

SUMMARY
Brown adipose tissue (BAT) thermogenic activity is tightly regulated by cellular redox status, but the underlying molecular mechanisms are incompletely understood. Protein S-nitrosylation, the nitric-oxide-mediated cysteine thiol protein modification, plays important roles in cellular redox regulation. Here we show that diet-induced obesity (DIO) and acute cold exposure elevate BAT protein S-nitrosylation, including UCP1. This thermogenic-induced nitric oxide bioactivity is regulated by S-nitrosoglutathione reductase (GSNOR; alcohol dehydrogenase 5 [ADH5]), a denitrosylase that balances the intracellular nitroso-redox status. Loss of ADH5 in BAT impairs cold-induced UCP1-dependent thermogenesis and worsens obesity-associated metabolic dysfunction. Mechanistically, we demonstrate that Adh5 expression is induced by the transcription factor heat shock factor 1 (HSF1), and administration of an HSF1 activator to BAT of DIO mice increases Adh5 expression and significantly improves UCP1-mediated respiration. Together, these data indicate that ADH5 controls BAT nitroso-redox homeostasis to regulate adipose thermogenesis, which may be therapeutically targeted to improve metabolic health.
Graphical abstract
In brief
Sebag et al. report that ADH5-mediated nitroso-redox homeostasis regulates brown adipose thermogenesis, and loss of HSF1-Adh5 activation leads to obesity-associated metabolic dysfunction.
INTRODUCTION
Brown adipose tissue (BAT) plays critical roles in converting intercellular and systemic lipid and glucose fuels directly into heat in response to thermogenetic challenge (Bartelt et al., 2011; Cannon and Nedergaard, 2004; Kajimura and Saito, 2014; Nedergaard et al., 2007; Saito et al., 2009; Townsend and Tseng, 2014). The key signaling molecule nitric oxide (NO) regulates BAT biology and physiology (Saha and Kuroshima, 2000) by increasing blood flow following noradrenergic stimulation (Kikuchi-Utsumi et al., 2002; Nagashima et al., 1994), promoting BAT hyperplasia through alteration of the proliferation and differentiation of brown adipocyte (BA) precursors (Nisoli et al., 1998) and influencing BA hypertrophy (Jankovic et al., 2017; Saha and Kuroshima, 2000). NO also modulates BAT lipid and glucose metabolism (Sansbury and Hill, 2014), sympathetic tone (Giordano et al., 2002; Nisoli et al., 1997), and mitochondrial biogenesis in response to acute thermogenic stimuli (Nisoli et al., 2003, 2004). Although basal levels of NO are critical for maintaining cellular and systemic homeostasis, excess NO production contributes to the development of numerous pathological metabolic disorders, including obesity (Chiarelli et al., 2000; Engeli et al., 2004; Jankovic et al., 2017; Noronha et al., 2005; Sansbury and Hill, 2014). However, the pathological role of BAT NO bioactivity in the context of obesity is largely unknown.
NO regulates cellular functions largely through cyclic guanosine monophosphate (cGMP)-dependent signaling, reactive oxygen species (ROS) and reactive nitrogen species (RNS) signal transduction pathways, and protein modifications (Stone and Marletta, 1996). In mice, diet-induced obesity (DIO) increases aberrant NO-mediated protein S-nitrosylation (SNO), leading to nitrosative stress that disrupts cellular homeostasis (Kaneki et al., 2007; Ovadia et al., 2011; Qian et al., 2018). We have recently shown that protein SNOs are elevated in the livers of obese patients compared to lean patients as well as in high-fat diet (HFD)-fed mice, resulting in impaired endoplasmic reticulum (ER) homeostasis and hepatic autophagy (Qian et al., 2018, 2019; Yang et al., 2015). In contrast, attenuating nitrosative stress by suppressing inducible NO synthase (iNOS) ameliorates obesity-associated insulin resistance in the liver, skeletal muscle and adipose tissue (Becerril et al., 2018; Perreault and Marette, 2001; Yang et al., 2015). With regard to pathophysiological studies in BAT, it has been demonstrated that in vitro, excessive generation of NO inhibits lipolysis in rat brown preadipocytes (Nisoli et al., 1998), while in vivo, ablation of iNOS improves the energy balance of ob/ob mice in part by stimulating thermogenesis in brown fat cells (Becerril et al., 2010). Although these studies suggest a link between pathological NO bioactivity and impaired BAT function, the contribution of aberrant cellular nitrosative signaling in obesity-associated BAT metabolic dysfunction remains uncharacterized.
Alcohol dehydrogenase 5 (ADH5; also named S-nitrosoglutathione reductase [GSNOR]) is the major cellular denitrosylase that regulates cellular NO availability by catalyzing and regulating the breakdown of SNOs in order to balance the intracellular thiol redox status (Barnett and Buxton, 2017; Benhar et al., 2009; Liu et al., 2001). In plants, ADH5 plays a critical role in modulating tolerance to both heat and cold stressors (Hussain et al., 2019; Leterrier et al., 2011; Lv et al., 2017). In mammals, dysregulation of ADH5 contributes to the pathogenesis of a diverse array of chronic diseases such as fatty liver disease, cardiovascular diseases and aging (Barnett and Buxton, 2017; Qian et al., 2018; Que et al., 2009; Rizza et al., 2018; Rizza and Filomeni, 2018; Sips et al., 2013; Tang et al., 2013). Notably, a recent study demonstrated that ADH5 regulates adipocyte differentiation (Cao et al., 2015). However, the role of ADH5-mediated protein denitrosylation during physiological cold adaptation or in the pathogenesis of obesity-associated BAT dysfunction remains unknown.
Here, we demonstrate that ADH5 is required for maintaining BAT metabolic homeostasis by playing a key role in balancing cellular NO bioactivity in brown adipocytes. Furthermore, we identified that dysregulation of an important proteostasis regulator, heat shock factor 1 (HSF1), leads to disruption of BAT ADH5 activation in the context of obesity. As such, targeting nitrosative stress in BAT by modulating the balance between ADH5 and iNOS actions may provide an avenue for the development of new therapies to improve metabolic health.
RESULTS
Obesity elevates protein SNO and decreases cellular denitrosylation in BAT
To determine the impact of NO bioactivity on BAT function in response to various metabolic stressors, we first investigated the effects of nutritional overload on protein SNO in BAT using immunohistochemistry and a tandem mass tag (TMT)-based SNO western blot analysis (Qu et al., 2014). Compared to mice fed a regular diet (RD), there was a significant increase in general protein SNO in BAT from mice fed a HFD (Figures 1A, 1B, S1A, and S1B), which was associated with elevated basal nitrite and nitrate (NOx) released into the supernatant from BAT explants (Figure 1C). This obesity-associated NO signaling status was correlated with a reduction in BAT mitochondrial uncoupling protein 1 (UCP1)-mediated oxygen consumption (Figure 1D) as measured by high-resolution respirometry (Porter, 2017). To investigate the NO bioactivity dynamics in response to acute cold challenge, we measured BAT SNO levels in wild-type (WT) mice acclimated from thermoneutrality (TN; 30°C) to room temperature (RT; 22°C) as well as to acute cold (4°C). In response to acute-cold challenge, BAT SNO was induced at 2 h, reduced at 6 h, and again increased at 24 h (Figures 1E, 1F, and S1C). Together, these data indicate that thermogenic programs induce protein SNO in BAT.
Figure 1. Diet-induced obesity (DIO) elevates nitrosative stress in brown adipose tissue (BAT).
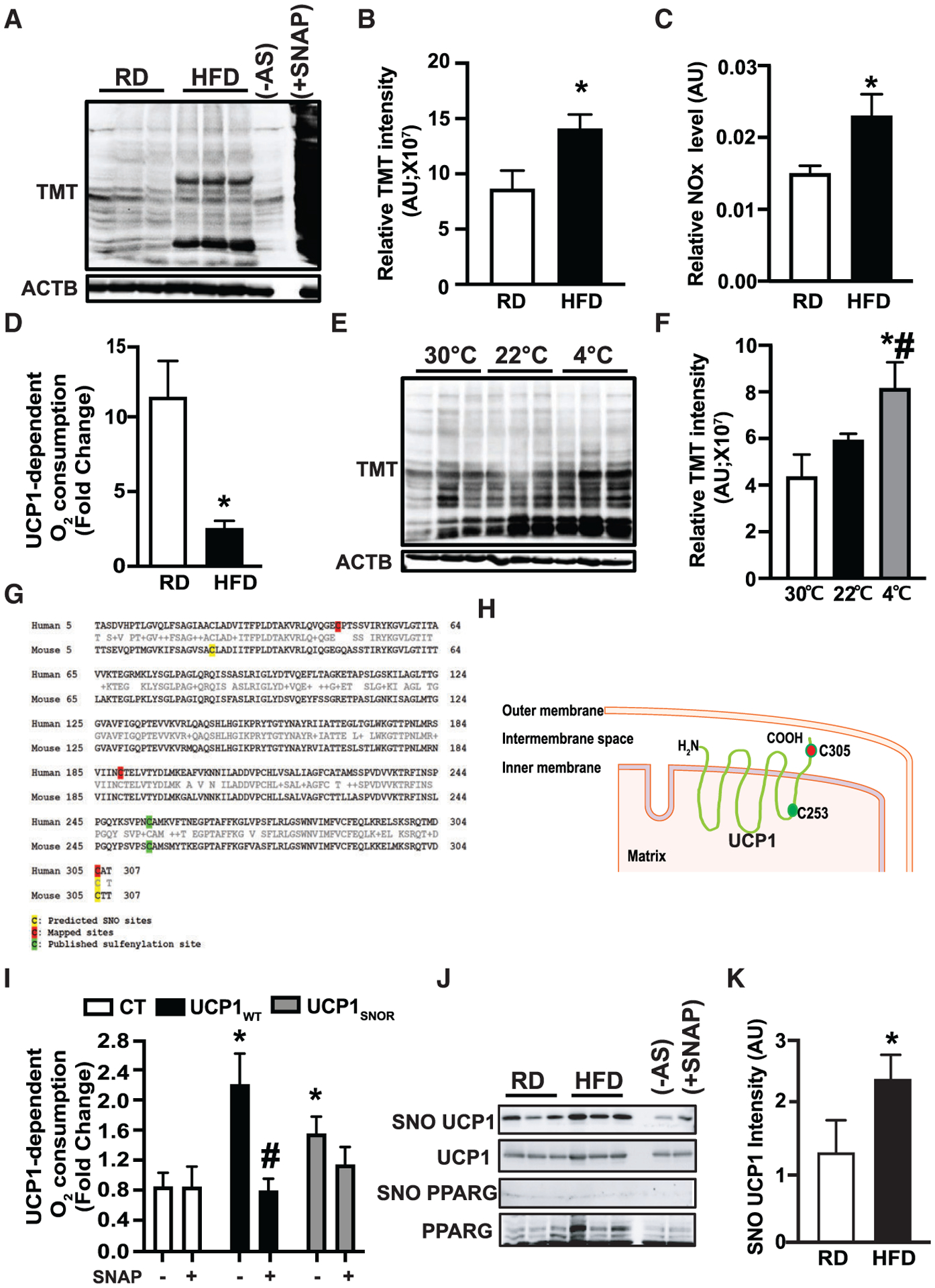
(A and B) Western blots and densitometric analysis of protein S-nitrosylation (SNO) in interscapular BAT (iBAT) from mice fed a regular diet (RD) or high-fat diet (HFD) for 10 weeks. (−) AS, no ascorbate (negative control for S-nitrosylation detection); (+) SNAP, positive control for SNO detection. n = 4–5 mice/group. Data were normalized to actin beta (ACTB) input.
(C) Relative nitrite/nitrate NOx release from supernatants of BAT explants isolated from mice on a RD (n = 8 mice) or HFD (n = 11 mice) for 10 weeks.
(D) High-resolution respirometry in fresh BAT from mice in (A) (n = 4 mice/group). The UCP1-mediated respiration was calculated and reported as fold change of GDP-inhibited leak respiration (1 mM) over baseline.
(E and F) Western blots and quantification of SNO in interscapular iBAT mice exposed to thermoneutrality (TN; 30°C for 1 week), room temperature (RT; 22°C), or cold (4°C, 24 h). n = 3–6 mice/group. Data were normalized to ACTB input.
(G and H) Predicted SNO sites on mouse UCP1 and mapped SNO sites on human UCP1.
(I) High-resolution respirometry in human brown adipocytes transiently transfected with control plasmid (CT; pcDNA-EGFP), UCP1WT, or UCP1C305A. Cells were treated with 1 μM isoproterenol (ISO) (2 h) in the presence or absence of 100 μM SNAP (2 h). The UCP1-mediated respiration was presented as fold change of GDP-inhibited leak (1 mM) over ISO-mediated baseline. n = 3 independent experiments.
(J and K) Representative western blots and densitometric analysis of S-nitrosylated proteins in BAT from mice on a RD and HFD for 8 weeks. In western blots, each lane represents an individual mouse. Data were normalized to UCP1 input.
All data are presented as means ± SEM. Asterisk indicates statistical significance compared to the RD groups (B–D and K), the 30°C group (F), and the CT groups (I). Number sign indicates statistical significance between 22°C and 4°C (F) and effect of SNAP in same cells (I). Statistical significance was determined by Student’s t test (B–D) and (K) and one-way ANOVA (F and I); p < 0.05).
Previous studies have shown that acute induction of thermogenesis in BAT generates physiologic levels of mitochondrial ROS, which in turn alters the redox status of UCP1, leading to activation of UCP1 (Chouchani et al., 2016). In contrast, studies have shown that excessive levels of ROS and RNS impair BAT mitochondrial function (Cui et al., 2019; Lee et al., 2020). Using a group-based prediction (GPS)-SNO prediction tool (Xue et al., 2010), we found that Cys25 and Cys305 are potential SNO sites on murine UCP1, while Cys305 was predicted as a potential SNO site on human UCP1 (Figure 1G). We then mapped SNO sites on human UCP1 by mass spectra analysis following SNO protein labeling by iodoacetyl tandem mass tag (iodoTMT) reagent and found that Cys47, Cys189, and Cys305 are targeted by SNO. Therefore, among these SNO sites, Cys305 is the common SNO site in both human and mouse UCP1 (Figures 1G and 1H). We then generated an SNO-resistant mouse UCP1 (C305A; UCP1SNOR) variant and its WT control (UCP1wt; Figure S1D). Isoproterenol (ISO)-mediated UCP1-dependent oxygen consumption was significantly reduced by the chemical NO donor S-nitroso-N-acetyl-DL-penicillamine (SNAP) in human BAT cells overexpressing UCP1wt (hTERT-hA41BAT-SVF) (Xue et al., 2015) (Figure 1I). However, the SNAP-mediated suppression of UCP1 uncoupling was significantly dampened in cells overexpressing UCP1SNOR (Figure 1I). We next determined whether UCP1 is targeted by SNO in mouse BAT in vivo using immunohistochemistry and biotin switch analyses. As shown in Figures 1J, 1K and S1E, not only is UCP1 targeted by SNO modification, but also SNO-UCP1 expression is significantly increased in BAT of mice fed a HFD (8 weeks on diet) compared to RD controls. However, we did not detect SNO of peroxisome proliferator activated receptor gamma (PPARG), an SNO target reported in mesenchymal stem cells (MSCs) (Cao et al., 2015), in BAT from either lean or obese mice (Figure 1J). Collectively, these data indicate a potential role for protein SNO in the pathogenesis of obesity-mediated BAT dysfunction.
ADH5 protects BAT against obesity-associated metabolic dysfunction
ADH5 modulates NO availability in the cell by catalyzing the breakdown of SNOs (Barnett and Buxton, 2017; Benhar et al., 2009; Liu et al., 2004). We found that ADH5 was significantly increased in BAT of WT RD mice challenged with acute cold (4°C, 24 h; Figure 2A). In contrast, in the context of obesity, Adh5 transcript and protein expression in BAT were significantly reduced after 12 weeks of HFD (Figures 2B and 2C). These data are consistent with a recent study demonstrating that obesity reduces cellular antioxidant capacity in BAT (Alcalá et al., 2017).
Figure 2. ADH5 protects metabolic dysfunction of BAT in the context of obesity.
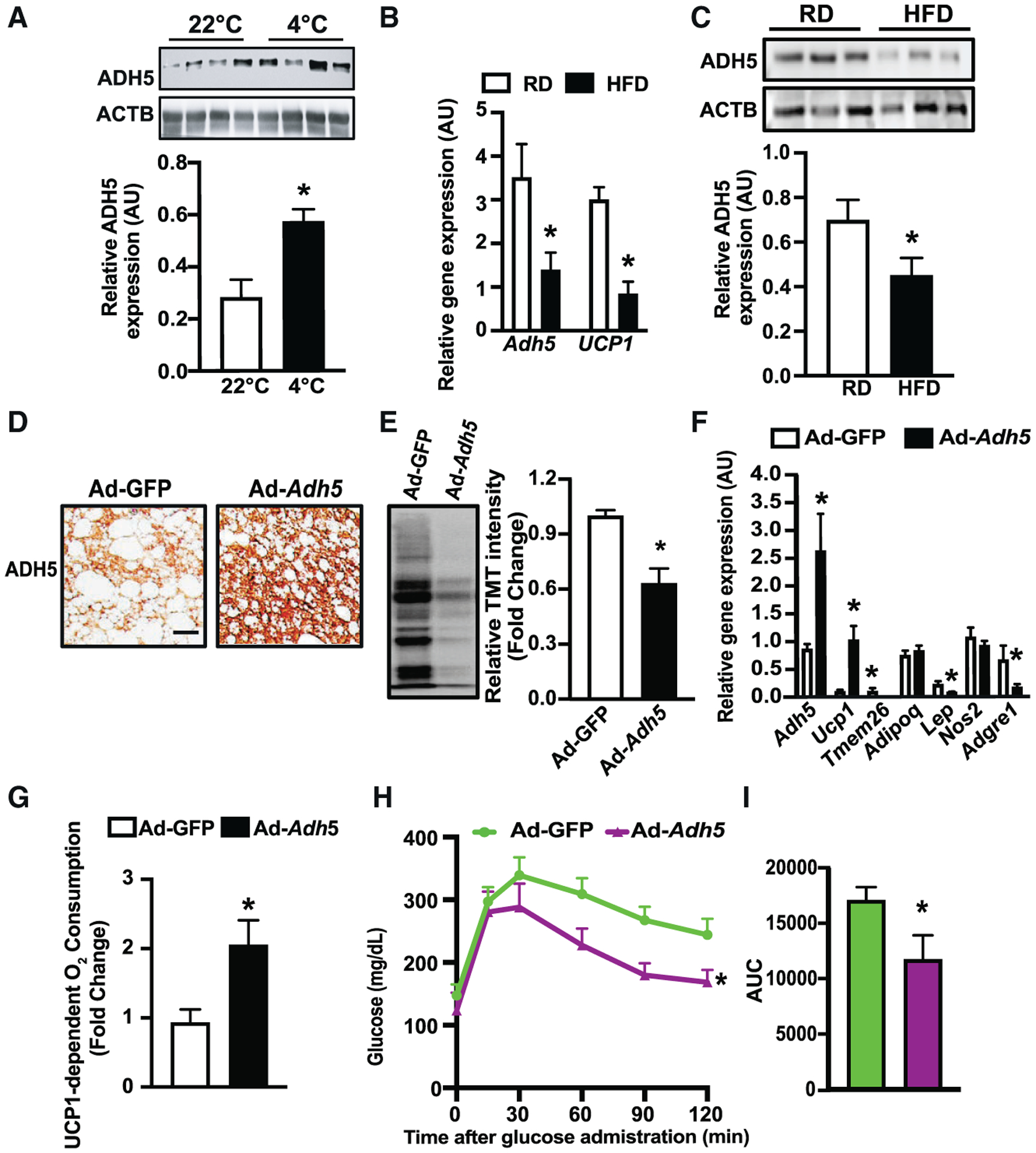
(A) Representative western blots and densitometric analysis of tested proteins in BAT from mice exposed to 22°C or 4°C for 24 hr. Data were normalized to expression of ACTB (n = 4 mice/group).
(B) Levels of mRNAs encoding the indicated genes in BAT from mice fed a RD (n = 3 mice) or HFD for 12 weeks (n = 5 mice), data were normalized to the expression of Hprt.
(C) Representative western blots and densitometric analyses in BAT from mice fed a RD or HFD for 12 weeks (n = 9–10 mice/group). Data were normalized to the expression of ACTB.
(D) Representative images (20×) of immunohistochemistry of ADH5 in BAT from mice on a HFD for 8 weeks followed by interscapular transduction of Adeno-GFP (n = 7 mice) or Adeno-Adh5 (n = 5 mice) (2.5 × 109 pfu/mouse) for additional 2 weeks. Scale bar, 10 μm.
(E) Representative western and quantification of protein SNO in iBAT from mice in (D). n = 4 mice/group.
(F) Levels of mRNAs encoding the indicated genes. Gene expression was normalized to Gapdh. n = 3–5 mice/group.
(G) UCP1-dependent O2 consumption rates in BAT from mice in (D). n = 4 mice/group.
(H) Glucose tolerance tests (GTTs) and (I). AUC in mice in (D).
All data are presented as means ± SEM. Asterisk indicates statistical significance compared to 22°C (A), the RD groups (B and C), and the Adeno-GFP group (E–I), as determined by Student’s t test; p < 0.05.
We next sought to determine whether gain of ADH5 function could improve BAT function in the context of obesity. To this end, we overexpressed Adh5 in BAT of adult mice using an adenovirus-mediated gene delivery approach via interscapular BAT injection (Balkow et al., 2016; Bartelt et al., 2018). Adenovirus-mediated Adh5 overexpression reduced general BAT SNO compared to controls (Figures 2D and 2E) which was concomitant with an increase in Ucp1 transcript and reduced expression of the tested adipogenic markers (Figure 2F). Furthermore, high-resolution respirometry showed that these effects were in parallel with an increase in UCP1-mediated mitochondrial respiration in HFD-fed mice (Figures S1F and 2G). Although the immunological properties of BAT are distinct from white adipose tissue (WAT) and are comparatively less pronounced during the early stage of obesity (van den Berg et al., 2017; Villarroya et al., 2018a, 2018b), pathological inflammatory signals derived from both brown adipocytes and BAT-resident immune cells have deleterious effects on BAT recruitment and activation (Cao et al., 2018). We found that overexpression of Adh5 significantly reduced expression of the macrophage marker F4/80 (Figures S1G and S1H), which was associated with improved obesity-associated glucose intolerance (Figures 2H and 2I). Systemic glycemic control is tightly regulated by interplays between organs, including the BAT-liver axis. Overexpression ADH5 in BAT improved hepatic insulin sensitivity in mice on a HFD but did not significantly alter the expression of hepatic transcripts involved in gluconeogenesis and lipogenesis (Figures S1I–S1K). Together, these data show a protective role for BAT-ADH5 against obesity-associated BAT dysfunction and systemic glucose intolerance.
ADH5 deficiency impairs metabolic function of BAT
To determine a BAT-specific ADH5 role, we crossed Adh5fl mice (Wei et al., 2011) with a Ucp1-Cre transgenic line to generate BAT ADH5 knockout (KO) (Adh5BKO) mice. ADH5 expression in the BAT from C57BL/6J, Adh5fl and Ucp1-cre mice was similar among all three genotypes (Figure S2A). However, at RT, BAT from Adh5BKO lean mice displayed a “white-like” phenotype, with increased expression of proinflammatory markers (Nos2 and Adgre1) and Lep but reduced expression of brown and beige adipose identity markers (Ucp1 and Tmem26) compared to Adh5fl mice (Figures 3A and 3B). Furthermore, Adh5 deletion in BAT significantly suppressed UCP1 protein expression (Figures 3C and 3D), which was associated with elevated general BAT SNO expression, increased SNO of UCP1 and enhanced NOx production (Figures 3E, 3F and S2B–S2D). Systemically, Adh5BKO mice had a comparable core body temperature with Adh5fl mice when housed at either RT or TN (Figure S2E). In addition, at RT, Adh5BKO and Adh5fl mice had similar body composition (Figure S2F) and whole-body metabolic profiles, as measured by a comprehensive lab animal monitoring system (CLAMS; Figures S2G and S2H).
Figure 3. Deletion of Adh5 impairs metabolic function of BAT.
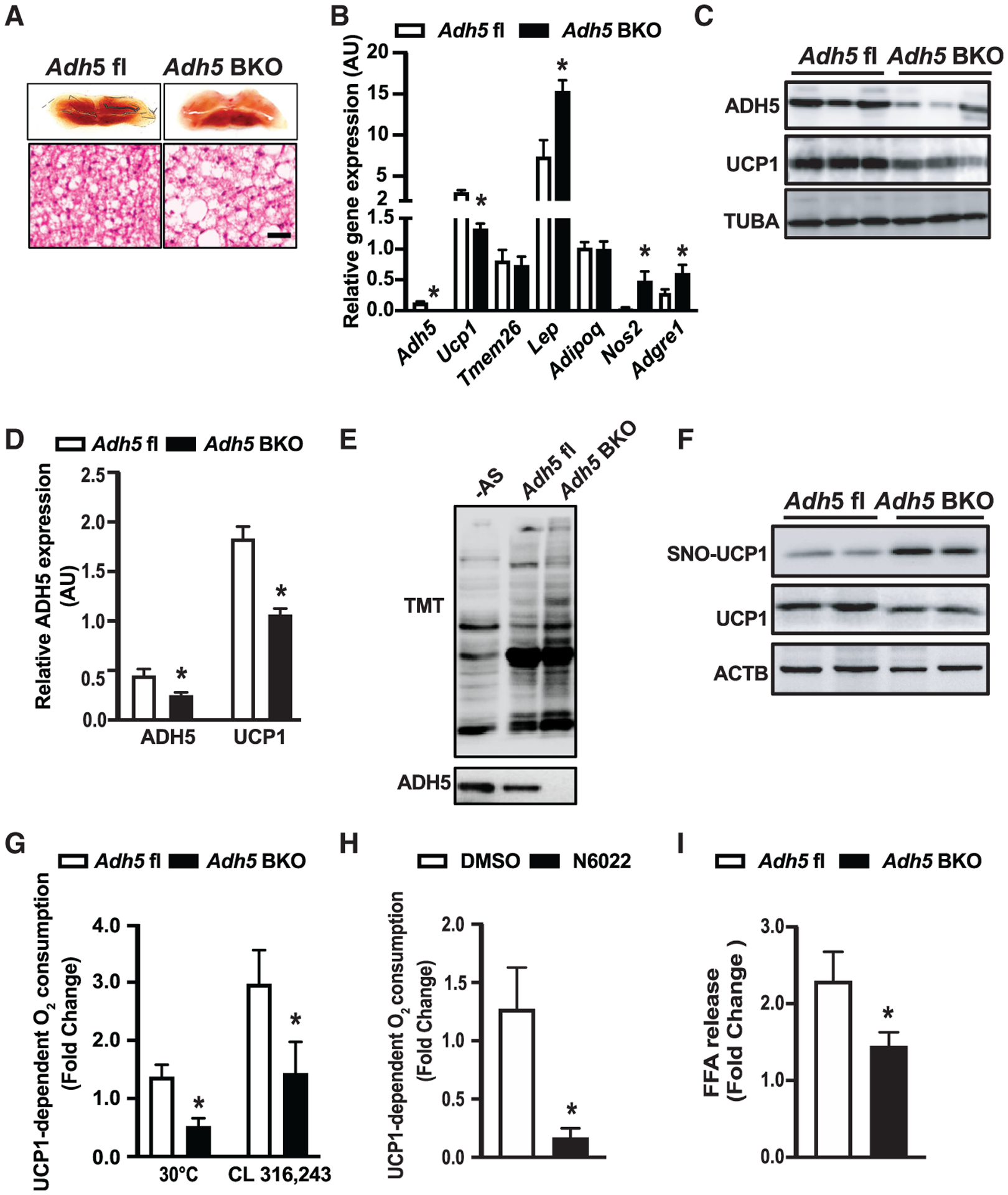
(A) Representative light microscopy and H&E images of BAT from Adh5fl or Adh5BKO mice raised at 22°C. Scale bar, 10 μm.
(B) Levels of mRNAs encoding the indicated genes in BAT from mice in (A). Expression is normalized to Gapdh. n = 3–5 mice/group.
(C and D) Representative western blots (C) and densitometric analysis (D) of ADH5 and UCP1 expression in BAT from Adh5fl and Adh5BKO mice. Expression is normalized to that of TUBA (TUBULIN). n = 4–5 mice/group.
(E and F) Representative western blots of general SNO (E) and S-nitrosylated UCP1 (F) in BAT from mice in (A).
(G and H) UCP1-dependent O2 consumption in BAT from Adh5fl and Adh5BKO mice maintained at 30°C (n = 3−4 mice/group) (G) or administrated with the β3-AR agonist CL316,243 (0.5 mg/kg; n = 4−5 mice/group) (H) in BAT from wild-type (WT) mice maintained at 30°C following treatment with the ADH5 inhibitor, N6022 (20 μM for 24 h), or vehicle (DMSO, 0.2%). n = 5 mice/group.
(I) Free fatty acid release measured in BAT explants from mice exposed to DMSO (0.1%) or ISO (1 μM; 1 h). Data were normalized to protein concentration. n = 5–6 mice/group.
All data are presented as means ± SEM. Asterisk indicates statistical significance compared to the Adh5fl groups (B, D, G, and I) and the vehicle group (H), as determined by Student’s t test; p < 0.05.
We next measured UCP1-mediated mitochondrial respiration in BAT fat pads from Adh5BKO mice and Adh5fl housed at TN or treated intraperitoneally (i.p.) with CL316,243, an adipocyte-specific β3-adrenergic agonist (Cannon and Nedergaard, 2011). As shown in Figure 3G, Adh5 deletion impaired UCP1-mediated mitochondrial respiration in both conditions. To confirm whether reduced mitochondrial respiration was attributed to defective ADH5 enzymatic activity, we excised BAT fat pads from WT mice fed a regular chow (housed at TN) and treated them ex vivo with an ADH5 chemical inhibitor, N6022 (Blonder et al., 2014). As shown in Figure 3H, pharmacological inhibition of ADH5 significantly decreased UCP1-mediated oxygen consumption. Consistent with our observation in the BAT, inhibition of ADH5 activity in a human BAT cell line (hTERT-hA41BAT-SVF) showed a reduced oxygen consumption rate (OCR) (Figures S2I and S2J). Finally, lipolysis induced by the nonselective β-agonist ISO was also significantly reduced in BAT explants from Adh5BKO mice compared to WT mice (Figure 3I).
ADH5 is required for cold-induced thermogenesis in BAT
BAT generates heat through non-shivering thermogenesis (Cannon and Nedergaard, 2004; Harms and Seale, 2013); thus, to investigate the functional role of BAT-specific ADH5 in cold-induced thermogenesis, Adh5fl and Adh5BKO mice weaned and raised at TN conditions were challenged with acute cold exposure (4°C for 24 h). Compared to littermate WT controls, both BAT and inguinal WAT (iWAT) from Adh5BKO mice displayed an increased proportion of large-sized adipocytes (Figure 4A) but reduced brown and beige-like markers (Figure S3A). These cellular alterations were correlated with significantly lowered core body temperature after either TN acclimation followed by cold exposure (Figure 4B) or after activation of the β-adrenergic receptor (AR) by the β-adrenergic agonist, norepinephrine (Figure 4C).
Figure 4. ADH5 is required for cold-induced BAT thermogenesis.
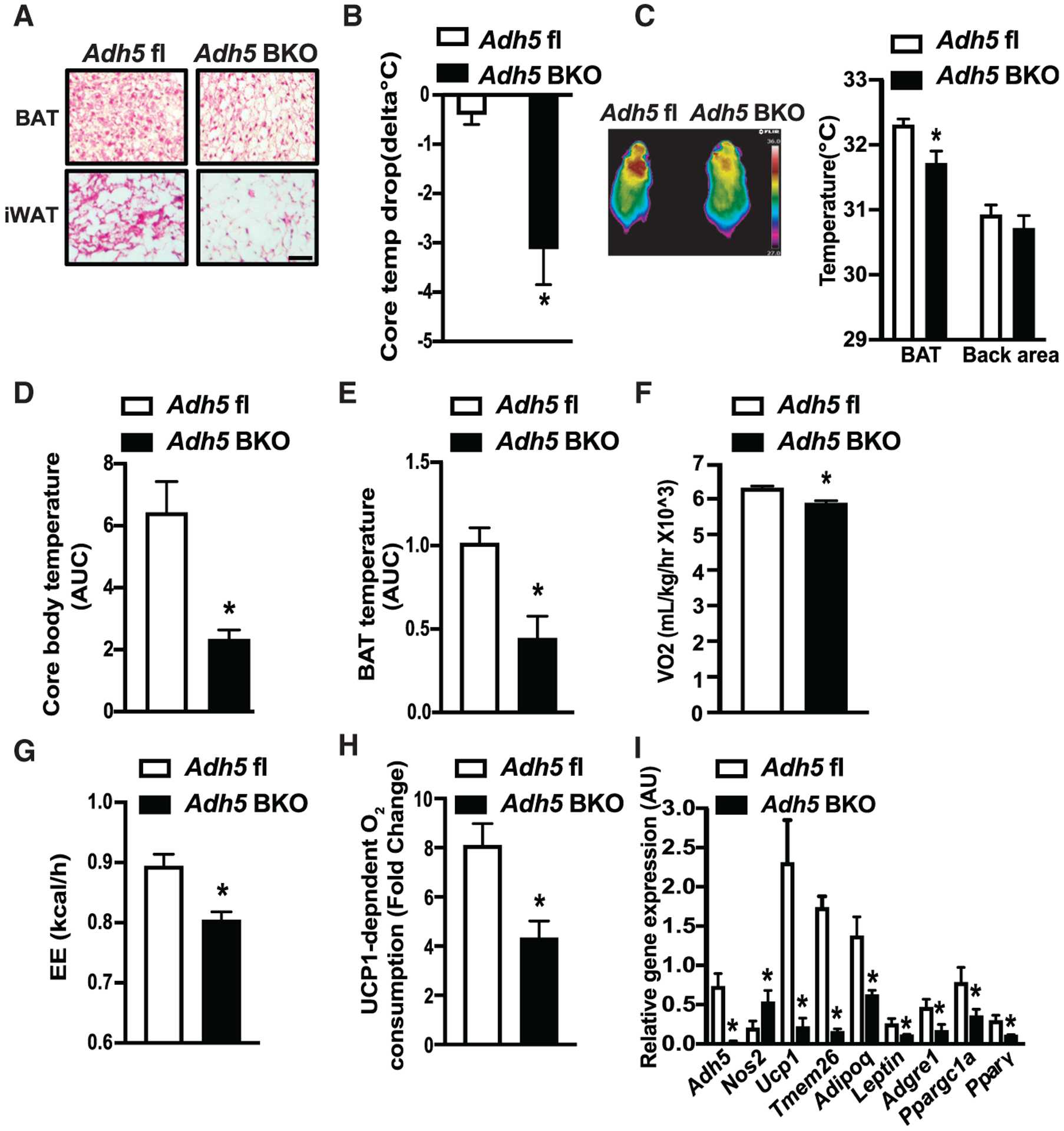
(A and B) Representative H&E images from BAT or iWAT (A) and cold tolerance from Adh5fl and Adh5BKO mice housed at 30°C following cold exposure to 4°C for 24 h (B). Data are presented as difference in core body temperature between 30°C and 4°C. n = 3–5 mice group. Scale bar, 100 μm.
(C) Representative high-resolution infrared images of body surface temperature of Adh5fl and Adh5BKO mice. Quantification of the images is shown by the side of image. n = 5 mice/group.
(D–G) Core temperature (AUC, area under the curve of dark cycles) (D), BAT temperature (AUC of dark cycles) (E), whole-body VO2 (F), and whole-body energy expenditure (EE) (G) measured in Adh5fl and Adh5BKO mice housed within a CLAMS at 4°C (n = 4–5 mice/group).
(H) UCP1-dependent O2 consumption in BAT from mice in (B). n = 4 mice/group.
(I) qPCR analysis measuring levels of mRNAs encoding the indicated genes in BAT from mice in (B). Expression was normalized to Gapdh.
All data are presented as means ± SEM. Asterisk indicates statistical significance compared to the Adh5fl group as determined by Student’s t test; p <0.05.
We then tested whether ADH5 in BAT is involved in the regulation of systemic thermogenic adaptation to cold exposure (4°C). Indirect mouse calorimetry analysis showed that cold exposure lowered both core and BAT temperature in Adh5BKO mice compared to WT controls (Figures 4D and 4E), which correlated with reduced whole-body oxygen consumption and energy expenditure (EE) (Figures 4F and 4G). Furthermore, in line with our observations from mice housed at TN (Figure 3F), Adh5BKO mice showed a significantly reduced cold-induced, UCP1-mediated oxygen consumption (Figure 4H). Finally, we found that compared to WT controls, loss of BAT Adh5 resulted in decreased expression of cold-induced genes that are normally activated as part of the thermogenic program (such as Ucp1, Pparg, and the cofactor Ppargc1a) (Figure 4I). Collectively, these results indicate that ADH5 is indispensable for adaptation of BAT to cold-induced thermogenesis.
It has been shown that Adh5-deficient MSCs exhibited impaired adipogenesis (Cao et al., 2015). We then asked if ADH5 regulates BAT recruitment by assessing brown preadipocyte differentiation using stromal vascular fraction (SVF) isolated from Adh5fl mice followed by transduction of adeno-Cre to delete Adh5. Mature (Pparg and Ucp1) and preadipocyte (Dlk1) markers, as well as lipid content, were comparable between the tested groups (Figures S3B and S3C). Similar results were also observed in human cells treated with the pharmacologic ADH5 inhibitor N6022 (Figures S3D and S3E). Together, our data indicate a regulatory role for ADH5 in modulation of BAT activation.
BAT ADH5 protects against obesity-associated metabolic dysfunction
Obesity suppresses BAT metabolic function (Blondin et al., 2015; Himms-Hagen, 1985; Leitner et al., 2017; Shimizu et al., 2014; Shimizu and Walsh, 2015) and increases immune cell infiltration and ROS production (Alcalá et al., 2017, 2019). To establish the direct regulation of ADH5 in the context of obesity, we evaluated BAT function and systemic metabolic homeostasis in Adh5BKO and Adh5fl mice fed a HFD. HFD-fed Adh5BKO mice and Adh5fl mice had comparable body weight and composition (Figure 5A). In addition, indirect mouse calorimetry demonstrated that activity, heat production and cumulative food intake were slightly decreased (yet did not reach statistical significance) in HFD-fed Adh5BKO mice (Figures S4A–S4C). However, Adh5 deficiency in BAT worsened obesity-associated glucose intolerance (Figures 5B and 5C), lowered whole-body oxygen consumption (Figure 5D) and significantly decreased UCP1-dependent oxygen consumption compared to explants from WT mice (Figure 5E).
Figure 5. Obesity impairs HSF1-mediated Adh5 activation leading to BAT metabolic dysfunction.

(A) Body composition of Adh5fl and Adh5BKO mice on a HFD (8 weeks), measured by nuclear magnetic resonance (NMR; n = 7 mice/group).
(B and C) GTT (B) and AUC (C) from Adh5fl and Adh5BKO mice on a RD or HFD. n = 11–12 mice/group.
(D and E) Whole-body VO2 (D) and HFD-mediated high-resolution respirometry analysis in BAT (E). n = 4–5 mice/group.
(F) Insulin signaling in BAT as in (A). p-AKT: AKTSer473. Densitometric analysis is shown under the blots, and data are normalized to ACTB. n = 3 mice/group.
(G) Activity of the Adh5 promoter in HEK293A cells 48 h after transfection. Control cells (CT), PGL4.10 vector. Data were normalized to Renilla luciferase with schematic representation of conserved HSF1-binding sequences identified in the promoter regions of Adh5 using Japan Automotive Software Platform and Architecture (JASPAR) on top.
(H) Representative western blots and densitometric analysis of nuclear HSF1 expression from BAT (RD or HFD for 16 weeks; n = 6–8 mice/group). Expression was normalized to Lamin A/C (LMNA).
(I) High-resolution respirometry analysis in BAT (HFD for 10 weeks) followed by interscapular treatment with vehicle (DMSO; n = 4 mice) or an HSF1 activator (1 mM; n = 7 mice) every other day for 2 weeks. UCP1-mediated respiration was calculated and reported as fold change of GDP-inhibited leak (1 mM) over baseline.
All data are presented as means ± SEM. Asterisk indicates statistical significance compared to the Adh5fl groups (C–E), the CT group (G), or the RD groups (H), and treatment effects in same type of mice (I). Number sign indicates genetic effects in mice with same treatment (C and I) and statistical significance compared to WT (G). Statistical significance was determined by one-way ANOVA (B, C, G, and I) and Student’s t test (D–F and H); p < 0.05.
Malfunction of UCP1 and enhanced NO bioactivity activates innate immune cells in BAT, leading to impaired glucose homeostasis (Bond et al., 2018). Here, we found a significant increase in HFD-mediated recruitment of F4/80-positive immune cells (Figures S4D and S4E), which was concomitant with a significant increase in the transcript levels of adipogenic markers (Lep and Adipoq), proinflammatory mediators (Tnfα and Il6), and the macrophage marker Adgre1 (Figure S4F). These changes in inflammatory markers were concomitant with worsened insulin signaling in BAT from Adh5BKO mice (Figure 5F), indicating that ADH5-brown adipocyte expression is pivotal in preserving a healthy immune-metabolic balance of BAT during DIO. Finally, although proper ER function and autophagy dynamics play important roles in BAT and beige adipocyte activation, as well as for adipose tissue maintenance (Altshuler-Keylin et al., 2016; Bartelt et al., 2018; Singh et al., 2009), we found comparable expression of tested markers in BAT (Figures S4G and S4H).
Obesity impairs HSF1-mediated activation of Adh5
The heat shock response (HSR) is a highly conserved physiological stress response to diverse stressors, including cold-induced adrenergic activation of BAT (Matz et al., 1996; Verma et al., 2020). In an in silico analysis, we found that HSF1 possesses binding elements at the Adh5 promoter (Figure 5G). To determine if HSF1 activates Adh5, we generated Adh5 luciferase reporter constructs with HSF1 occupancy sites (−1,683 to −1,671), as well as a control construct lacking this binding site. HSF1 activated the Adh5 promoter, which was abolished in cells expressing a mutant construct lacking this potential site (Figure 5G). We then examined BAT-nuclear expression of HSF1 from RD and HFD mice and found that DIO suppressed HSF1 nuclear translocation (Figure 5H). To explore the therapeutic potential of increasing HSF1 activity in BAT, we treated WT DIO mice with HSF1A (a pharmacological activator of HSF1) via interscapular BAT injection. HFD mice treated with HSF1A demonstrated increased expression of Hsf1, HSF1 target genes (Hsp90, Dnajb1 and Hsp27), as well as Adh5 and Ucp1 transcripts in BAT (Figure S5A). This increased Adh5 expression by HSF1 activation was associated with increased UCP1-dependent oxygen consumption in BAT fat pads compared to controls (Figure 5I). Furthermore, activation of mitochondrial respiration in BAT by HSF1 in the context of obesity was not altered in Adh5BKO mice (Figure 5I), indicating that ADH5 is a downstream factor in the HSF signaling cascade. Recently, Rizza et al. showed that ten-eleven translocation 1 (TET; demethylase) and ADH5 concomitantly declined with age in the brain (Rizza et al., 2018). To determine whether DNA methylation is involved in HSF1-mediated Adh5 induction in the context of obesity, we treated BAT explants from HFD-fed mice with a DNA methylation inhibitor, 5-Aza-2′-deoxycytidine (5aza) (Christman, 2002), in the presence or absence of HSF1A and examined 5aza inhibition efficiency (Figure S5B). As shown in Figure S5C, 5aza treatment slightly increased Adh5, but this did not reach statistical significance. Furthermore, while activation of HSF1 significantly increased Adh5 transcript in BAT from obese mice, this effect was not additive in samples treated with both HSF1 and 5aza. These findings suggest that hypermethylation of BAT may not directly modulate the HSF1-Adh5 axis. Together, these data indicate that activation of the HSF1-ADH5 axis protects BAT against obesity-associated metabolic dysfunction.
Previous studies have also shown that HSF1 is activated by cold challenge (Ma et al., 2015; Reinke et al., 2008; Sarge and Cullen, 1997). Indeed, we found that both cytosolic- and nuclear-localized HSF1 were significantly increased by 2 h, remaining elevated after 6 and 24 h of cold exposure (Figures S5D and S5E). Compared to HSF1 activation, cold-induced ADH5 was delayed, increasing at 6 and 24 h (Figures S5D and S5E). Together, these data indicate that cold-induced HSF1 activation leads to increased ADH5 over time, which together may provide a feedback mechanism to fine tune the SNO in BAT. To further assess the direct effects of β3-adrenergic signaling on HSF1-mediated BAT protein SNO, we stimulated freshly isolated BAT of WT mice with CL316,243 in the presence or absence of a chemical HSF1 inhibitor (KRIBB11) (Yoon et al., 2011), which significantly reduced the expression of HSF target genes (Figure S5F). Furthermore, CL316,243 elevated BAT general SNO was almost abolished by KRIBB11 (Figures S5G and S5H). Together, these data support the hypothesis that physiological BAT ADH5 and NO bioactivity are regulated, in part, by HSF1.
Inhibiting iNOS in BAT alleviates DIO-associated metabolic dysfunction
Excessive NO production by parenchymal cells is one of the key factors that drives immune cell infiltration and polarization in the context of obesity (Sansbury and Hill, 2014). We observed higher iNOS expression in BAT from mice on HFD, which is worsened by deletion of Adh5 (Figure 6A). To define the major cellular source of the iNOS induction, we isolated SVF from Adh5BKO and Adh5fl mice fed a HFD. While transcript levels of Adh5, Nos2, and Tnf in the SVF were comparable between genotypes, BAT-specific Adh5 deletion significantly increased expression of these markers in the BA fraction (Figures 6B and 6C), which was in agreement with a higher NOx production released from the BA fraction (Figure 6D). To further examine the direct role of NO bioactivity on BAT metabolic homeostasis in the context of obesity, we injected 1400W, a specific iNOS inhibitor (Garvey et al., 1997), into DIO mice intrascapularly. As shown in Figure 6E, treatment with 1400W decreased HFD-mediated NOx production and improved UCP1-mediated oxygen consumption regardless of the genotype (Figure 6F). Gene expression analyses further supported this notion, showing that 1400W treatment reduced proinflammatory markers and ameliorated Ucp1 in BAT from both Adh5fl and Adh5BKO mice on HFD (Figure 6G). Taken together, these data demonstrate that ADH5 deficiency in BAs propagates obesity-associated BAT metabolic dysfunction, and inflammation is attributable in part to the induction of iNOS-mediated NO signaling.
Figure 6. Inhibiting iNOS improves metabolic homeostasis in BAT from ADH5BKO mice.
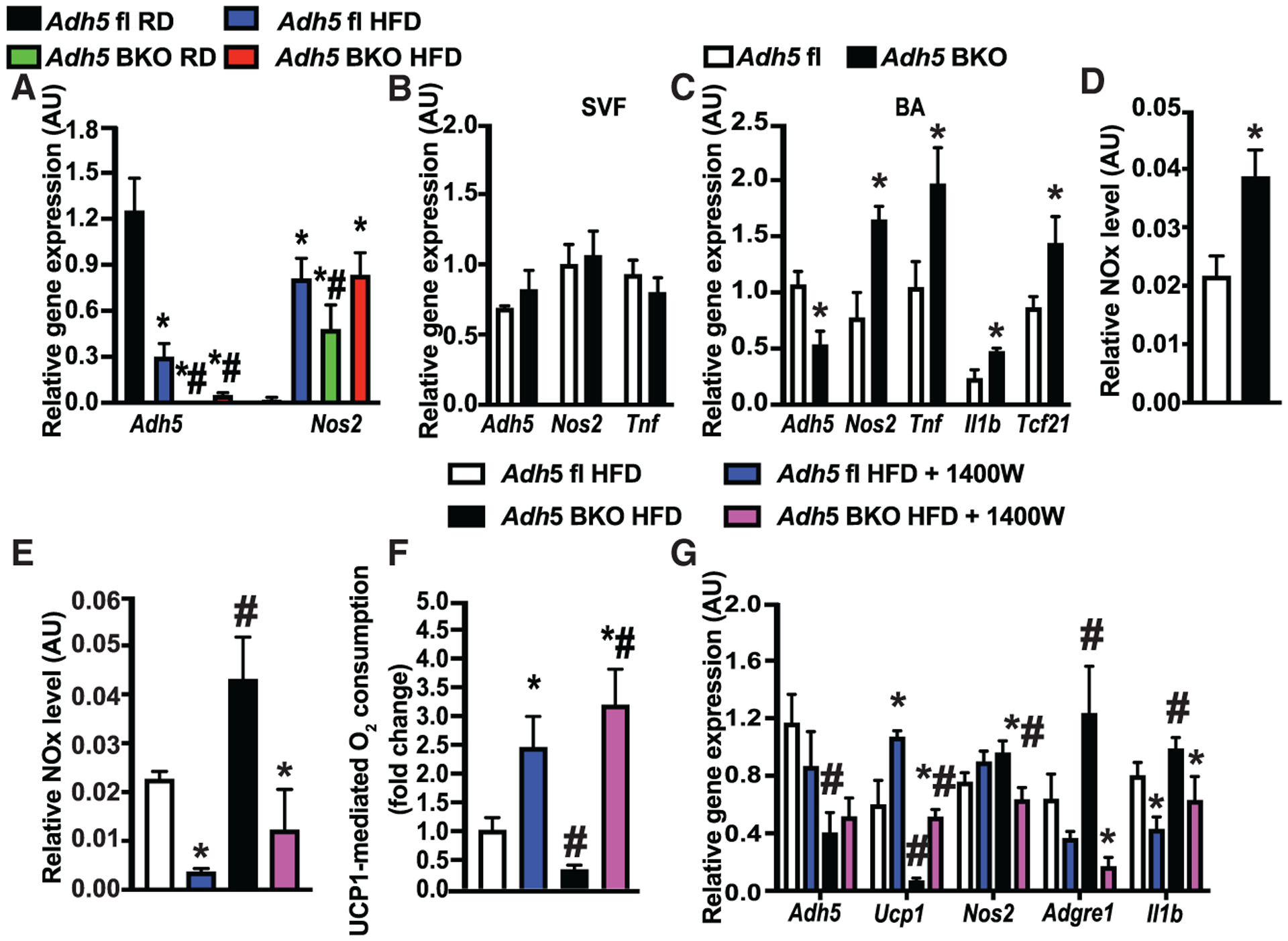
(A) qRT-PCR analysis measuring levels of mRNAs encoding the indicated genes in BAT from Adh5fl and Adh5BKO mice fed a RD or HFD for 16 weeks. n = 4 mice/group.
(B and C) qRT-PCR analysis measuring levels of mRNAs encoding the indicated genes from SVF (B), and mature brown adipocytes (C) isolated from brown adipose tissues of Adh5fl and Adh5BKO mice (n = 6 mice/group; 10 weeks HFD). Data were normalized to Gapdh.
(D) Total nitrite/nitrate NOx release from mature brown adipocytes from mice in (B) and (C). Data were normalized to protein level.
(E and F) Total nitrite/nitrate NOx release (E) and high-resolution respirometry analysis from BAT explants (F) in mice (fed a HFD for 12 weeks) interscapularly injected with DMSO control or 1400W (0.2 mg/kg) every 2 days for 3 weeks. n = 7–12 mice/group.
(G) qRT-PCR analysis measuring levels of mRNAs encoding the indicated genes in BAT from mice in (F). Expression was normalized to Gapdh.
All data are presented as mean ± SEM. Asterisk indicates statistical significance compared to the Adh5fl RD groups (A) or the Adh5fl group (C and D) and treatment effects in mice with same genotype (E–G). Number sign indicates genetic effects in mice with same treatments (A and E–G). Statistical significance was determined by two-way ANOVA followed by Tukey’s multiple comparison (A and E–G) and Student’s t test (B–D); p < 0.05.
DISCUSSION
ROS and RNS are redox mediators that regulate diverse aspects of cellular processes and functions (Di Meo et al., 2016). Acute induction of thermogenesis in BAT generates physiological levels of mitochondrial ROS, which in turn alters UCP1-thiol redox status and activates UCP1-dependent uncoupling (Chouchani et al., 2016; Mailloux et al., 2012). In contrast, under chronic metabolic-thermogenic conditions such as obesity, an excessive level of ROS is produced, which promotes mitochondrial damage in BAT (Cui et al., 2019; Kazak et al., 2017; Lee et al., 2020),. However, it is largely unknown whether and how NO bioactivity, another key redox mediator, modulates BAT function in health and disease. Our present study found that the protein SNO signaling cascade plays a key role in regulating BAT thermogenic function.
NO bioactivity is tightly regulated by the balance of NO availability and the capacity to remove it in part by denitrosylases (Benhar et al., 2009). For the former, two NOS isoforms, endothelial nitric oxide synthase (eNOS) and iNOS, are expressed in BAT (Engeli et al., 2004; Kikuchi-Utsumi et al., 2002), with temporal studies revealing that eNOS is expressed early during cold acclimation, whereas iNOS is elevated in the late phase (Otasevic et al., 2011). On the other hand, ADH5 (Cao et al., 2015) and thioredoxin reductase 1 (Ayyappan and Nagajyothi, 2017), both of which serve to denitrosylate and modify protein function (Nakamura and Lipton, 2016), are expressed in BAT. These data suggest that within BAT, there exists a fine-tuned equilibrium between protein SNO by NOS synthases and denitrosylases. However, the pathophysiological role of S-nitrosylative signaling in BAT has yet to be well defined.
Previous studies investigating the modulation of nitrosative signaling in the adipose tissue have involved deletion or inhibition of NOS expression and activity (Becerril et al., 2018; Fujimoto et al., 2005; Tsuchiya et al., 2007; Zanotto et al., 2016), thereby diminishing all forms of NO bioactivity while also impairing both cGMP and SNO-mediated signaling. Ultimately, these approaches to dissect the function of protein SNO in regulating BAT physiology are very challenging. Our study focused on the key denitrosylase, ADH5. ADH5 has been shown to play an important role in multiple cellular responses, including β-AR signaling, endocytosis, inflammation, angiogenesis, and apoptotic cell death (Barnett and Buxton, 2017). Here we investigated the hypothesis that ADH5 regulates BAT metabolic and thermogenic function by balancing intracellular NO bioactivity. Our study provides evidence that DIO increases BAT NO production and elevates general protein SNO, including UCP1; and ADH5 protects against impairment of UCP1-dependent oxygen consumption and glucose intolerance in obese mice. Furthermore, BAT-Adh5 deletion led to the dysregulation of thermogenic responses to both cold and direct β-AR3 activation.
In our study, we found that obesity promotes SNO of UCP1, which is augmented by Adh5 deletion in BAT. We further showed that SNO of UCP1 at C305 impairs BAT mitochondrial respiration (Figure 1I). It is important to note that sulfenylation of UCP1 at Cys253 is required for BAT function during thermogenesis (Chouchani et al., 2017) and is altered in aging (Xiao et al., 2020). Therefore, future studies focusing on delineating redox-mediated fine-tuning of post-transcriptional and post-translational UCP1 modifications generated by acute and chronic stress may provide additional insights into the thermogenic function of BAT in health and disease. Regarding the fate of SNO UCP1, it has been shown that SNO results in degradation of the key DNA repair protein O6-alkylguanine-DNA alkyltransferase (AGT) and tumor necrosis factor (TNF)-receptor-associated protein 1 (Trap1) by the ubiquitin-proteasome system in the liver (Rizza et al., 2016; Wei et al., 2010). Therefore, it is possible that obesity-associated downregulation of UCP1 may be in part attributed to SNO-UCP1 and consequential degradation. We also found that ADH5 expression positively correlates with the expression of Ucp1 transcript. SNO signaling modulates diverse cellular events, including controlling transcriptional programs and epigenic regulations (Hess et al., 2005; Nott et al., 2008; Mussbacher et al., 2019; Cao et al., 2015). Thus, it is possible that nitrosative stress alters the BAT transcriptome, including adipogenic markers and Ucp1 expression, in the context of obesity.
We recognize that the role of ADH5 in regulating BAT metabolic function could involve UCP1-independent actions. Multiple thermogenic regulators are tightly regulated by protein SNO in other organ systems. In the heart, two key proteins involved in β2-AR receptor regulation, GRK2 and ARRB2, are shown to be targeted by SNO (Whalen et al., 2007). In skeletal muscle, it has been shown that ADH5-KO mice exhibit an impaired inotropic response to ISO in part by reducing denitrosylation of RYR2 (Beigi et al., 2012; Stomberski et al., 2019). Of note, a recent study demonstrated that SNO of PPARG in MSCs reduces adipogenesis and increases osteoblastogenesis (Cao et al., 2015). Although we did not observe detectable SNO of PPARG in BAT or an alteration of Adh5 deletion on BA differentiation (Figures 1J, 1K, and S3B–S3E), we postulate that this regulation may be context dependent and is an outcome of whole-body deletion versus BAT-specific deletion.
Mechanistically, our study demonstrates that ADH5 is directly induced by HSF1, a master regulator of the HSR. However, studies have shown that HSF1 is also activated by cold challenge (Ma et al., 2015; Reinke et al., 2008; Sarge and Cullen, 1997). Here, we found the HSF1-ADH5 signaling cascade maintains metabolic homeostasis of BAT in obesity, supporting a previous study showing treatment with celastrol activates BAT-HSF1 and improves metabolic function (Ma et al., 2015). We also showed that the HSF1-Adh5-axis-mediated BAT protein SNO is, in part, through β3-AR signaling (Figures S5G and S5H). Ultimately, our study provides a mechanistic insight into how the HSF1-mediated transcriptional program controls cellular nitro-redox (SNO) signaling by modulating expression of Adh5. Another key feature of obesity-associated metabolic abnormalities is low-grade chronic inflammation in the adipose tissue, contributing to insulin resistance and metabolic abnormalities (Hotamisligil, 2006; Xu et al., 2003), as well as defects in BAT recruitment and activation (Bartelt et al., 2018; Cao et al., 2018). Conversely, supplementing with anti-inflammatory molecules (e.g., epigallocatechin gallate and berberine) increases BAT thermogenic activity (Okla et al., 2017). Accumulated evidence has also shown that an excess of NO bioactivity and nitrosative stress modulate transcription factor activities that directly regulate inflammatory and adipogenic signatures (Choi et al., 2016; Marshall et al., 2004; Ovadia et al., 2011; Shinozaki et al., 2014). Our study showed that Adh5 deletion increased Nos2 expression, NO production, and proinflammatory markers in the BA, all of which were attenuated by chemical inhibition of iNOS (via 1400W) in BAT (Figures 6A–6D). An important objective for future studies is to determine the impact of ADH5-mediated BA NO bioactivity on the immune signature, vascularization, and sympathetic tone, as well as remodeling in BAT, in health and disease.
Taken together, our study demonstrates that cellular nitrosative signaling is critical for BAT health. Identifying the molecular mechanisms underlying the immuno-metabolic function of BAT through ADH5-mediated nitro-redox signaling will help establish a framework for developing therapies to ameliorate obesity-mediated BAT dysfunction.
Limitations of the study
In our study, we used a germline Adh5 deletion approach; therefore, we could not rule out the possibility that the effect of ADH5 deficiency on systemic metabolic dysfunction is due to a maladaptive systemic response from other organs/tissues. Future studies will include an inducible Cre transgenic mouse line to assess the HSF1-Adh5 signaling axis both basally and after metabolic stress in adult mice. Furthermore, we found that a UCP1 SNO-resistant variant protects against chemical NO-donor-induced impairment of UCP1-mediated mitochondrial respiration in brown adipocytes. However, we recognize that nitrosative stress responses in vitro may differ from obesity-mediated effects in vivo. Future studies will focus on determining the impact of SNO of UCP1 at Cys305 in vivo.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Ling Yang (ling-yang@uiowa.edu).
Materials availability
Plasmids that were used for this study are available upon request. There are no other new materials generated in this manuscript.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse models
Animal care and experimental procedures were performed with approval from the University of Iowa’s Institutional Animal Care and Use Committee. Animals received humane care in compliance with the Guide for the Care and Use of Laboratory Animals and with the Principles of Laboratory Animal Care formulated by the National Society for Medical Research.
C57BL/6J mice (The Jackson Laboratory, 000664), UCP1-cre (The Jackson Laboratory, B6.FVB-Tg(UCP1-cre)1Evdr/J, 024670), and Adh5fl (Wei et al., 2011) mice were kept on a 12-hour light/dark cycle. Male mice used to generate the DIO model were placed on a 60% kCal high-fat diet (Research Diets, D12492) immediately after weaning (i.e., at 3 weeks of age). Adenovirus-Adh5 (Qian et al., 2018) and adenovirus-GFP were delivered to male mice interscapularly at a titer of 2.5×109 pfu/mouse (Bartelt et al., 2018). HSF1A (Millipore,1196723-93-9) and 1400W (Sigma, 214358-33-5) was interscapularly administrated at a dosage of 1 mM/mouse or 0.2 mg/kg respectively, every other day for 2 weeks. For cold-exposure and thermoneutrality experiments, male mice were singly housed in an environmental chamber during a 12-hour light cycle (Metabowl; Jencons Scientific). Mice were given access to standard 2920X Teklad Global Diet and water ad libitum while in cages. In diet-induced obesity mouse model, experiments were performed at 8–16 weeks of HFD feeding (age of 11–19 weeks) and age-matched normal chow fed mice controls. All tissues were harvested, frozen in liquid nitrogen, and kept at −80°C until processed.
Cell Culture
HEK293A cells (ATCC) were cultured in DMEM medium with 10% cosmic calf serum (CCS) and 1% penicillin-streptomycin. The hTERT A41hBAT-SVF was grown in BGM medium.
METHOD DETAILS
BAT fat pad and adipocytes treatment
Primary brown adipocytes were isolated from iBAT depots of Adh5fl mice as previously described (Gao et al., 2017; Ikeda et al., 2017). Briefly, adipose tissues were minced and digested in 1 mg/mL liberase (Roche, 5401119001) in KRBH buffer in a 37°C water bath with agitation for 30 minutes, followed by filtration through nylon mesh. The samples were centrifuged at 500×g for 10 minutes at 10°C. The floating mature adipocytes were collected and cultured in DMEM/F-12 containing 10% cosmic calf serum (CCS) and 1% penicillin-streptomycin (basal growth media, BGM). Cells were then grown for 5 days, transduced with Adeno-GFP (1×1012 pfu) or Adeno-ADH5 (1×1012 pfu) and differentiated in BGM containing 0.25 nM 3-Isobutyl-1-methylxanthine (IBMX, Sigma, I5879), 1 μM Rosiglitazone (Sigma, 557366-M), 0.125 nM Indomethacin (Sigma, 405268), 1 nM T3 (Sigma, T5516), Humulin R (Lily, Humulin R U-500, 0002-8501-01) and 2 μg/mL dexamethasone (Sigma, D4902) for up to 10 days with media change every other day. The hTERT A41hBAT-SVF was grown in BGM medium, followed by differentiation with or without 25 nM N6022 (Sigma, 1208315-24-5).
For HSF1 inhibition, BAT explants from WT mice were exposed to 37°C or 43°C with or without 2 μM KRIBB11 (Sigma, 385770) for 30 min. In addition, WT BAT explants were treated with 1 μM CL316,243 (Sigma, C5976) at 37°C in the presence or absence of 2 μM KRBI11 for 2 hours. For other experiments, isolated BAT explants (HFD-fed, 16 weeks) were transduced with Adeno-GFP or Adeno-Adh5 (1×1012 pfu) for 3 days. All tissues were harvested, frozen in liquid nitrogen, and kept at −80°C until processed.
Mutagenesis and transfection
The mouse UCP1 (UCP1WT) and the S-nitrosylation resistant (UCP1SNOR) variants were generated in a pcDNA3.1 backbone (Addgene, V790–20) using cDNA isolated from WT BAT. Site-directed mutagenesis was then done with the NEBuilder HiFi DNA assembly system (E5520S) following the manufacturer’s instructions. Primers used for cloning are listed in the Table S1.
The hTERT A41hBAT-SVF were transfected with either pcDNA-EGFP, UCP1WT or UCP1SNOR (2.5 μg/well, pcDNA3.1 backbone) using Lipofectamine 2000 reagent (ThermoFisher Scientific, L3000015) in Opti-MEM medium (ThermoFisher Scientific, 31985–062). At 24–48 hours post-transfection, cells were exposed to isoproterenol (ISO, 1 μM, Sigma, 54750-10-6) in the presence or absence of S-Nitroso-N-acetyl-DL-penicillamine (SNAP, 100 μM, Sigma, N3398) for 2 hours. Cells were then trypsinized, spun at 500xg for 5 minutes at 4°C and assessed for UCP1-dependent oxygen consumption (see below).
HEK293A cells (ATCC, CRL-1573) were transfected with DNA constructs (0.5 μg/well) using Lipofectamine 2000 reagent in Opti-MEM medium. At 48 hours post transfection, the activities of Firefly and Renilla luciferase were measured using the Luciferase Assay System (Promega, E1500) and Renilla Luciferase Assay System (Promega, E2810), separately.
Nitrite and nitrate assay
Briefly, BAT depots were excised, cut into small pieces and incubated in 24-well plates in BGM (without phenol red) for 24 hours. The supernatant was collected, then assayed for NOx using the Nitrate/Nitrite Colorimetric Assay Kit (Cayman Chemical, 780001) according to manufacturers’ instructions. Values were normalized to RNA content.
Quantitative Real-time RT-PCR
Total RNA was isolated using TRIzol reagent (Invitrogen,15-596-018) and reverse transcribed into cDNA using the iScript cDNA synthesis kit (Bio-Rad, 1708890). Quantitative real-time RT-PCR analysis was performed using SYBR Green (Invitrogen, KCQS00). Primer sequences are found in Table S1.
Western blot Analysis
Proteins were extracted from cells or tissues and subjected to SDS–polyacrylamide gel electrophoresis, as previously described (Yang et al., 2015). Membranes were incubated with primary antibodies at 1:1000 dilution (see STAR Methods); and then incubated with the appropriate secondary antibody conjugated with horseradish peroxidase (1:5000, Santa Cruz, sc-2005 or 1:5000, Cell Signaling Technology, 7074S). Signal was detected using the ChemiDoc Touch Imaging System (Bio-Rad), and densitometry analyses of western blot images were performed by using the Image Lab software (Bio-Rad).
Nuclear fractionation
The nuclear fractions were prepared as described previously (Yang et al., 2015). 30 μg BAT was homogenized in hypotonic buffer: 250 mM sucrose, 20 mM HEPES, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, and 1 mM EGTA followed by filtering through a 100 μm cell strainer. The cell lysates were centrifuged at 825 g for 20 minutes at 4°C. The pellets were dissolved in NE buffer (20 mM HEPES, pH 7.9, 1.5 mM MgCl2, 0.5 M NaCl, 0.2 mM EDTA, 20% glycerol and passed through a 32G needle. The lysate was further cleared by spinning at 18000 g for 15 minutes at 4°C.
S-nitrosylation detection
In situ detection of S-nitrosylated protein
The assay was performed as described with minor modifications (Thibeault et al., 2010). Briefly, frozen BAT sections were fixed with 3.5% paraformaldehyde, and then permeabilized with 0.1% Triton X-100 in PBS containing 1mM EDTA and 0.1 mM neocuproine. Biotin-switch assay was performed by first blocking free thiols using HENS buffer containing 20 mM MMTS at room temperature for 30 minutes. Then the S-nitrosylated proteins were labeled in HENS buffer with 0.4mM biotin-HPDP and 10 mM ascorbic acid for 1 hour. Biotinylated proteins were labeled using strepvidin conjugated with Alexa 488 (ThermoFisher Scientific, A32731). The sections were then subject to immunostaining for UCP1 using antibodies against UCP1 (1:1000, Abcam, ab10983) and secondary antibodies conjugated to Alexa-568 (Molecular Probes, A-11004). The images were observed using a Ziess 700 confocal or Leica fluorescence microscope. The images were quantified using ImarisColoc (Bitplane).
Detection of S-nitrosylation proteins
Total S-nitrosylated proteins in BAT was detected by S-nitrosylation Western Blot Kit (Thermo Fisher Scientific). Briefly, BAT was lysed in HEN buffer and 500 μg protein was used for each sample. Free cysteines were first blocked with MMTS for 20 minutes at 50°C. Following precipitation, using 4 × volume of cold acetone for 60 minutes at −20°C, S-nitrosylated cysteines were selectively labeled with iodoTMT reagent in the presence of 20 mM sodium ascorbate at RT for 2 hours. Samples were then separated by SDS-PAGE and blotted to membrane for detection of SNO proteins by the anti-TMT antibody.
Assessment of SNO of UCP1 by mass spectrometry
100 μg of recombinant human UCP1 (MyBioSource) was treated with or without of 200 μM SNAP for 15 minutes in dark at RT. The samples were then blocked with MMTS for 20 minutes at 50°C to block free cysteines, and labeled with iodoTMT reagent as described above. After acetone precipitation and desalt processes, mass spectrometry analysis was performed by the Taplin Biological Mass Spectrometry Facility at the Harvard Medical School using an Orbitrap Fusion mass spectrometer. MS/MS data were searched against the Uniprot mouse database using the SEQUEST algorithm.
ADH5 Activity assay
The ADH5 enzyme activity assay was performed as described previously (Liu et al., 2001). Briefly, 10 μg protein lysate was added to 200 μL of assay mix (20 mM Tris-Hcl, 200 μM NADH, and 0.5 mM EDTA). The kinetics of GSNO-dependent NADH consumption was measured in the absence or presence of 100 μM of GSNO by using a microplate reader (340 nm) at 37°C. The resulting ADH5 activity was expressed as nmol of NADH degraded per min/mg protein.
Oil Red O staining
Murine or human differentiated brown adipocytes were fixed with 4% PFA and stained with 0.3% Oil Red O solution (Sigma-Aldrich, O0625) and imaged using a Nikon microscope.
Immunohistochemistry and Immunofluorescence
For immunohistochemistry, iBAT depots were fixed with 4% PFA and sectioned at 5 μm thick, followed by deparaffinization and rehydration processes. Tissue sections were stained using H&E. The images were observed under a Nikon microscope (10x). For F4/80/UCP1 dual staining, sections were deparaffinized, blocked, and incubated overnight at 4°C with UCP1 (1:100) followed by secondary staining at RT (1 hour, 1:50). Slides were then washed and blocked prior to overnight staining with F4/80 antibody (1:100) followed by secondary staining for one hour at RT (1:50). Nuclei were stained with 4′,6-diamidino-2-phenylindole (1:2500, DAPI). Images were taken using a LSM 880 confocal microscope (Carl Zeiss) and analyzed with NIH ImageJ. For ADH5 staining, sections were incubated with primary antibody (1:100) followed by staining with a Vectastain ABC kit (Vector Laboratories) using Sigma Fast 3,3′-diaminobenzidine (Sigma-Aldrich) as the substrate. Images were taken by using a Nikon microscope.
Lipolysis assay
Lipolysis in BAT was measured as described previously (Bartelt et al., 2018). Briefly, BAT depots were excised and incubated in 24-well plates in DMEM with high glucose supplemented with 2% w/v FA-free BSA (FA-free BSA, Sigma, 10775835001). BAT was then incubated for 1 hour at 37°C before stimulation with 10 μM isoproterenol (ISO). Samples were taken at the indicated time points, and the supernatant was assayed for free fatty acid (FFA) using a FFA fluorometric kit (Cayman Chemical). The fatty acid release was normalized to RNA content.
Metabolic Phenotyping
Whole-body energy expenditure and body composition
Whole-body energy expenditure (VO2, VCO2), food intake, and locomotor activity were monitored using a Comprehensive Lab Animal Monitoring System (CLAMS, Columbus Instruments) at the Fraternal Order of Eagles Metabolic Phenotypic Core. For core temperature measurements, a thermistor was implanted subcutaneously within the abdominal cavity; for BAT temperature recording, a thermistor was implanted between BAT and the underlying muscle layer in the inter-scapular region prior. The temperature at each tissue was recorded by CLAMS Telemetry Temperature Transmitter system. Body composition was measured by using Bruker Minispecs (LF50).
Infrared imaging
Mice were injected with norepinephrine (0.3 mg/kg) via i.p. 15 minutes after injections, body surface temperature was imaged in fully awake, unrestrained mice using a high-resolution infrared camera (A655sc Thermal Imager; FLIR Systems, Inc.) as described (Zhu et al., 2014).
Respirometry of BAT
hTERT A41hBAT-SVF (2.5×105 cells) or freshly isolated iBAT (weighed for ~2 mg) was resuspended or minced in respiration buffer (2.5 mM glucose, 50 μM palmitoryl-L-carnitine hydrochloride, 2.5 mM malate, 120 mM NaCl, 4.5 mM KCl, 0.7 mM Na2HPO4, 1.5 mM NaH2PO4 and 0.5 mM MgCl2, pH 7.4). High-resolution O2 consumption was measured in 2 mL of buffer Z containing 0.5mM EDTA using the OROBOROS Oxygraph-2k (O2k; Oroboros Instruments, Innsbruck, Austria (Tyrrell et al., 2016). All respiration measurements were conducted at 37°C and a working range O2 of ~350–180 μM. Respiration was measured as follows: 5 mM malate + 10 mM pyruvate followed by 1 mM GDP (UCP1) and finally 0.5 mM adenosine diphosphate (ADP) (complex I OXPHOS substrate). Data shown correspond to subtraction of the oxygen flux observed after guanidine diphosphate (GDP) addition (1 μM, for UCP1) or 0.5 mM adenosine diphosphate (ADP, for Complex I) from that measured after the addition of 10mM pyruvate + 5mM malate and normalized to baseline flux. The graph shown corresponds to subtraction of the oxygen flux observed after addition of 1 μM GDP from that measured after addition of 10 mM pyruvate + 5 mM malate and normalized to baseline flux. For hTERT A41hBAT-SVF transfected with control or UCP1 constructs, the baseline was normalized to total protein content.
Primary brown adipocyte cellular respirometry
Cellular OCR of BAs was determined using a Seahorse XFe96 Analyzer. Adipocytes were plated and differentiated (for 6 days) in XF96 cell culture microplates. Basal respiration was determined to be the OCR in the presence of substrate alone. The respiratory rate was measured at 37°C in 8 replicates (independent wells). ATP-synthase-independent respiration was measured after injection of 1uM oligomycin, maximal respiration was then measured after addition of 1 μM FCCP and finally, leak respiration was measured after injection with 15uM rotenone/antimycin. All values were normalized to total protein content determined by BCA (Pierce).
Glucose tolerance test
Animals were fasted for 16 hours prior to GTT. Glucose tolerance was tested by measuring glucose concentration at different time points after an intraperitoneal (IP) glucose injection (0.8–1.25 g/kg body weight, 50% dextrose, Hospra Inc, 0409-6648-02) (Qian et al., 2018).
QUANTIFICATION AND STATISTICAL ANALYSIS
Results are expressed as the mean ± the standard error of the mean (SEM); n represents the number of individual mice (biological replicates) or individual experiments (technical replicates) as indicated in the figure legends. We performed the Shapiro-Wilk Normality test in experiments that have a relatively large sample size (n > 5) and found that these data pass the normality test (alpha = 0.05). Data were further analyzed with two-tailed Student’s and Welch’s t test for two-group comparisons or ANOVA for multiple comparisons. For both One-Way ANOVA and Two-Way ANOVA, Tukey’s post hoc multiple comparisons were applied as recommended by Prism. In all cases, GraphPad Prism (GraphPad Software Prism 8) was used for the calculations.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat anti-Mouse polyclonal IgG HRP-linked antibody | Santa Cruz | Cat$sc-2005; RRID:AB_631736 |
| Goat anti-rabbit IgG (H+L) Antibody, Alexa Fluor 488 | ThermoFisher Scientific | Cat#A32731; RRID:AB_2633280 |
| Goat anti-rabbit IgG (H+L) Antibody, Alexa Fluor 568 | Molecular Probes | Cat#A-11004; RRID:AB_2534072 |
| Goat anti-Rabbit IgG HRP-linked antibody | Cell Signaling | Cat#7074S, RRID:AB_2099233 |
| Mouse monoclonal anti-DDIT3 (L63F7) | Cell Signaling | Cat#2895; RRID:AB_2089254 |
| Mouse monoclonal anti-INSRbeta (C-19) | Santa Cruz | Cat#sc-711; RRID:AB_631835 |
| Mouse monoclonal anti-SQSTM1/P62 | Abnova | Cat#H00008878-M03; RRID:AB_1112274 |
| Rabbit monoclonal anti-ADGRE1/F4/80 (D2S9R) | Cell Signaling | CAT#70076; RRID:AB_2799771 |
| Rabbit monoclonal anti-ATG7 | Abgent | Cat#AP1813a; RRID:AB_636747 |
| Rabbit monoclonal anti-EIF2AK3(D11A8) | Cell Signaling | Cat#5683, RRID:AB_10841299 |
| Rabbit monoclonal anti-FOXO1 (C29H4) | Cell Signaling | Cat#76764; RRID:AB_2799887 |
| Rabbit monoclonal anti-HSPA5(C50B12) | Cell Signaling | Cat#3177S; RRID:AB_2119845 |
| Rabbit monoclonal anti-Phospho-AKT1 (Ser473) (D9E) | Cell Signaling | Cat#4060; RRID:AB_2315049 |
| Rabbit monoclonal anti-phospho-EIF2AK3 (Thr980) | Cell Signaling | Cat#3179S; RRID:AB_2095853 |
| Rabbit monoclonal anti-TUBA (11H10) | Cell Signaling | Cat#2125S; RRID:AB_2619646 |
| Rabbit polyclonal anti-ACTB(H-300) | Santa Cruz | Cat#sc-10731; RRID:AB_2223515 |
| Rabbit polyclonal anti-ADH5 | Abcam | Cat#91385; RRID:AB_2049142 |
| Rabbit polyclonal anti-AKT1 2/3 (H-136) | Santa Cruz | Cat#sc-8312; RRID:AB_671714 |
| Rabbit polyclonal anti-ATG5 | Novus | Cat#NB110-53818; RRID:AB_828587 |
| Rabbit polyclonal anti-Histone H3K27me3 Active Motif | ThermoFisher Scientific | Cat#39156; RRID:AB_2867260 |
| Rabbit polyclonal anti-HSF1 | Cell Signaling | Cat#4356S; RRID:AB_2120258 |
| Rabbit polyclonal anti-MAP1LC3B | Novus | Cat#NB600-1384; RRID:AB_669581 |
| Rabbit polyclonal anti-phospho-FOXO1 (Ser256) | Cell Signaling | Cat#9461; RRID:AB_329831 |
| Rabbit polyclonal anti-UCP1 | Abcam | Cat#ab10983; RRID:AB_2241462 |
| TMT Monoclonal Antibody (25D5) | ThermoFisher Scientific | Cat#90075; RRID:AB_10854708 |
| Bacterial and virus strains | ||
| Adeno-Adh5 | Dr. Ling Yang; Qian et al., 2018 | N/A |
| Adeno-GFP | Yang et al., 2015 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| 1400W | Sigma | Cat#214358-33-5 |
| 3-Isobutyl-1 -methylxanthine (IBMX) | Sigma | Cat#I5879 |
| 3,3′-Diaminobenzidine (DAB) Liquid Substrate System tetrahydrochloride | Sigma | Cat#D7304-1SET |
| 5-Aza-2′-Deoxycytidine (5aza) | Sigma | Cat#189825 |
| Adenosine 5′-diphosphate bis(cyclohexylammonium) salt (ADP) | Sigma | Cat#A4386 |
| CL316, 243 | Sigma | Cat#C5976 |
| DAPI (4′,6-diamidino-2-phenylindole) | ThermoFisher Scientific | Cat#D1306 |
| Dexamethasone | Sigma | Cat#D4902 |
| Guanosine 5′-diphosphate sodium salt (GDP) | Sigma | Cat#G7127 |
| HSF1A | Millipore | Cat#1196723-93-9 |
| Humulin R U-500 | Lily | Cat#0002-8501-01 |
| Indomethacin | Sigma | Cat#4025268 |
| Iodoacetyl Tandem Mass TagTM (iodoTMTTM) Reagents | ThermoFisher Scienticic | Cat#90102 |
| Isoproterenol | Sigma | Cat#54750-10-6 |
| KRIBB11 | Sigma | Cat#385570 |
| Liberase | Roche | Cat#5401119001 |
| Lipofectamine 2000 | ThermoFisher Scientific | Cat#L30000015 |
| Malate | Sigma | Cat#W237418 |
| N6022 | Sigma | Cat#1208315-24-5 |
| NDC 0409-6648-02 DEXTROSE | Hospira | Cat#0409-6648-02 |
| Recombinant human UCP1 | MyBiosource | Cat#MBS963514 |
| Rosiglitazone | Sigma | Cat#557366-M |
| S-Nitroso-N-acetyl-DL-penicillamine (SNAP) | Sigma | Cat#N3398 |
| Sodium pyruvate | Sigma | Cat#P5280 |
| Succinate | Sigma | Cat#W239607 |
| Critical commercial assays | ||
| Free Fatty Acid Fluorometric Assay Kit | Cayman Chemical | Cat#700310 |
| Luciferase Assay | Promega | Cat#E1500 |
| NBEBuilder HiFi DNA assembly system | NEB | Cat# E5520S |
| Nitrate/Nitrite Colorimetric Assay Kit | Cayman Chemical | Cat#780001 |
| Pierce S-Nitrosylation Western Blot Kit | ThermoFisher Scientific | Cat#90105 |
| Renilla Luciferase Assay | Promega | Cat#E2810 |
| VECTASTAIN® ABC-HRP Kit, Peroxidase (Standard) PK-4000 | Vector Laboratories | Cat#PK-4000 |
| Experimental models: Cell lines | ||
| Human: A41hBAT-SVF | ATCC | Cat#CRL-3385 |
| Human: HEK293A | ATCC | Cat#CRL-1573 |
| Mouse: Brown Adipocytes | Dr. Ling Yang | N/A |
| Mouse: Stromal Vascular Fraction | Dr. Ling Yang | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: Adh5BKO | This paper | N/A |
| Mouse; Adh5fl | Dr. Limin Liu; Wei et al.; 2011 | N/A |
| Mouse: C57BL/6J | The Jackson Laboratory | JAX: Cat#000664 |
| Mouse: FVB-Tg(UCP1-cre)1Evdr/J | The Jackson Laboratory | JAX: Cat#024670 |
| Oligonucleotides. See Table S1. | ||
| Recombinant DNA | ||
| Plasmid:C305AUCP1 | This paper | N/A |
| Plasmid:GFP | This paper | N/A |
| Plasmid:pcDNA3.1-IDH2-WT | Addgene | Cat#V790-20 |
| Plasmid:WTUCP1 | This paper | N/A |
| Software and algorithms | ||
| GraphPad Prism 9.20 | GraphPad Software, LLC. | https://www.graphpad.com/scientific-software/prism/ |
| Image Studio Lite | LI-COR Biosciences | https://www.licor.com/bio/image-studio-lite/ |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Other | ||
| 60%kCal high-fat diet | Research Diets | Cat#D12492 |
| Oil Red O staining | Sigma | Cat#O0625 |
| Opti-MEM | ThermoFisher Scientific | Cat#31985-062 |
| Teklad global soy protein-free extruded 2920X | Teklad Global Diet | N/A |
Highlights.
Thermogenesis induces BAT protein S-nitrosylation modification
ADH5 is required for maintaining BAT metabolic homeostasis
Diet-induced obesity suppresses BAT HSF1-mediated activation of Adh5
ADH5 overexpression ameliorates BAT metabolic dysfunction in the context of obesity
ACKNOWLEDGMENTS
L.Y., Z.Zhang, and S.C.S. designed the study and wrote the manuscript. S.C.S., Q.Q., Z.Zhang, M.L., M.H., Z.Zhu and W.L., performed the experiments. V.L., L.L., M.J.P., L.Z., and A.B. provided critical reagents and scientific suggestions on the manuscript. L.Y. conceived and supervised the study. Z.Zhang is supported by an American Heart Association predoctoral award (19PRE34380258). L.Y. is supported by an American Diabetes Association Innovative Basic Science Award (1-18-IBS-149) and NIH grant R01 DK108835-01A1.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.110003.
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
We worked to ensure sex balance in the selection of non-human subjects. We worked to ensure diversity in experimental samples through the selection of the genomic datasets. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
REFERENCES
- Alcalá M, Calderon-Dominguez M, Bustos E, Ramos P, Casals N, Serra D, Viana M, and Herrero L (2017). Increased inflammation, oxidative stress and mitochondrial respiration in brown adipose tissue from obese mice. Sci. Rep 7, 16082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcalá M, Calderon-Dominguez M, Serra D, Herrero L, and Viana M (2019). Mechanisms of Impaired Brown Adipose Tissue Recruitment in Obesity. Front. Physiol 10, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altshuler-Keylin S, Shinoda K, Hasegawa Y, Ikeda K, Hong H, Kang Q, Yang Y, Perera RM, Debnath J, and Kajimura S (2016). Beige Adipocyte Maintenance Is Regulated by Autophagy-Induced Mitochondrial Clearance. Cell Metab. 24, 402–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyappan JP, and Nagajyothi JF (2017). Diet Modulates Adipose Tissue Oxidative Stress in a Murine Acute Chagas Model. JSM Atheroscler. 2, 1030. [PMC free article] [PubMed] [Google Scholar]
- Balkow A, Hoffmann LS, Klepac K, Glöde A, Gnad T, Zimmermann K, and Pfeifer A (2016). Direct lentivirus injection for fast and efficient gene transfer into brown and beige adipose tissue. J. Biol. Methods 3, e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett SD, and Buxton ILO (2017). The role of S-nitrosoglutathione reductase (GSNOR) in human disease and therapy. Crit. Rev. Biochem. Mol. Biol 52, 340–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartelt A, Bruns OT, Reimer R, Hohenberg H, Ittrich H, Peldschus K, Kaul MG, Tromsdorf UI, Weller H, Waurisch C, et al. (2011). Brown adipose tissue activity controls triglyceride clearance. Nat. Med 17, 200–205. [DOI] [PubMed] [Google Scholar]
- Bartelt A, Widenmaier SB, Schlein C, Johann K, Goncalves RLS, Eguchi K, Fischer AW, Parlakgül G, Snyder NA, Nguyen TB, et al. (2018). Brown adipose tissue thermogenic adaptation requires Nrf1-mediated proteasomal activity. Nat. Med 24, 292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becerril S, Rodríguez A, Catalán V, Sáinz N, Ramírez B, Collantes M, Peñuelas I, Gómez-Ambrosi J, and Frühbeck G (2010). Deletion of inducible nitric-oxide synthase in leptin-deficient mice improves brown adipose tissue function. PLoS ONE 5, e10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becerril S, Rodríguez A, Catalán V, Méndez-Giménez L, Ramírez B, Sáinz N, Llorente M, Unamuno X, Gómez-Ambrosi J, and Frühbeck G (2018). Targeted disruption of the iNOS gene improves adipose tissue inflammation and fibrosis in leptin-deficient ob/ob mice: role of tenascin C. Int. J. Obes 42, 1458–1470. [DOI] [PubMed] [Google Scholar]
- Beigi F, Gonzalez DR, Minhas KM, Sun QA, Foster MW, Khan SA, Treuer AV, Dulce RA, Harrison RW, Saraiva RM, et al. (2012). Dynamic denitrosylation via S-nitrosoglutathione reductase regulates cardiovascular function. Proc. Natl. Acad. Sci. USA 109, 4314–4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benhar M, Forrester MT, and Stamler JS (2009). Protein denitrosylation: enzymatic mechanisms and cellular functions. Nat. Rev. Mol. Cell Biol 10, 721–732. [DOI] [PubMed] [Google Scholar]
- Blonder JP, Mutka SC, Sun X, Qiu J, Green LH, Mehra NK, Boyanapalli R, Suniga M, Look K, Delany C, et al. (2014). Pharmacologic inhibition of S-nitrosoglutathione reductase protects against experimental asthma in BALB/c mice through attenuation of both bronchoconstriction and inflammation. BMC Pulm. Med 14, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondin DP, Labbé SM, Noll C, Kunach M, Phoenix S, Guérin B, Turcotte EE, Haman F, Richard D, and Carpentier AC (2015). Selective Impairment of Glucose but Not Fatty Acid or Oxidative Metabolism in Brown Adipose Tissue of Subjects With Type 2 Diabetes. Diabetes 64, 2388–2397. [DOI] [PubMed] [Google Scholar]
- Bond LM, Burhans MS, and Ntambi JM (2018). Uncoupling protein-1 deficiency promotes brown adipose tissue inflammation and ER stress. PLoS ONE 13, e0205726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon B, and Nedergaard J (2004). Brown adipose tissue: function and physiological significance. Physiol. Rev 84, 277–359. [DOI] [PubMed] [Google Scholar]
- Cannon B, and Nedergaard J (2011). Nonshivering thermogenesis and its adequate measurement in metabolic studies. J. Exp. Biol 214, 242–253. [DOI] [PubMed] [Google Scholar]
- Cao Y, Gomes SA, Rangel EB, Paulino EC, Fonseca TL, Li J, Teixeira MB, Gouveia CH, Bianco AC, Kapiloff MS, et al. (2015). S-nitrosoglutathione reductase-dependent PPARg denitrosylation participates in MSC-derived adipogenesis and osteogenesis. J. Clin. Invest 125, 1679–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Zhu Q, Liu L, Glazier BJ, Hinkel BC, Liang C, and Shi H (2018). Global Transcriptome Analysis of Brown Adipose Tissue of Diet-Induced Obese Mice. Int. J. Mol. Sci 19, E1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarelli F, Cipollone F, Romano F, Tumini S, Costantini F, di Ricco L, Pomilio M, Pierdomenico SD, Marini M, Cuccurullo F, and Mezzetti A (2000). Increased circulating nitric oxide in young patients with type 1 diabetes and persistent microalbuminuria: relation to glomerular hyperfiltration. Diabetes 49, 1258–1263. [DOI] [PubMed] [Google Scholar]
- Choi MS, Jung JY, Kim HJ, Ham MR, Lee TR, and Shin DW (2016). S-nitrosylation of fatty acid synthase regulates its activity through dimerization. J. Lipid Res 57, 607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani ET, Kazak L, Jedrychowski MP, Lu GZ, Erickson BK, Szpyt J, Pierce KA, Laznik-Bogoslavski D, Vetrivelan R, Clish CB, et al. (2016). Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature 532, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani ET, Kazak L, and Spiegelman BM (2017). Mitochondrial reactive oxygen species and adipose tissue thermogenesis: Bridging physiology and mechanisms. J. Biol. Chem 292, 16810–16816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christman JK (2002). 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene 21, 5483–5495. [DOI] [PubMed] [Google Scholar]
- Cui X, Xiao W, You L, Zhang F, Cao X, Feng J, Shen D, Li Y, Wang Y, Ji C, and Guo X (2019). Age-induced oxidative stress impairs adipogenesis and thermogenesis in brown fat. FEBS J. 286, 2753–2768. [DOI] [PubMed] [Google Scholar]
- Di Meo S, Reed TT, Venditti P, and Victor VM (2016). Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev 2016, 1245049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engeli S, Janke J, Gorzelniak K, Böhnke J, Ghose N, Lindschau C, Luft FC, and Sharma AM (2004). Regulation of the nitric oxide system in human adipose tissue. J. Lipid Res 45, 1640–1648. [DOI] [PubMed] [Google Scholar]
- Fujimoto M, Shimizu N, Kunii K, Martyn JA, Ueki K, and Kaneki M (2005). A role for iNOS in fasting hyperglycemia and impaired insulin signaling in the liver of obese diabetic mice. Diabetes 54, 1340–1348. [DOI] [PubMed] [Google Scholar]
- Gao W, Kong X, and Yang Q (2017). Isolation, Primary Culture, and Differentiation of Preadipocytes from Mouse Brown Adipose Tissue. Methods Mol. Biol 1566, 3–8. [DOI] [PubMed] [Google Scholar]
- Garvey EP, Oplinger JA, Furfine ES, Kiff RJ, Laszlo F, Whittle BJ, and Knowles RG (1997). 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. J. Biol. Chem 272, 4959–4963. [DOI] [PubMed] [Google Scholar]
- Giordano A, Tonello C, Bulbarelli A, Cozzi V, Cinti S, Carruba MO, and Nisoli E (2002). Evidence for a functional nitric oxide synthase system in brown adipocyte nucleus. FEBS Lett. 514, 135–140. [DOI] [PubMed] [Google Scholar]
- Harms M, and Seale P (2013). Brown and beige fat: development, function and therapeutic potential. Nat. Med 19, 1252–1263. [DOI] [PubMed] [Google Scholar]
- Hess DT, Matsumoto A, Kim SO, Marshall HE, and Stamler JS (2005). Protein S-nitrosylation: purview and parameters. Nat. Rev. Mol. Cell Biol 6, 150–166. [DOI] [PubMed] [Google Scholar]
- Himms-Hagen J (1985). Defective brown adipose tissue thermogenesis in obese mice. Int. J. Obes 9 (Suppl 2), 17–24. [PubMed] [Google Scholar]
- Hotamisligil GS (2006). Inflammation and metabolic disorders. Nature 444, 860–867. [DOI] [PubMed] [Google Scholar]
- Hussain A, Yun BW, Kim JH, Gupta KJ, Hyung NI, and Loake GJ (2019). Novel and conserved functions of S-nitrosoglutathione reductase in tomato. J. Exp. Bot 70, 4877–4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda K, Kang Q, Yoneshiro T, Camporez JP, Maki H, Homma M, Shinoda K, Chen Y, Lu X, Maretich P, et al. (2017). UCP1-independent signaling involving SERCA2b-mediated calcium cycling regulates beige fat thermogenesis and systemic glucose homeostasis. Nat. Med 23, 1454–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankovic A, Korac A, Buzadzic B, Stancic A, Otasevic V, Ferdinandy P, Daiber A, and Korac B (2017). Targeting the NO/superoxide ratio in adipose tissue: relevance to obesity and diabetes management. Br. J. Pharmacol 174, 1570–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimura S, and Saito M (2014). A new era in brown adipose tissue biology: molecular control of brown fat development and energy homeostasis. Annu. Rev. Physiol 76, 225–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneki M, Shimizu N, Yamada D, and Chang K (2007). Nitrosative stress and pathogenesis of insulin resistance. Antioxid. Redox Signal 9, 319–329. [DOI] [PubMed] [Google Scholar]
- Kazak L, Chouchani ET, Stavrovskaya IG, Lu GZ, Jedrychowski MP, Egan DF, Kumari M, Kong X, Erickson BK, Szpyt J, et al. (2017). UCP1 deficiency causes brown fat respiratory chain depletion and sensitizes mitochondria to calcium overload-induced dysfunction. Proc. Natl. Acad. Sci. USA 114, 7981–7986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi-Utsumi K, Gao B, Ohinata H, Hashimoto M, Yamamoto N, and Kuroshima A (2002). Enhanced gene expression of endothelial nitric oxide synthase in brown adipose tissue during cold exposure. Am. J. Physiol. Regul. Integr. Comp. Physiol 282, R623–R626. [DOI] [PubMed] [Google Scholar]
- Lee JH, Go Y, Kim DY, Lee SH, Kim OH, Jeon YH, Kwon TK, Bae JH, Song DK, Rhyu IJ, et al. (2020). Isocitrate dehydrogenase 2 protects mice from high-fat diet-induced metabolic stress by limiting oxidative damage to the mitochondria from brown adipose tissue. Exp. Mol. Med 52, 238–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner BP, Huang S, Brychta RJ, Duckworth CJ, Baskin AS, McGehee S, Tal I, Dieckmann W, Gupta G, Kolodny GM, et al. (2017). Mapping of human brown adipose tissue in lean and obese young men. Proc. Natl. Acad. Sci. USA 114, 8649–8654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leterrier M, Chaki M, Airaki M, Valderrama R, Palma JM, Barroso JB, and Corpas FJ (2011). Function of S-nitrosoglutathione reductase (GSNOR) in plant development and under biotic/abiotic stress. Plant Signal. Behav 6, 789–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Hausladen A, Zeng M, Que L, Heitman J, and Stamler JS (2001). A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature 410, 490–494. [DOI] [PubMed] [Google Scholar]
- Liu L, Yan Y, Zeng M, Zhang J, Hanes MA, Ahearn G, McMahon TJ, Dickfeld T, Marshall HE, Que LG, and Stamler JS (2004). Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell 116, 617–628. [DOI] [PubMed] [Google Scholar]
- Lv X, Ge S, Jalal Ahammed G, Xiang X, Guo Z, Yu J, and Zhou Y (2017). Crosstalk between Nitric Oxide and MPK1/2 Mediates Cold Acclimation-induced Chilling Tolerance in Tomato. Plant Cell Physiol. 58, 1963–1975. [DOI] [PubMed] [Google Scholar]
- Ma X, Xu L, Alberobello AT, Gavrilova O, Bagattin A, Skarulis M, Liu J, Finkel T, and Mueller E (2015). Celastrol Protects against Obesity and Metabolic Dysfunction through Activation of a HSF1-PGC1α Transcriptional Axis. Cell Metab. 22, 695–708. [DOI] [PubMed] [Google Scholar]
- Mailloux RJ, Adjeitey CN, Xuan JY, and Harper ME (2012). Crucial yet divergent roles of mitochondrial redox state in skeletal muscle vs. brown adipose tissue energetics. FASEB J. 26, 363–375. [DOI] [PubMed] [Google Scholar]
- Marshall HE, Hess DT, and Stamler JS (2004). S-nitrosylation: physiological regulation of NF-kappaB. Proc. Natl. Acad. Sci. USA 101, 8841–8842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matz JM, LaVoi KP, and Blake MJ (1996). Adrenergic regulation of the heat shock response in brown adipose tissue. J. Pharmacol. Exp. Ther 277, 1751–1758. [PubMed] [Google Scholar]
- Mussbacher M, Stessel H, Pirker T, Gorren ACF, Mayer B, and Schrammel A (2019). S-nitrosoglutathione inhibits adipogenesis in 3T3-L1 preadipocytes by S-nitrosation of CCAAT/enhancer-binding protein β. Sci. Rep 9, 15403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagashima T, Ohinata H, and Kuroshima A (1994). Involvement of nitric oxide in noradrenaline-induced increase in blood flow through brown adipose tissue. Life Sci. 54, 17–25. [DOI] [PubMed] [Google Scholar]
- Nakamura T, and Lipton SA (2016). Protein S-Nitrosylation as a Therapeutic Target for Neurodegenerative Diseases. Trends Pharmacol. Sci 37, 73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard J, Bengtsson T, and Cannon B (2007). Unexpected evidence for active brown adipose tissue in adult humans. Am. J. Physiol. Endocrinol. Metab 293, E444–E452. [DOI] [PubMed] [Google Scholar]
- Nisoli E, Tonello C, Briscini L, and Carruba MO (1997). Inducible nitric oxide synthase in rat brown adipocytes: implications for blood flow to brown adipose tissue. Endocrinology 138, 676–682. [DOI] [PubMed] [Google Scholar]
- Nisoli E, Clementi E, Tonello C, Sciorati C, Briscini L, and Carruba MO (1998). Effects of nitric oxide on proliferation and differentiation of rat brown adipocytes in primary cultures. Br. J. Pharmacol 125, 888–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valerio A, Francolini M, Moncada S, and Carruba MO (2003). Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science 299, 896–899. [DOI] [PubMed] [Google Scholar]
- Nisoli E, Falcone S, Tonello C, Cozzi V, Palomba L, Fiorani M, Pisconti A, Brunelli S, Cardile A, Francolini M, et al. (2004). Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc. Natl. Acad. Sci. USA 101, 16507–16512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noronha BT, Li JM, Wheatcroft SB, Shah AM, and Kearney MT (2005). Inducible nitric oxide synthase has divergent effects on vascular and metabolic function in obesity. Diabetes 54, 1082–1089. [DOI] [PubMed] [Google Scholar]
- Nott A, Watson PM, Robinson JD, Crepaldi L, and Riccio A (2008). S-Nitrosylation of histone deacetylase 2 induces chromatin remodelling in neurons. Nature 455, 411–415. [DOI] [PubMed] [Google Scholar]
- Okla M, Kim J, Koehler K, and Chung S (2017). Dietary Factors Promoting Brown and Beige Fat Development and Thermogenesis. Adv. Nutr 8, 473–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otasevic V, Korac A, Buzadzic B, Stancic A, Jankovic A, and Korac B (2011). Nitric oxide and thermogenesis–challenge in molecular cell physiology. Front. Biosci. (Schol. Ed.) 3, 1180–1195. [DOI] [PubMed] [Google Scholar]
- Ovadia H, Haim Y, Nov O, Almog O, Kovsan J, Bashan N, Benhar M, and Rudich A (2011). Increased adipocyte S-nitrosylation targets anti-lipolytic action of insulin: relevance to adipose tissue dysfunction in obesity. J. Biol. Chem 286, 30433–30443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perreault M, and Marette A (2001). Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nat. Med 7, 1138–1143. [DOI] [PubMed] [Google Scholar]
- Porter C (2017). Quantification of UCP1 function in human brown adipose tissue. Adipocyte 6, 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Q, Zhang Z, Orwig A, Chen S, Ding WX, Xu Y, Kunz RC, Lind NRL, Stamler JS, and Yang L (2018). S-Nitrosoglutathione Reductase Dysfunction Contributes to Obesity-Associated Hepatic Insulin Resistance via Regulating Autophagy. Diabetes 67, 193–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Q, Zhang Z, Li M, Savage K, Cheng D, Rauckhorst AJ, Ankrum JA, Taylor EB, Ding WX, Xiao Y, et al. (2019). Hepatic Lysosomal iNOS Activity Impairs Autophagy in Obesity. Cell. Mol. Gastroenterol. Hepatol 8, 95–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Z, Meng F, Bomgarden RD, Viner RI, Li J, Rogers JC, Cheng J, Greenlief CM, Cui J, Lubahn DB, et al. (2014). Proteomic quantification and site-mapping of S-nitrosylated proteins using isobaric iodoTMT reagents. J. Proteome Res 13, 3200–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Que LG, Yang Z, Stamler JS, Lugogo NL, and Kraft M (2009). S-nitrosoglutathione reductase: an important regulator in human asthma. Am. J. Respir. Crit. Care Med 180, 226–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke H, Saini C, Fleury-Olela F, Dibner C, Benjamin IJ, and Schibler U (2008). Differential display of DNA-binding proteins reveals heat-shock factor 1 as a circadian transcription factor. Genes Dev. 22, 331–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizza S, and Filomeni G (2018). Role, Targets and Regulation of (de)nitrosylation in Malignancy. Front. Oncol 8, 334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizza S, Montagna C, Cardaci S, Maiani E, Di Giacomo G, Sanchez-Quiles V, Blagoev B, Rasola A, De Zio D, Stamler JS, et al. (2016). S-nitrosylation of the Mitochondrial Chaperone TRAP1 Sensitizes Hepatocellular Carcinoma Cells to Inhibitors of Succinate Dehydrogenase. Cancer Res. 76, 4170–4182. [DOI] [PubMed] [Google Scholar]
- Rizza S, Cardaci S, Montagna C, Di Giacomo G, De Zio D, Bordi M, Maiani E, Campello S, Borreca A, Puca AA, et al. (2018). S-nitrosylation drives cell senescence and aging in mammals by controlling mitochondrial dynamics and mitophagy. Proc. Natl. Acad. Sci. USA 115, E3388–E3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha SK, and Kuroshima A (2000). Nitric oxide and thermogenic function of brown adipose tissue in rats. Jpn. J. Physiol 50, 337–342. [DOI] [PubMed] [Google Scholar]
- Saito M, Okamatsu-Ogura Y, Matsushita M, Watanabe K, Yoneshiro T, Nio-Kobayashi J, Iwanaga T, Miyagawa M, Kameya T, Nakada K, et al. (2009). High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes 58, 1526–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansbury BE, and Hill BG (2014). Regulation of obesity and insulin resistance by nitric oxide. Free Radic. Biol. Med 73, 383–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarge KD, and Cullen KE (1997). Regulation of hsp expression during rodent spermatogenesis. Cell. Mol. Life Sci 53, 191–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu I, and Walsh K (2015). The Whitening of Brown Fat and Its Implications for Weight Management in Obesity. Curr. Obes. Rep 4, 224–229. [DOI] [PubMed] [Google Scholar]
- Shimizu I, Aprahamian T, Kikuchi R, Shimizu A, Papanicolaou KN, MacLauchlan S, Maruyama S, and Walsh K (2014). Vascular rarefaction mediates whitening of brown fat in obesity. J. Clin. Invest 124, 2099–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinozaki S, Chang K, Sakai M, Shimizu N, Yamada M, Tanaka T, Nakazawa H, Ichinose F, Yamada Y, Ishigami A, et al. (2014). Inflammatory stimuli induce inhibitory S-nitrosylation of the deacetylase SIRT1 to increase acetylation and activation of p53 and p65. Sci. Signal 7, ra106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Xiang Y, Wang Y, Baikati K, Cuervo AM, Luu YK, Tang Y, Pessin JE, Schwartz GJ, and Czaja MJ (2009). Autophagy regulates adipose mass and differentiation in mice. J. Clin. Invest 119, 3329–3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sips PY, Irie T, Zou L, Shinozaki S, Sakai M, Shimizu N, Nguyen R, Stamler JS, Chao W, Kaneki M, and Ichinose F (2013). Reduction of cardiomyocyte S-nitrosylation by S-nitrosoglutathione reductase protects against sepsis-induced myocardial depression. Am. J. Physiol. Heart Circ. Physiol 304, H1134–H1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stomberski CT, Hess DT, and Stamler JS (2019). Protein S-Nitrosylation: Determinants of Specificity and Enzymatic Regulation of S-Nitrosothiol-Based Signaling. Antioxid. Redox Signal 30, 1331–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone JR, and Marletta MA (1996). Spectral and kinetic studies on the activation of soluble guanylate cyclase by nitric oxide. Biochemistry 35, 1093–1099. [DOI] [PubMed] [Google Scholar]
- Tang CH, Wei W, Hanes MA, and Liu L (2013). Hepatocarcinogenesis driven by GSNOR deficiency is prevented by iNOS inhibition. Cancer Res. 73, 2897–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibeault S, Rautureau Y, Oubaha M, Faubert D, Wilkes BC, Delisle C, and Gratton JP (2010). S-nitrosylation of beta-catenin by eNOS-derived NO promotes VEGF-induced endothelial cell permeability. Mol. Cell 39, 468–476. [DOI] [PubMed] [Google Scholar]
- Townsend KL, and Tseng YH (2014). Brown fat fuel utilization and thermogenesis. Trends Endocrinol. Metab 25, 168–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya K, Sakai H, Suzuki N, Iwashima F, Yoshimoto T, Shichiri M, and Hirata Y (2007). Chronic blockade of nitric oxide synthesis reduces adiposity and improves insulin resistance in high fat-induced obese mice. Endocrinology 148, 4548–4556. [DOI] [PubMed] [Google Scholar]
- Tyrrell DJ, Bharadwaj MS, Jorgensen MJ, Register TC, and Molina AJ (2016). Blood cell respirometry is associated with skeletal and cardiac muscle bioenergetics: Implications for a minimally invasive biomarker of mitochondrial health. Redox Biol. 10, 65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg SM, van Dam AD, Rensen PC, de Winther MP, and Lut-gens E (2017). Immune Modulation of Brown(ing) Adipose Tissue in Obesity. Endocr. Rev 38, 46–68. [DOI] [PubMed] [Google Scholar]
- Verma N, Perie L, and Mueller E (2020). The mRNA levels of heat shock factor 1 are regulated by thermogenic signals via the cAMP-dependent transcription factor ATF3. J. Biol. Chem 295, 5984–5994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarroya F, Cereijo R, Gavaldà-Navarro A, Villarroya J, and Giralt M (2018a). Inflammation of brown/beige adipose tissues in obesity and metabolic disease. J. Intern. Med 284, 492–504. [DOI] [PubMed] [Google Scholar]
- Villarroya F, Cereijo R, Villarroya J, Gavaldà-Navarro A, and Giralt M (2018b). Toward an Understanding of How Immune Cells Control Brown and Beige Adipobiology. Cell Metab. 27, 954–961. [DOI] [PubMed] [Google Scholar]
- Wei W, Li B, Hanes MA, Kakar S, Chen X, and Liu L (2010). S-nitrosylation from GSNOR deficiency impairs DNA repair and promotes hepatocarcinogenesis. Sci. Transl. Med 2, 19ra13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Yang Z, Tang CH, and Liu L (2011). Targeted deletion of GSNOR in hepatocytes of mice causes nitrosative inactivation of O6-alkylguanine-DNA alkyltransferase and increased sensitivity to genotoxic diethylnitrosamine. Carcinogenesis 32, 973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen EJ, Foster MW, Matsumoto A, Ozawa K, Violin JD, Que LG, Nelson CD, Benhar M, Keys JR, Rockman HA, et al. (2007). Regulation of beta-adrenergic receptor signaling by S-nitrosylation of G-protein-coupled receptor kinase 2. Cell 129, 511–522. [DOI] [PubMed] [Google Scholar]
- Xiao H, Jedrychowski MP, Schweppe DK, Huttlin EL, Yu Q, Heppner DE, Li J, Long J, Mills EL, Szpyt J, et al. (2020). A Quantitative Tissue-Specific Landscape of Protein Redox Regulation during Aging. Cell 180, 968–983.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, and Chen H (2003). Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Invest 112, 1821–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Liu Z, Gao X, Jin C, Wen L, Yao X, and Ren J (2010). GPS-SNO: computational prediction of protein S-nitrosylation sites with a modified GPS algorithm. PLoS ONE 5, e11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue R, Lynes MD, Dreyfuss JM, Shamsi F, Schulz TJ, Zhang H, Huang TL, Townsend KL, Li Y, Takahashi H, et al. (2015). Clonal analyses and gene profiling identify genetic biomarkers of the thermogenic potential of human brown and white preadipocytes. Nat. Med 21, 760–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Calay ES, Fan J, Arduini A, Kunz RC, Gygi SP, Yalcin A, Fu S, and Hotamisligil GS (2015). METABOLISM. S-Nitrosylation links obesity-associated inflammation to endoplasmic reticulum dysfunction. Science 349, 500–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon YJ, Kim JA, Shin KD, Shin DS, Han YM, Lee YJ, Lee JS, Kwon BM, and Han DC (2011). KRIBB11 inhibits HSP70 synthesis through inhibition of heat shock factor 1 function by impairing the recruitment of positive transcription elongation factor b to the hsp70 promoter. J. Biol. Chem 286, 1737–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotto TM, Quaresma PGF, Guadagnini D, Weissmann L, Santos AC, Vecina JF, Calisto K, Santos A, Prada PO, and Saad MJA (2016). Blocking iNOS and endoplasmic reticulum stress synergistically improves insulin resistance in mice. Mol. Metab 6, 206–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Sierra A, Burnett CM, Chen B, Subbotina E, Koganti SR, Gao Z, Wu Y, Anderson ME, Song LS, et al. (2014). Sarcolemmal ATP-sensitive potassium channels modulate skeletal muscle function under low-intensity workloads. J. Gen. Physiol 143, 119–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.