

Abstract
The Wnt/β-catenin pathway initiates a signaling cascade that is critical in cell differentiation and the normal development of multiple organ systems. The reactivation of this pathway has been documented in experimental and human idiopathic pulmonary fibrosis, wherein Wnt/β-catenin activation has been implicated in epithelial-cell repair. Furthermore, the canonical ligand Wnt3a is known to induce myofibroblast differentiation; however, the role of noncanonical Wnt ligands remains unclear. This study showed significantly higher levels of Wnt11 expression in cells from both patients with idiopathic pulmonary fibrosis and bleomycin-treated mice, as well as in TGFβ-treated mouse lung fibroblasts. Moreover, Wnt11 induced myofibroblast differentiation as manifested by increased α-SMA (ACTA2) expression, which was similar to that induced by canonical Wnt3a/β-catenin signaling. Further investigation revealed that Wnt11 induction of α-SMA was associated with the activation of JNK (c-Jun N-terminal kinase)/c-Jun signaling and was inhibited by a JNK inhibitor. The potential importance of this signaling pathway was supported by in vivo evidence showing significantly increased levels of Wnt11 and activated JNK in the lungs of mice with bleomycin-induced pulmonary fibrosis. Interestingly, fibroblasts did not express canonical Wnt3a, but treatment of these cells with exogenous Wnt3a induced endogenous Wnt11 and Wnt5a, resulting in repression of the Wnt3a/β-catenin target gene Axin2. These findings suggested that the noncanonical Wnt induction of myofibroblast differentiation mediated by the JNK/c-Jun pathway might play a significant role in pulmonary fibrosis, in addition to or in synergy with canonical Wnt3a/β-catenin signaling. Moreover, Wnt3a activation of noncanonical Wnt signaling might trigger a switch from canonical to noncanonical Wnt signaling to induce myofibroblast differentiation.
Keywords: Wnt signaling, myofibroblast differentiation, ACTA2
The de novo emergence of myofibroblasts is a key feature of tissue repair and remodeling in response to injury (1, 2). In the fibrotic lung, these differentiated myofibroblasts from various precursors are localized in active fibrotic foci and are considered as a predominant cellular source of extracellular matrix and profibrogenic cytokines such as TGF-β (2–4). The persistence of the myofibroblast is believed to be crucial in the propagation of fibrosis with evolution to terminal end-stage fibrotic lung diseases such as idiopathic pulmonary fibrosis (IPF) (5, 6). The mechanism underlying the genesis of the myofibroblast is complex. The induction of α-SMA (ACTA2) is a common key element for determination of myofibroblast differentiation and a major player in contractile force production (1, 2). Although the TGF-β/Smad signaling induction of α-SMA has been extensively studied and established (7, 8), many aspects of the regulation of a-SMA involving other diverse cell-signaling pathways still remain elusive.
Wnt signaling is implicated in many cellular and physiological processes regulating cell proliferation, differentiation, migration, and patterning during development and tissue homeostasis (9, 10). For example, the canonical Wnt/β-catenin pathway initiates a signaling cascade that is critical in cell differentiation and the normal development of multiple organ systems, including the brain, intestines, skin, and lung (11). Recently, the reactivation of this pathway has been documented in experimental and human IPF (12–17). Several cellular functions, such as epithelial-cell proliferation and differentiation, epithelial-to-mesenchymal transition, and fibroblast migration and differentiation, have been linked to Wnt/β-catenin signaling (15, 18, 19). In lung epithelial cells, Wnt/β-catenin activation is reported to lead to a proliferative response and increased Wnt target-gene expression, consistent with its involvement in epithelial-cell repair (12, 20–22). On the other hand, participation of Wnt/β-catenin in induction of myofibroblast differentiation is expected to delay resolution and contribute to chronic lung pathologies, including fibrosis (12, 16, 23, 24). It has been suggested that sustained activation of the pathway may drive inflammation and fibrotic changes in the lung (12, 20, 25, 26). The underlying mechanism for sustained activation is not clear. In addition to this canonical Wnt signaling pathway, noncanonical Wnt signaling has recently also been implicated in cardiomyocyte differentiation and chronic lung pathologies (15, 27–29). Induced overexpression of Wnt11 in bone marrow–derived mesenchymal stem cells (MSCs) significantly increases the expression of cardiac differentiation markers and promotes MSC transdifferentiation into myocardial phenotypes by upregulating GATA-4 (27). Furthermore, Wnt11-overexpressing MSCs play a paracrine role in cardiac progenitor-cell differentiation to cardiomyocytes (30, 31). Moreover, TGFβ enhances mesenchymal gene expression in renal epithelial cells, which is associated with upregulation of Wnt11 (19). Another noncanonical component, Wnt5a, is reported to be increased in fibroblasts obtained from fibrotic lung tissue and is suggested to play a role in fibroblast expansion and survival by regulating fibroblast proliferation in a manner that is independent of canonical Wnt/β-catenin signaling (15). However, the precise role of noncanonical Wnt signaling in pulmonary fibrosis, especially in fibroblasts, is unclear.
In this study, we sought to investigate the possible role of the noncanonical ligand Wnt11 in lung myofibroblast differentiation and its potential significance in pulmonary fibrosis. Our findings suggested that noncanonical Wnt11/5a induced lung myofibroblast differentiation, which appeared to be mediated by the JNK (c-Jun N-terminal kinase)/c-Jun pathway. Moreover, induction of Wnt11 together with upregulation of JNK/c-Jun signaling in bleomycin (BLM)-induced pulmonary fibrosis suggested potential in vivo pathophysiological relevance. Treatment with the canonical ligand Wnt3a or TGFβ induced Wnt11/5a expression and myofibroblast differentiation in lung fibroblasts, which was inhibited by knockdown of Wnt11. Interestingly Wnt11 inhibited β-catenin target-gene expression, suggesting that this noncanonical ligand inhibits canonical Wnt signaling while inducing myofibroblast differentiation. These findings suggest a significant role for noncanonical Wnt signaling in mediating the myofibroblast differentiation induced by known ligands, including the canonical Wnt ligand Wnt3a.
Methods
Mice and BLM Model of Pulmonary Fibrosis
Six-week-old female C57BL/6 mice were purchased from The Jackson Laboratory. To induce pulmonary fibrosis, BLM (Blenoxane) was dissolved in sterile PBS and instilled endotracheally at a dose of 2 U/kg of body weight. Control groups received PBS only (five mice per group). At the indicated time points after BLM treatment, the mice were killed, and the lungs were harvested rapidly for tissue lysate preparation and fibroblast isolation. All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee at the University of Michigan.
Lung Fibroblast Isolation
Primary mouse lung fibroblasts and type II alveolar epithelial cells (AECIIs) were isolated from PBS- or BLM-treated mouse lung tissues and maintained as before (32, 33). Fibroblasts at passages 2–5 or primary AECIIs were used in the indicated experiments. Deidentified primary human lung fibroblasts (HLFs) isolated from control subjects or patients with IPF were kindly provided by Dr. Craig A. Henke (University of Minnesota) and were maintained in Dulbecco’s modified Eagle medium supplemented with 10% FBS (Sigma-Aldrich).
Fibroblast Treatment and siRNA Transfection
Fibroblasts at ∼85% confluence were treated with recombinant TGFβ (5 ng/ml), Wnt3a (200 ng/ml), Wnt11 (200 or 400 ng/ml), Wnt5a (200 or 400 ng/ml) (R&D Systems), or CHIR99021 (5μM) (Sigma-Aldrich) at the indicated time points. When indicated, 10μM SP600125 (c-JNK inhibitor) was added 24 hours before the treatment with these ligands. In fibroblast transfection experiments, predesigned siRNAs for Wnt5a, Wnt11, and β-catenin and the negative control siRNAs were purchased from Ambion, Inc. Primary cultured mouse lung fibroblasts were transfected with 100nM siRNA by using electroporation (Nucleofector II, Amaxa Biosystems). After 16 hours, recombinant Wnt3a, Wnt11, or TGFβ was added, and the cells were incubated for another 48 hours.
Quantitative PCR and Western Blotting
For quantitative PCR analysis, TaqMan primers/probes for human α-SMA (ACTA2), human or mouse Wnt3a (WNT3a or Wnt3a), Wnt11 (WNT11 or Wnt11), human Wnt5a (WNT5A or Wnt5a), human Axin2 (AXIN2), human β-catenin (CTNNB1), and 18s ribosomal RNA were purchased from Thermo Fisher Scientific. 18s RNA was used as internal control for normalization. One-step RT-PCR was performed on a GeneAmp 7500 sequence-detection system (Applied Biosystems). Results were calculated by using the 2−ΔΔCT method with the indicated control group as a calibrator and were expressed as the fold change relative to the respective control groups.
For Western blotting assays, anti–α-SMA (catalog number A2547; Sigma-Aldrich), anti-Wnt11 (catalog number BBP1-31406; Novus Biologicals, LLC), anti–β-catenin (catalog number 05-665; EMD Millipore), anti–JNK2/p-JNK (Thr183/Tyr185) (catalog numbers 9258 and 9255; Cell Signaling Technology), anti–c-Jun/p-c-Jun (Ser63) (catalog numbers 9165 and 9261, Cell Signaling Technology), and anti-GAPDH (catalog number G9295; Sigma-Aldrich) antibodies were used. All original full blots with multiple samples are provided in the data supplement.
Statistical Analysis
All data were expressed as the mean ± SD unless otherwise indicated. Differences between the means of various treatment and control groups were assessed for statistical significance by using ANOVA followed by post hoc analysis with Scheffé’s test. A P value of <0.05 was considered to indicate statistical significance.
Results
Wnt 11 Was Induced in Lung Fibroblasts during Injury and Fibrosis
Noncanonical Wnt ligand Wnt 11 induces bone marrow–derived MSC transdifferentiation into myocardial phenotypes (27). To explore its potential role in lung myofibroblast differentiation, Wnt11 was first examined in lung fibroblasts isolated from a mouse fibrosis model or from patients with IPF. The results showed that Wnt11 mRNA was significantly induced in the fibroblasts isolated from the lungs of mice undergoing BLM-induced pulmonary fibrosis relative to those in cells from control mouse lungs. Similarly, a greater than twofold Wnt11 induction was noted in HLFs isolated from patients with IPF relative to those from control subjects (Figure 1A). The induction of Wnt11 expression was also observed at the protein level in mouse fibrotic fibroblasts compared with controls, which paralleled the increase in α-SMA expression (Figure 1B). Furthermore, both Wnt11 expression and α-SMA expression were also induced by TGFβ treatment in vitro. These findings indicated that noncanonical ligand Wnt11 was induced in fibrotic lung fibroblasts, which was associated with myofibroblast differentiation during pulmonary fibrosis.
Figure 1.

Wnt11 induction in fibrotic fibroblasts. (A) Lung fibroblasts were isolated from control (Cont) subjects or patients with idiopathic pulmonary fibrosis (IPF) as well as from Cont mice or bleomycin (BLM)-injured mice. Total cellular RNA was analyzed for Wnt11 mRNA expression by using qPCR analysis. Wnt11 mRNA levels were calculated by using the 2−ΔΔCT method with 18s rRNA as an internal Cont for normalization and were shown as the fold change relative to their respective Conts. **P < 0.01 between the two indicated groups (N = 3). (B) The primary cultured mouse lung fibroblasts (MLFs) (passage 2) isolated as described in A were treated with 5 ng/ml TGFβ in vitro for 48 hours and were then analyzed for Wnt11 and α-SMA (ACTA2) protein expression by using Western blotting. The GAPDH signal was used as internal Cont. Representative blots from at least three separate experiments are shown. B-MLF = MLF isolated from BLM-treated mice; S-MLF = MLF isolated from PBS-treated mice; TGF = transforming growth factor.
Noncanonical Wnt Signaling Directly Promoted Myofibroblast Differentiation Independently of Canonical Wnt/β-Catenin Signaling
Because myofibroblast differentiation was a key event in fibrosis development, the effect of Wnt11 on α-SMA expression, an indicator of differentiation, was investigated. We found that recombinant Wnt11 was able to significantly induce α-SMA mRNA expression in HLFs. There was a twofold increase caused by treatment with Wnt11 compared with that in untreated HLFs, whereas canonical Wnt3a caused an approximately threefold induction, as expected (Figure 2A). The dose-dependent Wnt11 induction of α-SMA was confirmed by its significant increase at the protein level in both control and IPF HLFs (Figure 2B). The control HLFs showed a greater response to Wnt 11 treatment than the IPF HLFs. As positive controls, TGFβ and the canonical ligand Wnt3a also induced α-SMA expression in HLFs. Similarly, another noncanonical Wnt ligand, Wnt5a, was also able to induce α-SMA mRNA expression dose-dependently in control HLFs (Figure 2C). To determine whether or not the induction of α-SMA expression by noncanonical Wnt11 was dependent on canonical Wnt β-catenin signaling, the effect of β-catenin siRNA on Wnt11-induced α-SMA expression was examined. The results obtained by using quantitative PCR analysis at 16 hours after siRNA transfection showed a greater than 90% knockdown of β-catenin expression, which was also observed at the β-catenin protein level (Figure 2D). Wnt3a induction of α-SMA mRNA in control HLFs was essentially abolished by the β-catenin siRNA (Figure 2E). In contrast, the Wnt11-induced increase in α-SMA mRNA was not significantly affected by β-catenin knockdown. These observations at the mRNA level were confirmed by using Western blotting (Figures 2F and 2G). Thus, in addition to the canonical Wnt induction of myofibroblast differentiation, noncanonical ligands also promoted such differentiation independently of canonical β-catenin signaling.
Figure 2.
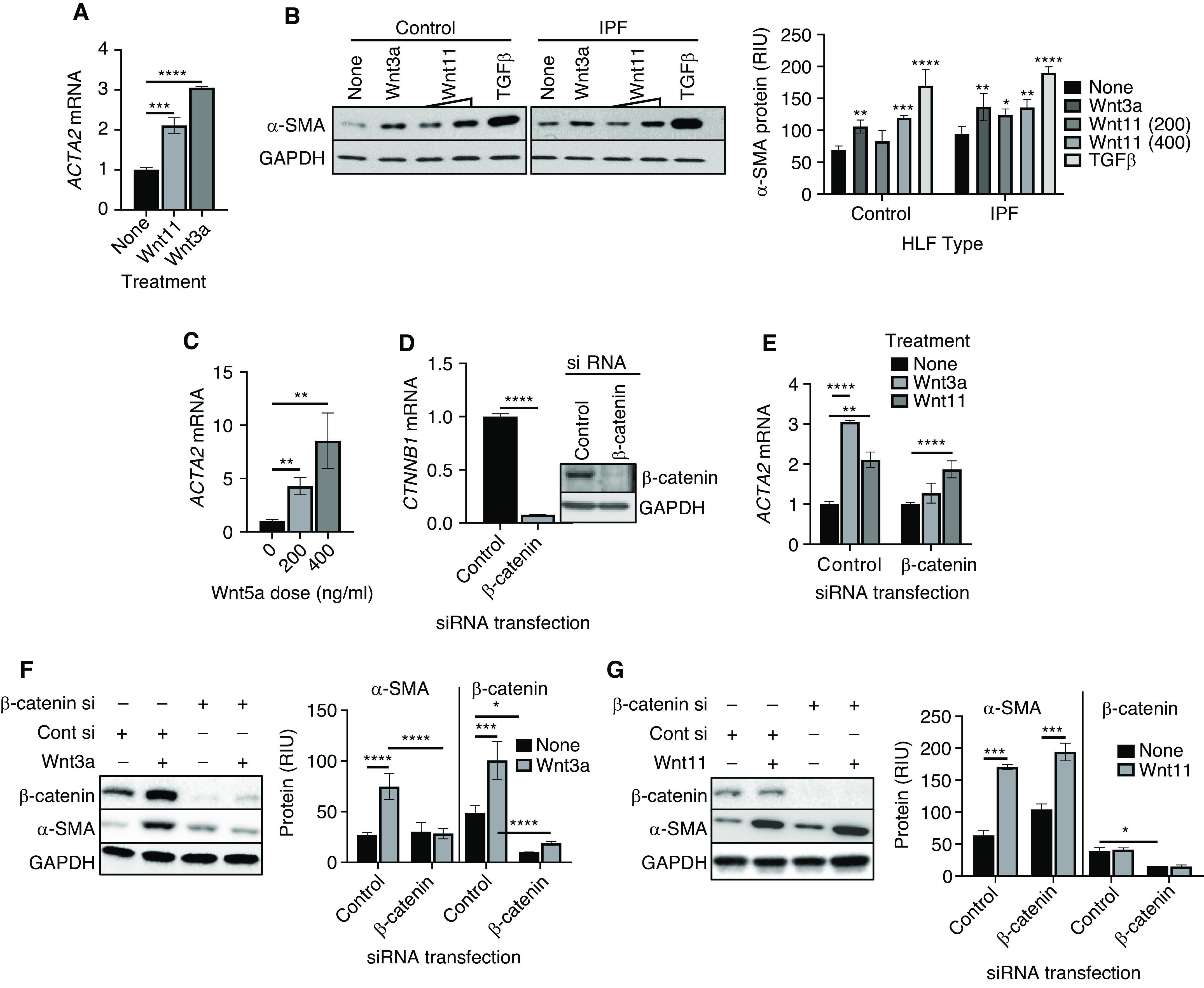
Wnt11 induction of myofibroblast differentiation is β-catenin independent. (A) Normal human lung fibroblasts (HLFs) treated with 200 ng/ml Wnt 3a or 400 ng/ml Wnt11 were analyzed for α-SMA (ACTA2) mRNA by using qPCR analysis, and the results were expressed as the fold change over that in untreated cells. (B) The protein level of α-SMA in HLFs isolated from Cont subjects or patients with IPF treated with Wnt3a (200 ng/ml), Wnt11 (200 and 400 ng/ml), or TGFβ (5 ng/ml) was assayed by using Western blotting. The GAPDH signal was used as internal Cont. The quantified protein expression level was expressed in RIU after normalization to GAPDH signals (bottom). Representative blots from at least three separate experiments are shown. (C) The mRNA level of ACTA2 was analyzed in HLFs treated with Wnt5a after 24 hours. (D) HLFs were transfected with Cont siRNA (Cont si) or siRNA against β-catenin (β-catenin si) as indicated, and the β-catenin mRNA (CTNNB1) and protein in the HLFs were analyzed by using qPCR (left panel) and Western blotting (right panel) analysis. (E) HLFs were treated with Wnt3a or Wnt11 after 16 hours of transfection with Cont si or β-catenin si for another 24 hours and were then analyzed for the ACTA2 gene. With the same treatment used in E, the proteins were analyzed by using Western blotting for HLF β-catenin and α-SMA protein after (F) Wnt3a treatment or (G) β-catenin si and/or Wnt11 treatment, with respective bar graphs summarizing the quantitative analyses. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 between the two indicated groups (N = 3 for all groups). RIU = relative integration units.
The JNK/c-Jun Pathway Mediated Wnt11-induced Myofibroblast Differentiation
Upon noncanonical Wnt stimulation, a number of signaling pathways are activated, including JNK/AP1-dependent planar cell polarity and the Wnt–calcium pathways (34). TGFβ-activated Wnt11 induced mesenchymal gene expression through the JNK pathway (19). To elucidate the signaling pathway that mediates Wnt11 induction of α-SMA, the role of the JNK/c-Jun pathway was studied. We found that Wnt11 was able to activate JNK through phosphorylation of JNK while affecting neither the total level of JNK (Figure 3A) nor the total level of β-catenin (data not shown). This Wnt11 activation of JNK was accompanied by increased phosphorylation of c-Jun, a JNK target protein (Figure 3B). Furthermore, treatment of HLFs with the JNK inhibitor sp600125 abolished c-Jun phosphorylation (Figure 3C), which also suppressed the Wnt11-induced increase of α-SMA expression (Figure 3D) without producing detectable cell toxicity. These findings together indicated that Wnt11 induced myofibroblast differentiation by activating the downstream JNK/c-Jun pathway.
Figure 3.
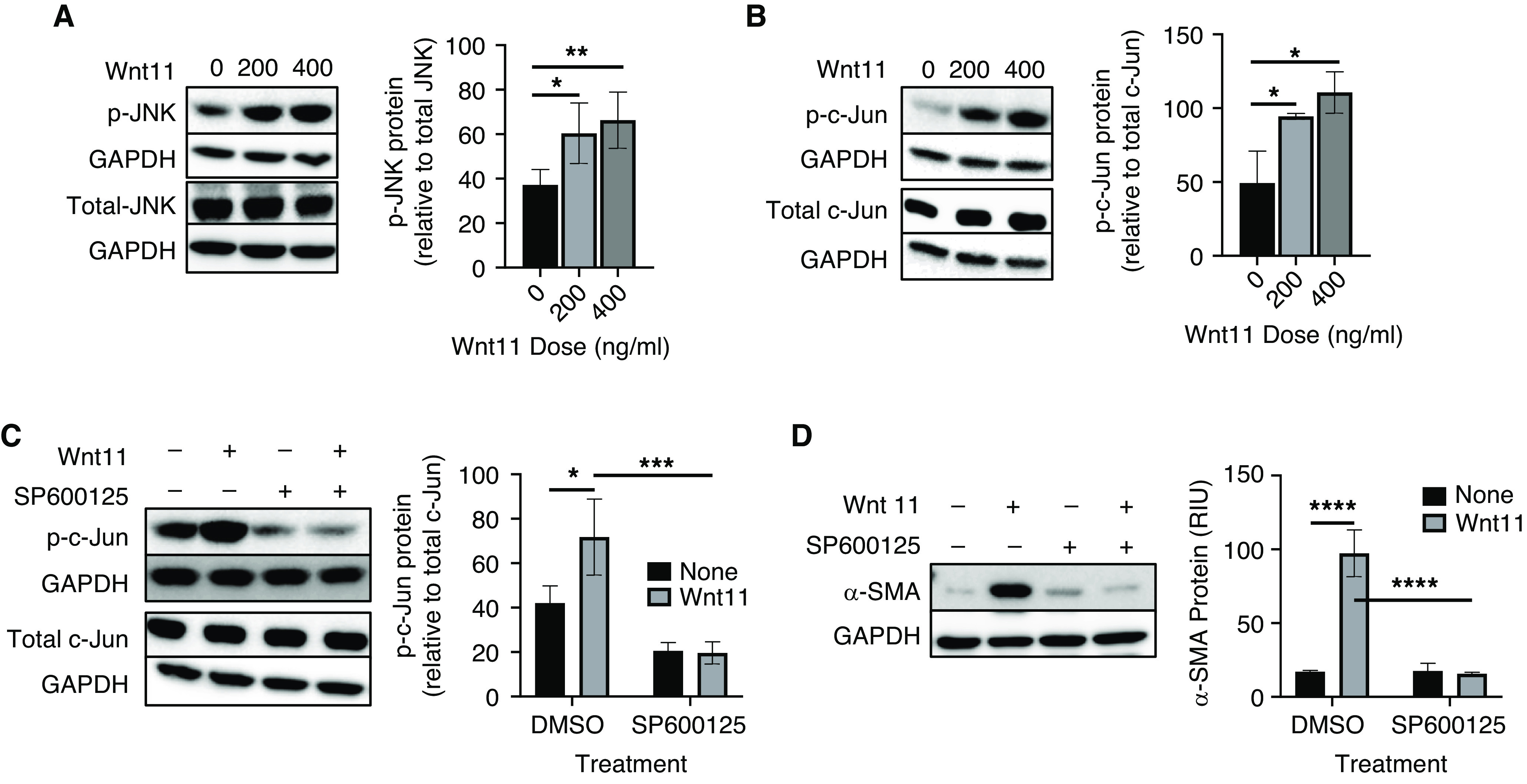
JNK(c-Jun N-terminal kinase)/c-Jun–mediated Wnt11 induction of myofibroblast differentiation. Normal HLFs treated with Wnt11 at the indicated doses were analyzed for total and phosphorylated (A) JNK and (B) c-Jun by using Western blotting. The quantified protein level of p-JNK or p-c-Jun was expressed as the percentage of total JNK or c-Jun after normalization to their respective GAPDH signals. Normal HLFs were also treated with 400 ng/ml Wnt11 in the presence of 10μM sp600125 (JNK inhibitor) and were then analyzed for total and phosphorylated (C) c-Jun as well as (D) α-SMA by using Western blotting. GAPDH signals were used as loading Conts for all the Western blots. Representative blots are shown. N = 4 in A and N = 3 in B–D. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Noncanonical Wnt Signaling Was Activated in BLM-induced Mouse Pulmonary Fibrosis
To evaluate the potential importance of Wnt11 signaling in lung injury and fibrosis, its expression was first analyzed in the lungs of mice with BLM-induced pulmonary fibrosis. The results revealed that the levels of both lung-tissue Wnt11 mRNA and lung-tissue Wnt11 protein were elevated in BLM-treated mice relative to control mice at Day 21 after treatment (Figures 4A and 4B). This increase in Wnt11 expression was accompanied by activation of JNK signaling, as evidenced by the increase in phosphorylated c-JNK (Figure 4C). Because TGF-β is implicated in fibrosis in part by promoting myofibroblast differentiation, we next performed an analysis to see whether Wnt11 mediated this response in lung fibroblasts. Consistent with previous observations (19), TGF-β induced Wnt11, which paralleled induction of α-SMA. This TGFβ induction of α-SMA was abolished when Wnt11 was knocked down at 24 hours after siRNA transfection (Figure 4D). However, the inhibitory effect of Wnt11 knockdown began to wane at 48 hours after siRNA transfection, whereas the effect of TGFβ alone continued to increase for up to 72 hours. Persistence of Wnt11 knockdown for up to 72 hours made it unlikely that this waning inhibition at later time points was due to declining transient knockdown of Wnt11 by the siRNA. The possibility that Wnt11-independent pathways assumed greater importance at later time points could not be ruled out. Nevertheless, these findings signified that Wnt11 might mediate, at least in part and especially during the early stages, the TGF-β induction of α-SMA and lung myofibroblast differentiation. Thus noncanonical Wnt induction of myofibroblast differentiation via the JNK/c-Jun pathway might play a role in vivo during BLM-induced pulmonary fibrosis. To identify possible cellular sources for Wnt ligands, mouse AECIIs and lung fibroblasts, two key cell types involved in lung development and repair, were analyzed for their expression of canonical and noncanonical Wnt ligands. The results showed distinct expression patterns between AECIIs and fibroblasts. In AECIIs, canonical Wnt3a mRNA was expressed at much lower levels than either noncanonical Wnt11 or noncanonical Wnt5a, whereas the Wnt11 mRNA level was more than sixfold higher than that of Wnt5a. In contrast, Wnt3a was undetectable in lung fibroblasts, and the Wnt11 mRNA level was ∼100-times lower than that of Wnt5a (Figure 4E), suggesting that canonical Wnt3a/β-catenin induction of myofibroblast differentiation in vivo might be mainly due to cross-talk between epithelial cells and adjacent fibroblasts.
Figure 4.
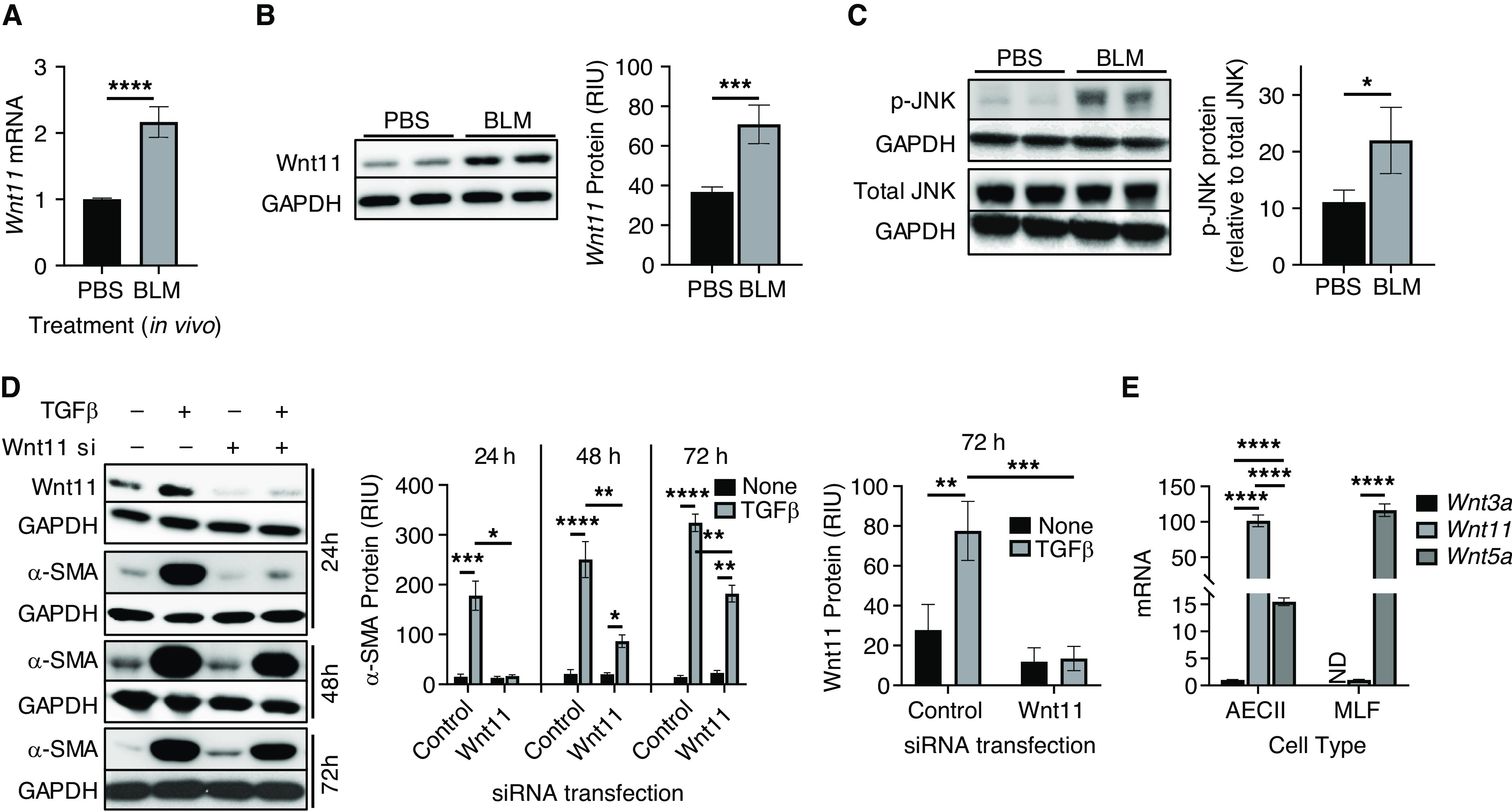
Noncanonical Wnt11 signaling was induced in mouse lung fibrosis. Whole-lung tissue RNA or protein lysates were collected at Day 21 after BLM or PBS treatment and were then analyzed for (A) Wnt11 mRNA by using qPCR analysis and for (B) protein by using Western blotting. (C) Total c-Jun and phosphorylated c-Jun in BLM-treated lung tissue were also assayed by using Western blotting. (D) MLFs were transfected with Cont si or Wnt11 siRNA (Wnt11 si), which was followed by TGFβ treatment. Wnt11 and α-SMA proteins were analyzed at 24, 48, and 72 hours after treatments by using Western blotting, and quantified protein levels normalized to GAPDH signals are shown in the bar graphs. (E) AECIIs and MLFs were analyzed for mRNA expression of Wnt3a, Wnt11, and Wnt5a by using qPCR analysis. N = 4 in A, and N = 3 in D and E. Representative blots are shown in B, C, and D from at least three separate experiments. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. AECII = type II alveolar epithelial cell; ND = not determined.
Endogenous Noncanonical Wnt Signaling Was Activated by Canonical Wnt3a/β-Catenin Signaling
Recent reports have suggested a possible switch between canonical and noncanonical Wnt signaling during disease processes (35–37). Moreover, it has been suggested that activation of β-catenin in alveolar epithelial cells at the early stages of injury may tend toward repair; however, sustained activation of the pathway could drive fibrotic changes instead (12). Interestingly, we found that recombinant Wnt3a was able to induce both the endogenous noncanonical Wnt ligand Wnt11 and Wnt5a expression in a time-dependent manner, with the rate of increase of the former being slower than that of the latter, in both control and IPF HLFs (Figures 5A and B). However, Wnt3a-induced Wnt11 expression in control HLFs was up to >16-fold higher by 48 hours after treatment, whereas such induction was greater than threefold in IPF HLFs. The effect of Wnt3a on Wnt5a mRNA expression was much lower than that of the Wnt11 induction. It reached a twofold increase after 48 hours of treatment in control HLFs, but only an ∼1.3-fold increase in IPF HLFs was observed (Figure 5B). To confirm mediation by canonical Wnt/β-catenin signaling, the effects of a GSK3 inhibitor, CHIR99021, was analyzed by using control HLFs. The results showed that CHIR99021 significantly augmented both Wnt11 and Wnt5a mRNA expression after 6 hours of treatment and reached >18-fold induction for Wnt 11 at 48 hours after treatment (Figure 5C), thus confirming the induction of noncanonical Wnt ligands by canonical Wnt signaling. The Wnt11 induction by Wnt3a was further inspected after the β-catenin gene was knocked down by siRNA in HLFs. The results showed that the high level of Wnt11 mRNA induction by Wnt3a treatment was essentially abrogated upon β-catenin knockdown, which was also observed for Wnt5a mRNA (Figure 5D). This Wnt3a induction of Wnt11 via β-catenin was also observed at Western blotting analysis at the protein level, which also was abolished (Figure 5E) when β-catenin was knocked down (Figure 2F). This was associated with the effects of β-catenin inhibition on α-SMA expression in HLFs (Figure 2F). These data provided evidence that endogenous noncanonical Wnt signaling may be activated as a consequence of the activation of canonical Wnt/β-catenin signaling.
Figure 5.
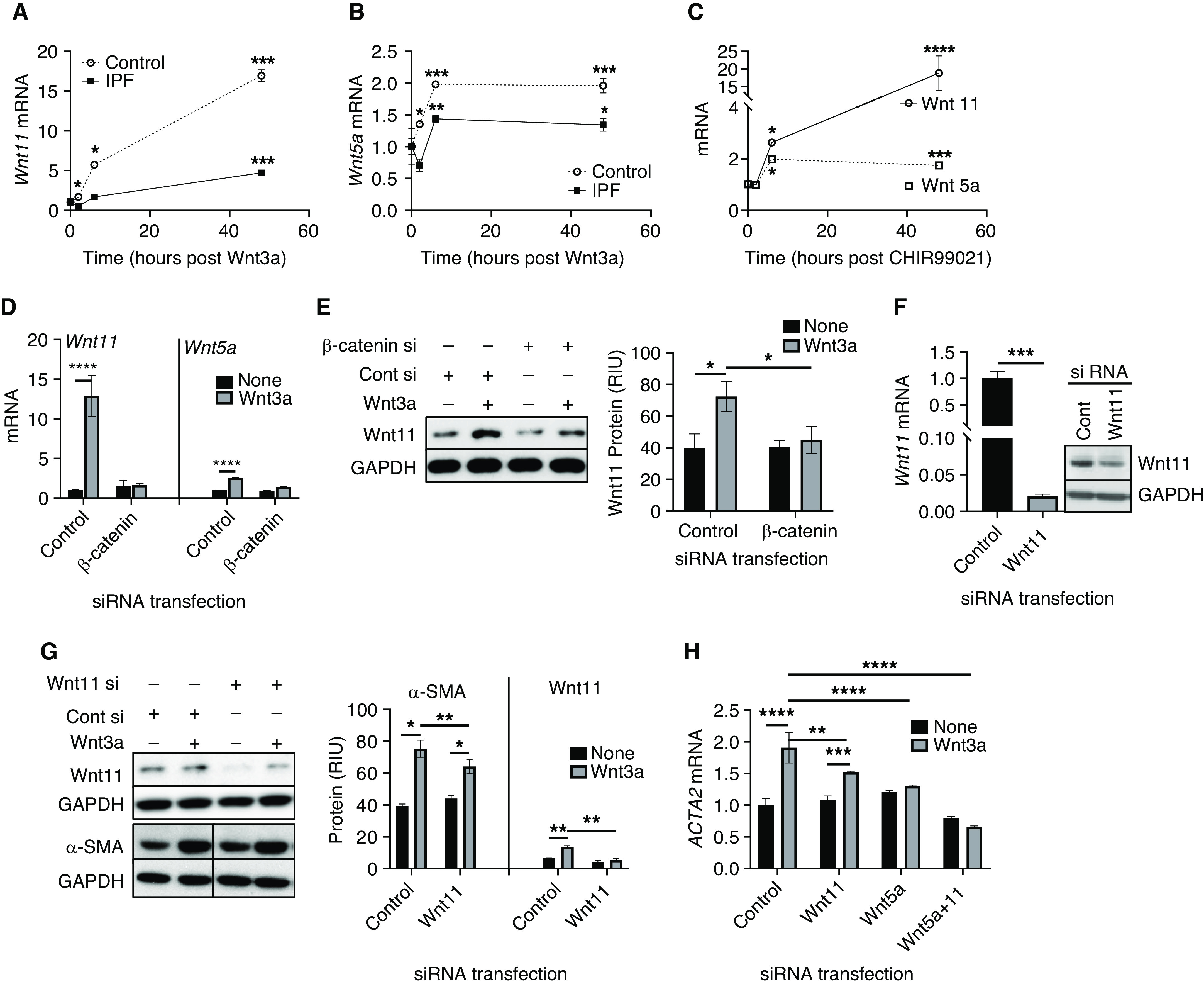
Noncanonical Wnt signaling was induced by canonical Wnt3a/β-catenin. HLFs isolated from patients with IPF or Cont subjects were treated with Wnt3a for 2, 6, or 48 hours. The mRNA level of (A) Wnt11 and (B) Wnt 5a in the treated HLFs were analyzed. (C) The Cont HLFs were treated with 5μM CHIR99021 and analyzed at the same time points that were used in A and B, and the Wnt11 and Wnt5a mRNA levels were analyzed by using qPCR analysis. The normal HLFs were transfected with Cont si or β-catenin si by using electroporation before the addition of Wnt3a. The cells were incubated for another 24 hours before harvesting for (D) the analysis of Wnt11 and Wnt5a mRNAs as well as for (E) the protein level of Wnt11. A representative gel from three separate experiments is shown in E. (F) The normal HLFs were transfected with Wnt11 si as described in D, which was followed by Wnt3a treatment. Wnt11 mRNA and protein were measured in transfected HLFs before Wnt3a treatment to confirm knockdown by the siRNA. A representative gel is shown as the inset. (G) The effects of Wnt3a with or without Wnt11 knockdown by using siRNA on the protein levels of α-SMA were analyzed by using Western blotting. Quantitative data are from three separated experiments, and a representative gel set is shown. (H) The α-SMA mRNA levels in Wnt3a-treated HLFs in the presence of Wnt11, Wnt 5a, or combined Wnt11 si and Wnt5a siRNA at 24 hours were analyzed by using qPCR analysis. N = 3 in all. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
To determine whether Wnt3a induction of α-SMA is also dependent on or mediated by Wnt11, the effect of Wnt11 knockdown before stimulation by Wnt3a was examined. Wnt11 was confirmed to be significantly decreased at both the mRNA and the protein level by siRNA (Figure 5F), without Wnt3a-induced β-catenin expression being affected (data not shown). The Wnt3a-induced β-catenin signaling activation was accompanied by an increase in α-SMA expression, which was reduced significantly by Wnt11 knockdown (Figures 5G and 5H). Because Wnt5a and Wnt11 are both required to induce Isl1 and Nkx2.5 gene expression in differentiating embryoid bodies during second heart field development (38), we also investigated whether Wnt5a also played a role. We found that the Wnt3a-induced increase in α-SMA mRNA was significantly diminished by knockdown of either Wnt 11 or Wnt5a or by knockdown of both simultaneously(Figure 5H). Thus, Wnt3a-induced myofibroblast differentiation was dependent on induction of endogenous noncanonical Wnt11 or 5a acting in an autocrine and paracrine (on adjacent fibroblasts) fashion.
Although Wnt3a induced the noncanonical Wnt ligands Wnt11 or 5a, the expression of Axin2, a direct downstream target of Wnt3a/β-catenin, was significantly inhibited by treatment with exogenous Wnt11 or Wnt5a (Figure 6A). To determine whether Wnt3a-induced endogenous Wnt11 or 5a also has an inhibitory effect on canonical signaling, we examined the effects of Wnt11 or 5a knockdown by siRNA on Wnt3a-induced Axin2 expression. The results showed that Wnt3a-induced Axin2 was further enhanced by Wnt11 or Wnt5a knockdown, with combined Wnt11/5a knockdown showing an even greater effect (Figure 6B). Such an effect would be consistent with a potential Wnt11/5a-dependent negative feedback loop for canonical Wnt3a signaling. These findings together suggested that noncanonical Wnt11 induced myofibroblast differentiation via the JNK signaling pathway while inhibiting the canonical Wnt3a/β-catenin signaling pathway. Furthermore, the reduction in Wnt3a-induced myofibroblast differentiation upon Wnt11 and/or Wnt5a knockdown suggested that the Wnt3a effect might be mediated by noncanonical ligands that were also induced by this canonical ligand.
Figure 6.

Noncanonical Wnt ligands inhibited Wnt3a-induced Axin2 expression. (A) The mRNA level of Axin2 was analyzed in HLFs treated with Wnt3a (200 ng/ml), Wnt11 (400 ng/ml), or Wnt5a (400 ng/ml) for 24 hours. (B) After 24 hours of treatment with Wnt3a without or with Wnt11 si, Wnt5a siRNA, or both, the HLF Axin2 mRNA was analyzed by using qPCR analysis. N = 3. **P < 0.01 and ****P < 0.0001.
Discussion
The differentiated myofibroblast is a key effector cell implicated in extracellular matrix deposition in tissue fibrosis. Understanding its origin and genesis will provide insight for developing therapeutic strategies for the termination or elimination of myofibroblast activation/accumulation (1, 2, 5, 6). Wnt signal reactivation and/or aberrant alterations have been implicated in chronic lung pathologies, including IPF (12). The results from this study suggested that both canonical and noncanonical Wnt signaling could induce myofibroblast differentiation but that they do so via different pathways. Activated noncanonical Wnt11 signaling induced human lung myofibroblast differentiation through the JNK/c-Jun pathway independently of the canonical signaling pathway, as it was not affected by β-catenin deficiency. However canonical Wnt3a-induced differentiation was dependent on β-catenin, which was also essential for induction of Wnt11 by this canonical ligand. It is worth noting that a shift from canonical to noncanonical Wnt signaling may occur during myofibroblast differentiation, as has been reported in the hepatic stellate-cell activation to a myofibroblast phenotype (39). Furthermore, this suggests that Wnt3a-induced differentiation may be mediated by induction of endogenous Wnt11 or 5a, which in turn had an inhibitory effect on canonical Wnt3a signaling in a negative feedback loop. This would be consistent with the reduced canonical pathway–induced differentiation in cells deficient in Wnt11 or 5a. Thus, the importance of β-catenin in Wnt3a induction of myofibroblast differentiation is due to its ability to induce the noncanonical ligands Wnt11 or 5a.
The Wnt proteins are involved in a wide range of biological processes that include cellular differentiation, proliferation, adhesion, survival, and aging (40). A large body of evidence demonstrates that Wnt signaling is reactivated in fibrotic diseases characterized by mesenchymal-cell differentiation (13–15, 19). Its chronic activation may contribute to lung epithelial-cell senescence associated progenitor exhaustion and reprogramming into the fibrotic phenotype (41). Although participation of canonical Wnt/β-catenin signaling pathway in chronic lung pathologies is well documented (16, 17), recent reports highlight that noncanonical Wnt signaling may also significantly contribute to fibrotic disorders (12, 18, 42, 43). The Wnt5b/Fzd8-mediated profibrotic response is independent of β-catenin (14). Increased noncanonical Wnt5a ligand expression mediates extracellular matrix deposition by pulmonary fibroblasts and protect these cells from oxidative stress–induced apoptosis in usual interstitial pneumonia (15). Wnt11 cooperates with TGFβ to induce mesenchymal gene expression in renal epithelial cells that is dependent on the JNK pathway (19). In the human epithelial cell line A549, overexpression of Wnt11 causes a decrease in E-cadherin and an increase in N-cadherin, whereas Wnt11 knockdown by siRNA has the opposite effects (44). Wnt11 plays an important role in cardiogenesis (40, 45, 46). It is sufficient to induce cardiomyogenic differentiation in bone marrow mononuclear cells by inducing heart-specific genes, including Nkx2.5, GATA-4, and β-myosin, and to markedly decrease the expression of pluripotent marker Oct-4 and Nanog (45). A number of downstream signaling pathways are activated upon noncanonical Wnt 5 or 11 stimulation, including the JNK/AP1-dependent planal cell polarity and Wnt–calcium pathways, leading to alterations in the cytoskeleton (34). As a subfamily of the MAPK pathways (47), the activated JNK pathway can phosphorylate many cytoplasmic substrates, such as cytoskeletal and mitochondrial proteins, in addition to regulating a variety of transcription factors, such as c-Jun, c-Fos, ATF-2, and AP-1 (activator protein 1). Subsequently, a wide range of cellular processes are triggered, including cell proliferation, apoptosis, motility, and metabolism (48–50). In the present study, Wnt11 activated JNK and its downstream target protein c-Jun through phosphorylation, and such activation was observed in the BLM-induced fibrotic lung, wherein myofibroblast differentiation is a key event in the fibrotic process. Activation of the JNK/c-Jun pathway has been reported in a preclinical model of pulmonary fibrosis (51, 52) and in human IPF (53). JNK inhibitor CC-930 attenuates collagen gene expression, peribronchiolar collagen deposition, and MMP-7 expression in the lung, BAL fluid, and serum in an animal model of pulmonary fibrosis (52). In addition, JNK inhibitor significantly inhibits CTGF and collagen I expression, as well as corneal scar formation (54). JNK signaling is activated in renal fibrosis and promotes tubular epithelial-cell production of proinflammatory and profibrotic mediators as well as tubular cell dedifferentiation toward a mesenchymal phenotype, perhaps through activation by the Wnt11/TGFβ pathways. In contrast, JNK inhibition decreases Wnt11/TGFβ induction of mesenchymal genes (19, 55). Similarly, JNK is also involved in the pathogenesis of hepatic fibrosis by activating hepatic stellate cells. CCl4-induced hepatic fibrosis is reduced in JNK-deficient animals or by use of a JNK inhibitor. Moreover, patients with chronic hepatitis C who display decreased fibrosis in response to the angiotensin receptor type 1 blocker losartan show decreased JNK phosphorylation (56). These studies suggest that activation of JNK signaling is involved in various organ injuries and fibrosis, in which JNK/c-Jun functions as a central molecular mediator. This provides potential common treatment strategies for these diverse fibrotic diseases (53), although JNK/c-Jun may not be the unique downstream signaling pathway for Wnt11 induction of α-SMA, as other signaling pathways such as the RhoA/ROCK (Rho kinase) pathway could also mediate Wnt11-driven α-SMA expression (57, 58).
The present study suggested that there might be a switch from canonical to noncanonical Wnt signaling that induces myofibroblast differentiation in human cells. In addition to the direct ability to induce myofibroblast differentiation by increasing α-SMA, Wnt3a also activated noncanonical Wnt signaling, perhaps by increasing endogenous Wnt11 and Wnt5a secretion in fibroblasts, a mechanism which was β-catenin dependent. Interestingly, noncanonical Wnt11 or Wnt5a inhibited the Wnt3a downstream target Axin2, which is indicative of inhibition of canonical signaling. Notably, various studies suggest that specific noncanonical WNT ligands are able to inhibit canonical Wnt/β-catenin signaling (41, 59–61). The possible switches from canonical to noncanonical Wnt signaling are implicated in different cellular processes (36, 37, 62, 63). The components of noncanonical Wnt 11 or 5a can suppress the activation of Wnt/β-catenin signaling; for example, Wnt5a plays an active role in the promotion of β-catenin degradation (15), Wnt11-mediated inhibition of canonical signaling in NIH3T3 cells is ligand-specific, and Wnt11 could effectively repress canonical signaling activated by Wnt1, Wnt3, or Wnt3a but not by Wnt7a or Wnt7b (64). A switch from canonical to noncanonical Wnt signaling mediates early differentiation of human neural stem cells. Although a canonical Wnt agonist (GSK3 inhibitor CHIR99021) caused activation of the noncanonical Wnt5a/Ca2+ pathway during retinal progenitor-cell self-renewal (65), Dkk1, a canonical Wnt antagonist, fails to block the effects of Wnt3a on neuronal gene expression, suggesting that these Wnt3a effects are due to activation of noncanonical signaling (35). In a mouse model of cigarette smoke–induced chronic obstructive pulmonary disease, increased Wnt5a levels in whole-lung homogenates are correlated with reduced expressions of β-catenin and the downstream target Axin2. The same study reveals that fibroblast-derived Wnt5a attenuates Wnt3a-induced LRP6 phosphorylation as well as β-catenin–dependent gene transcription (as assessed by using a TOP/FOP-flash assay) in alveolar epithelial cells, including human A549 and mouse MLE12 cells, a process partly mediated by PKC (18). During stem-cell aging–associated differentiation, elevated expression of Wnt5a in aged, long-term hematopoiesis causes a shift from canonical to noncanonical Wnt signaling in mice, which is manifested by depredation of nuclear β-catenin and decreased expression of its target gene Axin2, occurring via the Wnt5a–Cdc42 signaling axis (37). The Wnt 11 promoter contains two conserved Tcf/LEF binding sites, suggesting that canonical Wnt signals and GATA family members can regulate Wnt11 transcription directly (66, 67). Two reports indicate that Tcf-1 and/or Tcf-3 expression levels are changed during the Wnt-induced differentiation of mouse embryonic stem cells and human neural stem cells (35, 68).
During human lung myofibroblast differentiation, the paracrine role of the Wnt 11 ligand is independent of canonical Wnt/β-catenin, whereas endogenous Wnt 11 induction by Wnt 3a was depended on β-catenin signaling. The current observation is consistent with those of other studies showing that Wnt 11 is unable to activate canonical signaling in renal epithelial cells and NIH3T3 fibroblasts (19, 64). These findings implied that Wnt 11 effects might be dependent on the cellular context and microenvironments. Paracrine Wnt11 activation of the noncanonical pathway though JNK/c-Jun might represent an alternative Wnt function that is independent of β-catenin. The stimulation of the secreted signaling molecule Wnt 11 by TGF-β in the epithelial cells (19) was thus implicated to have a potential impact on the microenvironment and the adjacent fibroblasts/myofibroblasts in the lung interstitium through paracrine signaling or a paracrine mechanism. Wnt3a is induced in fibrotic alveolar epithelia (20) but was not detectable in lung fibroblasts. Wnt/β-catenin signaling is essential for stem-cell function, including the progenitor potential of AECIIs under homeostatic conditions, but its prolonged activation may lead to progenitor-cell senescence and reprogramming during continuous injury (41). It has been postulated that the injured epithelia may be the source of the Wnt3a that subsequently activates endogenous Wnt11 or 5a in adjacent fibroblasts. Compared with the mRNA levels of noncanonical Wnt11 and Wnt5a, the mRNA level of Wnt3a is lowest in AECIIs. This would be consistent with the fact that AECIIs with low endogenous Wnt/β-catenin activity exhibit an organoid-forming ability, which is reduced in epithelial progenitor cells from a mouse model of emphysema (69). These findings may also reflect the diverse and often opposing roles of canonical versus noncanonical Wnt signaling in lung epithelial progenitor cells versus mesenchymal cells upon injury and fibrosis.
Taken together, our studies provided evidence that both canonical Wnt signaling and noncanonical Wnt signaling participate in HLF differentiation into myofibroblasts, probably through different pathways, although the canonical pathway–induced differentiation may be mediated by the induction of noncanonical ligands. Noncanonical Wnt11 induction of myofibroblast differentiation mediated by the JNK/c-Jun pathway is Wnt3a/β-catenin independent. Although canonical Wnt3a was able to activate noncanonical Wnt11 signaling via β-catenin, the reverse was not observed; namely, Wnt11 failed to induce Wnt3a. Furthermore, Wnt11 or 5a blocked Wnt3a/β-catenin signaling by decreasing Axin2 expression. These findings indicated there might be a switch from canonical to noncanonical Wnt signaling, which may play a paracrine role in myofibroblast differentiation. Finally, noncanonical Wnt 11 and the downstream JNK/c-Jun pathway were activated in an animal model of pulmonary fibrosis as well as in fibrotic HLFs, suggesting their potential significance in myofibroblast differentiation during fibrosis in vivo.
Footnotes
Supported by the National Heart, Lung, and Blood Institute grants HL112880 and HL138417.
Author Contributions: T.L. and S.H.P. conceived and designed the study. T.L., F.G.D.L.S., M.H., and Z.W. performed the experiments. T.L. and S.H.P. analyzed data and wrote the manuscript.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2020-0499OC on June 8, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmoulière A, Varga J, et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol. 2012;180:1340–1355. doi: 10.1016/j.ajpath.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Phan SH. Genesis of the myofibroblast in lung injury and fibrosis. Proc Am Thorac Soc. 2012;9:148–152. doi: 10.1513/pats.201201-011AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang K, Gharaee-Kermani M, Jones ML, Warren JS, Phan SH. Lung monocyte chemoattractant protein-1 gene expression in bleomycin-induced pulmonary fibrosis. J Immunol. 1994;153:4733–4741. [PubMed] [Google Scholar]
- 4. Zhang K, Rekhter MD, Gordon D, Phan SH. Myofibroblasts and their role in lung collagen gene expression during pulmonary fibrosis: a combined immunohistochemical and in situ hybridization study. Am J Pathol. 1994;145:114–125. [PMC free article] [PubMed] [Google Scholar]
- 5. Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170:1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Phan SH. The myofibroblast in pulmonary fibrosis. Chest. 2002;122(6)(Suppl):286S–289S. doi: 10.1378/chest.122.6_suppl.286s. [DOI] [PubMed] [Google Scholar]
- 7. Hu B, Tack DC, Liu T, Wu Z, Ullenbruch MR, Phan SH. Role of Smad3 in the regulation of rat telomerase reverse transcriptase by TGFβ. Oncogene. 2006;25:1030–1041. doi: 10.1038/sj.onc.1209140. [DOI] [PubMed] [Google Scholar]
- 8. Hu B, Wu Z, Liu T, Ullenbruch MR, Jin H, Phan SH. Gut-enriched Krüppel-like factor interaction with Smad3 inhibits myofibroblast differentiation. Am J Respir Cell Mol Biol. 2007;36:78–84. doi: 10.1165/rcmb.2006-0043OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 10. MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kahn M. Can we safely target the WNT pathway? Nat Rev Drug Discov. 2014;13:513–532. doi: 10.1038/nrd4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Baarsma HA, Königshoff M. ‘WNT-er is coming’: WNT signalling in chronic lung diseases. Thorax. 2017;72:746–759. doi: 10.1136/thoraxjnl-2016-209753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lam AP, Herazo-Maya JD, Sennello JA, Flozak AS, Russell S, Mutlu GM, et al. Wnt coreceptor Lrp5 is a driver of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2014;190:185–195. doi: 10.1164/rccm.201401-0079OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Spanjer AI, Baarsma HA, Oostenbrink LM, Jansen SR, Kuipers CC, Lindner M, et al. TGF-β-induced profibrotic signaling is regulated in part by the WNT receptor Frizzled-8. FASEB J. 2016;30:1823–1835. doi: 10.1096/fj.201500129. [DOI] [PubMed] [Google Scholar]
- 15. Vuga LJ, Ben-Yehudah A, Kovkarova-Naumovski E, Oriss T, Gibson KF, Feghali-Bostwick C, et al. WNT5A is a regulator of fibroblast proliferation and resistance to apoptosis. Am J Respir Cell Mol Biol. 2009;41:583–589. doi: 10.1165/rcmb.2008-0201OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Königshoff M, Balsara N, Pfaff EM, Kramer M, Chrobak I, Seeger W, et al. Functional Wnt signaling is increased in idiopathic pulmonary fibrosis. PLoS One. 2008;3:e2142. doi: 10.1371/journal.pone.0002142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Königshoff M, Kramer M, Balsara N, Wilhelm J, Amarie OV, Jahn A, et al. WNT1-inducible signaling protein-1 mediates pulmonary fibrosis in mice and is upregulated in humans with idiopathic pulmonary fibrosis. J Clin Invest. 2009;119:772–787. doi: 10.1172/JCI33950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Baarsma HA, Skronska-Wasek W, Mutze K, Ciolek F, Wagner DE, John-Schuster G, et al. Noncanonical WNT-5A signaling impairs endogenous lung repair in COPD. J Exp Med. 2017;214:143–163. doi: 10.1084/jem.20160675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang P, Cai Y, Soofi A, Dressler GR. Activation of Wnt11 by transforming growth factor-β drives mesenchymal gene expression through non-canonical Wnt protein signaling in renal epithelial cells. J Biol Chem. 2012;287:21290–21302. doi: 10.1074/jbc.M112.357202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aumiller V, Balsara N, Wilhelm J, Günther A, Königshoff M. WNT/β-catenin signaling induces IL-1β expression by alveolar epithelial cells in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2013;49:96–104. doi: 10.1165/rcmb.2012-0524OC. [DOI] [PubMed] [Google Scholar]
- 21. Flozak AS, Lam AP, Russell S, Jain M, Peled ON, Sheppard KA, et al. β-Catenin/T-cell factor signaling is activated during lung injury and promotes the survival and migration of alveolar epithelial cells. J Biol Chem. 2010;285:3157–3167. doi: 10.1074/jbc.M109.070326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zemans RL, McClendon J, Aschner Y, Briones N, Young SK, Lau LF, et al. Role of β-catenin-regulated CCN matricellular proteins in epithelial repair after inflammatory lung injury. Am J Physiol Lung Cell Mol Physiol. 2013;304:L415–L427. doi: 10.1152/ajplung.00180.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Akhmetshina A, Palumbo K, Dees C, Bergmann C, Venalis P, Zerr P, et al. Activation of canonical Wnt signalling is required for TGF-β-mediated fibrosis. Nat Commun. 2012;3:735. doi: 10.1038/ncomms1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen S, McLean S, Carter DE, Leask A. The gene expression profile induced by Wnt 3a in NIH 3T3 fibroblasts. J Cell Commun Signal. 2007;1:175–183. doi: 10.1007/s12079-007-0015-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tanjore H, Degryse AL, Crossno PF, Xu XC, McConaha ME, Jones BR, et al. β-catenin in the alveolar epithelium protects from lung fibrosis after intratracheal bleomycin. Am J Respir Crit Care Med. 2013;187:630–639. doi: 10.1164/rccm.201205-0972OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Henderson WR, Jr, Chi EY, Ye X, Nguyen C, Tien YT, Zhou B, et al. Inhibition of Wnt/beta-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. Proc Natl Acad Sci USA. 2010;107:14309–14314. doi: 10.1073/pnas.1001520107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. He Z, Li H, Zuo S, Pasha Z, Wang Y, Yang Y, et al. Transduction of Wnt11 promotes mesenchymal stem cell transdifferentiation into cardiac phenotypes. Stem Cells Dev. 2011;20:1771–1778. doi: 10.1089/scd.2010.0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Newman DR, Sills WS, Hanrahan K, Ziegler A, Tidd KM, Cook E, et al. Expression of WNT5A in idiopathic pulmonary fibrosis and its control by TGF-β and WNT7B in human lung fibroblasts. J Histochem Cytochem. 2016;64:99–111. doi: 10.1369/0022155415617988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rydell-Törmänen K, Zhou XH, Hallgren O, Einarsson J, Eriksson L, Andersson-Sjöland A, et al. Aberrant nonfibrotic parenchyma in idiopathic pulmonary fibrosis is correlated with decreased β-catenin inhibition and increased Wnt5a/b interaction. Physiol Rep. 2016;4:e12727. doi: 10.14814/phy2.12727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zuo S, Jones WK, Li H, He Z, Pasha Z, Yang Y, et al. Paracrine effect of Wnt11-overexpressing mesenchymal stem cells on ischemic injury. Stem Cells Dev. 2012;21:598–608. doi: 10.1089/scd.2011.0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Koyanagi M, Haendeler J, Badorff C, Brandes RP, Hoffmann J, Pandur P, et al. Non-canonical Wnt signaling enhances differentiation of human circulating progenitor cells to cardiomyogenic cells. J Biol Chem. 2005;280:16838–16842. doi: 10.1074/jbc.M500323200. [DOI] [PubMed] [Google Scholar]
- 32. Huaux F, Liu T, McGarry B, Ullenbruch M, Phan SH. Dual roles of IL-4 in lung injury and fibrosis. J Immunol. 2003;170:2083–2092. doi: 10.4049/jimmunol.170.4.2083. [DOI] [PubMed] [Google Scholar]
- 33. Yang J, Wheeler SE, Velikoff M, Kleaveland KR, LaFemina MJ, Frank JA, et al. Activated alveolar epithelial cells initiate fibrosis through secretion of mesenchymal proteins. Am J Pathol. 2013;183:1559–1570. doi: 10.1016/j.ajpath.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Montcouquiol M, Crenshaw EB, III, Kelley MW. Noncanonical Wnt signaling and neural polarity. Annu Rev Neurosci. 2006;29:363–386. doi: 10.1146/annurev.neuro.29.051605.112933. [DOI] [PubMed] [Google Scholar]
- 35. Bengoa-Vergniory N, Gorroño-Etxebarria I, González-Salazar I, Kypta RM. A switch from canonical to noncanonical Wnt signaling mediates early differentiation of human neural stem cells. Stem Cells. 2014;32:3196–3208. doi: 10.1002/stem.1807. [DOI] [PubMed] [Google Scholar]
- 36. Bordonaro M, Tewari S, Cicco CE, Atamna W, Lazarova DL. A switch from canonical to noncanonical Wnt signaling mediates drug resistance in colon cancer cells. PLoS One. 2011;6:e27308. doi: 10.1371/journal.pone.0027308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Florian MC, Nattamai KJ, Dörr K, Marka G, Uberle B, Vas V, et al. A canonical to non-canonical Wnt signalling switch in haematopoietic stem-cell ageing. Nature. 2013;503:392–396. doi: 10.1038/nature12631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cohen ED, Miller MF, Wang Z, Moon RT, Morrisey EE. Wnt5a and Wnt11 are essential for second heart field progenitor development. Development. 2012;139:1931–1940. doi: 10.1242/dev.069377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kordes C, Sawitza I, Häussinger D. Canonical Wnt signaling maintains the quiescent stage of hepatic stellate cells. Biochem Biophys Res Commun. 2008;367:116–123. doi: 10.1016/j.bbrc.2007.12.085. [DOI] [PubMed] [Google Scholar]
- 40. Flaherty MP, Kamerzell TJ, Dawn B. Wnt signaling and cardiac differentiation. Prog Mol Biol Transl Sci. 2012;111:153–174. doi: 10.1016/B978-0-12-398459-3.00007-1. [DOI] [PubMed] [Google Scholar]
- 41. Lehmann M, Hu Q, Hu Y, Hafner K, Costa R, van den Berg A, et al. Chronic WNT/β-catenin signaling induces cellular senescence in lung epithelial cells. Cell Signal. 2020;70:109588. doi: 10.1016/j.cellsig.2020.109588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Heijink IH, de Bruin HG, Dennebos R, Jonker MR, Noordhoek JA, Brandsma CA, et al. Cigarette smoke-induced epithelial expression of WNT-5B: implications for COPD. Eur Respir J. 2016;48:504–515. doi: 10.1183/13993003.01541-2015. [DOI] [PubMed] [Google Scholar]
- 43. Januskevicius A, Vaitkiene S, Gosens R, Janulaityte I, Hoppenot D, Sakalauskas R, et al. Eosinophils enhance WNT-5a and TGF-β1 genes expression in airway smooth muscle cells and promote their proliferation by increased extracellular matrix proteins production in asthma. BMC Pulm Med. 2016;16:94. doi: 10.1186/s12890-016-0254-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bartis D, Csongei V, Weich A, Kiss E, Barko S, Kovacs T, et al. Down-regulation of canonical and up-regulation of non-canonical Wnt signalling in the carcinogenic process of squamous cell lung carcinoma. PLoS One. 2013;8:e57393. doi: 10.1371/journal.pone.0057393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Flaherty MP, Abdel-Latif A, Li Q, Hunt G, Ranjan S, Ou Q, et al. Noncanonical Wnt11 signaling is sufficient to induce cardiomyogenic differentiation in unfractionated bone marrow mononuclear cells. Circulation. 2008;117:2241–2252. doi: 10.1161/CIRCULATIONAHA.107.741066. [DOI] [PubMed] [Google Scholar]
- 46. Flaherty MP, Dawn B. Noncanonical Wnt11 signaling and cardiomyogenic differentiation. Trends Cardiovasc Med. 2008;18:260–268. doi: 10.1016/j.tcm.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Arthur JS, Ley SC. Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol. 2013;13:679–692. doi: 10.1038/nri3495. [DOI] [PubMed] [Google Scholar]
- 48. Johnson GL, Nakamura K. The c-jun kinase/stress-activated pathway: regulation, function and role in human disease. Biochim Biophys Acta. 2007;1773:1341–1348. doi: 10.1016/j.bbamcr.2006.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sabapathy K. Role of the JNK pathway in human diseases. Prog Mol Biol Transl Sci. 2012;106:145–169. doi: 10.1016/B978-0-12-396456-4.00013-4. [DOI] [PubMed] [Google Scholar]
- 50. Yang SH, Sharrocks AD, Whitmarsh AJ. MAP kinase signalling cascades and transcriptional regulation. Gene. 2013;513:1–13. doi: 10.1016/j.gene.2012.10.033. [DOI] [PubMed] [Google Scholar]
- 51. Alcorn JF, van der Velden J, Brown AL, McElhinney B, Irvin CG, Janssen-Heininger YM. c-Jun N-terminal kinase 1 is required for the development of pulmonary fibrosis. Am J Respir Cell Mol Biol. 2009;40:422–432. doi: 10.1165/rcmb.2008-0174OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. van der Velden JL, Ye Y, Nolin JD, Hoffman SM, Chapman DG, Lahue KG, et al. JNK inhibition reduces lung remodeling and pulmonary fibrotic systemic markers. Clin Transl Med. 2016;5:36. doi: 10.1186/s40169-016-0117-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wernig G, Chen SY, Cui L, Van Neste C, Tsai JM, Kambham N, et al. Unifying mechanism for different fibrotic diseases. Proc Natl Acad Sci USA. 2017;114:4757–4762. doi: 10.1073/pnas.1621375114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shi L, Chang Y, Yang Y, Zhang Y, Yu FS, Wu X. Activation of JNK signaling mediates connective tissue growth factor expression and scar formation in corneal wound healing. PLoS One. 2012;7:e32128. doi: 10.1371/journal.pone.0032128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Grynberg K, Ma FY, Nikolic-Paterson DJ. The JNK signaling pathway in renal fibrosis. Front Physiol. 2017;8:829. doi: 10.3389/fphys.2017.00829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kluwe J, Pradere JP, Gwak GY, Mencin A, De Minicis S, Osterreicher CH, et al. Modulation of hepatic fibrosis by c-Jun-N-terminal kinase inhibition. Gastroenterology. 2010;138:347–359. doi: 10.1053/j.gastro.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kumawat K, Koopmans T, Menzen MH, Prins A, Smit M, Halayko AJ, et al. Cooperative signaling by TGF-β1 and WNT-11 drives sm-α-actin expression in smooth muscle via Rho kinase-actin-MRTF-A signaling. Am J Physiol Lung Cell Mol Physiol. 2016;311:L529–L537. doi: 10.1152/ajplung.00387.2015. [DOI] [PubMed] [Google Scholar]
- 58. Toyama T, Lee HC, Koga H, Wands JR, Kim M. Noncanonical Wnt11 inhibits hepatocellular carcinoma cell proliferation and migration. Mol Cancer Res. 2010;8:254–265. doi: 10.1158/1541-7786.MCR-09-0238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mikels AJ, Nusse R. Purified Wnt5a protein activates or inhibits beta-catenin-TCF signaling depending on receptor context. PLoS Biol. 2006;4:e115. doi: 10.1371/journal.pbio.0040115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nemeth MJ, Topol L, Anderson SM, Yang Y, Bodine DM. Wnt5a inhibits canonical Wnt signaling in hematopoietic stem cells and enhances repopulation. Proc Natl Acad Sci USA. 2007;104:15436–15441. doi: 10.1073/pnas.0704747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Upadhyay M, Kuna M, Tudor S, Martino Cortez Y, Rangan P. A switch in the mode of Wnt signaling orchestrates the formation of germline stem cell differentiation niche in Drosophila. PLoS Genet. 2018;14:e1007154. doi: 10.1371/journal.pgen.1007154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Davis LA, Zur Nieden NI. Mesodermal fate decisions of a stem cell: the Wnt switch. Cell Mol Life Sci. 2008;65:2658–2674. doi: 10.1007/s00018-008-8042-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Keats EC, Dominguez JM, II, Grant MB, Khan ZA. Switch from canonical to noncanonical Wnt signaling mediates high glucose-induced adipogenesis. Stem Cells. 2014;32:1649–1660. doi: 10.1002/stem.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Maye P, Zheng J, Li L, Wu D. Multiple mechanisms for Wnt11-mediated repression of the canonical Wnt signaling pathway. J Biol Chem. 2004;279:24659–24665. doi: 10.1074/jbc.M311724200. [DOI] [PubMed] [Google Scholar]
- 65. Jin C, Ou Q, Li Z, Wang J, Zhang J, Tian H, et al. The combination of bFGF and CHIR99021 maintains stable self-renewal of mouse adult retinal progenitor cells. Stem Cell Res Ther. 2018;9:346. doi: 10.1186/s13287-018-1091-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Katoh M, Katoh M. Integrative genomic analyses of WNT11: transcriptional mechanisms based on canonical WNT signals and GATA transcription factors signaling. Int J Mol Med. 2009;24:247–251. doi: 10.3892/ijmm_00000227. [DOI] [PubMed] [Google Scholar]
- 67. Uysal-Onganer P, Kypta RM. Wnt11 in 2011: the regulation and function of a non-canonical Wnt. Acta Physiol (Oxf) 2012;204:52–64. doi: 10.1111/j.1748-1716.2011.02297.x. [DOI] [PubMed] [Google Scholar]
- 68. Osei-Sarfo K, Gudas LJ. Retinoic acid suppresses the canonical Wnt signaling pathway in embryonic stem cells and activates the noncanonical Wnt signaling pathway. Stem Cells. 2014;32:2061–2071. doi: 10.1002/stem.1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hu Y, Ng-Blichfeldt JP, Ota C, Ciminieri C, Ren W, Hiemstra PS, et al. Wnt/β-catenin signaling is critical for regenerative potential of distal lung epithelial progenitor cells in homeostasis and emphysema. Stem Cells. 2020;38:1467–1478. doi: 10.1002/stem.3241. [DOI] [PMC free article] [PubMed] [Google Scholar]