

Abstract
BACKGROUND AND AIMS:
Disease extent varies in ulcerative colitis (UC) from proctitis to left-sided colitis to pancolitis and is a major prognostic factor. When the extent of UC is limited there is often a sharp demarcation between macroscopically involved and uninvolved areas and what defines this or subsequent extension is unknown. We characterized the demarcation site molecularly and determined genes associated with subsequent disease extension.
METHODS:
We performed RNA sequence analysis of biopsy specimens from UC patients with endoscopically and histologically confirmed limited disease, of which a subset later extended. Biopsy specimens were obtained from the endoscopically inflamed upper (proximal) limit of disease, immediately adjacent to the uninvolved colon, as well as at more proximal, endoscopically uninflamed colonic segments.
RESULTS:
Differentially expressed genes were identified in the endoscopically inflamed biopsy specimens taken at each patient’s most proximal diseased site relative to healthy controls. Expression of these genes in the more proximal biopsy specimens transitioned back to control levels abruptly or gradually, the latter pattern supporting the concept that disease exists beyond the endoscopic disease demarcation site. The gradually transitioning genes were associated with inflammation, angiogenesis, glucuronidation, and homeodomain pathways. A subset of these genes in inflamed biopsy specimens was found to predict disease extension better than clinical features and were responsive to biologic therapies. Network analysis revealed critical roles for interferon signaling in UC inflammation and poly(ADP-ribose) polymerase 14 (PARP14) was a predicted key driver gene of extension. Higher PARP14 protein levels were found in inflamed biopsy specimens of patients with limited UC that subsequently extended.
CONCLUSION:
Molecular predictors of disease extension reveal novel strategies for disease prognostication and potential therapeutic targeting.
Keywords: UC, Disease Extension, Molecular, Interferon Signaling, PARP14
Ulcerative colitis (UC) is a progressive and disabling disease characterized by chronic inflammation of the colon.1 The extent of colonic involvement in UC is a defining feature with the Montreal classification distinguishing proctitis (E1), left-sided colitis (E2), and pancolitis (E3).2 Approximately half of UC cases present with proctitis (E1) at the time of diagnosis, with the remaining evenly split between E2 and E3.3,4 In patients with limited disease (E1 and E2), a demarcation zone can be identified that delineates endoscopically involved and uninvolved areas. However, UC is a dynamic disease with proximal disease extension occurring over time in approximately 30% of E1/E2 patients, with significant implications for prognosis and treatment.3 Extensive colitis is associated with increased need for aggressive therapy as well as higher rates of hospitalizations, surgery, and colorectal cancer.1
At present, very few factors have been identified that characterize UC demarcation and subsequent risk of extension. Burisch et al,3 in a Danish cohort, found that only extent at diagnosis (eg, E1 or E2) was a clinical predictor of disease extension after 7 years of follow-up, which was similar to a retrospective Asian study.5 Beyond the clinic, recent genome-wide genetic and gene expression6 data types have improved inflammatory bowel disease (IBD) subtyping.7 For example, Lee et al8 identified a circulating CD8+ T-cell transcriptional signature that subdivided patients with indistinguishable IBD into 2 subtypes, with each experiencing different disease courses. These types of signatures are continuing to be evaluated9 as well as being integrated into clinical care, with the first biomarker-stratified trial in IBD10 currently ongoing. Another study using mucosal transcriptomes from patients with treatment-naïve UC in The Predicting Response to Standardized Pediatric Colitis Therapy (PROTECT) study cohort revealed a corticosteroid response gene signature linked to mitochondrial dysfunction in active UC and higher anti-inflammatory ALOX15 expression during remission.11 Finally, integration of the molecular datatypes into network models has successfully revealed novel key driver genes (KDGs) associated with IBD pathology.12
Because the endoscopic line of demarcation is a striking clinical feature in some patients with limited UC, we aimed to characterize it at the molecular level to find potential markers of disease extension. Specifically, we measured gene expression changes in the inflamed and the adjacent and more proximally sampled uninflamed mucosa in a UC cohort with histologically and endoscopically confirmed limited disease. We then evaluated if expression of these genes was predictive of later disease extension.
Methods
The Mount Sinai Crohn’s and Colitis Registry
Gene expression data was obtained from a cross-sectional cohort (~1100 UC, Crohn’s disease [CD], and control patients) from the Mount Sinai Crohn’s and Colitis Registry (MSCCR) between December 2013 and September 201613 (Figure 1). Subjects were recruited from the outpatient and endoscopy units of the Mount Sinai Hospital. Institutional review board approval and informed consents were obtained. Study participants provided biopsy specimens during a colonoscopy planned as part of standard of care and allowed prospective clinical follow-up through electronic medical records. The biopsy specimens used for RNA extraction and sequencing were collected in RNAlater, and were generally sampled from 3 intestinal segments per patient, with the most endoscopically inflamed region being prioritized, followed by an adjacent (proximal) endoscopically noninflamed area, and then 1 additional section that was either colonic or ileal.
Figure 1.
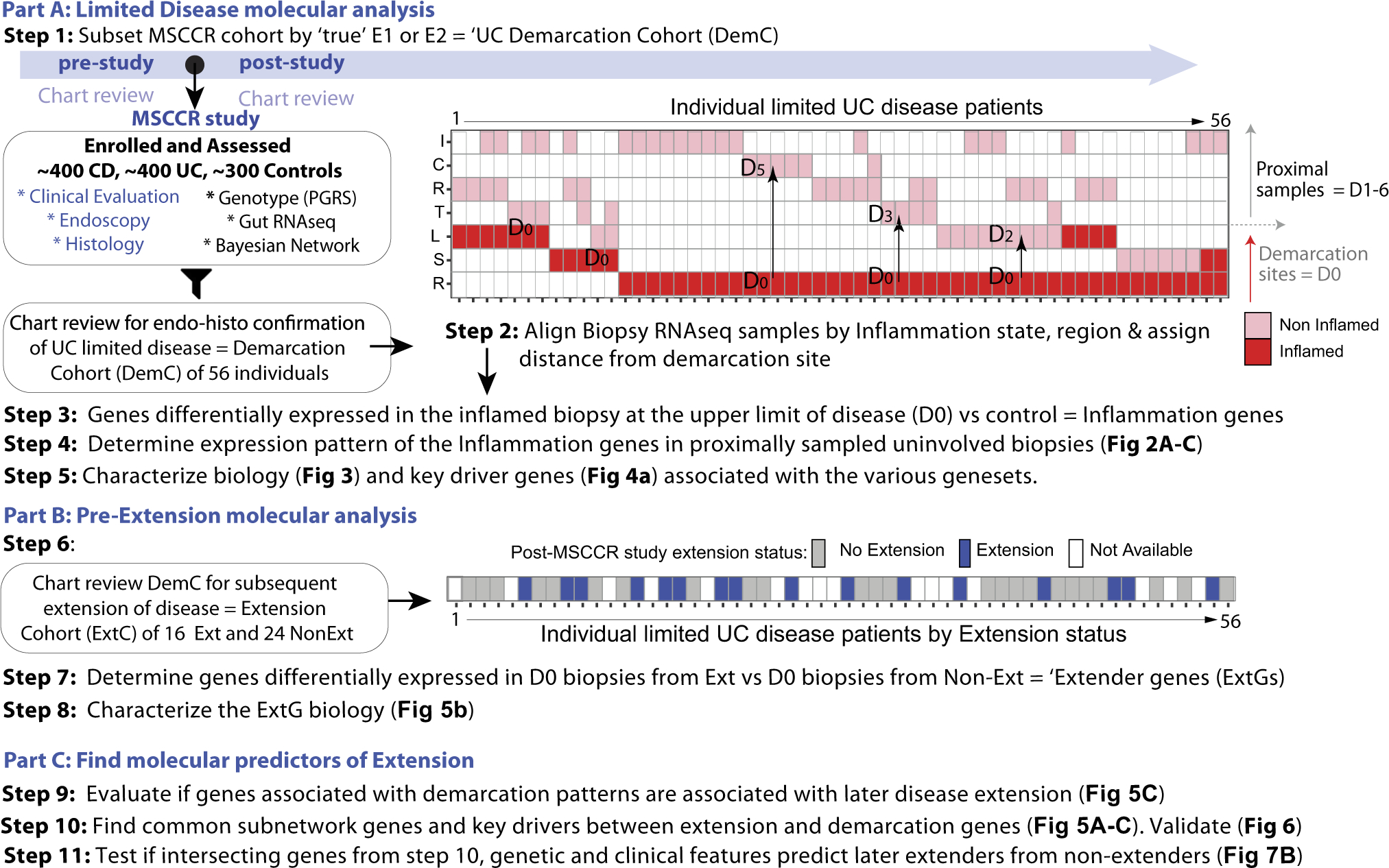
Study design. The MSCCR cohort was subset according to individuals with true limited disease, referred to as Demarcation Cohort (DemC). A schema summarizing the DemC biopsy specimens according to individual (X-axis), intestinal region (Y-axis), and inflammation status. I, ileum; C, cecum; R, rectum; T, transverse; L, left-side; S, sigmoid. The lower panel indicates which patients had known extension status. A “distance” was assigned between each pair of patient biopsy specimens. D0 tagged the most proximal inflamed biopsy and then a number of D1–D6 was assigned for the matching patient uninvolved biopsy depending on the proximal colonic segment it was sampled. Steps 3–5 summarize how molecular disease demarcation patterns were determined including KDGs. The DemC was further subset according to disease extension status (post-MSCCR study) evaluated through chart review. Steps 7 and 8 described the determination of genes associated with later extension and their biological processes. Part C involved integration of the demarcation and extender genes on a network level. Common genes were tested in models to predict later extension.
Characterization of the E1/E2 UC MSCCR Cohort
A subset of MSCCR patients with limited and active UC disease was selected based on chart review. We included patients that always had limited disease extent up until the time of MSCCR study enrollment and had at least 1 endoscopically defined inflamed and 1 proximal noninflamed biopsy specimen. Patients in endoscopic remission or those with more extensive endoscopic and/or histologic disease in the past were largely excluded. Patients with histologic inflammation in endoscopically normal tissue proximal to line of demarcation were also excluded. Fifty-four patients met these requirements. Two additional E2 patients had a previous E3 diagnosis but were included as post-study endoscopy follow-up information was available and a clustering analysis revealed they were not outliers (data not shown). The breakdown of the limited UC cohort at time of study endoscopy was E1 = 16 and E2 = 40 with clinical and demographic information summarized in Supplementary Table 1a. The Mayo endoscopic scoring was done at the time of the study endoscopy by the gastroenterologists, of whom all were IBD specialists and subsequently reviewed from the charts by 2 authors (R.U. and J.F.C.). The criteria for histologically normal mucosa were established by an expert pathologist (N.H.) to ensure limited disease and read as normal. Normal mucosa was characterized by preserved mucosal architecture and absence of inflammatory cell infiltration. Specifically, the crypts were parallel, of uniform tubular shape and width, and abutted on the muscularis mucosae. There was no excess of mononuclear inflammatory cells in the lamina propria and no neutrophil infiltration of the surface or crypt epithelium.
E1 and E2 patients were then classified as either extenders (ie, E1 to E2 or E3, or E2 to E3) or nonextenders (on later endoscopy) based on review of medical records (post-MSCCR visit) and were included if they had at least 1 visit post-study that included a colonoscopy. The number of patients we could subsequently determine disease status post-study, within a median follow-up time of 33.9 months, included 24 non-extenders and 16 extenders (Supplementary Table 1b).
Statistical Modeling of Proximal Disease Genes in the Limited UC MSCCR Cohort
Biopsy RNA was extracted as detailed previously13 (Supplementary Methods). To model gene expression changes near to, immediately adjacent to, and proximal to the site of demarcation, we assigned a ‘distance’ (D) to each biopsy within a patient according to the number of colonic segments proximal to the distal inflamed biopsy (designated D0). For example, the noninflamed biopsy specimen immediately adjacent to the most proximal sampled inflamed site was designated D1 (Figure 1). We then modelled the changes in gene expression from the most proximal site of inflammation (D0) to D6, including 461 non-IBD control noninflamed biopsy specimens (from 243 unique control patients) to account for regional changes in gene expression. Because our objective was to evaluate the changes as we move farther away from the site of inflammation, only differentially expressed genes (at FDR <0.05, | lgFCH | >1.5) between inflamed biopsy specimens (sampled from the upper limit of disease) and non-IBD controls were evaluated. A linear mixed-effect model was used with D (distance) as a fixed effect and covariates of IBD disease (UC/control), region (rectum, colon-nonrectum, ileum), and topical rectal medication use. Using the coefficients from this model, we determined 2 potential molecular patterns of demarcation genes, namely, sharply resolving (ResG) and slowly advancing (AdvG) gene sets.
To evaluate if use of rectal topical medication impacts the 2 molecular demarcation patterns, we ran a model including an interaction between rectal medication and distance to inflamed (D0) and identified those 2 sets of genes for patients using or not using medication.
Statistical Modeling to Determine Genes Associated With Subsequent UC Extension
Gene expression differences were also determined (unadjusted P value <.01) in the most proximal inflamed biopsy specimens of patients with limited UC between patients who subsequently extended as compared with those that did not (ExtG).
MSCCR Bayesian Gene Regulatory Network Generation, Subnetwork and KDG Analysis
A Bayesian gene regulatory network (BGRN)12,13 was generated from RNA sequence data generated on intestinal colonic inflamed and noninflamed biopsy specimens from the MSCCR UC cohort (Supplementary Methods). Subnetworks were generated for ResG, AdvG, or ExtG, by selecting the signature genes on the network and expanding out 1 or 2 layers (undirected) to include the nearest neighbor genes. Subnetworks were tested for enrichment to various pathways using the Fisher exact test and P values were adjusted using Benjamini-Hochberg procedure. KDG analysis, which identifies key or “master” driver genes (KDGs), was determined for ResG, AdvG, and ExtG gene sets.
Gene Set Variation Analysis
We estimated the overall expression activity of various gene signatures using gene set variation analysis (GSVA)14 (Supplementary Methods). The GSE7366115 series includes gene expression profiles from colonic biopsy specimens from patients with UC enrolled in 2 vedolizumab efficacy trials15 as well as 12 non-IBD and 23 patients before and 4–6 weeks after first infliximab treatment. Response at week 22 was defined as endoscopic mucosal healing.
Promoter Motif Enrichment Analysis
iRegulon (v1.3)16 was used within Cytoscape (v3.7.2) to predict transcription factors enriched in genes associated with subnetworks (Supplementary Methods).
Immunofluorescence Imaging
Formalin-fixed, paraffin-embedded intestinal biopsy specimens were available on a subset of the cohort: 6 healthy controls with 11 samples (6 rectum and 5 colon); 6 UC subsequent extenders with 13 samples (3 noninflamed colon, 1 noninflamed rectum, 4 inflamed colon, and 5 inflamed rectum); and 5 UC subsequent nonextenders with 9 samples (3 noninflamed colon, 4 inflamed colon, and 2 inflamed rectum). Immunofluorescence (IF) staining with PARP14 and anti-CD68 antibodies was performed. Expression of PARP14 in the cytoplasmic, nuclear, and total cellular compartments was quantified separately (Supplementary Methods).
Polygenic Risk Score
A polygenic risk score (PGRS) was generated using UK biobank IBD GWAS loci with P value cutoff of 1 × 10−8. MSCCR DNA genotype information was generated as described13 and available for 9 extenders and 18 nonextenders.
Predictive Modeling Approach to Identify Features That Predict UC Extension
The Elastic Net algorithm was used to build a predictor of UC extension based on clinical and molecular features alone or in combination (Supplementary Methods).
Results
Part A: Molecular Analysis of the E1/E2 UC Cohort
Molecular disease in limited UC extends beyond endoscopic and histologic disease demarcations.
RNA sequencing analysis was performed on 56 MSCCR individuals with limited UC confirmed both endoscopically and histologically. Compared with healthy control biopsy specimens, 2522 genes (1360 up- and 1162 down-regulated) were differentially expressed (@FDR < 0.05,|lgFCH| > 1.5, where lgFCH is log fold change; Supplementary Table 2) in inflamed biopsy specimens taken at the upper disease demarcation site (D0). When we plotted the expression of these genes according to their sampled location (Figure 1) we noted 2 main expression pattern changes (Figure 2A, Supplementary Table 2). One pattern showed genes abruptly transition back to near control levels in the immediately adjacent noninvolved tissue segment (D1) and they are referred to as ‘abruptly’ resolving genes (ResG). The second expression change pattern showed genes only gradually normalizing in expression along the several segments sampled above (D1–D6) the inflamed site and we refer to these genes as slowly advancing disease genes (AdvG). Statistical modeling of the expression changes was guided by these 2 patterns (Figure 2B and 2C).
Figure 2.
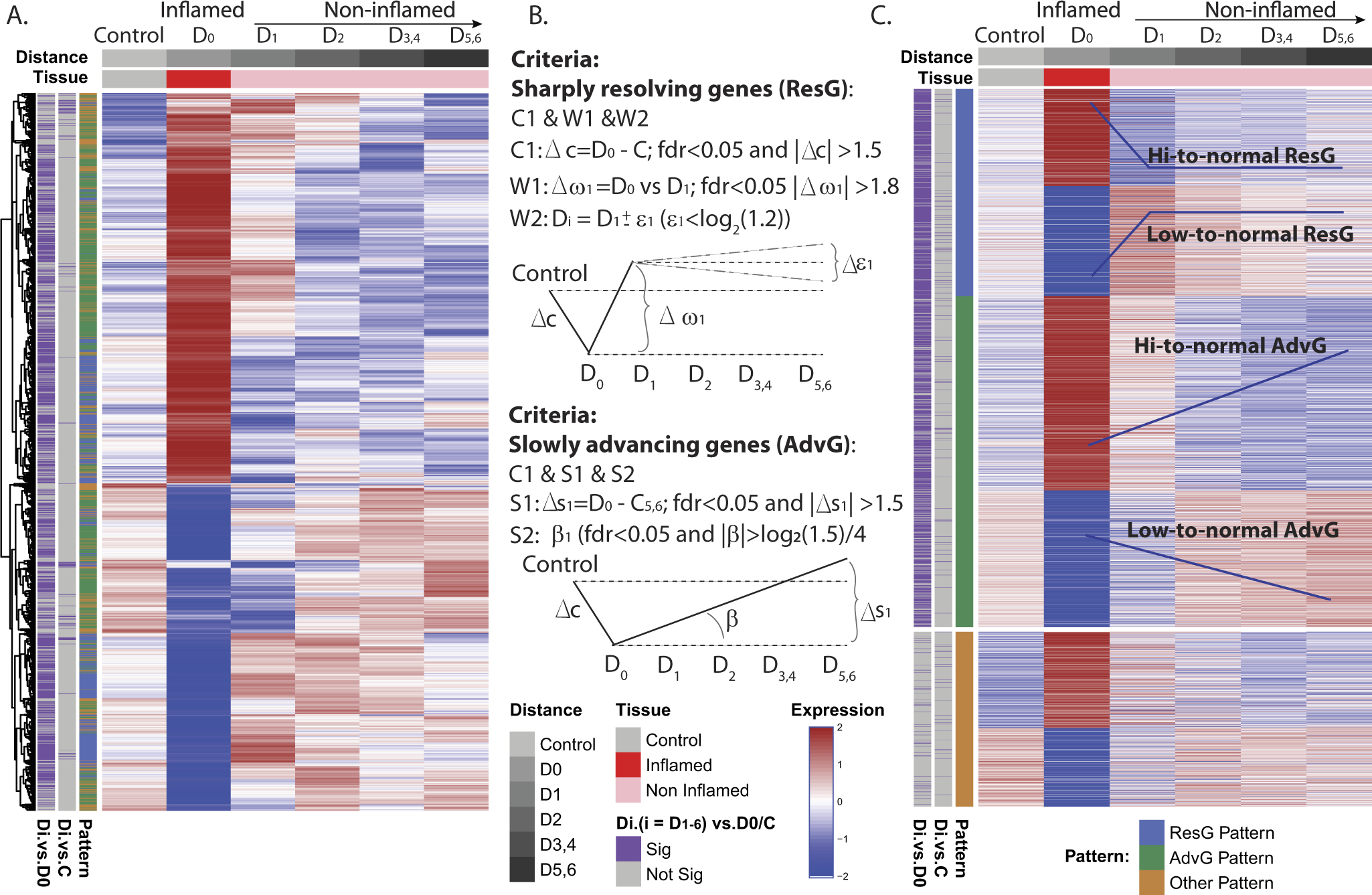
Characterization of the molecular expression patterns proximal to the site of demarcation. (A) Unsupervised clustering of the demarcation disease genes defined as those differentially expressed between D0 and control. The heatmap shows 2 potential patterns of expression changes. (B) Statistical modeling was used to identify genes of specific expression change patterns of either (1) “sharply resolving (ResG),” which have an abrupt change in expression between inflamed and immediate adjacent uninflamed biopsy and does not change in segments sampled more proximally and (2) slowly advancing (AdvG) molecular changes (either inclining or declining in expression) along the colonic segments proximally. (C) Reclustering of genes in B, supervised by the assigned demarcation pattern.
We next evaluated potential effects of common IBD medication use and found the most prominently used drugs were rectal medications (31 of 56 patients). Patients were stratified by rectal medication use or nonuse and we determined their medication use did not affect the expression of AdvG. A strong overlap was observed between genes identified using the full cohort versus the medication-use stratified groups (Supplementary Figure 1A–1C). In contrast, the overlap of ResG in patients stratified based on rectal medication use (Supplementary Figure 1D–1F) versus the nonmedicated group was very small. Two reasons for the poor overlap stemmed from the nonmedicated group displaying the following: (1) smaller fold-changes in gene expression between the inflamed (D0) biopsy and the first adjacent noninvolved biopsy (D1) and (2) that gene expression normalization in a subset of genes occurred at D2 instead of at D1 as in the medicated group (Supplementary Figure 2). Therefore, we subdivided the ResG according to genes that abruptly normalized at D1 (ResG@D1) or D2 (ResG@D2) (Supplementary Table 3).
The demarcation genes that transition from high-to-normal levels are inflammatory and angiogenic.
AdvG or ResG (@D1 or @D2) that were up-regulated in inflamed biopsy specimens (D0) relative to healthy controls were designated high-to-normal transitioning genes (Figure 3A, Supplementary Table 3) and were enriched in several inflammatory pathways. These included interferon gamma (IFNɣ) response, interleukin (IL)6-JAK-STAT3 signaling, tumor necrosis factor alpha (TNFα) signaling via nuclear factor KB, as well as pro-inflammatory IL17 and oncostatin M signaling (Figure 3B, Supplementary Table 4, Supplementary Figure 3A and 3B). Angiogenesis, epithelial-mesenchymal transition (EMT) and vasculature development were among the nonimmune-enriched functions (Figure 3B, Supplementary Figure 4A and 4B, Supplementary Tables 5 and 6). Cell type enrichment analysis using gene sets from single-cell RNA sequencing data showed enrichment of both ResG and AdvG genes with high-to-normal transitioning patterns in pathogenic colonic UC cell types,17 including inflammatory monocytes, inflammatory fibroblasts, and endothelial cells as well as in pathogenic ileal CD cell types,18 namely, inflammatory fibroblasts and ACKR1+ activated endothelial cells (Supplementary Figure 5). Scar-associated endothelial and mesenchymal cell types from cirrhotic human liver biopsy specimens19 were also significantly enriched in the ResG (Supplementary Figure 5). Two angiogenesis signature sets, 1 common to several different cancer types20 and another from a noncancerous retinopathy mouse model,21 were also significantly enriched in ResG and AdvG. These data suggest inflammation, fibrosis, and vascular remodeling are present despite normal endoscopic and histologic appearance.
Figure 3.
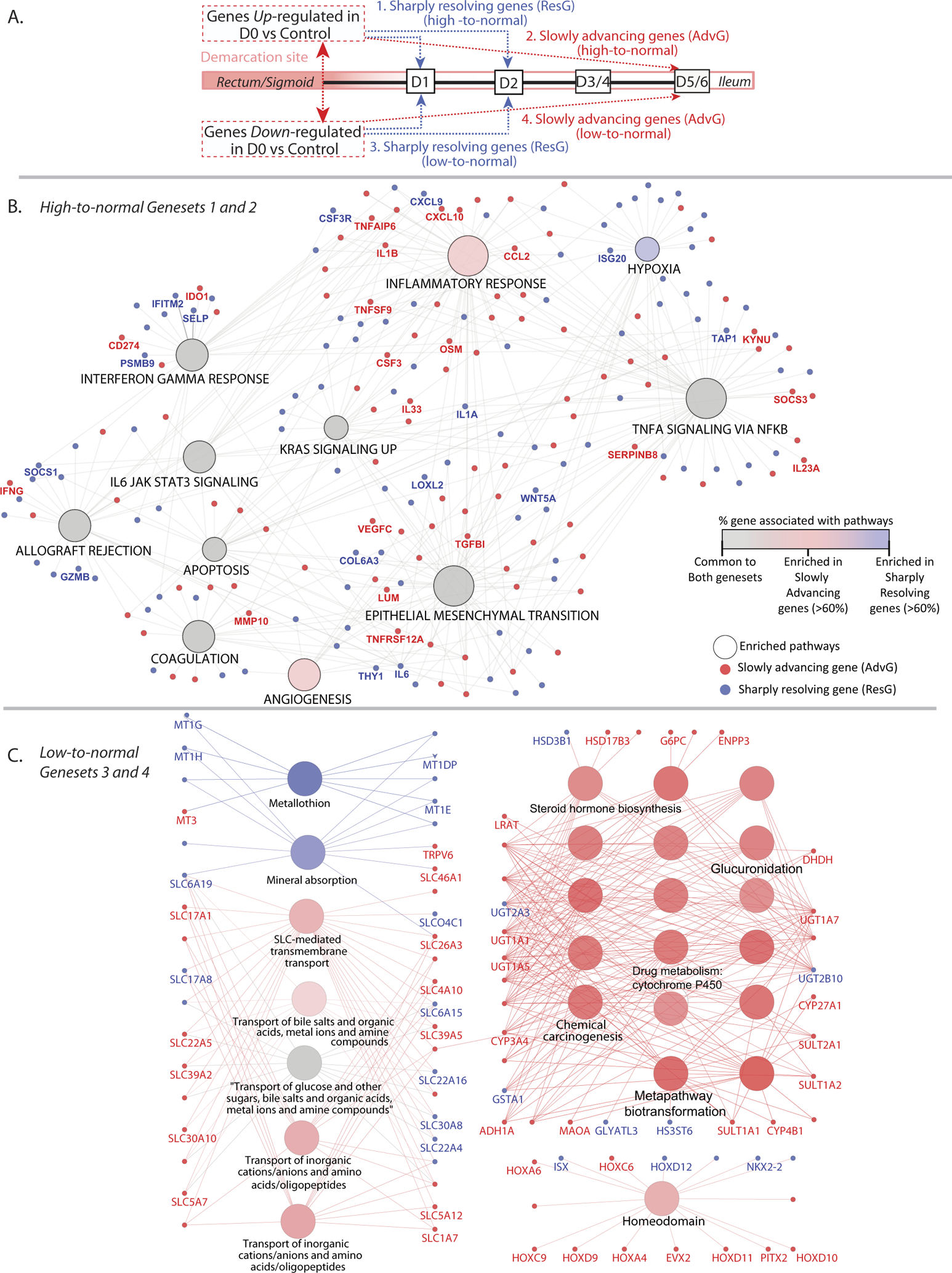
Pathways associated with demarcation genes. (A) Schematic summarizing the 2 main molecular demarcation expression patterns identified and the distinction as high-to-normal or low-to-normal. (B) Hallmark pathway enrichment analysis of the ResG or AdvG, with a high-to-normal change in expression. (C) Pathway enrichment analysis according to Kegg, Reactome, Bioplanet, and PFAM of the ResG or AdvG, with a low-to-normal change in expression. Nodes depict enriched terms and the coloring represents pathways in common (gray) or specific for AdvG (red) or ResG (blue). ResGs were queried as a single gene set (resolved @D1 or @D2). The edges connect genes to pathways with a subset labeled with gene names. Pathways shown are Benjamini-Hochberg adjusted P value <.05.
Demarcation genes with low-to-normal levels, are enriched in gut homeostasis functions.
AdvG or ResG that were down-regulated in the inflamed biopsy (D0) versus healthy controls were considered low-to-normal transitioning genes. Pathways found to be enriched included homeodomain-containing genes, drug metabolism processes, as well as solute-carrier–mediated transmembrane transport (Figure 3C, Supplementary Table 8). Accordingly, AdvG were enriched in absorptive or secretory epithelial cell types (Supplementary Figure 5), supporting epithelial cell loss or dysfunction beyond the endoscopic-histologic limit of disease.
We also investigated pathways specifically associated with the ResG@D2 gene set, containing genes that are delayed in resolution at D2 instead of D1 with nonmedication use. Functional enrichment using Kegg, Reactome, or BioPlanet databases revealed IBD-relevant terms, such as Th17 cell differentiation and Jak-STAT signaling (Supplementary Figure 6), consistent with expected beneficial effects due to the rectal medication use.
Bayesian network analysis reveals KDGs of the ResG and AdvG.
To determine the gene:gene regulatory relationships and potential KDGs underlying the ResG and AdvG, we searched for these gene sets within the context of the MSCCR UC_BGRN and applied subnetwork finding algorithms resulting in 3 subnetworks (Figure 4A left panel, Supplementary Table 9). Nearly all the genes were found in a single subnetwork of 1787 nodes (4.7-fold enrichment and P < 2.2−16), supporting a strong co-regulation (Supplementary Figure 7A). KDGs were then determined and summarized across the 3 subnetworks (Supplementary Figure 7B, Supplementary Table 10). A subnetwork associated with the top 10 shared KDGs is shown (Figure 4A right panel).
Figure 4.
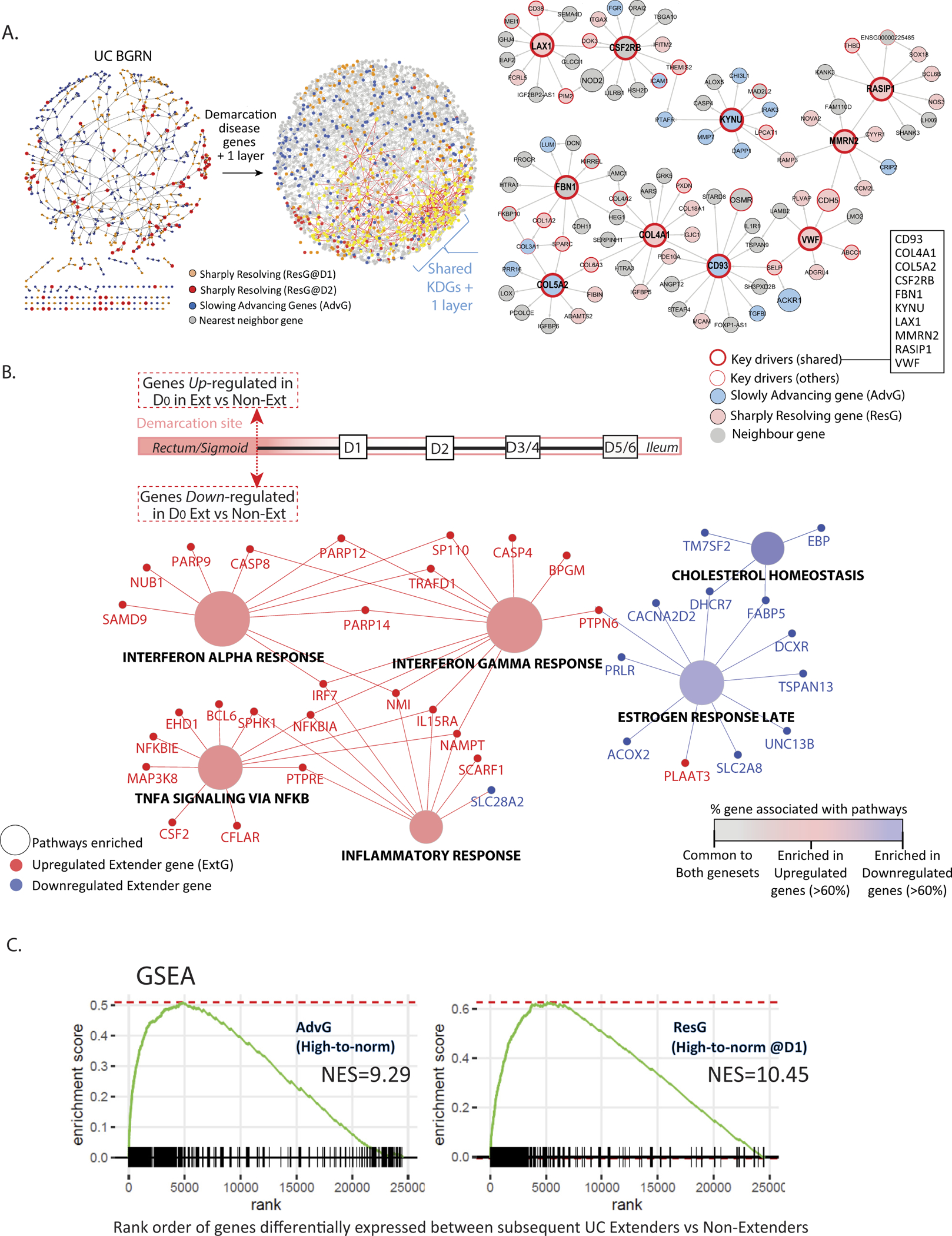
Network analysis of ResG and AdvG and pathway analysis of ExtG. (A) The UC_BGRN was queried for either AdvG, ResG@D1, or ResG@D2 and the resulting subnetworks without (left) or with (middle) 1 additional layer of nonsignature genes. Subnetwork associated with the 10 shared KDGs (black box and SF7B) and 1 additional layer gene (far right). (B) Hallmark pathway enrichment analysis (Benjamini-Hochberg adjusted P < .05) of the ~380 genes found differentially expressed (at unadjusted P < .01) in the inflamed biopsy specimens of patients that subsequently extended compared with those that did not (ExtG). Nodes depict the pathway and their coloring depicts those pathways commonly or specifically enriched for either up- (red) or down- regulated genes (blue). Edges connect the gene to pathway. (C) ExtG were ranked according to logFC differences between extenders vs. nonextenders and were tested for enrichment in ResG or AdvG. The trace of the enrichment scores is shown for 2 gene sets: AdvG (high-to-normal genes) and ResG (high-to-normal genes) with full results in SF9. The legend for Figure 3 contains explanation of gene sets. Normalized enrichment score (NES) and false discovery rate.
A subset of AdvG are differentially expressed in macroscopically uninvolved biopsy specimens relative to location-matched healthy control biopsy specimens.
We also determined if the AdvG were differentially expressed at uninflamed UC biopsy specimens (D1–D6) in comparison to region-matched healthy controls. At an unadjusted P < .05 several AdvG (377 genes) were differentially expressed when compared with controls (Supplementary Table 11). The pathway enrichments were also comparable with the complete AdvG set, including inflammatory and nonimmune pathways (Supplementary Figure 8), thus further supporting that, molecularly, disease extends proximal to the endoscopic and histologic border.
Part B: Molecular Analysis of UC Disease Extension
TNFɑ- and IFN-associated pathways distinguish subsequent UC extenders.
Genes associated with later disease extension were determined by comparing the most proximally inflamed biopsy specimens (D0) between subsequent extenders and nonextenders (ExtG). Given the limited sample size, an unadjusted P value cutoff of <.01 was used, resulting in 347 differentially expressed genes (Figure 4B, Supplementary Table 12a). Because even fewer genes (166) were identified comparing transcriptomes of the noninflamed regions between subsequent extenders and nonextenders (Supplementary Table 12b), we focused on the inflamed regions. Genes down-regulated in subsequent extenders were enriched in mainly cholesterol homeostasis and estrogen response pathways (Figure 4B, Supplementary Table 13). Pathways associated with genes having higher expression in subsequent extenders included mainly TNFɑ signaling via nuclear factor KB and IFNɑ/ɣ signaling. Because these latter pathway enrichments were similar to those observed with AdvG and ResG, we tested if the ExtG were also enriched in AdvG and ResG using gene set enrichment analysis, and they were, suggesting a common pathology (Figure 4C, Supplementary Figure 9).
We next investigated the ExtG within the UC_BGRN and isolated an ExtG-associated subnetwork. The ExtG subnetwork formed a single connected subnetwork of 2120 nodes when queried with the ResG- and AdvG-associated subnetworks (Figure 5A, Supplementary Table 14). The ResG- and AdvG-associated subnetworks were both found to be significantly enriched in the ExtG-associated subnetwork and testing the overlap of their KDGs was even more significant (Figure 5A, Supplementary Table 15). Thus genes associated with subsequent clinically defined extension and the molecular changes in the endoscopically noninvolved proximal colonic regions sampled from patients with limited UC appear to converge.
Figure 5.
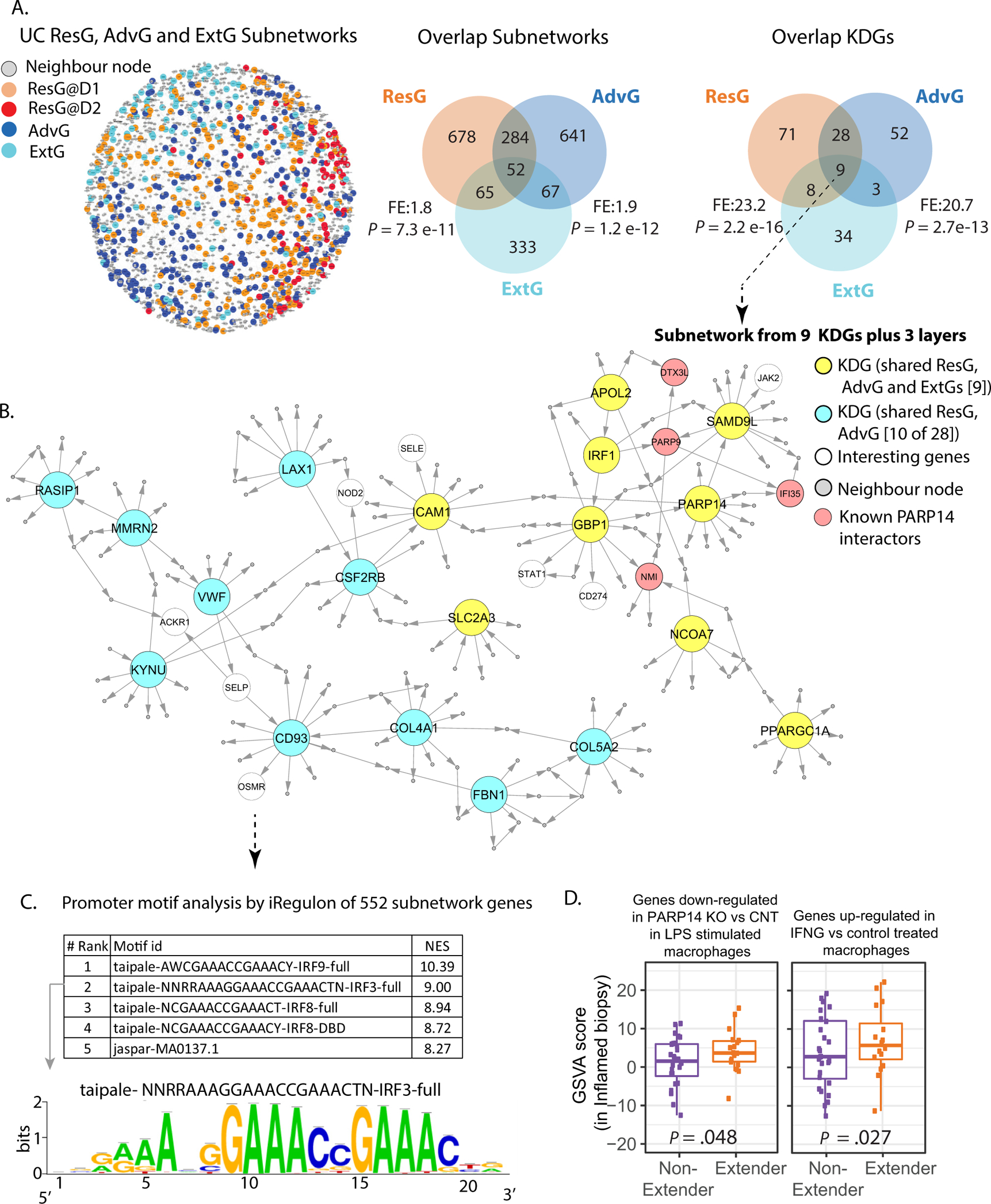
Network analysis of extender and demarcation genes. (A) ExtG, ResG, and AdvG were queried in the UC_BGRN and found to generate a single connected subnetwork of ~2000 genes with 1 layer of nonsignature genes. Venn diagrams comparing the demarcation and ExtG-associated subnetworks and KDGs with fold-enrichment and significance of overlap. (B) A subset of the network (552 total nodes) associated with the 9 shared KDGs between ExtG and demarcation gene sets (ST16). (C) iRegulon analysis using the genes’ promoters defined as 10 kb within the transcription start site (TSS). A summary of the top 5 motifs and their normalized enrichment score (ST17). (D) GSVA analysis of gene sets listed in the figure panel, using the inflamed biopsy expression data from the MSCCR patients with UC that subsequently extended vs. those that did not.
PARP14 is a KDG of the IFN-mediated UC disease pathology and later extension.
We investigated a subnetwork of 552 genes associated with the 9 shared KDGs (plus 3 layers) between ExtG and demarcation gene sets (Figure 5B, Supplementary Table 16). The top enriched intestinal gut cell type was inflammatory monocytes and a striking enrichment in IFN signaling defined by BioPlanet pathways and macrophage chemical perturbation gene sets22 was observed (Supplementary Figure 10). We also observed enrichment in an IFN-stimulated endothelial cell signature23 (Supplementary Figure 10). One of the KDGs identified was a mono-ADP-ribosylation gene known as PARP14. Macrophage PARP14 messenger RNA (mRNA) expression is known to be strongly induced by liposaccharide (LPS), IFN-B, and LPS + IFNɣ exposures. The expression of PARP14 is mainly within the cytosolic compartment in unstimulated cells. However, PARP14 is readily detectable in the nucleus after LPS stimulation, supporting its role in antimicrobial defense.24 PARP14 was shown previously to co-immunoprecipitate with IFN-stimulated gene (ISG)-encoded proteins, supporting PARP14 having transcriptional coregulatory activity. PARP14 binders included the ISG’s PARP9, DTX3L, PARP12, IFI35, NMI, and SQSTM124 as well as RNA Pol II, which affected its recruitment to promoters of IRF3-regulated genes. Interestingly, aside from SQSTM1 and RNA pol II, all these genes were found within 3 network layers of PARP14 (Figure 5B and data not shown), suggesting a similar function in the context of the gut and many had increased expression in extenders’ inflamed biopsy specimens versus nonextenders (Supplementary Table 12a). This subnetwork was also significantly enriched in genes dysregulated by PARP14 depletion in stimulated macrophages as well as in genes with IRF9 and IRF3 regulatory elements, supporting a KDG role for PARP14 (Figure 5C, Supplementary Figure 10, Supplementary Table 17). Supporting elevated PARP14 activity in patients with UC that eventually extend was higher GSVA scores of genes down-regulated with PARP14 knock-down in stimulated macrophages (Figure 5D) in inflamed biopsy specimens of extenders as compared with nonextenders. Supporting altered IFN signaling was higher GSVA scores of genes up-regulated in IFNɣ-treated macrophages, in the inflamed biopsy specimens of extenders as compared with nonextenders (Figure 5D). Overall, our analysis suggests that IFN signaling, PARP14 activity, and effects of its down-stream targets may modulate risk of subsequent disease extension.
Validation of nuclear PARP14 protein levels as associated with subsequent disease extension.
Inflammatory mechanisms have been associated with PARP14, however, its expression in colons of healthy controls or patients with UC is unknown. Figure 6A shows representative IF staining in colorectal biopsy specimens from non-UC controls of PARP14 (green) and CD68 (red). PARP14 expression is abundant both in the lamina propria (LP) and within the epithelial compartment at the apical surface (top) and at the base of the crypts (bottom). We next evaluated PARP14 expression levels in inflamed and uninflamed biopsy specimens in a subset of the patients with UC that subsequently extended (n = 6) compared with those that did not (n = 5). Based on our mRNA analyses, we predicted PARP14 protein levels to be higher in subsequent extenders compared with nonextenders. For this analysis we focused on the LP cells given our molecular analysis highlighted PARP14 in an immune context. Further, we quantified PARP14 expression with cytosolic, nuclear, and total cellular compartments separately. We found that the frequency of LP cells with high PARP14 nuclear staining was significantly higher in patients with UC that subsequently extended as compared with nonextenders in both uninflamed and inflamed colorectal biopsy specimens (Figure 6B and 6C). Although cytoplasmic and total cellular PARP14 levels were also significantly elevated in extenders compared with nonextenders in noninflamed biopsy specimens, this was a trend in the inflamed biopsy specimens (Supplementary Figure 11). Although for technical reasons we could not restrict our quantification of the LP cells to a specific cell type, we did assess if PARP14 could be found colocalized with a monocyte marker CD68 in UC-associated biopsy specimens. We observed some positive costaining (Figure 6B), supporting that PARP14 could be associated with macrophages in the gut, a cell type that has been reported to be enriched in inflamed UC.25
Figure 6.

Colorectal localization of PARP14 in non-UC controls, UC extenders, and nonextenders. PARP14 (green), CD68 (red), and nuclei (blue) staining in colorectal biopsy specimens. (A) Representative staining from non-UC controls. Magnified images of positive areas are shown in the second column. No primary and isotype controls are shown in the far right panel. (B) Representative staining of inflamed and uninflamed colorectal biopsy specimens from patients with UC with and without subsequent disease extension. Magnified areas with representative CD68+ cells with nuclear PARP14 staining in biopsy specimens from an extender are shown on the far right (arrows) without 4’,6-diamidino-2-phenylindole (DAPI). (C) The frequency of LP cells with high PARP14 nuclear staining is higher in extenders as compared with nonextenders in both uninflamed (top) and inflamed (bottom) colorectal biopsy specimens. High nuclear staining was defined as an average nuclear intensity greater than the median positive value in the non-UC controls. A 1-way analysis of variance was used to compare groups. *P < .05. Scale bar; 100 μm.
Part C: Integrating Clinical, Genetic, and Molecular Features to Predict UC Extension
We next compared commonly used clinical endpoints and the recently introduced PGRS between subsequent extenders and nonextenders. No clinical endpoints, such as extent at diagnosis or C-reactive protein levels, were significantly associated with disease extension (Supplementary Table 1b). Patients with a higher UC PGRS had a higher probability of subsequently extending (adjusted P < .1, Figure 7A). We evaluated if a combination of clinical features, including C-reactive protein, age of diagnosis, extent at diagnosis, and Mayo endoscopic score, could discriminate between subsequent extenders and nonextenders. Using a lasso algorithm, the most predictive feature was a model using age at diagnosis but this model had a predictive performance with an area under the curve (AUC) of 0.61, nearly indistinguishable from random chance (Figure 7B). The addition of PGRS did not improve the predictive performance (AUC = 0.62).
Figure 7.
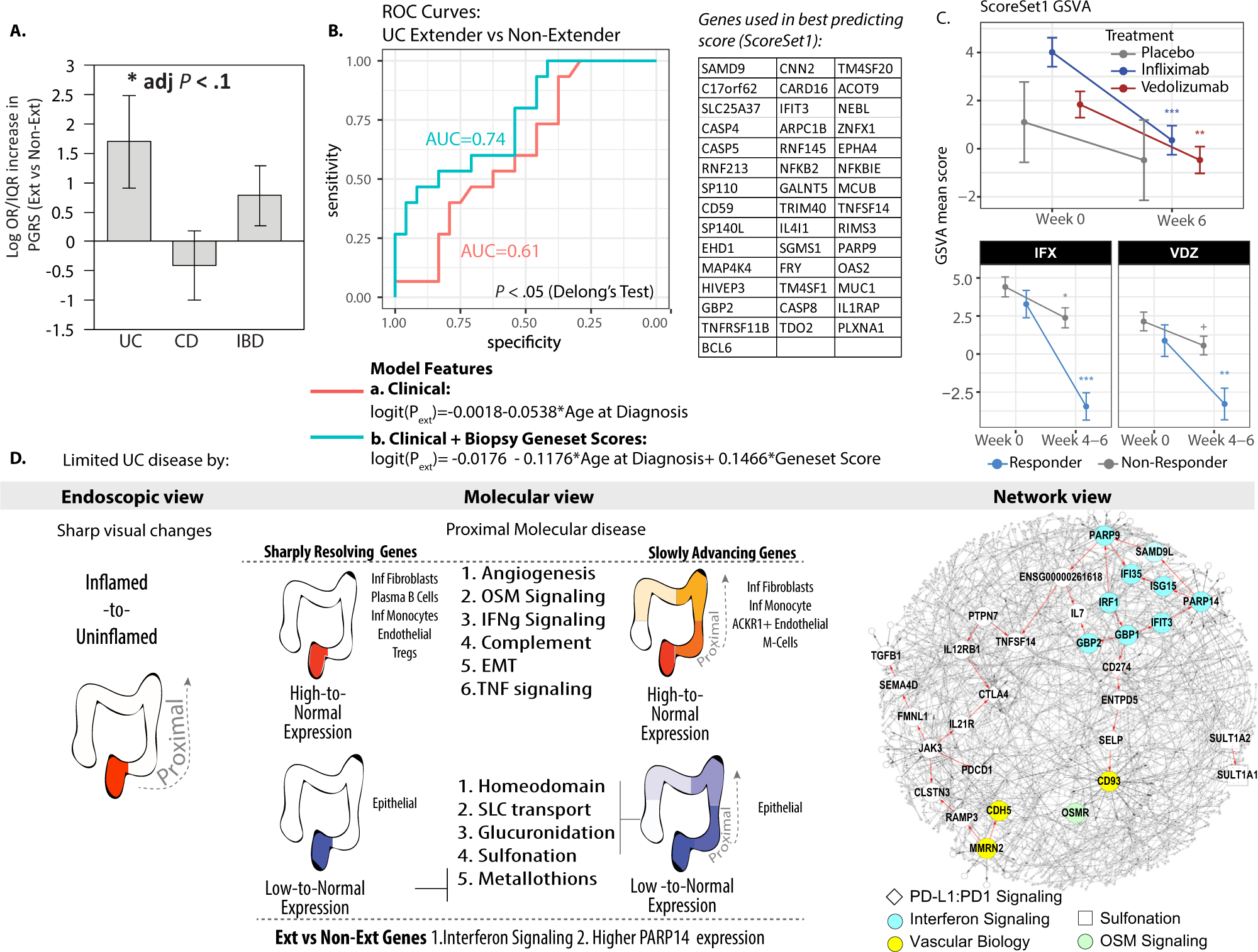
A subset of AdvG is predictive of extension. (A) Average PGRS of the MSCCR subcohort that subsequently extended vs. nonextenders. (B) Receiver operating characteristic curve for the prediction of extension in patients with limited UC. The red line summarizes the results of the best-performing model that uses clinical features and includes age at diagnosis only. The teal line summarizes the results of the model that showed that the inclusion of molecular scores to clinical features was more predictive than clinical features alone (1-sided P value = .049). The most predictive molecular feature was GSVA scores generated on the inflamed D0 biopsy specimens using genes found in common between the ExtG- and AdvG-associated subnetworks from Figure 5A (and Supplementary Table 18, Scoreset1). (C) GSVA mean score for Scoreset1 as determined in biopsy transcriptome data available from patients with UC (GSE73661) at baseline and after treatment with either vedolizumab (VDZ) or infliximab (IFX, upper panel) vs. placebo groups or according to treatment responders vs. nonresponders (lower panel). (D) Summary of limited UC by endoscopy, molecular, and network views. The granularity of information associated with molecular and network analysis can be appreciated as pathways and potential KDGs associated with the pathology are learned. +P < .1; *P < .05; **P < .01; ***P<.001.
Given this poor performance, we evaluated if molecular scores derived from the inflamed gut biopsy specimens of patients, taken before extension, could lead to a model with better predictive performance in combination with clinical features. Given their association with limited disease, we prioritized testing molecular scores derived from genes in the demarcation subnetwork analyses in Figure 5A (Supplementary Table 18), using GSVA. When the molecular scores were used in the lasso algorithm, 1 molecular set was identified that could discriminate subsequent extenders from nonextenders with an AUC of 0.74, a significantly higher performance than the model including clinical features only (P = .049, De Long test, Figure 7B). This best-performing molecular score (called ScoreSet1) included genes shared between the subnetwork of AdvG with ExtG. Patients with higher values in those scores had higher odds of being extensors (B = 0.147, log odds ratio), indicating a 50% increase in risk for patients with the top 10% score compared with those in the lower 10%.
We examined the genes associated with the ScoreSet1 and observed PARP9, a binding partner of the KDG, PARP14, was a member, suggesting that it may be one down-stream mediator of PARP14’s effect on colitis extension. ScoreSet1 genes were also related to TNFɑ signaling, thus we asked if anti-TNF or other biologic therapies may suppress expression of genes associated with UC extension. Using publicly available colonic transcriptome data,15 we observed that expression of ScoreSet1 genes was significantly decreased with either infliximab or vedolizumab treatment as well as in treatment responders compared with nonresponders (Figure 7C). Finally, we also observed higher expression of genes associated with patients with anti-TNF refractory UC26 in the inflamed biopsy specimens of subsequent extenders versus nonextenders (d = 3.95, P = .025).
Discussion
In this study we determined the molecular landscape at the boundary of the endoscopic-histologic–defined disease and at more proximal sites in patients with UC and probed whether these genes play a role in later disease extension. We statistically modeled expression changes in genes found dysregulated at the most upper limit of disease along the length of the gut, starting from the most proximally inflamed site from active E1 or E2 patients, moving segment by segment all the way to the ileum. We observed that gene expression changes were either abruptly resolved or gradually transitioned to normal control levels. Because the gradual extension genes indicate that disease extension has already begun, this suggests that molecular changes exist beyond the current endoscopic-histologic definition of UC demarcation. Importantly, subnetworks associated with the gradually transitioning genes, related to IFN signaling, exposed better predictors of disease extension than clinical indicators. Thus molecular expression levels may have utility in informing on clinical extension of disease. Finally, as scores generated from these molecular predictors of extension showed responsiveness to biologic therapy, our data suggest that anti-TNF therapy and vedolizumab could potentially suppress extension of disease in patients with limited UC.
An intriguing question is why the tissue beyond the line of demarcation “looks” normal, but our limited disease molecular analysis (Part A of the study) suggests it is not. One hypothesis stems from the cell type enrichments associated with the advancing genes, which were comparatively less extensive than those observed with abruptly resolving genes. The sharply resolving (high to normal) gene sets were indeed enriched in 16 different cell types. In contrast, the cell type enrichments in the advancing genes included only 8 cell types, some of which (eg, M cells) are known to express chemokines and play an important role in subsequent recruitment of immune cells to the gut mucosa.27 These observations might explain why a transcriptional signature of UC precedes overt macroscopic inflammation. What drives this cellular composition change, in sometimes an abrupt timeframe clinically, is still uncertain, however, the biology highlighted from our study can guide future studies.
Biological interpretation of the genes associated with limited UC was facilitated by our network analysis. Several KDGs we now implicate in clinically limited UC disease in this analysis have roles in vascular homeostasis (Figure 7D). The intestinal vascular endothelium barrier, defined as the region between the epithelial barrier and blood stream, is involved in regulation of immune cell transmigration, fluid homeostasis, and nutrient transport.28 IBD inflammation has been previously associated with altered angiogenesis but also a dysregulated vascular homeostasis with leaky vessels, edema, and irregular shaping, thus supporting our molecular observations.28,29 The identification of anti–endothelial cell antibodies in patients with UC30 also suggests an important role of the vascular endothelium in UC pathogenesis. Importantly, fibrogenic cells, such as the activated fibroblasts whose signature was detected in our analysis, can stand at the intersection between inflammation and angiogenesis and play a pivotal role in vascular remodeling.31 The KDGs CD93, CLEC14A, and THBD are members of the same C-type lectin domain group 14 family. Serum THBD is known to be elevated in patients with active UC, consistent with our gene expression data32 and soluble CD93 levels were shown to correlate with inflammation.33–35 Another KDG, MMRN2, is a ligand for CLEC14A and CD93.33,36 Interestingly, mice deficient in MMRN2 have impaired pericyte recruitment and increased vascular leakage in retinal vessels due to defects in CDH5 and adherens junctions.37 The MMRN2 subnetwork in our study contains CDH5 and was also significantly enriched in genes altered by CDH5 knockdown in mouse vascular endothelial cells38 (fold enrichment = 2.1, P = .003), thus supporting MMRN2:CDH5 molecular interactions also in UC. A recent study ties IFN signaling and vascular pathology by demonstrating that IFNɣ promotes IBD pathogenesis through disruption of endothelial CDH5 leading to vascular barrier dysfunction.28 Vessels without membrane CDH5 expression are more common in inflamed intestinal UC tissues compared with uninvolved tissues.28 Given our observations, we hypothesize that interactions between MMRN2, CD93, and CDH5 in the context of IFN signaling are altered both at the site of demarcation and beyond and could be possible new therapeutic avenues or biomarkers of UC disease.
IFN-signaling was identified as a key process at the intersection of genes associated with demarcation and subsequent extension determined in Part B of our study. This is consistent with literature showing extensive IFNɣ up-regulation in IBD and IFNɣ is an IBD GWAS locus.39,40 In our dataset, IFNɣ mRNA was identified as a gradually decreasing gene, but interestingly its expression changes in the subsequent proximal uninvolved biopsy segments reduced to levels even below those of healthy controls. Although the prominence of IFNɣ signaling in UC is well known, our network approaches highlight novel associations of IFN-associated genes in an IBD context, including PARP14 and PARP9. PARP9/14 have been previously associated with IFN signaling in nonintestinal tissues and co-regulate macrophage pro- and anti-inflammatory activation.24,41 PARP14 suppresses pro-inflammatory IFNɣ STAT1 signaling via ADP-ribosylation and activates the anti-inflammatory IL4-STAT6 pathway. PARP9 opposes PARP14’s effects through inhibiting its enzymatic function.41 PARP14 also has known nonenzymatic functionalities, namely, the regulation of the nuclear accumulation of a selected group of IFN-stimulated proteins on endotoxin stimulation in macrophages. Interestingly, this latter function is in line with our GSVA analysis using genes found down-regulated in LPS-stimulated PARP14-depleted macrophages. We observed higher expression of genes associated with reduced PARP14 expression in the inflamed UC biopsy specimens from subsequent extenders versus nonextenders, suggesting PARP14 activity is higher in subsequent extenders compared with nonextenders. In support of this molecular evidence, IF analysis showed higher PARP14 nuclear expression in inflamed biopsy specimens of patients that subsequently extended compared with those that did not. How higher PARP14 activity translates into potential risk of extension is uncertain, however, many ISG binding targets of PARP14, including PARP9, or PARP14’s ADP-ribosylation effects are candidates.24 For example, PARP9 in combination with another PARP14 ISG binding partner, DTX3L, has been shown to enhance IFN signaling by acting via a STAT1-associated component of type I IFN receptor signal transduction, thus promoting ISG expression in various cell types.42 Certainly, a balance in IFN signaling is important with deficiency leading to defects in immune system–mediated control of microbial pathogens but hyper-responsive IFN signaling linked to autoimmune diseases.43
A few challenges underscore our study’s strengths and weaknesses. We aimed to identify a cohort of patients with UC that were E1 and E2 at the time of their molecular characterization. Although our strict criteria, being endoscopic and histologic assessments with minimal inclusion of patients with limited disease due to remission, is a strength of our study, it is also a limitation because this significantly affected our sample size. Furthermore, we acknowledge that centralized reading of the endoscopy reports was not possible and could be a potential limitation given known discordances.44 However, all endoscopies were initially graded by IBD specialists and criteria for histologically normal mucosa were established by an expert IBD pathologist. The second challenge was the length of time for follow-up to identify which patients subsequently extended or remained limited in disease extent. Not all the patients had follow-up endoscopies within the Mount Sinai School of Medicine health system in addition to the rate of extension being relatively rare with only ~30% of patients with limited UC at diagnosis extending over 10 years.4 Despite this, we assessed extension status on 40 patients and found predictive markers. Finally, we were unable to test our molecular predictors in a replication cohort because we are unaware of any equivalent molecularly characterized cohort. In summary, molecular expression patterns identified at the site of demarcation and proximally, in combination with network approaches, has revealed novel aspects of the biology of UC, candidate biomarkers, and novel targets. We reveal gene signatures that as molecular scores in the inflamed biopsy of a patient with limited UC can predict disease extension with moderate accuracy (0.74) and better than available clinical metrics alone (Part C of study). Finally, from a clinical standpoint, our data potentially suggest that there is a subgroup of patients with limited UC (eg, those with higher-risk molecular features) who could benefit from earlier use of targeted biologic therapy to prevent disease extension.
Supplementary Material
Supplementary Figure 1. Effect of medication on proximal disease genes. A subset analysis was performed to evaluate the effect of rectal medication use on the expression of demarcation genes (DemG) with either the gradually advancing pattern (AdvG) (A) or the sharply reverting genes (RevG) (D). The Venn diagram shows the overlap of the signatures as determined either in the medicated or nonmedicated group and as compared with the analysis performed in the combined group. A scatter plot showing the correlation between effects on gene expression in rectal vs. nonrectal medication group (B and E). Violin plot summarizing the expression of AdvG (C) and the ResG (F) in inflamed and uninflamed biopsy specimens.
{kind=link}
Supplementary Figure 2. Clustering analysis reveals a delayed expression pattern. Heatmap summarizing the normalized expression levels of genes found differentially expressed in the medicated or nonmedicated users that have the sharply reverting expression change relative to D0. Two key patterns were recognized, a set of genes with a delayed gene expression resolution, occurring at D2 in the nonmedicated group compared with D1 in the medicated group. These genes were called ResG@D2 genes. The second pattern identifies genes whose expression was resolved at D1 with medication use but, in the nonmedicated group, these were resolved at D1 but with a smaller effect size compared with the medicated group.
{kind=link}
Supplementary Figure 3. Pathway enrichment analysis of demarcation disease genes that are elevated with inflammation that are slowly or sharply resolving. (A) KEGG, BioPlanet, and Reactome pathway enrichment analysis of proximal disease genes with high-to-normal changes in expression. Nodes depict the enriched terms and the coloring represents whether the pathways were commonly enriched (gray) in or specifically enriched in AdvG (red) or ResG (blue). Note, the ResG were queried as a single gene set (ie, either resolved @D1 or @D2). (B) A subset of the pathways from A are shown with edges connected to the genes belonging to the pathway terms. Only pathways that were significant with Bonferroni adjusted P < .05 were included.
{kind=link}
Supplementary Figure 4. Gene ontology (GO) pathway enrichment analysis of demarcation disease genes. Proximal disease genes that have a high-to-normal expression pattern change were queried for functional enrichment using the GO biological function database. (A) Nodes represent the significantly enriched pathways with node color representing pathways that are either commonly enriched for (grey) or more specifically enriched in ResG (blue) or AdvG (red). Edges connect pathway nodes with overlapping gene membership to show redundancy of terms. Only pathways that were significantly enriched with Bonferroni adjusted P < .05 are shown. (B) A subset of GO terms identified in panel A are highlighted with edges connected to the genes that are associated with the pathway and are found in either the ResG (blue node) or AdvG (red nodes) gene set.
{kind=link}
Supplementary Figure 5. Gut cell type enrichment analysis. (A) The demarcation gene sets, as defined in Figure 2, were queried against gene expression signatures curated from Smillie et al17 that associate with epithelial, immune, and stromal cell types as determined in colons of patients with UC. Fold enrichment and level of significance with Benjamini-Hochberg adjusted P values are represented, and only results found significant at P < .1 are shown. (B) Gene sets associated with a distinct coexpressed cellular module in CD inflamed biopsy specimens called GIMATs, which contained immunoglobulin G plasma cells, inflammatory mononuclear phagocytes, and activated T and stromal cells. (C) Gene sets associated with angiogenesis20,21 or (D) fibrosis disease models.19
{kind=link}
Supplementary Figure 6. Pathway enrichment analysis of demarcation disease genes that are sharply resolved at D2 due to the lack of medication. Demarcation disease genes that have either a low-to-normal or high-to-normal expression pattern change and are delayed in resolution at D2 instead of D1 with nonmedication use were queried for functional enrichment using KEGG, Reactome, or BioPlanet databases. Nodes represent the significantly enriched pathways with node color representing pathways that have overlapping gene members with the edges connecting genes associated with the pathway terms. The node size represents the level of significance. Only pathways that were significantly enriched with Bonferroni adjusted P <.05 are shown.
{kind=link}
Supplementary Figure 7. KDGs associated with proximal disease identified gene sets. Demarcation disease genes, either the ResG (sharply resolving) and AdvG (slow advancing) were queried in the UC BGRN and the most connected subnetworks were extracted allowing for 1 nearest neighboring gene to be included. The Venn diagram (A) shows the overlap of genes of the 3 gene set–associated subnetworks. KDA was performed for each demarcation gene–associated subnetwork of genes and then summarized according to the frequency of recurrence as a KDG across the 3 subnetworks tested. The bar plot includes genes found recurring in either 3 or 2 of 3 subnetworks tested and the color of the bar represents the subnetwork it is a predicted KDG for, either blue for AdvG, orange for RevG@D1, or red for RevG@D2. The table on the right summarizes those genes predicted to be KDGs in only 1 of 3 tested subnetworks.
{kind=link}
Supplementary Figure 8. Bioplanet pathway enrichments of the AdvG also found differentially expressed between uninvolved and healthy controls. An analysis was performed comparing transcriptomes between noninvolved biopsy specimens of the patients with limited UC with region-matched biopsy specimens collected from healthy controls. At a nominal P value <.05 a subset of the genes (~377) were found to be in common with the AdvG and these genes, either as up- or down-regulated gene sets, were queried for pathway enrichment analysis. BioPlanet Pathways that were found significantly enriched with Benjamini-Hochberg adjusted P < .1 are shown.
{kind=link}
Supplementary Figure 9. The disease genes are over-represented in genes associated with subsequent extension as determined using gene set enrichment analysis (GSEA). A subcohort analysis was performed comparing transcriptomes from inflamed biopsy specimens of patients that subsequently extended to those that did not (ExtG). The ExtG were then ranked according to log fold change differences between extenders vs. nonextenders and the rank order was tested for enrichment in genes associated with the demarcation disease gene sets (AdvG and ResG). The trace of the enrichment scores is shown in the figures for the various gene sets tested and the normalized enrichment score and associated FDR adjusted P values are shown in the inset of each panel. These data support significant enrichment of the ExtG with those that are altered in expression proximally.
{kind=link}
Supplementary Figure 10. Subnetwork associated with common KDGs between extender and demarcation gene sets are enriched in IFN signaling. A table summarizing the top enrichments of the shared KDG-associated subnetwork in Figure 5B but with 3 additional layers (~500 nodes in total). Fold enrichment and Benjamini-Hochberg adjusted P values for the Bioplanet; gut cell type; macrophage cytokine perturbation22; PARP14 knockout vs. wild-type RAW264 macrophage experiment24; and IFNalpha-stimulated human umbilical vein endothelial cells23 gene expression signature sets are shown.
{kind=link}
Supplementary Figure 11. Quantification of PARP14 in UC extender and UC nonextender colonic biopsy specimens. PARP14 and 4’6-diamidino-2-phenylindole nuclear staining in colorectal biopsy specimen was performed and used for quantification. The frequency of LP cells with high PARP14 nuclear, cellular (nuclear + cytoplasmic), or cytoplasmic staining was determined in controls and UC extenders and compared with nonextenders in both uninflamed (top row) and inflamed (bottom row) colorectal biopsy specimens. A 1-way analysis of variance was used to compare groups. *P < .05.
{kind=link}
WHAT YOU NEED TO KNOW.
BACKGROUND AND CONTEXT
In ulcerative colitis (UC), disease extent, which varies from proctitis to pancolitis, is a major prognostic factor. What defines the frequently observed sharp demarcation between macroscopically involved and uninvolved areas in limited UC and what is driving its subsequent extension in some patients are unknown.
NEW FINDINGS
We observed molecular changes beyond the current endoscopic-histologic definition of UC demarcation. Gene subnetworks associated with these genes were related to interferon signaling and uncovered better predictors of disease extension than clinical indicators. Poly(ADP-ribose) polymerase 14 (PARP14) levels were found to be associated with subsequent disease extension.
LIMITATIONS
The precise mechanisms mediating the effect of PARP14 on disease extension are unknown. A validation cohort of patients with UC limited disease including some with disease extension was not available.
IMPACT
This study provides one of the first insights regarding a key clinical characteristic of UC. Scores generated from the molecular predictors of extension showed responsiveness to biologic therapy suggesting that anti–tumor necrosis factor therapy and vedolizumab could potentially suppress extension of disease in patients with limited UC.
Acknowledgments
All sample processing was provided by Human Immune Monitoring Center at Icahn School of Medicine at Mount Sinai. We are grateful for assistance by Icahn School of Medicine IBD clinicians and patients who participated in the study.
Funding
The sampling of the Inflammatory Bowel Disease cohort (Crohn’s disease and ulcerative colitis) was jointly designed as part of the research alliance between Janssen Biotech, Inc. and The Icahn School of Medicine at Mount Sinai. M.S., R.K., B.L., A.D., L.P., H.I., W.W., R.H. (Ruiqi Huang), K.H., E.E.S., J.Z., M.C.D., B.E.S., A.K., and C.A. were partially funded as part of research alliance between Janssen Biotech and The Icahn School of Medicine at Mount Sinai. C.A., L.P., E.E.S., and G.W. were supported in part by The Leona M. and Harry B. Helmsley Charitable Trust. M.T. was supported by the Digestive Disease Research Foundation. R.C.U. is supported by a National Institutes of Health K23 Career Development Award (K23KD111995-01A1). This work was supported in part through the computational resources and staff expertise provided by Scientific Computing at the Icahn School of Medicine at Mount Sinai.
Conflicts of interest
M.S., R.K., B.L., A.D., L.P., W.W., R.H. (Ruiqi Huang), H.I., K.H., E.E.S., J.Z., M.C.D., B.E.S., A.K., and C.A. were partially funded as part of research alliance between Janssen Biotech and The Icahn School of Medicine at Mount Sinai. M.C., A.S., and C.B. are employees at Janssen Research and Development. J.R.F. is a former employee at Janssen Research and Development and is currently employed at Alnylam Pharmaceuticals. B.E.S. discloses the following: consulting fees from 4D Pharma, Abbvie, Allergan, Amgen, Arena Pharmaceuticals, AstraZeneca, Boehringer Ingelheim, Boston Pharmaceuticals, Capella Biosciences, Celgene, Celltrion Healthcare, EnGene, Ferring, Genentech, Gilead, Hoffmann-La Roche, Immunic, Ironwood Pharmaceuticals, Janssen, Lilly, Lyndra, MedImmune, Morphic Therapeutic, Oppilan Pharma, OSE Immunotherapeutics, Otsuka, Palatin Technologies, Pfizer, Progenity, Prometheus Laboratories, Redhill Biopharma, Rheos Medicines, Seres Therapeutics, Shire, Synergy Pharmaceuticals, Takeda, Target PharmaSolutions, Theravance Biopharma R&D, TiGenix, and Vivelix Pharmaceuticals; honoraria for speaking in CME programs from Takeda, Janssen, Lilly, Gilead, Pfizer, and Genetech; and research funding from Celgene, Pfizer, Takeda, Theravance Biopharma R&D, and Janssen. M.C.D. discloses consulting fees from Abbvie, Allergan, Amgen, Arena Pharmaceuticals, AstraZeneca, Boehringer Ingelheim, Celgene, Ferring, Genentech, Gilead, Hoffmann-La Roche, Janssen, Pfizer, Prometheus Biosciences, Takeda, and Target PharmaSolutions and research funding from Abbvie, Janssen, Pfizer, and Prometheus Biosciences Takeda. J.F.C. reports the following: receiving research grants from AbbVie, Janssen Pharmaceuticals, and Takeda; receiving payment for lectures from AbbVie, Amgen, Allergan, Inc., Ferring Pharmaceuticals, Shire, and Takeda; receiving consulting fees from AbbVie, Amgen, Arena Pharmaceuticals, Boehringer Ingelheim, Bristol Myers Squibb, Celgene Corporation, Eli Lilly, Ferring Pharmaceuticals, Galmed Research, Glaxo Smith Kline, Geneva, Iterative Scopes, Janssen Pharmaceuticals, Kaleido Biosciences, Landos, Otsuka, Pfizer, Prometheus, Sanofi, Takeda, and TiGenix; and holding stock options in Intestinal Biotech Development. E.E.S., K.H., A.K., and J.Z. are associated with Sema4. N.H. is a consultant for and has service agreements with Abbvie, Celgene, and Lilly USA. S.M. has received investigator-initiated grant funding from Takeda Pharma and Genentech and has served as consultant or paid speaker for Takeda Pharma, Genentech, Morphic, and Glaxo Smith Kline. S.M. and J.F.C. have an unrestricted, investigator-initiated grant from Takeda Pharmaceuticals to examine novel homing mechanisms to the gastrointestinal tract.
Abbreviations used in this paper:
- AdvG
advancing genes
- AUC
area under the curve
- BGRN
Bayesian gene regulatory network
- ExtG
extender genes
- FCH
fold change
- FDR
false discovery rate
- GSVA
gene set variation analysis
- IBD
inflammatory bowel disease
- IF
immunofluorescence
- IFN
interferon
- IL
interleukin
- ISG
interferon-stimulated gene
- KDG
key driver gene
- LP
lamina propria
- LPS
liposaccharide
- mRNA
messenger RNA
- MSCCR
Mount Sinai Crohn’s and Colitis Registry
- PARP14
poly(ADP-ribose) polymerase 14
- PGRS
polygenic risk score
- ResG
resolving genes
- TNF
tumor necrosis factor
- UC
ulcerative colitis
Footnotes
Supplementary Material
Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at http://doi.org/10.1053/j.gastro.2021.08.053.
References
- 1.Ungaro R, Mehandru S, Allen PB, et al. Ulcerative colitis. Lancet 2017;389:1756–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Silverberg MS, Satsangi J, Ahmad T, et al. Toward an integrated clinical, molecular and serological classification of inflammatory bowel disease: report of a Working Party of the 2005 Montreal World Congress of Gastroenterology. Can J Gastroenterol 2005;19(Suppl A):5A–36A. [DOI] [PubMed] [Google Scholar]
- 3.Burisch J, Ungaro R, Vind I, et al. Proximal disease extension in patients with limited ulcerative colitis: a Danish population-based inception cohort. J Crohns Colitis 2017;11:1200–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roda G, Narula N, Pinotti R, et al. Systematic review with meta-analysis: proximal disease extension in limited ulcerative colitis. Aliment Pharmacol Ther 2017;45:1481–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qiu Y, Chen B, Li Y, et al. Risk factors and long-term outcome of disease extent progression in Asian patients with ulcerative colitis: a retrospective cohort study. BMC Gastroenterol 2019;19:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Furey TS, Sethupathy P, Sheikh SZ. Redefining the IBDs using genome-scale molecular phenotyping. Nat Rev Gastroenterol Hepatol 2019;16:296–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marigorta UM, Denson LA, Hyams JS, et al. Transcriptional risk scores link GWAS to eQTLs and predict complications in Crohn’s disease. Nat Genet 2017; 49:1517–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee JC, Lyons PA, McKinney EF, et al. Gene expression profiling of CD8+ T cells predicts prognosis in patients with Crohn disease and ulcerative colitis. J Clin Invest 2011;121:4170–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gasparetto M, Payne F, Nayak K, et al. Transcription and DNA methylation patterns of blood-derived CD8(+) T cells are associated with age and inflammatory bowel disease but do not predict prognosis. Gastroenterology 2021;160:232–244.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parkes M, Noor NM, Dowling F, et al. PRedicting Outcomes For Crohn’s dIsease using a moLecular biomarkEr (PROFILE): protocol for a multicentre, randomised, biomarker-stratified trial. BMJ Open 2018;8: e026767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haberman Y, Karns R, Dexheimer PJ, et al. Ulcerative colitis mucosal transcriptomes reveal mitochondriopathy and personalized mechanisms underlying disease severity and treatment response. Nat Commun 2019; 10:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peters LA, Perrigoue J, Mortha A, et al. A functional genomics predictive network model identifies regulators of inflammatory bowel disease. Nat Genet 2017; 49:1437–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suarez-Farinas M, Tokuyama M, Wei G, et al. Intestinal inflammation modulates the expression of ACE2 and TMPRSS2 and potentially overlaps with the pathogenesis of SARS-CoV-2- related disease. Gastroenterology 2021;160:287–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanzelmann S, Castelo R, Guinney J. GSVA: gene se variation analysis for microarray and RNA-seq data. BMC Bioinformatics 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arijs I, De Hertogh G, Lemmens B, et al. Effect of vedolizumab (anti-α4β7-integrin) therapy on histological healing and mucosal gene expression in patients with UC. Gut 2018;67:43. [DOI] [PubMed] [Google Scholar]
- 16.Janky R, Verfaillie A, Imrichova H, et al. iRegulon: from a gene list to a gene regulatory network using large motif and track collections. PLoS Comput Biol 2014;10: e1003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smillie CS, Biton M, Ordovas-Montanes J, et al. Intraand inter-cellular rewiring of the human colon during ulcerative colitis. Cell 2019;178:714–730.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin JC, Chang C, Boschetti G, et al. Single-cell analysis of Crohn’s disease lesions identifies a pathogenic cellular module associated with resistance to Anti-TNF therapy. Cell 2019;178:1493–1508.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramachandran P, Dobie R, Wilson-Kanamori JR, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 2019;575:512–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Masiero M, Simoes FC, Han HD, et al. A core human primary tumor angiogenesis signature identifies the endothelial orphan receptor ELTD1 as a key regulator of angiogenesis. Cancer Cell 2013;24:229–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guarischi-Sousa R, Monteiro JS, Alecrim LC, et al. A transcriptome-based signature of pathological angiogenesis predicts breast cancer patient survival. PLoS Genet 2019;15:e1008482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xue J, Schmidt SV, Sander J, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014;40:274–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Indraccolo S, Pfeffer U, Minuzzo S, et al. Identification of genes selectively regulated by IFNs in endothelial cells. J Immunol 2007;178:1122–1135. [DOI] [PubMed] [Google Scholar]
- 24.Caprara G, Prosperini E, Piccolo V, et al. PARP14 controls the nuclear accumulation of a subset of type I IFN-inducible proteins. J Immunol 2018;200:2439–2454. [DOI] [PubMed] [Google Scholar]
- 25.Liu H, Dasgupta S, Fu Y, et al. Subsets of mononuclear phagocytes are enriched in the inflamed colons of patients with IBD. BMC Immunol 2019;20:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.West NR, Hegazy AN, Owens BMJ, et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat Med 2017;23:579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bigaeva E, Uniken Venema WTC, Weersma RK, et al. Understanding human gut diseases at single-cell resolution. Hum Mol Genet 2020;29:R51–R58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Langer V, Vivi E, Regensburger D, et al. IFN-gamma drives inflammatory bowel disease pathogenesis through VE-cadherin-directed vascular barrier disruption. J Clin Invest 2019;129:4691–4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Danese S, Sans M, de la Motte C, et al. Angiogenesis as a novel component of inflammatory bowel disease pathogenesis. Gastroenterology 2006;130:2060–2073. [DOI] [PubMed] [Google Scholar]
- 30.Aldebert D, Notteghem B, Reumaux D, et al. Anti-endothelial cell antibodies in sera from patients with inflammatory bowel disease. Gastroenterol Clin Biol 1995;19:867–870. [PubMed] [Google Scholar]
- 31.Lawrance IC, Rogler G, Bamias G, et al. Cellular and molecular mediators of intestinal fibrosis. J Crohns Colitis 2017;11:1491–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boehme MW, Autschbach F, Zuna I, et al. Elevated serum levels and reduced immunohistochemical expression of thrombomodulin in active ulcerative colitis. Gastroenterology 1997;113:107–117. [DOI] [PubMed] [Google Scholar]
- 33.Borah S, Vasudevan D, Swain RK. C-type lectin family XIV members and angiogenesis. Oncol Lett 2019; 18:3954–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malarstig A, Silveira A, Wagsater D, et al. Plasma CD93 concentration is a potential novel biomarker for coronary artery disease. J Intern Med 2011;270:229–236. [DOI] [PubMed] [Google Scholar]
- 35.Park HJ, Oh EY, Han HJ, et al. Soluble CD93 in allergic asthma. Sci Rep 2020;10:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khan KA, Naylor AJ, Khan A, et al. Multimerin-2 is a ligand for group 14 family C-type lectins CLEC14A, CD93 and CD248 spanning the endothelial pericyte interface. Oncogene 2017;36:6097–6108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pellicani R, Poletto E, Andreuzzi E, et al. Multimerin-2 maintains vascular stability and permeability. Matrix Biol 2020;87:11–25. [DOI] [PubMed] [Google Scholar]
- 38.Morini MF, Giampietro C, Corada M, et al. VE-cadherin-mediated epigenetic regulation of endothelial gene expression. Circ Res 2018;122:231–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Verma R, Verma N, Paul J. Expression of inflammatory genes in the colon of ulcerative colitis patients varies with activity both at the mRNA and protein level. Eur Cytokine Netw 2013;24:130–138. [DOI] [PubMed] [Google Scholar]
- 40.Jostins L, Ripke S, Weersma RK, et al. Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012;491:119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iwata H, Goettsch C, Sharma A, et al. PARP9 and PARP14 cross-regulate macrophage activation via STAT1 ADP-ribosylation. Nat Commun 2016;7:12849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Y, Mao D, Roswit WT, et al. PARP9-DTX3L ubiquitin ligase targets host histone H2BJ and viral 3C protease to enhance interferon signaling and control viral infection. Nat Immunol 2015;16:1215–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pott J, Stockinger S. Type I and III interferon in the gut: tight balance between host protection and immunopathology. Front Immunol 2017;8:258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gottlieb K, Daperno M, Usiskin K, et al. Endoscopy and central reading in inflammatory bowel disease clinical trials: achievements, challenges and future developments. Gut 2021;70:418–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Effect of medication on proximal disease genes. A subset analysis was performed to evaluate the effect of rectal medication use on the expression of demarcation genes (DemG) with either the gradually advancing pattern (AdvG) (A) or the sharply reverting genes (RevG) (D). The Venn diagram shows the overlap of the signatures as determined either in the medicated or nonmedicated group and as compared with the analysis performed in the combined group. A scatter plot showing the correlation between effects on gene expression in rectal vs. nonrectal medication group (B and E). Violin plot summarizing the expression of AdvG (C) and the ResG (F) in inflamed and uninflamed biopsy specimens.
Supplementary Figure 2. Clustering analysis reveals a delayed expression pattern. Heatmap summarizing the normalized expression levels of genes found differentially expressed in the medicated or nonmedicated users that have the sharply reverting expression change relative to D0. Two key patterns were recognized, a set of genes with a delayed gene expression resolution, occurring at D2 in the nonmedicated group compared with D1 in the medicated group. These genes were called ResG@D2 genes. The second pattern identifies genes whose expression was resolved at D1 with medication use but, in the nonmedicated group, these were resolved at D1 but with a smaller effect size compared with the medicated group.
Supplementary Figure 3. Pathway enrichment analysis of demarcation disease genes that are elevated with inflammation that are slowly or sharply resolving. (A) KEGG, BioPlanet, and Reactome pathway enrichment analysis of proximal disease genes with high-to-normal changes in expression. Nodes depict the enriched terms and the coloring represents whether the pathways were commonly enriched (gray) in or specifically enriched in AdvG (red) or ResG (blue). Note, the ResG were queried as a single gene set (ie, either resolved @D1 or @D2). (B) A subset of the pathways from A are shown with edges connected to the genes belonging to the pathway terms. Only pathways that were significant with Bonferroni adjusted P < .05 were included.
Supplementary Figure 4. Gene ontology (GO) pathway enrichment analysis of demarcation disease genes. Proximal disease genes that have a high-to-normal expression pattern change were queried for functional enrichment using the GO biological function database. (A) Nodes represent the significantly enriched pathways with node color representing pathways that are either commonly enriched for (grey) or more specifically enriched in ResG (blue) or AdvG (red). Edges connect pathway nodes with overlapping gene membership to show redundancy of terms. Only pathways that were significantly enriched with Bonferroni adjusted P < .05 are shown. (B) A subset of GO terms identified in panel A are highlighted with edges connected to the genes that are associated with the pathway and are found in either the ResG (blue node) or AdvG (red nodes) gene set.
Supplementary Figure 5. Gut cell type enrichment analysis. (A) The demarcation gene sets, as defined in Figure 2, were queried against gene expression signatures curated from Smillie et al17 that associate with epithelial, immune, and stromal cell types as determined in colons of patients with UC. Fold enrichment and level of significance with Benjamini-Hochberg adjusted P values are represented, and only results found significant at P < .1 are shown. (B) Gene sets associated with a distinct coexpressed cellular module in CD inflamed biopsy specimens called GIMATs, which contained immunoglobulin G plasma cells, inflammatory mononuclear phagocytes, and activated T and stromal cells. (C) Gene sets associated with angiogenesis20,21 or (D) fibrosis disease models.19
Supplementary Figure 6. Pathway enrichment analysis of demarcation disease genes that are sharply resolved at D2 due to the lack of medication. Demarcation disease genes that have either a low-to-normal or high-to-normal expression pattern change and are delayed in resolution at D2 instead of D1 with nonmedication use were queried for functional enrichment using KEGG, Reactome, or BioPlanet databases. Nodes represent the significantly enriched pathways with node color representing pathways that have overlapping gene members with the edges connecting genes associated with the pathway terms. The node size represents the level of significance. Only pathways that were significantly enriched with Bonferroni adjusted P <.05 are shown.
Supplementary Figure 7. KDGs associated with proximal disease identified gene sets. Demarcation disease genes, either the ResG (sharply resolving) and AdvG (slow advancing) were queried in the UC BGRN and the most connected subnetworks were extracted allowing for 1 nearest neighboring gene to be included. The Venn diagram (A) shows the overlap of genes of the 3 gene set–associated subnetworks. KDA was performed for each demarcation gene–associated subnetwork of genes and then summarized according to the frequency of recurrence as a KDG across the 3 subnetworks tested. The bar plot includes genes found recurring in either 3 or 2 of 3 subnetworks tested and the color of the bar represents the subnetwork it is a predicted KDG for, either blue for AdvG, orange for RevG@D1, or red for RevG@D2. The table on the right summarizes those genes predicted to be KDGs in only 1 of 3 tested subnetworks.
Supplementary Figure 8. Bioplanet pathway enrichments of the AdvG also found differentially expressed between uninvolved and healthy controls. An analysis was performed comparing transcriptomes between noninvolved biopsy specimens of the patients with limited UC with region-matched biopsy specimens collected from healthy controls. At a nominal P value <.05 a subset of the genes (~377) were found to be in common with the AdvG and these genes, either as up- or down-regulated gene sets, were queried for pathway enrichment analysis. BioPlanet Pathways that were found significantly enriched with Benjamini-Hochberg adjusted P < .1 are shown.
Supplementary Figure 9. The disease genes are over-represented in genes associated with subsequent extension as determined using gene set enrichment analysis (GSEA). A subcohort analysis was performed comparing transcriptomes from inflamed biopsy specimens of patients that subsequently extended to those that did not (ExtG). The ExtG were then ranked according to log fold change differences between extenders vs. nonextenders and the rank order was tested for enrichment in genes associated with the demarcation disease gene sets (AdvG and ResG). The trace of the enrichment scores is shown in the figures for the various gene sets tested and the normalized enrichment score and associated FDR adjusted P values are shown in the inset of each panel. These data support significant enrichment of the ExtG with those that are altered in expression proximally.
Supplementary Figure 10. Subnetwork associated with common KDGs between extender and demarcation gene sets are enriched in IFN signaling. A table summarizing the top enrichments of the shared KDG-associated subnetwork in Figure 5B but with 3 additional layers (~500 nodes in total). Fold enrichment and Benjamini-Hochberg adjusted P values for the Bioplanet; gut cell type; macrophage cytokine perturbation22; PARP14 knockout vs. wild-type RAW264 macrophage experiment24; and IFNalpha-stimulated human umbilical vein endothelial cells23 gene expression signature sets are shown.
Supplementary Figure 11. Quantification of PARP14 in UC extender and UC nonextender colonic biopsy specimens. PARP14 and 4’6-diamidino-2-phenylindole nuclear staining in colorectal biopsy specimen was performed and used for quantification. The frequency of LP cells with high PARP14 nuclear, cellular (nuclear + cytoplasmic), or cytoplasmic staining was determined in controls and UC extenders and compared with nonextenders in both uninflamed (top row) and inflamed (bottom row) colorectal biopsy specimens. A 1-way analysis of variance was used to compare groups. *P < .05.