

Abstract
Due to their intrinsic nuclease resistance, l-oligonucleotides are being increasingly utilized in the development of molecular tools and sensors. Yet, it remains challenging to synthesize long l-oligonucleotides, potential limiting future applications. Herein, we report straightforward and versitile approach to assemble long l-RNAs from two or more shorter fragments using T4 RNA ligase 1. We show that this approach is compatible with the assembly of several classes of functional l-RNA, which we highlight by generating a 124-nt l-RNA biosensor that functions in serum.
As chiral molecules, native d-DNA and d-RNA have enantiomers, referred to as mirror image l-DNA and l-RNA. Although l-oligonucleotides (ON) no longer exist in nature, they can be prepared synthetically in the laboratory, and from a biotechnology perspective, have several advantageous properties relative to their native counterparts.1 In particular, l-ONs are highly resistant to degradation by nucleases, providing them superior stability in harsh biological environments.2, 3 l-ONs also avoid potentially toxic off-target interactions with endogenous nucleic acids because ON of opposite chirality are incapable of forming contiguous WC base pairs with each other.2, 4 Furthermore, as enantiomers, d- and l-ON have the same physical properties in terms of duplex thermostability and hybridization kinetics4–6, making them identical from a design perspective.7 Due to these favourable properties, l-ON are being increasingly employed in biomedical applications, including the development of l-aptamers8, microarrays2, molecular sensors9, and live cell imaging.10–12
As a result of their bio-orthogonal nature, l-ON cannot be synthesized enzymatically using native proteins, such as DNA/RNA polymerases. Consequently, l-ONs are almost exclusively prepared using solid-phase phosphoramidite chemistry, which imposes a practical limit on their length and quality. This potentially limits the types of applications that can be accessed using this powerful nucleic acid analogue. To address this challenge, researchers have begun to explore alternative strategies to assemble l-ON. For example, the Joyce group developed a “cross-chiral” ribozyme that can catalyze the ligation of two or more l-RNAs, allowing for the assembly of long l-RNAs from several short, synthetically accessible fragments.13 Others have reported the chemical synthesis of d-amino acid versions of protein polymerases14, 15 and ligases16 and successfully demonstrated transcription and PCR amplification of long l-RNAs and l-DNAs, respectively. While promising, these synthetic approaches remain highly specialized and have proven difficult to scale, and thus, are not practical solutions for the average laboratory, especially if large quantities of long l-ON are desired. Therefore, there remains a need for general, straightforward strategies to assemble long l-ON.
For many applications of l-ON, preparation of the entire molecule using solely l-(deoxy)ribonucleotides may not be necessary. For example, l-aptamers (or Spiegelmers)17 often contain stem-loops or other structural domains that are not directly involved in ligand binding, and thus, could be potentially replaced by non-canonical nucleotides or chemical linkers.8, 18 From a synthetic perspective, these modifiable domains also serve as potential fragmentation sites whereby a long DNA/RNA molecule can be assembled through the joining of multiple fragments using non-native or chemical linkages, such as click chemistry.19 In the case of l-ON, we reasoned that such linkages could be made simply of native d-DNA/RNA, allowing two or more l-ON fragments to be joined through enzymatic ligation. On this basis, the goal of this study was to determine whether larger l-RNA molecules could be assembled via the enzymatic ligation of two shorter l-RNA pieces containing terminal d-ribonucleotides (Figure 1a).
Fig. 1.
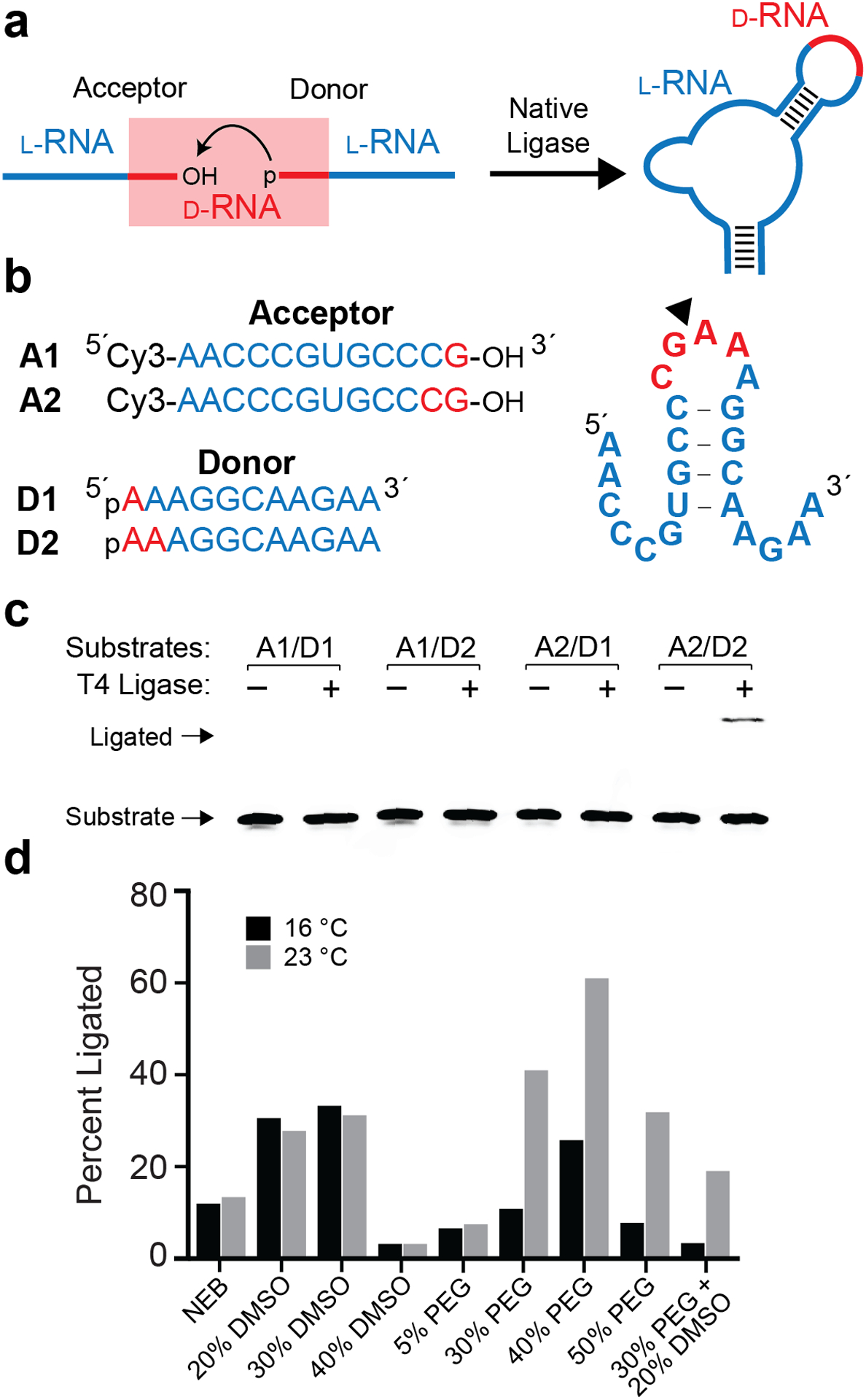
T4 RNA ligase-mediated assembly of l-RNA. (a) Schematic of the ligation strategy. l-RNA donor and acceptor strands containing 3′ and 5′ terminal d-RNA residues are ligated by T4 RNA ligase. (b) Sequences and secondary structure of the donor and acceptor substrates. Blue text: l-RNA; red text: d-RNA. (c) Pilot ligation experiments using different donor/acceptor combinations. Reactions contained 2 μM of indicated RNA substrates, T4 Buffer, and 25 U of T4 RNA ligase 1 and were incubated at 23 °C for 24 hours. The uncropped gel image is presented in Figure S5. (d) Reaction optimization. Conditions were the same as (c), with T4 Buffer substituted with the indicated additive.
To begin, we designed and synthesized a series of l-RNA donor and acceptor ligation substrates containing either one or two d-ribonucleotides on their 5′ and 3′ ends, respectively (Figure 1b and Table S1). The donor and acceptor substrates were designed to form an RNA hairpin structure with a five-nucleotide loop upon ligation. By exploiting the self-complementarity of stem-loops, this strategy eliminates the potential need for a splint oligonucleotide, thereby reducing synthetic burden and simplifying experimental setup. For this initial study, we focused on T4 RNA Ligase 1 due to its well-known ability to catalyze the joining of adjacent 3′ and 5′ “dangling” RNA ends, resulting in the formation of a hairpin.20 T4 RNA ligase 1 is also commonly used for 3′ end labeling of RNA using 3′,5′-bisphosphate (pNp) donors21, suggesting its potential compatibility with a small number of native d-ribonucleotides in our design. We determined the ligation efficiency of all four possible substrate combinations using T4 RNA Ligase 1 under reaction conditions recommended by the enzyme provider (T4 Buffer: 50 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 1 mM DTT, 20% PEG 8000, 10% DMSO) (Figure 1c). Only substrate combination A2/D2 showed a usable ligation yield (~11%) after 24 hours. A small amount of product (<1%) was detected for substrate combination A2/D1, whereas no ligation was observed with any combination including substrate A1. These results suggest that T4 RNA Ligase 1 requires a minimum of four terminal d-ribonucleotides – two on the acceptor and two on the donor – in order to efficiently ligate two strands of l-RNA.
Focusing on substrate combination A2/D2, we next sought to optimize ligation yields by varying both the composition and temperature of the ligation reaction mixture. In particular, additives such as dimethyl sulfoxide (DMSO) and polyethylene glycol (PEG) are commonly used in ligation reactions to increase efficiency.22 Increasing the amount of DMSO in T4 Buffer from 10% up to 30% resulted in the formation of ~3-fold more ligated product for both temperatures tested (Figure 1d). However, increasing the amount of DMSO further (40%) almost completely inhibited the reaction, which is likely due to disruption of base pairing interactions between the donor and acceptor strands.23 Likewise, increasing the amount of PEG from 20% up to 40% led to a substantial increase in product formation, reaching ~60% yield at 23 °C after 24 hours (Figure 1d). Further increasing the amount of PEG to 50% impeded the reaction. Interestingly, increasing the amount of both PEG and DMSO simultaneously resulted in lower ligation yields than when either component was added separately. Based on these observations, we selected T4 Buffer containing 40% PEG and 23 °C as the optimal ligation reaction condition. We acknowledge that the ligation efficiency may vary significantly depending on the sequence and secondary structure of the donor and acceptor strands, especially at the ligation junction.24 Indeed, a preliminary screen of terminal nucleotides on the donor strand (D2) revealed that a 5′-rC may be optimal, although all nucleotides were well tolerated (Figure S1). Moreover, a substrate pair not capable of forming a hairpin was ligated with similar efficiency as the A2/D2 pair. These data further demonstrate the generality of this approach, and indicate that there is room for future optimization.
As a proof-of-principle for assembling a functional l-RNA molecule using this approach, we assembled an l-RNA version of the minimal hammerhead ribozyme (l-HRz) from two shorter pieces (Figure 2a and Table S1).25 The ligation junction was placed at the center of a 5′-GAAA-3′ tetraloop at the end of the H2 stem, the sequence of which is nonessential for catalysis.26 l-HRz was configured for trans-cleavage of a fluorescently labeled l-RNA substrate (l-S), a common strategy employed for biosensor development.27 An appreciable ligation yield (>30%) was obtained between the donor and acceptor strands, with an isolated yield of ~5% following gel purification (Figure S2a and Table S2). Assembly of full-length l-HRz was confirmed by mass-spectrometry (Figure S2b). Importantly, we also prepared an all-d-RNA version of the identical ribozyme construct (d-HRz) and its substrate (d-S) by chemical synthesis to allow for a direct comparison between the two enantiomers of HRz. As shown in Figure 2b, both configurations of HRz performed similarly, cleaving ~60% of the substrate after 30 minutes. These results show that inverting the stereochemistry of 5′-GAAA-3′ tetraloop (i.e. incorporation of four d-nucleotides) has a minimal effect on l-HRz function. In contrast, the non-ligated donor and acceptor strands failed to cleave a significant fraction of l-S (Figure 2b, split), demonstrating the importance of the intact tetraloop on HRz stability and catalysis.
Fig. 2.
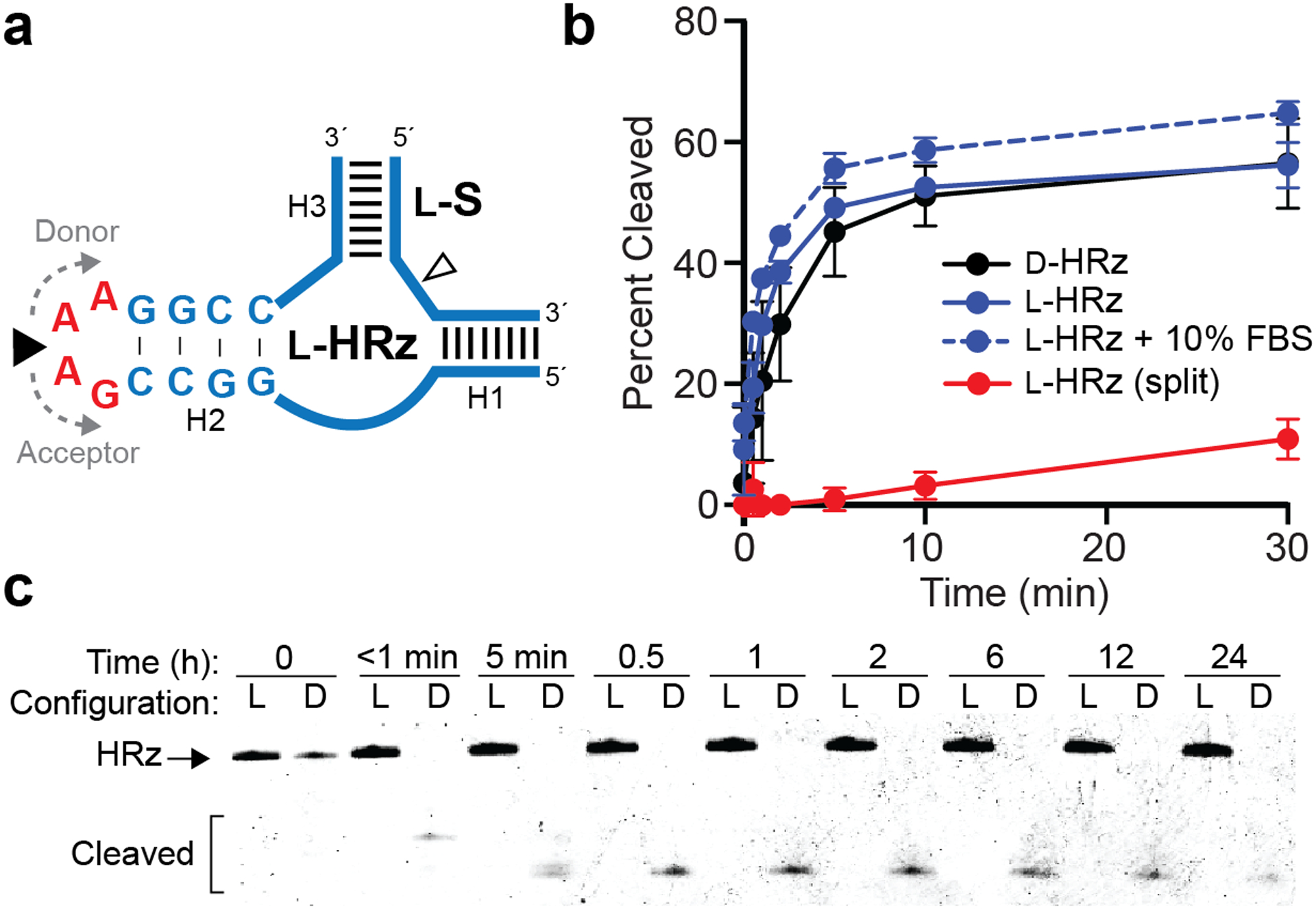
Ligation of a functional l-RNA ribozyme. (a) Schematic illustration of the hammerhead ribozyme (HRz) and substrate (S). Blue text/lines: l-RNA; red text: d-RNA. The ligation junction and RNA cleavage site are indicated by closed and open arrows, respectively. Sequences of all strands are listed in Table S1. (b) Relative kinetics of d- and l-HRz cleavage reactions. Each data point represents the mean ± S.D. (n = 3). (c) Stability of d- and l-HRz in 10% FBS. The uncropped gel image is presented in Figure S6.
To examine the generality of this ligation strategy, we also assembled a 46-nt l-RNA aptamer, l-6–4t, from two shorter pieces (Figure 3a and Table S1). l-6–4t binds tightly to the trans-activation responsive (TAR) element of HIV-1 RNA.18 As with l-HRz, the ligation junction was positioned at the center of a 5′-GAAA-3′ tetraloop, which resulted in a ligation yield of nearly 40% during T4 RNA ligase-mediated assembly (Figure S3 and Table S2). We determined the Kd of ligated l-6–4t (l-6–4tLIG) by EMSA and compared it side-by-side to an all l-RNA version prepared as a single piece by solid-phase synthesis (l-6–4t). As shown in Figure 3b, l-6–4tLIG binds d-TAR RNA with a Kd of 346 ± 35 nM, which is significantly higher than its all l-RNA counterpart. One possible explanation is that inverting the stereochemistry of the tetraloop mitigates its stabilizing effects on the underlying stem28, thereby reducing the overall structural stability of l-6–4tLIG relative to the all-l-RNA version. However, binding of l-6–4tLIG to TAR RNA required ligation, as the non-ligated donor and acceptor fragments failed to bind TAR (split). Given that the same d-RNA tetraloop and ligation junction was used to assemble l-HRz with minimal impact on functionality (Figure 2b), these results suggest that inverted nucleotides can affect functional RNAs in different ways, and in some cases, may require optimization. Nevertheless, the combined results clearly demonstrate that T4 RNA ligase can be used to assemble functional l-RNA aptamers and ribozymes.
Fig. 3.
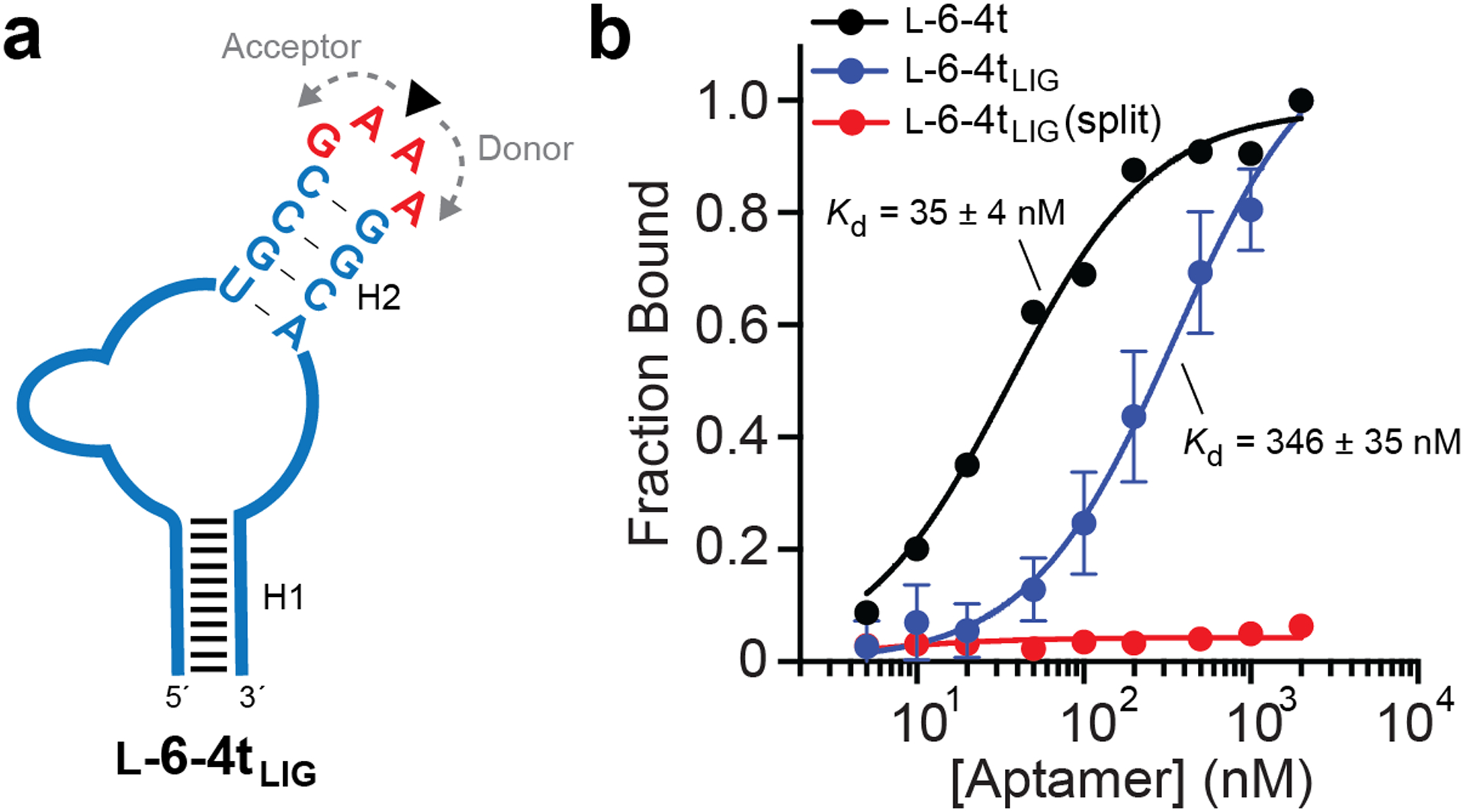
Ligation of a functional l-RNA aptamer. (a) Schematic of the l-6–4t l-RNA aptamer. The ligation junction is indicated by the arrow. (b) Saturation plot for binding of either l-6–4t or l-6–4tLIG to TAR RNA. Kd values reported as mean ± S.D. (n = 3)
A key feature of l-ON is their intrinsic resistance to nuclease digestion, which has been exploited for various biomedical applications.1 However, we realize that incorporation of multiple d-nucleotides within a larger l-RNA could introduce a point of vulnerability into an otherwise nuclease resistant polymer, potentially undermining the utility of our approach. In order to demonstrate the compatibility of our enzymatically ligated l-RNAs with harsh biological environments, we investigated the behavior of l-HRz (Figure 2a) in the presence of 10% fetal bovine serum (FBS). Despite the presence of four contiguous d-ribonucleotides, no detectable cleavage was observed for l-HRz after 24 hours in the presence of 10% FBS, as determined by gel electrophoresis (Figure 2c). This is consistent with prior work showing that chimeric d/l-oligonucleotides are stable in human serum.29 In contrast, d-HRz was completely degraded within a few seconds under the same condition. Importantly, l-HRz remained catalytically active in 10% FBS (Figure 2b). In fact, the fraction of l-S cleaved by l-HRz in the presence of 10% FBS closely mirrored cleavage in the absence of FBS. Thus, despite introducing a potential vulnerability into the nuclease resistant backbone, these results demonstrate that incorporation of several d-ribonucleotides into a functional l-RNA does not significantly impede its performance and lifetime within harsh biological environments.
The primary motivation behind this work was to facilitate synthesis of long l-RNA molecules that are not otherwise accessible using current solid-phase synthesis methods. To demonstrate the utility of native RNA ligation to generate long l-RNAs, we assembled an l-RNA version of a 124-nt long theophylline biosensor (HRzTheo) previously reported by You et al. (Figure 4a).30 The sensor comprises a theophylline-activated hammerhead ribozyme and an unfolded form of the fluorogenic aptamer Broccoli. Upon binding theophylline, the ribozyme undergoes self-cleavage and releases Broccoli, which subsequently folds and becomes fluorescent. In addition to its length, this target was chosen because it carefully integrates several classes of functional RNA, making it a good model system to demonstrate direct stereochemical inversion of a complex RNA-based sensor design. Importantly, both theophylline and DFHBI-1T, the fluorogenic dye bound by Broccoli, are achiral molecules, and therefore must interact identically with d-RNA and l-RNA versions of the sensor. Similar to the examples above, we positioned the ligation junction in the middle of a 5′-GAAA-3′ tetraloop, in this case on stem H3 of the hammerhead ribozyme. This resulted in a 62-nt acceptor strand and a 62-nt donor strand, each of which contained two d-ribonucleotides on their 3′ and 5′ ends, respectively (Figure 4a). Despite its large size and extensive secondary structures, a ligation yield of ~40% (~7% isolated yield) was achieved (Figure S4a and Table S2). As a control, we also prepared an all-d-RNA version of HRzTheo by in vitro transcription. When tested side-by-side, both D and L versions of HRzTheo behaved similarly, resulting in ~5–6-fold increase in self-cleavage in the presence of 200 μM theophylline (Figure 4b). Increased fluorescence was also observed for both stereoisomers in the presence of theophylline (Figure 4c). Importantly, the l-RNA version of HRzTheo was fully functional is 10% FBS, whereas the original d-RNA version was fully degraded during the experiment (Figure S4b). This further demonstrates that use of l-RNA (and assembly thereof) represents a straightforward strategy for preparing nuclease resistant versions of common RNA-based biosensors designs.
Fig. 4.
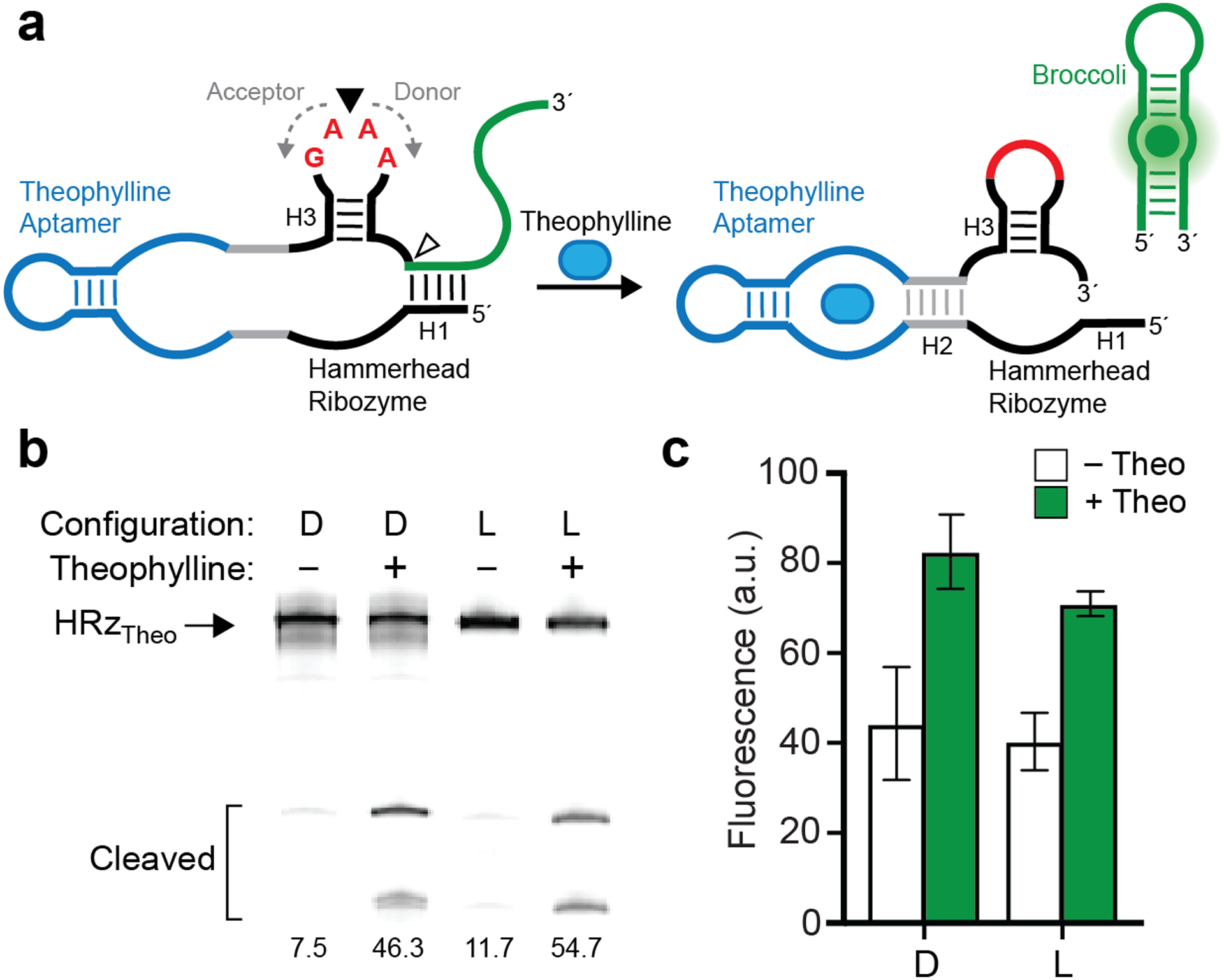
Assembly and operation of a 124-nt l-RNA biosensor. (a) Schematic illustration of the theophylline biosensor (HRzTheo) based on the hammerhead ribozyme and broccoli aptamer. (b) Theophylline-induced self-cleavage of d- or l-HRzTheo. Percent cleaved is indicated below each lane. Uncropped gel image is presented in Figure S7. (c) Fluorescence activation of d- or l-HRzTheo in the presence of theophylline. Each data point represents the mean ± S.D. (n = 3).
In summary, we demonstrated that two l-RNA molecules containing terminally positioned d-ribonucleotides can be efficiently ligated using T4 RNA ligase 1, providing a straightforward strategy for assembling long (>100-nt) l-RNAs that are not easily accessible through purely synthetic methods. We showed that this approach is compatible with the assembly of several classes of functional l-RNA, including aptamers and ribozymes, and that the internally position d-ribonucleotides used for ligation do not make the polymer more susceptible to nuclease degradation. In most cases, the ligation junction and requisite d-ribonucleotides were position within a 5′-GNRA-3′ type tetraloop, a structural motif commonly found in native RNA molecules and is often engineered into unnatural RNAs designed in the laboratory. Thus, while some optimization may be required, we anticipate that this approach can be used to obtain suitable mirror-image versions of a broad swath of functional nucleic acids. Indeed, in order to demonstrate the applicability of this approach for assembling a long l-RNA molecule, we prepared the l-RNA version of a 124-nt theophylline-specific fluorescent biosensor that was fully operational in serum. The ability to prepare nuclease resistant versions of RNA-based sensors and other devices with minimal design consideration, and at scalable quantities, greatly expands the utility of such technologies for biomedical applications in molecular sensing and imaging. Finally, although this work focused on the ligation of l-RNA, we expect that these methods can be readily applied to l-DNA, as well as other modified nucleic acids, including xeno nucleic acids (XNAs), many of which are not compatible with common synthetic methods.
Supplementary Material
Footnotes
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Young BE, Kundu N and Sczepanski JT, Chem. Eur. J, 2019, 25, 7981–7990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hauser NC, Martinez R, Jacob A, Rupp S, Hoheisel JD and Matysiak S, Nucleic Acids Res, 2006, 34, 5101–5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Urata H, Ogura E, Shinohara K, Ueda Y and Akagi M, Nucleic Acids Res, 1992, 20, 3325–3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoehlig K, Bethge L and Klussmann S, PLOS ONE, 2015, 10, e0115328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Urata H, Shinohara K, Ogura E, Ueda Y and Akagi M, J. Am. Chem. Soc, 1991, 113, 8174–8175. [Google Scholar]
- 6.Szabat M, Gudanis D, Kotkowiak W, Gdaniec Z, Kierzek R and Pasternak A, PLoS ONE, 2016, 11, e0149478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kabza AM, Young BE and Sczepanski JT, J. Am. Chem. Soc, 2017, 139, 17715–17718. [DOI] [PubMed] [Google Scholar]
- 8.Vater A and Klussmann S, Drug Discov. Today, 2015, 20, 147–155. [DOI] [PubMed] [Google Scholar]
- 9.Feagin TA, Olsen DPV, Headman ZC and Heemstra JM, J. Am. Chem. Soc, 2015, 137, 4198–4206. [DOI] [PubMed] [Google Scholar]
- 10.Zhong W and Sczepanski JT, ACS Sens, 2019, 4, 566–570. [DOI] [PubMed] [Google Scholar]
- 11.Ke G, Wang C, Ge Y, Zheng N, Zhu Z and Yang CJ, J. Am. Chem. Soc, 2012, 134, 18908–18911. [DOI] [PubMed] [Google Scholar]
- 12.Cui L, Peng R, Fu T, Zhang X, Wu C, Chen H, Liang H, Yang CJ and Tan W, Anal. Chem, 2016, 88, 1850–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sczepanski JT and Joyce GF, Nature, 2014, 515, 440–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang M, Jiang W, Liu X, Wang J, Zhang B, Fan C, Liu L, Pena-Alcantara G, Ling J-J, Chen J and Zhu TF, Chem, 2019, 5, 848–857. [Google Scholar]
- 15.Pech A, Achenbach J, Jahnz M, Schülzchen S, Jarosch F, Bordusa F and Klussmann S, Nucleic Acids Res, 2017, 45, 3997–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weidmann J, Schnölzer M, Dawson PE and Hoheisel JD, Cell Chem. Biol, 2019, 26, 645–651. [DOI] [PubMed] [Google Scholar]
- 17.Klussmann S, Nolte A, Bald R, Erdmann VA and Fürste JP, Nat. Biotechnol, 1996, 14, 1112–1115. [DOI] [PubMed] [Google Scholar]
- 18.Sczepanski JT and Joyce GF, J. Am. Chem. Soc, 2013, 135, 13290–13293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.El-Sagheer AH and Brown T, Acc. Chem. Res, 2012, 45, 1258–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Depmeier H, Hoffmann E, Bornewasser L and Kath-Schorr S, ChemBioChem, 2021, DOI: 10.1002/cbic.202100161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.England TE and Uhlenbeck OC, Nature, 1978, 275, 560–561. [DOI] [PubMed] [Google Scholar]
- 22.Song Y, Liu KJ and Wang T-H, PLOS ONE, 2014, 9, e94619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee J, Vogt CE, McBrairty M and Al-Hashimi HM, Anal. Chem, 2013, 85, 9692–9698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhuang F, Fuchs RT, Sun Z, Zheng Y and Robb GB, Nucleic acids research, 2012, 40, e54–e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pley HW, Flaherty KM and McKay DB, Nature, 1994, 372, 68–74. [DOI] [PubMed] [Google Scholar]
- 26.Ruffner DE, Stormo GD and Uhlenbeck OC, Biochemistry, 1990, 29, 10695–10702. [DOI] [PubMed] [Google Scholar]
- 27.Felletti M and Hartig JS, WIREs RNA, 2017, 8, e1395. [DOI] [PubMed] [Google Scholar]
- 28.Bevilacqua PC and Blose JM, Annu. Rev. Phys. Chem, 2008, 59, 79–103. [DOI] [PubMed] [Google Scholar]
- 29.Damha MJ, Giannaris PA and Marfey P, Biochemistry, 1994, 33, 7877–7885. [DOI] [PubMed] [Google Scholar]
- 30.You M, Litke JL, Wu R and Jaffrey SR, Cell Chem. Biol, 2019, 26, 471–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.