

Abstract
Background:
Current drug treatments have limited efficacy and intolerable side effects in advanced-to-end-stage Parkinson’s disease (advPD). D1 agonists have the potential to provide symptomatic benefit, but there are no data on safety and feasibility in this population.
Methods:
A two-week, randomized, double blind, crossover phase Ib study was performed in advPD patients to compare standard-of-care (SoC) carbidopa/levodopa with PF-06412562, a selective D1/D5 dopamine receptor partial agonist. Each week, there was a Day 1 baseline evaluation with overnight levodopa washout, then treatment on Days 2 and 3 with either SoC or PF-06412562 (split dose 25+20 mg), followed by discharge on Day 4. Primary endpoints were safety and tolerability. Secondary endpoints were global clinical impression of change (GCI-C) rated by clinicians and caregivers.
Results:
Eight advPD patients and their caregivers consented to participate and six were randomized (average disease duration: 22 y). None withdrew voluntarily. One participant with baseline Day 1 dehydration, pre-renal kidney injury, and autonomic dysfunction experienced symptomatic and serious hypotension after receiving PF-06412562 in Week 1 and was discontinued from the study. All other adverse events were rated mild (PF-06412562: n=1, SoC: n=0), moderate (PF-06412562: n=1, SoC: n=1), or severe but non-serious (PF-06412562: n=3, SoC: n=2). No clinically meaningful laboratory changes were observed. Among the five participants who completed the study, GCI-C favored PF-06412562 in two per clinicians’ and four participants per caregivers’ rating.
Conclusions:
PF-06412562 was tolerated in advPD patients. This study provides the feasibility for future safety and efficacy studies in this population with unmet needs.
Trial Registration#:
Keywords: D1 dopamine receptor, dopamine D1 agonists, advanced Parkinson’s disease, levodopa, safety, feasibility
Introduction
The dopamine precursor levodopa has provided marked symptomatic relief and prolonged life expectancy in Parkinson’s disease (PD) [1], but with progressive loss of dopaminergic neurons, conversion of levodopa to dopamine becomes less efficient/predictable. Coupled with “off-target” effects [2], there is loss of responsiveness and/or inability to tolerate levodopa. Newer levodopa formulations are somewhat helpful, but do not address the fundamental challenges and often render patients wheelchair-/bed-bound [3, 4]. Theoretically, this could be overcome by mimicking dopamine neurotransmission directly, using a class of drug commonly called “dopamine agonists” to activate postsynaptic dopamine receptors. Indeed, there are many “dopamine agonists” (e.g., pramipexole, ropinirole, etc.) that are or have been approved. In actuality, all approved “dopamine agonists” are misnamed because they are more selective for the dopamine D2-like subfamily (especially the D2 and D3) [5], but not D1-like (D1 and D5) subfamily. Although they reduce motor fluctuations and levodopa-induced dyskinesia in early PD, they are much less efficacious than levodopa [6], and cause intolerable side effects such as hallucinations and confusion that are linked to D2-like receptor stimulation in a significant fraction of advanced-to-end stage PD (advPD) [7–9].
The distribution of dopamine receptors in the basal ganglia is known to be highly segregated. Unlike D2-receptors, D1 receptors are expressed almost solely on postsynaptic striatal GABAergic neurons of the direct pathway that we hypothesized were of specific importance in treating PD [10]. Because the striatal GABAergic cells do not degenerate as do nigral dopamine neurons, we hypothesized that D1-like agonists could be highly efficacious antiparkinson drugs [11, 12]. The first generation of selective full D1 agonists (dihydrexidine and ABT-431) led to proof-of-concept data by their profound antiparkinsonian effects in both MPTP-treated non-human primates [13] and PD patients [14–16].
Recent discovery of orally available D1 agonists [17], the second generation of D1 agonists, has overcome many pharmacokinetic limitations of the first generation compounds [6, 18] and has the potential to impact clinical practice. Promisingly, one of these second generation D1 agonists is in early Phase III trials. The focus of the ongoing D1 trials is not advPD, but earlier-stage patients who still respond to levodopa. This choice is understandable because advPD patients are medically complex, and to our knowledge, have never been the subject of a controlled interventional trial despite their extremely poor quality of life and their unmet needs.
The following lines of preclinical evidence suggest that D1 agonists will be beneficial for advPD patients. First, the D1 agonist dihydrexidine was more effective in severely parkinsonian MPTP non-human primates than in moderately affected animals [19]. Second, this full D1 agonist caused profound antiparkinson effects in two MPTP-treated primates unresponsive to levodopa or the D2 agonist bromocriptine [20]. The availability of efficacious orally available second generation D1 agonists with excellent pharmacokinetics (e.g., PF-06649751 and PF-06412562 [21]) made it both essential and imperative to determine if studies of D1 agonists can be conducted in patients with advPD, and to assess whether advanced patients/family were willing and able to participate.
Based on pharmacokinetic considerations, we chose to use PF-06412562 rather than a related compound PF-06649751. Although the latter has somewhat higher D1 intrinsic activity, it is currently dosed via a tapering over several weeks to achieve full therapeutic levels [22], whereas PF-06412562 showed acute antiparkinsonian effects in 13 PD patients and was well-tolerated at a 50 mg oral dose (t½ 6.4 h) split into 30 and 20 mg doses given four hours apart, eliminating the need for gradual tapering [23]. To provide proof-of-concept that it is feasible to study D1/5 agonists in advPD, we conducted a randomized, double blind, crossover, phase Ib, mixed methodology, clinical trial evaluating PF-06412562 versus standard-of-care (SoC) carbidopa/levodopa in advPD patients.
Methods
Trial drug
PF-06412562 (Figure 1) is a non-catechol, D1/5 partial agonist (KI 95 nM for human D1 receptor; adenylate cyclase EC50 580 nM; Emax 44%) The drug and matched placebo were provided by Pfizer. Based on a previous study showing safety and tolerability of a 50 mg oral split dose of PF-06412562, [23], we chose a slightly lower total daily dosage of 45 mg (split 25 and 20 mg) for advPD patients. Sinemet (carbidopa/levodopa, 25/100 mg, encapsulated) was used as reference, with SoC dose based on each participant’s pre-trial total levodopa usage.
Figure 1.
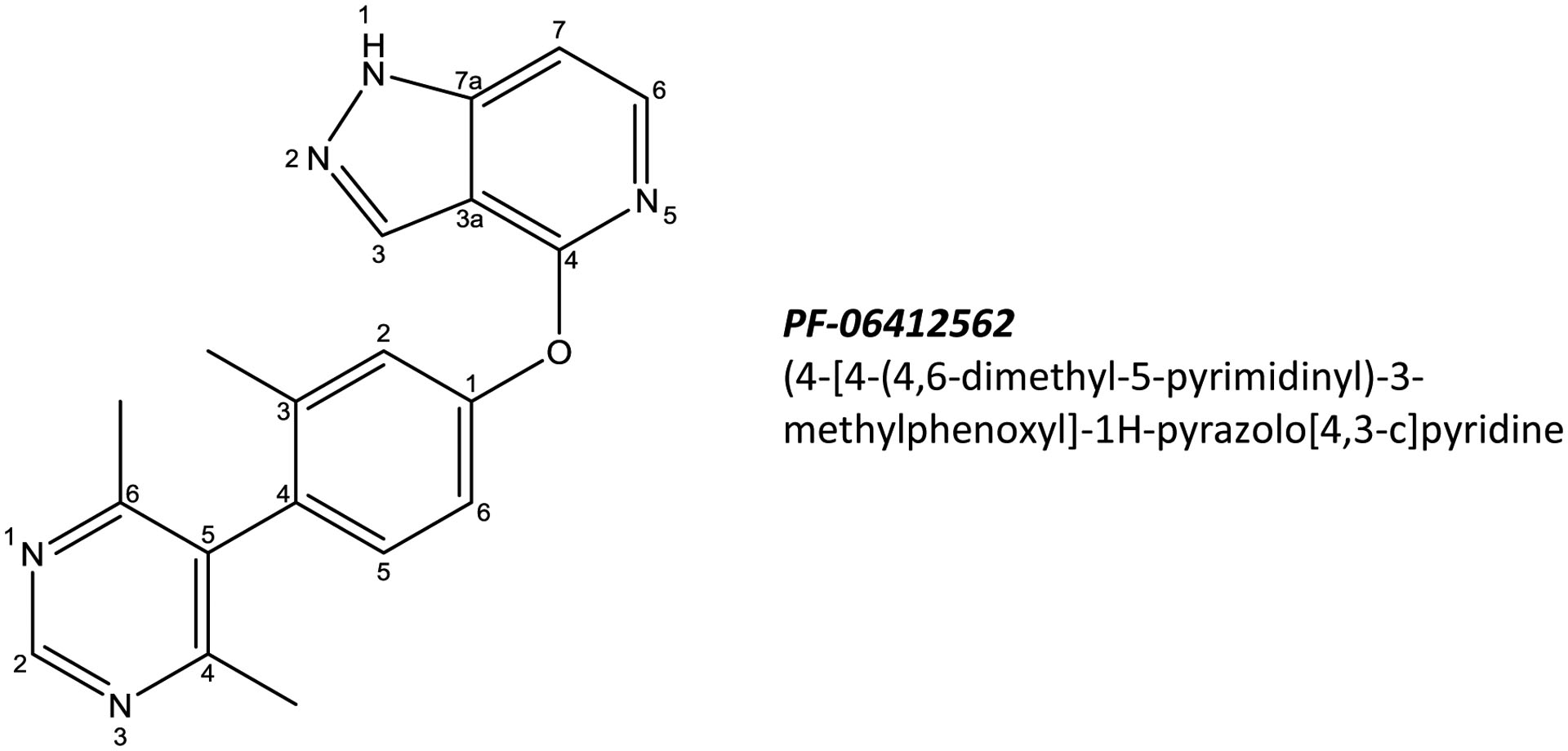
Chemical structure of PF-06412562: (4-[4-(4,6-dimethyl-5-pyrimidinyl)-3-methylphenoxyl]-1H-pyrazolo[4,3-c]pyridine
Trial oversight
This study was conducted at Penn State Health in compliance with the ethical principles of the Declaration of Helsinki and the guidelines for Good Clinical Practice issued by the International Conference on Harmonization. It was reviewed and approved by the US Food and Drug Administration and PSH Institutional Review Board. All participants and/or their caregivers provided signed informed consent. The Data Safety Board (DSB) reviewed safety data after each participant completed the study. The investigators attest to the fidelity of the trial protocol and the authors vouch for the completeness and accuracy of the data. The authors, funder (Pfizer), and current licensee of study drug (Cerevel) agreed to the manuscript submission.
Subjects
Participants were recruited between August 2018-January 2019 from the PSH Movement Disorders Clinic or via a local PD support group. The inclusion and exclusion criteria are detailed in Supplemental Table S1. All met published diagnostic criteria [24], had disease duration >15 years since diagnosis, and were Hoehn & Yahr Stage >IV, even when taking levodopa or when deep brain stimulation was turned on. After consenting, participants and their caregiver were admitted to the Clinical Research Center (CRC) for four days during two consecutive weeks. On Day 1, clinical history, vital signs, blood work [complete blood count (CBC) and chemistry (Na, K, HCO3, Ca, Mg, AST, ALT, BUN and creatinine)], urinalysis, and a 12-lead electrocardiogram (ECG) were obtained to serve as baseline and assess for significant and/or unstable medical conditions.
If systolic blood pressure (SBP) was <100 mm Hg with evidence of dehydration (BUN/creatinine ratio >20) on Day 1, or SBP was <90 mm Hg with clinical symptoms (dizziness, fainting) on Days 2–4, normal saline was given intravenously until SBP reached >100 mm Hg. Since patients might have been under-medicated during the trial, they were given Sinemet (for parkinsonian symptoms) if patient and/or caregiver deemed that he/she needed it. In addition, acetaminophen (for pain), ondansetron (for nausea), and diphenhydramine (for allergies) were given throughout the study when needed.
Trial design
All study procedures, including a 1-month follow-up phone call, were conducted between August 2018-April 2019. Eligible participants were randomized to either PF-06412562 (Sequence A) or carbidopa/levodopa (Sequence B) in Week 1 (Figure 2) by PSH Investigational Drug Service (IDS) personnel using a 1:1 random allocation sequence (detailed in Supplemental Text S1). Participants, caregivers, and investigators were blinded to sequence assignment.
Figure 2. Schematic of the overall design of the study.
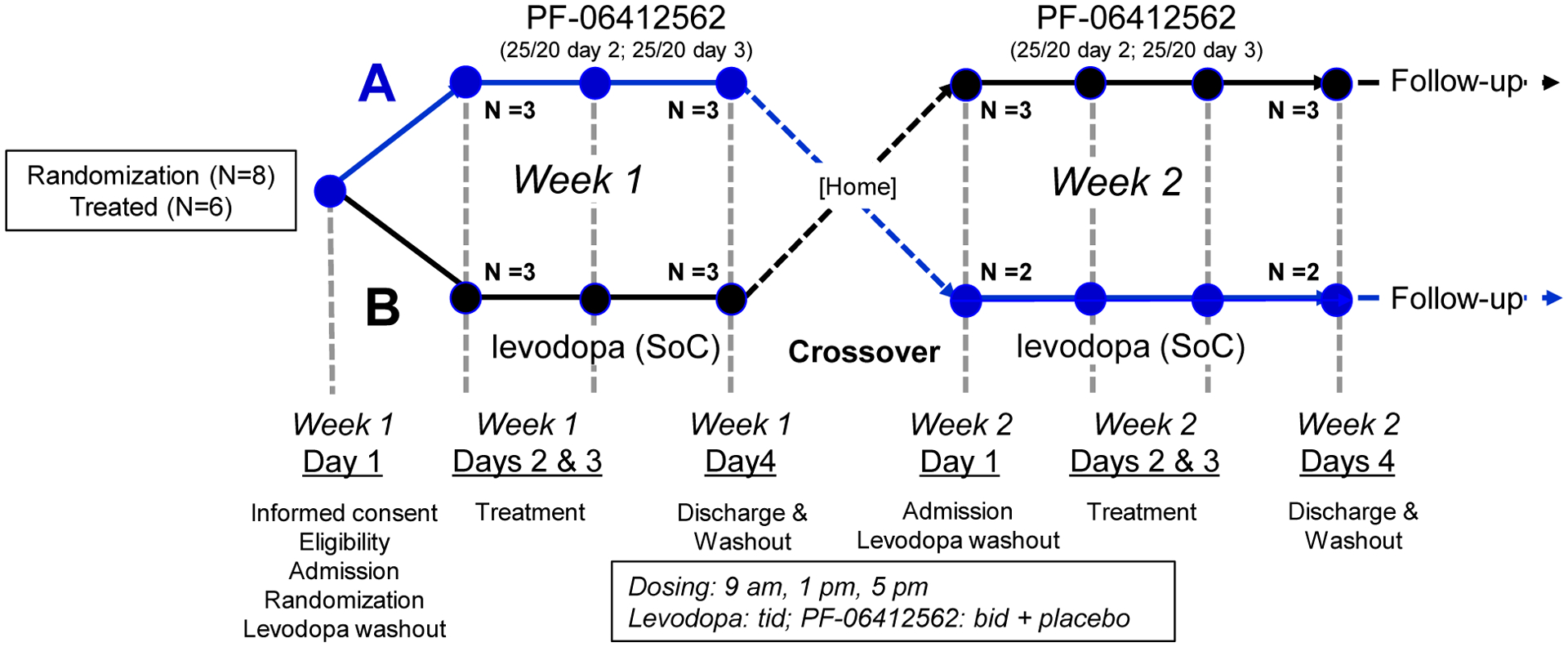
Subjects were randomized to receive PF-06412562 followed by levodopa (Sequence A, top) or levodopa followed by PF-06412562 (Sequence B). PF-06412562 (5 mg) or placebo tablets were provided by Pfizer. Sinemet (carbidopa/levodopa, 25/100 mg) tablets were encapsulated to preserve study blind. The bottom part of the schematic shows the events that occurred on each day. Abbreviations: SoC = Standard of Care. Some subjects received a fourth dose of levodopa if that was required according to their pretrial regimen.
Following baseline evaluation and overnight pre-trial levodopa/dopamine agonist washout on Day 1, participants assigned Sequence A received PF-06412562 (25 mg at ~09:00 h and 20 mg 4 h later) on Days 2 and 3 during Week 1, along with placebo (cellulose-filled capsule). During Week 2, these participants received encapsulated Sinemet (25/100 mg) 3–4 times (depending on pretrial regimen) 4 h apart on Days 2–3, along with placebo (Supplemental Table S2). The participants assigned Sequence B received carbidopa/levodopa in Week 1 and PF-06412562 in Week 2. On Day 4, all participants resumed pre-trial treatment and were discharged after demonstrating no significant complications. Blood levels of PF-06412562 were determined blindly (detailed in Supplemental Text S2).
Primary endpoints
The primary outcome was safety and tolerability of PF-06412562. Quantitative assessments included: vital signs, MDS-Unified PD Rating Scale dyskinesia scale-IV (MDS-UPDRS-IV), Columbia Suicide Severity Rating Scale (CSSRS), and routine safety tests (CBC, electrolytes, liver and kidney functions, ECG) at baseline and Day 4. Qualitative data included reports from participants, caregivers, nursing staff, and investigative team members.
Since hypotension was an adverse event observed with the first generation D1 agonists such as dihydrexidine [25], and also with usage of approved D2 agonists, supine blood pressure (BP) and heart rate (HR) were measured on Day 1 after admission (~16:00 h), on Days 2 and 3 at baseline prior to medication (~06:30 h), and at two and six hours after drug administration. In addition to supine vitals, cardiovascular autonomic testing was performed at these time points to assess cardiac sympathetic and parasympathetic tone. Although the original protocol included measurement of orthostatic vitals before and after drug administration, some of our advPD patients were unable to tolerate sitting or standing for prolonged periods of time at baseline (pre-test drug), and thus orthostatic measurements were not obtained.
For cardiovascular autonomic testing, participants were asked to rest in the supine position for at least 10 min, followed by 15 min of continuous BP and HR recordings. BP was measured continuously with finger photoplethysmography (Finometer FMS) and intermittently with an arm cuff via oscillometric methods (Philips SureSigns VS3). HR was measured using continuous three-lead ECG (Cardiocap/5, GE Healthcare). At the end of the 15-min recording period, subjects underwent respiratory sinus arrhythmia (HR response to deep breathing; 6 breaths/minute; normal response, maximum to minimum HR ratio >1.2) and cold pressor (hand in ice water for 1 min; normal response, increase in SBP >20 mm Hg) tests to assess the integrity of cardiac parasympathetic and sympathetic nerves, respectively. Resting HR variability, a measure of parasympathetic tone, was assessed in all subjects from continuous ECG recordings using time-domain spectral analysis methods [standard deviation of the average normal-to-normal intervals (SDNN) [26, 27].
Secondary endpoints
Two validated clinical global impression scales were modified a priori to capture both clinician and caregiver quantitative assessments of severity or change. For clinician assessment, the Clinician Global Impression of Severity (MD-CGI-S) first was used to assess baseline disease severity of the participant on Day 1. Second, the Clinician Global Impression of Change (MD-CGI-C) assessed clinical change from baseline (Day 1) using a 7-point Likert scale at two-hours post-dose. Lastly, the Overall Clinician Global Impression of Change Summary (MD-CGI-CS, Supplemental Table S3) is an overall clinician assessment of the entire week at the end of Day 3 using the same 7-point Likert scale. For caregivers, the scales were adapted from the clinician assessment tool as detailed in Supplemental Table S4. First, the Caregiver Global Impression of Severity (CG-CGI-S) was administered on Day 1 based on caregiver knowledge of the participant’s disease at home. Second, the Caregiver Global Impression of Change (CG-CGI-C) was assessed on the evening of Days 2–3 based on caregiver’s observation. Since Day 2 may be more confounded by excitement/noise/environment changes, we pre-determined that the evaluation performed at the end of Day 3 would be used for final analyses.
Statistical analysis
Primary endpoint analyses included all randomized subjects. Adverse events were summarized by MedDRA (ver. 21.2) criteria and severity (mild/moderate/severe). Vital signs and cardiovascular autonomic testing results were summarized using descriptive statistics. Laboratory results were evaluated immediately by physicians for clinical significance, and summarized at baseline and day-of-discharge. Secondary endpoint analyses included subjects who completed both study weeks. Data were summarized by treatment arms using descriptive statistics.
Results
Participants baseline clinical characteristics
A total of eight participants consented for the study and underwent a screening visit. Two were excluded due to unstable medical condition (ECG changes), or the need for medication adjustment after the screening visit. Six participants (detailed in Table 1) were on stable medication for greater than 60 days, and then were randomized to the study. All participants were in Hoehn and Yahr Stage >IV and not able to walk independently, two (001 and 008) were wheelchair/bed-bound and needed total assistance in transfer, four others (003, 004, 006, 007) required wheelchairs for transportation, and walkers, along with human assistance, for short distance walking or transfer.
Table 1.
Demographic, clinical history, and baseline data for the randomized participants
| ID | Sex | Age of diagnosis | Disease duration | key medical, surgery, progression history | Current medications |
|---|---|---|---|---|---|
| 001 | M | 37 y | 19 y | Pallidotomy at 46 y Wheelchair use at 62 y PEG at 63 y |
DA drugs: Parcopa Non-DA drugs: rivastigmine transdermal |
| 003 | M | 60 y | 18 y | PD at 60 y STN-DBS at 70 y Walker/wheelchair at 77 y |
DA drugs: Sinemet R & CR Non-DA drugs: vitamin B12 |
| 004 | F | 56 y | 15 y | STN-DBS at 62 y Walker use at 68 y Wheelchair use at 70 y |
DA drugs: Sinemet R & CR, Rytary, Zelapar, Mirapex Non DA Drugs: dexlansoprazole, melatonin, midodrine, donepezil, memantine, clozapine, rimantadine, methylphenidate, venlafaxine fludrocortisdone |
| 006 | F | 58 y | 19 y | Walker use at 76 y Wheelchair at 77 |
DA drugs: Rytary, Sinemet Non-DA drugs: gabapentin, donepezil, lorazepam, quetiapine, melatonin, tramadol |
| 007 | M | 53 y | 23 y | Cane use at 73 y Wheelchair use at 75 y |
DA drugs: Rytary, rasagiline Non-DA drugs: donepezil |
| 008 | M | 42 y | 31 y | On L-dopa until 70 y Bed-bound at 70 y |
DA drugs: none* Non-DA drugs: none |
L-dopa discontinued due to severe drowsiness.
Abbreviations:F: Female; M: Male; Y: years; STN: Subthalamic nucleus; DBS: Deep brain stimulation; PEG: Percutaneous endoscopic gastrostomy; DA: Dopaminergic; Sinemet: carbidopa/levodopa; R: regular release; CR: controlled release.
One participant (001) had a history of pallidotomy that helped reduce levodopa-induced dyskinesia, and percutaneous endoscopic gastrostomy to assist nutrition status. Two participants (003 and 004) had DBS to the subthalamic nucleus, but remained Hoehn and Yahr Stage >IV even with stimulation on. The participant (008) with the longest disease duration of 31 y, benefited from levodopa and other anti-PD drugs earlier in his course of disease. He, however, experienced severe drowsiness,u and was unarousable without clear cause while on anti-PD medication including levodopa. His family elected to stop all medications including levodopa three years prior to this trial. He had erratic sleep-awake cycles prior to the study and no meaningful communication, but was able to swallow food when his mouth was stimulated.
Three participants (001, 006, 008) had severe baseline dehydration (SBP <100 and BUN/creatinine >20), with one (006) showing signs of pre-renal kidney failure [anuria, creatinine=1.34, estimated glomerular filtration rate (GFR)=38]. They were all treated with IV hydration on Day 1 (baseline) of each week per our pre-determined study procedures (see Table 2). Baseline vital signs and cardiovascular autonomic testing were obtained on Day 1 following any hydration procedures. There were no apparent overall differences in baseline vital signs or cardiovascular autonomic data between testing periods (Figure 3).
Table 2:
Administration of IV fluid (0.9% saline) and/or rescue levodopa usage for each participant
| Study ID | Levodopa (SoC) | PF-06412562 | ||||
|---|---|---|---|---|---|---|
| Day 1 | Days 2–3 | Day 4 | Day 1 | Days 2–3 | Day 4 | |
| Baseline | Test | Discharge | Baseline | Test | Discharge | |
| 001 | 0.9 % saline 500 mL | --- | --- | 0.9 % saline 500 mL | --- | --- |
| 003 | --- | --- | --- | --- | --- | --- |
| 004 | --- | Half tab of Sinemet R 25/100 |
One tab of Sinemet R 25/100 |
--- | --- | --- |
| 006 | 0.9 % saline 500 mL |
0.9 % saline 500 mL |
0.9% saline 500 mL |
Trial terminated by safety board | ||
| 007 | --- | --- | --- | --- | --- | --- |
| 008 | 0.9 % saline 1000 mL | --- | --- | 0.9 % saline 1,000 mL | --- | --- |
Figure 3. Vital signs and cardiovascular autonomic test results.
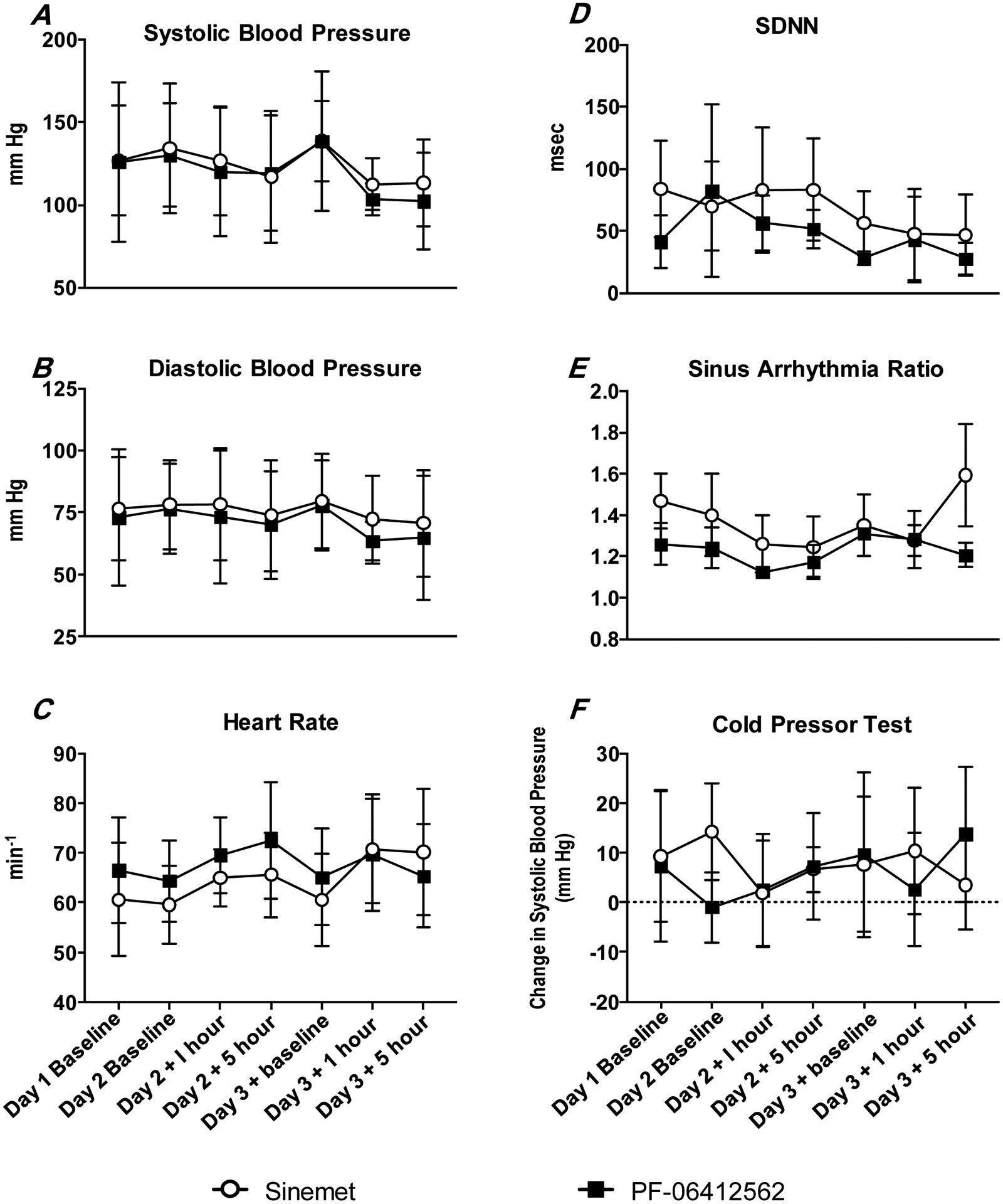
There were no apparent overall differences in baseline vital signs or cardiovascular autonomic data between testing periods.
All participants completed cold pressor testing, and four participants (003, 004, 006, 007) were able to follow breathing commands for sinus arrhythmia testing. Of these participants, three had abnormal cardiac sympathetic responsiveness (001, 006, 007: SBP changes: 9, −3 and −5 mm Hg, respectively), and one had abnormal cardiac parasympathetic responsiveness (006, sinus arrhythmia ratio=1.12), at baseline. One participant (006) with baseline impairment of cardiac sympathetic and parasympathetic nerve activity also exhibited reduced resting HR variability (SDNN=15 msec) when compared to the other participants (SDNN=37–106 msec).
Primary endpoints
No participants withdrew from the study voluntarily. No clinically meaningful laboratory test abnormalities were observed in either treatment periods. During the D1 period, blood PF-06412562 concentrations were 424 ng/mL (SD=73; range 305–498 ng/mL), and were not detectable during the levodopa period (<0.55 ng/mL). One participant (004) required Sinemet 25/100 rescue on both Days 2 (1/2 tab) and 3 (one tab) for feeling more “off” during the SoC/levodopa period, but not the PF-06412562 period (Table 2). Most adverse events were rated as mild or moderate (Table 3).
Table 3.
Adverse events reported according to Grades, System Organ Class, and Preferred Terms
| System Organ Class and Preferred Terms | Grade 1 | Grade 2 | Grade 3 | Grade 4 or 5 | ||||
|---|---|---|---|---|---|---|---|---|
| Drug assignments** | LD | PF | LD | PF | LD | PF | LD | PF |
| Subjects completed | N=5 | N=6 | N=5 | N=6 | N=5 | N=6 | N=5 | N=6 |
| A. All events including Day 1 and one month after discharge | ||||||||
| Any adverse event * | 0 | 1 | 1 | 1 | 2 | 4 | 0 | 0 |
| Any serious adverse event | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Psychiatric disorders | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 |
| Anxiety | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Restlessness | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| Insomnia | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Confusion | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| Hallucinations | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| Vascular/Automatic disorders | 0 | 1 | 0 | 0 | 2 | 3 | 0 | 0 |
| Hypotension | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 |
| Hypertension | 0 | 0 | 0 | 0 | 2 | 2 | 0 | 0 |
| Gastrointestinal disorders | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| Gastrointestinal pain | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| Musculoskeletal disorders | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| Pain | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| Nervous System disorders | 1 | 0 | 0 | 2 | 0 | 0 | 0 | 0 |
| Headache | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| Fall | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Dizziness | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| B. Events after test drug each week (including Days 2 and 3, and after discharge) | ||||||||
| Any adverse event * | 0 | 0 | 0 | 2 | 2 | 3 | 0 | 0 |
| Any serious adverse event | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Psychiatric disorders | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 0 |
| Anxiety | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Restlessness | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| Insomnia | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Confusion | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| Hallucinations | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| Vascular/autonomic disorders | 0 | 0 | 0 | 0 | 2 | 3 | 0 | 0 |
| Hypotension | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Hypertension | 0 | 0 | 0 | 0 | 2 | 2 | 0 | 0 |
| Gastrointestinal disorders | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Gastrointestinal pain | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Musculoskeletal disorders | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| Pain | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| Nervous System disorders (total) | 1 | 0 | 0 | 2 | 0 | 0 | 0 | 0 |
| Headache | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| Fall | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Dizziness | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
Number of patients with an adverse event, summarized for each treatment group.
LD = levodopa/carbidopa; PF = PF-06412562; PT = preferred term; SOC = system organ class. MedDRA version used was 22.01
For each PT, adverse event per patient are counted only once according to the most severe grade. AEs were assessed for severity using the Common Terminology Criteria for Adverse Events (CTCAE) with Grade 1–5 classification (1:mild; 2:moderate; 3:severe; 4: life-threatening; 5:death)
There were no significant differences in BP or HR, with a similar mild to moderate hypotension and reflex tachycardia observed following PF-06412562 or levodopa administration (Figure 3). Three participants (003, 004, 008) with normal baseline cardiac sympathetic responsiveness did not change following PF-06412562 or levodopa. Similarly, three participants (003, 004, 007) with normal cardiac parasympathetic responsiveness at baseline showed no differences in response to PF-06412562 versus levodopa. There were also no significant differences in HR variability between drugs.
One participant (006) who exhibited severe sympathetic impairment and mild parasympathetic impairment at baseline experienced symptomatic hypotension and was treated with IV hydration on Day 3 of the PF-06412562 week. The participant was discharged on Day 4 with improved laboratory results (creatinine=0.98, BUN=24, and estimated GFR=55), normal ECG findings, and appeared clinically stable. The participant, however, required emergency room visits at 1, 13, 15, and 24 days and subsequent hospitalizations to treat hypotension after Week 1 discharge. Although the participant and caregiver wanted to proceed to study period 2, we followed the recommendation of the DSB to discontinue their participation.
Secondary endpoints
As seen in Table 4, no subjects were rated worse (negative scores) in clinical global impression of change than baseline during PF-06412562 periods by either clinicians or caregivers, whereas one clinician and three caregivers rated subjects worse during the SoC/levodopa period. Overall caregiver scores favored PF-06412562 in four participants (001, 003, 004, 007) and levodopa in one participant (008) (Table 3). Clinician impression of change scores were more diverse, with two (003, 004) favoring PF-06412562, two (001 and 008) levodopa, and one (007) equivocal.
Table 4.
The overall clinical impression of change at the end of Day 3
| Participants’ Study ID | Levodopa (SoC) period | PF-06412562 period | ||||
|---|---|---|---|---|---|---|
| Week* | Clinician** | Caregiver*** | Week | Clinician | Caregivers | |
| 001 | 1 | 2 | 8 | 2 | 1 | 22 |
| 003 | 2 | 0 | −1 | 1 | 3 | 27 |
| 004 | 1 | −1 | −4 | 2 | 3 | 21 |
| 007 | 1 | 0 | −4 | 2 | 0 | 13 |
| 008 | 2 | 2 | 32 | 1 | 0 | 2 |
Week when the drug was administered/tested
Clinician rating on a 7 point Likert scale (−3=marked worsening; 0=no change; 3=marked improvement) (see details in Supplemental Table S3)
Caregiver rating is the sum [−51 (marked worsening) to[51 (marked improvement)] of 17 items listed in Supplemental Table S4 with each item on a 7 point Likert scale as above.
One participant (008) was ‘awakened’ during Week 2 when given Sinemet. After extensive discussions with the research ethicist, we disclosed this finding to the family for their consideration. The family decided to resume Sinemet, but the ‘awakened’ state did not recur.
Discussion
Recent therapeutic development for PD has focused on disease modification [28–30], but unfortunately remaisn unsuccessful. Current symptomatic treatments have allowed many PD patients to have life expectancies 15 years or more after diagnosis [31], although quality-of-life decreases with continuing disease progression. Due to limited pharmacological options, management of advPD patients transitions to one of support and palliative measures [32]. As the first drug trial targeting PD patients in advanced-to-end-stages of disease, this study elucidated many of the challenges, as well as potential solutions. Equally important, this study demonstrated the feasibility of studying this population, and provided preliminary data that PF-06412562, a partial D1/5 agonist, was tolerated in a small cohort of advPD patients.
Although past and ongoing clinical trials have avoided inclusion of patients with advPD, this study showed that patients and their family are willing to participate in drug trials. Between July 16, 2018 (IRB approval) to the end of study enrollment on December 30, 2019, we enrolled eight subjects at a single site, and received additional request for participation after the enrollment deadline. This enthusiasm and willingness for participation underscores the need for better drug treatment options for this population.
No participants withdrew from the study voluntarily. and PF-06412562 was well tolerated in advPD patients. This is consistent with previous clinical trials of milder-stage PD [23]. Based on results of prior studies with other agents, a main focus was assessment of potential for hypotension. Indeed, the only selective D1 agonist approved clinically, fenoldopam, is indicated for emergency acute hypertensive crises. Fenoldopam, however, does not pass the blood brain barrier, and therefore is not centrally active [33, 34]. Although having antiparkinson effects, the first generation full D1 agonist dihydrexidine has been associated with hypotension requiring intensive care unit admission for some subjects [25]. We suspected that this side effect may have been due, in part, to rapid intravenous administration for the study [25], as slower subcutaneous infusion of dihydrexidine was well tolerated, achieved measurable blood levels [35], permitting use in studies of cognition efficacy in psychiatric patients [36, 37].
All current dopamine replacement therapy lowers BP somewhat, and in some patients precipitates orthostatic hypotension [38]. It was thought that dopamine receptor stimulation induces orthostatic hypotension by venous and arteriolar dilation. Consistent with this notion, we found similar BP lowering (Figure 3) in both the PF-06412562 and levodopa arms even in the supine position, an effect greater on the second treatment day (Day 3). Unfortunately, we were not able to determine if PF-06412562 affects orthostatic BP, as we originally intended, since some participants (e.g. subjects 001 and 008) were not able to sit/stand for prolonged periods of time. Future studies therefore may need to incorporate head-up tilt table testing to evaluate the degree and tolerability of BP drops. Nonetheless, PF-06412562 was well-tolerated with symptomatic hypotension only observed in one participant (006) with pre-existing dehydration and acute pre-renal kidney injury.
In the current study, we found that half of participants (3/6) had normal cardiac sympathetic integrity, and 3/4 of participants who completed sinus arrhythmia testing had intact cardiac parasympathetic integrity. There were no differences in cardiac sympathetic and parasympathetic tone or responsiveness, suggesting PF-06412562 or levodopa did not adversely impact cardiovascular autonomic function. Of note, the participant (006) who developed symptomatic hypotension had the most significant sympathetic and parasympathetic dysfunction. This suggests that screening for cardiovascular autonomic function will be valuable in future trials. In addition, to protect against any potential cardiovascular vasodilatory effects, we speculate that pre-treatment of advPD patients with therapies to increase sympathetic tone (such as the norepinephrine prodrug droxidopa) may be useful in future studies with PF-06412562 or other D1-like agonists.
Since dehydration is common in advPD patients, we did not exclude advPD patients with dehydration in the current study. Instead, we put into place pre-determined study criteria to protect this subset of patients against potential serious cardiovascular outcomes such as symptomatic hypotension. We recognize this may alter resting cardiovascular tone and drug responsiveness. We did not observe any apparent differences, however, in vital signs or cardiac sympathetic and parasympathetic tone between participants who did or did not receive hydration support. Of note, the participant (006) who developed symptomatic hypotension also had pre-renal kidney dysfunction. Future studies may need to consider including baseline renal dysfunction, even secondary to dehydration, as an exclusion criterion (Table 2).
A secondary goal of our study was to provide pilot data for possible efficacy of D1 agonists in advPD. Because of the complex, multi-domain dysfunction of advPD patients, we chose to include a caregiver evaluation to improve the possibility of detecting meaningful changes. A single clinician rater was not feasible throughout the study, so the data are from two clinician raters and one caregiver for each of the participants. The overall results from the caregiver assessments favored PF-06412562 over levodopa, but the clinicians’ impressions found no difference. Although one would predict that expert clinicians would provide more objective evaluations, the clinicians’ scores may suffer from inter-rater variability in judging the severity of such advPD patients. Conversely, the caregivers were experienced with the subject they evaluated, and might be more sensitive to changes. Future studies should emphasize the use of a single clinician rater for a specific participant as much as possible, and consider integrating caregiver evaluations as an integral part of data collection.
In summary, although physicians, patients, and caregivers have recognized the many unmet needs of advPD patients for decades, these patients essentially have been excluded from therapeutic clinical trials. This study provides proof-of-concept that therapeutic drug trials are feasible in advPD, and that D1/5 agonists are tolerated. Because its potential to provide benefit in advPD, future studies are warranted to include a larger sample size, longer treatment duration, additional safety measurements (such orthostatic BP with tilt table), and patient-centric meaningful outcome assessments that include caregivers’ perspectives.
Supplementary Material
Acknowledgements
We express deep gratitude to all of the patients and their caregivers for participating in the study. The completion of the study was made possible by an exceptional team including Shirlynn Mottilla, Nancy Handley, Brenda Hershey-Fell, Lyndsey Houser, Carrie Criley, Patricia Cain, Sue Kocher, Lisa Zeno, Alison Enimpah, Kelly Hoffman, Lori Martin, Brynn Vanderveer, Dina Angello, Kiley Klock, Fiona Fortman, Sandra Franz, Crystal DeMedici, Sarah Debold, and Natalya Knapp. We thank Margaret Hopkins and Dr. K. Randy Young for assisting with the qualitative and mixed methods analysis; Dr. Andrew Foy for consulting on cardiology and ECG questions raised during screening and the conduct of the project; Sara Marlin for serving as our data monitor; and Dr. Gary Thomas for serving on the Data Safety Board (DSB). In addition, we thank Dr. Jennifer McCormick, research ethicist, for providing critical input on our decision to unblind participant 008. This work was supported by a grant from Pfizer Central Research who also supplied test drug and placebo. This work also was supported in part by the National Institute of Neurological Disorders and Stroke and Parkinson’s Disease Biomarker Program (NS060722, NS082151 to XH), and the Penn State National Center for Advancing Translational Sciences (UL1 TR002014). All analyses, interpretations, and conclusions are those of the authors and not the research sponsors.
Potential Conflicts of Interest
Xuemei Huang: Received funding from the National Institutes of Health (R01 ES019672, U01 NS082151, U01 NS112008), the Michael J. Fox Foundation, the Michael J. Fox Foundation for Parkinson’s Research, Alzheimer’s Association, Alzheimer’s Research UK, and the Weston Brain Institute, Bristol Myers Squibb/Biogen, Pfizer, and the Department of Defense.
Mechelle M. Lewis: Received funding from the National Institutes of Health (R01 ES019672, U01 NS082151, U01 NS112008), the Michael J. Fox Foundation, the Michael J. Fox Foundation for Parkinson’s Research, Alzheimer’s Association, Alzheimer’s Research UK, and the Weston Brain Institute, Bristol Myers Squibb/Biogen, Pfizer, and the Department of Defense.
Lauren Jodi Van Scoy: Received funding from the National Institute of Nursing Research Institute (R21 NR017259, 2R01 NR012757-06), the John and Wauna Francis/Francis Family Foundation, Canadian Institute of Health Research, Association for Clinical Pastoral Education.
Sol De Jesus: Has received funding from the National Institute for Neurological Disorders and Stroke (NINDS), Bristol Myers Squibb, Pfizer, as well as consultant fees from Medtronic Inc. and Boston Scientific.
Paul J. Eslinger Received funding from the National Institutes of Health (R01 ES019672), Alzheimer’s Therapeutic Research Institute, National Institute of Aging, Alzheimer’s Association, Canadian Institutes of Health Research, National Institute on Aging.
Amy C. Arnold Received funding from the National Institutes of Health (R00 HL122507 and UL1TR002014), Dysautonomia International, and the Central Society for Clinical and Translational Research. Served as a medical consultant for the Vaccine Injury Compensation Program for the Department of Health and Human Services.
Amanda J. Miller Received funding from the American Heart Association.
Julio Fernandez-Mendoza: Received funding from the National Institutes of Health (R01 MH118308, R01 HL136587), American Heart Association (14SDG19830018), Penn State University Grace Woodward Grant for Collaborative Research in Engineering and Medicine, and Freiburg-Penn State Collaboration Development Program.
Bethany Snyder Has no conflict of interest to disclose.
William Harrington Has no conflict of interest to disclose.
Ada Wang Has no conflict of interest to disclose.
Lan Kong: Received funding from the National Institutes of Health (R01 ES019672, U01 NS082151, U01 NS112008), the Michael J. Fox Foundation for Parkinson’s Research, Alzheimer’s Association, Alzheimer’s Research UK, and the Weston Brain Institute.
Xi Wang Has no conflict of interest to disclose.
Marielle Delnomdedieu Is a Pfizer employee who worked on the PD program.
Sridar Duvvuri Was an employee and shareholder of Pfizer, Inc at the time of study design and initiation. He is now an employee of Cerevel Therapeutics
Susan E Mahoney Is a Pfizer employee who worked on the PD program
David Gray Was an employee and shareholder of Pfizer, Inc at the time of study design and initiation. He is now an employee of Cerevel Therapeutics
Richard B. Mailman: Received funding from the National Institutes of Health (R01 NS105471). Patents related to D1 dopamine agonists as therapeutic agents (assigned to University foundations). Past D1-related grant, consulting, and travel from Pfizer Central Research. Unrelated to this study: Past expert witness or consultant to several law firms and US Department of Justice. Past consultant to Jazz Pharma (only publishing costs).
Footnotes
Drs. Huang (PI) and Mailman declared a potential conflict of interest (COI) due to existing patents related due to the discovery or use of D1 agonists, although neither has any financial interest that are affected by the results of this study. Drs. Huang and Mailman did not participate in consenting subjects, were not involved with the Data Safety Board (DSB, composed of three investigators and three clinicians), and did not participate in data analysis until the data were locked. Dr. Huang worked closely with Drs. De Jesus (a movement disorder specialist) and Van Scoy (a pulmonary and critical care physician) to provide the best care for the participants throughout the study. Drs. Huang or De Jesus provided blinded ratings for the clinician global impression of change based on their availability.
References:
- [1].LeWitt PA, Fahn S (2016) Levodopa therapy for Parkinson disease: A look backward and forward. Neurology 86, S3–12. [DOI] [PubMed] [Google Scholar]
- [2].Barone P (2010) Neurotransmission in Parkinson’s disease: beyond dopamine. Eur J Neurol 17, 364–376. [DOI] [PubMed] [Google Scholar]
- [3].Fox S, Gannon E, Cashell A, Kernohan WG, Lynch M, McGlade C, O’Brien T, O’Sullivan SS, Sweeney C, Timmons S (2015) Survey of Health Care Workers Suggests Unmet Palliative Care Needs in Parkinson’s Disease. Mov Disord Clin Pract 2, 142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lex KM, Larkin P, Osterbrink J, Lorenzl S (2018) A Pilgrim’s Journey-When Parkinson’s Disease Comes to an End in Nursing Homes. Front Neurol 9, 1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Millan MJ, Maiofiss L, Cussac D, Audinot V, Boutin JA, Newman-Tancredi A (2002) Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. I. A multivariate analysis of the binding profiles of 14 drugs at 21 native and cloned human receptor subtypes. Journal of Pharmacology and Experimental Therapeutics 303, 791–804. [DOI] [PubMed] [Google Scholar]
- [6].Mailman RB, Huang X (2007) Dopamine receptor pharmacology. Handb Clin Neurol 83, 77–105. [DOI] [PubMed] [Google Scholar]
- [7].Varanese S, Birnbaum Z, Rossi R, Di Rocco A (2011) Treatment of advanced Parkinson’s disease. Parkinsons Dis 2010, 480260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Latt MD, Lewis S, Zekry O, Fung VSC (2019) Factors to Consider in the Selection of Dopamine Agonists for Older Persons with Parkinson’s Disease. Drugs Aging 36, 189–202. [DOI] [PubMed] [Google Scholar]
- [9].Shulman LM, Minagar A, Rabinstein A, Weiner WJ (2000) The use of dopamine agonists in very elderly patients with Parkinson’s disease. Movement Disorders 15, 664–668. [DOI] [PubMed] [Google Scholar]
- [10].Huang X, Lawler CP, Lewis MM, Nichols DE, Mailman RB (2001) D1 dopamine receptors. Int. Rev. Neurobiol 48, 65–139. [DOI] [PubMed] [Google Scholar]
- [11].Mailman RB, Nichols DE (1998) Dopamine D1 receptor agonists as antiparkinson drugs. Trends in Pharmacological Sciences 19, 255–256. [DOI] [PubMed] [Google Scholar]
- [12].Mailman R, Huang X, Nichols DE (2001) Parkinson’s disease and D1 dopamine receptors. Curr Opin Investig Drugs 2, 1582–1591. [PubMed] [Google Scholar]
- [13].Taylor JR, Lawrence MS, Redmond DE Jr., Elsworth JD, Roth RH, Nichols DE, Mailman RB (1991) Dihydrexidine, a full dopamine D1 agonist, reduces MPTP-induced parkinsonism in monkeys. Eur J Pharmacol 199, 389–391. [DOI] [PubMed] [Google Scholar]
- [14].Michaelides MR, Hong Y, DiDomenico S Jr., Asin KE, Britton DR, Lin CW, Williams M, Shiosaki K (1995) (5aR,11bS)-4,5,5a,6,7,11b-hexahydro-2-propyl-3-thia-5-azacyclopent-1- ena[c]-phenanthrene-9,10-diol (A-86929): a potent and selective dopamine D1 agonist that maintains behavioral efficacy following repeated administration and characterization of its diacetyl prodrug (ABT-431). J Med Chem 38, 3445–3447. [DOI] [PubMed] [Google Scholar]
- [15].Rascol O, Blin O, Thalamas C, Descombes S, Soubrouillard C, Azulay P, Fabre N, Viallet F, Lafnitzegger K, Wright S, Carter JH, Nutt JG (1999) ABT-431, a D1 receptor agonist prodrug, has efficacy in Parkinson’s disease. Ann. Neurol 45, 736–741. [DOI] [PubMed] [Google Scholar]
- [16].Rascol O, Nutt JG, Blin O, Goetz CG, Trugman JM, Soubrouillard C, Carter JH, Currie LJ, Fabre N, Thalamas C, Giardina WW, Wright S (2001) Induction by dopamine D1 receptor agonist ABT-431 of dyskinesia similar to levodopa in patients with Parkinson disease. Arch Neurol 58, 249–254. [DOI] [PubMed] [Google Scholar]
- [17].Davoren JE, Nason D, Coe J, Dlugolenski K, Helal C, Harris AR, LaChapelle E, Liang S, Liu Y, O’Connor R, Orozco CC, Rai BK, Salafia M, Samas B, Xu WJ, Kozak R, Gray D (2018) Discovery and Lead Optimization of Atropisomer D1 Agonists with Reduced Desensitization. Journal of Medicinal Chemistry 61, 11384–11397. [DOI] [PubMed] [Google Scholar]
- [18].Mailman RB, Huang X, Nichols DE (2001) Dopamine D1 full agonists and Parkinson’s disease. Current Opinion in Investigational Drugs 2 (11), 1582–1591. [PubMed] [Google Scholar]
- [19].Goulet M, Madras BK (2000) D-1 dopamine receptor agonists are more effective in alleviating advanced than mild parkinsonism in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated monkeys. Journal of Pharmacology and Experimental Therapeutics 292, 714–724. [PubMed] [Google Scholar]
- [20].Mailman RB, Yang Y, Huang X (2020) D1, not D2, dopamine receptor activation can improve levodopa unresponsive parkinsonism. Eur J Pharmacol, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gray DL, Allen JA, Mente S, O’Connor RE, DeMarco GJ, Efremov I, Tierney P, Volfson D, Davoren J, Guilmette E, Salafia M, Kozak R, Ehlers MD (2018) Impaired beta-arrestin recruitment and reduced desensitization by non-catechol agonists of the D1 dopamine receptor. Nat Commun 9, 674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sohur US, Gray DL, Duvvuri S, Zhang Y, Thayer K, Feng G (2018) Phase 1 Parkinson’s Disease Studies Show the Dopamine D1/D5 Agonist PF-06649751 is Safe and Well Tolerated. Neurol Ther 7, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Papapetropoulos S, Liu W, Duvvuri S, Thayer K, Gray DL (2018) Evaluation of D1/D5 Partial Agonist PF-06412562 in Parkinson’s Disease following Oral Administration. Neurodegener Dis 18, 262–269. [DOI] [PubMed] [Google Scholar]
- [24].Hughes AJ, Daniel SE, Ben-Shlomo Y, Lees AJ (2002) The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain 125, 861–870. [DOI] [PubMed] [Google Scholar]
- [25].Blanchet PJ, Fang J, Gillespie M, Sabounjian L, Locke KW, Gammans R, Mouradian MM, Chase TN (1998) Effects of the full dopamine D1 receptor agonist dihydrexidine in Parkinson’s disease. Clin Neuropharmacol 21, 339–343. [PubMed] [Google Scholar]
- [26].Arnold AC, Okamoto LE, Gamboa A, Black BK, Raj SR, Elijovich F, Robertson D, Shibao CA, Biaggioni I (2016) Mineralocorticoid Receptor Activation Contributes to the Supine Hypertension of Autonomic Failure. Hypertension 67, 424–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Miller T, Marshall E (1980) Suppressor cell regulation of cell-mediated immune responses in renal infection in vitro modulation of suppressor cell activity. J Clin Invest 66, 621–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kuramoto L, Cragg J, Nandhagopal R, Mak E, Sossi V, de la Fuente-Fernandez R, Stoessl AJ, Schulzer M (2013) The nature of progression in Parkinson’s disease: an application of non-linear, multivariate, longitudinal random effects modelling. PLoS One 8, e76595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Jennings D, Siderowf A, Stern M, Seibyl J, Eberly S, Oakes D, Marek K, Investigators P (2017) Conversion to Parkinson Disease in the PARS Hyposmic and Dopamine Transporter-Deficit Prodromal Cohort. JAMA Neurol 74, 933–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Liepelt-Scarfone I, Brandle B, Yilmaz R, Gauss K, Schaeffer E, Timmers M, Wurster I, Brockmann K, Maetzler W, Van Nueten L, Streffer JR, Berg D (2017) Progression of prodromal motor and non-motor symptoms in the premotor phase study - 2-year follow-up data. Eur J Neurol 24, 1369–1374. [DOI] [PubMed] [Google Scholar]
- [31].Cilia R, Cereda E, Klersy C, Canesi M, Zecchinelli AL, Mariani CB, Tesei S, Sacilotto G, Meucci N, Zini M, Ruffmann C, Isaias IU, Goldwurm S, Pezzoli G (2015) Parkinson’s disease beyond 20 years. J Neurol Neurosurg Psychiatry 86, 849–855. [DOI] [PubMed] [Google Scholar]
- [32].Lokk J, Delbari A (2012) Clinical aspects of palliative care in advanced Parkinson’s disease. BMC Palliat Care 11, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Oparil S, Aronson S, Deeb GM, Epstein M, Levy JH, Luther RR, Prielipp R, Taylor A (1999) Fenoldopam: a new parenteral antihypertensive: consensus roundtable on the management of perioperative hypertension and hypertensive crises. Am J Hypertens 12, 653–664. [DOI] [PubMed] [Google Scholar]
- [34].Tumlin JA, Dunbar LM, Oparil S, Buckalew V, Ram CV, Mathur V, Ellis D, McGuire D, Fellmann J, Luther RR (2000) Fenoldopam, a dopamine agonist, for hypertensive emergency: a multicenter randomized trial. Fenoldopam Study Group. Acad Emerg Med 7, 653–662. [DOI] [PubMed] [Google Scholar]
- [35].George MS, Molnar CE, Grenesko EL, Anderson B, Mu Q, Johnson K, Nahas Z, Knable M, Fernandes P, Juncos J, Huang X, Nichols DE, Mailman RB (2007) A single 20 mg dose of dihydrexidine (DAR-0100), a full dopamine D1 agonist, is safe and tolerated in patients with schizophrenia. Schizophr Res 93, 42–50. [DOI] [PubMed] [Google Scholar]
- [36].Rosell DR, Zaluda LC, McClure MM, Perez-Rodriguez MM, Strike KS, Barch DM, Harvey PD, Girgis RR, Hazlett EA, Mailman RB, Abi-Dargham A, Lieberman JA, Siever LJ (2015) Effects of the D1 dopamine receptor agonist dihydrexidine (DAR-0100A) on working memory in schizotypal personality disorder. Neuropsychopharmacology 40, 446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mu Q, Johnson K, Morgan PS, Grenesko EL, Molnar CE, Anderson B, Nahas Z, Kozel FA, Kose S, Knable M, Fernandes P, Nichols DE, Mailman RB, George MS (2007) A single 20 mg dose of the full D1 dopamine agonist dihydrexidine (DAR-0100) increases prefrontal perfusion in schizophrenia. Schizophr Res 94, 332–341. [DOI] [PubMed] [Google Scholar]
- [38].Johns DW, Ayers CR, Carey RM (1984) The dopamine agonist bromocriptine induces hypotension by venous and arteriolar dilation. J Cardiovasc Pharmacol 6, 582–587. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.