

Abstract
Human rhinovirus (RV) is a major risk factor for chronic obstructive pulmonary disease (COPD) and asthma exacerbations. The exploration of RV pathogenesis has been hampered by a lack of disease-relevant model systems. We performed a detailed characterization of host responses to RV infection in human lung tissue ex vivo and investigated whether these responses are disease relevant for patients with COPD and asthma. In addition, impact of the viral replication inhibitor rupintrivir was evaluated. Human precision-cut lung slices (PCLS) were infected with RV1B with or without rupintrivir. At Days 1 and 3 after infection, RV tissue localization, tissue viability, and viral load were determined. To characterize host responses to infection, mediator and whole genome analyses were performed. RV successfully replicated in PCLS airway epithelial cells and induced both antiviral and proinflammatory cytokines such as IFNα2a, CXCL10, CXCL11, IFN-γ, TNFα, and CCL5. Genomic analyses revealed that RV not only induced antiviral immune responses but also triggered changes in epithelial cell–associated pathways. Strikingly, the RV response in PCLS was reflective of gene expression changes described in patients with COPD and asthma. Although RV-induced host immune responses were abrogated by rupintrivir, RV-triggered epithelial processes were largely refractory to antiviral treatment. Detailed analysis of RV-infected human PCLS and comparison with gene signatures of patients with COPD and asthma revealed that the human RV PCLS model represents disease-relevant biological mechanisms that can be partially inhibited by a well-known antiviral compound and provide an outstanding opportunity to evaluate novel therapeutics.
Keywords: rhinovirus, human lung, epithelial response, asthma, COPD
Viral infections are a major trigger of acute respiratory diseases, with human rhinovirus (RV) being the most prominent pathogen producing common cold symptoms (1). In healthy individuals, RV infections are usually self-limiting to the upper respiratory tract. In contrast, in patients with asthma or chronic obstructive pulmonary disease (COPD), underlying pathological changes such as disrupted epithelial layers, ciliary dysfunction, and mucus hypersecretion facilitate transmission of the virus from the upper to the lower respiratory tract, leading to more severe, prolonged infections (2). Respiratory viruses have been detected in up to 80% of patients with asthma and severe acute exacerbations and in 22–64% of patients with exacerbating COPD (2, 3). In experimentally infected patients with asthma and COPD, RV both augmented and prolonged symptoms, including cough, shortness of breath, wheezing, and airflow obstruction (4–6). In addition, RV infection resulted in secondary bacterial infections in 60% of infected patients with COPD (7).
RV is a small, nonenveloped positive single-strand RNA virus (8) infecting respiratory epithelial cells as the primary host cells (3, 9). The infected airway epithelium rapidly elicits an immune response by activating signaling pathways via Toll-like receptors (TLRs) and RIG-I-like receptors (RLRs). This activation results in secretion of antiviral and proinflammatory cytokines (e.g., type I and type III IFN [9–11], IL-1β, and IL-8 [8]). Collectively, these proteins restrict viral replication, promote apoptosis of infected cells, and recruit innate and adaptive immune cells (12–14).
Although current standard-of-care therapies are effective in reducing respiratory symptoms in patients with COPD and asthma, they are less effective in virus-induced disease exacerbations. Therefore, there is a clear unmet need for novel therapeutics that can address virus-induced pathophysiological mechanisms, in particular those that contribute to disease progression in patients with COPD and asthma. To discover new therapeutic targets, an increased understanding of the underlying disease mechanisms is needed. Given the important role of both the airway epithelium and immune cells as drivers of the antiviral response, we used RV infection of human precision-cut lung slices (PCLS) to start addressing these complex interactions in the natural lung microenvironment.
In this study, we provide, for the first time, an in-depth characterization of the host response after successful RV infection of human lower respiratory tract tissue ex vivo using whole genome analysis. Moreover, we compare the RV response observed in lung slices with gene signatures of bronchial brushings from patients with COPD and asthma and identified significant overlap. The antiviral compound rupintrivir inhibited disease-relevant immune responses but left RV-induced epithelial cell changes unaffected. We propose that RV infection of human lung slices may provide a unique opportunity to evaluate novel therapeutics. Some of the results of these studies have been previously reported in the form of abstracts (15, 16).
Methods
Human PCLS Sample Acquisition and Preparation
Human lung lobes were obtained from male and female patients (average age 63 ± 8 years) undergoing surgery (for details, see Table E1 in the data supplement). The human biological samples were sourced ethically, and their research use was in accordance with the terms of the informed consent under Ethics Committee-approved protocol 2701–2015 (Medical School Hannover, Germany). PCLS were prepared as previously described (17).
PCLS Infection and Antiviral Treatment
Human RV serotype 1B (RV1B) was obtained from the Health Protection Agency (National Collection of Pathogenic Viruses #624). Virus replication and purification were performed as previously described (18). PCLS were infected with 1 × 106 IU/mL RV1B by inoculation at 33°C and 5% CO2. Controls were sham-infected with ultraviolet (UV)-inactivated virus or incubated with medium only. After 2 hours, the supernatant was replaced by fresh medium and PCLS cultured for 1 or 3 days. PCLS were treated with 100 nM rupintrivir (viral 3C protease inhibitor; Axon Medchem BV) or respective medium control (with 0.006% DMSO). Tissue viability was determined using lactate dehydrogenase (LDH) cytotoxicity assay and LIVE/DEAD staining (Life Technologies) as described previously (17).
Immunofluorescence Staining
PCLS were fixed overnight in paraformaldehyde at 4°C, and immunohistochemical stainings were performed as detailed in the data supplement. Briefly, PCLS were stained for double-stranded RNA (dsRNA) to detect replicating virus and pan-cytokeratin to visualize airway epithelial cells. Imaging analysis was performed with confocal laser scanning microscope LSM510 Meta (Zeiss) and IMARIS 7.6.0 software (Bitplane Scientific Software).
Immunohistochemistry Staining
Paraformaldehyde-fixed PCLS were embedded in paraffin. Subsequent sections were stained for Krt5 and digitized with the Hamamatsu S-210 (Hamamatsu), and whole slide images were analyzed using Visiopharm Software.
Mediator Analyses
Mediator analysis was performed from supernatant or PCLS lysed with 1% Triton X-100/PBS using MSD multiplex (MesoScaleDiscovery) and ELISA (R&D DuoSet). Total protein content was analyzed with BCA Protein Assay Kit (Thermo Fisher Scientific).
RNA Isolation and Whole Genome Transcriptomic Analysis
Total RNA was isolated from flash-frozen PCLS, and RNA quantity and quality were determined by spectrophotometry and Agilent 2100 Bioanalyzer. All samples had acceptable RNA integrity numbers (range, 7.1–9.6; average, 8.7). Total RNA was converted into double-stranded cDNA and amplified, and HG‐U133_Plus_2.0 microarrays (Affymetrix) were performed. CEL files were submitted to the National Center for Biotechnology Information Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) available under accession number GSE137905. Data were quality-checked, normalized, and analyzed as described in detail in the data supplement.
For comparisons of COPD and asthma datasets, published data (19, 20) were extracted from the GEO accession viewer (GSE37147 and GSE76226, respectively) and processed using the same methods as applied to our internal microarray dataset, as detailed in the data supplement.
Statistics
Data in figures are given as median with interquartile range, as detailed in figure legends. Statistical analysis was performed by Mann-Whitney or the false discovery rate method, as detailed in the data supplement.
Results
RV Is Capable of Infecting PCLS and Elicits Robust Antiviral Host Responses
In RV-infected PCLS, colocalization of dsRNA and pan-cytokeratin immunohistochemistry staining demonstrated active replication of RV in airway epithelial cells (Figures 1A–1D). Infection of airway epithelium was observed to occur in patches instead of homogeneous distribution, with occasional changes in epithelial cell morphology and detachment of cells observed at the spots of infection (Figures 1A and 1B). Tissues inoculated with UV-inactivated RV or noninfected tissue were negative for dsRNA (data not shown). RV infection did not induce a significant cytopathic effect, as the majority of the tissue remained intact and viable up to 72 hours after infection as demonstrated by LIVE/DEAD staining (Figure E1A). The intact tissue structure was further confirmed by hematoxylin and eosin staining, which showed a good preservation of the tissue architecture, including the bronchial epithelium (Figure E2). Despite this, increased LDH release was observed in infected PCLS (Figure E1B), which could indicate RV-induced epithelial damage at the spots of infection. However, as no obvious tissue damage was detected by LIVE/DEAD and hematoxylin and eosin stainings of PCLS cross-sections, LDH could be primarily released from cells located in the peripheral regions of the slices that may be more susceptible to infection.
Figure 1.

Rhinovirus infects airway epithelium and elicits antiviral and proinflammatory host immune responses. (A–D) Replication of rhinovirus (RV) 1B in viable human lung tissue was detected by dsRNA staining (yellow) (A, B, and D), and localization within AEC was confirmed by staining of pan-cytokeratin (red) (A, C, and D). Three-dimensional images: scale bars: A, 100 μm; and B–D, 15 μm. (E) Proinflammatory and antiviral cytokines were detected by human 7-plex tissue culture kit from MesoScaleDiscovery after RV infection with and without antiviral treatment (I, 100 nM rupintrivir) as compared with Med and UV-RV. Cytokine release from lung tissue is displayed in ng cytokine/mg whole protein content. Box and whisker plots show median, 25th and 75th percentiles, and minimum and maximum values, showing all data points. *P < 0.05, **P < 0.005, and ***P < 0.001 according to linear mixed-effects model and false discovery rate method as detailed in the online supplement. N = 5–6 donors, with duplicate precision-cut lung slices per condition. AEC = airway epithelial cells; dsRNA = double-stranded RNA; I = inhibitor; Med = medium; UV-RV = ultraviolet-inactivated RV control.
RV infection resulted in the activation of epithelial cells as well as immune cells. Resident immune cells, such as macrophages, T cells, and B cells, are present in PCLS, as confirmed by immunohistochemistry staining of uninfected tissue (Figure E3). Analyses of mediator release showed a significant induction of IFN-α, CXCL10, and CXCL11 in RV-infected PCLS compared with uninfected controls at 24 hours and 72 hours after infection, indicating a strong induction of a type I IFN-driven antiviral response (Figure 1E). Proinflammatory mediators, including IFN-γ, TNF-α, and CCL5, were also significantly upregulated in RV-infected PCLS compared with controls (Figure 1E). The antiviral compound rupintrivir was able to significantly suppress the RV-induced release of these antiviral and proinflammatory cytokines at both time points (Figure 1E). In contrast, the proinflammatory mediators IL-1β and IL-6 were only minimally induced or not induced by RV and were not altered by rupintrivir treatment at either time point (Figure E4).
Whole Genome Analyses of RV-induced Responses in Human PCLS
Hierarchical clustering and principal component analyses of whole genome transcriptomic data demonstrated a clear separation between gene expression profiles observed in RV-infected PCLS and medium-treated PCLS, demonstrating a robust response to RV across all donors despite some donor-to-donor variability (Figures 2A and 2B). RV infection regulated a large number of genes when compared with medium (upregulated: 3,582 probes corresponding with 2,202 genes; downregulated: 3,842 probes corresponding with 2,611 genes; fold change ⩾ 1.5; uncorrected P ⩽ 0.05) (Figure 2C). Although some genes were regulated at both time points, specific genes showed a clear time dependency, as they were regulated at either 24 hours or 72 hours after infection (Figure 2C). Figure E5 shows that a large proportion of transcripts with a P value of less than 0.05 passed false discovery rate correction (Figure E5A) with virtually no overlap between upregulated and downregulated probes (Figure E5B), indicating robustness of the dataset. A full list of differentially expressed transcripts is provided in the data supplement (Table E2).
Figure 2.
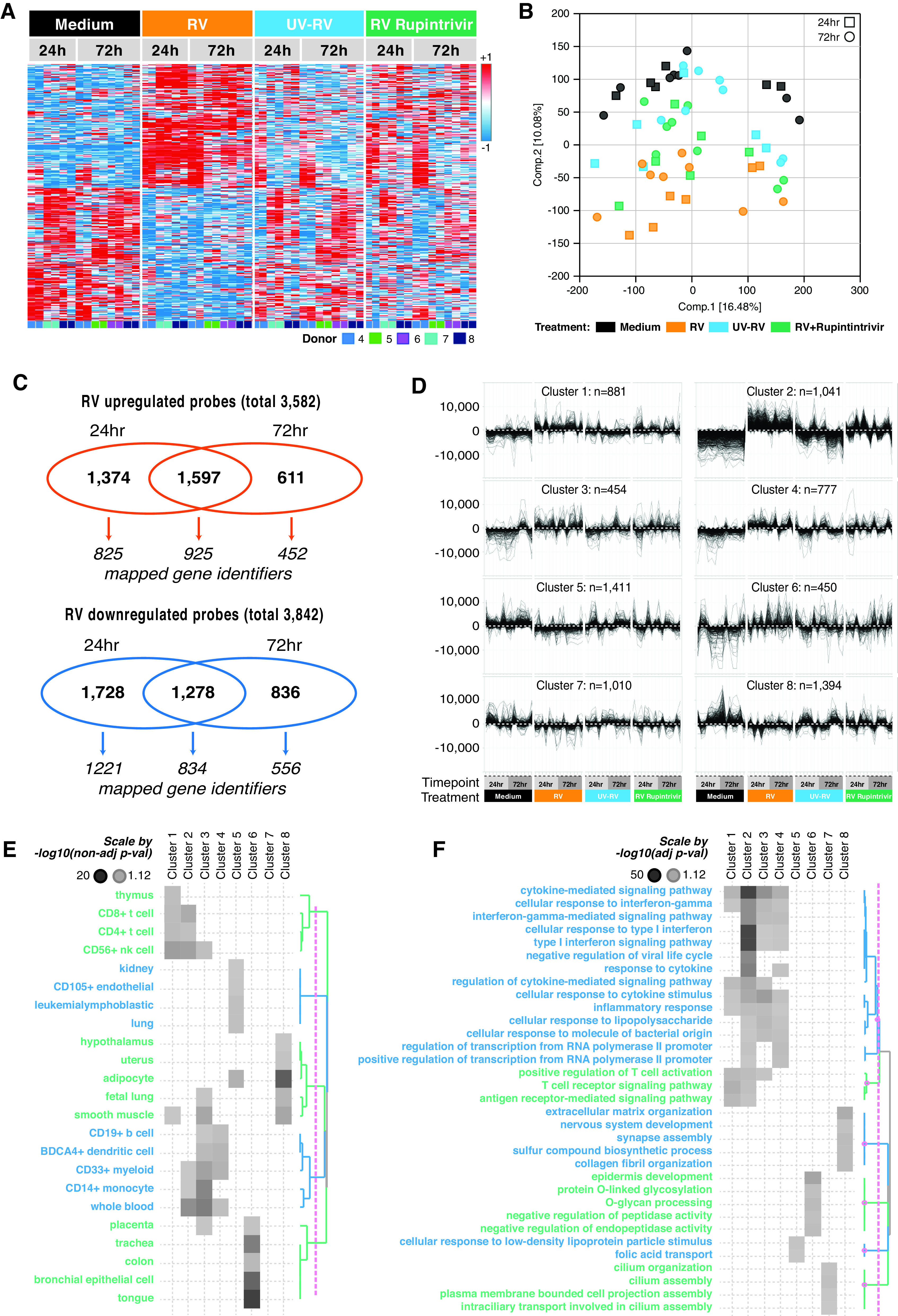
Whole genome expression analysis reveals RV-induced immune and epithelial cell responses. (A and B) Distinct clustering of RV-infected samples versus Med and UV-RV is displayed in heat map of differentially expressed genes of Med (black) versus RV (orange), UV-RV (blue), and RV + rupintrivir (green) (A) and principal component analysis of Med (black), RV (orange), UV-RV (blue), RV + rupintrivir (green) (B). (C) Microarray analysis of differentially regulated genes showed prominent response of lung tissue to RV infection ex vivo. Venn diagram showing the distribution of differentially upregulated (>1.5-fold change; P < 0.05) and downregulated genes (<1.5-fold change; P < 0.05) by RV infection. (D) Regulated probes were clustered into eight partitioning around medoids (PAM) clusters. (E and F) Row dendrogram of clusters presents involvement of PAM clusters in biological functions (E) and pathways (F). N = 3 donors for 24 h after infection and N = 4 donors for 72 h after infection, with two technical replicates each (representing individual analysis of two separate precision-cut lung slices per condition).
Partitioning around medoids clustering was chosen to identify genes with similar transcriptional profiles across time points and treatment groups. This resulted in eight clusters that resolved signatures associated with specific cell types (Figure 2D). Human Gene Atlas annotations illustrated that T cell– and natural killer cell–associated genes were mostly enriched in Clusters 1 and 2, and myeloid cell–associated genes were mostly enriched in Clusters 3 and 4, while epithelial cell–associated and related organ-associated genes were represented in Cluster 6 (Figure 2E). This cluster aligned with tissues such as the kidney, uterus, and placenta, as they are rich in epithelial and stromal cells and thus share molecular pathways regulating tissue homeostasis and cellular responses. Clusters 5 and 8 contained genes associated with endothelial cells and adipocytes, respectively. No significant cell type enrichment was detected in Cluster 7.
When gene ontology biological processes analyses were applied on each cluster, type I and II IFN responses were most prominently represented in Cluster 2 and, to a smaller extent, in Clusters 3 and 4, suggesting a strong antiviral immune response by epithelial and immune cells represented in these clusters (Figure 2F). Cytokine- and inflammation-related signatures were detected across the immune response–related Clusters 1–4, whereas T-cell signaling pathways converged mostly toward Clusters 1 and 2. Consistent with the Human Gene Atlas database, epithelial function–relevant pathways were represented in Cluster 6 and included epidermis development, O-linked glycosylation, and endopeptidase activity. Furthermore, this analysis revealed ciliation pathways in Cluster 7 (Figure 2F).
Ex Vivo RV-induced Gene Signatures Overlap with Asthma and COPD Disease Data Sets
The purpose of this project was to study the acute response of “healthy” lower respiratory tract human lung tissue to RV infection ex vivo. However, on the basis of our observations that not only antiviral immune responses but also epithelial repair processes were triggered by RV in PCLS ex vivo, we were interested in exploring potential overlap with similar processes present in chronic lung diseases such as COPD and asthma. Therefore, we compared the observed RV-induced gene signatures in PCLS ex vivo with disease signatures from patients with COPD and asthma. Gene expression profiles of bronchial brushings from patients with COPD and asthma generated by Steiling and colleagues (20) or Kuo and colleagues (19) were derived from the GEO database, and differentially expressed genes were calculated and overlaid with the RV-induced PCLS gene signature. These analyses demonstrated a large overlap of COPD and asthma disease-relevant genes with RV-induced PCLS responses (52%, 107 of 204 COPD-relevant genes; 52%, 109 of 210 asthma-relevant genes) (Figure 3A). Figure 3B visualizes the fold change of RV-regulated genes in PCLS at 24 hours versus 72 hours after infection and their respective overlap with COPD or asthma disease-relevant genes. The 107 COPD-relevant genes that overlapped with RV-regulated genes in PCLS demonstrated mostly similar directionality (Figure 3B, left), whereas observations for the 109 asthma-relevant genes upregulated by RV were more mixed (Figure 3B, right). Complete lists of overlapping genes can be found in the data supplement (Table E3).
Figure 3.
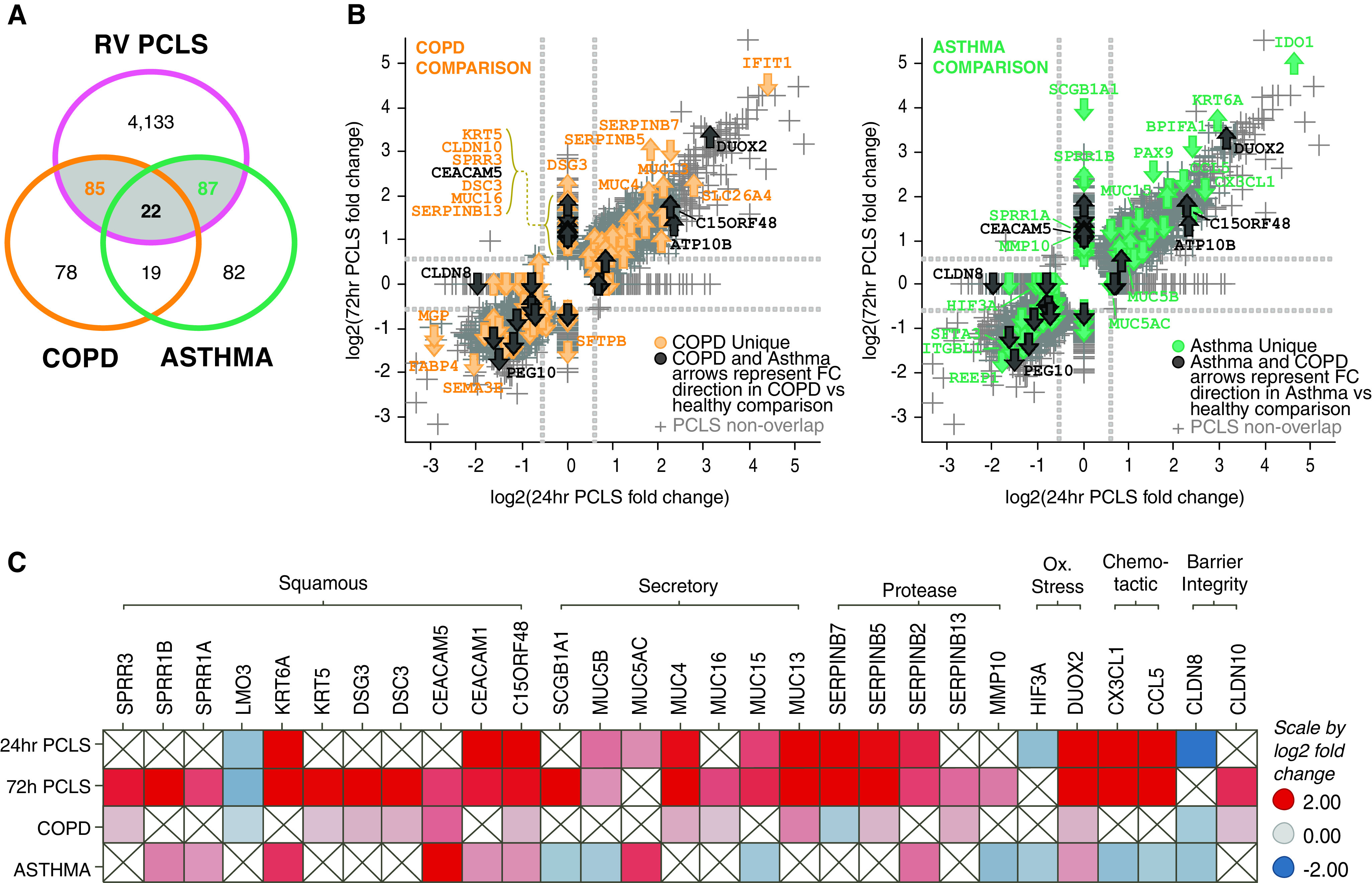
RV-induced gene signatures in precision-cut lung slices (PCLS) overlap with chronic obstructive pulmonary disease (COPD) and asthma gene signatures. Gene signatures from RV-infected PCLS (significant at either 24 h or 72 h time point with >1.5-fold change; P < 0.05) were compared with gene expression data sets from patients with COPD (GSE37147) (20) and asthma (GSE76226) (19). (A) Venn diagram illustrating that ∼50% of the disease-relevant genes (107 of 204 in COPD and 109 of 210 in asthma) were induced by RV in PCLS. (B) All genes significantly regulated in PCLS were plotted versus COPD-regulated (left) or asthma-regulated (right) genes in scatter plots. Gray markers represent every probe of the PCLS data set. Colored arrows highlight overlapping genes, thus indicating genes that were significantly regulated in PCLS by RV and were regulated in the respective disease data set (yellow = COPD [left]; green = asthma [right]; black = COPD and asthma). The direction of the arrow represents direction of regulation in the disease data set versus healthy control subjects (arrow up = upregulated; arrow down = downregulated). (C) Heatmap showing comparative analysis of selected overlapping genes grouped along disease-relevant biological processes and their regulation in PCLS (24 h and 72 h time points), COPD and asthma data set (red = upregulated; blue = down-regulated). N = 3 donors for 24 h after infection and N = 4 donors for 72 h after infection, with duplicate PCLS per condition.
Interestingly, the disease-relevant RV-regulated gene signatures contained transcripts reflective of epithelial repair or metaplastic processes that were also regulated in COPD (e.g., KRT5, SPRR3, DSC3, DSG3, LMO3 and CEACAM5) or in asthma (e.g., SPRR1A, SPRR1B, KRT6A, and CEACAM1/5) (Figure 3C).
Moreover, disease-relevant RV-induced genes included transmembrane mucins (e.g., MUC4, MUC13, and MUC16 for COPD and MUC15 for asthma). Secreted factors from epithelial cells, including MUC5AC, MUC5B, and SCGB1A1, were also among the disease-relevant RV-regulated genes overlapping with asthma. Furthermore, regulators of proteolytic activity (Serpin family members B5, B7, B13 in COPD and SERPINB2 and MMP10 in asthma), barrier integrity (CLDN10 in COPD and CLND8 in both COPD and asthma), and oxidative stress (DUOX2 in both COPD and asthma and HIF3A in asthma) were detected among disease-relevant RV-regulated gene signatures (Figure 3C). Together, these data demonstrate that RV stimulation of PCLS not only provides a model in which to evaluate inflammatory endpoints but also provides an excellent opportunity to explore disease-relevant epithelial responses.
To further confirm our findings on the RV-induced regulation of epithelial repair processes, IHC staining of the basal cell marker Krt5 was performed in PCLS from three additional donors after RV infection compared with uninfected control samples. Krt5 was expressed in basal cell regions of differentiated pseudostratified epithelium and at higher concentrations in squamous cells in undifferentiated areas of the epithelium (Figures 4A and 4B). Interestingly, Krt5 expression was also observed in terminal bronchioles (Figure 4B). Overall, automated quantification demonstrated a significant increase of Krt5 in RV-infected samples compared with uninfected control samples in all three donors (Figure 4C), which correlated with the concentrations of IP-10 induced in these donors (Figure 4D).
Figure 4.

RV-induced increase of Krt5 protein expression. (A and B) Immunohistochemistry staining of Krt5 was performed in 4-μm thin sections of paraffin-embedded PCLS previously infected with RV (A) or Med control (B). Krt5 is shown as red staining with blue hematoxylin counterstain for nuclei. Scale bars, 50 μm. Arrowheads indicate regions with differentiated airway epithelium and low Krt5 expression, and arrows highlight areas with squamous shape in undifferentiated areas of the epithelium with high Krt5 expression. Triangle indicates Krt5 expression in terminal bronchioles. (C) Quantification of Krt5 expression (positive per negative pixel count) was performed by whole slide analysis using Visiopharm Software. (D) IP-10 release was measured 72 h after infection by MesoScaleDiscovery for the same donors. (C and D) Symbols represent donor-specific individual values of fold change Krt5-positive cells per tissue area (C) or IP-10 release (D) in Medium versus RV-infected PCLS for N = 3 donors with duplicate samples each. *P ⩽ 0.05 using Student’s t test.
Rupintrivir Inhibits RV-induced Inflammation Whereas Epithelial Responses are Largely Refractory
To evaluate the impact of viral replication inhibition on RV-induced responses, we tested the viral 3C protease inhibitor rupintrivir in ex vivo infected PCLS. As shown in Figure 1E, rupintrivir demonstrated a nearly complete inhibition of the RV-induced antiviral and proinflammatory mediator release. In-depth analyses of the rupintrivir effect on the transcriptomics profile, however, revealed that only a fraction of the RV-regulated genes was responsive to rupintrivir treatment (at 24 h: 26%, 1,532 of 5,977; at 72 h: 22%, 954 of 4,322) (Figure 5A). Using our previously defined partitioning around medoids clusters, we determined the percentage of rupintrivir-affected probes per cluster (Figure 5B). Interestingly, on average, 36% of probes in the immune response–related Clusters 1–4 were rupintrivir inhibited (Figure 5B), which constituted, on average, 78% of the RV-regulated probes affected by rupintrivir. In contrast, only, on average, 12% of probes in the epithelial function–related Clusters 6 and 7 were rupintrivir sensitive, equating to only, on average, 6% of RV-regulated rupintrivir-inhibited probes (Figure 5B). Moreover, when rupintrivir-affected probes were mapped to the disease-relevant RV-induced gene signatures, we observed that a large proportion of these genes were refractory to rupintrivir treatment (73%, 102 of 140 at 24 h; 80%, 108 of 135 at 72 h; Figure 5C). This included epithelial metaplastic responses and secreted factors, transmembrane mucins, and regulators of proteolytic activity and barrier integrity. Interestingly, genes associated with oxidative stress, such as DUOX2 and HIF3A, were rupintrivir sensitive (Figure 5C).
Figure 5.
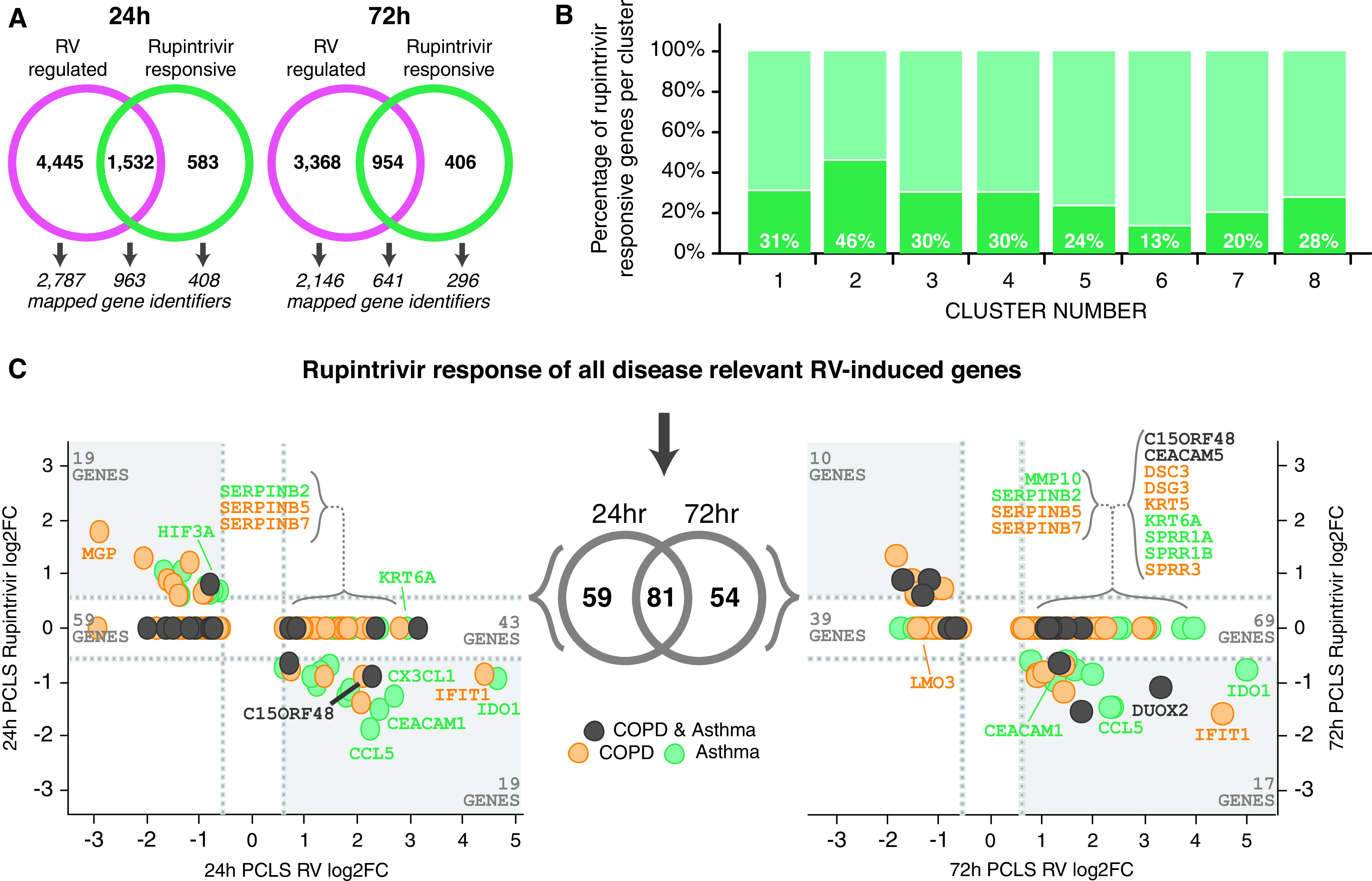
Differential effect of antiviral treatment on RV-induced disease-relevant responses in immune and epithelial cell clusters. The effect of antiviral treatment with rupintrivir on RV-induced gene signatures was analyzed and compared for the identified PAM clusters. (A) Venn diagram illustrating that only a fraction of RV-induced gene expression profiles was affected by antiviral treatment with rupintrivir. (B) Differential responsiveness to rupintrivir treatment was observed in the eight identified PAM clusters (30–46% in immune cell Clusters 1–4 and 13% in epithelial Cluster 6). (C) Disease-relevant genes (yellow = COPD, green = asthma, and black = COPD and asthma) that were significantly upregulated by RV in PCLS at 24 h (left) and 72 h (right) were plotted versus the effect of rupintrivir treatment. In each graph, RV-induced rupintrivir-responsive genes are located in the lower right quadrant (e.g., CCL5, CX3CL1, and IFIT1), whereas RV-induced genes unaffected by rupintrivir are located in the nonsignificant fold change region above (e.g., SERPINs, KRT6A, and CEACAM5). N = 3 donors for 24 h after infection and N = 4 donors for 72 h after infection, with duplicate PCLS per condition.
In summary, our data demonstrate that rupintrivir effectively inhibits inflammatory mediator release and oxidative stress pathways but does not affect epithelial barrier responses to RV infection.
Discussion
RV infection is frequently discussed as the “missing link” in the pathogenesis of COPD (21). In this study, we demonstrated that RV infection in viable human lung slices ex vivo triggered both immune and epithelial cell responses, which showed a significant overlap with disease signatures observed in patients with COPD and asthma (19, 20). Surprisingly, disease-relevant immune, but not epithelial, cell responses were inhibited by antiviral treatment, pointing toward their role in disease progression and their potential as a novel target.
Viral respiratory tract infections induce profound inflammatory and antiviral responses that are mediated by both epithelial and immune cells. Experimental models reflecting this complex reaction are essential for in-depth characterization of the host response. Human PCLS, a complex ex vivo tissue model, are able to reflect early events of respiratory diseases (17, 22, 23). Therefore, we used PCLS to characterize the host response to RV infection in “healthy” tissue and mapped the observed patterns to gene signatures of patients with COPD and asthma to evaluate whether our ex vivo lung model could be used to study biological processes triggered by RV that might also be relevant in these disease conditions. This comparison has limitations, and our results cannot be directly translated to the clinical situation, as the tissue we were able to use was mainly derived from “healthy” (i.e., tumor-free) tissue from cancer resections. Lung tissue resections from patients with asthma were not available, and technical challenges precluded preparation of PCLS from end-stage disease COPD tissue that presented with massive emphysema. However, the purpose of this study was to characterize the acute response to lower respiratory tract RV infection, and we observed a robust response to RV across all donors, enabling an in-depth characterization of the involved biological processes. Nevertheless, we found a surprisingly large overlap of RV-triggered differential expressed genes in PCLS ex vivo with those from COPD and asthma data sets. As the publications on those patient data sets do not include any information on potential virus exacerbation of these patients, we cannot fully exclude that some of the overlap we observe might be due to a virus infection those patients might have experienced before. On the other hand, we sourced PCLS from lung surgeries mainly from cancer resection, but because of mandatory data protection rules we have no additional information on potential previous treatments, history of smoking, or the potential COPD status of those patients with cancer, which could also contribute to the overlap we observed. Nevertheless, the overlapping signature was not merely a baseline condition, but RV infection specifically triggered the observed processes robustly across all donors, also in those with other disease backgrounds. This strongly indicates that the observed overlap is not only a coincidence but that specific processes driven by RV infection are related to processes present in COPD and asthma. This opens up new opportunities to study RV-driven processes in the ex vivo lung model that are also relevant for COPD and asthma. As expected, epithelial cell responses were particularly well reflected, as the COPD and asthma gene signatures were sourced from epithelial brushes (19, 20). A recent study investigating gene expression profiles in nasal epithelial cells from patients with asthma challenged with RV16 in vivo found predominantly IFN responses in the top-regulated genes (24). These genes were also upregulated in our RV-infected PCLS, demonstrating the capability of this ex vivo model to reflect the in vivo observed innate antiviral response to RV infection.
We also observed clear differences between the RV-triggered response in PCLS and the compared disease data sets. For example, chemokines CCL5 and CX3CL1, both upregulated in RV-infected PCLS, were downregulated in the asthma data. IFIT, strongly upregulated by RV in PCLS, was downregulated in the COPD data. These differences could be the result of an impaired antiviral immune response, which has been discussed widely in asthma and COPD (5, 25).
The immune cell response to RV infection was associated with myeloid, natural killer, and T-cell clusters and linked to IFN-mediated signaling, release of proinflammatory and antiviral cytokines, and activation of immune cells. Small airway inflammation with increased numbers of inflammatory cells is a classic feature of COPD, with lymphocytes and macrophages especially driving the inflammatory response (26). RV infection has been reported to elicit increased concentrations of inflammatory cytokines such as IL-6, IL-8, TNF-α, CXCL10, and CCL5 in COPD (21). PCLS with intact small airways and resident immune cells are able to reflect this inflammatory immune response, as demonstrated in our study by the RV-triggered release of cytokines, including TNF-α, IL-1β, CCL5, CXCL10, and IFN-γ.
We used UV-radiated virus as additional control to discriminate immune responses triggered by recognition of the virus itself via pathogen-associated molecular pattern versus processes depending on active virus replication. The UV-inactivated, replication-defective virus can be detected by endosomal and cytoplasmic pattern recognition receptors (i.e., TLR7/8- and RIG-I–recognizing single-stranded RNA) and induce cytokine release, especially from macrophages (9). In contrast, TLR3 and MDA-5 recognize dsRNA, which is only present if the virus replicates, and are both necessary to mount a full-blown antiviral immune response (9).
As tissues of adults were used in this study, the contribution of preexisting cellular and humoral immunity remains a possibility. The observed time frame and missing connection to lymphoid system precludes de novo activation of naive RV serotype-specific T cells in lung tissue. Regulation of T cell–specific genes observed in Clusters 1 and 2 (lymphoid-associated genes) implies that tissue-resident T cells mounted a recall response. CD4- and CD8-specific T-cell responses develop as consequences of RV infection (8, 27). In line with this, the T-cell activation marker IL-2 mRNA was significantly upregulated by RV in our study. We have previously shown that T cells are present in PCLS and react with a strong recall response to influenza virus antigen (28). CD8+ T-lymphocyte numbers were increased in small airways and correlated with severity of airflow obstruction in patients with COPD (21, 26, 29). In asthma, the shift toward a Th2-type driven immune response contributes to the impaired innate antiviral host response (30) and thus promotes virus-induced disease progression. CD8+ T cells secreting type 2 and other proinflammatory cytokines were reported to be enriched in severe eosinophilic asthma (31). Thus, in both diseases, a (re)activation of cytotoxic T cells by RV infection might contribute to a dysbalanced immune response, augmenting inflammation and tissue damage and thereby perpetuating disease pathology.
Immune cell responses in the lung inevitably depend on cross-talk with airway epithelial cells as the main entry site for invading respiratory viruses. As expected, recognition of RV induced a strong antiviral epithelial response and secretion of mediators recruiting and activating immune cells. Surprisingly, in addition, genes associated with repair processes such as epithelial proliferation, hyperplasia, mucus hypersecretion, and alteration of barrier integrity were detected in the epithelial cell–associated gene cluster. It is likely that these changes result from cellular death and regeneration of the epithelium. However, although this is part of the normal healing process, it can be deleterious in disease conditions. In COPD and asthma, the existing dysfunction and virus-induced repair of the epithelial layer could mutually amplify one another.
In COPD, structural changes of the airway epithelium include squamous metaplasia and goblet and basal cell hyperplasia, which can persist in the small airways as the site of airflow obstruction (32–34). RV infection of PCLS induced genes associated with epithelial repair processes, which were also upregulated in the COPD data (20). These included upregulation of adhesion molecules such as CEACAM1 and CEACAM5. CEACAMs are involved in the regulation of various cellular functions, such as differentiation, proliferation, and apoptosis, and have been described to be associated with squamous cell metaplasia in COPD (32). Pathogens can use these molecules for adhesion and to exert immunosuppressive functions enabling mucosal colonization. Upregulation of CEACAMs has been linked to RV-16 infection in human nasal epithelial cell cultures (35) and to Moraxella catarrhalis and nontypable Haemophilus influenza infection of bronchial epithelial cells (bacterial pathogens associated with COPD exacerbation) (36). Thus, RV-induced upregulation of CEACAMs might provide a link between infection and squamous cell metaplasia. Interestingly, a recent study reported IFN-γ–induced inflammatory responses in airway epithelial cells via CEACAM1 and PI3K signaling, promoting cellular growth and proliferation, linking epithelial transition from inflammation to cancer (37).
The presence of repair processes in the airway epithelium were further substantiated by the basal cell marker Krt5, which was consistently upregulated not only at the gene expression level but also the protein level in RV-infected PCLS. This suggests that RV infection is sufficient to induce epithelial damage and an epithelial repair response that involves upregulation of Krt5. This is in line with reports showing KRT5+ basal cells to be essential for lung regeneration by repopulating the damaged alveolar parenchyma upon influenza infection (38, 39). A recent study also described upregulation of several squamous cell metaplasia– and mucus hypersecretion–related gene modules during exacerbations of asthma (40). Interestingly, upregulation of Krt5 was also observed in the COPD data set.
RV infection has been described to impair barrier integrity (e.g., in human nasal as well as airway epithelial cell cultures) by disrupting tight junctions and adherens junctions (41–43). Although the expression of most tight junction proteins was not significantly altered upon RV infection in PCLS at this early stage, the tight junction–sealing protein claudin-8 was significantly downregulated not only in PCLS but also in both asthma and COPD data, indicating a loss of tight junction integrity. Interestingly, RV infection of airway epithelial cells of children with asthma resulted in sustained disruption of tight junctions and the loss of epithelial barrier integrity, whereas this was transient in healthy control subjects (44).
On the basis of the frequently discussed potential role of RV promoting COPD and asthma disease progression, antivirals could be a beneficial therapy for these patients (45, 46). We used the 3C protease inhibitor rupintrivir to evaluate the effect of antiviral treatment during acute RV infection in PCLS. Strikingly, although rupintrivir was beneficial in subduing the virus-induced inflammatory immune response, the epithelial response associated with squamous, basal, and goblet cell hyperplasia was largely refractory to antiviral treatment. This suggests that these disease-relevant epithelial processes, despite being initially triggered by RV infection, might progress autonomously. Current antiviral and antiinflammatory therapeutics are unable to revert the RV-triggered pathological epithelial processes. Consequently, epithelial cells and pathological repair and remodeling processes emerge as potential targets for novel therapeutics. Recently, bronchial cells derived from club cell progenitors, driving epithelial mesenchymal transition and remodeling, have been suggested as novel targets for the treatment of chronic lung diseases (47). Basal cells that are susceptible to virus infection and subsequently can alter epithelial differentiation (48, 49) might represent another interesting target.
In conclusion, with the in-depth analysis of RV-infected human PCLS presented here, combined with the comparative analysis of patient gene signatures of patients with COPD and asthma, we demonstrated that the human RV PCLS model reflects disease-relevant biological mechanisms and provides an outstanding opportunity to evaluate novel therapeutics.
Acknowledgments
Acknowledgment
The authors thank the Fraunhofer ITEM PCLS team as well as Gina Franke, Sabine Schild, and Emma Spies for excellent technical assistance. The authors thank Susann Dehmel for excellent support concerning confocal microscopy imaging. On behalf of The U-BIOPRED Study Group with input from the U-BIOPRED Patient Input Platform and patient representatives from the Ethics Board and Safety Management Board.
Footnotes
A complete list of the U-BIOPRED study group may be found in the data supplement.
The U-BIOPRED consortium receives funding from the European Community and from the European Federation of Pharmaceutical Industries and Associations as an Innovative Medicines Initiative Joint Undertaking funded project (IMI JU 115010).
Author Contributions: S.W. conceived and designed the study, performed part of the experiments, analyzed data, interpreted results, and drafted and finalized the manuscript. S.B. performed part of the experiments, analyzed data, interpreted results, and drafted and critically revised the manuscript. H.O. designed and performed experiments, performed confocal microscopy imaging, analyzed data, and contributed to the draft the manuscript. N.N.B. and K.A.S. analyzed data and interpreted results. M.G.L. performed statistical analysis. D.S. performed immunohistological analysis. G.W., P.Z., H.-G.F., and L.W. provided human lung samples. P.B. and D.J. provided expert pathological assessment of human lung material. E.M.H. analyzed data and interpreted results and drafted and critically revised the manuscript. A.B. and K.S. supervised the project and contributed to conception of the study, interpretation of results, and writing of the manuscript. All authors approved the final version of the manuscript.
This article has a data supplement, which is accessible from this issue’s table of content online at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2020-0337OC on June 28, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
Contributor Information
Collaborators: on behalf of the U-BIOPRED Study Group
References
- 1. Peltola V, Waris M, Osterback R, Susi P, Hyypia T, Ruuskanen O. Clinical effects of rhinovirus infections. J Clin Virol. 2008;43:411–414. doi: 10.1016/j.jcv.2008.08.014. [DOI] [PubMed] [Google Scholar]
- 2. Papi A, Bellettato CM, Braccioni F, Romagnoli M, Casolari P, Caramori G, et al. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am J Respir Crit Care Med. 2006;173:1114–1121. doi: 10.1164/rccm.200506-859OC. [DOI] [PubMed] [Google Scholar]
- 3. Hewitt R, Farne H, Ritchie A, Luke E, Johnston SL, Mallia P. The role of viral infections in exacerbations of chronic obstructive pulmonary disease and asthma. Ther Adv Respir Dis. 2016;10:158–174. doi: 10.1177/1753465815618113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jackson DJ, Johnston SL. The role of viruses in acute exacerbations of asthma. J Allergy Clin Immunol. 2010;125:1178–1187, quiz 1188–1189. doi: 10.1016/j.jaci.2010.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mallia P, Message SD, Gielen V, Contoli M, Gray K, Kebadze T, et al. Experimental rhinovirus infection as a human model of chronic obstructive pulmonary disease exacerbation. Am J Respir Crit Care Med. 2011;183:734–742. doi: 10.1164/rccm.201006-0833OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Message SD, Laza-Stanca V, Mallia P, Parker HL, Zhu J, Kebadze T, et al. Rhinovirus-induced lower respiratory illness is increased in asthma and related to virus load and Th1/2 cytokine and IL-10 production. Proc Natl Acad Sci USA. 2008;105:13562–13567. doi: 10.1073/pnas.0804181105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mallia P, Footitt J, Sotero R, Jepson A, Contoli M, Trujillo-Torralbo MB, et al. Rhinovirus infection induces degradation of antimicrobial peptides and secondary bacterial infection in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;186:1117–1124. doi: 10.1164/rccm.201205-0806OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kennedy JL, Turner RB, Braciale T, Heymann PW, Borish L. Pathogenesis of rhinovirus infection. Curr Opin Virol. 2012;2:287–293. doi: 10.1016/j.coviro.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Makris S, Johnston S. Recent advances in understanding rhinovirus immunity. F1000 Res. 2018;7:F1000. doi: 10.12688/f1000research.15337.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bartlett NW, Slater L, Glanville N, Haas JJ, Caramori G, Casolari P, et al. Defining critical roles for NF-κB p65 and type I interferon in innate immunity to rhinovirus. EMBO Mol Med. 2012;4:1244–1260. doi: 10.1002/emmm.201201650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Khaitov MR, Laza-Stanca V, Edwards MR, Walton RP, Rohde G, Contoli M, et al. Respiratory virus induction of alpha-, beta- and lambda-interferons in bronchial epithelial cells and peripheral blood mononuclear cells. Allergy. 2009;64:375–386. doi: 10.1111/j.1398-9995.2008.01826.x. [DOI] [PubMed] [Google Scholar]
- 12. Contoli M, Message SD, Laza-Stanca V, Edwards MR, Wark PA, Bartlett NW, et al. Role of deficient type III interferon-lambda production in asthma exacerbations. Nat Med. 2006;12:1023–1026. doi: 10.1038/nm1462. [DOI] [PubMed] [Google Scholar]
- 13. Kohlmeier JE, Cookenham T, Roberts AD, Miller SC, Woodland DL. Type I interferons regulate cytolytic activity of memory CD8(+) T cells in the lung airways during respiratory virus challenge. Immunity. 2010;33:96–105. doi: 10.1016/j.immuni.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Padovan E, Spagnoli GC, Ferrantini M, Heberer M. IFN-alpha2a induces IP-10/CXCL10 and MIG/CXCL9 production in monocyte-derived dendritic cells and enhances their capacity to attract and stimulate CD8+ effector T cells. J Leukoc Biol. 2002;71:669–676. [PubMed] [Google Scholar]
- 15. Obernolte H, Braubach P, Jonigk D, Beinke S, Belyaev NN, Lennon M, et al. U-BIOPRED. Transcriptomic analyses reveal anti-viral responses of epithelial cells and multiple immune cell types in HRV infected human lung tissue. Eur Respir J. 2017;50:PA4126. [Google Scholar]
- 16. Obernolte H, Braubach P, Jonigk D, Krüger M, Warnecke G, Pfennig O, et al. U-BIOPRED. Anti-viral and inflammatory host response to rhinovirus is induced in vital ex vivo human lung tissue and reduced by anti-viral treatments. Am J Respir Crit Care Med. 2017;195:A4040. [Google Scholar]
- 17. Neuhaus V, Danov O, Konzok S, Obernolte H, Dehmel S, Braubach P, et al. Assessment of the Cytotoxic and Immunomodulatory Effects of Substances in Human Precision-cut Lung Slices. J Vis Exp. 2018:57042. doi: 10.3791/57042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rochlitzer S, Hoymann HG, Muller M, Braun A. No exacerbation but impaired anti-viral mechanisms in a rhinovirus-chronic allergic asthma mouse model. Clinical Sci (Lond) 2014;126:55–65. doi: 10.1042/CS20130174. [DOI] [PubMed] [Google Scholar]
- 19. Kuo CS, Pavlidis S, Loza M, Baribaud F, Rowe A, Pandis I, et al. U-BIOPRED Project Team‡. A Transcriptome-driven Analysis of Epithelial Brushings and Bronchial Biopsies to Define Asthma Phenotypes in U-BIOPRED. Am J Respir Crit Care Med. 2017;195:443–455. doi: 10.1164/rccm.201512-2452OC. [DOI] [PubMed] [Google Scholar]
- 20. Steiling K, van den Berge M, Hijazi K, Florido R, Campbell J, Liu G, et al. A dynamic bronchial airway gene expression signature of chronic obstructive pulmonary disease and lung function impairment. Am J Respir Crit Care Med. 2013;187:933–942. doi: 10.1164/rccm.201208-1449OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Linden D, Guo-Parke H, Coyle PV, Fairley D, McAuley DF, Taggart CC, Kidney J. Respiratory viral infection: a potential “missing link” in the pathogenesis of COPD. European Respir Rev. 2019;28:180063. doi: 10.1183/16000617.0063-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Danov O, Jiménez Delgado SM, Obernolte H, Seehase S, Dehmel S, Braubach P, et al. Human lung tissue provides highly relevant data about efficacy of new anti-asthmatic drugs. PLoS One. 2018;13:e0207767. doi: 10.1371/journal.pone.0207767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gerckens M, Alsafadi HN, Wagner DE, Lindner M, Burgstaller G, Königshoff M. Generation of Human 3D Lung Tissue Cultures (3D-LTCs) for Disease Modeling. J Vis Exp. 2019 doi: 10.3791/58437. [DOI] [PubMed] [Google Scholar]
- 24. Ravi A, Chang M, van de Pol M, Yang S, Aliprantis A, Thornton B, et al. U-BIOPRED Study Group. Rhinovirus-16 induced temporal interferon responses in nasal epithelium links with viral clearance and symptoms. Clin Exp Allergy. 2019;49:1587–1597. doi: 10.1111/cea.13481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hilzendeger C, da Silva J, Henket M, Schleich F, Corhay JL, Kebadze T, et al. Reduced sputum expression of interferon-stimulated genes in severe COPD. Int J Chron Obstruct Pulmon Dis. 2016;11:1485–1494. doi: 10.2147/COPD.S105948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Caramori G, Casolari P, Barczyk A, Durham AL, Di Stefano A, Adcock I. COPD immunopathology. Semin Immunopathol. 2016;38:497–515. doi: 10.1007/s00281-016-0561-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gern JE, Galagan DM, Jarjour NN, Dick EC, Busse WW. Detection of rhinovirus RNA in lower airway cells during experimentally induced infection. Am J Respir Crit Care Med. 1997;155:1159–1161. doi: 10.1164/ajrccm.155.3.9117003. [DOI] [PubMed] [Google Scholar]
- 28. Neuhaus V, Schwarz K, Klee A, Seehase S, Förster C, Pfennig O, et al. Functional testing of an inhalable nanoparticle based influenza vaccine using a human precision cut lung slice technique. PLoS One. 2013;8:e71728. doi: 10.1371/journal.pone.0071728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350:2645–2653. doi: 10.1056/NEJMoa032158. [DOI] [PubMed] [Google Scholar]
- 30. Contoli M, Ito K, Padovani A, Poletti D, Marku B, Edwards MR, et al. Th2 cytokines impair innate immune responses to rhinovirus in respiratory epithelial cells. Allergy. 2015;70:910–920. doi: 10.1111/all.12627. [DOI] [PubMed] [Google Scholar]
- 31. Hilvering B, Hinks TSC, Stöger L, Marchi E, Salimi M, Shrimanker R, et al. Synergistic activation of pro-inflammatory type-2 CD8+ T lymphocytes by lipid mediators in severe eosinophilic asthma. Mucosal Immunol. 2018;11:1408–1419. doi: 10.1038/s41385-018-0049-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rigden HM, Alias A, Havelock T, O’Donnell R, Djukanovic R, Davies DE, et al. Squamous Metaplasia Is Increased in the Bronchial Epithelium of Smokers with Chronic Obstructive Pulmonary Disease. PLoS One. 2016;11:e0156009. doi: 10.1371/journal.pone.0156009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shaykhiev R. Emerging biology of persistent mucous cell hyperplasia in COPD. Thorax. 2019;74:4–6. doi: 10.1136/thoraxjnl-2018-212271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Willemse BW, Postma DS, Timens W, ten Hacken NH. The impact of smoking cessation on respiratory symptoms, lung function, airway hyperresponsiveness and inflammation. Eur Respir J. 2004;23:464–476. doi: 10.1183/09031936.04.00012704. [DOI] [PubMed] [Google Scholar]
- 35. Wang JH, Kwon HJ, Jang YJ. Rhinovirus enhances various bacterial adhesions to nasal epithelial cells simultaneously. Laryngoscope. 2009;119:1406–1411. doi: 10.1002/lary.20498. [DOI] [PubMed] [Google Scholar]
- 36. Klaile E, Klassert TE, Scheffrahn I, Müller MM, Heinrich A, Heyl KA, et al. Carcinoembryonic antigen (CEA)-related cell adhesion molecules are co-expressed in the human lung and their expression can be modulated in bronchial epithelial cells by non-typable Haemophilus influenzae, Moraxella catarrhalis, TLR3, and type I and II interferons. Respir Res. 2013;14:85. doi: 10.1186/1465-9921-14-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhu Y, Song D, Song Y, Wang X. Interferon gamma induces inflammatory responses through the interaction of CEACAM1 and PI3K in airway epithelial cells. J Transl Med. 2019;17:147. doi: 10.1186/s12967-019-1894-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ray S, Chiba N, Yao C, Guan X, McConnell AM, Brockway B, et al. Rare SOX2+ Airway Progenitor Cells Generate KRT5+ Cells that Repopulate Damaged Alveolar Parenchyma following Influenza Virus Infection. Stem Cell Reports. 2016;7:817–825. doi: 10.1016/j.stemcr.2016.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zuo W, Zhang T, Wu DZ, Guan SP, Liew AA, Yamamoto Y, et al. p63(+)Krt5(+) distal airway stem cells are essential for lung regeneration. Nature. 2015;517:616–620. doi: 10.1038/nature13903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Altman MC, Gill MA, Whalen E, Babineau DC, Shao B, Liu AH, et al. Transcriptome networks identify mechanisms of viral and nonviral asthma exacerbations in children. Nat Immunol. 2019;20:637–651. doi: 10.1038/s41590-019-0347-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sajjan U, Wang Q, Zhao Y, Gruenert DC, Hershenson MB. Rhinovirus disrupts the barrier function of polarized airway epithelial cells. Am J Respir Crit Care Med. 2008;178:1271–1281. doi: 10.1164/rccm.200801-136OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Waltl EE, Selb R, Eckl-Dorna J, Mueller CA, Cabauatan CR, Eiwegger T, et al. Betamethasone prevents human rhinovirus- and cigarette smoke- induced loss of respiratory epithelial barrier function. Sci Rep. 2018;8:9688. doi: 10.1038/s41598-018-27022-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yeo NK, Jang YJ. Rhinovirus infection-induced alteration of tight junction and adherens junction components in human nasal epithelial cells. Laryngoscope. 2010;120:346–352. doi: 10.1002/lary.20764. [DOI] [PubMed] [Google Scholar]
- 44. Looi K, Buckley AG, Rigby PJ, Garratt LW, Iosifidis T, Zosky GR, et al. Effects of human rhinovirus on epithelial barrier integrity and function in children with asthma. Clin Exp Allergy. 2018;48:513–524. doi: 10.1111/cea.13097. [DOI] [PubMed] [Google Scholar]
- 45. Casanova V, Sousa FH, Stevens C, Barlow PG. Antiviral therapeutic approaches for human rhinovirus infections. Future Virol. 2018;13:505–518. doi: 10.2217/fvl-2018-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mirabelli C, Scheers E, Neyts J. Novel therapeutic approaches to simultaneously target rhinovirus infection and asthma/COPD pathogenesis. F1000 Res. 2017;6:1860. doi: 10.12688/f1000research.11978.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Brasier AR. Therapeutic targets for inflammation-mediated airway remodeling in chronic lung disease. Expert Rev Respir Med. 2018;12:931–939. doi: 10.1080/17476348.2018.1526677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jakiela B, Brockman-Schneider R, Amineva S, Lee WM, Gern JE. Basal cells of differentiated bronchial epithelium are more susceptible to rhinovirus infection. Am J Respir Cell Mol Biol. 2008;38:517–523. doi: 10.1165/rcmb.2007-0050OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Persson BD, Jaffe AB, Fearns R, Danahay H. Respiratory syncytial virus can infect basal cells and alter human airway epithelial differentiation. PLoS One. 2014;9:e102368. doi: 10.1371/journal.pone.0102368. [DOI] [PMC free article] [PubMed] [Google Scholar]