1 (101.7) |
| I / σI | 3.6 (1.3) | 13.1 (0.7) | 14.9 (0.5) | 10.1 (2.1) |
| Completeness (%) | 99.6 (87.1) | 97.8 (69.3) | 99.9 (99.1) | 94.8 (90.1) |
| Redundancy | 3.3 (3.3) | 15.4 (2.0) | 26.9 (26.6) | 6.7 (6.5) |
| Refinement | ||||
| No. reflections | 40292 (2737) | 30761 (1526) | 44759 (4617) | 32041/(1688) |
| Rwork / Rfree | 23.8/28.8 | 18.8/21.4 | 22.3/26.3 | 21.4/27.3 |
| No. atoms | ||||
| Protein | 10642 | 2198 | 5176 | 5381 |
| Ligand/ion | 88 | 0 | 315 | 46 |
| Water | 0 | 219 | 170 | 14 |
| B-factors | ||||
| Protein | 71.9 | 37.1 | 91.0 | 64.9 |
| Ligand/ion | 120.0 | 155.5 | 58.8 | |
| Water | 43.8 | 94.3 | 57.5 | |
| R.m.s. deviations | ||||
| Bond lengths (Å) | 0.005 | 0.013 | 0.009 | 0.005 |
| Bond angles (°) | 0.77 | 1.29 | 1.16 | 0.70 |
Extended Data Table 2 |. HDX peptides.
Sequence of each peptic peptide and corresponding peptide ID number for Extended Fig 3. Residue number is based on (UniprotID: Q9UM73).
| Peptide ID # | Residue # | Amino Acid Sequence |
|---|---|---|
| 1 | 690–707 | ASGPHGPTQAQCNNAYQN |
| 2 | 710–724 | LSVEVGSEGPLKGIQ |
| 3 | 711–722 | SVEVGSEGPLKG |
| 4 | 711–724 | SVEVGSEGPLKGIQ |
| 5 | 711–725 | SVEVGSEGPLKGIQI |
| 6 | 714–722 | VGSEGPLKG |
| 7 | 714–725 | VGSEGPLKGIQI |
| 8 | 718–725 | GPLKGIQI |
| 9 | 723–733 | IQIWKVPATDT |
| 10 | 725–733 | IWKVPATDT |
| 11 | 734–751 | YSISGYGAAGGKGGKNTM |
| 12 | 734–751 | YSISGYGAAGGKGGKNTMM |
| 13 | 734–752 | SISGYGAAGGKGGKNTMM |
| 14 | 736–752 | ISGYGAAGGKGGKNTMM |
| 15 | 737–752 | SGYGAAGGKGGKNTMM |
| 16 | 753–760 | RSHGVSVL |
| 17 | 761–771 | GIFNLEKDDML |
| 18 | 764–770 | NLEKDDM |
| 19 | 766–771 | EKDDML |
| 20 | 773–780 | ILVGQQGE |
| 21 | 773–789 | ILVGQQGEDACPSTNQL |
| 22 | 775–789 | VGQQGEDACPSTNQL |
| 23 | 790–798 | IQKVCIGEN |
| 24 | 814–825 | WAGGGGGGGGAT |
| 25 | 826–841 | YVFKMKDGVPVPLIIA |
| 26 | 827–841 | VFKMKDGVPVPLIIA |
| 27 | 828–841 | FKMKDGVPVPLIIA |
| 28 | 829–840 | KMKDGVPVPLII |
| 29 | 829–841 | KMKDGVPVPLIIA |
| 30 | 833–838 | GVPVPL |
| 31 | 868–889 | LGLNGNSGAAGGGGGWNDNTSL |
| 32 | 869–889 | GLNGNSGAAGGGGGWNDNTSL |
| 33 | 871–889 | NGNSGAAGGGGGWNDNTSL |
| 34 | 890–895 | LWAGKS |
| 35 | 938–950 | IGGNAASNNDPEM |
| 36 | 951–957 | DGEDGVS |
| 37 | 951–958 | DGEDGVSF |
| 38 | 959–964 | ISPLGI |
| 39 | 959–965 | ISPLGIL |
| 40 | 959–967 | ISPLGILYT |
| 41 | 964–970 | ILYTPAL |
| 42 | 966–977 | YTPALKVMEGHG |
| 43 | 984–1019 | YLNCSHCEVDECHMDPESHKVICFCDHGTVLAEDGV |
| 44 | 1017–1026 | DGVSCIVSPT |
Extended Data Table 3 |. Binding analysis of GRD and ALKAL mutants.
N.B., no binding detected, signal less than 5% of WT at highest concentration. See Methods for details.
| KD (nM) | SD | Relative KD | ||
|---|---|---|---|---|
| GRD | WT | 329 | 13 | 1 |
| ΔEGF | 35250 | 10560 | 107 | |
| charged C-terminal loop | E859R | N.B. | - | - |
| E974R | 1410 | 147 | 4.2 | |
| E978R | 6110 | 906 | 19 | |
| E974L/E978Y | N.B. | - | - | |
| uncharged C-terminal loop | Y966R | 5125 | 290 | 16 |
| T967R | N.B. | - | - | |
| L970R | N.B. | - | - | |
| V972R | N.B. | - | - | |
| ALKAL | R123E | 391 | 20 | 1.2 |
| R133E | N.B. | - | - | |
| R140E | 2947 | 240 | 9.0 |
Supplementary Material
Acknowledgments:
We thank Claudio Alarcon, Yansheng Liu, Jonathan Abraham and their laboratories for valuable discussions, as well as members of the Klein, Lemmon, and Schlessinger laboratories. Our thanks to Andrey Reshetnyak, David Puleo and Jyotidarsini Mohanty for their contributions.
Funding:
This work was supported by the NIH, NIGMS grant R35 GM122485 (to M.A.L.), and NCI grant R01 CA248532 (to D.E.K.).
This work is based in part upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). The Pilatus 6M detector on 24-ID-C beam line is funded by a NIH-ORIP HEI grant (S10 RR029205). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. GM/CA@APS has been funded in whole or in part with Federal funds from the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. The Eiger 16M detector at GM/CA-XSD was funded by NIH grant S10 OD012289.
Footnotes
Competing interests: J.S. is a member of the SAB of Celldex. D.A. is an employee of Celldex Therapeutics.
Supplementary Information is available for this paper.
Data Availability
The refined structural protein models and corresponding structure–factor amplitudes have been deposited under PDB accession codes 7LS0 (human ALK GRD-ALKAL fusion complex), 7LRZ (human ALK GRD), 7LIR (worm ALK GRD), and 7MK7 (ALKAL2-AD).
References
- 1.Weiss JB et al. Anaplastic lymphoma kinase and leukocyte tyrosine kinase: functions and genetic interactions in learning, memory and adult neurogenesis. Pharmacol Biochem Behav 100, 566–574, doi: 10.1016/j.pbb.2011.10.024 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Orthofer M et al. Identification of ALK in Thinness. Cell 181, 1246–1262 e1222, doi: 10.1016/j.cell.2020.04.034 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Hallberg B & Palmer RH Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat Rev Cancer 13, 685–700, doi: 10.1038/nrc3580 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Carpenter EL et al. Antibody targeting of anaplastic lymphoma kinase induces cytotoxicity of human neuroblastoma. Oncogene 31, 4859–4867, doi: 10.1038/onc.2011.647 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mosse YP et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 455, 930–935, doi: 10.1038/nature07261 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trigg RM & Turner SD ALK in Neuroblastoma: Biological and Therapeutic Implications. Cancers (Basel) 10, doi: 10.3390/cancers10040113 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Borenas M et al. ALK ligand ALKAL2 potentiates MYCN-driven neuroblastoma in the absence of ALK mutation. EMBO J 40, e105784, doi: 10.15252/embj.2020105784 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reshetnyak AV et al. Augmentor alpha and beta (FAM150) are ligands of the receptor tyrosine kinases ALK and LTK: Hierarchy and specificity of ligand-receptor interactions. Proc Natl Acad Sci U S A 112, 15862–15867, doi: 10.1073/pnas.1520099112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guan J et al. FAM150A and FAM150B are activating ligands for anaplastic lymphoma kinase. Elife 4, e09811, doi: 10.7554/eLife.09811 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lemmon MA & Schlessinger J Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134, doi: 10.1016/j.cell.2010.06.011 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loren CE et al. A crucial role for the Anaplastic lymphoma kinase receptor tyrosine kinase in gut development in Drosophila melanogaster. EMBO Rep 4, 781–786, doi: 10.1038/sj.embor.embor897 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang H et al. Deorphanization of the human leukocyte tyrosine kinase (LTK) receptor by a signaling screen of the extracellular proteome. Proc Natl Acad Sci U S A 111, 15741–15745, doi: 10.1073/pnas.1412009111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reshetnyak AV et al. Identification of a biologically active fragment of ALK and LTK-Ligand 2 (augmentor-alpha). Proc Natl Acad Sci U S A 115, 8340–8345, doi: 10.1073/pnas.1807881115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qin LY et al. Discovery of 7-(3-(piperazin-1-yl)phenyl)pyrrolo[2,1-f][1,2,4]triazin-4-amine derivatives as highly potent and selective PI3Kdelta inhibitors. Bioorg Med Chem Lett 27, 855–861, doi: 10.1016/j.bmcl.2017.01.016 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Youn SJ et al. Construction of novel repeat proteins with rigid and predictable structures using a shared helix method. Sci Rep 7, 2595, doi: 10.1038/s41598-017-02803-z (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holm L DALI and the persistence of protein shape. Protein Sci 29, 128–140, doi: 10.1002/pro.3749 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eck MJ & Sprang SR The structure of tumor necrosis factor-alpha at 2.6 A resolution. Implications for receptor binding. J Biol Chem 264, 17595–17605, doi: 10.2210/pdb1tnf/pdb (1989). [DOI] [PubMed] [Google Scholar]
- 18.Warkentin E et al. A rare polyglycine type II-like helix motif in naturally occurring proteins. Proteins 85, 2017–2023, doi: 10.1002/prot.25355 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Crick FH & Rich A Structure of polyglycine II. Nature 176, 780–781 (1955). [DOI] [PubMed] [Google Scholar]
- 20.Dunne M et al. Salmonella Phage S16 Tail Fiber Adhesin Features a Rare Polyglycine Rich Domain for Host Recognition. Structure 26, 1573–1582 e1574, doi: 10.1016/j.str.2018.07.017 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Vadas O, Jenkins ML, Dornan GL & Burke JE Using Hydrogen-Deuterium Exchange Mass Spectrometry to Examine Protein-Membrane Interactions. Methods Enzymol 583, 143–172, doi: 10.1016/bs.mie.2016.09.008 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Sano R et al. An antibody-drug conjugate directed to the ALK receptor demonstrates efficacy in preclinical models of neuroblastoma. Sci Transl Med 11, doi: 10.1126/scitranslmed.aau9732 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tate JG et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res 47, D941–D947, doi: 10.1093/nar/gky1015 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ishihara T et al. HEN-1, a secretory protein with an LDL receptor motif, regulates sensory integration and learning in Caenorhabditis elegans. Cell 109, 639–649, doi: 10.1016/s0092-8674(02)00748-1 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Englund C et al. Jeb signals through the Alk receptor tyrosine kinase to drive visceral muscle fusion. Nature 425, 512–516, doi: 10.1038/nature01950 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Lee HH, Norris A, Weiss JB & Frasch M Jelly belly protein activates the receptor tyrosine kinase Alk to specify visceral muscle pioneers. Nature 425, 507–512, doi: 10.1038/nature01916 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Murray PB et al. Heparin is an activating ligand of the orphan receptor tyrosine kinase ALK. Sci Signal 8, ra6, doi: 10.1126/scisignal.2005916 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Reshetnyak A e. a. Mechanism for the activation of the anaplastic lymphoma kinase receptor. Nature (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jenni S, Goyal Y, von Grotthuss M, Shvartsman SY & Klein DE Structural Basis of Neurohormone Perception by the Receptor Tyrosine Kinase Torso. Mol Cell 60, 941–952, doi: 10.1016/j.molcel.2015.10.026 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Klein DE, Stayrook SE, Shi F, Narayan K & Lemmon MA Structural basis for EGFR ligand sequestration by Argos. Nature 453, 1271–1275, doi: 10.1038/nature06978 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vonrhein C, Blanc E, Roversi P & Bricogne G Automated structure solution with autoSHARP. Methods Mol Biol 364, 215–230, doi: 10.1385/1-59745-266-1:215 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132, doi: 10.1107/S0907444904019158 (2004). [DOI] [PubMed] [Google Scholar]
- 33.McCoy AJ et al. Phaser crystallographic software. J Appl Crystallogr 40, 658–674, doi: 10.1107/S0021889807021206 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langer G, Cohen SX, Lamzin VS & Perrakis A Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat Protoc 3, 1171–1179, doi: 10.1038/nprot.2008.91 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liebschner D et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D Struct Biol 75, 861–877, doi: 10.1107/S2059798319011471 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morin A et al. Collaboration gets the most out of software. Elife 2, e01456, doi: 10.7554/eLife.01456 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patil K, Jordan EJ, Park JH, Suresh K, Smith CM, Lemmon AA, Mossé YP, Lemmon MA, and Radhakrishnan R. Computational Studies of Anaplastic Lymphoma Kinase Mutations Reveal Common Mechanisms of Oncogenic Activation. PNAS in press; (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grimm JB et al. A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat Methods 12, 244–250, 243 p following 250, doi: 10.1038/nmeth.3256 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kabsch W Xds. Acta Crystallogr D Biol Crystallogr 66, 125–132, doi: 10.1107/S0907444909047337 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Winn MD et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67, 235–242, doi: 10.1107/S0907444910045749 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adams PD et al. The Phenix software for automated determination of macromolecular structures. Methods 55, 94–106, doi: 10.1016/j.ymeth.2011.07.005 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The refined structural protein models and corresponding structure–factor amplitudes have been deposited under PDB accession codes 7LS0 (human ALK GRD-ALKAL fusion complex), 7LRZ (human ALK GRD), 7LIR (worm ALK GRD), and 7MK7 (ALKAL2-AD).