

Abstract

Despite the advances in the synthesis of mechanically interlocked molecules, a generally applicable approach to interlocked natural products, such as lasso peptides, is yet to be formulated. While amino acid sequences have been introduced into several rotaxanes, the key structural components have always been dictated by the method used for supramolecular preorganization. In this work, we report the use of an ester-functionalized, aromatic δ-amino acid as the central covalent templating unit in the synthesis of both a [2]catenane and a [2]rotaxane from the same multimacrocyclic intermediate. This represents a key step toward future synthetic peptide-based interlocked products.
Keywords: rotaxanes, catenanes, mechanically interlocked molecules, macrocycles, templated synthesis, lasso peptides
Lasso peptides are ribosomally derived natural products. Their linear polypeptide precursor chain is post-translationally folded and ring-closed by multiple enzymes into a mechanically interlocked molecule (MIM).1 Although additional cross-linkages can exist between portions of the peptide backbone, in their simplest form, these compounds can be classified as [1]rotaxanes, in which the macrocycle and axle are connected by a “loop”.2−4 Ever since the milestone elucidation of the structure of microcin J25, a number of analogous compounds have been discovered. Some of these showed potent biological activities, improved ability to penetrate the cell membrane, and a high stability toward metabolic breakdown, making them attractive targets in the development of next-generation antibiotics.4−7 Remarkably, almost 20 years after their discovery, only a few attempts toward synthetic lasso peptide-like structures have been disclosed. Several [2]rotaxanes featuring a crown ether macrocycle and an ammonium ion thread were grafted with short amino acid sequences, resulting in [1]rotaxanes that mimic the loop region of a lasso peptide.8 Ammonium ions were also used to template the formation of [1]rotaxanes by self-entanglement or from a polyether macrocycle bearing amide functional groups.9,10 In both cases, poly-Gly sequences were installed in the loop region. To achieve an entirely peptidic rotaxane macrocycle, poly-l-ProGly cyclic peptides with cation binding properties were combined with diammonium threads and capped with bulky stoppers.11
Alternatively to ammonium ions, peptide sequences themselves have also been used to template the formation of an interlocking macrocycle onto a thread, even allowing the controlled release of a bioactive pentapeptide.12−14 However, the limited number and variety of supramolecular strategies employed in the synthesis of peptidic interlocked products is in stark contrast with the otherwise extensive development of MIM synthesis.15 This urges the consideration of atypical approaches to MIMs, such as covalent templating. Interestingly, the only total synthesis of a lasso peptide known to date took advantage of such covalent interactions; however, the unique strategy has not been reported for the synthesis of any other interlocked product, thus remaining an isolated and unverified case.16
To address the synthesis of so-called impossible MIMs, inaccessible by supramolecular templating,17 we embarked on the development of alternative covalent approaches. Inspired by the landmark directed synthesis of a catenane by Schill,18 we recently prepared a [2]catenane by templated backfolding.19 In both strategies, a ketal group was used to reversibly bind the separate fragments and enforce a perpendicular geometry between them before the key macrocyclizations. This preorganization is a strict requirement for the formation of the desired interlocked products.20,21 In addition to the backfolding macrocyclization, we developed a rotaxane synthesis based on terephthalic acid, which was proposed as a template by Schill and first employed successfully by Höger.22,23 In this case, perpendicularity is achieved via a sterically congested phenol terephthalate ester bond. In this work, we combined these two fundamentally different covalent template-directed strategies, resulting in multimacrocyclic intermediate 1 (Figure 1). This features an isophthalate δ-amino acid central template (in blue), equivalent in length to a dipeptide unit or the side chain of glutamic acid and bearing an additional carboxylic group. The template is included into one of the interlocking fragments via two directionally aligned amide bonds. It connects to the other (in red) via a phenol ester and two acid-labile benzylic tethers (in black), combining elements from our rotaxane and backfolding macrocyclization strategies, respectively. From 1, either a precatenane or a prerotaxane (sometimes referred to as pretzelane and [1]rotaxane, respectively)24 can be obtained by clipping or capping of the thread (in blue). Cleavage of the temporary ester and benzylic linkages then liberates [2]catenane 2 or [2]rotaxane 3 and restores the amide and side-chain groups of the amino acid template, yielding an approach that is effectively traceless on one of the interlocking fragments.
Figure 1.

Schematic plan of the synthesis of a [2]catenane (2) and a [2]rotaxane (3) from a common multimacrocyclic intermediate (1) via either clipping or capping approaches.
Multimacrocyclic intermediate 1 is prepared in a convergent way by introducing the macrocycle (in red) as two separate fragments, 4 and 5, onto thread fragment 6 (Scheme 1). All three building blocks 4–6 were prepared on a gram scale (see Supporting Information). The construction of the multicyclic architecture starts with transesterification of methyl ester 6 with phenol 5 via a perfluorophenol ester intermediate. The following deprotection of the o-hydroxyl benzylic tethers to give 7 allows the introduction of the remaining half of the macrocycle. This is done via a direct transesterification with diperfluorophenol ester 4, giving macrolactone 8 in 77% yield. The 31-membered macrocycle is then clipped around the template via a copper-catalyzed azide–alkyne cycloaddition reaction to give multimacrocyclic intermediate 1 in 78% yield. Starting from the thread structure, key intermediate 1 is accessed in a sequence of just six steps, all proceeding in good to high yields.
Scheme 1. Synthesis of Multimacrocyclic Intermediate 1.
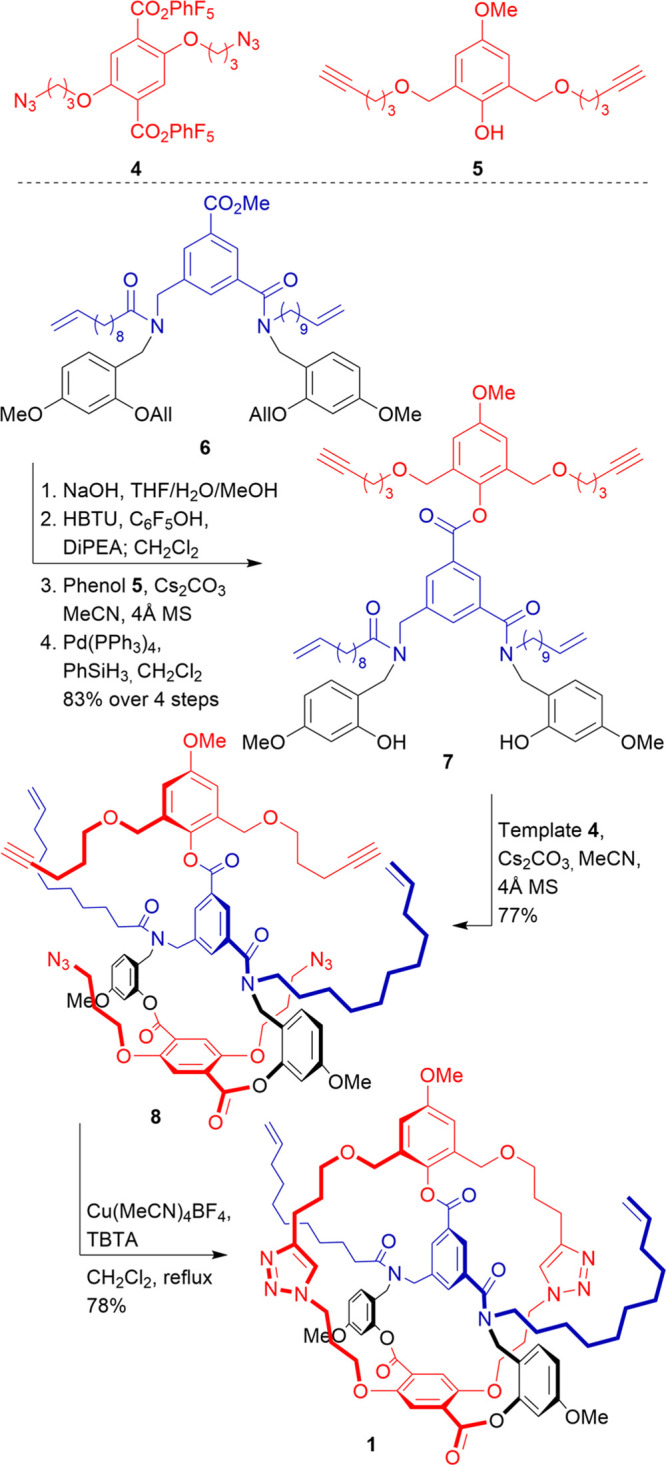
First, compound 1 was transformed into a [2]catenane. Precatenane 9 was obtained by a Grubbs-type ring-closing metathesis (RCM) reaction, followed by hydrogenation of the newly formed internal alkene to remove E/Z isomerism (Scheme 2). Despite our best efforts, RCM required high temperatures and proceeded sluggishly. Although some starting material could be recovered and recycled, the yield was mediocre, likely due to an insufficient length of the terminal alkene bearing fragments (vide infra). Subsequent conversion of precatenane 9 to [2]catenane 10 was accomplished with surprising ease: mild solvolytic conditions at room temperature were sufficient to break all three temporary ester linkages within hours. This was unexpected, as in our previous efforts liberation of the interlocked species was always made difficult by a catenand effect.25,26 Notably, at this stage, no noninterlocked fragments were observed, and 10 was obtained in high yield after purification, suggesting that compound 1 is formed solely in the expected conformation. In order to restore the peptide-like amide bonds of the template, the benzylic tethers were cleaved under acidic conditions. Despite competing cleavage of the benzylic ethers in the phenol macrocycle (in red), the final [2]catenane 2 could be obtained in 68% yield.
Scheme 2. Synthesis of [2]Catenane 2.
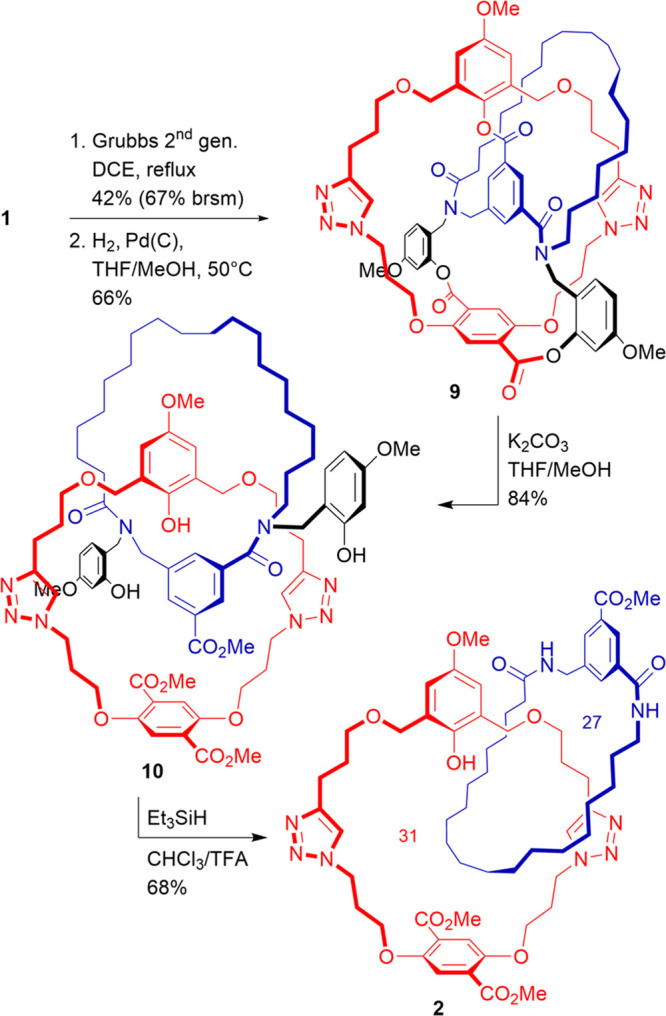
After the successful synthesis of [2]catenane, our next goal was to convert key intermediate 1 to prerotaxane 12 (Scheme 3). This was done via a Grubbs-type cross-metathesis reaction with alkene-functionalized stopper unit 11.20 Use of this electron-poor coupling partner in excess gave the desired product in 76% yield, with only trace amounts of competing RCM product 9 being observed. The higher yield and milder conditions required to obtain 12 as compared to 9 corroborate our hypothesis on the insufficient size of the catenane 27-membered macrocycle (in blue). Subsequent liberation of [2]rotaxane 13 proceeded in good yield and under mild conditions, while cleavage of the benzylic appendages to give 3 was complicated by the same side reactions observed for [2]catenane 2.
Scheme 3. Synthesis of [2]Rotaxane 3.

The identity of both compounds 2 and 3 was confirmed by mass spectrometry and by comparison of their 1H NMR spectra with those of equimolar mixtures of the respective noninterlocked components (Figure 3). Regarding [2]catenane 2, the most striking difference is the significant splitting of nearly all signals from the 31-membered macrocycle (in red). In principle, [2]catenane 2 possesses the same symmetry properties as Sauvage’s “rubber glove” system, resulting in two enantiomorphous conformers that interconvert via rotation of the terephthalic unit (Figure 2).27 This conformational exchange follows a path devoid of achiral intermediates, analogous to the inversion of a rubber glove by turning it inside out. In addition, computational modeling of structure 2 (see Supporting Information) suggests a tendency of all aromatic rings of the 31-membered macrocycle to lie parallel to its cavity, potentially resulting in two local conformations for each one of them. This could explain the observed splitting of aromatic signals h and b, while the “rubber glove” symmetry would result in diastereotopic environments for the two l protons.
Figure 3.

(I) 1H NMR spectrum of an equimolar mixture of the noninterlocked components of 2. (II) 1H NMR spectrum of [2]catenane 2. (III) Schematic representation of the proposed pirouetting-derived enantiomeric conformers of 3. (IV) 1H NMR spectrum of an equimolar mixture of the noninterlocked components of 3. (V) 1H NMR spectrum of [2]rotaxane 3. (VI) Schematic representation of the proposed translational isomers of 3. All NMR spectra were recorded in CDCl3 at a concentration of ∼13 mM at 500 MHz. Signals are color coded based on their respective component. The blue arrows in the schemes represent the directionality of the template amide bonds.
Figure 2.

Enantiomorphous conformations of [2]catenane 2 according to the “topological rubber glove” model. The conformers are related by the mirror plane marked with a dashed line and interconverted by rotation of the terephthalic unit.
Furthermore, aliphatic signals d, e, f, g, and k display similar or stronger splitting, suggesting the presence of significantly different diastereotopic environments. In addition, variable-temperature 1H NMR experiments (see Supporting Information) reveal coalescence of the aromatic ring signals between 70 and 110 °C in DMSO-d6, which is incompatible with the expected rate of rotation of some of the smaller aromatic rings. Our proposed explanation is a hindered pirouetting of the 31-membered macrocycle within the 27-membered macrocycle due to the presence of two tetrasubstituted aromatic rings, as depicted in Figure 3(III). As a result of this, one side of the 31-membered macrocycle lies within the cavity of the 27-membered macrocycle, likely experiencing a significantly different chemical environment compared to the opposite side. Only upon heating beyond 70 °C, pirouetting of the 31-membered macrocycle occurs at such a rate that the chemical shifts of protons on both sides of the ring are averaged. This is compatible with the steric hindrance of the terephthalic and phenolic groups, as a simple p-tBu phenyl unit was observed to completely prevent slippage of a 24-membered polyether macrocycle at room temperature28 and a di-m-tBu phenyl stopper has been used to form stable rotaxanes with a 26-membered macrocycle.29 A fascinating consequence is that 2 possesses co-conformational “topological” chirality, due to the directionality imparted to one of the macrocycles by the amide bonds.30 Combined with the “rubber glove” chirality, at least two diastereoisomers of [2]catenane 2 would be expected, but either their NMR spectra are too similar to be resolved or rotation of the terephthalic unit occurs too rapidly at room temperature.
The resonances of the 27-membered macrocycle (in blue) show interesting differences, as well: aliphatic signals A, B, F, and H, together with the majority of signals around 1.2 ppm shift significantly upfield, likely due to the aromatic ring current effects within the 31-membered macrocycle.31 In addition, signals A, B, and F appear to split into an asymmetrical pattern. These likely correspond to two co-conformations of the 27-membered macrocycle within the 31-membered macrocycle, with either one of the amide groups hydrogen bonding with the phenol or triazole groups, indicated by the downfield shift of amide proton C. It should be noted that these conformers are unlikely to be the origin of the large splitting in the macrocycle signals, as these have a 1:1 ratio and only coalesce at high temperature, whereas the only barrier to this conformational exchange appears to be the amide hydrogen bonding.
Contrary to what is observed for [2]catenane 2, most signals relative to the thread component (in blue) of [2]rotaxane 3 appear to split. This can be explained by a hindered movement of the 31-membered macrocycle along the axle, as depicted in Figure 3(VI). The amide bond directionality in the thread fragment results in the formation of two isomers, each with its unique set of peaks. This is most clearly visible for the thread template signals D. Moving away from the central template, the two sides of the thread become nearly indistinguishable, except for the presence or absence of the encircling macrocycle. As a result, trivial thread signals A, B, F, H, J, K, and N only have two discernible counterparts in the spectrum of [2]rotaxane 3: one with a little shift relative to the noninterlocked component and another one upfield of the first. This shift is caused by the ring current effects exerted by the surrounding macrocycle and can only affect one side of the thread at a time. In contrast, the average chemical shift of the template and stopper units, D and P, are barely affected. This indicates that the macrocycle occupies two favored positions on the thread, corresponding to the unhindered aliphatic regions.
Aside from steric hindrance, hydrogen bonding is likely to play a role in this equilibrium. In fact, the significant downfield shift of NH protons C, G, and M (∼0.8 ppm for all four) suggests a substantial interaction with the triazole (peak h shifts by 0.3 ppm) and phenol groups.29,32 Notably, the NH signals appear to split in two sets, with only one being affected by the macrocycle, consistent with the fact that this can only interact with one side of the thread at a time.
As expected, increasing the temperature causes peaks D to coalesce into a single set of three in DMSO-d6 (see Supporting Information), as the rate of the shuttling motion of the macrocycle increases. However, aromatic ring signals b, h, and l and those of their methyl substituents, a and m, already appear as single peaks in DMSO-d6 at room temperature, suggesting their splitting in CDCl3 is the result of a different process. In this case, it is the “rubber glove” symmetry of the terephthalic unit and a hindered rotation of the triazole and phenol rings that causes the splitting, which is notably absent for the nonaromatic protons of the macrocycle. Due to the hindered shuttling motion, one face of the 31-membered macrocycle is directly adjacent to the template unit, while the other is next to one of the stoppers, resulting in completely different chemical environments. This helps to explain why at room temperature this effect is only observed for [2]rotaxane 3 and not for [2]catenane 2, as in the latter the differences between the environments on the two faces are far more subtle.
In conclusion, we were able to synthesize both a [2]catenane and a [2]rotaxane from a common multimacrocyclic intermediate. These complex structures, featuring translational isomerism or co-conformational chirality, were obtained in a convergent synthesis, requiring a relatively brief number of steps, with most key reactions proceeding in good yield. All covalent templating interactions were based on a dipeptide-like unit and took advantage of functional groups and motifs which are commonly found in peptides. This showcases the potential for the application of this strategy to the synthesis of lasso peptide analogues with closer resemblance to their target structure. Work is in progress to replace the isophthalic moiety by a glutamic, aspartic, or dipeptide moiety as covalent templates and the further incorporation of peptide fragments with the aim to develop a general route to peptidic MIMs.
Acknowledgments
The authors would like to thank The Netherlands Organisation for Scientific Research (NWO-CW, ECHO Grant No. 711.012.007 to J.H.v.M). E. Zuidinga and Dr. A. Ehlers (University of Amsterdam) are acknowledged for mass spectrometry and NMR assistance.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsorginorgau.1c00017.
Synthetic protocols, experimental conditions, and full characterization of new compounds (PDF)
Author Contributions
S.P. performed the synthetic experiments. S.P. and J.H.v.M. developed the methodology. J.H.v.M. supervised the research. S.P., J.H.v.M., and S.I.J. wrote the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Wang X.-W.; Zhang W.-B. Chemical Topology and Complexity of Protein Architectures. Trends Biochem. Sci. 2018, 43 (10), 806–817. 10.1016/j.tibs.2018.07.001. [DOI] [PubMed] [Google Scholar]
- Niemyska W.; Dabrowski-Tumanski P.; Kadlof M.; Haglund E.; Sułkowski P.; Sulkowska J. I. Complex Lasso: New Entangled Motifs in Proteins. Sci. Rep. 2016, 6 (1), 36895. 10.1038/srep36895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Gómez H.; Tulla-Puche J. Lasso Peptides: Chemical Approaches and Structural Elucidation. Org. Biomol. Chem. 2018, 16 (28), 5065–5080. 10.1039/C8OB01304G. [DOI] [PubMed] [Google Scholar]
- Hegemann J. D.; Zimmermann M.; Xie X.; Marahiel M. A. Lasso Peptides: An Intriguing Class of Bacterial Natural Products. Acc. Chem. Res. 2015, 48 (7), 1909–1919. 10.1021/acs.accounts.5b00156. [DOI] [PubMed] [Google Scholar]
- Rosengren K. J.; Clark R. J.; Daly N. L.; Göransson U.; Jones A.; Craik D. J. Microcin J25 Has a Threaded Sidechain-to-Backbone Ring Structure and Not a Head-to-Tail Cyclized Backbone. J. Am. Chem. Soc. 2003, 125 (41), 12464–12474. 10.1021/ja0367703. [DOI] [PubMed] [Google Scholar]
- Maksimov M. O.; Pan S. J.; James Link A. Lasso Peptides: Structure, Function, Biosynthesis, and Engineering. Nat. Prod. Rep. 2012, 29 (9), 996. 10.1039/c2np20070h. [DOI] [PubMed] [Google Scholar]
- Rowe S. M.; Spring D. R. The Role of Chemical Synthesis in Developing RiPP Antibiotics. Chem. Soc. Rev. 2021, 50 (7), 4245–4258. 10.1039/D0CS01386B. [DOI] [PubMed] [Google Scholar]
- Saito F.; Bode J. W. Synthesis and Stabilities of Peptide-Based [1]Rotaxanes: Molecular Grafting onto Lasso Peptide Scaffolds. Chem. Sci. 2017, 8 (4), 2878–2884. 10.1039/C7SC00021A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young M. J.; Akien G. R.; Evans N. H. An Amide Hydrogen Bond Templated [1]Rotaxane Displaying a Peptide Motif – Demonstrating an Expedient Route to Synthetic Mimics of Lasso Peptides. Org. Biomol. Chem. 2020, 18 (27), 5203–5209. 10.1039/D0OB01190H. [DOI] [PubMed] [Google Scholar]
- Clavel C.; Fournel-Marotte K.; Coutrot F. A pH-Sensitive Peptide-Containing Lasso Molecular Switch. Molecules 2013, 18 (9), 11553–11575. 10.3390/molecules180911553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aucagne V.; Leigh D. A.; Lock J. S.; Thomson A. R. Rotaxanes of Cyclic Peptides. J. Am. Chem. Soc. 2006, 128 (6), 1784–1785. 10.1021/ja057206q. [DOI] [PubMed] [Google Scholar]
- Leigh D. A.; Murphy A.; Smart J. P.; Slawin A. M. Z. Glycylglycine Rotaxanes—The Hydrogen Bond Directed Assembly of Synthetic Peptide Rotaxanes. Angew. Chem., Int. Ed. Engl. 1997, 36 (7), 728–732. 10.1002/anie.199707281. [DOI] [Google Scholar]
- Lane A. S.; Leigh D. A.; Murphy A. Peptide-Based Molecular Shuttles. J. Am. Chem. Soc. 1997, 119 (45), 11092–11093. 10.1021/ja971224t. [DOI] [Google Scholar]
- Fernandes A.; Viterisi A.; Coutrot F.; Potok S.; Leigh D. A.; Aucagne V.; Papot S. Rotaxane-Based Propeptides: Protection and Enzymatic Release of a Bioactive Pentapeptide. Angew. Chem., Int. Ed. 2009, 48 (35), 6443–6447. 10.1002/anie.200903215. [DOI] [PubMed] [Google Scholar]
- Sluysmans D.; Stoddart J. F. The Burgeoning of Mechanically Interlocked Molecules in Chemistry. Trends Chem. 2019, 1 (2), 185–197. 10.1016/j.trechm.2019.02.013. [DOI] [Google Scholar]
- Chen M.; Wang S.; Yu X. Cryptand-Imidazolium Supported Total Synthesis of the Lasso Peptide BI-32169 and Its D -Enantiomer. Chem. Commun. 2019, 55 (23), 3323–3326. 10.1039/C8CC10301A. [DOI] [PubMed] [Google Scholar]
- Hannam J. S.; Lacy S. M.; Leigh D. A.; Saiz C. G.; Slawin A. M. Z.; Stitchell S. G. Controlled Submolecular Translational Motion in Synthesis: A Mechanically Interlocking Auxiliary. Angew. Chem. 2004, 116 (25), 3322–3326. 10.1002/ange.200353606. [DOI] [PubMed] [Google Scholar]
- Schill G.; Lüttringhaus A. The Preparation of Catena Compounds by Directed Synthesis. Angew. Chem., Int. Ed. Engl. 1964, 3 (8), 546–547. 10.1002/anie.196405461. [DOI] [Google Scholar]
- Pilon S.; Jørgensen S. I.; van Maarseveen J. H. [2]Catenane Synthesis via Covalent Templating. Chem. - Eur. J. 2021, 27 (7), 2310–2314. 10.1002/chem.202004925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steemers L.; Wanner M. J.; Lutz M.; Hiemstra H.; van Maarseveen J. H. Synthesis of Spiro Quasi[1]Catenanes and Quasi[1]Rotaxanes via a Templated Backfolding Strategy. Nat. Commun. 2017, 8, 15392. 10.1038/ncomms15392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schill G.; Beckmann W.; Vetter W. Statistische Synthesen von Rotaxanen. Chem. Ber. 1980, 113 (3), 941–954. 10.1002/cber.19801130314. [DOI] [Google Scholar]
- Schill G.Catenanes, Rotaxanes, and Knots; Elsevier, 1971; Vol. 22. [Google Scholar]
- Schweez C.; Shushkov P.; Grimme S.; Höger S. Synthesis and Dynamics of Nanosized Phenylene-Ethynylene-Butadiynylene Rotaxanes and the Role of Shape Persistence. Angew. Chem. 2016, 128 (10), 3389–3394. 10.1002/ange.201509702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter C.; Mohry A.; Sobanski A.; Vögtle F. [1]Rotaxanes and Pretzelanes: Synthesis, Chirality, and Absolute Configuration. Chem. - Eur. J. 2000, 6 (9), 1674–1682. . [DOI] [PubMed] [Google Scholar]
- Albrecht-Gary A. M.; Saad Z.; Dietrich-Buchecker C. O.; Sauvage J. P. Interlocked Macrocyclic Ligands: A Kinetic Catenand Effect in Copper (I) Complexes. J. Am. Chem. Soc. 1985, 107 (11), 3205–3209. 10.1021/ja00297a028. [DOI] [Google Scholar]
- Godt A. Non-Rusty [2]Catenanes with Huge Rings and Their Polymers. Eur. J. Org. Chem. 2004, 2004 (8), 1639–1654. 10.1002/ejoc.200300503. [DOI] [Google Scholar]
- Chambron J.-C.; Sauvage J.-P.; Mislow K.; De Cian A.; Fischer J. A [2]Catenane and a [2]Rotaxane as Prototypes of Topological and Euclidean Molecular “Rubber Gloves". Chem. - Eur. J. 2001, 7 (19), 4085–4096. . [DOI] [PubMed] [Google Scholar]
- Ashton P. R.; Baxter I.; Fyfe M. C. T.; Raymo F. M.; Spencer N.; Stoddart J. F.; White A. J. P.; Williams D. J. Rotaxane or Pseudorotaxane? That Is the Question!. J. Am. Chem. Soc. 1998, 120 (10), 2297–2307. 10.1021/ja9731276. [DOI] [Google Scholar]
- Lahlali H.; Jobe K.; Watkinson M.; Goldup S. M. Macrocycle Size Matters: “Small” Functionalized Rotaxanes in Excellent Yield Using the CuAAC Active Template Approach. Angew. Chem., Int. Ed. 2011, 50 (18), 4151–4155. 10.1002/anie.201100415. [DOI] [PubMed] [Google Scholar]
- Jamieson E. M. G.; Modicom F.; Goldup S. M. Chirality in Rotaxanes and Catenanes. Chem. Soc. Rev. 2018, 47 (14), 5266–5311. 10.1039/C8CS00097B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehnbom A.; Hall M. B.; Gladysz J. A. Origin of Shielding and Deshielding Effects in NMR Spectra of Organic Conjugated Polyynes. Org. Lett. 2019, 21 (3), 753–757. 10.1021/acs.orglett.8b03990. [DOI] [PubMed] [Google Scholar]
- Hori S.; Yamauchi K.; Kuroki S.; Ando I. Proton NMR Chemical Shift Behavior of Hydrogen-Bonded Amide Proton of Glycine-Containing Peptides and Polypeptides as Studied by Ab Initio MO Calculation. Int. J. Mol. Sci. 2002, 3 (8), 907–913. 10.3390/i3080907. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.