

Abstract
Mesenchymal stem cells (MSCs) have natural immunoregulatory functions that have been explored for medicinal use as a cell therapy with limited success. A phase Ib study was conducted to evaluate the safety and immunoregulatory mechanism of action of MSCs using a novel ex vivo product (SBI‐101) to preserve cell activity in patients with severe acute kidney injury. Pharmacological data demonstrated MSC‐secreted factor activity that was associated with anti‐inflammatory signatures in the molecular and cellular profiling of patient blood. Systems biology analysis captured multicompartment effects consistent with immune reprogramming and kidney tissue repair. Although the study was not powered for clinical efficacy, these results are supportive of the therapeutic hypothesis, namely, that treatment with SBI‐101 elicits an immunotherapeutic response that triggers an accelerated phenotypic switch from tissue injury to tissue repair. Ex vivo administration of MSCs, with increased power of testing, is a potential new biological delivery paradigm that assures sustained MSC activity and immunomodulation.
Keywords: adult stem cells, cellular therapy, clinical trials, cytokines, immunotherapy, lymphocytes, monocyte, MSCs
Mesenchymal stem cells (MSCs) have natural immunoregulatory functions that have been explored for medicinal use as a cell therapy with limited success. A phase 1b study was conducted to evaluate the safety and immunoregulatory mechanism of action of MSCs using a novel ex vivo product (SBI‐101) to preserve cell activity in patients with severe acute kidney injury (AKI).
Lessons learned
Ex vivo mesenchymal stem cell (MSC) therapy is feasible without any signs of significant safety concerns.
MSC‐secreted factors are detectable after manufacturing, shipping, and product use as a powerful demonstration of a viable cell product.
MSCs respond in a patient‐specific manner as a new paradigm in precision immunotherapy.
A systems biological pharmacodynamic response was observed at multiple physiological levels and included systemic signs of cytokine switching, peripheral immune cell dynamic changes, and local kidney injury marker reductions.
A stratification approach that clustered biological families of biomarkers together demonstrated statistically relevant responses to ex vivo MSC therapy.
Significance statement
Conventional administration of mesenchymal stem cells (MSCs) by intravascular routes has shown low persistence of cells, potentially shortening their therapeutic window. In the cell therapy product described here, SBI‐101, MSCs are immobilized in an ex vivo drug delivery device that circulates the patient's blood akin to a dialysis procedure. Use of this ex vivo cell therapeutic device controls, for first time, the exposure of a patient to MSCs and their immunomodulating secreted factors. Data from this phase I/II trial support the therapeutic hypothesis that ex vivo delivery of MSC‐secreted factors leads to immunomodulation, reprogramming of immune cells, and subsequent protection of kidney injury.
1. INTRODUCTION
Dysregulated, immune‐mediated inflammation is a core component of many conditions that include acute kidney injury (AKI), burn injury, acute respiratory distress syndrome, acute liver failure, sepsis, and coronavirus disease 2019 (COVID‐19). 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 All these conditions have been associated with high morbidity and mortality rates. 9 , 10 A concert of cytokines and components of the innate immune system are the initial mediators of inflammation. 11 , 12 , 13 , 14 Under normal circumstances, these mediators neutralize the original insult and, with a subsequent adaptive immune response, restore homeostasis. 15 , 16 , 17 In critical illnesses, these innate mediators of inflammation may become dysregulated, driving a “cytokine storm” and eliciting an abnormal adaptive immune response that augments and propagates inflammation systemically. 18 , 19 , 20 , 21 A second phase of acute inflammation, mediated primarily by tissue stromal and immune cells, is triggered by the initial inflammatory response and can last for days to weeks. After initial activation of neutrophils and monocytes, a second wave of intraparenchymal infiltration by adaptive immune cells, such as effector memory T cells, mitigates further tissue destruction and ideally begins a tissue remodeling process and accelerates tissue repair. These infiltrating cells are primed by systemic factors before encountering a local tissue microenvironment containing activated macrophages and tissue cytokines. The acute inflammatory response also incorporates counterregulatory components, such as recruited regulatory T cells and induced anti‐inflammatory cytokines (eg, interleukin [IL]‐10). 22 , 23 , 24 These mediators are key regulators in the phenotypic switch from tissue injury to tissue repair. Thus, rebalancing this multifocal inflammatory response may be a potential approach to restoring underlying organ function after severe injury.
Mesenchymal stem cells (MSCs) secrete several types of molecules (eg, lipids, cytokines, chemokines, and exosomes) that collectively modulate an immune cell response to inflammation. 25 , 26 , 27 Through a multitude of in vitro and animal studies, they have been shown to module just about every circulating immune cell but are well characterized for their ability to inhibit T‐cell activation, induce regulatory T cells, promote T helper cell type 1 (Th1)/type 2 (Th2) switching, and polarize monocytes/macrophages toward a regulatory M2 phenotype. 28 After decades of MSC research, an allogeneic MSC product finally received marketing approval in 2018 for the local treatment of complex perianal fistulas in Crohn's disease. 29 More recently, systemic administration of allogeneic MSC treatment in pediatric patients with acute graft vs host disease showed a significant improvement in survival rate compared with historical controls and a sustained therapeutic effect at 6 months after treatment (NCT02336230), although no therapeutic biomarkers were clearly associated with the response. 30 In addition, the potential applicability of these cells to treat sequalae of COVID‐19 infection has introduced a new indication for MSCs in systemic immunoregulation. 31 , 32 , 33 Nonetheless, conventional administration of MSCs by intravascular routes has shown low persistence of cells and may have the potential to induce clotting events. 34 These pharmacological barriers manifested by a short and uncontrolled exposure of a patient's blood to MSCs and their secreted factors have likely contributed to the lack of efficacy in several clinical studies with the intravascular administration of MSCs. 34 , 35 , 36 , 37
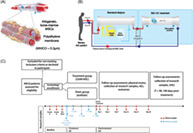
To control viability and exposure to MSCs and their secreted factors, while simultaneously reducing safety risks of thromboembolic events and pulmonary injury due to systemic infusion, SBI‐101 was developed as an ex vivo MSC product that immobilizes allogenic bone marrow‐derived MSCs, at a target cell number, on the extracapillary space of a hollow fiber hemofiltration device. Circulating blood cells passing through the hollow fibers are exposed to MSC‐secreted factors through a high pore size plasmapheresis membrane (Figure 1A). This configuration enables controlled, extended exposure of the full MSC dose and facilitates cross‐membrane, bidirectional communication between the MSCs and the patient's blood circulating in the intracapillary space of the filter. SBI‐101 recapitulates the in vivo three‐dimensional microenvironment of MSCs, that is, pericytes in capillary vessels. 38 SBI‐101 is designed to operate in the context of extracorporeal therapy and can be used in tandem with other ex vivo devices such as renal replacement therapy (RRT) used in AKI (Figure 1B).
FIGURE 1.
Schematic illustration of SBI‐101‐01. A, Schematic of cell membrane interaction with blood flow. B, Extracorporeal circuit with integrated SBI‐101. The configuration enables in situ monitoring via extracapillary sampling. C, Study design. D, Sampling for exploratory endpoint analysis. AE, adverse event; AKI, acute kidney injury; AKI‐D, dialysis‐dependent acute kidney injury; CRRT, continuous renal replacement therapy; MSC, mesenchymal stem cell; MWCO, molecular weight cutoff; PD, pharmacodynamic; PK, pharmacokinetic; SAE, serious adverse event; T, time
Comprehensive preclinical testing of SBI‐101 in vitro 39 , 40 and in animal models of disease 41 , 42 , 43 has shown that the product is safe and efficacious in various models of systemic inflammatory disease. We hypothesized that MSC immunoregulation via SBI‐101 may be of therapeutic value in dialysis‐dependent AKI (AKI‐D), wherein initial kidney parenchymal cell loss is exacerbated by a local and systemic inflammatory response. To evaluate this hypothesis, a phase I/II, multicenter, randomized, sham‐controlled, double‐blind study of SBI‐101 in subjects with AKI receiving continuous renal replacement therapy (CRRT) was initiated, and an interim analysis of the group receiving an active dose (250 × 106 cells per SBI‐101) was compared with the sham‐treated group (NCT03015623).
2. MATERIALS AND METHODS
2.1. Study design
This study (SBI‐101‐01, NCT 03015623, IND 17204) was designed as a multicenter (conducted in 12 centers in the United States), randomized, double‐blind, sham‐controlled, multiple ascending dose, parallel‐group study investigating the safety, tolerability, and pharmacology of SBI‐101 in adult subjects with AKI‐D while on CRRT. The design of the study is shown in Figure 1C. Sixteen subjects were meant to be randomized, in a 2:1 ratio, to an active low dose of SBI‐101 (250 × 106 MSCs) or to a sham (no MSCs). However, the study randomized 12 patients to active treatment and four to sham treatment because the protocol allowed replacement of subjects to the same treatment allocation who do not complete a minimum of 12 hours of treatment. Use of a sham device reduces bias in interpreting data, particularly during evaluation of safety parameters possibly related to the study drug. The study captured general and specialized safety assessments and pharmacological parameters in plasma and urine.
The study population to be treated in this study included those who had been diagnosed with AKI of any etiology not specifically excluded in the exclusion criteria and had been, in the Investigator's opinion, stable for at least 12 hours after commencement of CRRT and were likely to require CRRT for an additional 48 hours. Complete inclusion and exclusion criteria have been previously described. 44 All subjects or legally acceptable representatives provided written informed consent. The study population for analysis included the Safety Analysis Set, which consisted of all subjects who signed the informed consent and were exposed to the investigational agent.
2.2. Investigational agent and its administration
The investigational agent was SBI‐101. Allogeneic MSCs are the active ingredient (drug substance) in SBI‐101 (Figure 1A). The MSCs are derived from the bone marrow of a healthy donor and processed into a master cell bank and working cell banks, in compliance with Good Manufacturing Practice standards for biologics and cellular therapies. SBI‐101 (drug product) is a combination of the allogeneic cells and a Food and Drug Administration‐approved hollow fiber plasma separator. Cells are immobilized onto the extracapillary surface of the polyethylene hollow fibers. The hollow fibers are contained within a transparent housing.
The sham is the hollow fiber plasma separator but devoid of cells.
SBI‐101 was administered in sequence with a NxStage System One (NxStage Medical Inc., Lawrence, Massachusetts) or Baxter Prismaflex system (Baxter, Deerfield, Illinois), each configured for CRRT (Figure 1B). To be part of the per protocol analysis set, subjects must have remained on SBI‐101 therapy or sham for at least 12 and up to 24 hours (exclusive of rinse‐back procedures). Subjects receiving less than 12 hours of treatment may have been replaced. All subjects exposed to SBI‐101 or sham were evaluated in the Treated Analysis Set.
SBI‐101 was integrated with the NxStage System One or Baxter Prismaflex system immediately after priming the NxStage System One circuit or Baxter Prismaflex circuit and before removing air bubbles from the lines. At the end of treatment, the subject's blood was returned prior to the subject's removal from the CRRT circuit.
2.3. Assessments of safety
All safety analyses were performed on the Safety Analysis Set. Subjects who were randomized but did not receive SB‐101 were considered screen failures. Study populations for analysis included the following: (a) Safety Analysis Set: The Safety Analysis Set consisted of all subjects who signed the informed consent and were exposed to SB‐101. All safety analyses were performed on the Safety Analysis Set. Subjects who were randomized but did not receive SB‐101 were considered screen failures. (b) Treated Analysis Set: The Treated Analysis Set consisted of all subjects who received SB‐101 or sham control, whether or not they completed the full 12 hours of treatment. All analyses of efficacy and exploratory endpoints were performed on the Treated Analysis Set. (c) Per Protocol Set: The Per Protocol Set consisted of all subjects who received SB‐101 for a minimum of 12 hours and had no important protocol deviations. The safety and tolerability measurements included adverse events, physical examination findings, vital signs, electrocardiograms, and laboratory tests. Results were presented separately by treatment phase (pre‐CRRT/investigational therapy, during CRRT/Investigational therapy, on CRRT/off investigational therapy, off CRRT/investigational therapy) and by group and treatment group. Sample collection was performed at clinical sites as previously described. 44
2.4. Assessment of efficacy and exploratory endpoints
All analyses of efficacy and exploratory endpoints were performed on the Per Protocol Set. Two additional sham‐treated subjects whose treatments ran for 10.5 and 3 hours were also included since this short time of perfusion with the empty filter should not affect efficacy and pharmacological analysis. Hence, four patients were considered in each group.
Samples were collected as shown in Figure 1C. Sample testing using a multiplex immunoassay method for proteins detected in the extracapillary compartment of the product or in‐patient plasma samples were collected in a plasma preparation tube (362 788; BD Biosciences, San Jose, California) performed at Eve Technologies (Calgary, Canada) (Table S3). Flow cytometry testing of patient leukocytes was performed by ReachBio (Spokane, Washington). Blood was collected in CPT tubes (362 753; BD Biosciences), and peripheral blood mononuclear cells (PBMCs) were isolated and cryopreserved at a concentration of 2 million per milliliter. Six panels were used for each sample (Table S4).
2.5. Analysis
SBI‐101 (active) dose groups were compared against the sham group using unpaired, nonparametric analyses for data not normally distributed, a Wilcoxon rank sum test for ordinal outcomes, and Fisher's exact test for binary outcomes. Within‐dose comparisons (using the subject as his or her own control) were evaluated using a paired t test for continuous endpoints and using McNemar's paired comparison test for changes from baseline for binary outcomes. An exact binomial test was used to compare overall rates for safety, tolerability, and 28‐day survival outcomes relative to a predetermined acceptability standard. All statistical tests were two‐sided with a .05 significance level.
3. RESULTS
3.1. Demographics and safety
A total of 16 subjects were enrolled in the study: 12 in the SBI‐101 active (250 × 106 MSCs) group and 4 in the sham group (study design is depicted in Figure 1C; other information such as inclusion and exclusion criteria has been previously described 44 ). The primary cause of AKI‐D in 13 of the 16 subjects enrolled was ischemia reperfusion injury (10 active and 3 sham). In the remaining three subjects, sepsis was the cause of AKI‐D (two active and one sham). Among the 13 subjects with ischemia reperfusion injury, nine subjects had major surgery (cardiac, vascular, abdominal) leading to AKI‐D (six active and three sham). The demographics of subjects in the study were well balanced for age, gender, and body mass index within the constraints of the numbers of subjects in each group (Table S1).
The Per Protocol Set included six subjects (four active and two sham) who received SBI‐101 for a minimum of 12 hours and had no significant protocol deviations. The remaining 10 participants, 8 of 12 (67%) in the active group and 2 of 4 (50%) in the sham group, were each disconnected from SBI‐101 treatment prior to 12 hours of treatment, although all continued with RRT as per standard of care. Most SBI‐101 discontinuations were due to circuit clotting. Although not uncommon in clinical practice, 28 the frequency of circuit clotting in this study significantly limited the number of subjects evaluable for treatment effects. The protocol did not require anticoagulation, but principal investigators had the option to administer anticoagulation if clinically indicated. Notably, the four Per Protocol Set subjects in the active arm all received anticoagulation during treatment and successfully completed the full 24 hours of treatment.
There were no serious and unexpected suspected adverse reactions or unanticipated adverse device effects reported during the study. The mortality rate was consistent with what has been reported in the literature for AKI. 45 No deaths or serious adverse event were considered related to the investigational agent as determined by the principal investigators, the Safety Committee, and the Data Safety Monitoring Board. Patient deaths were attributed to baseline conditions or complications experienced during their hospital stay, not unexpected in a critically ill population with significant comorbidities. Based on the data available to date, no overt safety signals have been identified. Pharmacokinetic (PK) and pharmacodynamic (PD) analyses was performed on subjects in the Per Protocol Set who were treated with low‐dose SBI‐101 for 24 hours (n = 4) or sham‐treated subjects (n = 4). Table S1 summarizes subjects and their treatment allocations, including exposure time to SBI‐101 and baseline measurements of inflammation and kidney failure.
3.2. Confirmation of SBI‐101 bioactivity prior to treatment
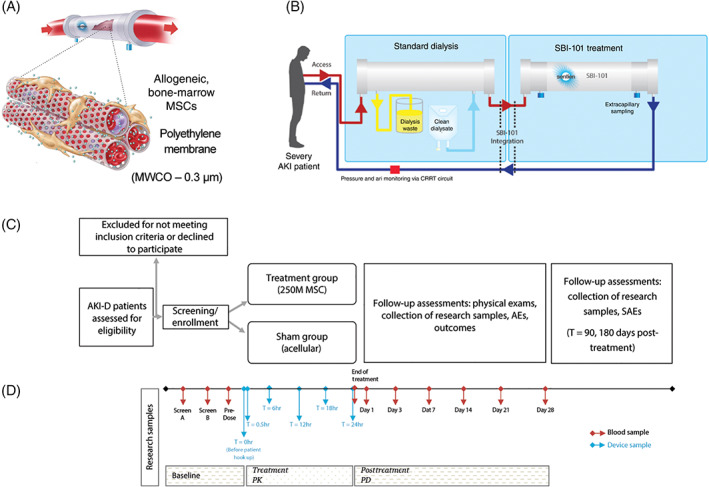
SBI‐101 uniquely allows for sampling to assure the activity and consistency of the cell therapy product by indirectly measuring cell‐specific factors. A sample from the extracapillary (EC) space was taken just prior to participant integration and was multiplexed to evaluate if MSC‐secreted function was still intact after product manufacturing and distribution to sites. Analytes with significant differences between active SBI‐101 (n = 11) and sham (n = 3) are shown in Figure 2A. Fractalkine, platelet‐derived growth factor AA (PDGF‐AA), transforming growth factor (TGF)‐β1, fibroblast growth factor 2 (FGF‐2), growth‐regulated oncogene (GRO) alpha (C‐X‐C motif chemokine ligand [CXCL] 1), IL‐6, IL‐7, IL‐8, monocyte chemoattractant protein (MCP)‐1 (C‐C motif chemokine ligand [CCL] 2), vascular endothelial growth factor A (VEGF‐A), and TGF‐β2 were all found to be present in SBI‐101 containing 250 × 106 MSCs, indicating that cells are active and secreting proteins between the time they are seeded in SBI‐101 and when SBI‐101 is integrated to the patient. Not surprisingly, all these factors have been previously described as present in the MSC secretome. 25 , 46 , 47 All factors were then compared with viability at inoculation to investigate the potential correlation of cell viability and analyte levels at product baseline. High correlations were found between viability and levels of MCP‐1 (CCL2), TGF‐β1, and VEGF‐A (Figure 2B). All together, these results provide important insights into the quality and consistency of SBI‐101 manufacturing and establish a baseline for characterizing the response of MSCs to patient‐specific inflammatory profiles.
FIGURE 2.
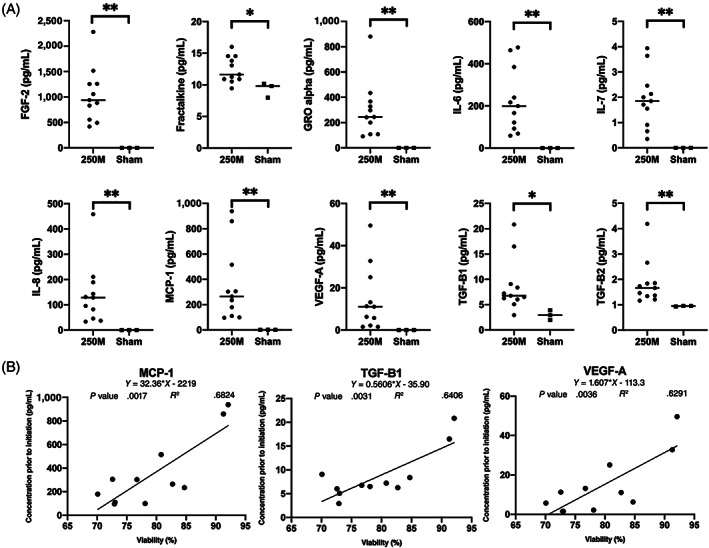
Mesenchymal stem cell (MSC) factors measured in the extracapillary space of SBI‐101 prior to initiation of therapy. A, Extracapillary samples collected from SBI‐101 prior to patient therapy displayed significant increased levels of cytokine and growth factor secretion in the low‐dose (250 × 106 MSCs) SBI‐101 (n = 11) compared with sham (n = 3). Fractalkine, TGF‐β1 (*P < .05), FGF‐2, GRO alpha (C‐X‐C motif chemokine ligand 1), IL‐6, IL‐7, IL‐8, MCP‐1 (C‐C motif chemokine ligand 2 [CCL2]), VEGF‐A, and TGF‐β2 (**P < .01) were detected in the seeded SBI‐101 devices. B, Correlation of MSC viability at seeding of SBI‐101 to that of MCP‐1 (CCL2), TGF‐β1, and VEGF‐A were high with calculated R 2 values of 0.68, 0.64, and 0.63, respectively. Data are presented as scatter plots with means ± SD. Nonparametric Mann‐Whitney unpaired test was used for statistical analysis (*P < .05; **P < .01). 250M, 250 × 106 MSCs; FGF‐2, fibroblast growth factor 2; GRO alpha, growth‐regulated oncogene alpha; IL, interleukin; MCP‐1, monocyte chemoattractant protein 1; TGF, transforming growth factor; VEGF‐A, vascular endothelial growth factor A
3.3. PK observations of patient‐specific MSC activity
Pharmacokinetics is typically known as the study of the bodily absorption, distribution, metabolism, and excretion of a drug, simplistically described as “what the body does to the drug.” Assessment of pharmacokinetics in cellular therapies is problematic because the “drug,” (ie, MSCs) cannot be retrieved and assayed after infusion. In SBI‐101, MSCs are immobilized in an extracorporeal cartridge so that the drug is in a concentrated location that can be assayed and even retrieved after patient use. Sampling the extracapillary space in SBI‐101 allows for direct measurement of plasma ultrafiltrate during treatment and hence offers a unique way of interrogating how the body is affecting the drug. Measurements of MSC activity from within SBI‐101 assessed during treatment (30 minutes, 6 hours, 12 hours, 18 hours, and 24 hours). Several MSC‐secreted markers increased over time compared with sham (Figure 3A). MSCs within SBI‐101 were secreting proteins in a dynamic fashion, including IL‐6, IL‐8, MCP‐3 (CCL7), and epithelial‐derived neutrophil‐interacting protein 78 (CXCL5). Given the measurement of human MSC factors in a background of human patient plasma, the source of these proteins cannot be definitively established. However, the delta between active SBI‐101 therapy and sham devices as a negative control provides confidence that these factors were likely produced by MSCs. Importantly, factors enriched in the EC space during treatment did not necessarily equate to increased plasma levels of such factors. This is well illustrated by IL‐6, a factor that clearly increased inside SBI‐101 during treatment, whereas participant plasma levels were decreased after treatment (Figure S1). These results suggest that SBI‐101 creates a local, concentrated environment of MSC‐secreted factors that are exposed to circulating immune cells within the device, much like an artificial tissue environment.
FIGURE 3.
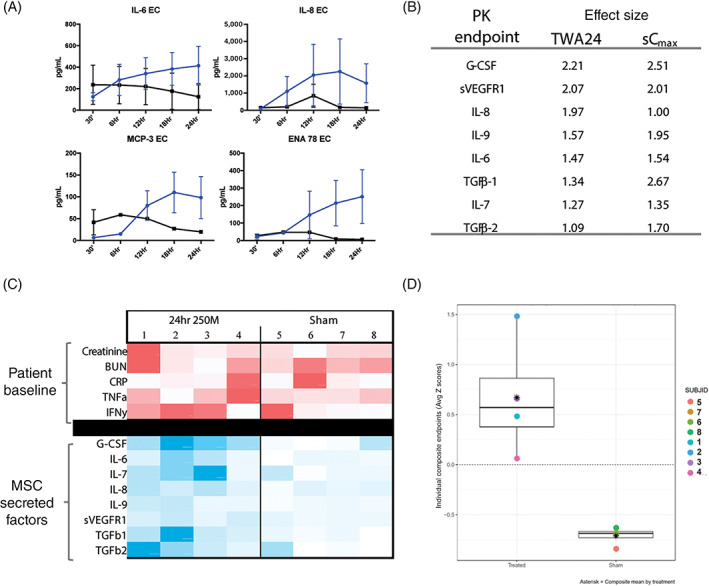
Pharmacokinetic analysis of SBI‐101 treatment shows sustained and subject‐specific exposure to mesenchymal stem cell (MSC)–secreted factors (n = 4 per group). A, Multiplex immunoassay measurements of MSC‐secreted factors in SBI‐101 (blue) compared with sham (black). B, PK biomarkers with effect sizes greater than one (absolute value). C, Participant‐specific analysis of MSC‐secreted factors as a function of patient baseline phenotype. A heatmap was created using measurements of baseline inflammatory and kidney function markers (red) and TWA24 for the secreted factors during therapy (blue). Comparative analysis of intensities was calculated within each row with darker colors representing larger values. D, Standardized TWA24 Z‐scores in treated and sham subjects for PK composite: G‐CSF, IL‐6, IL‐7, IL‐8, IL‐9, sVEGFR1. Differences between sham and treated subjects can be easily detected as the full range of Z‐scores observed among treated patients was positive. 250M, 250 × 106 MSCs; BUN, blood urea nitrogen; CRP, C‐reactive protein; EC, extracapillary; ENA 78, epithelial‐derived neutrophil‐interacting protein 78; G‐CSF, granulocyte colony‐stimulating factor; IFNγ, interferon γ; IL, interleukin; MCP‐3, monocyte chemoattractant protein 3; PK, pharmacokinetic; sCmax, maximum concentration; sVEGFR1, soluble vascular endothelial growth factor receptor 1; TGF, transforming growth factor; TNFα, tumor necrosis factor α; TWA24, time‐weighted average over 24‐hour treatment period
Each biomarker collected from the EC space of SBI‐101 during the study was quantified using traditional pharmacokinetic methods to identify a maximum concentration (C max) or the time‐weighted average (TWA). Because it was hypothesized that SBI‐101 had the potential for modulating multiple biological pathways, these two metrics were chosen to provide an enhanced understanding of changes in potential biomarkers. C max represented a maximum concentration of biomarker observed over the complete dosing interval. TWA24 provided an average concentration of a given biomarker in SBI‐101 over a 24‐hour treatment period.
Effect sizes and Z‐scores were used to characterize differences between active and sham groups for each PK biomarker. Biomarkers were identified as “contenders” (i.e., of potential relevance) if the effect size was shown to be greater than one for both C max and TWA. The top contenders with the highest effect sizes for pharmacokinetics are shown in Figure 3B. Not surprisingly, these were factors typically associated with the MSC secretome: granulocyte colony‐stimulating factor (G‐CSF), soluble vascular endothelial growth factor receptor 1 (sVEGFR1), IL‐8, IL‐9, IL‐6, TGF‐β1, IL‐7, and TGF‐β2. Of note, neither metric of exposure (TWA24 and C max) appeared to be substantially more sensitive to differences between the active and sham groups. This finding could be due to small sample sizes and truncated sampling schemes rather than physiological phenomena. Given that TWAs carry information from repeated within participant samples, whereas C max is obtained from a single measure and is highly influenced by sample collection time, preferential focus was placed on TWA. The TWA24 for each of the factors listed above was analyzed in a heatmap showing TWA24 values for each patient (Figure 3C). A heatmap was also created with baseline plasma levels for creatinine, blood urea nitrogen, C‐reactive protein, tumor necrosis factor α (TNFα), and interferon γ (IFNγ) for each participant. Put together, each participant background was unique with differing levels of disease and inflammation. Separately, the MSC response also showed a unique profile for each participant, suggestive of a “personalized” therapy. Overall, these results suggest that SBI‐101 therapy is working as hypothesized, where (a) known MSC factors are being secreted continuously in SBI‐101 and (b) MSCs are dynamically responsive to participant‐specific inflammatory signals. In addition, PK analysis also verified a panel of factors that were increased in active SBI‐101 treatment, which can be followed in future clinical studies. Indeed, a putative composite PK endpoint comprising G‐CSF, IL‐6, IL‐7, IL‐8, IL‐9, and sVEGFR1 was identified, showing a clear separation between TWA24 in the active vs sham groups (Figure 3D), which could be used in subsequent studies to further understand PK/PD relations. These data strongly support SBI‐101 as a combinatorial biological delivery system, controlling the exposure of multiple cytokines and chemokines in a local systemic blood compartment.
3.4. PD multicytokine modulation after MSC therapy
Molecular and cellular biomarkers were tested as exploratory endpoints to study the PD changes associated with SBI‐101 treatment. Peripheral blood sampling was performed during screen A, screen B, predose baseline, and post‐treatment days 1, 3, 7, 14, 21, and 28. The start of PD exposure was defined as 24 hours after initiation of treatment. PD areas under the curve (AUCs) characterized the total change in exposure at discrete intervals, starting with day 1, and were increased incrementally up until the end of sample collection on day 28. PD TWAs represented the TWA change in concentration over the same collection intervals. Differences between TWAs and AUCs were not substantive; however, TWAs allowed pooling of endpoints that may have had different collection periods and were therefore used preferentially over AUCs. Although assessments continued beyond 7 days of treatment, the most dynamic period occurred in the immediate week following treatment; although AUCs and TWAs were generated that represented greater periods of time, the earlier assessments were therefore considered most relevant with regard to detecting PD effects. As before, effect sizes and Z‐scores were used to characterize differences between active and sham groups for each PD biomarker. Absolute values of effect sizes were generated for each biomarker. A biomarker was considered a contender if the absolute value for the effect size was greater than or equal to one for both ratios of log‐transformed values over baseline and TWA. A list of “top contender” PD cellular and molecular biomarkers is shown in Table S2.
Known pro‐inflammatory markers such as TNFα and IFNγ remained low or decreased in the active group, whereas corresponding values in the sham group continued to increase (Figure 4A). For known anti‐inflammatory markers IL‐10 and TGF‐β1, the reciprocal response occurred, suggesting that the presence of SBI‐101 has an inflammatory mediating response (Figure 4B). Concordantly, the ratio of IL‐10 to TNFα showed a high effect size that was above one in every assessment through day 7 (Table S2). All these changes are consistent with MSC biology. 48 , 49 , 50 Decrease in TNFα levels at day 3 after treatment inversely correlated with time to treatment (Figure S2), suggesting that treating patients earlier in their injury may show stronger PD effects and ultimately better therapeutic efficacy. Overall, molecular biomarker analysis was consistent with the therapeutic hypothesis and MSC mechanism of action. Trends appeared to be somewhat transient, and further analysis with higher doses and with increased number of subjects after day 7 after treatment will be needed to further evaluate the duration of PD effect.
FIGURE 4.
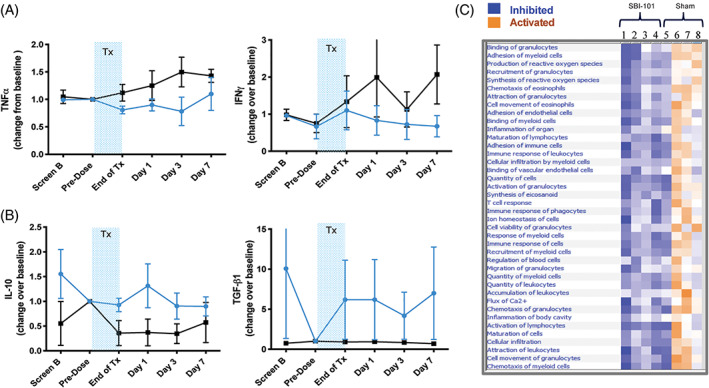
Pharmacodynamic effects in plasma of patients after treatment with SBI‐101. Plasma measurements of, A, pro‐inflammatory markers TNFα and IFNγ and, B, anti‐inflammatory markers IL‐10, TGF‐β1 in treated (blue) and sham patients (black). Values represent the mean at each time point of the change from baseline (predose values or screen B in the case predose was not available) for each biomarker ± SEM (n = 4 per group). C, Unsupervised pathway analysis of patient response at day 3 normalized to baseline. Disease and function analysis comparison heatmap was sorted by hierarchical clustering based on sample activation score. The activation score (Z‐score) predicts direction of potential regulators assessing the match of observed and predicted upstream or downstream processes. Z‐score serves as both a significance measure and a predictor for the activation state of the regulator (orange, +/activation, blue, −/inhibition). IFNγ, interferon γ; IL‐10, interleukin‐10; TGF, transforming growth factor; TNFα, tumor necrosis factor α; Tx, treatment
To examine these exploratory data from a systems biology perspective, pathway analysis was performed on the analyte data collected from the plasma of subjects in both groups. Analytes at day 3 were normalized to predose baseline levels and entered into the Ingenuity Pathway Analysis software. The comparison analysis for subjects with active SBI‐101 treatment (observations 1‐4) vs sham treatment (observations 5‐8) was performed using high stringency conditions (using cutoff −1.2 [down] and +1.2 [up]) and based on trend and Z‐score, and a resulting heatmap was generated (Figure 4C). Results showed a distinct clustering of SBI‐101‐treated compared with sham‐treated subjects. SBI‐101‐treated subject profiles suggested broad spectrum inhibition of immune‐mediated pathways (blue), whereas sham‐treated subjects showed most of those same pathways as activated (orange). This unbiased analysis supports the notion that SBI‐101 is modulating the immune system broadly, a key feature of cell therapy that is distinct from single‐factor agents. Intriguingly, one subject profile in the sham group (patient 5 in Figure 4B) resembled the SBI‐101‐treated subjects more so than the other sham‐treated subjects. After unblinding the study, we learned that this subject presented with septic shock that, during the treatment period, was in a resolving phase. Anecdotally, this control group subject thus provided the profile of a patient with AKI with naturally resolving systemic inflammation from sepsis. This profile was similar to those treated with SBI‐101, supporting the hypothesis that SBI‐101, through broad immunomodulation, is resolving inflammation and accelerating healing and tissue repair.
3.5. Peripheral immune cell dynamics and associated chemokines are altered after MSC therapy
In addition to measuring molecular biomarkers, PBMCs from each subject were collected for immunophenotypic analysis. Changes were observed in different populations and subsets (Table S2), with robust changes at the level of monocytes. Preclinical testing of SBI‐101 in animal models suggested that monocytes might be decreased with treatment, consistent with findings from other groups. 51 Here, quantification of each monocyte subpopulation following clinical application of SBI‐101 suggests that this finding is also consistent in humans. Both classic and intermediate populations decreased, whereas the nonclassic population did not change (Figure 5A). This decrease in monocyte populations manifested in a pharmacodynamic response with known decreases in monocyte chemoattractants MCP‐1 (CCL2), MCP‐2 (CCL8), and MCP‐3 (CCL7) over time and with greater effect than in the sham group (Figure 5B). These results were also consistent with pathway analysis focused on monocyte‐related diseases showing inhibition with low‐dose SBI‐101 treatment monocyte‐related pathways (Figure S3). Specific subsets of B cells (unswitched memory and marginal zone) were found to be increased after treatment, and other trends in T‐cell, natural killer cell, and dendritic cell subsets were also noted (Table S2). These preliminary findings will need to be further validated with larger sample size and supporting blood differential test results.
FIGURE 5.
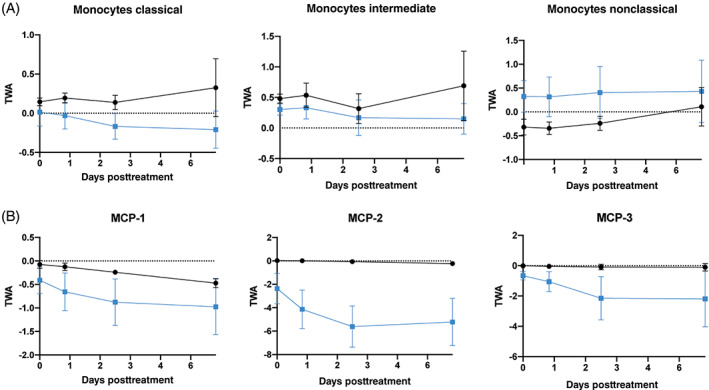
SBI‐101 effect on circulating monocytes. A, Flow cytometric analysis of different monocyte populations in patient peripheral blood mononuclear cells after treatment with SBI‐101 (blue) or sham (black). B, Quantification of monocyte chemoattractants MCP‐1 (C‐C motif chemokine ligand [CCL] 2), MCP‐2 (CCL8), and MCP‐3 (CCL7). Results are represented as TWAs ± SEM (n = 4 per group). MCP, monocyte chemoattractant protein; TWA, time‐weighted average
3.6. Stratification of SBI‐101 responses based on composite endpoints reveals multifocal therapy
A systems biology view of molecular and cellular PD biomarkers with high effect sizes illustrates a therapy that affects both the adaptative and innate immune system, increasing Th2 anti‐inflammatory markers (eg, IL‐10, TNFα, IL‐13) while decreasing pro‐inflammatory or Th1 cytokines (IFNγ, TNF‐related apoptosis‐inducing ligand [TRAIL], and TNFα) (Figure 6A). Importantly, certain kidney injury markers (kidney injury molecule 1 [KIM‐1] and osteoactivin) were also found to be decreased with an effect size greater than one. In order to enhance the detectability of effect sizes, pooling of PD endpoints into composite endpoints was also performed. Although PD composites were enhanced via purely mathematical approaches, the most robust effect sizes were observed when composites were created based on biological validity (Figure 6B). The effect sizes over time of all six PD groupings are plotted in Figure 6C. Pooling PD biomarkers to form aggregate endpoints increased the observed effect sizes for differences between the active and sham groups. Two groups, the monocyte/macrophage group and the “high effect size” group, had effect sizes greater than four for the first 3 days after treatment, indicating that the onset of effect was rapid and highly detectable. Moreover, the differentiation from sham was sustained in both PD groupings for the full 28 days following treatment.
FIGURE 6.
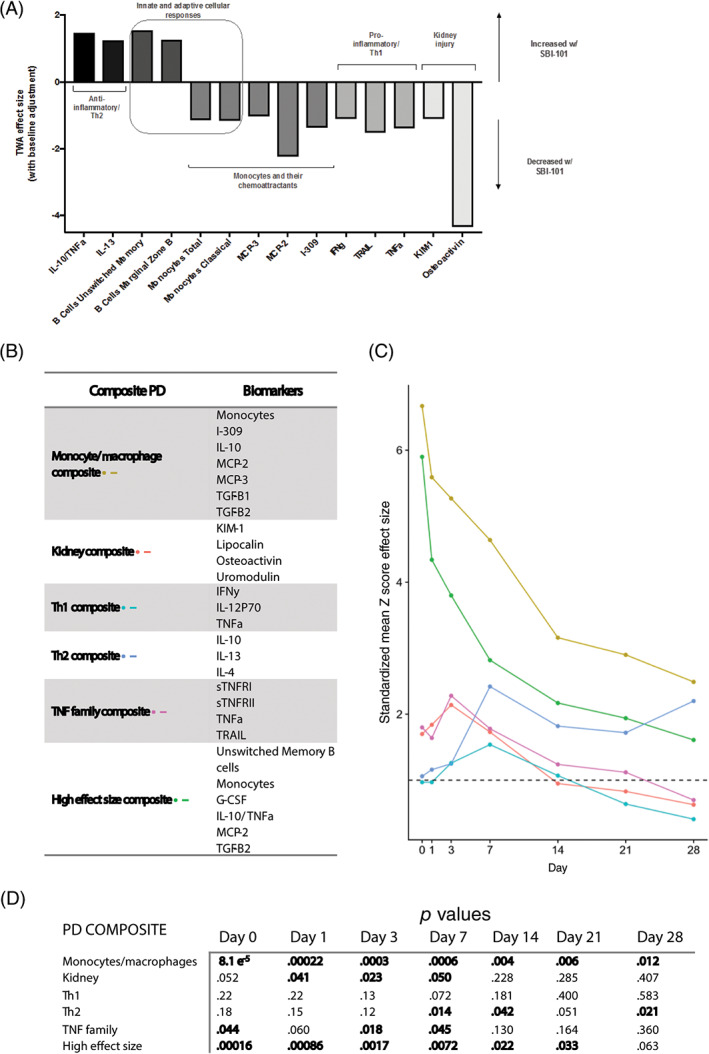
Effect sizes of individual and composite pharmacodynamic markers. A, SBI‐101 induced changes in both molecular and cellular parameters with effect sizes greater than one. B, Composite groupings were created based on both the magnitude of effect size and the scientific plausibility of each combination. C, Composite scores were generated for each subject. Composite Z‐scores were the average Z‐score across markers within a composite group, for each subject and time point. Composite endpoints were generated for each treatment group by averaging the individual composite Z‐scores in each group. Finally, composite endpoints were used to generate composite effect sizes, calculated as the difference between treated and control composite endpoints divided by the pooled SD. D, Values of P from a two‐tailed t test performed for each group at each observation day after treatment. G‐CSF, granulocyte colony‐stimulating factor; I‐309, inflammatory cytokine I‐309; IFNγ, interferon γ; IL, interleukin; KIM‐1, kidney injury molecule 1; MCP, monocyte chemoattractant protein; PD, pharmacodynamic; sTNFR, soluble TNF receptor; TGF, transforming growth factor; Th1, T helper cell type 1; Th2, T helper cell type 2; TNF, tumor necrosis factor; TRAIL, TNF‐related apoptosis‐inducing ligand; TWA, time‐weighted average
Although the largest effect sizes were observed within the first 7 days following the end of treatment, the P values for each composite endpoint (Figure 6D) showed some significant differences between the active and sham groups up to day 28. In fact, P values for the monocyte/macrophage group were significant between active and sham at every time point measured. The “high effect size group” that comprises a mixture of B‐cell subsets, monocytes, G‐CSF, IL‐10: TNFα, MCP‐2 (CCL8), and TGF‐β2 also showed statistical significance up to day 21. A composite based on the TNFα family members (soluble TNF receptors I and II, TNFα, and TRAIL) decreased significantly in active subjects on day 0 (end of 24 hours of treatment), day 3, and day 7, supporting the therapeutic hypothesis that SBI‐101 therapy reduces TNFα levels in the subject. Furthermore, a composite made of IL‐10, IL‐13, and IL‐ 4 was significantly increased at day 17 and day 14, suggesting that there may have been a switch from a Th1 to a Th2 type of response. Finally, a composite consisting of kidney‐related markers such as KIM‐1, lipocalin, osteoactivin, and uromodulin was found to be statistically significant between active and sham groups at day 0 up to day 3 and day 7 (P = .05), suggestive of kidney repair.
Overall, PD effects as measured by individual molecular and cellular biomarkers as well as by composite endpoints were consistent with MSC biology and were indicative of a switch from a pro‐inflammatory to an anti‐inflammatory response that leads to accelerated organ healing and repair. These data also supported the hypothesis that ex vivo MSC therapy via SBI‐101 modulates both adaptive and innate immune cells, as evidenced by changes at the levels of both B and T cells as well as monocytes and dendritic cells. Further studies are needed to increase the sample size and investigate the impact of higher doses of MSCs.
4. DISCUSSION
AKI‐D has a reported mortality rate of 50% to 70%. 52 , 53 Current treatments, such as RRT, are supportive and fail to address the underlying inflammatory processes. Inflammation is an important contributor to mortality and resistance to treatment among patients with AKI. 2 , 3 , 54 In AKI‐D, as in many other critical injuries, no single disease‐targeted therapy exists. Here we describe preliminary data from our phase I/II study in AKI‐D where we tested a novel cell therapy product designed to regulate systemic inflammation and promote organ repair.
The therapeutic hypothesis of SBI‐101 is that broad reprogramming of an immune response associated with systemic inflammation restores homeostasis and accelerates tissue repair, leading to a beneficial clinical outcome. SBI‐101 is designed to behave as a self‐sensing and regulating “drug” delivery product where the pharmacological load consists of a complex secretome of cytokines and related molecules. Sampling directly from an ex vivo MSC compartment prior to the patient's connection enabled the first characterization of a complex MSC secretome in humans, which consisted of molecules such as fractalkine, PDGF‐AA, TGF‐β1, FGF‐2, GRO alpha (CXCL1), IL‐6, IL‐7, IL‐8, MCP‐1 (CCL2), VEGF‐A, and TGF‐β2. Further analysis of these types of samples, for example, at the level of extracellular vesicles (EVs), will continue to contribute to product consistency and product characterization. Using a miniaturized version of SBI‐101 ex vivo, MSC‐derived EVs have been shown to be altered in size in the presence of inflammatory stimuli. 40 The same study showed that MSCs in the bioreactor modified PBMC‐secreted factor milieu both directly through cell‐secreted factors and indirectly via altered EV characteristics. The role of EVs in the mechanism of action of ex vivo MSC therapy requires further studies.
The study enrolled subjects with severe and life‐threatening medical conditions. A substantial adverse event profile was expected for such a critically ill population with significant comorbidities. No serious adverse events were considered related to SBI‐101, but a substantial number of subjects did not complete 24 hours of therapy because of discontinuations, mostly secondary to clotting. Since anticoagulation is routinely used in CRRT, Sentien Biotechnologies is now mandating anticoagulation therapy for all future enrolled subjects.
At this early stage of development, the primary objective of the study was to assess the safety and tolerability of SBI‐101 in patients with AKI‐D. With a small sample size, the study was not powered for clinical efficacy. The small sample size coupled with the complexity and seriousness of the patients' current health conditions significantly contributed to the limitations of this early data set. As this study continues, efforts (such as requiring anticoagulation therapy for study subjects and potentially harmonizing enrollment based on severity of disease upon randomization) will be important to evaluate treatment effect on standard AKI endpoints.
Pharmacological data were obtained from four treated and four sham subjects, which allowed an initial PK/PD analysis. Results demonstrated that SBI‐101 is active; known MSC‐secreted factors were measured well above sham levels during the 24 hours of treatment. The secretome measured during treatment was found to have a different composition from patient to patient. The differences in composition did not correlate with baseline variability of each product. Comparison of gene expression profiles of the resulting MSCs with control MSCs prior to treatment would corroborate these secretome data. Unfortunately, because of the biohazard nature of the device after treatment, we were not able to recover the product for post hoc cellular analysis. These data support a core component of the therapeutic hypothesis for SBI‐101, namely, that the crosstalk between the drug substance (MSCs) and the subject's blood cells leads to MSCs sensing and reacting to their environment 55 throughout SBI‐101 treatment. Furthermore, given the close proximity of MSCs to immune cells within the device, short‐lived factors such as lipids that cannot diffuse far can still remain bioactive.
Unlike the variability observed in the PK response, PD effects were fairly conserved across the treated subjects despite the low number. The hallmarks of MSC biology were indeed observed in our study, as demonstrated by decreases in TNFα levels and increases in IL‐10 levels. 48 The reduction of Th1 cytokines (such as TNFα and IFNγ) and increase in Th2 cytokines (such as IL‐10 and IL‐13) suggested a shift from a pro‐ to an anti‐inflammatory state, which was also accompanied by the reduction in kidney injury markers such as KIM‐1. In addition, the composite endpoint made up of TNF family members showed a high effect size. This also supports the potential benefit of SBI‐101 on the kidney itself, as the activation of the TNFα pathway is known to mediate tubular cell injury and contributes toward tubular cell loss in AKI. 56 , 57 Of note, elevated PK markers did not necessarily correspond to an observed pharmacodynamic increase of the same marker, as can be seen by the levels of IL‐6 measured in SBI‐101 and in the patient's plasma after treatment. This speaks to the hypothesis underlying the multifaceted approach of MSC therapy, which, in stark contrast to single‐agent therapies, targets many biological pathways concomitantly.
Immunomodulation at a molecular level was observed in conjunction with cellular changes. The most striking effect noted was a decrease in monocytes along with a decrease in monocyte and macrophage chemoattractants such as MCP‐1 (CCL2), MCP‐2 (CCL8), and MCP‐3 (CCL7). Consistent with other clinical reports, these findings have been recently proposed as potential predictive markers of MSC response. 58 Interestingly, nonclassic monocytes, which are known to be patrolling, noninflammatory cells, did not decrease with SBI‐101 treatment. The reduction of pro‐inflammatory monocytes in circulation could be the result of diminished recruitment of monocytes from the bone marrow or the migration of these cells to the injured tissues. If these monocytes indeed migrated to the injured tissues, as previously reported when MSCs were administered in a sepsis model, 59 it is plausible that they would differentiate preferentially into M2 macrophages because of the increased levels of TGF‐β and Th2 cytokines also observed in the current study. Unlike the most predominant M1 pro‐inflammatory macrophages, M2 macrophages promote healing and tissue regeneration in AKI. 60
Although changes in T cells were expected, they did not appear to be significant or robust at this initial dose level. On the other hand, B cells, specifically unswitched memory B cells, did seem to be increased with SBI‐101 treatment. The effect of MSCs on B cells is still controversial, 61 but the observed result is consistent with in vitro studies using a small scaled SBI‐101, where CD19‐positive cells consistently go up upon ex vivo MSC therapy. 40 More recently, a study on COVID‐19 showed that the cytokine storm, and specifically the elevation of TNFα, may inhibit the formation of germinal centers resulting in the reduction of the number of memory B cells necessary to develop long‐term immunity. 62 Hence it is plausible that by reducing TNFα and other Th1 cytokine levels, SBI‐101 allows for an expanded B‐cell response.
The immunoprofiling data will be better understood once more patients are treated. The small number of subjects was a major limitation of the current study and was the motivation for using effect sizes to assess pharmacological changes. Another major limitation was the lack of white blood cell counts and differentials for all the patients, which made it difficult to look at the effect of SBI‐101 on certain immune populations, for example, neutrophils. This information will be more efficiently collected going forward, as will other data related to baseline indicators of severity of disease (eg, sequential organ failure assessment scores) to aid the assessment of clinical outcomes. Finally, this study has only assessed one dose (250 × 106 MSCs). Higher doses (eg, 500 or 750 × 106 cells per SBI‐101) may enhance the efficacy of SBI‐101 and prolong the PD effects relative to those observed in lower doses.
SBI‐101 represents a novel ex vivo environment in which human MSCs interact in close proximity with the blood system without introducing MSCs directly into a patient. This may be advantageous to mitigate risks of MSC‐induced instant blood‐mediated inflammatory reactions and other potential safety concerns. 63 , 64 In addition, dosing and fitness of the MSCs can have an impact on potency. 47 Hence this alternative mode of delivery of MSCs assures a controlled exposure of blood to a specified number of viable, recovered MSCs over a specified time without administering MSCs directly to a patient, thus providing a longer, directly measurable therapeutic window with an enhanced safety profile. Because it is designed to address the regulation of programmed transitions in the phenotype of immune effector cells as an integrated systems biology problem, SBI‐101 is uniquely positioned to serve as an effective therapy to modulate the innate and reprogram the adaptive immune responses. Thus, SBI‐101 has potential for broad applicability to treat diseases with a high unmet medical need caused by dysregulated, immune‐mediated inflammation.
5. CONCLUSION
We conclude that the ex vivo MSC therapy broadly reprogrammed the molecular and cellular peripheral immune compartment in patients with systemic inflammation. Pharmacological data and systems biology analysis provided supporting evidence for therapeutic hypothesis, namely, that treatment with SBI‐101 elicits an immunotherapeutic response that triggers an accelerated phenotypic switch from tissue injury to tissue repair.
conflict of interest
S.N., B.O., A.A., N.V., A.T., E.L., A.B., C.G., and R.N.B. are/were employees and are equity shareholders of Sentien Biotechnologies. B.P. and B.M. are equity shareholders and inventors of the technology with licensed patents to Sentien for commercialization. M.G.A. is Principal Investigator for contracted institutional research with Sentien, GlaxoSmithKline, REATA, Bristol‐Myers Squibb, Alnyalm. J.R. and K.U. declared research grant Sentien Biotechnologies.
author contributions
M.S., N.K., M.A, J.R., K.U.,: clinical integration/conduction, data analysis and review, manuscript preparation; S.N., B.O., A.A., N.V.,: data analysis and review, manuscript preparation; A.T.: clinical integration, conceptualization, data analysis and review, manuscript preparation; A.B.: conceptualization, data analysis and review; E.L., C.M., B.M., B.P.: conceptualization, manuscript preparation, funding acquisition; R.N.B.: conceptualization, data analysis and review, manuscript preparation, funding acquisition.
Supporting information
Table S1: Demographic Characteristics (Treated Set)
Table S2: Effect sizes of molecular and cellular PD Biomarkers
Table S3: Multiplex Panels
Table S4: Flow Cytometry Panels
Materials and Methods: Exploratory endpoint analysis
Fig. S1 IL‐6 measurements in SBI‐101 treated participants subjects.
IL‐6 was quantified in the plasma of each participant (n = 4, green, brown, blue and pink) pre and post treatment as well as in samples taken directly from the SBI‐101 device during the 24 hours of treatment. Despite an increase in detectable IL‐6 in the MSC microenvironment during treatment, plasma levels of IL‐6 seem to decrease post treatment.
Fig. S2 Time to treatment inversely correlates with TNF‐α reduction.
TNF‐ α was measure in plasma of subjects receiving SBI‐101 therapy at baseline and at day 3 post treatment. The delta between the two time points was calculated and plotted against time from diagnosis of AKI to integration of SBI1‐10 (IP‐ Investigational product). Strong correlation (r2 = 0.9) indicates that patients that receive SBI‐101 soon after their diagnosis may be more likely to have larger reductions of pro‐inflammatory cytokines such as TNF‐ α.
Fig. S3 Comparison Analysis of Monocyte‐related Diseases and Functions of 250 M SBI‐101 Treated and Sham. Disease and Function Analysis Comparison Heatmap was sorted by Hierarchical Clustering based on sample Activation Score. The activation score (Z‐score) predicts direction of gene regulation of potential regulators assessing the match of observed and predicted upstream or downstream processes. Z‐score serves as both a significance measure and a predictor for the activation state of the regulator (orange, +/activation, blue, −/inhibition). Monocyte‐related Diseases and Functions are highlighted.
acknowledgments
We are grateful to all the study participants. We are also very thankful to the staff at the Novel Cell Therapy Lab, Cell Manipulation Core Facility at Dana‐Farber Cancer Institute. We would like to acknowledge Payal Garg and Katrina MacNeil for their contribution throughout the study, as well as all the staff at the participating sites. This work was partially funded by the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases (5R44DK085766). The clinical trial conducted in this study is titled “A Study of Cell Therapy for Subjects with Acute Kidney Injury Who Are Receiving Continuous Renal Replacement Therapy.” ClinicalTrials.gov Identifier: NCT03015623.
Swaminathan M, Kopyt N, Atta MG, et al. Pharmacological effects of ex vivo mesenchymal stem cell immunotherapy in patients with acute kidney injury and underlying systemic inflammation. STEM CELLS Transl Med. 2021;10(12):1588-1601. doi: 10.1002/sctm.21-0043
Funding information National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases, Grant/Award Number: 5R44DK085766
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
references
- 1. Alberti C, Brun‐Buisson C, Goodman SV, et al. Influence of systemic inflammatory response syndrome and sepsis on outcome of critically ill infected patients. Am J Respir Crit Care Med. 2003;168:77‐84. [DOI] [PubMed] [Google Scholar]
- 2. Burne‐Taney MJ, Kofler J, Yokota N, Weisfeldt M, Traystman RJ, Rabb H. Acute renal failure after whole body ischemia is characterized by inflammation and T cell‐mediated injury. Am J Physiol Renal Physiol. 2003;285:F87‐F94. [DOI] [PubMed] [Google Scholar]
- 3. Bonventre JV, Zuk A. Ischemic acute renal failure: an inflammatory disease? Kidney Int. 2004;66:480‐485. [DOI] [PubMed] [Google Scholar]
- 4. Lederer JA, Rodrick ML, Mannick JA. The effects of injury on the adaptive immune response. Shock. 1999;11:153‐159. [DOI] [PubMed] [Google Scholar]
- 5. Rangel‐Frausto MS, Pittet D, Costigan M, Hwang T, Davis CS, Wenzel RP. The natural history of the systemic inflammatory response syndrome (SIRS). A prospective study. JAMA. 1995;273:117‐123. [PubMed] [Google Scholar]
- 6. Catanzaro M, Fagiani F, Racchi M, Corsini E, Govoni S, Lanni C. Immune response in COVID‐19: addressing a pharmacological challenge by targeting pathways triggered by SARS‐CoV‐2. Signal Transduct Target Ther. 2020;5:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Giamarellos‐Bourboulis EJ, Netea MG, Rovina N, et al. Complex immune dysregulation in COVID‐19 patients with severe respiratory failure. Cell Host Microbe. 2020;27:992‐1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Siddiqi HK, Mehra MR. COVID‐19 illness in native and immunosuppressed states: a clinical‐therapeutic staging proposal. J Heart Lung Transplant. 2020;39:405‐407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Comstedt P, Storgaard M, Lassen AT. The Systemic Inflammatory Response Syndrome (SIRS) in acutely hospitalised medical patients: a cohort study. Scand J Trauma Resusc Emerg Med. 2009;17:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Proctor MJ, McMillan DC, Horgan PG, et al. Systemic inflammation predicts all‐cause mortality: a Glasgow inflammation outcome study. PLoS One. 2015;10:e0116206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cook AD, Christensen AD, Tewari D, McMahon SB, Hamilton JA. Immune cytokines and their receptors in inflammatory pain. Trends Immunol. 2018;39:240‐255. [DOI] [PubMed] [Google Scholar]
- 12. Gullestad L, Ueland T, Vinge LE, Finsen A, Yndestad A, Aukrust P. Inflammatory cytokines in heart failure: mediators and markers. Cardiology. 2012;122:23‐35. [DOI] [PubMed] [Google Scholar]
- 13. Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol. 2014;14:329‐342. [DOI] [PubMed] [Google Scholar]
- 14. Shachar I, Karin N. The dual roles of inflammatory cytokines and chemokines in the regulation of autoimmune diseases and their clinical implications. J Leukoc Biol. 2013;93:51‐61. [DOI] [PubMed] [Google Scholar]
- 15. Varga G, Foell D. Anti‐inflammatory monocytes‐interplay of innate and adaptive immunity. Mol Cell Pediatr. 2018;5:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cronkite DA, Strutt TM. The regulation of inflammation by innate and adaptive lymphocytes. J Immunol Res. 2018;2018:1467538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barnig C, Levy BD. Innate immunity is a key factor for the resolution of inflammation in asthma. Eur Respir Rev. 2015;24:141‐153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aziz M, Jacob A, Yang WL, Matsuda A, Wang P. Current trends in inflammatory and immunomodulatory mediators in sepsis. J Leukoc Biol. 2013;93:329‐342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Delano ML, Ward PA. The immune system's role in sepsis progression, resolution, and long‐term outcome. Immunol Rev. 2016;274:330‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Horiguchi H, Loftus TJ, Hawkins RB, et al. Innate immunity in the persistent inflammation, immunosuppression, and catabolism syndrome and its implications for therapy. Front Immunol. 2018;9:595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yiu HH, Graham AL, Stengel RF. Dynamics of a cytokine storm. PLoS ONE. 2012;7:(10):e45027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gandolfo MT, Jang HR, Bagnasco SM, et al. Foxp3+ regulatory T cells participate in repair of ischemic acute kidney injury. Kidney Int. 2009;76(7):717‐729. [DOI] [PubMed] [Google Scholar]
- 23. Kinsey GR, Li L, Okusa MD. Inflammation in acute kidney injury. Nephron Exp Nephrol. 2008;109:e102‐e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim MG, Koo TY, Yan JJ, et al. IL‐2/anti‐IL‐2 complex attenuates renal ischemia‐reperfusion injury through expansion of regulatory T cells. J Am Soc Nephrol. 2013;24:1529‐1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kyurkchiev D, Bochev I, Ivanova‐Todorova E, et al. Secretion of immunoregulatory cytokines by mesenchymal stem cells. World J Stem Cells. 2014;6:552‐570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fontaine MJ, Shih H, Schafer R, et al. Unraveling the mesenchymal stromal cells' paracrine immunomodulatory effects. Transfus Med Rev. 2016;30:37‐43. [DOI] [PubMed] [Google Scholar]
- 27. Prockop DJ, Oh JY. Mesenchymal stem/stromal cells (MSCs): role as guardians of inflammation. Mol Ther. 2012;20:14‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Joannidis M, Oudemans‐van Straaten HM. Clinical review: patency of the circuit in continuous renal replacement therapy. Crit Care. 2007;11:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hoogduijn MJ, Lombardo E. Mesenchymal stromal cells anno 2019: dawn of the therapeutic era? Concise review. Stem Cells Translational Medicine. 2019;8:1126‐1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. U.S. Food and Drug Administration, Center for Drug Evaluation and Research FDA Briefing Document: Oncologic Drugs Advisory Committee (ODAC) Meeting, Session on Product Characterization (AM Session), August 13, 2020. BLA 125706, Remestemcel‐L. Applicant. Mesoblast, Inc. Washington, DC: Author.
- 31. Zumla A, Wang FS, Ippolito G, et al. Reducing mortality and morbidity in patients with severe COVID‐19 disease by advancing ongoing trials of mesenchymal stromal (stem) cell (MSC) therapy ‐ achieving global consensus and visibility for cellular host‐directed therapies. Int J Infect Dis. 2020;96:431‐439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Qu W, Wang Z, Hare JM, et al. Cell‐based therapy to reduce mortality from COVID‐19: systematic review and meta‐analysis of human studies on acute respiratory distress syndrome. Stem Cells Translational Medicine. 2020;9:1007‐1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tang L, Jiang Y, Zhu M, et al. Clinical study using mesenchymal stem cells for the treatment of patients with severe COVID‐19. Front Med. 2020;14:664‐673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Caplan H, Olson SD, Kumar A, et al. Mesenchymal stromal cell therapeutic delivery: translational challenges to clinical application. Front Immunol. 2019;10:1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Eggenhofer E, Benseler V, Kroemer A, et al. Mesenchymal stem cells are short‐lived and do not migrate beyond the lungs after intravenous infusion. Front Immunol. 2012;3:297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Swaminathan M, Stafford‐Smith M, Chertow GM, et al. Allogeneic mesenchymal stem cells for treatment of AKI after cardiac surgery. J Am Soc Nephrol. 2018;29:260‐267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Levy O, Kuai R, Siren EMJ, et al. Shattering barriers toward clinically meaningful MSC therapies. Sci Adv. 2020;6:eaba6884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Caplan AI. New MSC: MSCs as pericytes are sentinels and gatekeepers. J Orthop Res. 2017;35:1151‐1159. [DOI] [PubMed] [Google Scholar]
- 39. Li M, Khong D, Chin LY, Singleton A, Parekkadan B. Therapeutic delivery specifications identified through compartmental analysis of a mesenchymal stromal cell‐immune reaction. Sci Rep. 2018;8:6816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Allen A, Vaninov N, Li M, et al. Mesenchymal stromal cell bioreactor for ex vivo reprogramming of human immune cells. Sci Rep. 2020;10:10142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. van Poll D, Parekkadan B, Cho CH, et al. Mesenchymal stem cell‐derived molecules directly modulate hepatocellular death and regeneration in vitro and in vivo. Hepatology. 2008;47:1634‐1643. [DOI] [PubMed] [Google Scholar]
- 42. Parekkadan B, van Poll D, Suganuma K, et al. Mesenchymal stem cell‐derived molecules reverse fulminant hepatic failure. PLoS One. 2007;2:e941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Elman JS, Li M, Wang F, Gimble JM, Parekkadan B. A comparison of adipose and bone marrow‐derived mesenchymal stromal cell secreted factors in the treatment of systemic inflammation. J Inflamm. 2014;11:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Miller BLK, Garg P, Bronstein B, et al. Extracorporeal stromal cell therapy for subjects with dialysis‐dependent acute kidney injury. Kidney Int Rep. 2018;3:1119‐1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kee YK, Kim D, Kim SJ, et al. Factors associated with early mortality in critically ill patients following the initiation of continuous renal replacement therapy. J Clin Med. 2018;7:334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kehl D, Generali M, Mallone A, et al. Proteomic analysis of human mesenchymal stromal cell secretomes: a systematic comparison of the angiogenic potential. NPJ Regen Med. 2019;4:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Galipeau J, Sensébé L. Mesenchymal Stromal Cells: Clinical Challenges and Therapeutic Opportunities. Cell Stem Cell. 2018;22(6):824‐833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pittenger MF, Discher DE, Péault BM, Phinney DG, Hare JM, Caplan AI. Mesenchymal stem cell perspective: cell biology to clinical progress. NPJ Regen Med. 2019;4:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Milwid JM, Ichimura T, Li M, et al. Secreted factors from bone marrow stromal cells upregulate IL‐10 and reverse acute kidney injury. Stem Cells Int. 2012;2012:392050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gao F, Chiu SM, Motan DAL, et al. Mesenchymal stem cells and immunomodulation: current status and future prospects. Cell Death Dis. 2016;7:e2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mezey E, Mayer B, Németh K. Unexpected roles for bone marrow stromal cells (or MSCs): a real promise for cellular, but not replacement, therapy. Oral Diseases. 2010;16(2):129‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tolwani A. Continuous renal‐replacement therapy for acute kidney injury. N Engl J Med. 2012;367:2505‐2514. [DOI] [PubMed] [Google Scholar]
- 53. Christie E, Pannu N. Dialysis and acute kidney injury: current evidence. Semin Dial. 2014;27:154‐159. [DOI] [PubMed] [Google Scholar]
- 54. Rabb H, Griffin MD, McKay DB, et al. Inflammation in AKI: current understanding, key questions, and knowledge gaps. J Am Soc Nephrol. 2013;27:371‐379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bernardo ME, Fibbe WE. Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell. 2013;13:392‐402. [DOI] [PubMed] [Google Scholar]
- 56. Cunningham PN, Dyanov HM, Park P, Wang J, Newell KA, Quigg RJ. Acute renal failure in endotoxemia is caused by TNF acting directly on TNF receptor‐1 in kidney. J Immunol. 2002;168:5817‐5823. [DOI] [PubMed] [Google Scholar]
- 57. Linkermann A, Chen G, Dong G, Kunzendorf U, Krautwald S, Dong Z. Regulated cell death in AKI. J Am Soc Nephrol. 2014;25:2689‐2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhang Z, Wilson NA, Chinnadurai R, et al. Autologous mesenchymal stromal cells prevent transfusion‐elicited sensitization and upregulate transitional and regulatory B cells. Transplant Direct. 2018;4:e387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Németh K, Leelahavanichkul A, Yuen PST, et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)‐dependent reprogramming of host macrophages to increase their interleukin‐10 production. Nat Med. 2009;15:42‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lee SA, Noel S, Sadasivam M, Hamad ARA, Rabb H. Role of immune cells in acute kidney injury and repair. Nephron. 2017;137:282‐286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Franquesa M, Hoogduijn MJ, Bestard O, et al. Immunomodulatory effect of mesenchymal stem cells on B cells. Front Immunol. 2012;3:212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kaneko N, Kuo HH, Boucau J, et al. Loss of Bcl‐6‐expressing T follicular helper cells and the absence of germinal centers in COVID‐19. Cell. 2020;183(1):143‐157.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. O'Rourke B, Nguyen S, Tilles AW, et al. Mesenchymal stromal cell delivery via an ex vivo bioreactor preclinical test system attenuates clot formation for intravascular application. Stem Cells Translational Medicine. 2021;10:883‐894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Moll G, Ankrum JA, Kamhieh‐Milz J, et al. Intravascular mesenchymal stromal/stem cell therapy product diversification: time for new clinical guidelines. Trends Mol Med. 2019;25:149‐163. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Demographic Characteristics (Treated Set)
Table S2: Effect sizes of molecular and cellular PD Biomarkers
Table S3: Multiplex Panels
Table S4: Flow Cytometry Panels
Materials and Methods: Exploratory endpoint analysis
Fig. S1 IL‐6 measurements in SBI‐101 treated participants subjects.
IL‐6 was quantified in the plasma of each participant (n = 4, green, brown, blue and pink) pre and post treatment as well as in samples taken directly from the SBI‐101 device during the 24 hours of treatment. Despite an increase in detectable IL‐6 in the MSC microenvironment during treatment, plasma levels of IL‐6 seem to decrease post treatment.
Fig. S2 Time to treatment inversely correlates with TNF‐α reduction.
TNF‐ α was measure in plasma of subjects receiving SBI‐101 therapy at baseline and at day 3 post treatment. The delta between the two time points was calculated and plotted against time from diagnosis of AKI to integration of SBI1‐10 (IP‐ Investigational product). Strong correlation (r2 = 0.9) indicates that patients that receive SBI‐101 soon after their diagnosis may be more likely to have larger reductions of pro‐inflammatory cytokines such as TNF‐ α.
Fig. S3 Comparison Analysis of Monocyte‐related Diseases and Functions of 250 M SBI‐101 Treated and Sham. Disease and Function Analysis Comparison Heatmap was sorted by Hierarchical Clustering based on sample Activation Score. The activation score (Z‐score) predicts direction of gene regulation of potential regulators assessing the match of observed and predicted upstream or downstream processes. Z‐score serves as both a significance measure and a predictor for the activation state of the regulator (orange, +/activation, blue, −/inhibition). Monocyte‐related Diseases and Functions are highlighted.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.