

Abstract
The mechanisms by which DNA interstrand cross-links (ICLs) are repaired in mammalian cells are unclear. Studies in bacteria and yeasts indicate that both nucleotide excision repair (NER) and recombination are required for their removal and that double-strand breaks are produced as repair intermediates in yeast cells. The role of NER and recombination in the repair of ICLs induced by nitrogen mustard (HN2) was investigated using Chinese hamster ovary mutant cell lines. XPF and ERCC1 mutants (defective in genes required for NER and some types of recombination) and XRCC2 and XRCC3 mutants (defective in RAD51-related homologous recombination genes) were highly sensitive to HN2. Cell lines defective in other genes involved in NER (XPB, XPD, and XPG), together with a mutant defective in nonhomologous end joining (XRCC5), showed only mild sensitivity. In agreement with their extreme sensitivity, the XPF and ERCC1 mutants were defective in the incision or “unhooking” step of ICL repair. In contrast, the other mutants defective in NER activities, the XRCC2 and XRCC3 mutants, and the XRCC5 mutant all showed normal unhooking kinetics. Using pulsed-field gel electrophoresis, DNA double-strand breaks (DSBs) were found to be induced following nitrogen mustard treatment. DSB induction and repair were normal in all the NER mutants, including XPF and ERCC1. The XRCC2, XRCC3, and XRCC5 mutants also showed normal induction kinetics. The XRCC2 and XRCC3 homologous recombination mutants were, however, severely impaired in the repair of DSBs. These results define a role for XPF and ERCC1 in the excision of ICLs, but not in the recombinational components of cross-link repair. In addition, homologous recombination but not nonhomologous end joining appears to play an important role in the repair of DSBs resulting from nitrogen mustard treatment.
DNA interstrand cross-linking agents such as the nitrogen mustards, mitomycin C, cisplatin, and psoralen are widely used in cancer chemotherapy and phototherapy and are thought to exert their cytotoxic effects by preventing efficient DNA replication and transcription. Since interstrand cross-links (ICLs) affect both strands of DNA, repair of these lesions presents a special problem. In Escherichia coli and Saccharomyces cerevisiae, the repair of ICLs depends on both nucleotide excision repair (NER) and homologous recombination (10, 11, 28, 40, 58). In S. cerevisiae, the repair of psoralen-photoinduced DNA ICLs involves a double-strand break (DSB) intermediate resulting partially from NER incisions, and these DSBs are repaired by homologous recombination (16, 28, 40, 41). In contrast, recent studies from this laboratory have demonstrated the occurrence of NER-independent DSBs in yeast cells following treatment with nitrogen mustard (42). In common with psoralen cross-links, these DSB intermediates are repaired by homologous recombination (42).
In mammalian systems, ICL repair is poorly understood. However, the isolation and characterization of mutant cell lines with extreme sensitivities to cross-linking agents provide mechanistic clues. For example, Chinese hamster ovary (CHO) cells with mutations in the XPF and ERCC1 genes, involved in the NER pathway, are extremely sensitive to cross-linking agents, whereas other available NER mutants are only slightly sensitive (1, 15, 26). This suggests that XPF and ERCC1 play a central role in cross-link repair (1, 15, 26). The XPF and ERCC1 proteins form a heterodimer with a structure-specific endonuclease activity and are responsible for 5′ incisions on the damaged strand during NER (5, 7, 48). ERCC1 is homologous to yeast Rad10 (57), which is complexed with the yeast homologue of XPF, Rad1 (2, 3). This heterodimer is involved in two processes in yeast cells, NER and a recombination subpathway, single-strand annealing (SSA) (17, 19). In SSA, the resected 3′-single-strand ends of a DSB are joined through regions of 60 to 90 bp of homology (52), and the resulting overhangs are cleaved by the Rad1-Rad10 heterodimer (19, 20, 27). It has been suggested that the extreme sensitivity of XPF and ERCC1 mutants to cross-linking agents could be due to dual defects in SSA recombination as well as in NER (55).
A second class of cross-link-hypersensitive mutants are the irs1 and irs1SF cell lines, originally isolated on the basis of sensitivity to ionizing radiation (22, 31). The human genes that complement irs1 and irs1SF are XRCC2 and XRCC3, respectively, both of which encode members of an emerging family of Rad51-related proteins that participate in homologous recombination (39). These cell lines are only moderately sensitive to X-rays, γ radiation, and UV radiation but are extremely sensitive to DNA cross-linking agents (8). Interestingly, although they are sensitive to ionizing radiation, irs1 and irs1SF cells show no defects in the repair of radiation-induced DSBs (9, 22, 32, 53). However, recent studies have shown that both the XRCC2 (30) and XRCC3 (45) genes are involved in the repair of I-SceI endonuclease-induced DNA DSBs by homologous recombination. In contrast, nonhomologous end joining (NHEJ) mutants defective in the XRCC4, XRCC5, XRCC6, and XRCC7 genes have shown hypersensitivity to ionizing radiation but also show defects in the repair of radiation-induced DSBs (8, 12, 44, 46). Unlike XRCC2 and XRCC3 mutants, NHEJ mutants do not show extreme sensitivity to cross-linking agents (8). Collectively these studies show a divergence between the types of DSBs repaired by NHEJ and homologous recombination.
In this study we have investigated the roles of NER and both homologous and nonhomologous recombination in the repair of nitrogen mustard (mechlorethamine [HN2]) induced ICLs in mammalian cells. We confirm the hypersensitivity of ERCC1, XPF, XRCC2, and XRCC3 mutant CHO cells to HN2 and show a correlation between this hypersensitivity and a defect in the “unhooking” of ICLs in the ERCC1- and XPF defective cells but not in the recombination mutants XRCC2 and XRCC3. Using pulsed field gel electrophoresis (PFGE) we show that DSBs are induced following treatment with HN2 and that the XRCC2 and XRCC3 mutants are severely impaired in the repair of these DSBs. However, an XRCC5 mutant exhibited normal DSB repair kinetics. These observations define a role for XPF and ERCC1 in the incision step but not in the recombinational steps of cross-link repair and indicate that homologous recombination, but not NHEJ, is required for the repair of the DSBs induced by this cross-linking drug.
MATERIALS AND METHODS
Cell lines and culture conditions.
The cell lines used in this study are listed in Table 1. The AA8, UV23, UV42, UV61, and UV96 cell lines were obtained from M. Stefanini, and UV135 was purchased from the American Type Culture Collection (ATCC number CRL-1867). The V79, irs1, irs1SF, CHO-K1, and xrs5 cell lines were kindly provided by J. Thacker. All cell lines were grown as a monolayer in F12-Ham-HEPES medium (Sigma, Poole, U.K.) supplemented with 2 mM glutamine and 10% fetal calf serum (FCS). Cells were grown at 37°C in a 5% CO2 humidified incubator. Trypsin-versine solution was used for detaching cells. Nondividing cells were grown in confluent cultures, and the cell cycle status was confirmed by fluorescence-activated cell sorting analysis.
TABLE 1.
Chinese hamster cell lines used in this study
| Mutant cell line | Parent cell line | Defective gene |
|---|---|---|
| UV23 | AA8 | XPB |
| UV42 | AA8 | XPD |
| UV47 | AA8 | XPF |
| UV61 | AA8 | CSB |
| UV96 | AA8 | ERCC1 |
| UV135 | AA8 | XPG |
| irs1 | V79 | XRCC2 |
| irs1SF | AA8 | XRCC3 |
| xrs5 | CHO-K1 | XRCC5 |
Chemicals.
Analytical-grade mechlorethamine (nitrogen mustard [HN2]) and 2-dimethylaminoethylchloride hydrochloride 99% (HN1) were purchased from Sigma.
Cytotoxicity assay.
Cytotoxicity was assessed using the sulforhodamine B (SRB) assay (49). A total of 3 × 103 cells were seeded into each well of 96-well flat-bottomed microtiter plates in a volume of 100 μl and incubated at 37°C overnight. The desired concentrations of HN2 were prepared in culture medium (without FCS) immediately before use. Medium in wells was removed, and 100 μl of drug-medium mixture was added. Six replicates were used for each drug concentration. Plates were incubated for 1 h at 37°C. Following drug treatment, the medium was replaced with 200 μl of fresh complete medium, and the plates were incubated for 3 days at 37°C. The medium in the wells was removed, and 100 μl of ice-cold 10% (wt/vol) trichloroacetic acid was added to fix the cells. The plates were incubated at 4°C for 20 min and then washed four times with water. Cells were stained with 100 μl of 0.4% (wt/vol) SRB–1% acetic acid per well for 20 min at room temperature. Unbound dye was removed by five washes in 1% acetic acid, and plates were dried overnight at room temperature. The dye was solubilized by the addition of 100 μl of 10 mM Tris base into each well. Plates were left at room temperature for 20 min, and the optical density (OD) at 540 nm was determined using a Titretech 420 microtiter plate reader equipped with Titresoft II software (Flow Laboratories). Fraction of control A540 was calculated from the following equation: fraction of control A540 = OD of drug-treated wells/OD of untreated control. Finally, the mean fraction of control A540 figures for each drug concentration with standard deviations was calculated.
Determination of DNA interstrand cross-linking using the comet assay.
The modification of the comet assay to measure DNA interstrand cross-linking has recently been described in detail (50). Exponentially growing cells were treated with the desired concentrations of HN2 in FCS-free medium for 1 h at 37°C. The medium was replaced with fresh complete medium and incubated for the required postincubation time. Cells were then trypsinized, diluted to a density of 2.5 × 104 cells/ml, and kept on ice. All drug-treated samples plus one control were subjected to 12.5 Gy of X-irradiation on ice, and an unirradiated control was included. Microscope slides were precoated with 1% (wt/vol) type-IA agarose, and 0.5 ml of cells was mixed with 1 ml of 1% (wt/vol) type-VII agarose and spread over a precoated slide in duplicate. A coverslip was added, and the agarose was allowed to solidify. Coverslips were removed, and slides were placed in lysis solution (100 mM disodium EDTA, 2.5 M NaCl, 10 mM Tris-HCl [pH 10.5]) containing 1% Triton X-100 at 4°C and incubated for 1 h in the dark. Slides were subsequently washed with ice-cold water for 15 min, and this was repeated three times. The slides were then transferred to an electrophoresis tank containing ice-cold alkaline solution (50 mM NaOH, 1 mM disodium EDTA [pH 12.5]) and incubated for 45 min in the dark. Electrophoresis was carried out for 25 min at 18 V (0.6 V/cm) and 250 mA in the dark. Slides were removed, and 1 ml of neutralizing solution (0.5 M Tris-HCl [pH 7.5]) was added and incubated for 10 min. Each slide was rinsed twice with 1 ml of phosphate-buffered saline (PBS) and allowed to dry overnight at room temperature. Slides were stained with propidium iodide (2.5 μg/ml), and comets were analyzed using a Nikon DIAPHOT TDM inverted epifluorescent microscope (consisting of a high-pressure mercury vapor light source, a 580-nm dichromic mirror, 510- to 560-nm excitation filter, and 590-nm barrier filter) at ×20 magnification. Fifty cells were analyzed per slide using Komet Assay software (Kinetic Imaging, Liverpool, U.K.).
The degree of DNA interstrand cross-linking present in a drug-treated sample was determined by comparing the tail moment of the irradiated drug-treated samples with irradiated untreated samples and unirradiated untreated samples (50). The level of interstrand cross-linking is proportional to the decrease in the tail moment in the irradiated drug-treated sample compared to the irradiated untreated control. The decrease in tail moment is calculated by the formula % decrease in tail moment (DTM) = [1 − (TMdi − TMcu)/(TMci − TMcu)] × 100, where TMdi is the mean tail moment of the drug-treated, irradiated sample, TMci is the mean tail moment of the irradiated control sample, and TMcu is the mean tail moment of the unirradiated control sample. The unhooking of DNA interstrand cross-links was expressed as percent unhooking, which was calculated using the formula
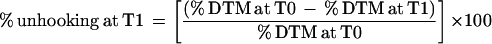 |
where T0 is the time immediately following drug treatment and T1 is the postincubation time in drug-free medium.
Analysis of DSBs by PFGE.
Cells growing in a monolayer were treated with HN2 or HN1 for 1 h, washed with 10 ml of PBS, and incubated with fresh medium for the required repair time. Cells were trypsinized, 3 × 106 cells were harvested, and PFGE plugs were prepared using the Bio-Rad Mammalian CHEF Genomic Plug Kit, as instructed by the manufacturer. PFGE was performed with a 0.7% gel (Pulse Field Certified agarose; Bio-Rad) in 0.25× Tris-borate-EDTA buffer using a Biometra Rotaphor type V apparatus. Electrophoresis runs were for 120 h at 14°C with the following parameters: interval 5,000 to 1,000 s log, angle 110 to 100° linear, and voltage 50 to 45 V linear. On completion, gels were stained with ethidium bromide (2 μg/ml) for 1 h, destained overnight with water, and photographed. Semiquantitative data were obtained by measuring the absolute integrated OD of each lane using Gel Pro Analyser (Media Cybernetics) and calculating the percentage of DNA released from the DNA plug.
RESULTS
Nitrogen mustard sensitivity of NER and recombination-defective cell lines.
To confirm the roles of NER and recombination in the repair of ICLs in mammalian cells, the sensitivity to the bifunctional alkylating agent nitrogen mustard (HN2) of Chinese hamster cell lines defective in these pathways was determined. The results are consistent with those obtained by other workers (1, 8, 15, 26) using a variety of cross-linking agents, implying that these observations are of general significance to the question of cross-link repair.
Among the mutant cell lines deficient in an NER process (Fig. 1), UV47 and UV96, defective in XPF and ERCC1, respectively, were highly sensitive (>15-fold) to HN2 compared to their isogenic parent cell line AA8. In contrast, UV135, defective in XPG, which is responsible for the 3′ incision in NER, was only slightly sensitive (<2-fold). Similarly, the UV23 and UV42 cell lines, bearing mutations in the XPB and XPD helicases, respectively, were also only slightly sensitive to HN2. Surprisingly, the UV61 CSB mutant cell line, involved in the transcriptional coupling of NER, was slightly resistant to HN2 compared to its isogenic parent.
FIG. 1.

Survival of NER mutants, XPB mutant UV23, XPD mutant UV42, XPF mutant UV47, CSB mutant UV61, ERCC1 mutant UV96, XPG mutant UV135, and the parent cell line AA8 following 1 h of exposure to increasing concentrations of HN2. All results are means of at least three independent experiments, and error bars show the standard error of the mean.
Figure 2 shows the sensitivities of XRCC (recombination) mutants to HN2. The irs1 (XRCC2 mutant) and irs1SF (XRCC3 mutant) lines derived from V79 and AA8, respectively, were extremely sensitive to HN2 (12-fold and 26-fold, respectively). In contrast, the NHEJ-defective cell line xrs5, bearing a mutation in the XRCC5 gene product, was only slightly sensitive to HN2 (1.5-fold) compared to its parent cell line CHO-K1.
FIG. 2.

Survival of homologous-recombination mutants. (A) XRCC2 mutants (irs1) and the wild-type cell line V79. (B) XRCC3 mutant (irs1SF) and wild-type AA8 cell line. (C) NHEJ XRCC5 mutant (xrs5) and the wild-type cell line CHO-K1. Survival was measured after 1 h of treatment with increasing concentrations of HN2. All results are means of at least three independent experiments, and error bars show the standard error of the mean.
Unhooking of ICLs in CHO repair-defective cell lines.
To investigate whether the extreme sensitivities of the XPF, ERCC1, XRCC2, and XRCC3 mutants are due to a defect in the incision step of cross-link repair, the efficiency of unhooking of HN2-induced cross-links in these mutants and the less sensitive XPB, XPD, XPG, and XRCC5 mutants was compared with that of their isogenic parents. Cross-linking was measured at the single-cell level using a modified version of the comet assay. This assay allows the initial cross-link incisions on one strand, releasing the covalent linkage of the two strands to be followed. Prior to cell lysis, samples receive a dose of X-rays to induce random DNA strand breakage. The presence of ICLs retards the migration of the irradiated DNA during electrophoresis, resulting in a reduced tail moment compared to the unirradiated control. Figure 3 shows the decrease in tail moment compared to the irradiated control due to the presence of cross-links induced following treatment with increasing concentrations of HN2. The ability of cells to unhook cross-linking can therefore be observed as an increase in tail moment following a repair period in drug-free medium.
FIG. 3.

Percent decrease in tail moment following 1 h of treatment of AA8 cells with increasing doses of HN2. Results are means of three individual experiments, and error bars show standard error of the mean.
Figure 4A illustrates the unhooking efficiency of XPF, ERCC1, XPB, and XPG mutants compared to that of their parent AA8. The XPB and XPG mutants showed unhooking kinetics indistinguishable from those of AA8. These cells were all able to unhook approximately 65% of the cross-links by 24 h and more than 85% by 48 h. In contrast, and in agreement with their extreme sensitivity, the XPF and ERCC1 mutants were highly defective in the unhooking of HN2 cross-links. Less than 15% of the cross-links were unhooked after 48 h in drug-free medium.
FIG. 4.

Efficiency of unhooking of ICLs following 1 h of treatment with 16 μM HN2. (A) NER mutants XPB mutant UV23, XPG mutant UV135, ERCC1 mutant UV96, XPF mutant UV47, and the AA8 wild-type (WT) cell line. (B) Homologous-recombination mutant XRCC2 mutant (irs1) and wild-type (WT) cell line V79. (C) XRCC3 mutant (irs1SF) and wild type (WT) cell line AA8.
The unhooking kinetics of the recombination mutants is shown in Fig. 4B and C. Although the XRCC2 and XRCC3 mutants are highly sensitive to HN2, they displayed unhooking kinetics similar to those of their isogenic parents, indicating a lack of correlation between their hypersensitivity and the ability to unhook cross-links. Attempts to measure unhooking kinetics in the xrs5 line were unsuccessful because a high level of background DSBs was observed, which obscured the results of this assay.
Evidence for the occurrence of DSBs following treatment with HN2.
It has been clearly demonstrated that in S. cerevisiae, the repair of ICLs, including those induced by HN2, involves the formation of DSBs (28, 40, 42). However the formation of DSB intermediates during the repair of cross-links in mammalian systems has not been systematically explored. To address this, exponentially growing and confluent cell cultures were treated with a range of doses of HN2 from 0 to 32 μM for 1 h, and DSB formation was assessed using PFGE.
As shown in Fig. 5A, DSBs were formed following 1 h of treatment with HN2 in a dose dependent manner. In exponentially growing cells, release of low-molecular-weight DNA from the high-molecular-weight DNA plugs as a result of DSBs was evident at doses as low as 4 μM. DSB induction in nondividing cells was significantly less efficient than in dividing cells, as has been shown previously for S. cerevisiae (42). Semiquantitative analysis of the percentage of DNA released into the gel from the plugs (Fig. 5B) showed that following treatment with 16 μM HN2 for 1 h, approximately 80% of DNA is released from dividing cell plugs, compared to only 10% in nondividing cell plugs. In order to rule out that DSBs were the result of a DNA-degrading activity of HN2, genomic DNA embedded in agarose plugs was treated with the same doses of HN2 (0 to 32 μM) and analyzed by PFGE. No DSBs were induced, even at much higher effective doses, when cellular DNA was exposed to HN2 (data not shown), indicating that these breaks are a result of cellular activities during the processing of HN2-induced DNA damage. To investigate whether these DSBs arise due to the processing of ICLs and are not due to the processing of monoadducts, induction of DSBs by HN2 and HN1 was compared. HN1 is a monofunctional alkylating agent, which can form N-alkylpurine monoadducts but not ICLs. Comparison of the alkylation potential of HN2 and HN1 using the 4-(4′-nitrobenzyl)pyridine assay (51) indicated that at equimolar doses, both agents alkylate DNA to a similar extent (data not shown). As shown in Fig. 5C, in exponentially growing cells no DSBs were observed following treatment with an equimolar dose of HN1 (16 μM). Even at a 100 μM dose, HN1 failed to induce DSBs (data not shown). These observations indicate that induction of DSBs requires the presence of ICLs.
FIG. 5.

(A) Induction of DSBs in dividing and nondividing parental V79 cells determined by PFGE. Cells were treated with 0, 4, 16, or 32 μM HN2 for 1 h, embedded in agarose plugs as described in Materials and Methods, and run on PFGE gels. Release of DNA from plugs indicates the presence of DNA DSBs. (B) Semiquantitative analysis of the percentage of DNA released from plugs following HN2 treatment. (C) Comparison of DSB induction following HN1 and HN2 treatment of dividing V79 cells with 0 and 16 μM for 1 h.
Homologous recombination repairs DSBs resulting from HN2 exposure in mammalian cells.
To examine the pathways involved in the repair of DSBs resulting from HN2 exposure, DSB repair was followed in both homologous recombination- and NHEJ-defective cell lines. The homologous-recombination mutants XRCC2 and XRCC3 were severely impaired in the repair of these DSBs (Fig. 6A). Again, treatment of cells with HN1 indicated that this agent fails to induce any DSBs during the 24-h repair period, demonstrating that monoadducts do not induce DSBs with delayed kinetics (data not shown). Semiquantitative analysis of the gels shown in Fig. 6A (Fig. 6B) indicated that within 24 h, complete DSB repair had occurred for the parent cell lines. In contrast, with the XRCC2 and XRCC3 mutants, more than 60% of DNA was still released from the plugs after 24 h, indicating that little repair of DSBs had occurred. These results imply a direct relationship between the extreme sensitivities of XRCC2 and XRCC3 mutants to HN2 and inability to repair DSBs resulting from HN2 exposure. Interestingly, the CHO-K1 parent cell line consistently repaired DSBs more rapidly than the AA8 and V79 parent cell lines (Fig. 6A and B and 7A and B). The XRCC5 mutant, defective in the NHEJ pathway, showed DSB repair kinetics indistinguishable from that of its isogenic parent cell line CHO-K1 (Fig. 6A and B). Consistent with sensitivity data, these results suggest that HNEJ is not involved in the repair of DSBs resulting from HN2 exposure.
FIG. 6.

(A) Induction and repair of DSBs in V79 wild-type (WT) cell line and XRCC mutants XRCC2 (irs1), XRCC3 (irs1SF), CHO-K1 parent cell line, and XRCC5 (xrs5). Exponentially growing cells were treated with 16 μM HN2 for 1 h and subsequently allowed to repair in fresh medium for 4, 8, or 24 h. Control cells (C) were treated with drug-free medium. Samples were then analyzed by PFGE. (B) Semiquantitative analysis of the percentage of DNA released from plugs in the gel shown in panel A.
NER-independent origin and repair of DSBs resulting from HN2.
It is clear from Fig. 1, and from other studies (4, 11, 28, 40, 42), that XPF and ERCC1 are involved in the processing of ICLs. Therefore, to investigate whether NER activities are required for the induction of ICL-associated DSBs, the formation of DSBs in mutant cells was investigated. Surprisingly, as shown in Fig. 7A and B, all mutants tested, including XPF and ERCC1, induced DSBs indistinguishably from their AA8 parent. These results indicate that the DSBs are not a result of incision activities associated with NER. It is also evident from Fig. 7 that the repair of DSBs in ERCC1 and XPF is normal. This suggests that the extreme sensitivity of these mutants is entirely the result of a defect in the unhooking step of ICL repair and not a dual defect in excision repair and recombination, as has been suggested (47, 55, 59).
FIG. 7.

(A) Induction and repair of DSBs in AA8 wild-type (WT) cell line and NER mutants XPF (UV47), XPG (UV135), XPB (UV23), and ERCC1 (UV96). Exponentially growing cells were treated with 16 μM HN2 for 1 h and subsequently allowed to repair in fresh medium for 4, 8, or 24 h. Control cells (C) were treated with drug-free medium. Samples were then analyzed by PFGE. (B) Semiquantitative analysis of the percentage of DNA released from plugs from the gel shown in panel A.
DISCUSSION
The precise mechanism by which mammalian cells eliminate ICLs remains largely unknown, but it is thought that, as in S. cerevisiae and E. coli, both NER and recombination are involved. The results presented here provide evidence that XPF and ERCC1 act in the incision step and XRCC2 and XRCC3 act in a DSB repair recombination step during the elimination of HN2-induced ICLs but that ERCC1 and XPF are not involved in the recombination arm of repair, as has been suggested previously (47, 55, 59).
Unhooking step of ICL repair.
In agreement with previous reports (1, 15, 26), we confirm that CHO cells defective in XPF and ERCC1 are highly sensitive to HN2 but found that the XPB, XPD, and XPG mutants are only slightly sensitive. Consistent with their extreme sensitivities, XPF and ERCC1 cells were defective in the unhooking of HN2-induced ICLs. In addition to ICLs, HN2 induces much more abundant but less toxic monoadducts. The slight sensitivity of XPB, XPD, and XPG to HN2 seems likely to be due to their involvement in the elimination of these lesions, since they are clearly proficient in the initiation of cross-link excision. Indeed, a requirement for these components in the excision of monoadducts produced by the closely related nitrogen mustard melphalan has recently been reported (24). As expected, the XRCC mutants, including the highly sensitive XRCC2 and XRCC3 lines, had normal unhooking kinetics. Therefore, of the components examined, XPF and ERCC1 are the only mammalian NER factors required to produce the incisions that release an ICL.
Bessho et al. (4) have reported that during the repair of psoralen ICLs by CHO cell extracts, the NER system excises 22- to 28-nucleotide oligomers from the 5′ side of the cross-link. If such an activity is responsible for removal of cross-links in vivo, then the XPB, XPD, and XPG mutants would be expected to show hypersensitivity to cross-linking agents and an inability to unhook cross-links. Our results do not favor such an incision activity in the repair of ICLs. The same workers recently demonstrated that the gap generated by removal of this oligomer is filled by a futile DNA synthesis reaction (43). In 90% of the cases, this repair patch terminates at a nick adjacent to the cross-link, and in 10% of the cases ligation occurs. In both cases the cross-link remains. They also demonstrated that in the presence of replication protein A (RPA), the XPF-ERCC1 heterodimer could act as a 3′ to -5′ exonuclease on cross-linked DNA with high processivity. This exonucleolytic digestion either terminates immediately past the cross-link or can continue to the 5′ terminus of the linear duplex substrates, completely removing one strand (43). This is consistent with our results, since it does not require other factors involved in NER apart from ERCC1 or XPF, and it results in the unhooking of ICLs in intact cells.
Presence of a DSB intermediate in the processing of an ICL.
Previous studies have clearly demonstrated that DSBs are generated at both psoralen and HN2 ICLs in dividing yeast cells (28, 40, 42). We now provide evidence for the dose-dependent induction of DSBs following HN2 treatment of mammalian cells. DSBs were not induced following treatment of cells with the monofunctional agent HN1, indicating that ICLs are required for the induction of DSBs. The ICL-associated DSBs were repaired rapidly, with the majority disappearing by 8 h and complete repair at 24 h. These DSBs arise as a result of cellular activity and are not due to DNA degradation by HN2, since DSBs were not observed when genomic DNA was treated. We have also observed the induction of DSBs following treatment of cells with other clinically used nitrogen mustard derivatives such as melphalan (data not shown). Therefore, DSBs are probably a common intermediate following treatment of dividing cells with the nitrogen mustard family of cross-linking anticancer drugs. Results obtained with yeast cells indicate that a necessary role for recombination might be restricted to S phase, since nondividing haploid yeast cells do not lose significant viability even when homologous recombination is completely disabled (in rad52 disruptants) (42). In these quiescent cells, the activity of polymerase ζ (REV3 gene) appears to be important for processing ICL intermediates. Therefore, we suggest that DSBs are not an obligate intermediate in the repair of ICLs, but are nevertheless inevitably generated during the in vivo replication of cross-linked DNA.
A proposed origin for these DSBs is via incision by NER components or as a result of the initiation of recombination. However, the normal induction of DSBs in all the repair defective mutants examined makes either of these possibilities unlikely. Furthermore, the maximum yield of DSBs was detected immediately after drug treatment, when very little ICL repair would have occurred. Our observation that the induction of DSBs following HN2 treatment is more efficient in dividing cells implies that a likely origin of these breaks is the processing of arrested replication forks. This pathway is currently poorly understood in eukaryotes (13, 21), although these events are well described for E. coli (13). Finally, there are reports of a novel human chromatin-associated DNA endonuclease complex which is involved in the repair of DNA ICLs (33, 34, 35, 36). Although the contribution of such an activity to the repair of ICLs in whole cells is not known, it is conceivable that these factors may play a part in the origin of the DSBs observed following HN2 treatment.
Repair of the DSBs resulting from HN2 treatment.
Homologous recombination is the predominant mechanism by which ICL-associated DSBs are repaired in S. cerevisiae. Both the RAD52 and RAD54 genes are essential for this, as is the activity of Mre11 (42). A rad51 disruptant of this organism displayed only a partial defect in ICL-associated DSB repair, and the contribution of NHEJ is very minor and can only be detected when RAD52 is disrupted (42). In mammalian cells, both the NHEJ and homologous recombination pathways play a role in the repair of DSBs. The contribution of the former is well established (reviewed in reference 44), but the importance of the latter is just emerging, and it appears that the pathway employed depends on the nature of the DSB substrate. DSBs resulting from ionizing radiation treatment are primarily repaired by NHEJ (44), whereas those resulting from I-SceI endonuclease cleavage are repaired by XRCC2- and XRCC3-dependent homologous recombination (30, 45).
The identification of mammalian cell lines sensitive to ionizing radiation has facilitated the isolation of genes involved in mammalian DSB repair. Of nine XRCC complementation groups, the cell lines in the XRCC4 to XRCC7 groups are defective in a DNA ligase IV-interacting protein, Ku86, Ku70, and DNA-Pkcat, respectively, components of the NHEJ apparatus (44, 46). These are all highly sensitive to ionizing radiation and are all defective in the repair of ionizing radiation-induced DSBs (29; reviewed in references 44 and 46). In agreement with previous reports (22, 31), the results of this study confirm that the XRCC2 and XRCC3 mutants are highly sensitive to HN2 but the NHEJ mutant XRCC5 is only slightly sensitive. Consistent with these observations, the XRCC2 and XRCC3 mutants showed impaired repair of DSBs resulting from HN2 exposure, but the XRCC5 mutant repaired these DSBs efficiently. These results confirm that in mammalian cells, the particular pathway responsible for the processing of DSBs depends strongly on the nature of the break. The XRCC2 and XRCC3 mutants did not show a complete defect in DSB repair, suggesting that there is redundancy in the repair of different types of DSBs. In this respect, the roles played by XRCC2 and XRCC3 in homologous recombination are currently only partly understood. Both XRCC2 and XRCC3 show limited homology to human Rad51 (39). They are both involved in maintaining chromosome stability during cell division (14), and it has been shown that mammalian Rad51 induces the formation of subnuclear foci in response to DNA damage by ionizing radiation, UV irradiation, methylmethanesulfate, and cisplatin (6, 25). Interestingly, in the absence of XRCC3, Rad51 foci are not formed, suggesting that XRCC3 plays a role in stabilization of Rad51 and these foci during DNA repair (6), and two-hybrid studies have also shown that human Rad51 protein and XRCC3 protein interact directly (39). Collectively these findings suggest that XRCC2 and XRCC3 may act as accessory factors for Rad51 in homologous recombination.
The presence of several Rad51-like genes in mammals (reviewed in reference 54) suggests the presence of subpathways of recombination which involve different sets of protein factors. Human and mouse Rad52 show only 30% identity with the S. cerevisiae factor, and studies in mouse and chicken cells have shown that the phenotype of RAD52 deficient vertebrate cells clearly differs from that of S. cerevisiae (reviewed in reference 56). Human and mouse Rad54 are about 50% identical to S. cerevisiae Rad54, and their function is conserved in mammalian cells (reviewed in reference 56). In comparison to XRCC2 and XRCC3 mutants, rad54−/− mouse cells show only a small degree of sensitivity to mitomycin C and no UV sensitivity (18). This again suggests the existence of subpathways of mammalian homologous recombination, and different proteins may contribute differentially to the repair of various types of DNA lesions, including cross-links.
Model for cross-link repair in mammalian cells.
Combining the results presented in this study and those of several other significant recent papers (4, 23, 37, 38, 41, 42, 43) leads us to propose a model for the repair of ICLs in mammalian cells. Any model must account for the following observations: (i) DSBs are induced in response to ICLs, but this is dependent on factors mainly found in dividing cells; (ii) homologous recombination, but not NHEJ, is required for the repair of these DSBs; (iii) the novel observation of Mu et al. that only XPF and ERCC1 are required for ICL unhooking, where the XPF-ERCC1 heterodimer (in the presence of RPA) acts as a 3′-to-5′ exonuclease able to digest DNA past a cross-link (43); (iv) XPF and ERCC1 are not required for the repair of ICL-associated DSBs; and (v) the strand exchanges stimulated by ICLs require XPF, ERCC1, XRCC2, XRCC3, and RPA to proceed efficiently (38). A proposed model incorporating DSB formation and ICL repair is outlined in Fig. 8. In dividing cells, replication forks encountering ICLs are inactivated, and a DSB is generated by an unknown mechanism. The DSB initiates homologous recombination, which involves strand invasion mediated by XRCC2 and XRCC3; this is an early event which may precede the ERCC1-XPF excision event. Migration may be stalled at the site of a cross-link, since it may require cross-link excision to proceed further. Alternatively, it may extend past the cross-link, which has been demonstrated for RuvAB branch migration past a site-specific psoralen cross-link in E. coli (23). The XPF and ERCC1 heterodimer unhooks the cross-link via its 3′-to-5′ exonucleolytic activity (43). The gap generated as a result of XPF-ERCC1-RPA digestion is filled using the invading strand as the template. The incised cross-link moiety is subsequently removed in a second XPF-ERCC1 excision event, and the recombination intermediates are resolved. We concur with Li et al. (38) that a strong candidate for the recombinational event is break-induced replication (BIR), for the following reasons. First, using a mammalian cell-free assay to measure DNA synthesis induced by the presence of a single psoralen ICL, these workers demonstrated that homology-independent DNA synthesis (BIR does not require significant homology) occurs on both the damaged plasmid and a second undamaged plasmid in response to cross-links (38). The XPF, ERCC1, XRCC2, XRCC3, RPA, and PCNA but not XPA, XPC, and XPG gene products were required for cross-link induced DNA incorporation (37, 38). Second, BIR is largely Rad51 independent in S. cerevisiae, and ICL-associated DSB repair does not require Rad51 in either this yeast or mammalian cell extracts (38, 41, 42).
FIG. 8.

Proposed model for the repair of ICLs in mammalian cells. In dividing cells, the arrest of replication forks near the site of cross-links generates DSBs. These DSBs initiate XRCC2- and XRCC3-dependent recombination. The XPF-ERCC1 heterodimer unhooks the cross-links through its 3′-to-5′ exonuclease activity in the presence of RPA. The gap generated is then filled, using the invading strand as the template. Second incision activity by the XPF-ERCC1 heterodimer then completely removes the unhooked cross link adduct. The gap generated is then filled using newly synthesized complementary strand as the template.
The origins of cross-link-induced DSBs are currently not known and require further investigation. It appears likely that in both yeast and mammalian cells they are the product of activities which act to process a stalled replication fork. This is an area of intense interest (13, 21), and although the biochemical details of the processes involved are emerging in E. coli, very little is known about these events in eukaryotes. A detailed understanding of molecular events at stalled replication forks will be required to develop a more detailed picture of the cellular processes acting to eliminate cross-links in higher organisms.
ACKNOWLEDGMENTS
We thank M. Stefanini for providing AA8, UV23, UV42, UV47, UV61, and UV96 and J. Thacker for providing V79, CHO-K1, irs1, irs1SF, and xrs5 cell lines.
This work was supported by the Cancer Research Campaign Programme grant SP2000/0402 and with a Ph.D. studentship to I. U. De Silva from the Clinical Research and Development Committee, University College London.
REFERENCES
- 1.Andersson B S, Sadeghi T, Siciliano M J, Legarski R, Murray D. Nucleotide excision repair genes as determinants of cellular sensitivity to cyclophosphamide analogs. Cancer Chemother Pharmacol. 1996;38:406–416. doi: 10.1007/s002800050504. [DOI] [PubMed] [Google Scholar]
- 2.Bailly V, Sommers C H, Sung P, Prakash L, Prakash S. Specific complex formation between proteins encoded by the yeast DNA repair and recombination genes RAD1 and RAD10. Proc Natl Acad Sci USA. 1992;89:8273–8277. doi: 10.1073/pnas.89.17.8273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bardwell L, Cooper A J, Freidberg E C. Stable and specific association between the yeast recombination and DNA repair proteins Rad1 and Rad10 in vitro. Mol Cell Biol. 1992;12:3041–3049. doi: 10.1128/mcb.12.7.3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bessho T, Mu D, Sancar A. Initiation of DNA interstrand cross-link repair in humans: the nucleotide excision repair system makes dual incisions 5′ to the cross-linked base and removes a 22- to 28-nucleotide-long damage-free strand. Mol Cell Biol. 1997;17:6822–6830. doi: 10.1128/mcb.17.12.6822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bessho T, Sancar A, Thompson L H, Thelen M P. Reconstitution of human excision nuclease with recombinant XPF-ERCC1 complex. J Biol Chem. 1997;272:3833–3837. doi: 10.1074/jbc.272.6.3833. [DOI] [PubMed] [Google Scholar]
- 6.Bishop D K, Ear U, Bhattacharyya A, Calderone C, Beckett M, Weichselbaum R R, Shinohara A. Xrcc3 is required for assembly of Rad51 complexes in vivo. J Biol Chem. 1998;273:21482–21488. doi: 10.1074/jbc.273.34.21482. [DOI] [PubMed] [Google Scholar]
- 7.Brookman K W, Lamerdin J E, Thelen M P, Hwang M, Reardon J T, Sancar A, Zhou Z Q, Walter C A, Parris C N, Thompson L H. ERCC4 (XPF) encodes a human nucleotide excision repair protein with eukaryotic recombination homologues. Mol Cell Biol. 1996;16:6553–6562. doi: 10.1128/mcb.16.11.6553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caldecott K, Jeggo P. Cross-sensitivity of γ-ray-sensitive hamster mutants to cross-linking agents. Mutat Res. 1991;255:111–121. doi: 10.1016/0921-8777(91)90046-r. [DOI] [PubMed] [Google Scholar]
- 9.Cheong N, Wang Y, Jackson M, Iliakis G. Radiation-sensitive irs mutants rejoin DNA double-strand breaks with efficiency similar to that of parental V79 cells but show altered response to radiation-induced G2 delay. Mutat Res. 1992;274:111–122. doi: 10.1016/0921-8777(92)90058-b. [DOI] [PubMed] [Google Scholar]
- 10.Cole R S, Sinden R R. Repair of cross-linked DNA in Escherichia coli. Basic Life Sci. 1975;5B:487–495. doi: 10.1007/978-1-4684-2898-8_10. [DOI] [PubMed] [Google Scholar]
- 11.Cole R S. Repair of DNA containing interstrand cross-links in Escherchia coli: sequential excision and recombination. Proc Natl Acad Sci USA. 1973;70:1064–1068. doi: 10.1073/pnas.70.4.1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collins A R. Mutant rodent cell lines sensitive to ultraviolet light, ionizing radiation, and cross-linking agents: a comprehensive survey of genetic and biochemical characteristics. Mutat Res. 1993;293:99–118. doi: 10.1016/0921-8777(93)90062-l. [DOI] [PubMed] [Google Scholar]
- 13.Cox M M, Goodman M F, Kreuzer K N, Sherratt D J, Sandler S J, Marians K J. The importance of repairing stalled replication forks. Nature. 2000;404:37–41. doi: 10.1038/35003501. [DOI] [PubMed] [Google Scholar]
- 14.Cui X, Brenneman M, Meyne J, Oshimura M, Goodwin E H, Chen D J. The XRCC2 and XRCC3 repair genes are required for chromosome stability in mammalian cells. Mutat Res. 1999;434:75–88. doi: 10.1016/s0921-8777(99)00010-5. [DOI] [PubMed] [Google Scholar]
- 15.Damia G, Imperatori L, Stefanini M, D'Incalci M. Sensitivity of CHO mutant cell lines with specific defects in nucleotide excision repair to different anti-cancer agents. Int J Cancer. 1996;66:779–783. doi: 10.1002/(SICI)1097-0215(19960611)66:6<779::AID-IJC12>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 16.Dardalhon M, Averbeck D. Pulse-field gel electrophoresis analysis of the repair of psoralen plus UVA induced DNA photoproducts in Saccharomyces cerevisiae. Mutat Res. 1995;336:49–60. doi: 10.1016/0921-8777(94)00037-7. [DOI] [PubMed] [Google Scholar]
- 17.Davies A A, Friedberg E C, Tomkinson A E, Wood R D, West S C. Role of the Rad1 and Rad10 proteins in nucleotide excision repair and recombination. J Biol Chem. 1995;270:24638–24641. doi: 10.1074/jbc.270.42.24638. [DOI] [PubMed] [Google Scholar]
- 18.Essers J, Hendriks R W, Swagemakers S M, Troelstra C, de Wit J, Bootsma D, Hoeijmakers J H, Kanaar R. Disruption of mouse RAD54 reduces ionizing radiation resistance and homologous recombination. Cell. 1997;89:187–193. doi: 10.1016/s0092-8674(00)80199-3. [DOI] [PubMed] [Google Scholar]
- 19.Fishman-Lobell J, Haber J E. Removal of nonhomologous DNA ends in double-strand break recombination: the role of the yeast ultraviolet repair gene RAD1. Science. 1992;258:480–484. doi: 10.1126/science.1411547. [DOI] [PubMed] [Google Scholar]
- 20.Fishman-Lobell J, Rudin N, Haber J E. Two alternative pathways of double strand break repair that are kinetically separable and independently modulated. Mol Cell Biol. 1992;12:1292–1303. doi: 10.1128/mcb.12.3.1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flores-Rozas H, Kolodner R D. Links between replication, recombination and genome instability in eukaryotes. Trends Biochem Sci. 2000;25:196–200. doi: 10.1016/s0968-0004(00)01568-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fuller L F, Painter R B. A Chinese hamster ovary cell line hypersensitive to ionizing radiation and deficient in repair replication. Mutat Res. 1988;193:109–121. doi: 10.1016/0167-8817(88)90041-7. [DOI] [PubMed] [Google Scholar]
- 23.George H, Kuraoka I, Nauman D A, Kobertz W R, Wood R D, West S C. RuvAB-mediated branch migration does not involve extensive DNA opening within the RuvB hexamer. Curr Biol. 2000;10:103–106. doi: 10.1016/s0960-9822(00)00296-7. [DOI] [PubMed] [Google Scholar]
- 24.Grant D F, Bessho T, Reardon J T. Nucleotide excision repair of melphalan monoadducts. Cancer Res. 1998;58:5196–5200. [PubMed] [Google Scholar]
- 25.Haaf T, Golub E I, Reddy G, Radding C M, Ward D C. Nuclear foci of mammalian Rad51 recombination protein in somatic cells after DNA damage and its localization in synaptonemal complexes. Proc Natl Acad Sci USA. 1995;92:2298–2302. doi: 10.1073/pnas.92.6.2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoy C A, Thompson L H, Mooney C L, Salazar E P. Defective DNA cross-link removal in Chinese hamster cell mutants hypersensitive to bifunctional alkylating agents. Cancer Res. 1985;45:1737–1743. [PubMed] [Google Scholar]
- 27.Ivanov E L, Harber J E. RAD1 and RAD10 but not other excision repair genes are required for double-strand break-induced recombination in Saccharomyces cerevisiae. Mol Cell Biol. 1995;15:2245–2251. doi: 10.1128/mcb.15.4.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jachymczyk W J, von Borstel R C, Mowat M R, Hastings P J. Repair of interstrand cross-links in DNA of Saccharomyces cerevisiae requires two systems for DNA repair: the RAD3 system and the RAD51 system. Mol Gen Genet. 1981;182:196–205. doi: 10.1007/BF00269658. [DOI] [PubMed] [Google Scholar]
- 29.Jeggo P A, Kemp L M. X-ray-sensitive mutants of Chinese hamster ovary cell line isolation and cross-sensitivity to other DNA-damaging agents. Mutat Res. 1983;112:313–327. doi: 10.1016/0167-8817(83)90026-3. [DOI] [PubMed] [Google Scholar]
- 30.Johnson R D, Liu N, Jasin M. Mammalian XRCC2 promotes the repair of DNA double strand breaks by homologous recombination. Nature. 1999;401:397–399. doi: 10.1038/43932. [DOI] [PubMed] [Google Scholar]
- 31.Jones N J, Cox R, Thacker J. Isolation and cross-sensitivity of X-ray sensitive mutants of V79-4 hamster cells. Mutat Res. 1987;183:279–86. doi: 10.1016/0167-8817(87)90011-3. [DOI] [PubMed] [Google Scholar]
- 32.Jones N J, Stewart S A, Thompson L H. Biochemical and genetic analysis of the Chinese hamster mutants irs1 and irs2 and their comparison to cultured ataxia telangiectasia cells. Mutagenesis. 1990;5:15–23. doi: 10.1093/mutage/5.1.15. [DOI] [PubMed] [Google Scholar]
- 33.Lambert M W, Lambert W C. DNA repair and chromatin structure in genetic diseases. Prog Nucleic Acid Res Mol Biol. 1999;63:257–310. doi: 10.1016/s0079-6603(08)60725-4. [DOI] [PubMed] [Google Scholar]
- 34.Lambert M W, Fenkart D, Clarke M. Two DNA endonuclease activities from normal human and xeroderma pigmentosum chromatin active on psoralen plus ultraviolet light treated DNA. Mutat Res. 1988;193:65–73. doi: 10.1016/0167-8817(88)90008-9. [DOI] [PubMed] [Google Scholar]
- 35.Lambert M W, Tsongalis G J, Lambert W C, Hang B, Parrish D D. Correction of the DNA repair defect in Fanconi anemia complementation groups A and D cells. Biochem Biophys Res Commun. 1997;230:587–591. doi: 10.1006/bbrc.1996.6008. [DOI] [PubMed] [Google Scholar]
- 36.Lambert M W, Tsongalis G J, Lambert W C, Hang B, Parrish D D. Defective DNA endonuclease activities in Fanconi anemia cells, complementation groups A and B. Mutat Res. 1992;273:57–71. doi: 10.1016/0921-8777(92)90050-d. [DOI] [PubMed] [Google Scholar]
- 37.Li L, Peterson C A, Zhang X, Legerski R J. Requirement for PCNA and RPA in interstrand crosslink-induced DNA synthesis. Nucleic Acids Res. 2000;28:1424–1427. doi: 10.1093/nar/28.6.1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li L, Peterson C A, Lu X, Wei P, Legerski R J. Interstrand cross-links induce DNA synthesis in damaged and undamaged plasmids in mammalian cell extracts. Mol Cell Biol. 1999;19:5619–5630. doi: 10.1128/mcb.19.8.5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu N, Lamerdin J E, Tebbs R S, Schild D, Tucker J D, Shen M R, Brookman K W, Siciliano M J, Walter C A, Fan W, Narayana L S, Zhou Z Q, Adamson A W, Sorensen K J, Chen D J, Jones N J, Thompson L H. XRCC2 and XRCC3, new human Rad51 family members, promote chromosome stability and protect against DNA cross-links and other damages. Mol Cell. 1998;1:783–793. doi: 10.1016/s1097-2765(00)80078-7. [DOI] [PubMed] [Google Scholar]
- 40.Magana-Schwencke N, Henriques J A, Chanet R, Moustacchi E. The fate of 8-methoxypsoralen photoinduced crosslinks in nuclear and mitochondrial yeast DNA: comparison of wild type and repair deficient strains. Proc Natl Acad Sci USA. 1982;79:1722–1726. doi: 10.1073/pnas.79.6.1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malkova A, Ivanov E L, Haber J E. Double-strand break repair in the absence of RAD51 in yeast: a possible role for break-induced replication. Proc Natl Acad Sci USA. 1996;93:7131–7136. doi: 10.1073/pnas.93.14.7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McHugh P J, Sones W R, Hartley J A. Repair of intermediate structures produced at DNA interstrand cross-links in Saccharomyces cerevisiae. Mol Cell Biol. 2000;20:3425–3433. doi: 10.1128/mcb.20.10.3425-3433.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mu D, Bessho T, Nechev L V, Chen D J, Harris T M, Hearst J E, Sancar A. DNA interstrand cross-links induce futile repair synthesis in mammalian cell extracts. Mol Cell Biol. 2000;20:2446–2454. doi: 10.1128/mcb.20.7.2446-2454.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pastink A, Lohman P H M. Repair consequences of double-strand breaks in DNA. Mutat Res. 1999;428:141–156. doi: 10.1016/s1383-5742(99)00042-3. [DOI] [PubMed] [Google Scholar]
- 45.Pierce A J, Johnson R D, Thompson L H, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rathmell W K, Chu G. Mechanisms for DNA double-strand break repair in eukaryotes. In: Nickoloff J A, Hoekstra M F, editors. DNA damage and repair. 2. DNA repair in higher eukaryotes. Totowa, N.J: Humana Press; 1998. pp. 299–315. [Google Scholar]
- 47.Sargent R G, Rolig R L, Kilburn A E, Adair G M, Wilson J H, Nairn R S. Recombination-dependent deletion formation in mammalian cells deficient in the nucleotide excision repair gene ERCC1. Proc Natl Acad Sci USA. 1997;94:13122–13127. doi: 10.1073/pnas.94.24.13122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sijbers A M, de Laat W J, Ariza R R, Biggerstaff M, Wei Y F, Moggs J G, Carter K C, Shell B K, Evans E, de Jong M C, Rademakers S, de Rooij J, Jaspers N G, Hoeijmakers J H, Wood R D. Xerodoma pigmentosum group F caused by a defect in a structure-specific DNA repair endonuclease. Cell. 1996;86:811–822. doi: 10.1016/s0092-8674(00)80155-5. [DOI] [PubMed] [Google Scholar]
- 49.Skehan P, Storung R, Scudiero R, Monks A, McMahon J, Vistica D, Warren J T, Bokesch H, Kenney S, Boyd M R. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 50.Spanswick V J, Hartley J M, Ward T H, Hartley J A. Measurement of drug-induced DNA interstrand crosslinking using the single-cell gel electrophoresis (Comet) assay. In: Brown R, Boger-Brown U, editors. Methods in molecular medicine. 28: cytotoxic drug resistance mechanisms. Totowa, N.J: Humana Press; 1999. pp. 143–154. [DOI] [PubMed] [Google Scholar]
- 51.Spears C P. Nucleophilic selectivity ratios of model and clinical alkylating agents by 4-(4′-nitrobenzyl) pyridine competition. Mol Pharmacol. 1981;19:496–504. [PubMed] [Google Scholar]
- 52.Sugawara N, Haber J E. Characterization of double-strand break-induced recombination: homology requirements and single-stranded DNA formation. Mol Cell Biol. 1992;12:563–575. doi: 10.1128/mcb.12.2.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thacker J, Ganesh A N. DNA-break repair, radioresistance of DNA synthesis, and campothecin sensitivity in the radiation-sensitive irs mutants: comparison to ataxia-telangiectasia cells. Mutat Res. 1990;235:49–58. doi: 10.1016/0921-8777(90)90057-c. [DOI] [PubMed] [Google Scholar]
- 54.Thacker J. The role of homologous recombination processes in the repair of severe forms of DNA damage in mammalian cells. Biochemie. 1999;81:77–85. doi: 10.1016/s0300-9084(99)80041-8. [DOI] [PubMed] [Google Scholar]
- 55.Thompson L H. Evidence that mammalian cells possess homologous recombinational repair pathways. Mutat Res. 1996;363:77–88. doi: 10.1016/0921-8777(96)00008-0. [DOI] [PubMed] [Google Scholar]
- 56.Thompson L H, Schild D. The contribution of homologous recombination in preserving genome integrity in mammalian cells. Biochemie. 1999;81:87–105. doi: 10.1016/s0300-9084(99)80042-x. [DOI] [PubMed] [Google Scholar]
- 57.Van Duin M, de Wit J, Odijk H, Westerveld A, Yasui A, Koken H M, Hoeijmakers J H, Bootsma D. Molecular characterization of the human excision repair gene ERCC1: cDNA cloning and amino acid homology with the yeast DNA repair gene RAD10. Cell. 1986;44:913–923. doi: 10.1016/0092-8674(86)90014-0. [DOI] [PubMed] [Google Scholar]
- 58.Van Houten B, Gamper H, Holbrook S R, Hearst J E, Sancar A. Action mechanism of ABC excision nuclease on a DNA substrate containing a psoralen crosslink at a defined position. Proc Natl Acad Sci USA. 1986;83:8077–8081. doi: 10.1073/pnas.83.21.8077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weeda G, Donker I, de Wit J, Morreau H, Janssens R, Vissers C J, Nigg A, van Steeg H, Bootsma D, Hoeijmakers J H J. Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Curr Biol. 1997;7:427–439. doi: 10.1016/s0960-9822(06)00190-4. [DOI] [PubMed] [Google Scholar]