

Abstract
Chronic obstructive pulmonary disease (COPD) is a multisystemic respiratory disease that is associated with progressive airway and pulmonary vascular remodeling due to the increased proliferation of bronchial smooth muscles cells (BSMCs) and pulmonary arterial smooth muscle cells (PASMCs) and the overproduction of extracellular matrix (e.g., collagen). Cigarette smoke (CS) and several mediators, such as PDGF (platelet-derived growth factor) and IL-6, play critical roles in COPD pathogenesis. HDAC6 has been shown to be implicated in vascular remodeling. However, the role of airway HDAC6 signaling in pulmonary vascular remodeling in COPD and the underlying mechanisms remain undetermined. Here, we show that HDAC6 expression is upregulated in the lungs of patients with COPD and a COPD animal model. We also found that CS extract (CSE), PDGF, and IL-6 increase the protein levels and activation of HDAC6 in BSMCs and PASMCs. Furthermore, CSE and these stimulants induced deacetylation and phosphorylation of ERK1/2 and increased collagen synthesis and BSMC and PASMC proliferation, which were outcomes that were prevented by HDAC6 inhibition. Inhibition of ERK1/2 also diminished the CSE-, PDGF-, and IL-6–caused elevation in collagen levels and cell proliferation. Pharmacologic HDAC6 inhibition with tubastatin A prevented the CS-stimulated increases in the thickness of the bronchial and pulmonary arterial wall, airway resistance, emphysema, and right ventricular systolic pressure and right ventricular hypertrophy in a rat model of COPD. These data demonstrate that the upregulated HDAC6 governs the collagen synthesis and BSMC and PASMC proliferation that lead to airway and vascular remodeling in COPD.
Keywords: COPD, HDAC6, cigarette smoke, PDGF, IL-6
Chronic obstructive pulmonary disease (COPD) is a severe inflammatory lung disease with high morbidity and mortality (1). Pathologic processes in COPD involve chronic bronchitis, emphysema, inflammation, and remodeling of the airways and lung vasculature (2). Excessive alterations and a disproportional deposition of extracellular matrix proteins, including collagen, and increased proliferation of bronchial smooth muscle cells (BSMCs) and pulmonary arterial smooth muscle cells (PASMCs) lead to the thickening of the walls and narrowing of the lumens of airways and pulmonary arteries (PAs) that are responsible for the development of airway obstruction and pulmonary hypertension (PH) in COPD (3–5). Cigarette smoke (CS) exposure is the most significant causative factor for COPD, but several other mediators, including growth factors and inflammatory cytokines, have been identified to be involved in the pathogenesis of COPD (6–9). CS exposure governs remodeling of the small airway by increasing the production of procollagen and growth factors such as PDGF (platelet-derived growth factor) (10). We have reported that PDGF stimulates collagen synthesis and the proliferation of smooth muscle cells (SMCs) and is a crucial player in pulmonary vascular remodeling (11–13). IL-6 is an important proinflammatory cytokine and is involved in the inflammatory processes in the airways and pulmonary vasculature of patients with COPD (14). High levels of serum IL-6 are associated with impaired lung function and poor clinical outcomes in patients with COPD (15, 16). IL-6 is also mitogenic for airway SMCs, leading to the thickening of airway walls, and contributes to the proliferation of vascular cells in pulmonary vascular remodeling (17, 18). However, the precise signaling mechanism for how these factors contribute to the development of COPD has not yet been established.
HDAC6 is a class II HDAC and regulates the acetylation status of several cytosolic proteins that play crucial roles in the proliferation and migration of cancer cells and other cells, including fibroblasts, endothelial cells, and SMCs (19–21). HDAC6 inhibition has been shown to exhibit antitumor and antiinflammation properties (22, 23). Protein expression of HDAC6 is upregulated in cardiac fibrosis tissues, and inhibition of HDAC6 abrogated the proliferation of cardiac fibroblasts (20). Overexpression of HDAC6 increased the proliferation and migration of vascular SMCs; in contrast, inhibition of HDAC6 prevented these processes and decreased neointima formation after vascular injury (24). Furthermore, inhibition of HDAC6 blunted CS-induced exacerbation of acute lung injury in vivo and endothelial cell (EC) barrier dysfunction in vitro (25). More importantly, HDAC6 is overexpressed in human and experimental models of pulmonary arterial hypertension (PAH), and inhibition of HDAC6 decreases vascular remodeling in a rodent PAH model (21). In addition, knockout of HDAC6 results in protection against chronic hypoxia–induced PAH (21). However, the role of airway HDAC6 signaling in pulmonary vascular remodeling in COPD and the underlying mechanisms remain undetermined.
In the present study, we demonstrate that increases in the protein expression and relative activity of HDAC6 contribute to the deacetylation and phosphorylation/activation of ERK1/2 in the BSMCs and PASMCs of the lungs of rats with CS-induced COPD. We showed that inhibition of HDAC6 diminishes the CS extract (CSE)-, PDGF- and IL-6–induced ERK deacetylation and phosphorylation, collagen synthesis, and BSMC and PASMC proliferation. ERK1/2 inhibition also abolished increases in collagen protein levels and cell proliferation stimulated by CSE and these mediators. More importantly, our results reveal that the HDAC6 inhibitor tubastatin A (Tub A) prevents airway and pulmonary vascular remodeling in a COPD animal model. Inhibition of HDAC6 might be a novel therapeutic target for the intervention and treatment of CS-induced COPD.
Methods
An extended Materials and Methods section is included in the data supplement.
Human Tissues
Lung sections from patients with COPD were obtained from the Lung Tissue Research Consortium, and lung sections from control subjects with normal lungs were obtained from the PH Breakthrough Initiative. For the clinical information of the patients, see Table E1 in the data supplement).
COPD Model
Sprague-Dawley rats were exposed to CS and room air for 16 weeks in a whole-body smoke inhalation system. The specific HDAC6 inhibitor Tub A was given at the 14th week of CS exposure. The dose of Tub A was 25 mg/kg, which was given intraperitoneally once daily for the last 3 weeks, as previously reported (21). After 16 weeks, echocardiography was performed, and the lung mechanics, PH status, and extent of airway and pulmonary vascular remodeling were assessed.
Echocardiography
Transthoracic echocardiography was completed by using the Visual Sonics Ultrasound Vevo2100 System. The cardiac output, tricuspid annulus plain systolic excursion, PA velocity–time integral and PA acceleration time were measured.
Assessment of Lung Mechanics
Lung mechanical properties were measured by performing intubation and connecting the trachea to the FlexiVent system (SCIREQ). Airway resistance, inspiratory capacity, and respiratory system compliance were assessed.
Measurement of PH and Histologic Analysis
Right ventricular systolic pressure was assessed by using a pressure transducer (AD Instruments). The Fulton index (right ventricular hypertrophy) was calculated as the ratio of the right ventricular weight to the weight of the left ventricle and the septum [right ventricular weight/(left ventricular weight + septal weight)]. The lung sections were stained with hematoxylin and eosin and then evaluated for bronchial wall thickness, emphysema, and pulmonary arterial wall thickness.
Immunohistochemistry
Double immunostaining of HDAC6/α-actin, collagen-I/α-actin, p-ERK1/2/α-actin, and total ERK1/2/α-actin was performed on lung tissue slides from control subjects with normal lungs, patients with COPD, and a COPD rat model, and slides were examined by using a Zeiss LSM 980 confocal microscope.
Preparation of CSE Solution
University of Kentucky 3R4F research cigarettes were continuously “smoked” by using vacuum pressure. The smoke was drawn through 30 ml of PBS by applying a vacuum to the vessel containing the PBS. Each cigarette was smoked for 5 minutes, and three cigarettes for each 30 ml of PBS were used to generate a CSE solution (27).
Cell Culture
Human primary BSMCs and PASMCs were cultured according to the supplier’s instructions. Human BSMCs and PASMCs were originally isolated from the PAs of five subjects with normal lungs (three males and two females, 11–59 years of age). The cell authentication has been established by the supplier. Before all experiments, the third- to seventh-passage cells were equilibrated in growth factor– and serum-free medium for 24 hours.
HDAC6 Activity Assay
HDAC6 activity was measured by using the fluorogenic acetylated peptide as a substrate.
Determination of Changes in the Acetylation Status of ERK1/2
Cell lysates were immunoprecipitated with acetyl-lysine affinity beads. The precipitates were analyzed through Western blotting by using an antibody against total ERK1/2.
Study Approval
All animal protocols were approved by the Augusta University Institutional Animal Care and Use Committee. A waiver of informed consent for the use of human lung samples was approved by the Augusta University Institutional Review Board.
Statistical Analysis
Results are shown as means ± SEMs for n experiments. One-way ANOVA followed by a Tukey-Kramer post hoc test and an unpaired Student’s t test (two-tailed) were used to determine the significance of differences between groups. P < 0.05 was considered to indicate statistical significance.
Results
Increased Expression Levels of HDAC6 in Lungs of Human Patients with COPD and a COPD Rat Model
To investigate the role of HDAC6 in COPD, we examined the protein levels of HDAC6 in lungs from patients with COPD and a COPD rat model. The double-immunofluorescence staining showed that HDAC6 protein was significantly increased in the smooth muscle layer of the bronchi and pulmonary arterioles of the lungs from patients with COPD compared with normal lungs (Figures 1A and 1B). HDAC6 protein levels were also higher in the smooth muscle layer of the bronchi and pulmonary arterioles of the lungs of rats exposed to CS than in those of rats exposed to room air (Figures 1C and 1D). To further quantify the HDAC6 expression, Western blot analysis of lung tissue homogenates was performed. As shown in Figures 1E and 1F, in agreement with the immunofluorescence results, the Western blot results confirmed the elevated expression levels of HDAC6 in the CSE-induced COPD animal model (Figures 1E and 1F). Despite of the increased protein expression of HDAC6, CS did not decrease the levels of acetylated α-tubulin, an HDAC6 substrate, in the rat lung homogenates. However, inhibition of HDAC6 by Tub A was associated with elevated acetylation of α-tubulin (Figure E11).
Figure 1.
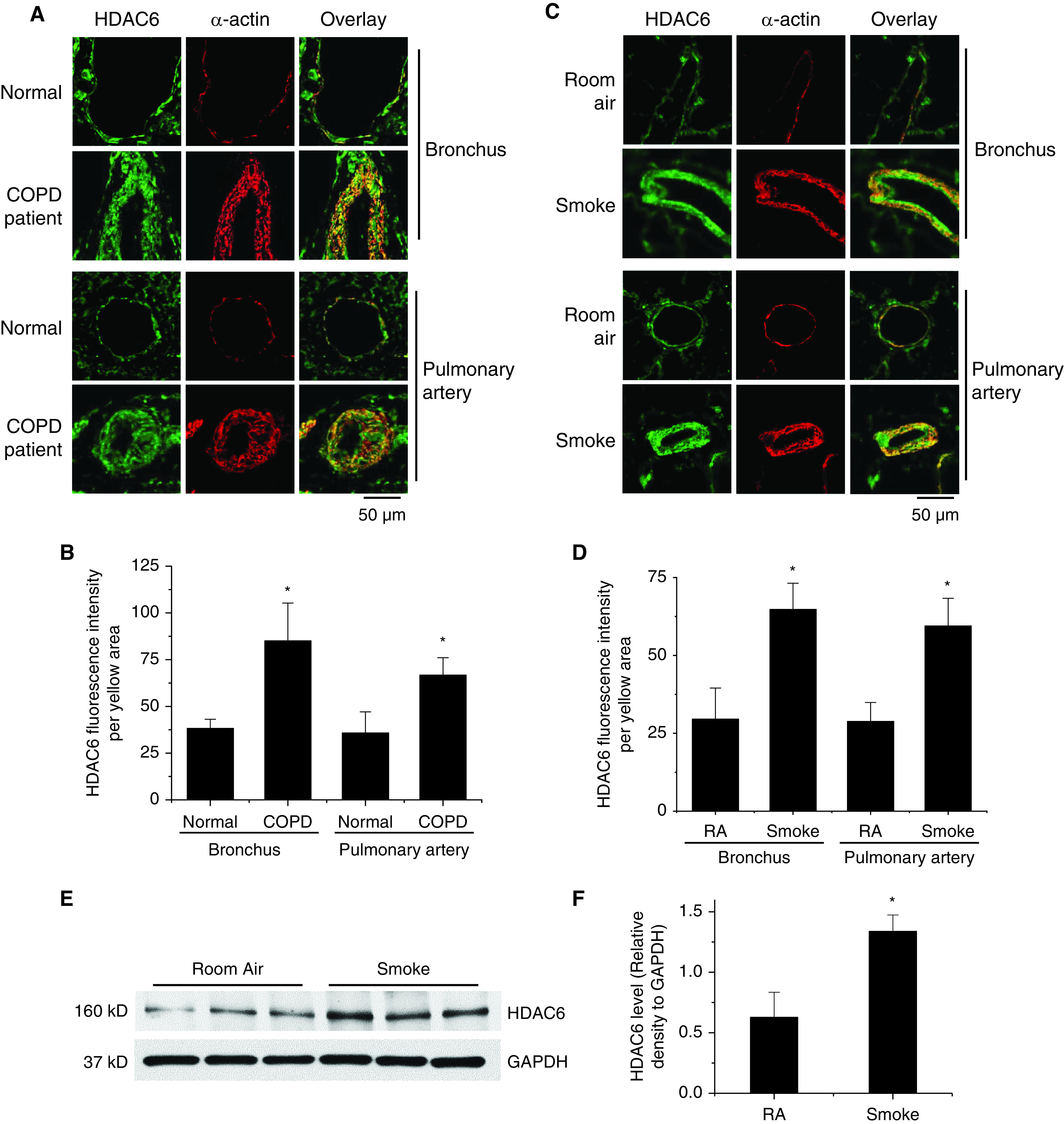
Higher levels of HDAC6 are expressed in the smooth muscle of the bronchi and pulmonary arterioles of the lungs from patients with chronic obstructive pulmonary disease (COPD) and from a COPD rat model. (A and B) Human lung slides were double-stained for α-actin (red) and HDAC6 (green). (A) Representative images of lung tissues from 8 patients with COPD and 8 control (Cntr) subjects with normal lungs. (B) Bar graph showing changes in HDAC6 fluorescence intensity in the airway and pulmonary arterial smooth muscle. *P < 0.05 versus Cntr subjects with normal lungs. (C–F) Eight-week-old male Sprague-Dawley rats were exposed to cigarette smoke (CS) for 16 weeks. At the beginning of the 14th week, CS-exposed rats were injected with tubastatin A (Tub A) (25 mg/kg, i.p.) or vehicle 5 d/wk for 3 weeks. Lung slides were double-stained for α-actin (red) and HDAC6 (green) for the bronchus and pulmonary artery (PA). (C) Representative images. (D) Bar graphs showing changes in HDAC6 fluorescence intensity in the airway and pulmonary arterial smooth muscle. (E and F) HDAC6 protein in lung homogenates from a COPD rat model were measured by using Western blots (WBs). The blots are representative immunoblots of five independent experiments. Results are expressed as means ± SEs; n = 5. Scale bars, 50 μm. *P < 0.05 versus RA. RA = room air.
CSE, PDGF, and IL-6 Induce Activation and Expression of HDAC6 in BSMCs and PASMCs
To determine whether CSE and mediators such as PDGF and IL-6 cause HDAC6 activation and increases in the protein levels of HDAC6, BSMCs and PASMCs were treated with or without CSE (1%), PDGF-BB (10 ng/ml), and IL-6 (10 ng/ml) for 30 minutes to 24 hours. As shown in Figure E1, incubation of BSMCs and PASMCs with CSE, PDGF, and IL-6 for 30 minutes significantly increased HDAC6 activity (Figures E1A–E1F). Furthermore, incubation of BSMCs and PASMCs with these mediators for 24 hours markedly increased HDAC6 activity and intracellular HDAC6 protein levels (Figures E1G–E1L).
CSE and Mediators Such as PDGF and IL-6 Induce Deacetylation and Phosphorylation of ERK1/2
To determine the effect of CSE, PDGF, and IL-6 on the acetylation status and phosphorylation/activation of ERK1/2, BSMCs were incubated with and without CSE (0.5–1%), PDGF-BB (10–30 ng/ml), and IL-6 (10–30 ng/ml) for 30 minutes. As shown in Figure E2, PDGF-BB markedly decreased the acetylation levels of ERK1/2. Moreover, incubation of BSMCs with these stimulants caused dose-dependent decreases in ERK1/2 acetylation and increases in phosphorylation/activation of the enzymes (Figures 2 and E3).
Figure 2.
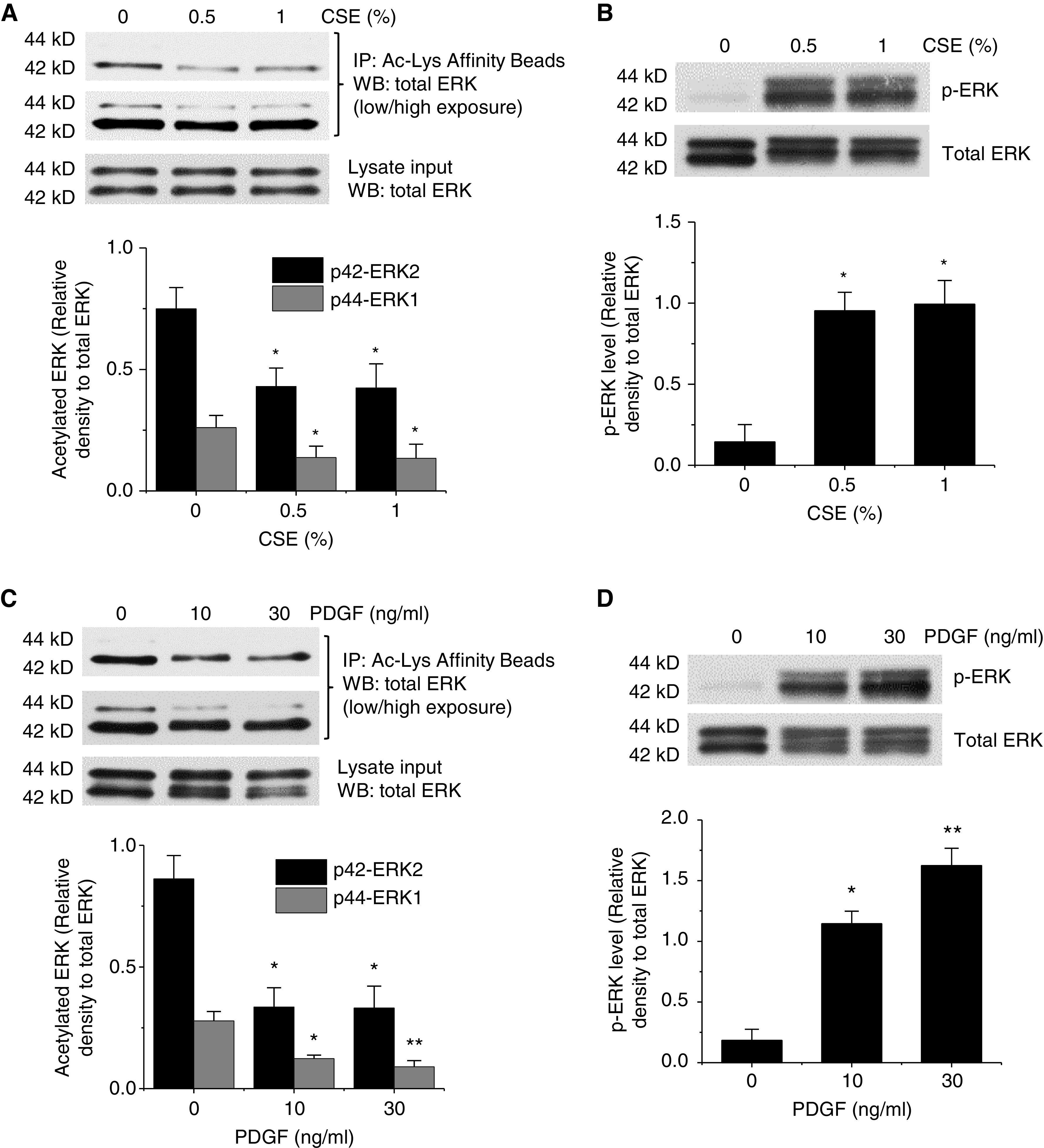
CS extract (CSE) and PDGF-BB (platelet-derived growth factor BB) induce ERK1/2 deacetylation and phosphorylation/activation of the enzymes. (A–D) Human bronchial smooth muscle cells (BSMCs) were incubated with and without CSE (0.5–1%) and PDGF-BB (10–30 ng/ml) for 30 minutes, after which the acetylation level of ERK1/2 (A and C) and the protein levels of p-ERK and total ERK (B and D) were measured. The blots are representative immunoblots of three independent experiments. Bar graphs depicting changes in acetylated ERK1/2 and in p-ERK levels are shown. Results are expressed as means ± SEs; n = 3. *P < 0.05 and **P < 0.01 versus Cntr (0). Cntr (0) indicates that the concentration of CSE or PDGF was 0% or 0 ng/ml in A and B as well as C and D. Ac-Lys = acetyl-lysine.
CSE, PDGF, and IL-6 Increase Collagen Synthesis and BSMC and PASMC Proliferation
To examine whether CSE and mediators such as PDGF and IL-6 stimulate collagen synthesis and cell proliferation, BSMCs and PASMCs were treated with CSE (1%), PDGF-BB (10 ng/ml), and IL-6 (10 ng/ml) for 24 hours. As shown in Figure 3, incubation of the cells with these mediators caused significant increases in the protein levels of collagen-I (Figures 3A and 3D) and in the BSMC and PASMC proliferation (Figures 3B, 3C, 3E, and 3F).
Figure 3.
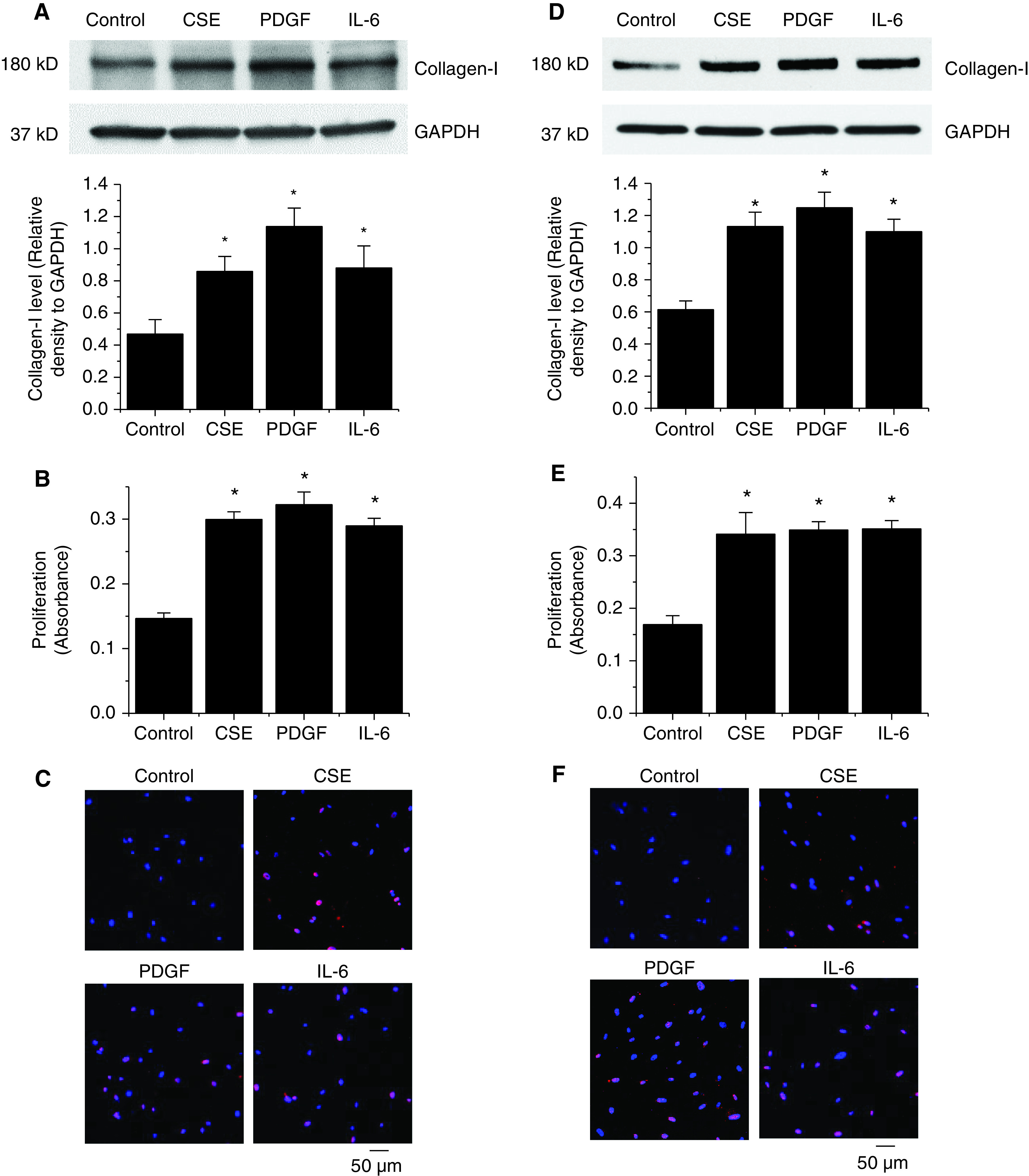
CSE, PDGF, and IL-6 induce collagen synthesis and BSMC and pulmonary arterial smooth muscle cell (PASMC) proliferation. (A–F) Human BSMCs (A–C) and human PASMCs (D–F) were incubated with CSE (1%), PDGF-BB (10 ng/ml), and IL-6 (10 ng/ml) for 24 hours, after which the intracellular collagen-I protein levels were measured by using WBs (A and D), and the levels of cell proliferation were measured by using a bromodeoxyuridine assay (B and E) and Ki67 staining (C and F). In A and D, the blots are representative immunoblots of four independent experiments. Bar graphs show changes in collagen-I levels in A and D and in proliferation in B and E. In C and F, representative images show labeling of proliferative Ki67-positive cells (red) and DAPI (blue). Results are expressed as means ± SEs; n = 4. Scale bars, 50 μm. *P < 0.05 versus Cntr.
Inhibition of HDAC6 Prevents CSE-, PDGF-BB–, and IL-6–induced Deacetylation and Phosphorylation/Activation of ERK1/2
To study whether CSE-, PDGF-, and IL-6–induced deacetylation and phosphorylation of ERK1/2 is caused by HDAC6 activation, BSMCs and PASMCs were incubated with and without CSE (1%), PDGF-BB (10 ng/ml), and IL-6 (10 ng/ml) in the absence and presence of the HDAC6 inhibitor Tub A (1μM) for 30 minutes. As shown in Figures 4 and E4, treatments of BSMCs and PASMCs with CSE and these mediators caused remarkable reductions in ERK1/2 acetylation and increases in ERK1/2 phosphorylation. More importantly, Tub A abolished CSE-, PDGF-BB–, and IL-6–induced increases in deacetylation and phosphorylation of ERK1/2, indicating that HDAC6 regulates the ERK activation induced by CSE and these mediators in BSMCs and PASMCs.
Figure 4.
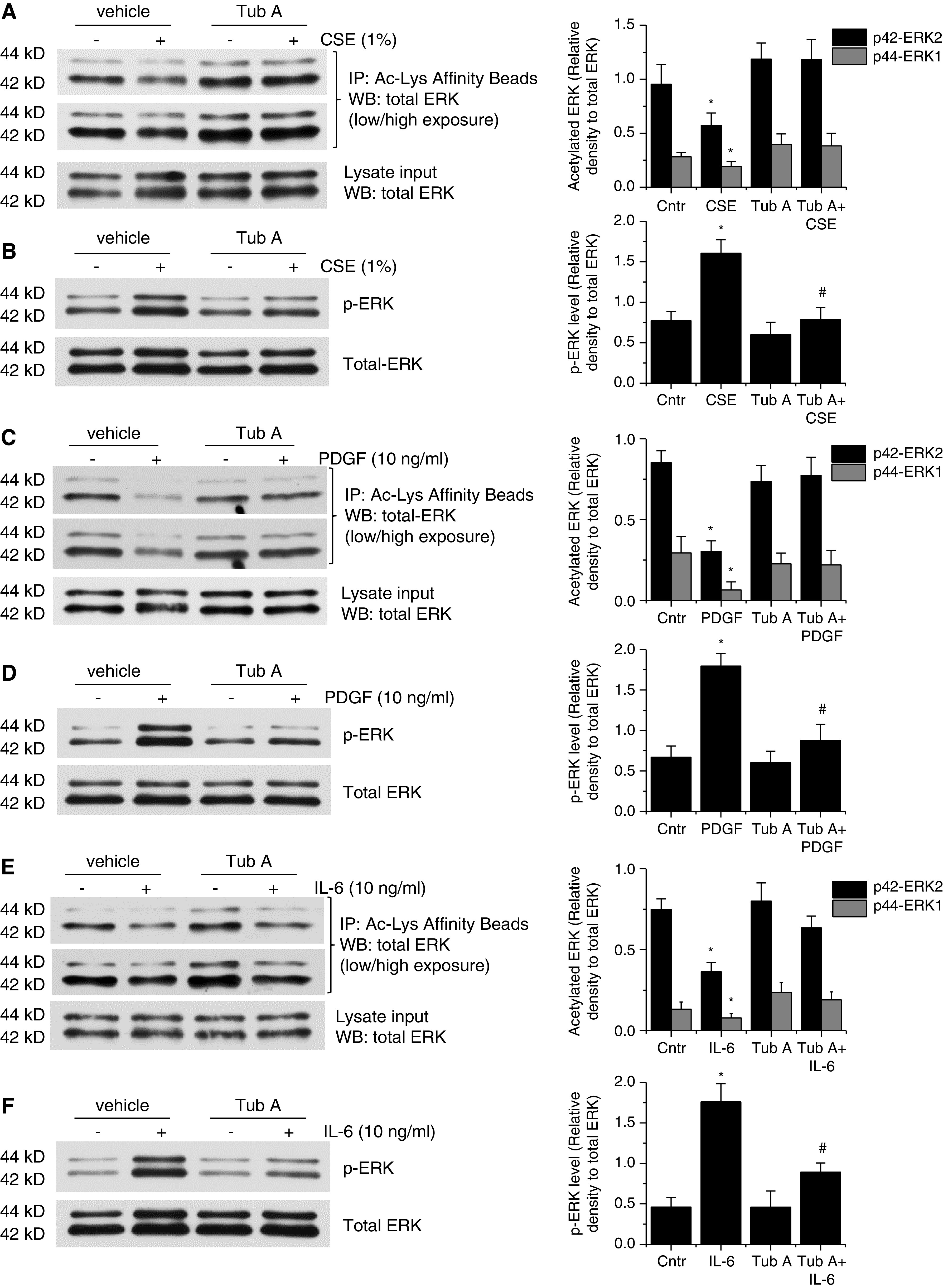
Inhibition of HDAC6 prevents CSE-, PDGF-, and IL-6–induced deacetylation and phosphorylation/activation of ERK1/2 in BSMCs. (A–F) Human BSMCs were incubated with CSE (1%), PDGF-BB (10 ng/ml), and IL-6 (10 ng/ml) in the presence and absence of the HDAC6 inhibitor Tub A (1μM) for 30 minutes, after which the acetylation level of ERK1/2 (A, C, and E) and the protein levels of p-ERK and total ERK (B, D, and F) were measured. The blots are representative immunoblots of three independent experiments. Bar graphs depict changes in acetylated ERK1/2 in A, C, and E and in p-ERK levels in B, D, and F. Results are expressed as means ± SEs; n = 3. *P < 0.05 versus Cntr. #P < 0.05 versus CSE, PDGF, and IL-6.
Inhibition of HDAC6 Reduces CSE-, PDGF-BB–, and IL-6–stimulated Collagen Synthesis and BSMC and PASMC Proliferation
To investigate the role of HDAC6 in CSE-, PDGF-, and IL-6–induced collagen synthesis and cell proliferation, BSMCs and PASMCs were incubated with and without CSE (1%), PDGF-BB (10 ng/ml), and IL-6 (10 ng/ml) in the absence and presence of the HDAC6 inhibitor Tub A (1μM) for 24 hours. We found that Tub A diminished the increases in collagen-I protein levels and cell proliferation induced by CSE and these mediators in BSMCs and PASMCs (Figures 5A–5D, E5A–E5D, E6A–E6D, and E7A). Moreover, inhibition of HDAC6 prevented the CSE-, PDGF-, and IL-6–induced increases in the number of viable BSMCs and PASMCs (Figure E12). Furthermore, knockdown of HDAC6 reduced collagen-I protein levels and completely abrogated CSE-, PDGF-BB–, and IL-6–induced increases in collagen-I protein levels and BSMC and PASMC proliferation (Figures 5E–5H, E5E–E5H, E6E–E6H, and E7B). These data indicate that HDAC6 mediates the collagen synthesis and BSMC and PASMC proliferation induced ty CSE and these mediators.
Figure 5.
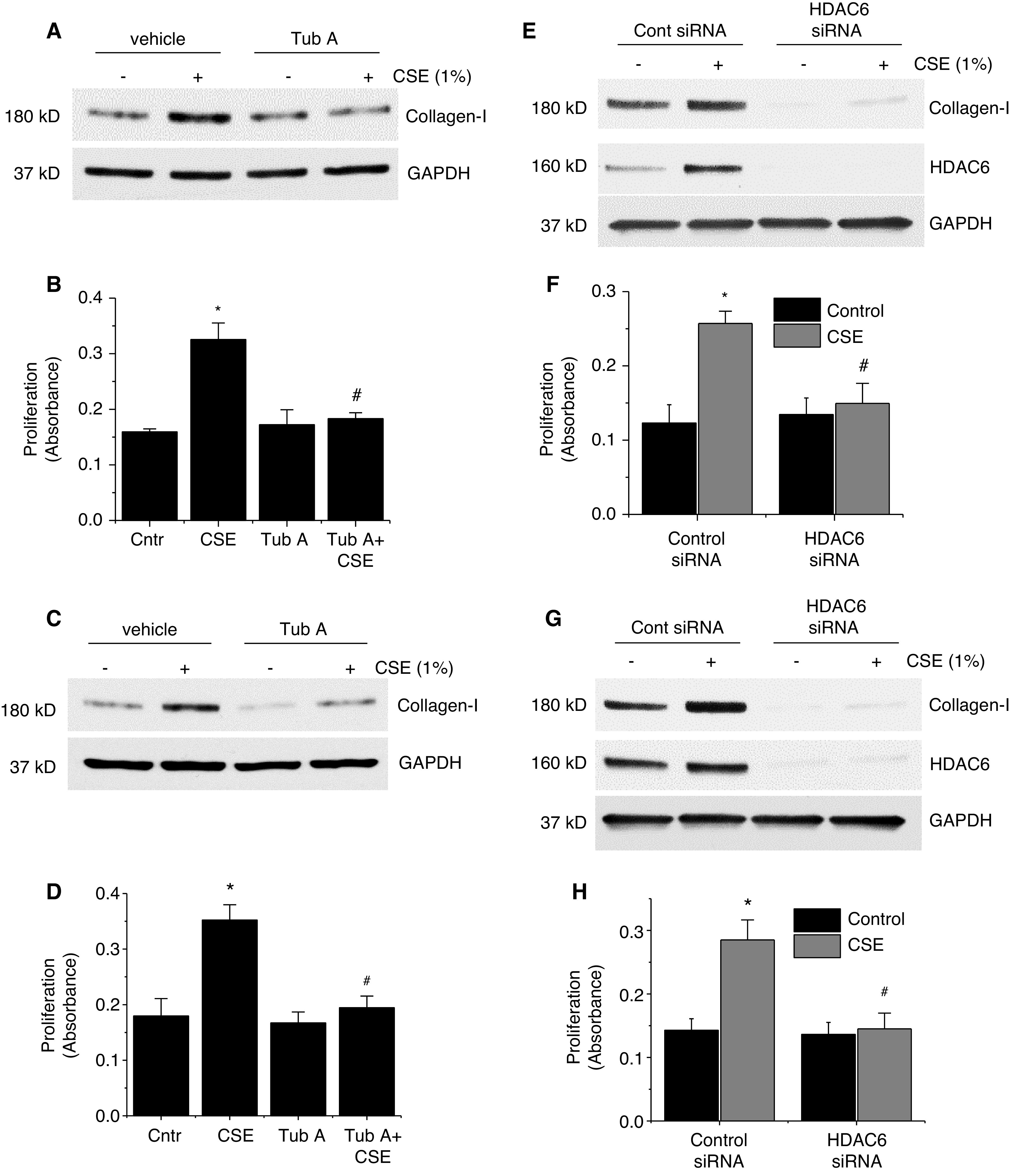
Inhibition of HDAC6 prevents CSE-induced collagen synthesis and BSMC and PASMC proliferation. (A–D) Human BSMCs (A and B) and human PASMCs (C and D) were incubated with CSE (1%) in the absence and presence of the HDAC6 inhibitor Tub A (1μM) for 24 hours, after which the collagen-I protein levels (A and C) and cell proliferation (B and D) were measured. (E–H) Human BSMCs (E and F) and human PASMCs (G and H) were transfected with siRNAs against HDAC6 or Cntr siRNA, respectively. After 72 hours, the cells were incubated with CSE (1%) for 24 hours, and then the protein levels of collagen-I and HDAC6 (in E and G) and the level of cell proliferation (in F and H) were measured. In A, C, E, and G, the blots are representative immunoblots of four independent experiments. In B, D, F, and H, bar graphs depict changes in cell proliferation. Results are expressed as means ± SEs; n = 4. *P < 0.05 versus Cntr and #P < 0.05 versus CSE. Cont = control.
Inhibition of ERK 1/2 Prevents CSE-, PDGF-BB–, and IL-6–induced Collagen Synthesis and BSMC and PASMC Proliferation
To determine whether CSE-, PDGF-, and IL-6–induced elevation of the collagen-I protein levels and cell proliferation occurs via an HDAC6–ERK1/2 signaling mechanism, ERK1/2 expression was knocked down in BSMCs and PASMCs, and cells were treated with CSE and these mediators for 24 hours. As shown in Figure E8, knockdown of ERK1/2 abolished the increases in collagen synthesis and BSMC and PASMC proliferation caused by CSE, PDGF-BB, and IL-6.
HDAC6 Inhibition by Tub A Suppresses Airway Remodeling and Elevation in Airway Resistance in a CS-induced COPD Model
To assess whether HDAC6 inhibition is able to prevent the development of COPD, we tested the effect of the HDAC6 inhibitor Tub A in a rat model of CS-induced COPD. After 13 weeks of CS exposure, rats exposed to CS were injected with Tub A (25 mg/kg, i.p.) or vehicle 5 d/wk for 3 weeks. As shown in Figure 6, administration of the HDAC6 inhibitor Tub A attenuated the increases in bronchial wall thickening (Figures 6A and 6B), airway resistance (Figures 6C), inspiratory capacity (Figure 6D), and the mean linear intercept (Figures 6E and 6F) caused by 16 weeks of CS exposure. Moreover, Tub A markedly reduced the elevation in protein levels of collagen-I and p-ERK1/2 in the smooth muscle of the bronchi of the lungs (Figures E9A, E9B, E10A, and E10B) and in the lung homogenates (Figures E9E, E9F, E10C, and E10D) of rats exposed to CS. In addition, we observed a trend toward increased respiratory system compliance in the CS-exposed rats compared with the room air–exposed rats (room air: 1.22 ± 0.09 vs. CS: 1.37 ± 0.10), which was attenuated by the HDAC6 inhibitor Tub A (CS: 1.37 ± 0.10 vs. CS + Tub A: 1.24 ± 0.11). However, there were no significant differences between the groups due to the high variation among the subjects. Together, these data demonstrate that the HDAC6 inhibitor Tub A attenuates the development of CS-induced airway remodeling and emphysema in a rat COPD model.
Figure 6.
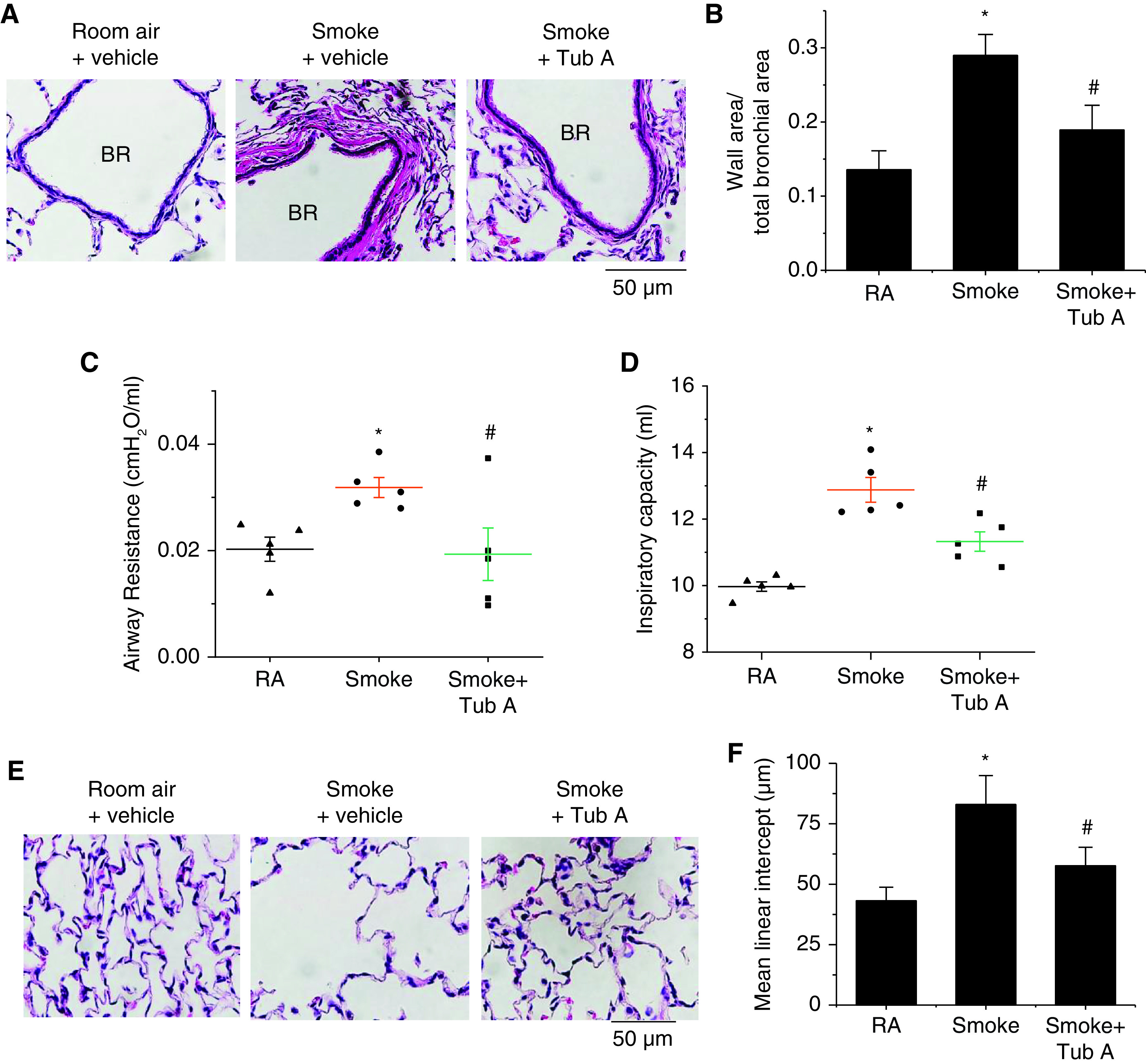
Tub A attenuates airway remodeling and elevation in airway resistance in a CS-induced COPD model. Eight-week-old male Sprague-Dawley rats were exposed to CS for 16 weeks. At the beginning of the 14th week, CS-exposed rats were injected with Tub A (25 mg/kg, i.p.) or vehicle 5 d/wk for 3 weeks, and then airway remodeling and airway resistance were assessed. (A and E) Representative images of lung sections of rats exposed to RA or CS. (B–D and F) Bar graphs depict changes in the ratio of the bronchial wall area to the total bronchial area (B), changes in airway resistance (C), changes in the inspiratory capacity (D), and changes in the mean linear intercept (F). Results are expressed as means ± SEs; n = 5. Scale bars, 50 μm. *P < 0.05 versus RA and #P < 0.05 versus smoke. BR = bronchus.
HDAC6 Inhibition by Tub A Prevents the Development of Pulmonary Vascular Remodeling and PH in a CS-induced COPD Model
Exposure to CS for 16 weeks caused significant increases in the pulmonary arterial wall thickness (Figures 7A and 7B), right ventricular systolic pressure (Figure 7C), Fulton index (Figure 7D), and protein levels of collagen-I (Figures E9C and E9D) and p-ERK1/2 (Figures E10A and E10B) in the smooth muscle of the pulmonary arterioles of rat lungs, and administration of the HDAC6 inhibitor Tub A attenuated these changes. Furthermore, echocardiography revealed that Tub A attenuated the diminishment of the cardiac output (Figure 7E), tricuspid annulus plain systolic excursion (Figure 7F), PA velocity–time integral (Figure 7G), and PA acceleration time (Figure 7H) in CS-exposed rats. Together, these results suggest that the inhibition of HDAC6 reduces pulmonary vascular remodeling and improves PH and right ventricular function in CS-induced COPD.
Figure 7.
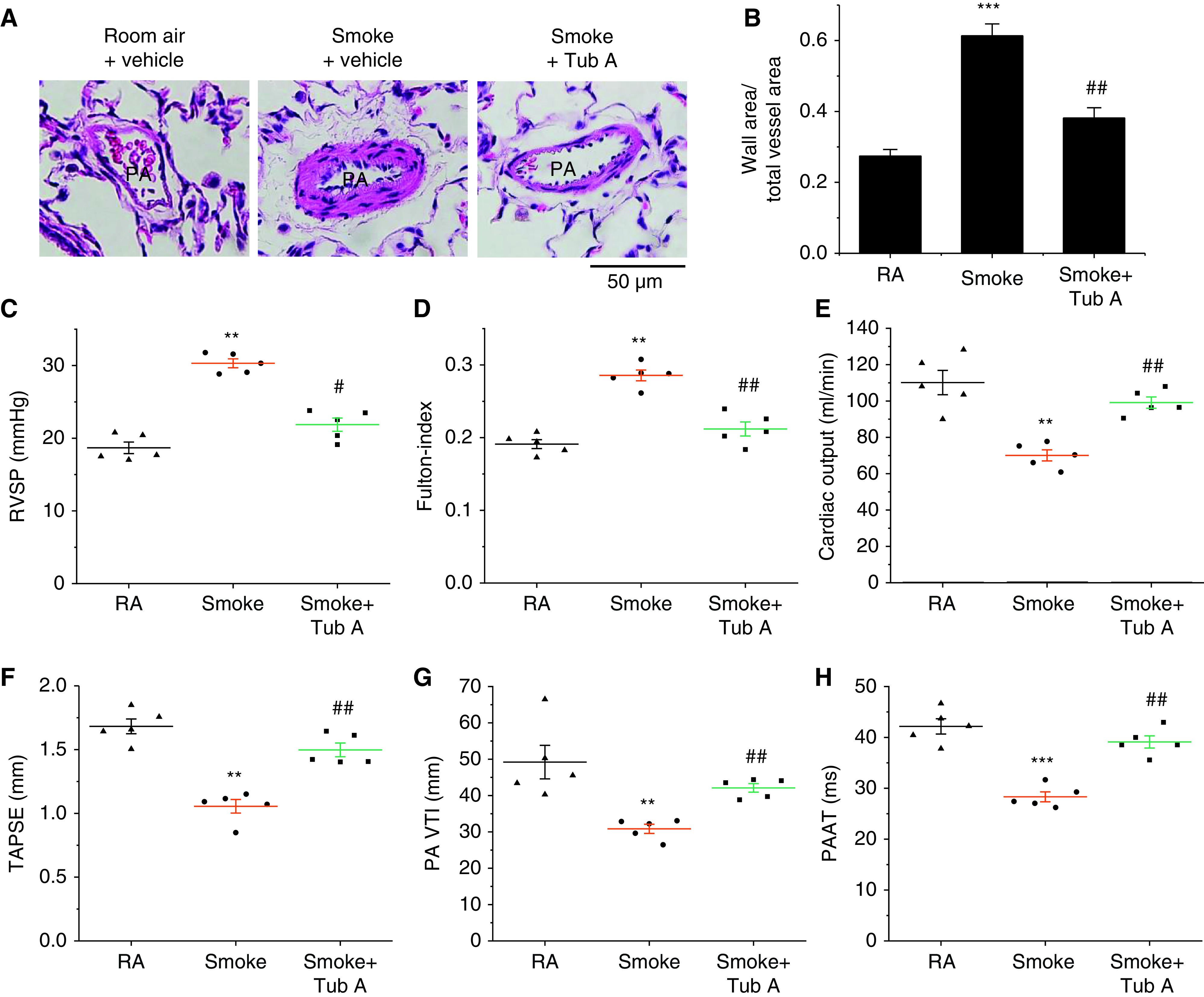
Tub A mitigates the progression of pulmonary vascular remodeling and pulmonary hypertension in a CS-induced COPD model. Eight-week-old male Sprague-Dawley rats were exposed to CS for 16 weeks. At the beginning of the 14th week, CS-exposed rats were injected with Tub A (25 mg/kg, i.p.) or vehicle 5 d/wk for 3 weeks, and then echocardiographic findings, the extent of pulmonary vascular remodeling, and the extent of pulmonary hypertension were assessed. (A) Representative images of lung sections of rats exposed to RA or CS. (B) Changes in the ratio of the vessel wall area to the total vessel area. (C) Changes in RVSP. (D) Changes in the Fulton index [right ventricular weight/(left ventricular weight + septal weight)]. (E–H) Echocardiographic cardiac output, TAPSE, PA VTI, and PAAT results. Results are expressed as means ± SEs; n = 5. **P < 0.01; ***P < 0.001 vs. RA. #P < 0.05; ##P < 0.01 vs. Smoke. PAAT = PA acceleration time; TAPSE = tricuspid annulus plain systolic excursion; RVSP = right ventricular systolic pressure; VTI = velocity–time integral.
In addition, changes in body-weight gain were observed (Figure E13). From the third week of CS exposure, body-weight gain was significantly decreased in the CS-exposed rats compared with the rats exposed to room air. After 12 weeks of CS exposure, CS-exposed rats showed a mild declining trend in body-weight gain, whereas the room air–exposed rats had a continuous increase in body weight. During the 3 weeks of Tub A treatment (between Weeks 13 and 16), inhibition of HDAC6 attenuated the decreases in body-weight gain in CS-exposed rats.
Discussion
We previously reported that ERK1/2 signaling contributes to the elevation of collagen production and BSMC and PASMC proliferation in the airway and to pulmonary vascular remodeling in COPD and that inhibition of ERK1/2 attenuates PH and the thickening of the SMC layer of pulmonary arterioles in the lungs of rats in a Sugen/hypoxia PAH model (11, 26). In the present study, we further studied the mechanism for CS-induced ERK1/2 activation in BSMCs and PASMCs in CS-induced COPD. We found that phosphorylation/activation of ERK1/2 is associated with deacetylation induced by HDAC6, which contributes to the collagen synthesis and BSMC and PASMC proliferation induced by CSE and mediators such as PDGF and IL-6.
In COPD, the components of CS induce inflammation and excessive cell proliferation in the airway and lungs (6, 28). CSE stimulates the proliferation of airway SMCs and extracellular matrix deposition, leading to airway remodeling (29, 30). CSE also induces PASMC proliferation via PDGF activation, contributing to the pulmonary vascular remodeling that leads to PH (31). In addition, the PAs of rats exposed to CS have higher expression levels of PDGF and PDGFRβ (PDGF receptor β). Inhibition of the PDGF receptor blocked the CSE-induced proliferation of PASMCs, indicating that PDGF signaling plays an important role in CS-induced PH (32). Cigarette smoking is also correlated with increased IL-6 expression (33). IL-6 signaling contributes to pulmonary vascular remodeling by promoting the proliferation and survival of PASMCs (34). The signaling mechanism responsible for the CSE-, PDGF-, and IL-6–caused elevation of cell proliferation and collagen production in the airways and for the vascular remodeling of COPD is poorly understood. Our previous studies show that ERK1/2 signaling contributes to the elevation in collagen synthesis and in BSMC and PASMC proliferation in CS-induced COPD (26). Phosphorylation of the threonine and tyrosine residues of ERK1/2 is responsible for the activation of the enzymes (35). In the present study, we revealed that both ERK1 and ERK2 are acetylated in BSMCs and PASMCs. We also found that CSE, PDGF, and IL-6 induce deacetylation of ERK1/2, causing increases in the phosphorylation/activation of ERK1/2. More importantly, inhibition of HDAC6 by using Tub A prevented the decreases in the acetylation levels of ERK 1/2 and the increases in the phosphorylation of the enzymes evoked by CSE and mediators such as PDGF and IL-6. Tub A also diminished the increases in ERK1/2 phosphorylation in BSMCs and PASMCs in COPD rat lungs. In addition, we found that silencing of ERK1/2 by siRNA abolished the CSE-, PDGF-, and IL-6–induced collagen synthesis and BSMC and PASMC proliferation. Collectively, these results indicate that HDAC6 regulates the CSE-, PDGF-, and IL-6–induced increases in the protein levels of collagen-I and BSMC and PASMC proliferation via the deacetylation and phosphorylation/activation of ERK1/2.
Acetylation and phosphorylation are the two major posttranslational modifications. The cross-talk between acetylation and phosphorylation regulates the function of proteins. Our data suggest that HDAC6 deacetylates ERK and increases ERK phosphorylation and activation induced by CSE, PDGF, and IL-6. We speculate that deacetylation of ERK may cause protein conformational changes that make the phosphorylation site more accessible. Indeed, it has been shown that inhibition of HDACs regulates protein phosphorylation in the heart via inducing phosphatase gene expression or activating constitutively expressed phosphatases (36). We cannot exclude that HDAC6 may deacetylate other kinases or phosphatases that regulate ERK phosphorylation and activation.
HDAC6 is a cytoplasmic deacetylase that acts as a master regulator coordinating several cellular processes, including the proliferation, migration, and survival of a variety of cell types (37, 38). In our present study, we found that HDAC6-dependent deacetylation and phosphorylation/activation of ERK1/2 contribute to the CSE-, PDGF-, and IL-6–induced collagen synthesis and BSMC and PASMC proliferation. HDAC6 is upregulated in several diseases and participates in the pathologic processes seen in cancer, cardiovascular disorders, and lung disorders (39–42). We found that the smooth muscle layer of the bronchi and PAs of the lungs of patients with COPD, the lungs of rats with COPD, and the lung homogenates of rats with COPD have higher expression levels of HDAC6. Here, we showed that CSE, PDGF, and IL-6 significantly increased the activity and protein levels of HDAC6 in BSMCs and PASMCs. We also found that inhibition of HDAC6 with the specific inhibitor Tub A or with knockdown of HDAC6 by siRNA diminishes the increases in collagen-I protein levels and BSMC and PASMC proliferation induced by CSE, PDGF, and IL-6. More importantly, administration of the HDAC6 inhibitor Tub A attenuated the elevation of the protein levels of collagen-I in the smooth muscle of the bronchi and pulmonary arterioles and lung homogenates of CS-exposed rats. Moreover, inhibition of HDAC6 by using Tub A suppressed increases in the thickness of the bronchial and pulmonary vascular walls, increases in airway resistance, and PH in a CS-induced COPD model. These observations indicate that HDAC6 has crucial role in the COPD-associated collagen synthesis and SMC proliferation in the airway and pulmonary vasculature.
Notably, our data show that HDAC6 protein levels are similarly increased in BSMCs and PASMCs in patients with COPD and COPD rats, suggesting that this COPD animal model can reproduce the pathologic alterations of human COPD, in which HDAC6 is a critical molecule. Interestingly, we found that administration of the HDAC6 inhibitor Tub A alleviated the CS-induced increases in the mean linear intercept of the lungs and the in inspiratory capacity in a COPD rat model, suggesting that inhibition of HDAC6 suppresses CS-induced emphysema. It has been shown that CS exacerbates acute lung injury in vivo and increases EC barrier dysfunction in vitro and that these outcomes are blunted by the inhibition of HDAC6 (25). Others reported that selective inhibition of HDAC6 by using Tub A blocks TNF-α–induced caspase 3 activation and lung endothelial barrier disruption and prevents endotoxin-induced pulmonary edema (41, 43). Therefore, we speculate that CS exposure may also activate HDAC6 in the alveolar regions, which results in emphysema and increased inspiratory capacity.
In conclusion, our study demonstrates that CSE-, PDGF-, and IL-6–induced overexpression and activation of HDAC6 play a critical role in airway and pulmonary vascular remodeling in the development of COPD associated with CS. HDAC6 mediates deacetylation and activation of ERK1/2 and leads to collagen synthesis and BSMC and PASMC proliferation in COPD (Figure E14). Targeting HDAC6 might be an attractive therapeutic approach for COPD.
Footnotes
Supported by Foundation for the National Institutes of Health grant R01 HL134934 (Y.S.), VA Merit Review Award BX002035 (Y.S.), and American Heart Association Career Development Award 18CDA34110225 (L.K.).
Author Contributions: Conception, design, and experiment: Y.S., W.H., A.K.-K., and L.K. Analysis and interpretation: Y.S, W.H., and L.K. Drafting the manuscript for important intellectual content: Y.S., A.D.V., and L.K.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2020-0520OC on July 19, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Collaborators GBDCRD. GBD Chronic Respiratory Disease Collaborators. Prevalence and attributable health burden of chronic respiratory diseases, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Respir Med. 2020;8:585–596. doi: 10.1016/S2213-2600(20)30105-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Berg K, Wright JL. The pathology of chronic obstructive pulmonary disease: progress in the 20th and 21st centuries. Arch Pathol Lab Med. 2016;140:1423–1428. doi: 10.5858/arpa.2015-0455-RS. [DOI] [PubMed] [Google Scholar]
- 3. Grzela K, Litwiniuk M, Zagorska W, Grzela T. Airway remodeling in chronic obstructive pulmonary disease and asthma: The role of matrix metalloproteinase-9. Arch Immunol Ther Exp (Warsz) 2016;64:47–55. doi: 10.1007/s00005-015-0345-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sakao S, Voelkel NF, Tatsumi K. The vascular bed in COPD: pulmonary hypertension and pulmonary vascular alterations. Eur Respir Rev. 2014;23:350–355. doi: 10.1183/09059180.00007913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sand JM, Leeming DJ, Byrjalsen I, Bihlet AR, Lange P, Tal-Singer R, et al. High levels of biomarkers of collagen remodeling are associated with increased mortality in COPD: results from the ECLIPSE study. Respir Res. 2016;17:125. doi: 10.1186/s12931-016-0440-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Laniado-Laborín R. Smoking and chronic obstructive pulmonary disease (COPD): parallel epidemics of the 21 century. Int J Environ Res Public Health. 2009;6:209–224. doi: 10.3390/ijerph6010209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kovacs L, Su Y. Redox-dependent calpain signaling in airway and pulmonary vascular remodeling in COPD. Adv Exp Med Biol. 2017;967:139–160. doi: 10.1007/978-3-319-63245-2_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. de Boer WI, Alagappan VK, Sharma HS. Molecular mechanisms in chronic obstructive pulmonary disease: potential targets for therapy. Cell Biochem Biophys. 2007;47:131–148. doi: 10.1385/cbb:47:1:131. [DOI] [PubMed] [Google Scholar]
- 9. Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2016;138:16–27. doi: 10.1016/j.jaci.2016.05.011. [DOI] [PubMed] [Google Scholar]
- 10. Churg A, Tai H, Coulthard T, Wang R, Wright JL. Cigarette smoke drives small airway remodeling by induction of growth factors in the airway wall. Am J Respir Crit Care Med. 2006;174:1327–1334. doi: 10.1164/rccm.200605-585OC. [DOI] [PubMed] [Google Scholar]
- 11. Kovacs L, Han W, Rafikov R, Bagi Z, Offermanns S, Saido TC, et al. Activation of calpain-2 by mediators in pulmonary vascular remodeling of pulmonary arterial hypertension. Am J Respir Cell Mol Biol. 2016;54:384–393. doi: 10.1165/rcmb.2015-0151OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cai P, Kovacs L, Dong S, Wu G, Su Y. BMP4 inhibits PDGF-induced proliferation and collagen synthesis via PKA-mediated inhibition of calpain-2 in pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2017;312:L638–L648. doi: 10.1152/ajplung.00260.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kovacs L, Cao Y, Han W, Meadows L, Kovacs-Kasa A, Kondrikov D, et al. PFKFB3 in smooth muscle promotes vascular remodeling in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2019;200:617–627. doi: 10.1164/rccm.201812-2290OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rincon M, Irvin CG. Role of IL-6 in asthma and other inflammatory pulmonary diseases. Int J Biol Sci. 2012;8:1281–1290. doi: 10.7150/ijbs.4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wei J, Xiong XF, Lin YH, Zheng BX, Cheng DY. Association between serum interleukin-6 concentrations and chronic obstructive pulmonary disease: a systematic review and meta-analysis. PeerJ. 2015;3:e1199. doi: 10.7717/peerj.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Agustí A, Edwards LD, Rennard SI, MacNee W, Tal-Singer R, Miller BE, et al. Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) Investigators. Persistent systemic inflammation is associated with poor clinical outcomes in COPD: a novel phenotype. PLoS One. 2012;7:e37483. doi: 10.1371/journal.pone.0037483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gosens R, Roscioni SS, Dekkers BG, Pera T, Schmidt M, Schaafsma D, et al. Pharmacology of airway smooth muscle proliferation. Eur J Pharmacol. 2008;585:385–397. doi: 10.1016/j.ejphar.2008.01.055. [DOI] [PubMed] [Google Scholar]
- 18. Hassoun PM, Mouthon L, Barberà JA, Eddahibi S, Flores SC, Grimminger F, et al. Inflammation, growth factors, and pulmonary vascular remodeling. J Am Coll Cardiol. 2009;54:S10–S19. doi: 10.1016/j.jacc.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 19. Kaliszczak M, Trousil S, Åberg O, Perumal M, Nguyen QD, Aboagye EO. A novel small molecule hydroxamate preferentially inhibits HDAC6 activity and tumour growth. Br J Cancer. 2013;108:342–350. doi: 10.1038/bjc.2012.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tao H, Yang JJ, Hu W, Shi KH, Li J. HDAC6 promotes cardiac fibrosis progression through suppressing RASSF1A expression. Cardiology. 2016;133:18–26. doi: 10.1159/000438781. [DOI] [PubMed] [Google Scholar]
- 21. Boucherat O, Chabot S, Paulin R, Trinh I, Bourgeois A, Potus F, et al. HDAC6: A novel histone deacetylase implicated in pulmonary arterial hypertension. Sci Rep. 2017;7:4546. doi: 10.1038/s41598-017-04874-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Woan KV, Lienlaf M, Perez-Villaroel P, Lee C, Cheng F, Knox T, et al. Targeting histone deacetylase 6 mediates a dual anti-melanoma effect: enhanced antitumor immunity and impaired cell proliferation. Mol Oncol. 2015;9:1447–1457. doi: 10.1016/j.molonc.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vishwakarma S, Iyer LR, Muley M, Singh PK, Shastry A, Saxena A, et al. Tubastatin, a selective histone deacetylase 6 inhibitor shows anti-inflammatory and anti-rheumatic effects. Int Immunopharmacol. 2013;16:72–78. doi: 10.1016/j.intimp.2013.03.016. [DOI] [PubMed] [Google Scholar]
- 24. Wu H, Cheng XW, Hu L, Takeshita K, Hu C, Du Q, et al. Cathepsin S activity controls injury-related vascular repair in mice via the TLR2-mediated p38MAPK and pI3K-AKT/p-HDAC6 signaling pathway. Arterioscler Thromb Vasc Biol. 2016;36:1549–1557. doi: 10.1161/ATVBAHA.115.307110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Borgas D, Chambers E, Newton J, Ko J, Rivera S, Rounds S, et al. Cigarette smoke disrupted lung endothelial barrier integrity and increased susceptibility to acute lung injury via histone deacetylase 6. Am J Respir Cell Mol Biol. 2016;54:683–696. doi: 10.1165/rcmb.2015-0149OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhu J, Kovacs L, Han W, Liu G, Huo Y, Lucas R, et al. Reactive oxygen species-dependent calpain activation contributes to airway and pulmonary vascular remodeling in chronic obstructive pulmonary disease. Antioxid Redox Signal. 2019;31:804–818. doi: 10.1089/ars.2018.7648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Su Y, Han W, Giraldo C, De Li Y, Block ER. Effect of cigarette smoke extract on nitric oxide synthase in pulmonary artery endothelial cells. Am J Respir Cell Mol Biol. 1998;19:819–825. doi: 10.1165/ajrcmb.19.5.3091. [DOI] [PubMed] [Google Scholar]
- 28. Pezzuto A, Citarella F, Croghan I, Tonini G. The effects of cigarette smoking extracts on cell cycle and tumor spread: novel evidence. Future Sci OA. 2019;5:FSO394. doi: 10.2144/fsoa-2019-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guan P, Cai W, Yu H, Wu Z, Li W, Wu J, et al. Cigarette smoke extract promotes proliferation of airway smooth muscle cells through suppressing C/EBP-α expression. Exp Ther Med. 2017;13:1408–1414. doi: 10.3892/etm.2017.4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vogel ER, VanOosten SK, Holman MA, Hohbein DD, Thompson MA, Vassallo R, et al. Cigarette smoke enhances proliferation and extracellular matrix deposition by human fetal airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2014;307:L978–L986. doi: 10.1152/ajplung.00111.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xing AP, Du YC, Hu XY, Xu JY, Zhang HP, Li Y, et al. Cigarette smoke extract stimulates rat pulmonary artery smooth muscle cell proliferation via PKC-PDGFB signaling. J Biomed Biotechnol. 2012;2012:534384. doi: 10.1155/2012/534384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xing AP, Hu XY, Shi YW, Du YC. Implication of PDGF signaling in cigarette smoke-induced pulmonary arterial hypertension in rat. Inhal Toxicol. 2012;24:468–475. doi: 10.3109/08958378.2012.688885. [DOI] [PubMed] [Google Scholar]
- 33. Koo JB, Han JS. Cigarette smoke extract-induced interleukin-6 expression is regulated by phospholipase D1 in human bronchial epithelial cells. J Toxicol Sci. 2016;41:77–89. doi: 10.2131/jts.41.77. [DOI] [PubMed] [Google Scholar]
- 34. Tamura Y, Phan C, Tu L, Le Hiress M, Thuillet R, Jutant EM, et al. Ectopic upregulation of membrane-bound IL6R drives vascular remodeling in pulmonary arterial hypertension. J Clin Invest. 2018;128:1956–1970. doi: 10.1172/JCI96462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Roskoski R., Jr ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res. 2012;66:105–143. doi: 10.1016/j.phrs.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 36. Habibian J, Ferguson BS. The crosstalk between acetylation and phosphorylation: emerging new roles for HDAC inhibitors in the heart. Int J Mol Sci. 2018;20:102. doi: 10.3390/ijms20010102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang SL, Zhu HY, Zhou BY, Chu Y, Huo JR, Tan YY, et al. Histone deacetylase 6 is overexpressed and promotes tumor growth of colon cancer through regulation of the MAPK/ERK signal pathway. OncoTargets Ther. 2019;12:2409–2419. doi: 10.2147/OTT.S194986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang M, Urabe G, Little C, Wang B, Kent AM, Huang Y, et al. HDAC6 regulates the MRTF-A/SRF axis and vascular smooth muscle cell plasticity. JACC Basic Transl Sci. 2018;3:782–795. doi: 10.1016/j.jacbts.2018.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lemon DD, Horn TR, Cavasin MA, Jeong MY, Haubold KW, Long CS, et al. Cardiac HDAC6 catalytic activity is induced in response to chronic hypertension. J Mol Cell Cardiol. 2011;51:41–50. doi: 10.1016/j.yjmcc.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li T, Zhang C, Hassan S, Liu X, Song F, Chen K, et al. Histone deacetylase 6 in cancer. J Hematol Oncol. 2018;11:111. doi: 10.1186/s13045-018-0654-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yu J, Ma Z, Shetty S, Ma M, Fu J. Selective HDAC6 inhibition prevents TNF-α-induced lung endothelial cell barrier disruption and endotoxin-induced pulmonary edema. Am J Physiol Lung Cell Mol Physiol. 2016;311:L39–L47. doi: 10.1152/ajplung.00051.2016. [DOI] [PubMed] [Google Scholar]
- 42. Kovacs L, Kovacs-Kasa A, Verin AD, Fulton D, Lucas R, Su Y. Histone deacetylases in vascular permeability and remodeling associated with acute lung injury. Vessel Plus. 2018;2:15. doi: 10.20517/2574-1209.2018.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yu J, Ma M, Ma Z, Fu J. HDAC6 inhibition prevents TNF-α-induced caspase 3 activation in lung endothelial cell and maintains cell-cell junctions. Oncotarget. 2016;7:54714–54722. doi: 10.18632/oncotarget.10591. [DOI] [PMC free article] [PubMed] [Google Scholar]