

Abstract
The great success of chimeric antigen receptor T (CAR-T)-cell therapy in B-cell malignancies has significantly promoted its rapid expansion to other targets and indications, including T-cell malignancies and acute myeloid leukemia. However, owing to the life-threatening T-cell hypoplasia caused by CD7-CAR-T cells specific cytotoxic against normal T cells, as well as CAR-T cell-fratricide caused by the shared CD7 antigen on the T-cell surface, the clinical application of CD7 as a potential target for CD7+ malignancies is lagging. Here, we generated CD7ΔT cells using an anti-CD7 nanobody fragment coupled with an endoplasmic reticulum/Golgi retention domain and demonstrated that these cells transduced with CD7-CAR could prevent fratricide and achieve expansion. Additionally, CD7ΔCD7-CAR-T cells exhibited robust antitumor potiential against CD7+ tumors in vitro as well as in cell-line and patient-derived xenograft models of CD7-positive malignancies. Furthermore, we confirmed that the antitumor activity of CD7-CAR-T cells was positively correlated with the antigen density of tumor cells. This strategy adapts well with current clinical-grade CAR-T-cell manufacturing processes and can be rapidly applied for the therapy of patients with CD7+ malignancies.
Keywords: Humanized CD7 nanobody, ER/Golgi CD7 retention, fratricide-resistant CD7 CAR-T, adoptive immunotherapy
Introduction
T-lymphoblastic leukemia (T-ALL) is an aggressive disease that accounts for 25% of adult and 15% of pediatric cancers [1]. Although clinical outcomes have significantly improved for pediatric ALL patients under the intervention of chemotherapy, 20% of these patients and 50% of adults ultimately succumb to this disease [2,3]. Patients diagnosed with T-ALL frequently present with unfavorable symptoms accompanied by a higher risk of relapse. Nelarabine, the only drug approved for the treatment of relapsed and refractory T-ALL (r/r T-ALL), has a complete remission rate of only 20% to 30%. Additionally, the sustainable clinical development of notch-1 inhibitors targeting the commonly mutated variants of T-ALL is limited due to their high incidence of gastrointestinal toxicity and low response rates [4]. Meanwhile, recently, cyclin D3 was shown to play a key role in the induction of T-ALL, thus, prompting a study on cyclin D-cyclin-dependent kinase-4/6 inhibitors, namely abemaciclib and palbociclib, however, these drugs only exhibited moderate activity in preclinical research warranting further clinical investigation [5].
Major clinical responses have been reported following the application of CD19-targeting CAR-T cells for refractory/recurrent B-lineage malignancies [6-13]. Yescarta and Kymriah, two autologous CD19-targeting CAR-T cells, have been approved by the Food and Drug Administration (FDA) [14-16]. Meanwhile, B-lineage malignancies have safer and effective therapeutic targets (targeting CD19, CD20, and CD22), whereas T-ALL demonstrates higher malignant-tumor heterogeneity. Moreover, target selection and safety represent key issues primarily due to their shared target-antigen spectrum between tumor cells and T cells, which not only leads to CAR-T-cell internecine killing [17,18] but also to T-cell underdevelopment and even life-threatening T-cell immunodeficiency.
Currently, the primary targets of T-ALL-related CAR-T-cell research include CD7, CD5, and CD1a. CD5-specific CAR-T-cell therapy has resulted in serious adverse effects due to the upregulation of intercellular adhesion molecules by 4-1BB, which increases the stability of immune synapses [19]. Additionally, CD5-CAR-T cell-based therapy coupled with CD28 exhibit limited tumor clearance due to transient persistence. Moreover, the expression profile of CD5 is limited on T-cell tumors. These findings indicate that CD5 might not be suitable for further clinical applications. CD1a was also restricted due to its cortical T-ALL attribution [20-22]. CD7 is also a primary marker of T-cell malignancies [23,24], although studies indicate that it does not appear to critically contribute to T-cell development or function according to results following CD7 knockout in mice [25,26]. Furthermore, the expression of CD7 remains high according to analysis of human bone marrow samples at each therapeutic stage [18]. Therefore, CD7 might represent a potent target for acute myeloid leukemia (AML) and T-ALL immunotherapy.
In the early stage of this study, we successfully developed an anti-CD7 nanobody (CD7Nb) that was generated by screening a phage-display library obtained from camel B-cells immunized with CD7+ Jurkat cells. Further outcomes from studies employing immunotoxin-loaded CD7Nb and CD7-CAR-NK92MI cells provided a solid foundation for the exploration of CD7-CAR-T cells [27-29]. However, lack of persistent immunogenicity caused by immunotoxins, as well as the presence of allogeneic NK92MI cell characteristics, restrict the clinical application of CD7-CAR-T cells.
Here, we verified five CD7-CAR-T-cell structures that included monovalent and bivalent CD7Nbs produced by either camel B-cells (VHH6) or their humanized structures (HuVHH6) and different costimulatory domains of inducible T-cell co-stimulator (ICOS)/4-1BB, 4-1BB, and CD28. An optimal structure was screened for further CD7-CAR-T research through activation of CD7-CAR-Jurkat cells. CAR-T cells with CD7 blockade, induced via an intracellular retention motif, were fratricide-resistant. Further we investigated the anti-tumor potential of CAR-T cells in vitro and in vivo and explored the key elements affecting the biological activity of CAR-T cells. Therefore, these fratricide-resistant CD7-CAR-T cells address gaps in adoptive immunotherapy for T-ALL/T-cell lymphoblastic lymphoma (T-LBL) and potentially other CD7+ tumors, such as those associated with AML.
Materials and methods
Cell cultures
Acute T-ALL CCRF-CEM and acute T-cell leukemia Jurkat and the human erythroleukemia cell line HEL were maintained in RPMI-1640 medium. All cells were cultured according to standard cell culture conditions. To construct Luc+-GFP+-CCRF-CEM and Luc+-GFP+-HEL cells, a lentiviral vector co-encoding luciferase and green fluorescent protein (GFP) was utilized for transduction. Using flow cytometry (Agilent Technologies, Santa Clara, CA, USA), single cells selected from CCRF-CEM, and HEL-GFP-luciferase-transduced populations were used to generate a cell line. A GFP lentiviral vector co-constructed with CD7 was used for the transduction of HEK293T cells to obtain CD7+-GFP+-293T cells.
Peripheral blood mononuclear cells (PBMCs) derived from healthy volunteers were obtained by density gradient centrifugation and plated with 2×106 cells/mL in 24-well plates in TexMACS and subsequently activated with TransAct (Miltenyi Biotec, Bergisch Gladbach, Germany) containing human recombinant interleukin (IL)-7 (155 IU/mL) and human recombinant IL-15 (290 IU/mL). Two days later, activated T cells were used for the different lentiviral transductions. Primary T-ALL cells were a gift from Dr Xuejun Zhu.
Generation of CD7 blockade and CD7 CAR-T cells
CD7-specific antibody fragments were derived from monovalent and bivalent CD7Nbs produced by either camel B cells (VHH6) or their humanized structures (HuVHH6). The CD7Nbs were coupled with the CD8α signal peptide, while the transmembrane domain, and different costimulatory domains were derived from ICOS/4-1BB, 4-1BB, CD28, and CD3ζ. The bivalent CD7Nbs derived from camel B cells were joined to an endoplasmic reticulum (ER)/Golgi retention-signaling domain for the construction of CD7 blockade variants. All sequences were subcloned into the lentiviral vector. Virus particles were generated in suspension-293T cells (domesticated from 293T cells) and by ultracentrifugation, yielding transducing titers >1×108 units/mL.
Determination of CAR-T-cell specificity, gene expression, and cell-marker profile
CAR+ was detected with the Alexa Fluor 647-labeled goat anti-human Fc antibody (Jackson ImmunoResearch, West Grove, PA, USA) or the anti-truncated epidermal growth factor receptor antibody (iCarTAB) at a dilution of 1:1000. The CD7 blockade T-cell ratio was detected using an allophycocyanin (APC)- or fluorescein isothiocyanate (FITC)-conjugated anti-human CD7 antibody (BD Biosciences, San Jose, CA, USA). CD4 PerCP, CD3 FITC, CD8 APC, CCR7 FITC, CD45RA APC, CD69 APC, CD25 APC, and programmed cell death-1 (PD-1) APC were obtained from BD Biosciences. Cell staining was performed using a NovoCyte flow cytometer (Agilent Technologies).
To confirm that CD7 blockade specifically blocked the expression of CD7 molecules on the membrane surface, Jurkat cells were transduced with CD7 blockade and incubated with purified CD7Nb at different antibody concentrations ranging from extremely low to very high (0.61 ng/μL to 1250 ng/μL) for 1 h, after which the Alexa Flour 647-conjugated anti-human Fc antibody was added. Subsequently, a test for CD7-blockade specificity was performed prior to flow cytometric analysis.
Cell activation, cell cytotoxicity, and cytokine production
Jurkat cells transduced with different structures of CD7-CAR were incubated with the CD7+ cell line CCRF-CEM, which was pre-labeled with carboxyfluorescein succinimidyl ester (CFSE). After 24 h, cells were collected for staining with CD69 APC, CD25 APC, and PD1 APC, respectively.
In a cytotoxicity assay, suspension-cultured target cells labeled with CFSE (10 μM), and effective T cells were plated into a 24-well round-bottomed plate at various effector: target (E:T) ratios. After 24 h, 7-aminoactinomycin D and Annexin V were added to discriminate dead and apoptotic cells by flow cytometry (Multi Sciences Biotech, Hangzhou, China).
For repetitive tumor challenge, primary T-ALL cells and effective T cells were incubated at a 1:5 ratio. Quantitative primary T-ALL cells were added every 48 h, and CD7 expression on primary T-ALL cells and T-cell subtypes were analyzed by flow cytometry every 24 h after the addition of tumor cells.
To assess adherence to target cells and resulting cytotoxicity, target cells (CD7+-GFP+-293T) were plated in a 96-well plate overnight. On day 2, different E: T-cell ratios were added for further dynamic cell-growth monitoring via real-time cell analysis (RTCA).
To assess cytokine production, incubation supernatants of cell-activation and cytotoxicity assays were harvested and quantified using the indicated cytometric bead array kit (CBA; BD Biosciences) of interferon (IFN)-γ/granzyme B/IL-2/tumor necrosis factor (TNF)-α according to product instructions.
In vivo CCRF-CEM, HEL and patient-derived T-ALL xenograft (PDX) models
Six- to eight-week-old B-NDG mice (Biocytogen, Wakefield, MA, USA) were bred and maintained at the animal facility of Soochow University. Luc+-GFP+-CCRF-CEM cells (1×105 cells/mouse) were intravenously (i.v.) injected, followed 2 days later by infusion of CD7-CAR T cells (3×106/mouse, i.v.) or CD7 blockade T cells. Group of mice receiving phosphate-buffered saline (PBS) were used as controls. To analyze levels of blood platelets in mice after CAR-T-cell infusion, 250 μL of blood was collected by orbital bleeding. Before termination of the study, tissues were collected for CD7 immunohistochemical (IHC) staining.
For HEL cell-derived xenograft (CDX) models, 5×105 Luc+-GFP+-HEL cells were i.v. injected, followed 3 days later by infusion of CD7 blockade T or CD7-CAR-T cells (1×107/mouse, i.v.). Control mice received PBS instead of T cells. Body weight and IVIS imaging presented after D-luciferin intraperitoneal (i.p.) injection were monitored at different time points.
For the PDX model, 2×106 primary T-ALL cells were i.v. injected into B-NDG mice and propagated for 21 days (median survival). Following euthanasia, the spleens were harvested, sectioned, ground, filtered, and cultured for 2 days to harvest single T-ALL cells. After 3 days, T-ALL cells were re-injected into B-NDG mice, followed by infusion with CD7-CAR-T or CD7 blockade T cells. ALL mice received IL-2 (50,000 IU) daily for 2 weeks starting from the CAR-T-cell infusion date. Tissues were fixed in 4% paraformaldehyde for IHC analysis. Expansion of CAR in vivo was determined using quantitative polymerase chain reaction (qPCR) analysis.
qPCR
ReliaPrep gDNA miniprep system kit (Promega, Madison, WI, USA) was used for DNA extraction from peripheral blood. The primers used are listed in Table 1. qPCR was then performed using the QuantStudio DX real-time quantitative PCR system (Life Technologies, Carlsbad, CA, USA). DNA concentration was determined using a standard curve, and gene copies were calculated using the following formula: [(copies/μL)/(μg/μL) = copies/μg].
Table 1.
Sequences of primer/probe for CAR expansion
| Primer/Probe | Sequence |
|---|---|
| CD7-CAR-F | 5’-CTCTTCAGGGCGCTGTTGTAGT-3’ |
| CD7-CAR-R | 5’-CCTGCACCTTTAGCGGAGTGT-3’ |
| CD7-CAR-P | 5’-TCGCTGCCCCAAATCACTGCGA-3’ |
IHC staining
IHC assays were performed according to manufacturer instructions. Briefly, tissue sections were incubated at 60°C for 1 h to retrieve antigenicity. After dewaxing, dyeing, and sealing, the slides were scanned using an OLYMPUS CX23. Reagents used for HE and IHC assays included hematein (Beyotime, Beijing, China), anti-human CD7, biotin-labeled secondary antibody (Beyotime), avidin-labeled horseradish peroxidase (Beyotime), and 3,3’-diaminobenzidine (Beyotime).
Statistical analysis
For in vitro studies, data were assessed with GraphPad Prism 6 software (San Diego, CA, USA). The specific analyses performed are presented in the individual figure legends. Significance was calculated by Student’s t test. For three or more group comparisons, Sidak’s/Tukey’s multiple comparison tests were used. For in vivo studies, Mice survival was analyzed by the log-rank (Mantel-Cox) test.
Results
Design and optimize of CD7-CAR
We designed different structures of CD7-CAR containing either (1) monovalent or bivalent CD7Nbs derived from camel or humanized B cells were joined to the costimulatory domains of ICOS and 4-1BB (CD137) and CD3ζ; (2) monovalent or bivalent CD7Nbs derived from camel B cells were linked to the signaling domains of CD28 and CD3ζ (Figure 1A). Lentiviral transduction of these constructs in Jurkat cells caused different degrees of CD69, CD25, and PD-1 activation after incubation with CD7+ CCRF-CEM cells (Figure 1B). In coordination with cytokine secretion (Figure 1C), we screened the appropriate structure (PA3-17) for further analysis.
Figure 1.

Design, optimization, and screening of CD7-CAR derived from the CD7Nbs. A. Schematic of different CD7-CAR structures. B. Ratio of different activation markers on Jurkat cells transduced with different anti-CD7-CARs that were pre-incubated with or without CD7+ CCRF-CEM cells at a 1:1 ratio for 24 h. C. Cytokine secretion assessed by CBA kits. Levels of granzyme B and IL-2 in supernatants.
Abrogation of CD7 expression on T-cell surface prevents CAR-T-cell fratricide
As previously reported [17,18,30], CD7-CAR-T cells suffered fratricide due to CD7 co-expression on normal T cells. To create CD7 blockade we adopted the strategy described by Mamonkin [18] using the ER retention domain KDEL to link bivalent CD7Nbs developed in our laboratory (Figure 2A). CD7Nbs combine with the synthetic CD7 antigen and target the ER/Golgi via the retention motif, which prevents membrane expression of CD7 [31,32]. Results showed that CD7 surface expression on T cells was successfully abrogated (Figure 2B). By expressing CD7-CAR in combination with CD7 retention in the ER, T-cell viability was markedly improved and achieved expansion in vitro (Figure 2C).
Figure 2.
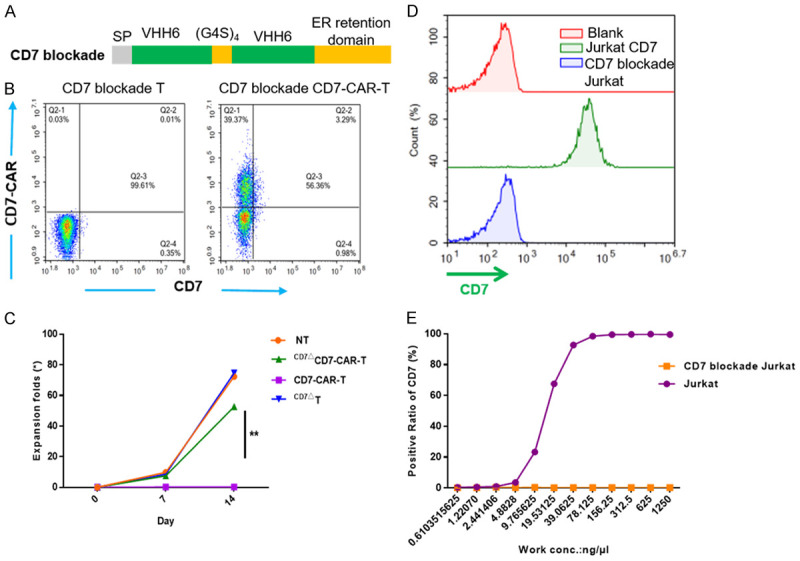
CD7ΔCD7-CAR-T cells are fratricide-resistant after CD7 blockade and CD7 blockade was specific targeting to CD7. A. Schematic of the CD7 blockade structure. B. Data illustrate the ratio of CD7 blockade or CAR+ in activated T cells by flow cytometry. CAR was detected using a goat anti-human IgG Fc antibody. C. Fold expansion of T cells and CD7-CAR-T cells transduced with or without CD7 blockade and measured at different time points by cell counting. Data represent three independent experiments (n=3 donors each). D. Flow cytometric analysis showing the expression of CD7 in blank and CD7 blockade Jurkat cells. E. The standard s-shaped curve shows different binding ratios of blank and CD7 blockade Jurkat cells after incubation with the humanized anti-CD7 nanobody under serially-diluted concentrations for 1 h, followed by incubation with the Alexa Flour 647-labeled anti-human Fc antibody.
To confirm that CD7 blockade specifically blocked CD7, we transduced the CD7 blockade antigen into Jurkat cells (Figure 2D). After culture with CD7Nbs at different concentrations, we found that CD7 blockade Jurkat cells (CD7ΔJurkat) did not bind CD7Nbs, whereas Jurkat cells showed dose-independent binding (Figure 2E).
CD7ΔCD7-CAR-T cells eradicate tumor cells in a CD7-specific and antigen density dependent manner
We then transduced CD7 antigen into HEK-293T cells (Figure 3A). CD7ΔCD7-CAR-T cells exhibited cytolysis against 293T-CD7 cells according to RTCA monitoring, indicating that CD7ΔCD7-CAR-T cells specifically target CD7 (Figure 3B). Additionally, CD7ΔCD7-CAR-T cells were cytolytic to primary T-ALL cells, which had been propagated in B-NDG mice. Furthermore, we found that cytokine secretion was also dependent on E:T ratios (Figure 3C and 3D).
Figure 3.

CD7ΔCD7-CAR-T cells exert specific cytotoxicity to CD7+ cells in an antigen-density-dependent manner in vitro. A. Flow cytometry diagram showing the overexpression of CD7 in HEK-293T cells. GFP was used as the detection marker following its co-expression with the CD7 antigen. B. Normalized cell index of 293T-CD7 cells incubated with or without CD7ΔCD7-CAR-T cells. Data were acquired by RTCA. C. The cytolysis activity of CD7ΔCD7-CAR-T cells was evaluated with primary leukemia cells from patients with T-ALL, non-transduced T (NT), and CD7ΔT cells (each with a different CD7 MFI) at a 1:2, 1:1, 2:1, and 4:1 E:T ratios for a 24 h incubation assay. The mean (±SD) of triplicate experiments are shown. The graph shows statistical analysis of CD7 MFI in primary T-ALL and NT cells. D. Bars represent the mean (±SD) of cytokine secretion which was assessed with a CBA kit by flow cytometry.
In addition to malignant cells, CD7ΔCD7-CAR-T cells might induce T-cell aplasia following infusion due to CD7 expression on normal T cells. Indeed, we observed specific cytotoxicity against mature T cells; however, this effect was significantly weaker than that observed against CD7+ tumor cells. Concurrent with the cytotoxicity, cytokine release was relatively lower relative to that elicited by CD7+ tumor cells (Figure 3C and 3D). As expected, CD7ΔCD7-CAR-T cells showed no cytolysis to CD7 blockade T cells, demonstrating that cytolysis was CD7-specific (Figure 3C and 3D). Compared with the cytotoxicity against mature T cells, we believe CD7ΔCD7-CAR-T cells showed a preference for attacking T-ALL cells, which have a higher CD7 abundance according to mean fluorescence intensity (MFI) (Figure 3C).
CD7ΔCD7-CAR-T cells manufactured from T-ALL patients are strongly cytotoxic against CD7+ tumor cells
To further assess potential clinical applications, we sorted healthy T cells from T-ALL patients using CD4 and CD8 microbeads and cultured them accordingly. Surface expression of CD7 in CCRF-CEM and HEL cells is shown in Figure 4A. The resulting CAR-T cells showed potent biological activity against CCRF-CEM and HEL cells (Figure 4B and 4C), with CCRF-CEM cell cytotoxicity greater than that in HEL cells, which was partially due to the expression and density of the CD7 antigen (Figure 4A). These results demonstrated that CD7Nb-derived CD7-CAR-T cells derived from T-ALL patients showed specific cytotoxicity against CD7+ tumor cells.
Figure 4.

CD7ΔCD7-CAR-T cells derived from primary T-ALL patients show robust cytotoxicity against CD7+ cells. A. Representative flow cytometric analysis show the CD7 expression in HEL and CCRF-CEM cell lines. B. Tumor cells (HEL) were pre-labeled with CFSE and plated in 24-well plates at an E:T ratio of 1:1 with different effector cells for 24 h. The mean ± SD of triplicate experiments are shown. C. Cytolysis of CD7ΔCD7-CAR-T cells against CCRF-CEM cells. ****P<0.0001.
CAR-T cells retain cytotoxic potential after tumor re-challenge
As previously reported, the target antigen CD19 was significantly downregulated following introduction of CD19-CAR-T cells. In our study, we found that after co-incubation of CD7-CAR-T cells with primary T-ALL cells, CD7 expression on primary T-ALL cells was downregulated, in a co-incubation time-dependent manner (Figure 5B). During the process, the proportion of CAR+ remained unaltered (Figure 5C). We then analyzed T-cell subtypes and found that the subtypes in the CAR-T-cell group changed from primary naive/stem memory to effector memory following repeated antigen stimulation (Figure 5D). During tumor re-challenge, CD7+ T-ALL cells remained effectively eliminated (Figure 5E), suggesting the persistence of CAR-T cells. Furthermore, it is possible that during the process of CAR-T cell-mediated cytotoxicity, para-pathways became engaged (e.g., cytokines) resulting in the targeting and destruction of tumor cells.
Figure 5.

CAR-T cells retain cytotoxic potential after tumor re-challenge. A. Schematic of repetitive tumor-challenge assay. Effector cells were co-cultured with primary T-ALL cells (5,000 T cells; 25,000 primary T-ALL cells; E:T=1:5) and re-challenged with 5000 T-ALL cells every other day. B. Statistical analysis of CD7 antigen on primary T-ALL cells at different re-challenge points. C. CAR+ retains similar levels in T cells after several rounds of co-incubation with primary T-ALL cells. D. Frequency of naïve-like (Tscm/naive), effector memory RA+ (TemRA+), effector memory (Tem) and central memory (Tcm) T cells after re-challenge with primary T-ALL cells, ****P<0.0001. E. T-ALL cells were nearly all undergoing apoptosis after tumor re-challenge (n=3 replicates per data point).
CD7ΔCD7-CAR-T cells exert anti-leukemic activity in a CCRF-CEM CDX model
Figure 6A shows the process of determining the infusion dose of GFP+ CCRF-CEM cells in the CDX model. Based on the data, we engrafted 6- to 8-week-old female B-NDG mice i.v. with 1×105 Luc+-GFP+-CCRF-CEM cells. Two days later, 1×106 CD7ΔCD7-CAR-T or CD7ΔT cells were injected in a single i.v. dose (Figure 6C). CD7ΔCD7-CAR-T cells protected against leukemia progression and prolonged mouse survival (Figure 6D). Additionally, as shown in Figure 6B, tumor cells accumulated in lungs, livers, spleens and bone marrow; the number of leukemic cells in these regions decreased dramatically after administration of CD7ΔCD7-CAR-T cells (Figure 7). Moreover, leukemia progressed in mice administered PBS or CD7ΔT cells, resulting in death at 20 or 21 days. We also confirmed that platelet counts in the CAR-T groups remained at the same level as those in healthy mice (Figure 6E).
Figure 6.

CD7ΔCD7-CAR-T cells exert anti-leukemic activity in a CCRF-CEM CDX model. A. B-NDG mice were injected i.v. with 0.2×105, 1×105, or 5×105 Luc+-GFP+-CCRF CEM cells, respectively (n=3), with 1×105 CCRF-CEM cells/mouse used as a control. The median survival of the mice was 19, 20, and 25 days, respectively. B. Flow cytometric analysis of the tissue distribution of tumor cells (CD45+GFP+) in mice. CCRF-CEM cells were mainly distributed in BM, lung, and liver, and a small number of tumor cells infiltrated the spleen and peripheral blood. BM, bone marrow. C. Schematic of the experiment. B-NDG mice were infused i.v. with 3×106 CD7-CAR-T cells or control 2 days after a single i.v. injection of 1×105 Luc+-GFP+-CCRF CEM cells. Mice injected with PBS were used as controls. D. The survival of mice are analyzed by Mantel-Cox log-rank test and shown with Kaplan-Meier curves and **P<0.01. E. Statistical analysis of platelet counts (PLT) in different treatment groups. CONT, healthy mice.
Figure 7.

Representative images of CD7 IHC of lungs, livers, BMs, and spleens from different groups in the T-ALL CDX model. CD7 infiltration was effectively relieved in the CAR-T-cell group. Magnification: 400×. Scar bar: 20 μm. BM, bone marrow; CONT, healthy mice.
CD7ΔCD7-CAR-T cells are protective from tumor progression in an AML CDX model
We then determined the antitumor capacity in the Luc+-GFP+-HEL model in B-NDG mice (Figure 8A). Following i.v. injection of CD7ΔCD7-CAR-T cells at day 3 after tumor engraftment, CD7ΔT cells or PBS were also injected as a control. We observed significantly reduced disease progression in the CAR-T group (Figure 8B). Additionally, we noted that the tumor burden evaluated by IVIS imaging was effectively alleviated after CAR-T cells treatment (Figure 8C and 8D), and that treated mice displayed no substantial weight loss as compared with control mice (Figure 8E).
Figure 8.

CD7 blockade and CD7-CAR-T cells exhibit antitumor activity in an AML (HEL) CDX model. A. Schematic of the experiment. Luc+-GFP+-HEL cells were infused i.v. in 15 B-NDG mice at 5×105 cells/mouse. Cohorts were randomized, and mice received 1×107 CD7-blockade and CAR-T cells, with mice treated with PBS used for the controls. B. The survival of mice are analyzed by Mantel-Cox log-rank test and shown with Kaplan-Meier curves and. **P<0.01. C. AML cell growth evaluated using the IVIS system after CD7-CAR-T-cell infusion. D. Statistic analysis of the burden of Luc+ tumor cells in different groups. E. Body weight was monitored throughout the experiment. NS, not significant.
CD7ΔCD7-CAR-T cells exert an antitumor capacity in T-ALL PDX model
To further consolidate the antitumor effect of CAR-T cells, a T-ALL PDX model was established via propagation of leukemia cells in B-NDG mice (Figure 10A). We further confirmed the immune phenotype of leukemic cells collected from mice after expansion, with surface expression of CD45, CD34, and CD7 and lack of CD4, CD33, CD8, CD3, CD123, and CD276 expression (Figure 9A). Of note, the primary T-ALL cells were injected into the mice for propagation as they are unable to expand ex vivo. In contrast to the distribution of CCRF-CEM in mice, most of the T-ALL cells accumulated in the bone marrow, liver, lung, spleen, and peripheral blood (Figure 9B). Moreover, mice infused with CD7ΔT cells exhibited leukemia progression (median survival: 26 days). By contrast, mice receiving CD7ΔCD7-CAR-T cells exhibited reversed tumor burden (Figure 10B), and CD7-CAR showed specific expansion in the CD7ΔCD7-CAR-T cells group (Figure 10C). Furthermore, tumor cell infiltration was significantly alleviated in CD7ΔCD7-CAR-T cells (Figure 10E), and splenomegaly was effectively relieved (Figure 10D).
Figure 10.

CD7ΔCD7-CAR-T cells exert an antitumor capacity in T-ALL PDX model. A. General outline of the experiment. Primary T-ALL cells, previously propagated in B-NDG mice, were injected i.v. at 2×106 cells/mouse in B-NDG mice. Half of the mice received i.v. injection of CD7ΔT cells, and the remaining half mice received CD7ΔCD7-CAR-T cells (1.2×106), as well as IL-2 (50,000 IU/mouse) administered i.p. daily for 2 weeks. B. The survival of mice are analyzed by Mantel-Cox log-rank test and shown with Kaplan-Meier curves. **P<0.01. C. qPCR of CAR copies in peripheral blood at different time points after treatment. D. Spleens of treated (CD7ΔCD7-CAR-T cells) and untreated (CD7ΔT cells) mice. E. Representative images of CD7 IHC staining in different tissues after CAR-T-cell treatment. Compared with the group receiving CD7ΔT cells, infiltration of CD7 was significantly reduced in the group receiving CAR-T cells. Magnification: 400×. Scar bar: 20 μm. BM, bone marrow.
Figure 9.

Phenotype and distribution of T-ALL cells propagated from B-NDG mice. A. Flow cytometric analysis of primary T-ALL cells to determine the immune phenotype after propagation in B-NDG mice. B. Different from the tissue distribution of CCRF-CEM cells, primary T-ALL cells (CD45+CD7+) dominantly infiltrated peripheral blood, BM, spleen, lung, and liver. BM, bone marrow.
Discussion
Although the success of adoptive cell therapy based on CARs in cancer treatment is inspiring, extending the application scope of CAR-T cells to treat T-ALL/T-LBL is impeded due to the target antigens being shared between CAR-T cells and T-lineage tumor cells [18,33-36]. Accordingly, two major obstacles need to be overcome to allow for further therapeutic application of CAR-T cells for T-ALL/T-LBL: 1) CAR-T-cell self-antigen-driven fratricide, which leads to the manufacture failure; and 2) the pan T-cell antigens targeted by CAR-T cells may induce T-cell underdevelopment, eventually leading to life-threatening immunodeficiency [18,33,35,36].
In our present research, we applied a strategy employing CD7 blockade using a CD7Nb, which is vital for retention efficiency and to allow binding and anchoring of CD7 molecules on the ER/Golgi through specific retention motifs. The affinity of the CD7Nb is within the nanomolar range [27]. Additionally, the CD7ΔT cells continued to expand by 80-fold, similar to normal cultured T cells, and CD7-CAR-T cells also showed viable expansion in vitro, suggesting that redirecting CAR-T cells against the CD7 antigen did not cause self-targeting/fratricide. The strategy bypasses sophisticated genome-editing-based disruption of target antigens to avoid self-targeting/fratricide and avoid the risk of off-target effects, which can be a common outcome from applying gene-editing technology. Furthermore, studies show that CRISPR/CAS9 system may induce a p53-mediated DNA-damage response, cause large deletions, and select p53-inactivating mutations. These studies highlighted the challenges associated with future clinical research of CAS9 technology [37-39].
Considering that the CD7-CAR-T-cell cytotoxic function is antigen-density-dependent, it remains a possibility that the low level of CD7 on normal T cells (according to the low MFI reported here) would allow them to evade CAR-T attack. Additionally, approximately 10% of T cells in vivo (usually in the form of PBMCs) are CD7- [40,41], which can help the body reshape the immune system of patients. It is possible that the endogenous CD3/T-cell receptor complex of infused CD7-CAR-T cells may contribute to the reconstitution of a healthy broad T-cell repertoire. Moreover, for T-ALL patients with high tumor risk, to maximize the success of allogeneic hematopoietic stem cell (HSC) transplantation, our technology can be used as an adjuvant therapy to reduce minimal residual disease [34,42]. In fact, in the final clinical application, we predict that the general immune function of blocked T cells would be unaffected. If the infusion of CAR-T cells is followed by infusion of autologous CD7 blockade T cells at the appropriate time, this would also aid re-establishment of a new immune system. A similar process occurs in CAR-T cells that target CD33. A previous study showed that infusion of autologous CD33-knockout HSCs overcome impairment of the hematopoietic system caused by the myelotoxicity due to CD33-CAR-T-cell infusion [43]. CD7 is thought to be associated with poor prognosis in AML and expressed in approximately 30% of AML cells. Because of the intensity of CD7, it may be used as a combination target with other AML target antigens to provide a new treatment strategy.
Previous studies indicate that although downregulation of CD7 occurs in mice following infusion of CD7-CAR-T cells, it does not compromise the ability of CD7-CAR-T cells in eliminating leukemia cells, indicating that CAR-T cells persistence, as well as antigen density, was also a pivotal element for relapse [17]. Similarly, we found that CD7-CAR-T cells retained tumor-clearance activity after antigen re-challenge. Of course, other factors act synergistically with the biological function of CAR-T cells. For example, due to differential metabolic profiling, 4-1BB co-stimulation presents slower but longer-lasting T-cell activation rather than CD28 [44,45]. Moreover, ICOS co-stimulation shows longer persistence and maintains a core molecular signature characteristic of T helper 17 cells among CD4+ T cells [46,47].
In conclusion, this study demonstrates the feasibility and efficacy of CD7ΔCAR-T cells for the immunotherapy of T-lineage tumors and AML, as well as applications for other tumors with surface expression of CD7. A phase I pilot clinical trial (ClinicalTrials.gov number, NCT04004637) is ongoing to further test the safety and efficiency of CD7-directed CAR-T cells in patients diagnosed with T-ALL/T-LBL and AML. Additionally, the ER/Golgi retention approach described here enables CAR redirection to other T-cell antigens.
Acknowledgements
We thank all members of PersonGen. Anke for their technical assistance. This work was supported by The Natural Science Foundation of China (grant nos. 81872431 and 31471283), The National Key R&D Program of China (2016YFC1303403), Priority Academic Program Development of Jiangsu Higher Education Institutions, and The Collaborative Innovation Major Project (grant no. XYXT- 2015304), the Six Talent Peaks Project in Jiangsu Province (no. SWYY-CXTD-010), and The Natural Science Foundation of The Jiangsu Higher Education Institutions of China (General Program, grant no. 19KJD320003). This study was also supported by The Project of State Key Laboratory of Radiation Medicine and Protection, Soochow University (no. GNZ1201803).
Disclosure of conflict of interest
None.
References
- 1.Vadillo E, Dorantes-Acosta E, Pelayo R, Schnoor M. T cell acute lymphoblastic leukemia (T-ALL): new insights into the cellular origins and infiltration mechanisms common and unique among hematologic malignancies. Blood Rev. 2018;32:36–51. doi: 10.1016/j.blre.2017.08.006. [DOI] [PubMed] [Google Scholar]
- 2.Karrman K, Johansson B. Pediatric T-cell acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2017;56:89–116. doi: 10.1002/gcc.22416. [DOI] [PubMed] [Google Scholar]
- 3.Marks DI, Rowntree C. Management of adults with T-cell lymphoblastic leukemia. Blood. 2017;129:1134–1142. doi: 10.1182/blood-2016-07-692608. [DOI] [PubMed] [Google Scholar]
- 4.Andersson ER, Lendahl U. Therapeutic modulation of Notch signalling--are we there yet? Nat Rev Drug Discov. 2014;13:357–378. doi: 10.1038/nrd4252. [DOI] [PubMed] [Google Scholar]
- 5.Carroll WL, Aifantis I, Raetz E. Beating the clock in T-cell acute lymphoblastic leukemia. Clin Cancer Res. 2017;23:873–875. doi: 10.1158/1078-0432.CCR-16-2825. [DOI] [PubMed] [Google Scholar]
- 6.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, Qu J, Wasielewska T, He Q, Fink M, Shinglot H, Youssif M, Satter M, Wang Y, Hosey J, Quintanilla H, Halton E, Bernal Y, Bouhassira DC, Arcila ME, Gonen M, Roboz GJ, Maslak P, Douer D, Frattini MG, Giralt S, Sadelain M, Brentjens R. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra225. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, Milone MC, Levine BL, June CH. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes MS, Sherry RM, Yang JC, Kammula US, Devillier L, Carpenter R, Nathan DA, Morgan RA, Laurencot C, Rosenberg SA. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN, Steinberg SM, Stroncek D, Tschernia N, Yuan C, Zhang H, Zhang L, Rosenberg SA, Wayne AS, Mackall CL. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, Mahnke YD, Melenhorst JJ, Rheingold SR, Shen A, Teachey DT, Levine BL, June CH, Porter DL, Grupp SA. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG, Lindgren CG, Lin Y, Pagel JM, Budde LE, Raubitschek A, Forman SJ, Greenberg PD, Riddell SR, Press OW. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood. 2012;119:3940–3950. doi: 10.1182/blood-2011-10-387969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, Sommermeyer D, Melville K, Pender B, Budiarto TM, Robinson E, Steevens NN, Chaney C, Soma L, Chen X, Yeung C, Wood B, Li D, Cao J, Heimfeld S, Jensen MC, Riddell SR, Maloney DG. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126:2123–2138. doi: 10.1172/JCI85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers GD, Qayed M, De Moerloose B, Hiramatsu H, Schlis K, Davis KL, Martin PL, Nemecek ER, Yanik GA, Peters C, Baruchel A, Boissel N, Mechinaud F, Balduzzi A, Krueger J, June CH, Levine BL, Wood P, Taran T, Leung M, Mueller KT, Zhang Y, Sen K, Lebwohl D, Pulsipher MA, Grupp SA. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378:439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T, Lin Y, Timmerman JM, Stiff PJ, Friedberg JW, Flinn IW, Goy A, Hill BT, Smith MR, Deol A, Farooq U, McSweeney P, Munoz J, Avivi I, Castro JE, Westin JR, Chavez JC, Ghobadi A, Komanduri KV, Levy R, Jacobsen ED, Witzig TE, Reagan P, Bot A, Rossi J, Navale L, Jiang Y, Aycock J, Elias M, Chang D, Wiezorek J, Go WY. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377:2531–2544. doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park JH, Riviere I, Gonen M, Wang X, Senechal B, Curran KJ, Sauter C, Wang Y, Santomasso B, Mead E, Roshal M, Maslak P, Davila M, Brentjens RJ, Sadelain M. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. 2018;378:449–459. doi: 10.1056/NEJMoa1709919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gomes-Silva D, Srinivasan M, Sharma S, Lee CM, Wagner DL, Davis TH, Rouce RH, Bao G, Brenner MK, Mamonkin M. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood. 2017;130:285–296. doi: 10.1182/blood-2017-01-761320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Png YT, Vinanica N, Kamiya T, Shimasaki N, Coustan-Smith E, Campana D. Blockade of CD7 expression in T cells for effective chimeric antigen receptor targeting of T-cell malignancies. Blood Adv. 2017;1:2348–2360. doi: 10.1182/bloodadvances.2017009928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mamonkin M, Mukherjee M, Srinivasan M, Sharma S, Gomes-Silva D, Mo F, Krenciute G, Orange JS, Brenner MK. Reversible transgene expression reduces fratricide and permits 4-1BB costimulation of CAR T cells directed to T-cell malignancies. Cancer Immunol Res. 2018;6:47–58. doi: 10.1158/2326-6066.CIR-17-0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burger R, Hansen-Hagge TE, Drexler HG, Gramatzki M. Heterogeneity of T-acute lymphoblastic leukemia (T-ALL) cell lines: suggestion for classification by immunophenotype and T-cell receptor studies. Leuk Res. 1999;23:19–27. doi: 10.1016/s0145-2126(98)00133-7. [DOI] [PubMed] [Google Scholar]
- 21.Niehues T, Kapaun P, Harms DO, Burdach S, Kramm C, Körholz D, Janka-Schaub G, Göbel U. A classification based on T cell selection-related phenotypes identifies a subgroup of childhood T-ALL with favorable outcome in the COALL studies. Leukemia. 1999;13:614–617. doi: 10.1038/sj.leu.2401382. [DOI] [PubMed] [Google Scholar]
- 22.van Grotel M, Meijerink JP, van Wering ER, Langerak AW, Beverloo HB, Buijs-Gladdines JG, Burger NB, Passier M, van Lieshout EM, Kamps WA, Veerman AJ, van Noesel MM, Pieters R. Prognostic significance of molecular-cytogenetic abnormalities in pediatric T-ALL is not explained by immunophenotypic differences. Leukemia. 2008;22:124–131. doi: 10.1038/sj.leu.2404957. [DOI] [PubMed] [Google Scholar]
- 23.Yeoh EJ, Ross ME, Shurtleff SA, Williams WK, Patel D, Mahfouz R, Behm FG, Raimondi SC, Relling MV, Patel A, Cheng C, Campana D, Wilkins D, Zhou X, Li J, Liu H, Pui CH, Evans WE, Naeve C, Wong L, Downing JR. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell. 2002;1:133–143. doi: 10.1016/s1535-6108(02)00032-6. [DOI] [PubMed] [Google Scholar]
- 24.Vodinelich L, Tax W, Bai Y, Pegram S, Capel P, Greaves MF. A monoclonal antibody (WT1) for detecting leukemias of T-cell precursors (T-ALL) Blood. 1983;62:1108–1113. [PubMed] [Google Scholar]
- 25.Lee DM, Staats HF, Sundy JS, Patel DD, Sempowski GD, Scearce RM, Jones DM, Haynes BF. Immunologic characterization of CD7-deficient mice. J Immunol. 1998;160:5749–5756. [PubMed] [Google Scholar]
- 26.Bonilla FA, Kokron CM, Swinton P, Geha RS. Targeted gene disruption of murine CD7. Int Immunol. 1997;9:1875–1883. doi: 10.1093/intimm/9.12.1875. [DOI] [PubMed] [Google Scholar]
- 27.Tang J, Li J, Zhu X, Yu Y, Chen D, Yuan L, Gu Z, Zhang X, Qi L, Gong Z, Jiang P, Yu J, Meng H, An G, Zheng H, Yang L. Novel CD7-specific nanobody-based immunotoxins potently enhanced apoptosis of CD7-positive malignant cells. Oncotarget. 2016;7:34070–34083. doi: 10.18632/oncotarget.8710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.You F, Wang Y, Jiang L, Zhu X, Chen D, Yuan L, An G, Meng H, Yang L. A novel CD7 chimeric antigen receptor-modified NK-92MI cell line targeting T-cell acute lymphoblastic leukemia. Am J Cancer Res. 2019;9:64–78. [PMC free article] [PubMed] [Google Scholar]
- 29.Yu Y, Li J, Zhu X, Tang X, Bao Y, Sun X, Huang Y, Tian F, Liu X, Yang L. Humanized CD7 nanobody-based immunotoxins exhibit promising anti-T-cell acute lymphoblastic leukemia potential. Int J Nanomedicine. 2017;12:1969–1983. doi: 10.2147/IJN.S127575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gomes-Silva D, Atilla E, Atilla PA, Mo F, Tashiro H, Srinivasan M, Lulla P, Rouce RH, Cabral JMS, Ramos CA, Brenner MK, Mamonkin M. CD7 CAR T cells for the therapy of acute myeloid leukemia. Mol Ther. 2019;27:272–280. doi: 10.1016/j.ymthe.2018.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jackson MR, Nilsson T, Peterson PA. Identification of a consensus motif for retention of transmembrane proteins in the endoplasmic reticulum. EMBO J. 1990;9:3153–3162. doi: 10.1002/j.1460-2075.1990.tb07513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Munro S, Pelham HR. A C-terminal signal prevents secretion of luminal ER proteins. Cell. 1987;48:899–907. doi: 10.1016/0092-8674(87)90086-9. [DOI] [PubMed] [Google Scholar]
- 33.Cooper ML, Choi J, Staser K, Ritchey JK, Devenport JM, Eckardt K, Rettig MP, Wang B, Eissenberg LG, Ghobadi A, Gehrs LN, Prior JL, Achilefu S, Miller CA, Fronick CC, O’Neal J, Gao F, Weinstock DM, Gutierrez A, Fulton RS, DiPersio JF. An “off-the-shelf” fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia. 2018;32:1970–1983. doi: 10.1038/s41375-018-0065-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maciocia PM, Wawrzyniecka PA, Philip B, Ricciardelli I, Akarca AU, Onuoha SC, Legut M, Cole DK, Sewell AK, Gritti G, Somja J, Piris MA, Peggs KS, Linch DC, Marafioti T, Pule MA. Targeting the T cell receptor beta-chain constant region for immunotherapy of T cell malignancies. Nat Med. 2017;23:1416–1423. doi: 10.1038/nm.4444. [DOI] [PubMed] [Google Scholar]
- 35.Mamonkin M, Rouce RH, Tashiro H, Brenner MK. A T-cell-directed chimeric antigen receptor for the selective treatment of T-cell malignancies. Blood. 2015;126:983–992. doi: 10.1182/blood-2015-02-629527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rasaiyaah J, Georgiadis C, Preece R, Mock U, Qasim W. TCRalphabeta/CD3 disruption enables CD3-specific antileukemic T cell immunotherapy. JCI Insight. 2018;3:99442. doi: 10.1172/jci.insight.99442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Charlesworth CT, Deshpande PS, Dever DP, Camarena J, Lemgart VT, Cromer MK, Vakulskas CA, Collingwood MA, Zhang L, Bode NM, Behlke MA, Dejene B, Cieniewicz B, Romano R, Lesch BJ, Gomez-Ospina N, Mantri S, Pavel-Dinu M, Weinberg KI, Porteus MH. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat Med. 2019;25:249–254. doi: 10.1038/s41591-018-0326-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haapaniemi E, Botla S, Persson J, Schmierer B, Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med. 2018;24:927–930. doi: 10.1038/s41591-018-0049-z. [DOI] [PubMed] [Google Scholar]
- 39.Ihry RJ, Worringer KA, Salick MR, Frias E, Ho D, Theriault K, Kommineni S, Chen J, Sondey M, Ye C, Randhawa R, Kulkarni T, Yang Z, McAllister G, Russ C, Reece-Hoyes J, Forrester W, Hoffman GR, Dolmetsch R, Kaykas A. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat Med. 2018;24:939–946. doi: 10.1038/s41591-018-0050-6. [DOI] [PubMed] [Google Scholar]
- 40.Reinhold U, Abken H, Kukel S, Moll M, Müller R, Oltermann I, Kreysel HW. CD7- T cells represent a subset of normal human blood lymphocytes. J Immunol. 1993;150:2081–2089. [PubMed] [Google Scholar]
- 41.Aandahl EM, Sandberg JK, Beckerman KP, Tasken K, Moretto WJ, Nixon DF. CD7 is a differentiation marker that identifies multiple CD8 T cell effector subsets. J Immunol. 2003;170:2349–2355. doi: 10.4049/jimmunol.170.5.2349. [DOI] [PubMed] [Google Scholar]
- 42.Leung W, Pui CH, Coustan-Smith E, Yang J, Pei D, Gan K, Srinivasan A, Hartford C, Triplett BM, Dallas M, Pillai A, Shook D, Rubnitz JE, Sandlund JT, Jeha S, Inaba H, Ribeiro RC, Handgretinger R, Laver JH, Campana D. Detectable minimal residual disease before hematopoietic cell transplantation is prognostic but does not preclude cure for children with very-high-risk leukemia. Blood. 2012;120:468–472. doi: 10.1182/blood-2012-02-409813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim MY, Yu KR, Kenderian SS, Ruella M, Chen S, Shin TH, Aljanahi AA, Schreeder D, Klichinsky M, Shestova O, Kozlowski MS, Cummins KD, Shan X, Shestov M, Bagg A, Morrissette JJD, Sekhri P, Lazzarotto CR, Calvo KR, Kuhns DB, Donahue RE, Behbehani GK, Tsai SQ, Dunbar CE, Gill S. Genetic inactivation of CD33 in hematopoietic stem cells to enable CAR T cell immunotherapy for acute myeloid leukemia. Cell. 2018;173:1439–1453. e19. doi: 10.1016/j.cell.2018.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng Z, Wei R, Ma Q, Shi L, He F, Shi Z, Jin T, Xie R, Wei B, Chen J, Fang H, Han X, Rohrs JA, Bryson P, Liu Y, Li QJ, Zhu B, Wang P. In vivo expansion and antitumor activity of coinfused CD28- and 4-1BB-engineered CAR-T cells in patients with B cell leukemia. Mol Ther. 2018;26:976–985. doi: 10.1016/j.ymthe.2018.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr, Patel PR, Guedan S, Scholler J, Keith B, Snyder NW, Blair IA, Milone MC, June CH. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. 2016;44:380–390. doi: 10.1016/j.immuni.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 46.Guedan S, Chen X, Madar A, Carpenito C, McGettigan SE, Frigault MJ, Lee J, Posey AD Jr, Scholler J, Scholler N, Bonneau R, June CH. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood. 2014;124:1070–1080. doi: 10.1182/blood-2013-10-535245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guedan S, Posey AD Jr, Shaw C, Wing A, Da T, Patel PR, McGettigan SE, Casado-Medrano V, Kawalekar OU, Uribe-Herranz M, Song D, Melenhorst JJ, Lacey SF, Scholler J, Keith B, Young RM, June CH. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight. 2018;3:e96976. doi: 10.1172/jci.insight.96976. [DOI] [PMC free article] [PubMed] [Google Scholar]