

Abstract
Purpose:
B-cell maturation antigen (BCMA)-targeted chimeric antigen receptor (CAR) T-cells (CART-BCMA) are a promising treatment for relapsed/refractory multiple myeloma (r/rMM). We evaluated the safety and feasibility of bridging radiation (RT) in subjects treated on a phase 1 trial of CART-BCMA.
Experimental Design:
Twenty-five r/rMM subjects were treated in 3 cohorts with two doses of CART-BCMA cells +/− cyclophosphamide. We retrospectively analyzed toxicity, response, and CART manufacturing data based on RT receipt.
Results:
Thirteen subjects received no RT <1 year before CART infusion (Group A). Eight subjects received RT <1 year before CART infusion (Group B) with median time from RT to apheresis of 114 days (range 40–301). Four subjects received bridging-RT (Group C) with a median dose of 22 Gy and time from RT to infusion of 25 days (18–35). Group C had qualitatively lower rates of grade 4 (G4) hematologic toxicities (25%) vs. A (61.5%) and B (62.5%). G3–4 neurotoxicity occurred in 7.7%, 25%, and 25% in Group A, B, and C, respectively. G3–4 CRS was observed in 38.5%, 25%, 25% in Group A, B, and C, respectively. Partial response or better was observed in 54%, 38%, and 50% of Group A, B, and C, respectively. RT administered <1 year (p=0.002) and <100 days (p=0.069) before apheresis was associated with lower in vitro proliferation during manufacturing; however, in vivo CART-BCMA expansion appeared similar across groups.
Conclusions:
Bridging-RT appeared safe and feasible with CART-BCMA therapy in our r/rMM patients, though larger future studies are needed to draw definitive conclusions.
Keywords: multiple myeloma, chimeric antigen receptor, CAR T-cell, BCMA, radiation, radiation oncology
Introduction:
Multiple myeloma (MM) remains incurable. Although immunomodulatory drugs (IMIDs), proteasome inhibitors (PIs), and monoclonal antibodies (mAbs) have prolonged survival(1,2), most patients relapse due to drug resistance(3–6). Chimeric antigen receptor (CAR) T-cell therapy can yield durable responses in patients with advanced hematologic malignances(7–10). B-cell maturation antigen (BCMA), a member of the tumor necrosis factor receptor superfamily, is a rational target for MM due to its highly selective expression in plasma cells. Early results of BCMA-specific CAR T-cells in heavily pre-treated patients with relapsed and/or refractory MM (r/rMM) have demonstrated impressive overall response rates (ORRs) of 64%−95% and induction of minimal residual disease (MRD)-negative complete responses (CRs)(11–15).
Patients with r/rMM often require radiation therapy (RT) to palliate symptomatic disease. Apart from its local effects, RT has systemic immunomodulatory effects. RT has been found to upregulate major histocompatibility complex class I (MHC-I) expression, give rise to novel peptides, and increase presentation of tumor-associated antigens(16–18). RT can also facilitate homing of antigen-specific T-cells and possibly counteract the immunosuppressive tumor microenvironment(16,19). Thus, RT has the potential to combat the following challenges of CART therapy: 1) cancer evasion by poorly antigenic tumor, 2) optimization of CAR T-cell trafficking to tumor, and 3) the immunoinhibitory tumor microenvironment. In fact, preclinical data suggest that RT conditioning may promote susceptibility to CART therapy and decrease antigen-negative tumor relapse(20).
The efficacy of CART therapies comes with risk of toxicity from immune activation, namely cytokine release syndrome (CRS) and neurotoxicity. Current anti-BCMA CART trials have cited CRS rates (any grade) of >60% with grade 3 CRS reaching up to 40%(11–13,21). Neurotoxicity (any grade) has been observed in approximately 20–30% of patients, with most cases being self-limited or responsive to steroids(11–13,21). Nonetheless, severe CRS and neurotoxicity can occur and are potentially fatal(11–13,21).
Additional considerations for CAR T-cells include the fitness of patients’ endogenous T-cells and lymphodepletion. Our institution conducted a phase 1 trial evaluating autologous T-cells expressing a fully human BCMA-specific CAR containing CD3ζ and 4–1BB signaling domains (CART-BCMA) in r/rMM patients, with and without lymphodepleting chemotherapy. Investigators showed a higher frequency of CD8+ naïve or early memory T-cells and a higher ratio of CD4+ to CD8+ T-cells (CD4/CD8 ratio) in the premanufacturing leukapheresis product were associated with better clinical response and more likely to be found in patients with minimal exposure to systemic therapy(22). Lymphodepletion was also found to enhance the clinical benefit of CART therapy(12,23–25).
Furthermore, while CART manufacturing protocols on average range from 8–12 days, vein to vein time can take 3–4 weeks, preventing prompt treatment of aggressive disease. In fact, 8–14% of apheresed patients in 2 recent trials never received CAR T-cells due to rapid progression, and 88% of patients in the KarMMA trial required bridging therapy (i.e. treatment between leukapheresis and CART infusion)(12,13). In some cases, patients may require RT as a bridge to CART infusion for fast relief of symptomatic lesions or maintenance of performance status. However, available literature on peri-CART RT is limited to small, single institution experiences evaluating CD19-directed CART therapy in non-Hodgkin lymphoma (26–28). Herein, we present the first report looking at safety and feasibility of bridging-RT and pre-apheresis RT in r/rMM patients who received CART-BCMA therapy.
Methods and Materials:
The University of Pennsylvania conducted a phase I, single-center, open-label study (NCT02546167) investigating the safety and efficacy of CART-BCMA in patients with r/rMM after failing at least 3 prior regimens, or 2 prior regimens if dual-refractory to a PI or IMID. Twenty-five subjects were treated in 3 cohorts: cohort 1, 1×108 to 5×108 CART-BCMA cells alone; cohort 2, cyclophosphamide (Cy) 1.5 g/m2 plus 1×107 to 5×107 CART-BCMA cells; cohort 3, Cy 1.5 g/m2 plus 1×108 to 5×108 CART-BCMA cells. A fully human BCMA-specific CAR construct containing CD3ζ and 4–1BB signaling domains was used. Leukapheresis was performed after enrollment following a 2 week washout from prior myeloma therapy (4 weeks for mAbs). Anti-myeloma therapy could resume during manufacturing until 2 weeks prior to first CART infusion. CART-BCMA cells were administered over 3 days in 3 dose fractions (10% of dose on day 0, 30% on day 1, and 60% on day 2). The 30% or 60% dose could be held if subjects developed signs of cytokine release syndrome (CRS). Cy was administered 3 days prior to first CART-BCMA infusion. This study was conducted in accordance with the Declaration of Helsinki, and written consent was obtained from all subjects. Full eligibility criteria and protocol methods have been previously published(12). Correlative studies, analyzed here in the current context, including the qPCR- and flow cytometry-based detection of CAR T-cells, flow cytometry for T-cell subsets, and levels of soluble BCMA (solBCMA) were previously reported(12).
The institutional review board at the University of Pennsylvania approved this retrospective study of subjects enrolled on the trial described above. RT plans were accessed to confirm treatment dates, treatment location, total dose delivered, fractionation, modality, and technique. Since the duration of how long RT affects the local environment and immune system is not well-established, patient grouping was intended to be inclusive, accounting for radiation recall (a tissue reaction that occurs in previously irradiated area after administration of systemic therapy) and variable recovery time of different lymphocytes. Radiation recall has been observed in patients with a long interval (~1 year) between the end of RT and delivery of immunotherapy and restoration of particular lymphocyte subsets post-RT has been shown to take 1 year or longer(29,30). Thus, subjects were characterized into 3 groups: Group A, no RT within 1 year before apheresis (n=13); Group B, RT within 1 year before apheresis (n=8); Group C, bridging-RT defined as RT delivered after apheresis but before CART infusion (n=4). Data from the parent trial were reviewed to document patient baseline demographics, clinicopathologic features, prior therapies, CART manufacturing details, and clinical outcomes. Toxicity data was collected from time of Cy-based lymphodepletion for cohort 2 and 3 or first CART-BCMA infusion for cohort 1. Toxicity was graded per CTCAE version 4.0 with the exception of CRS, which was graded per the University of Pennsylvania CRS Grading System(12). Myeloma responses were scored by the updated International Myeloma Working Group (IMWG) criteria(3).
Statistical analysis is primarily descriptive to due small sample sizes in each group and the pilot nature of the parent trial. Kaplan-Meier analysis was used to estimate progression-free (PFS), overall survival (OS), and associated median survival times with follow-up defined relative to first CART-BCMA infusion (designated Day 0). Associations between binary endpoints (e.g., receipt of RT) and continuous variables were assessed using the Wilcoxon rank-sum test. Kruskal-Wallis test was used when more than 2 groups were compared simultaneously. Exact 2-sided p-values are reported when applicable. A p-value <0.05 was considered significant. Analysis was performed using RStudio version 1.3.1056 and GraphPad Prism 9.
Results:
Patient Groups: No RT <1 year before apheresis (A), RT <1 year before apheresis (B), and Bridging-RT (C)
Twenty-five subjects receiving CART-BCMA between November 2015 and December 2017 for r/rMM were identified. Four subjects received bridging-RT (Group C) ≤35 days before CART infusion, 8 subjects received RT <1 year before apheresis (Group B), and 13 subjects had no RT <1 year before apheresis (Group A). Within Group A, 3 subjects had a history of remote RT before apheresis (range, 853–3452 days) and 10 subjects never had RT. Twenty-one subjects received all 3 planned CART-BCMA does fractions while 4 subjects (1 in Group A, 2 in Group B, and 1 in Group C) received 40% of planned CART-BCMA dose due to early CRS.
Baseline characteristics per group are summarized in Table 1. Each group was heavily pretreated with a median of 7 prior lines of therapy and high rates of prior autologous stem cell transplant. At least 1 high-risk cytogenetic abnormality was detected in all subjects in Group A and B and 3 of 4 subjects in Group C; Baseline tumor burden was high across all groups with comparable ranges of myeloma cells on bone marrow biopsy (Table 1) and serum concentrations of solBCMA prior to cyclophosphamide lymphodepletion (p=0.58, sFig1A). Extramedullary disease, a poor prognostic indicator, was absent in Group A but common in Group B (63%) and Group C (50%).
Table 1:
Subject characteristics for Group A (no RT <1 year prior to apheresis), Group B (RT <1 year prior to apheresis), and Group C (bridging-RT).
| Patient Characteristics | |||
|---|---|---|---|
| Group A (n=13) | Group B (n=8) | Group C (n=4) | |
| Median (range) or % | Median (range) or % | Median (range) or % | |
| Age, years | 58 (44–73) | 59 (47–75) | 57 (51–63) |
| Sex, M / F | 62% / 38% | 50% / 50% | 100% / 0% |
| Cohort 1 / 2 / 3 | 23% / 15% / 62% | 75% / 0% / 25% | 0% / 50% / 50% |
| Median time from diagnosis, years | 4.1(1.8–14.5) | 4.8 (1.8–8.8) | 4.3 (2.3–9.4) |
| High-risk cytogeneticsa | 100% | 100% | 75% |
| Del17p or TP53 mutation | 77% | 63% | 50% |
| Prior lines of therapy, no. | 7 (4–13) | 7 (3–10) | 7 (6–7) |
| Len / Bort / Pom / Carf / Dara, % exposed | 100% / 100% / 100% / 92% / 77% | 100% / 100% / 88% / 88% / 63% | 100% / 100% / 75%/ 100% / 100% |
| Len / Bort / Pom / Carf / Dara, % refractory | 85% / 92% / 100% / 69% / 69% | 75% / 88% / 75% / 75% / 63% | 50% / 75%/ 75% / 100% / 100% |
| Dual-/Triple-class/Quad-/ Penta-refractoryb | 100% / 69% / 69% / 54% | 88% / 50% / 38% / 25% | 100% / 100% / 25% / 25% |
| Prior autologous SCT | 92% | 100% | 100% |
| Extramedullary disease | 0% | 63% | 50% |
| Bone marrow plasma cells | 60% (13–90) | 85% (0–95) | 27% (3–80) |
| ALC pre-apheresis, x103/μl | 0.80 (0.40–1.30) | 0.65 (0.30–1.80) | 1.00 (0.27–1.50) |
| % Lymphocytes pre-apheresis | 25 (8–39) | 21 (11–36) | 22 (7–33) |
| Absolute CD3+ T cell count pre-apheresis, cell/μL | 538 (295–1513)c | 295 (151–1529) | 507 (376–1554) |
| Baseline LDH, U/l | 162 (75–315) | 169 (112–385) | 196 (130–308) |
| Baseline serum creatinine, mg/dl | 0.93 (0.64–1.83) | 1.0 (0.83–2.87) | 0.76 (0.55–1.77) |
| Baseline hemoglobin, g/dl | 8.9 (6.9–15.2) | 9.0 (6.9–12.2) | 9.3 (8.8–12.0) |
| Baseline platelets, x103/μl | 137 (25–221) | 152 (13–193) | 124 (41–316) |
Complex karyotype includes gain 1q, deletion 17p, t(14;16), and/or t(4;14).
Dual-refractory = refractory to both 1 PI and 1 IMID; Triple-class refractory = refractory to at least 1 PI, 1 IMID, and 1 CD38 antibody (usually daratumumab); Quad-refractory = refractory to 2 PIs and 2 IMIDs; Penta-refractory = refractory to 2 PIs, 2 IMIDs, and daratumumab.
Subjects 1 and 2 in Group A did not have pre-apheresis T-cell counts done (n=11). Normal range, 900–3245 cells/μL.
SCT, stem cell transplant; LDH, lactate dehydrogenase; ALC, absolute lymphocyte count.
Clinical Outcomes: Response rates, overall survival, and progression-free survival
Median follow-up for the subjects was 16.3 months (range, 0.8–42.1). At time of data cutoff, 18 subjects had expired, and 7 were still alive. OS was not significantly different between Group A, B, and C (median 667, 295, and 264 days, respectively; p=0.082) (Fig1A). At time of data cutoff, 2 subjects (subjects 19, 33), 1 from Group A and 1 from Group B, remained progression-free at 728 and 623 days (roughly 30 and 20 months), respectively; all remaining subjects had progressed. PFS was not significantly different between Group A, B, and C (median 64, 71, and 94 days, respectively; p=0.86) (Fig1B).
Figure 1: Clinical outcomes between Group A, B, and C using Kaplan-Meier plot with log-rank test based on group.
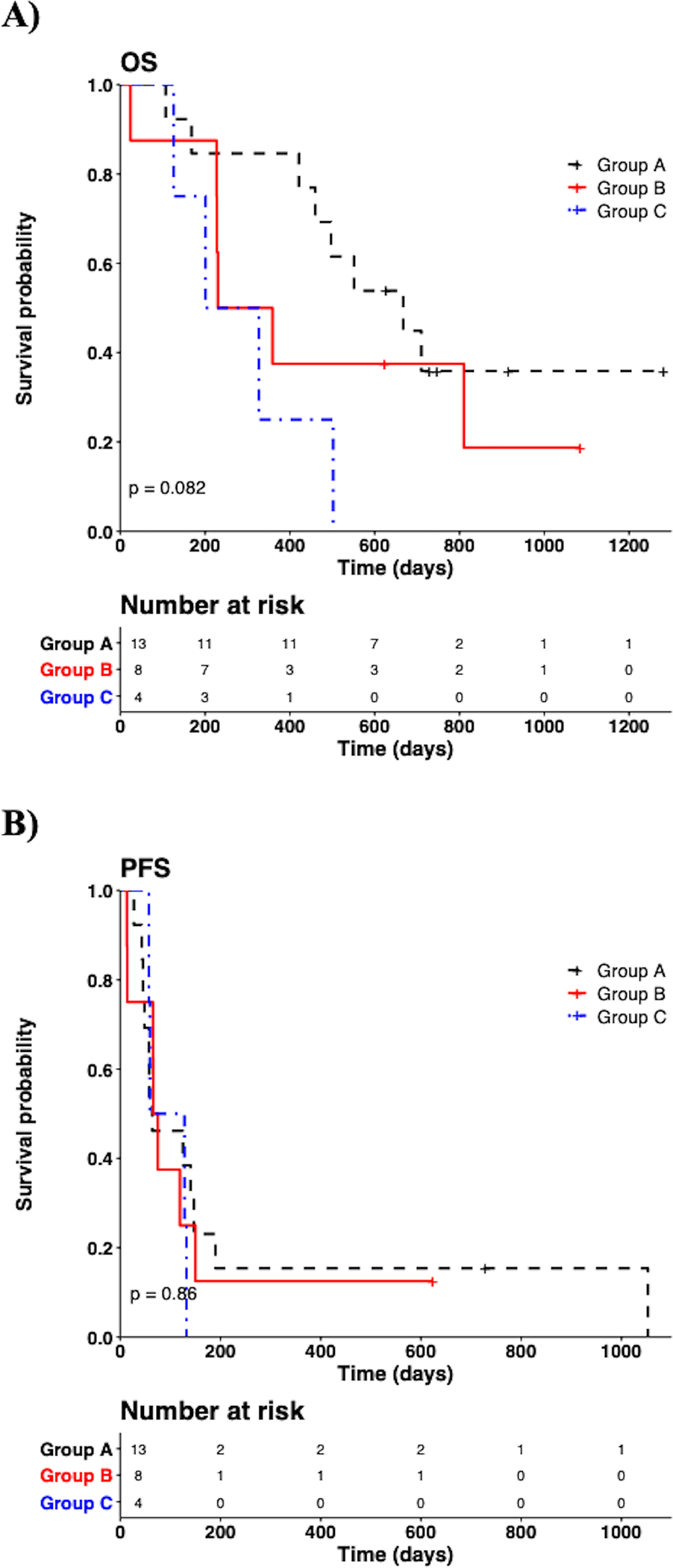
A) Overall survival (OS), (p=0.08). B) Progression-free survival (PFS), (p=0.86).
Conventional univariate analysis to compare treatment response between groups was not performed due to the limited sample size. Responses (minimal response [MR] or better) were observed in 9 of 13 subjects (69%) in Group A, 4 of 8 subjects (50%) in Group B, and 4 of 4 subjects (100%) in Group C. Objective responses (partial response [PR] or better) were confirmed in 54% in Group A, 38% in Group B, and 50% in Group C (Table 2).
Table 2: Patient responses for Group A (no RT <1 year prior to apheresis), Group B (RT <1 year prior to apheresis), and Group C (bridging-RT).
CR, complete response; MR, minimal response; PD, progressive disease; sCR, stringent complete response; SD, stable disease; VGPR, very good partial response.
| Patient Responses | |||
|---|---|---|---|
| Group A (n=13) | Group B (n=8) | Group C (n=4) | |
| n (%) | n (%) | n (%) | |
| sCR | 1 (8) | 0 (0) | 0 (0) |
| CR | 1 (8) | 0 (0) | 0 (0) |
| VGPR | 3 (23) | 2 (25) | 0 (0) |
| PR | 2 (15) | 1 (13) | 2 (50) |
| MR | 2 (15) | 1 (13) | 2 (50) |
| SD | 4 (31) | 2 (25) | 0 (0) |
| PD | 0 (0) | 2 (25) | 0 (0) |
| ≥MR | 9 (69) | 4 (50) | 4 (100) |
| ≥PR | 7 (54) | 3 (38) | 2 (50) |
| Ongoing Response | 1 (8) | 1 (13) | 0 (0) |
Initial results of the parent trial revealed a lower dose of CART-BCMA cells, as delivered in cohort 2, led to the lowest response rate(12). Thus, we compared cohort 2 subjects who did and did not receive bridging-RT to examine if bridging-RT affected response. Subject 13 and subject 16, who underwent bridging-RT, had the two largest in vivo CART expansions in cohort 2 (Fig2A). Cohort 2 subjects without bridging-RT (n=3) had no response whereas those with bridging-RT (n=2) had either MR or PR, with disease improvement both in and out of the RT field (Fig2B).
Figure 2: In vivo expansion of CART-BCMA cells and radiographic response in cohort 2 subjects who received bridging RT (Group C).
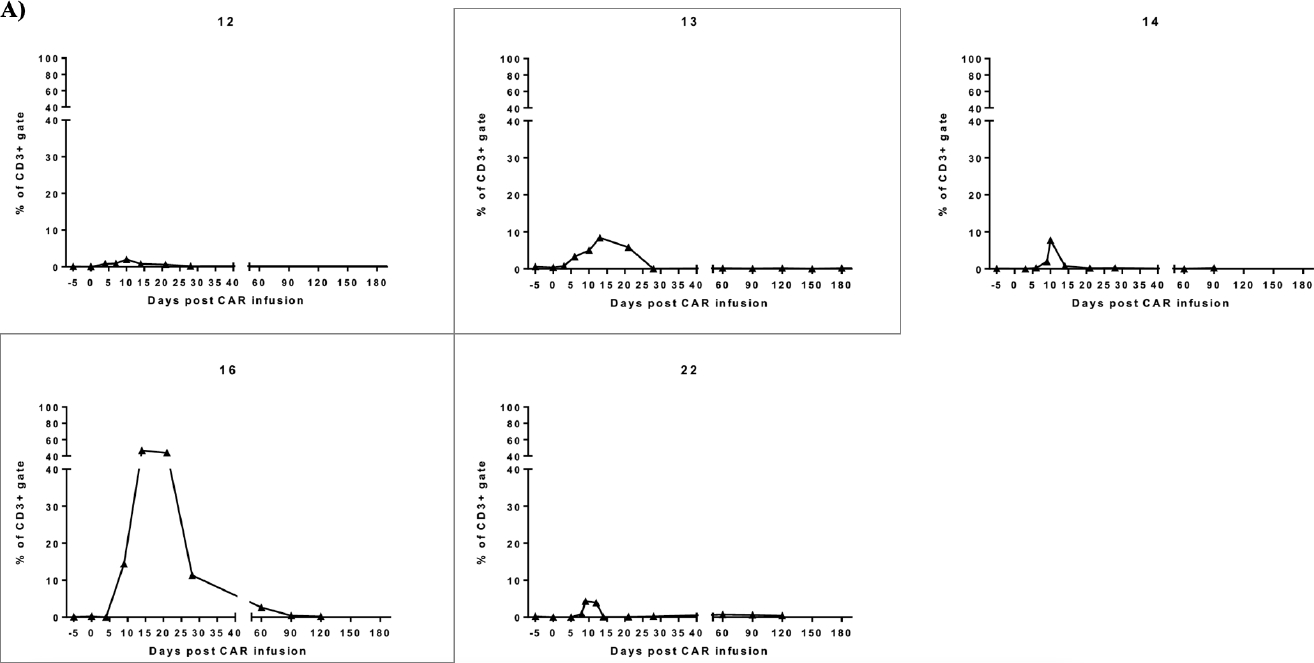
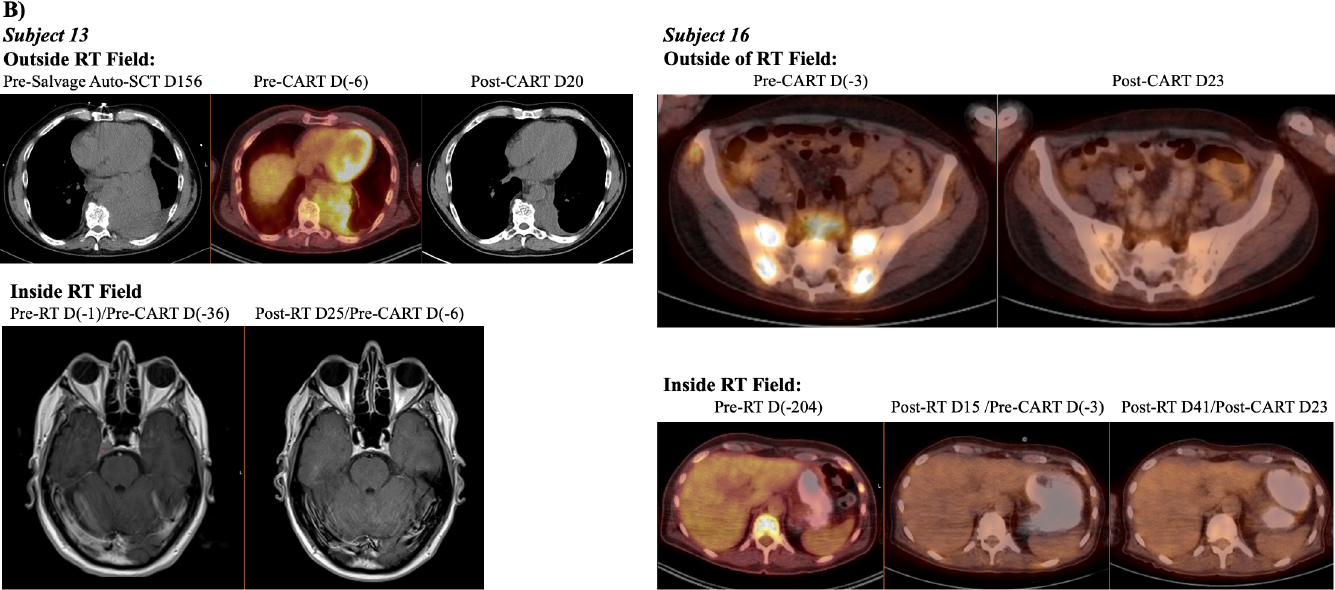
A) Previously published line plots show the frequency of CAR+ T-cells within the peripheral blood(12). CD3+ population was assessed by flow cytometry for each subject in cohort 2 (cyclophosphamide 1.5g/m2 plus 1×107 to 5×107 CART-BCMA cells). Subject 13 and 16 who received bridging-RT had the largest expansions in cohort 2 which translated to minimal and partial response, respectively. Remaining cohort 2 subjects (12, 14, and 22) had no response to CART-BCMA therapy. B) PET/CT images from Subject 13 show a thoracic plasmacytoma outside the RT field before CART-BCMA therapy and salvage autologous stem cell transplant (auto-SCT). The plasmacytoma showed partial resolution after auto-SCT and further resolution after CART. MRI images show interval resolution of right cavernous sinus plasmacytoma after RT to skull base and prior to CART. Subject 16 showed interval resolution of a T12 lesion after RT to T7-L1 and before CART. Sacral lesions outside of the RT fields showed interval resolution post CART.
Bridging-RT indications and radiation treatment planning characteristics in Group B and C:
Although no explicit criteria mandated referral to a radiation oncologist, patients in need of immediate symptomatic relief were referred for bridging-RT. Indications included severe/refractory bone pain (n=2) or functional deficits from local tumors (n=2). One subject was experiencing progressive diplopia and eye discomfort due to bilateral plasmacytomas of the orbital bones. Another patient presented with cranial nerve VI palsy caused by a progressive plasmacytoma of the ipsilateral cavernous sinus. Two subjects had severe bone pain requiring hospitalization for pain control and/or high risk for pathologic fracture.
Radiation dose, fractionation, and treatment fields were at the discretion of the treating radiation oncologists. Median bridging-RT dose in Group C was 22 Gy (range, 8–30 Gy in 3–8 Gy fractions) (sTable 1). Bridging-RT was started a median of 19 days after apheresis (range, 15–22) and completed a median of 25 days before CART infusion (range, 18–35). The radiation modality used was 3-dimensional conformal RT (3DCRT). Treatment sites included skull base (n=1), thoracic and lumbar spine (n=1), bilateral hips (n=1), and bilateral orbits (n=1).
Median RT dose in Group B was 25 Gy (range, 6–40 Gy in 2–8 Gy fractions) (sTable 1). RT was started a median of 77 days and 114 days before apheresis (range, 15–268) and CART infusion (range, 40–301) respectively. The radiation modalities used were 3DCRT to the head and pelvis (n=5), electrons to sternum and ribs (n=2), and intensity modulated RT (IMRT) in a re-irradiation case to the left maxillary sinus (n=1).
Bridging systemic therapy was delivered in 84% of subjects (Group A: n=11, Group B: n=6, Group C: n=4). Concurrent systemic therapy was administered in 2 subjects in Group C and 4 subjects Group B (sTable 1).
Bridging-RT and CART-BCMA-related toxicities:
Grade 1 (G1) and G2 RT-toxicities were observed in 1 of 4 Group C subjects. Overall, G1 and G2 RT side effects consisted of fatigue (n=1) and alopecia (n=1). No subject experienced pain flare, and no ≥G3 RT-toxicities occurred.
Rates of toxicities related to CART-BCMA were analyzed per group (Table 3). In Group C, 1 subject (25%) experienced G4 hematologic toxicities compared to 8 (62%) and 5 (63%) subjects in Group B and A respectively. Group A experienced the highest rate of G3 hematologic toxicities at 77% (n=10) compared to 25% (n=2) in Group B and 50% (n=2) in Group C. G2 hematologic toxicities were comparable between Group A (23%) and C (25%) and slightly higher in Group B (38%). No ≥G3 gastrointestinal (GI), infectious, or liver-related toxicities were observed in Group C. In contrast, Group A reported G3–4 liver-related toxicities in 3 subjects (23%) and G4 infections in 1 subject (8%); Group B reported G3 GI toxicities in 1 subject (13%) and G3–4 infections in 3 subjects (38%). G2 infections were equal between Group A and B at 38% and slighter lower in Group C at 25%.
Table 3: Toxicity rates in Group A (no RT <1 year prior to apheresis), Group B (RT <1 year prior to apheresis), and Group C (bridging-RT).
n (%), number of subjects (frequency of toxicity within group). CRS, cytokine release syndrome; LFTs, liver function tests.
| Toxicity | Group A (n=13) | Group B (n=8) | Group C (n=4) |
|---|---|---|---|
| n (%) | n (%) | n (%) | |
| Neurotoxicity | |||
| Grade 2 | 2 (15) | 0 (0) | 0 (0) |
| Grade 3 | 1 (8) | 2 (25) | 1 (25) |
| Grade 4 | 0 (0) | 2 (25) | 0 (0) |
| CRS | |||
| Grade 2 | 6 (46) | 5 (63) | 2 (50) |
| Grade 3 | 5 (39) | 2 (25) | 1 (25) |
| Grade 4 | 0 (0) | 1 (13) | 0 (0) |
| Hematologic | |||
| Grade 2 | 3 (23) | 3 (38) | 1 (25) |
| Grade 3 | 10 (77) | 2 (25) | 2 (50) |
| Grade 4 | 8 (62) | 5 (63) | 1 (25) |
| Gastrointestinal | |||
| Grade 2 | 3 (23) | 0 (0) | 1(25) |
| Grade 3 | 0 (0) | 1(13) | 0 (0) |
| Grade 4 | 0 (0) | 0 (0) | 0 (0) |
| Abnormalities in LFTs | |||
| Grade 2 | 1 (8) | 1 (13) | 0 (0) |
| Grade 3 | 2 (15) | 0 (0) | 0 (0) |
| Grade 4 | 1 (8) | 0 (0) | 0 (0) |
| Infections: | |||
| Grade 2 | 5 (38) | 3 (38) | 1 (25) |
| Grade 3 | 0 (0) | 2 (25) | 0 (0) |
| Grade 4 | 1 (8) | 1 (13) | 0 (0) |
The rates of neurotoxicity and CRS, including high grade events, did not appear to differ between the groups. G2–4 neurotoxicity was recorded in 23% (n=3) in Group A, 50% (n=4) Group B, and 25% (n=1) in Group C. G3 neurotoxicity was observed in 1 subject (8%), 2 subjects (25%), and 1 subject (25%) in the Group A, B, and C respectively. G4 neurotoxicity was observed in 2 subjects (25%) in Group B only. Overall CRS rates were high. G2–4 CRS was experienced by 85% (n=11) in Group A, 100% (n=8) in Group B, and 75% (n=3) in Group C. Specifically, G2 CRS was reported in 6 subjects (46%) in Group A, 5 subjects (63%) in Group B, and 2 subjects (50%) in Group C. G3 CRS, however, was slightly higher in Group A (39% vs. 25% in Group B and C). Only Group B had 1 subject (12.5%) with G4 CRS.
CART-BCMA manufacturing details and characteristics at peak expansion in peripheral blood:
To assess the impact of pre-apheresis RT on CART-BCMA manufacturing, we analyzed characteristics of CART product before, during, and after completion of manufacturing (sTable 2). Groups A, B, and C were similar across the following parameters: absolute lymphocyte count pre-apheresis, percent lymphocytes pre-apheresis, frequency of CD3+ cells in the pre- and post-manufacturing products, CD4/CD8 T-cell ratio in the pre- and post-manufacturing product, and frequency of naïve or early memory CD8+ T-cells in the pre-manufacturing product (%CD45RO-CD27+ of the CD8+ population) (Fig3A; sTable 2). In vitro fold expansion was significantly lower in Group B (14.7) vs. Group A (38.9) but not Group B vs. Group C (27.2) (p=0.0065). Given that bridging-RT should not affect manufacturing, we performed a parallel analysis of subjects combined from Group A and C vs. those from Group B; likewise, Group B had significantly lower in vitro proliferation (fold expansion, p=0.0015) while remaining parameters were similar (sTable 2). Since most peripheral T-cell recovery occurs within the first 3–4 months following focal RT(30,31), manufacturing details were also compared between subjects who did (n=6) and did not (n=19) receive RT within 100 days preceding apheresis (Fig3B). Fold expansion showed a similar reduction that trended towards significance in subjects who received RT <100 days before apheresis (fold expansion 20.3 vs. 31.7, p=0.069). Despite these potential differences, the minimum target goal of CART-BCMA cells was successfully manufactured from all apheresed subjects, and engraftment was seen in all subjects as well.
Figure 3: Manufacturing and peripheral blood characteristics of CART-BCMA relative to RT receipt.
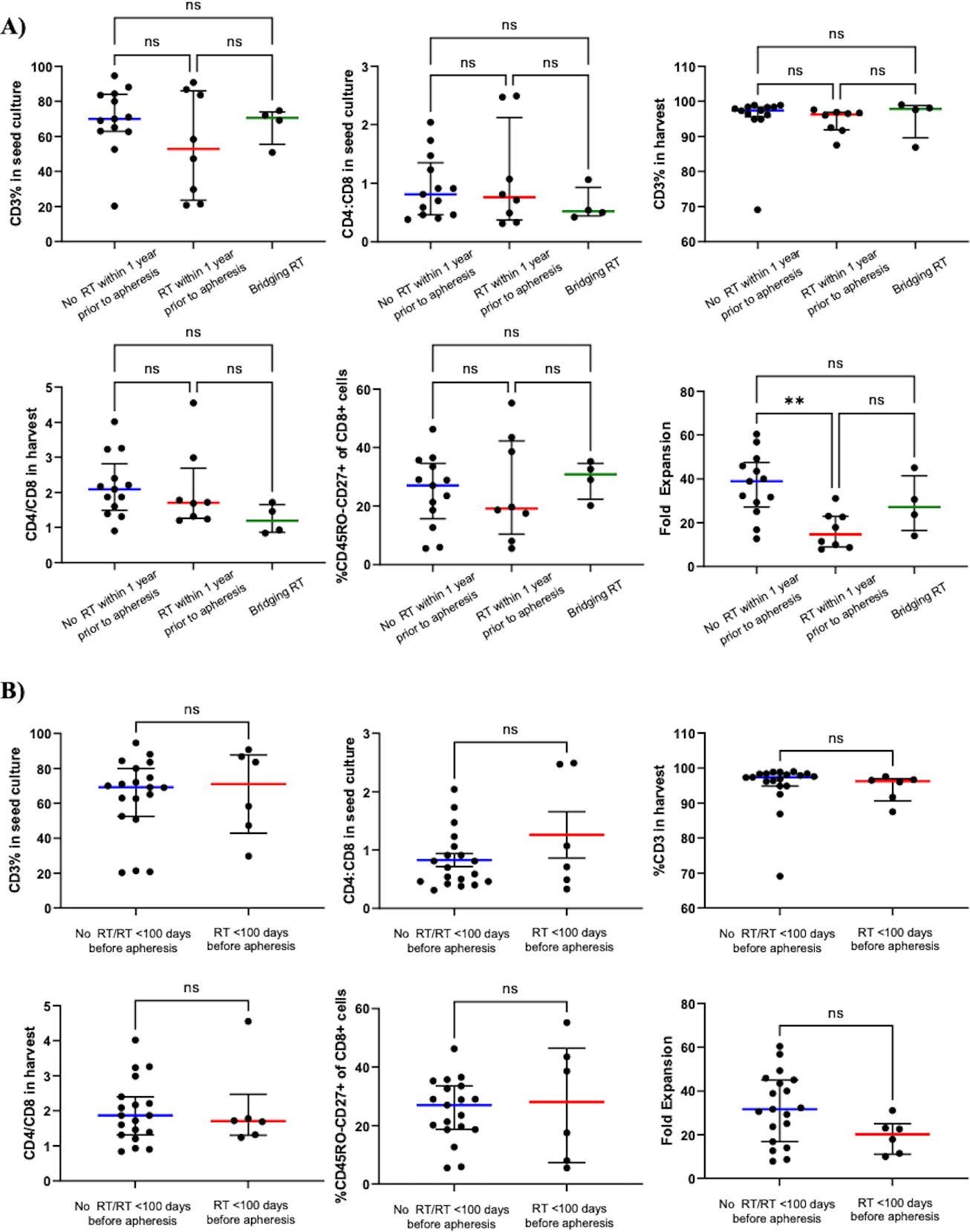
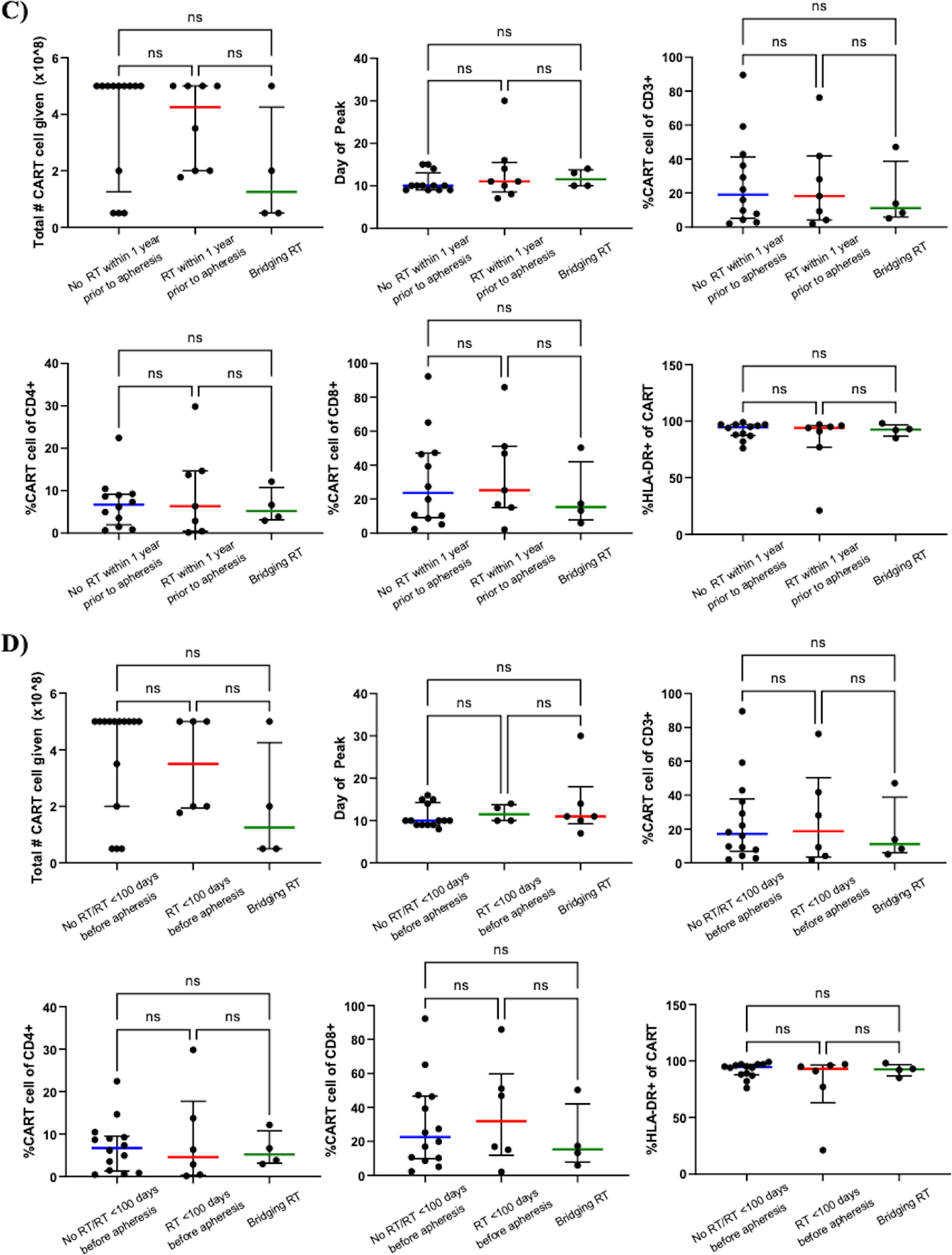
Panels A and C compare parameters of manufacturing and peripheral blood at peak expansion between Group A (no RT <1 year before apheresis; n=13), Group B (RT <1 year before apheresis; n=8), and Group C (bridging-RT defined as RT delivered after apheresis but before CART infusion; n=4), respectively. A) Group B had significantly lower fold expansion (p=0.0065) compared to Group A. B) A similar trend was observed between subjects who received RT <100 days before apheresis (n=6) relative to those who did not (No RT/RT <100 days before apheresis; n=19) (fold expansion, p =0.069). Percentage of CD8+ T-cells with the CD45RO-CD27+ phenotype in the pre-manufacturing product was comparable between Group A, B, and C (A) as well as between No RT within 100 days before apheresis and RT within 100 days before apheresis (B). All other manufacturing parameters were similar between Group A, B, and C and not associated with RT delivered closer to time of apheresis. Characteristics of peripheral blood CART-BCMA cells at peak expansion between Group A, B, and C (C) and between No RT/RT <100 days before apheresis (n=15), RT <100 days before apheresis (n=6), and bridging-RT (n=4) (D) were similar. The frequency of total CD3+, CD3+CD4+, CD3+CD8+, and CD8+CD45RO-CD27+ cells was assessed by flow cytometry before (“in seed culture”) and after (“at harvest”) manufacturing. The frequency of CART cells within CD3+, CD4+, and CD8+ populations and activation status at peak expansion (as measured by % of CART cells expressing HLA-DR) were also assessed by flow cytometry. Lines represent median values with interquartile ranges. The peak expansion of subject 9 in Group B was determined by qPCR since CAR+ cells were not detectable by flow cytometry. The peak for subject 34 in Group A could not be determined due to lack of sample between days 10–21. Subjects 1 (Group A), 3 (Group B), 15 (Group B), and 25 (Group C) received 40% of planned CART-BCMA dose due to early cytokine release syndrome. No RT/RT <100 days before apheresis, no history of RT or no RT <100 days preceding apheresis; %CD45RO-CD27+ of CD8+ cells, naïve or early memory CD8+ T-cells in the premanufacturing product.
At peak expansion of CAR T-cells, peripheral blood characteristics were compared between groups (sTable 2). There was no statistically significant difference in the number of CART-BCMA cells received between Group A, B, and C (Fig3C). The median day of peak CART expansion was consistent between groups, ranging from 10 to 11.5 days. At time of peak expansion, the percentage of CAR+ cells within the CD3+, CD4+, and CD8+ populations and the frequency of CAR+CD3+ cells expressing HLA-DR, an activated phenotype, were similar across groups. Although the manufactured product in all groups consisted of predominantly CD4+ T-cells, circulating CART-BCMA cells were largely CD8+ and highly activated (median % HLA-DR+ of CAR+ population for Group A, 94.5; B, 94; C, 92.5). Parallel comparisons between subjects who received bridging-RT (Group C, n=4), RT within 100 days preceding apheresis (n=6), and no RT within 100 days preceding apheresis (n=15) revealed no association between RT receipt/timing and the peripheral blood profile of CART-BCMA cells at peak expansion (Fig3D). A similar analysis of peak expansion based on qPCR measurement of CAR transgene copy number also failed to reveal any differences between groups based on prior RT exposure (sFig2).
To assess whether RT affects persistence of CART-BCMA cells, qPCR data from peripheral blood at day 28 (D28) after CART-infusion were analyzed based on RT receipt. Subjects who received no/remote RT (Group A) vs. RT <1 year before CART infusion (Group B and C) had no statistically significant difference in CAR transgene copy number at D28 (median 5199 vs. 731 copies/μg genomic DNA, p=0.46) (sFig3A). RT delivered closer to CART infusion did not significantly impact D28 persistence; subjects who received RT within 100 days before apheresis or the bridging period (n=10) vs. subjects who received no RT/remote RT outside this window had comparable CAR transgene copy numbers at D28 (median 732 vs. 3478 copies/μg DNA, p=0.55) (sFig3B). Similarly, no difference was found between subjects who received bridging-RT (n=4), RT <100 days before apheresis (n=6), or no/remote RT (n=14) (median 3102, 732, and 3478 copies/μg DNA, p=0.55) (sFig3C).
Prior analysis revealed that peak blood CART-BCMA expansion, as measured by qPCR, was significantly associated with a higher frequency of CD45RO-CD27+CD8+ T-cells in the pre-manufacturing leukapheresis product as well as a partial response or better(12). Reanalysis showed that levels of CART BCMA cells at peak expansion and D28 were in fact associated with any clinical response (median 5302 copies/μg DNA at peak expansion for ≥MR vs. 39519 for <MR, p=0.0028; median 5947 copies/μg DNA at D28 for ≥MR vs. 67 for <MR, p=0.0042) (sFig4A, sFig4C). Within responders (≥MR), RT did not impact in vivo levels of CART-BCMA cells at peak expansion (sFig4B) or D28 (sFig4D).
Discussion:
In this report, we sought to characterize the toxicity and disease outcomes of CART-BCMA therapy given either with or without bridging-RT in patients with r/rMM. Moreover, we report the profile of CART-BCMA manufacturing products as well as peripheral blood CART expansion in those who received RT within 1 year before apheresis, 100 days before apheresis and the bridging period. Bridging-RT was not associated with excessive RT- or CART-related toxicities and did not appear to significantly alter clinical outcomes (OS, PFS). It’s worth noting that although OS curves did not meet statistical significance, the bridging-RT group did appear to have poorer survival (p=0.08). The ALC pre-apheresis, percentage of lymphocytes pre-apheresis, CD4/CD8 ratio, and frequency of naïve or early memory CD8+ T-cells (CD45RO-CD27+CD8+) in the premanufactured product did not correlate with RT receipt within 1 year or 100 days before apheresis. However, those who received RT in the year prior to apheresis had less robust in vitro T-cell expansion during CAR T-cell manufacturing compared to those who did not (Group B vs. Group A+C, p=0.0015). We also observed a trend, which did not reach statistical significance, towards lower in vitro expansion in the subjects receiving RT within 100 days prior to apheresis (p=0.069). Of note, 75% of subjects who received RT in the year prior to apheresis completed RT within 100 days before apheresis. This suggests RT delivered 100 days prior to leukapheresis may negatively impact in vitro proliferation of seeded cells during manufacturing. Depending on the irradiated volume, sufficient T-cell recovery after RT may take at least 3–4 months to maintain the in vitro proliferative capacity of apheresed cells. Alternatively, these particular subjects may have had higher tumor burden which may negatively impact T-cell biology even before apheresis. Although solBCMA levels post-apheresis prior to cyclophosphamide administration (or simply first CART infusion for cohort 1 which received no lymphodepletion) did not significantly differ between groups and was not associated with RT exposure within 100 days before apheresis (sFig1), Group B had a higher level (885 ng/mL vs. 204 ng/mL in Group A and 151 ng/mL in Group C, p=0.58). This suggests that baseline tumor burden may have been similar in Group A and C and higher in Group B, and our study was not powered to investigate this relationship. We unfortunately did not collect solBCMA levels at the time of apheresis.
The parent trial showed that in vitro fold expansion correlated with in vivo CART-BCMA peak expansion, and in turn, in vivo CART-BCMA peak expansion correlated with clinical responses(12). These findings suggest ex vivo proliferative capacity may predict for in vivo activity. Although RT <1 year preceding apheresis was associated with lower in vitro fold expansion, which did not correlate with decreased in vivo peak expansion, it had numerically lower CAR transgene copy number (median 6109 copies/μg DNA) relative to No RT <1 year before apheresis (37,946 copies/μg DNA) and bridging-RT (17,528 copies/μg DNA) (sFig2A). Once again, our study was not powered to examine such a relationship.
We believe this study has significant value given the recent FDA approval of the idecabtagene vicleucel (ide-cel). This study is among the first to explore the outcomes of patients who have received CART-BCMA therapy with bridging RT or RT within 1 year prior to apheresis. As more patients with relapsed/refractory myeloma receive ide-cel, new clinical situations will arise on when and how to best offer bridging-RT to those needing palliation during the manufacturing period. Our report, although imperfect, provides the first characterization of the safety and feasibility of bridging-RT with BCMA-specific CAR T-cells, which to the best of our knowledge, has not been described in the literature apart from rare case-reports.
Limitations of this study largely result from its retrospective nature and small sample size, which preclude definitive conclusions. Specifically with respect to the association we noted between pre-apheresis RT and manufacturing characteristics, the need for pre-apheresis RT may simply be a marker of other factors, such as more intensive systemic treatment, which may explain slower in vitro T-cell expansion. Though we did not identify obvious differences in the baseline characteristics of our groups apart from the presence of extramedullary disease, the small sample size precluded a formal multivariable analysis; moreover, there may be confounding variables we did not analyze, such as differences in systemic therapy over the year prior to apheresis. Nonetheless, we did our best to characterize the groups and showed they were fairly balanced in regards to following prognostic factors: high-risk cytogenetics, number of prior lines of therapy, renal function, LDH, and prior autologous stem cell transplant. Since subjects were prohibited from receiving treatment during the 2-week washout prior to apheresis, we are unable to explore the effects of RT completed within this window on CART-BCMA manufacturing. However, given that RT is lymphodepleting, conventional wisdom points to avoiding its delivery within the few weeks preceding apheresis to ensure adequate ALC.
Similarly, none of the subjects received RT within 20 days following administration of CART-BCMA therapy (sTable 3). Therefore, we cannot assess if RT delivered within this early post-CART period can augment CART efficacy in patients who demonstrate progression after infusion as indicated by further rise in M-spike or new/growing radiographic lesions. In a case report, palliative RT delivered between day 6 and 20 following anti-BCMA CART therapy (coinciding with time of peak CART expansion) in a MM patient led to a synergistic abscopal effect and expansion of new T-cell receptor (TCR) clones, mediating the eradication of substantial tumor burden, including extraosseous lesions(32). This anecdote supports exploration of peri-CART therapy RT, which perhaps may be particularly useful for patients with extramedullary disease where responses are typically less durable, even after CART therapy(33).
Moreover, our analysis was limited to a cohort of r/rMM patients treated at a single institution with a single CAR T-cell product. The effects of bridging-RT or pre-apheresis RT on toxicity and manufacturing of other anti-BCMA CART products, e.g. ide-cel and ciltacabtagene autoleucel (cilta-cel)(11,21), requires future investigation. However, given that ide-cel and cilta-cel both feature a 4–1BB co-stimulatory moiety like our CART-BCMA construct, bridging-RT may be safe with these products.
Medical oncologists may be hesitant to recommend bridging-RT before CART-infusion due to concerns of adding RT-related toxicity to those of lymphodepletion and CART therapy. In this study, we palliatively treated the skull base, thoracic and lumbar spine, bilateral hips, and bilateral orbits. Notably, we observed mild acute RT-related toxicity, predominantly G1 fatigue, despite 2 subjects receiving concurrent chemotherapy. Similarly, patients with relapsed/refractory aggressive B-cell lymphoma who received bridging-RT prior to CD19-CAR T-cells experienced no significant acute toxicity in 3 recent series(26–28). Another concern with bridging therapy, either RT or systemic, is marrow suppression. Routine lymphodepletion chemotherapy can cause protracted cytopenias. So far, published data on RT-related toxicity appear proportional to the dose and size of RT fields used without exacerbation by CART therapy. Nonetheless, larger clinical studies are needed before we can draw conclusions.
Another potential concern is whether bridging-RT may worsen CART-associated toxicities. In our study, CRS and neurotoxicity rates appeared unaffected by bridging-RT. All 3 subjects with severe neurotoxicity (≥G3) had high tumor burden (2 with extramedullary disease) as well as G3 or G4 CRS. Studies of CD19-directed CART therapy in acute lymphocytic leukemia and lymphomas support the notion that tumor burden correlates with CRS severity(34–37). Therefore, bridging-RT may temper the severity of CRS or neurotoxicity by debulking systemic treatment-refractory myeloma. Furthermore, RT has an immune-priming potential to amplify immunomodulatory therapies via exposure of neoantigens. Taken together with the observation that incremental increases in the conditioning intensity improve engraftment and clinical outcomes (25,38), it is possible that modifying the lymphodepletion (e.g. adding low dose total body irradiation to cyclophosphamide plus fludarabine) may augment the activity of BCMA-directed CAR T-cells while reducing risk of CART-related toxicities.
How RT influences CART toxicity and efficacy via immunologic mechanisms requires further elucidation. Since CARs lead to MHC-independent T-cell activation, RT may synergize with CART therapy via several potential mechanisms: 1) RT may increase the migration and effector functions of CAR T-cells(20,39,40); 2) cytokines secreted by CAR T-cells in response to RT may prime endogenous T-cells to mount an abscopal-like response(32); and (3) RT-induced apoptosis of cancer cells leading to the release of antigens that are ultimately presented by antigen-presenting cells may stimulate broader T-cell responses (i.e. both CAR T-cell and endogenous T-cell clonal expansions)(17). We hope this study informs subsequent investigations into radiation as an adjunct to CART therapy.
Conclusions:
In our case series of r/rMM patients treated with CART-BCMA cells, bridging-RT seems safe without worsening rates of severe CRS, neurotoxicity, or hematologic toxicity. The time to peak expansion, the activation of CART-BCMA cells, or the frequency of CART-BCMA cells within CD3+, CD4+, and CD8+ populations in vivo at peak expansion appeared similar between those who did and did not receive bridging-RT. Our data suggest RT delivered <100 days before apheresis may negatively impact in vitro proliferation of T-cells during manufacturing. However, larger numbers are required to show significant associations. Future prospective trials investigating the impact of bridging-RT on anti-BCMA CAR T-cell response, long-term efficacy, and toxicity in r/rMM patients are warranted.
Supplementary Material
Translational Relevance:
Chimeric antigen receptor (CAR) T-cells are promising new therapies for hematologic malignancies. Neurotoxicity and cytokine release syndrome (CRS) are serious, potentially fatal toxicities associated with CAR T-cell (CART) therapy. In this study, we did not observe an association between bridging-radiation therapy (RT) and CRS, neurotoxicity, or hematologic toxicity in patients with relapsed/refractory multiple myeloma (r/rMM). We also showed that patients receiving bridging-RT achieved similar in vivo peak expansion and persistence of anti-BCMA CAR T-cells at day 28 post-infusion. However, larger prospective trials investigating RT with anti-BCMA CART therapy are needed to draw definitive conclusions and may further optimize the safety and long-term efficacy of this novel cellular treatment in r/rMM.
Acknowledgements:
We acknowledge and appreciate the assistance of J. Finklestein, F. Nazimuddin, C. Bartoszek, T. Mikheeva, S. Rajkumar, and B. Menchel for sample processing, I. Kulikovskaya, M. Gupta and A. Kim for qPCR analyses, D. Ambrose, L. Tian, and H. Parakandi for flow cytometry analyses, F. Chen and N. Koterba for Luminex cytokine analyses, V. Gonzalez and Y. Tanner for data management and quality control, and A. Lamontagne, A. Brennan, A. Malykhin, members of the Clinical Cell and Vaccine Production Facility for cell manufacturing and testing. We acknowledge the medical and nursing staff of the Apheresis Unit at the Hospital of the University of Pennsylvania for their care and management of patients undergoing leukapheresis. This work was supported by a sponsored research agreement between the University of Pennsylvania and Novartis, as well as NIH grant 1P01CA214278.
Disclosures: A.D.C. reports research funding from Novartis, research funding and personal fees from Bristol-Meyers Squibb, and personal fees from Celgene, Kite Pharma, Janssen, Seattle Genetics, Oncopeptides, Takeda, Array Biopharma, Genentech/Roche, Astrazeneca and GlaxoSmithKline. A.L.G. reports research funding from Novartis, Janssen, and Tmunity, and personal fees from Janssen and GlaxoSmithKline. J.J.M. receives research funding from Novartis, IASO Biotherapeutics, and Kite Pharma, a Gilead company. J.J.M. served as a speaker for Novartis and Johnson & Johnson, consults for Simcere of America, Shanghai Unicar Therapy, Johnson & Johnson, Poseida, Allogene, and IASO Biotherapeutics, and is on the Medical and Scientific Advisory Board of IASO Biotherapeutics. Furthermore, J.J.M. holds patents related to CAR T-cell manufacturing and biomarkers. S.F.L. reports research funding from Novartis, Tmunity, Carisma, and consultancy fees from Kite. A.M. reports research funding from Merck. E.A.S. reports research funding from AbbVie and personal fees from AbbVie, Celgene, Takeda, Janssen, and Amgen. B.L.L. reports personal fees from Avectas, Incysus/IN8bio, Novartis, Vycellix, Immuneel, Ori Biotech, Patheon/ThermoFisher and Viral Vector Services, and is a scientific founder of Tmunity Therapeutics, for which he has founders stock but no income. C.H.J. reports research funding from Novartis, and he is a scientific founder of Tmunity Therapeutics, for which he has founders stock but no income. M.C.M. report research funding from Novartis. All remaining authors have no disclosures. A.D.C. S.F.L, B.L.L., A.L.G., E.A.S., C.H.J., and M.C.M. hold or have pending patents related to intellectual property licensed by the University of Pennsylvania to Novartis.
Research Funding:
This retrospective study is based on a clinical trial (NCT02546167) funded by a sponsored research agreement between the University of Pennsylvania and Novartis to develop chimeric antigen receptor (CAR) T-cells for therapeutic use. Novartis provided the B cell maturation antigen CAR construct but did not dictate study design, conduct, or analysis. Some funds for correlative analyses were provided by NIH grant 1P01CA214278. A.L.G. acknowledges support from a Scholar in Clinical Research award from the Leukemia and Lymphoma Society.
Footnotes
Meeting Presentation Information: Earlier version of this work was accepted for quick pitch oral presentation at American Society of Radiation Oncology 2020.
References
- 1.Chim CS, Kumar SK, Orlowski RZ, Cook G, Richardson PG, Gertz MA, et al. Management of relapsed and refractory multiple myeloma: novel agents, antibodies, immunotherapies and beyond. Leukemia 2018;32(2):252–62 doi 10.1038/leu.2017.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goldschmidt H, Ashcroft J, Szabo Z, Garderet L. Navigating the treatment landscape in multiple myeloma: which combinations to use and when? Ann Hematol 2019;98(1):1–18 doi 10.1007/s00277-018-3546-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kumar SK, Lee JH, Lahuerta JJ, Morgan G, Richardson PG, Crowley J, et al. Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: a multicenter international myeloma working group study. Leukemia 2012;26(1):149–57 doi 10.1038/leu.2011.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nijhof IS, van de Donk N, Zweegman S, Lokhorst HM. Current and New Therapeutic Strategies for Relapsed and Refractory Multiple Myeloma: An Update. Drugs 2018;78(1):19–37 doi 10.1007/s40265-017-0841-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sonneveld P Management of multiple myeloma in the relapsed/refractory patient. Hematology Am Soc Hematol Educ Program 2017;2017(1):508–17 doi 10.1182/asheducation-2017.1.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhutani M, Foureau DM, Atrash S, Voorhees PM, Usmani SZ. Extramedullary multiple myeloma. Leukemia 2020;34(1):1–20 doi 10.1038/s41375-019-0660-0. [DOI] [PubMed] [Google Scholar]
- 7.Makita S, Imaizumi K, Kurosawa S, Tobinai K. Chimeric antigen receptor T-cell therapy for B-cell non-Hodgkin lymphoma: opportunities and challenges. Drugs Context 2019;8:212567 doi 10.7573/dic.212567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med 2018;378(5):439–48 doi 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med 2017;377(26):2531–44 doi 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med 2015;7(303):303ra139 doi 10.1126/scitranslmed.aac5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berdeja JG, Madduri D, Usmani SZ, Singh I, Zudaire E, Yeh T-M, et al. Update of CARTITUDE-1: A phase Ib/II study of JNJ-4528, a B-cell maturation antigen (BCMA)-directed CAR-T-cell therapy, in relapsed/refractory multiple myeloma. Journal of Clinical Oncology 2020;38(15_suppl):8505– doi 10.1200/JCO.2020.38.15_suppl.8505. [DOI] [Google Scholar]
- 12.Cohen AD, Garfall AL, Stadtmauer EA, Melenhorst JJ, Lacey SF, Lancaster E, et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J Clin Invest 2019;129(6):2210–21 doi 10.1172/JCI126397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N Engl J Med 2019;380(18):1726–37 doi 10.1056/NEJMoa1817226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brudno JN, Maric I, Hartman SD, Rose JJ, Wang M, Lam N, et al. T Cells Genetically Modified to Express an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma. J Clin Oncol 2018;36(22):2267–80 doi 10.1200/JCO.2018.77.8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Madduri D, Berdeja JG, Usmani SZ, Jakubowiak A, Agha M, Cohen AD, et al. CARTITUDE-1: Phase 1b/2 Study of Ciltacabtagene Autoleucel, a B-Cell Maturation Antigen-Directed Chimeric Antigen Receptor T Cell Therapy, in Relapsed/Refractory Multiple Myeloma. Blood 2020;136(Supplement 1):22–5 doi 10.1182/blood-2020-136307. [DOI] [Google Scholar]
- 16.Shabason JE, Minn AJ. Radiation and Immune Checkpoint Blockade: From Bench to Clinic. Semin Radiat Oncol 2017;27(3):289–98 doi 10.1016/j.semradonc.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 17.Flynn JP, O’Hara MH, Gandhi SJ. Preclinical rationale for combining radiation therapy and immunotherapy beyond checkpoint inhibitors (i.e., CART). Transl Lung Cancer Res 2017;6(2):159–68 doi 10.21037/tlcr.2017.03.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med 2006;203(5):1259–71 doi 10.1084/jem.20052494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, et al. Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell 2013;24(5):589–602 doi 10.1016/j.ccr.2013.09.014. [DOI] [PubMed] [Google Scholar]
- 20.DeSelm C, Palomba ML, Yahalom J, Hamieh M, Eyquem J, Rajasekhar VK, et al. Low-Dose Radiation Conditioning Enables CAR T Cells to Mitigate Antigen Escape. Mol Ther 2018;26(11):2542–52 doi 10.1016/j.ymthe.2018.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Munshi NC, Larry D. Anderson J, Shah N, Jagannath S, Berdeja JG, Lonial S, et al. Idecabtagene vicleucel (ide-cel; bb2121), a BCMA-targeted CAR T-cell therapy, in patients with relapsed and refractory multiple myeloma (RRMM): Initial KarMMa results. Journal of Clinical Oncology 2020;38(15_suppl):8503– doi 10.1200/JCO.2020.38.15_suppl.8503. [DOI] [Google Scholar]
- 22.Garfall AL, Dancy EK, Cohen AD, Hwang WT, Fraietta JA, Davis MM, et al. T-cell phenotypes associated with effective CAR T-cell therapy in postinduction vs relapsed multiple myeloma. Blood Adv 2019;3(19):2812–5 doi 10.1182/bloodadvances.2019000600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maus MV, June CH. Making Better Chimeric Antigen Receptors for Adoptive T-cell Therapy. Clin Cancer Res 2016;22(8):1875–84 doi 10.1158/1078-0432.CCR-15-1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 2011;118(18):4817–28 doi 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Turtle CJ, Hanafi LA, Berger C, Hudecek M, Pender B, Robinson E, et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med 2016;8(355):355ra116 doi 10.1126/scitranslmed.aaf8621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wright CM, LaRiviere MJ, Baron JA, Uche C, Xiao Y, Arscott WT, et al. Bridging Radiation Therapy Before Commercial Chimeric Antigen Receptor T-Cell Therapy for Relapsed or Refractory Aggressive B-Cell Lymphoma. Int J Radiat Oncol Biol Phys 2020;108(1):178–88 doi 10.1016/j.ijrobp.2020.05.014. [DOI] [PubMed] [Google Scholar]
- 27.Sim AJ, Jain MD, Figura NB, Chavez JC, Shah BD, Khimani F, et al. Radiation Therapy as a Bridging Strategy for CAR T Cell Therapy With Axicabtagene Ciloleucel in Diffuse Large B-Cell Lymphoma. Int J Radiat Oncol Biol Phys 2019;105(5):1012–21 doi 10.1016/j.ijrobp.2019.05.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pinnix CC, Gunther JR, Dabaja BS, Strati P, Fang P, Hawkins MC, et al. Bridging therapy prior to axicabtagene ciloleucel for relapsed/refractory large B-cell lymphoma. Blood Adv 2020;4(13):2871–83 doi 10.1182/bloodadvances.2020001837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deutsch E, Besse B, Le Pavec J, Le Pechoux C, Botticella A, Ammari S, et al. Can radiation-recall predict long lasting response to immune checkpoint inhibitors? Radiother Oncol 2021;154:125–7 doi 10.1016/j.radonc.2020.09.037. [DOI] [PubMed] [Google Scholar]
- 30.Velardi E, Tsai JJ, van den Brink MRM. T cell regeneration after immunological injury. Nat Rev Immunol 2020. doi 10.1038/s41577-020-00457-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang XB, Wu DJ, Chen WP, Liu J, Ju YJ. Impact of radiotherapy on immunological parameters, levels of inflammatory factors, and clinical prognosis in patients with esophageal cancer. J Radiat Res 2019;60(3):353–63 doi 10.1093/jrr/rrz006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith EL, Mailankody S, Staehr M, Wang X, Senechal B, Purdon TJ, et al. BCMA-Targeted CAR T-cell Therapy plus Radiotherapy for the Treatment of Refractory Myeloma Reveals Potential Synergy. Cancer Immunol Res 2019;7(7):1047–53 doi 10.1158/2326-6066.CIR-18-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang B, Liu J, Zhao W-H, Chen Y-X, Cao X-M, Yang Y, et al. Chimeric Antigen Receptor T Cell Therapy in the Relapsed or Refractory Multiple Myeloma with Extramedullary Disease--a Single Institution Observation in China. Blood 2020;136(Supplement 1):6– doi 10.1182/blood-2020-140243.32614958 [DOI] [Google Scholar]
- 34.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014;371(16):1507–17 doi 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hay KA, Hanafi LA, Li D, Gust J, Liles WC, Wurfel MM, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood 2017;130(21):2295–306 doi 10.1182/blood-2017-06-793141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang J, Hu Y, Yang S, Wei G, Zhao X, Wu W, et al. Role of Fluorodeoxyglucose Positron Emission Tomography/Computed Tomography in Predicting the Adverse Effects of Chimeric Antigen Receptor T Cell Therapy in Patients with Non-Hodgkin Lymphoma. Biol Blood Marrow Transplant 2019;25(6):1092–8 doi 10.1016/j.bbmt.2019.02.008. [DOI] [PubMed] [Google Scholar]
- 37.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 2014;6(224):224ra25 doi 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol 2008;26(32):5233–9 doi 10.1200/JCO.2008.16.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weiss T, Weller M, Guckenberger M, Sentman CL, Roth P. NKG2D-Based CAR T Cells and Radiotherapy Exert Synergistic Efficacy in Glioblastoma. Cancer Res 2018;78(4):1031–43 doi 10.1158/0008-5472.CAN-17-1788. [DOI] [PubMed] [Google Scholar]
- 40.Weiss T, Schneider H, Silginer M, Steinle A, Pruschy M, Polic B, et al. NKG2D-Dependent Antitumor Effects of Chemotherapy and Radiotherapy against Glioblastoma. Clin Cancer Res 2018;24(4):882–95 doi 10.1158/1078-0432.CCR-17-1766. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.