

Abstract
Smoking and human immunodeficiency virus 1 (HIV-1) infection are risk factors for chronic obstructive pulmonary disease (COPD), which is among the most common comorbid conditions in people living with HIV-1. HIV-1 infection leads to persistent expansion of CD8+ T cells, and CD8+ T cell–mediated inflammation has been implicated in COPD pathogenesis. In this study, we investigated the effects of HIV-1 infection and smoking on T-cell dynamics in patients at risk of COPD. BAL fluid, endobronchial brushings, and blood from HIV-1 infected and uninfected nonsmokers and smokers were analyzed by flow cytometry, and lungs were imaged by computed tomography. Chemokines were measured in BAL fluid, and CD8+ T-cell chemotaxis in the presence of cigarette smoke extract was assessed in vitro. HIV-1 infection increased CD8+ T cells in the BAL fluid, but this increase was abrogated by smoking. Smokers had reduced BAL fluid concentrations of the T cell–recruiting chemokines CXCL10 and CCL5, and cigarette smoke extract inhibited CXCL10 and CCL5 production by macrophages and CD8+ T-cell transmigration in vitro. In contrast to the T cells in BAL fluid, CD8+ T cells in endobronchial brushings were increased in HIV-1–infected smokers, which was driven by an accumulation of effector memory T cells in the airway mucosa and an increase in tissue-resident memory T cells. Mucosal CD8+ T-cell numbers inversely correlated with lung aeration, suggesting an association with inflammation and remodeling. HIV-1 infection and smoking lead to retention of CD8+ T cells within the airway mucosa.
Keywords: chronic obstructive pulmonary disease, alveolar macrophages, chemokines, BAL, lymphocytes
Clinical Relevance
Human immunodeficiency virus 1 (HIV-1) promotes recruitment of CD8+ T cells to the lung, whereas smoking inhibits their migration into the airspaces. Consequently, there is an increase in effector memory CD8+ T cells and resident memory CD8+ T cells in the airway mucosa of HIV-1–infected smokers. These findings suggest a potential mechanism for the accelerated development of chronic obstructive pulmonary disease in patients living with HIV-1.
People living with human immunodeficiency virus 1 (PLHIV) are at increased risk of chronic obstructive pulmonary disease (COPD) (1, 2), which is one of the most common comorbid conditions in this population (3). Smoking is the major risk factor for COPD (4–6), and PLHIV are nearly twice as likely to smoke than the general population (7). However, numerous studies suggest that human immunodeficiency virus 1 (HIV-1) is an independent risk factor for COPD (1, 2, 8).
Lung inflammation mediated by CD8+ T cells is a critical component of COPD pathogenesis (9–14). CD8+ T cells are increased in the pulmonary vasculature, lung interstitium, and airways of patients with COPD compared with healthy control subjects (12, 13). In particular, T cells that express the chemokine receptors CCR5 and CXCR3, which mark IFN-γ–producing T cells, are increased in the lungs of patients with COPD and correlate with the severity of disease (15, 16).
HIV-1 infection leads to robust activation and progressive dysfunction of CD8+ T cells (17, 18), and HIV-1–induced CD8+ T-cell activation has been postulated as a mechanism for the accelerated development of COPD in PLHIV (19). Despite viral suppression with combination antiretroviral therapy (cART), PLHIV experience persistent expansion of circulating and alveolar CD8+ T cells and a reduced CD4/CD8 T-cell ratio, which is associated with immune activation at mucosal sites and non–acquired immunodeficiency syndrome–related morbidity, including COPD (20, 21). Previous studies of CD8+ T cells in the lungs of PLHIV included primarily smokers without comparisons with uninfected nonsmokers. Thus, the relative effects of HIV-1 and smoking on T-cell dynamics in the lung remain unclear.
To characterize immunologic events early in COPD development, we studied T-cell dynamics in the lungs of individuals with HIV-1 infection and chronic tobacco smoke exposure before the onset of airway obstruction or emphysema. We recently demonstrated that these subjects have an increase in pulmonary perfusion heterogeneity, which reflects the vascular dysfunction that occurs early in COPD pathogenesis (22–24). We found that although HIV-1 infection increases recruitment of CD8+ T cells to the lung, smoking reduces production of T cell–recruiting chemokines from alveolar macrophages. Consequently, CD8+ T cells are retained in the airway mucosa of HIV-1–infected smokers. Mucosal CD8+ T-cell numbers inversely correlate with lung aeration as measured by chest computed tomography (CT), suggesting that they promote inflammation and remodeling within the lung. Thus, our data identify a novel mechanism by which HIV-1 and chronic tobacco smoke exposure may act synergistically to accelerate the development of COPD in PLHIV.
Methods
Study Design
The objective of this study was to evaluate the effects of HIV-1 and smoking on T cells in the lungs of human subjects at risk for COPD. Inclusion and exclusion criteria and the study protocol are provided in the data supplement. We recruited HIV-1–uninfected nonsmokers (n = 25), HIV-1–uninfected smokers (n = 14), HIV-1–infected nonsmokers (n = 18), and HIV-1–infected smokers (n = 16) (see Table E1 in the data supplement). Smokers were defined as current smokers with ⩾100 lifetime cigarettes. Nonsmokers were defined as never-smokers or former smokers. Former smokers were required to have been smoke-free for ⩾5 years before enrollment. HIV-1–infected subjects were virally suppressed through cART for ⩾12 months. Eligible subjects underwent bronchoscopy and phlebotomy. Some subjects also underwent CT imaging of the lungs. These subjects were previously shown to have pulmonary perfusion heterogeneity consistent with preclinical COPD (24). The study was approved by the Partners Healthcare Institutional Review Board. Subjects gave their written informed voluntary consent before inclusion in the study.
Bronchoscopy
BAL fluid was obtained from the right middle lobe and lingula by washing each with saline (25). A cytology brush was then repeatedly passed around the circumference of the lavaged bronchus to obtain endobronchial mucosal samples.
Chest CT
CT imaging and analysis details are provided in the data supplement. Hounsfield units (HU) less than −950 were used to calculate the percentage of low-attenuation area. The fraction of gas content (Fgas) in the lung was calculated as Fgas = (tissue density − density value of the voxel)/(tissue density − density of air), where tissue density = 0 HU and the density of air = −1000 HU. For example, a mean Fgas of 0.7 indicates that 70% of the total lung volume is gas and the remaining 30% is a combination of blood and tissue.
Flow Cytometry
Cells were stained with fluorescence-conjugated antibodies (Table E2) and fixable viability dye, run on an LSR Fortessa system (BD Biosciences), and analyzed by using FlowJo software (BD Biosciences).
Cell Culture and Chemotaxis
Monocyte-derived macrophages (MDMs) (26) were exposed to cigarette smoke extract (CSE) (Murty Pharmaceuticals) before co-culture with peripheral blood mononuclear cells in a Transwell assay and were incubated with CXCL10 (R&D Systems), CCL5 (R&D Systems), anti–human CXCR3 antibody (CXCR3-173, BioLegend), or an IgG isotype control antibody (HTK888, BioLegend).
Quantitative Real-Time PCR
RNA was extracted (RNAeasy, Qiagen), quantified, transcribed into cDNA (Applied Biosciences), and normalized against a housekeeping gene (27). Primer sequences are summarized in Table E3.
Chemokine Analysis
Chemokines were measured with a Milliplex panel (EMD Millipore) by using a 30-fold–concentrated BAL supernatant on a Luminex-200 system and were analyzed by using xPONENT 3.1 software (Luminex Corp.) (25). CXCL10 and CCL5 were measured by using an ELISA (R&D Systems).
Statistical Analysis
Data are expressed as the median and interquartile range. Nonparametric one-way ANOVA (Kruskal-Wallis test) with the Dunn’s test for multiple comparisons or the Mann-Whitney test were used to compare medians (GraphPad Prism, GraphPad Software). A Wilcoxon signed rank test was used to compare paired data between two groups. Spearman’s correlation coefficients were used to assess the strength of associations between predefined variables. Differences were considered statistically significant when the P value was determined to be <0.05 on the basis of two-sided test results.
Results
Subject Characteristics
Subjects were well matched across groups (Table E1). Blood CD4+ T-cell numbers were not different in HIV-1–infected subjects compared with uninfected subjects (Table E1). The viral load and years on cART were similar among HIV-1–infected subjects, and pack-years were similar among smokers. All subjects had normal pulmonary function, but uninfected nonsmokers had a higher forced expiratory volume in 1 second and forced vital capacity than HIV-1–infected smokers. The median HU and percentage of low-attenuation area on chest CT images were similar across groups.
Smokers Have Reduced BAL Fluid T Cells
We first compared T cells in peripheral blood with those in the BAL fluid. There were no differences in CD4+ T-cell numbers in peripheral blood across groups (Figures E1A and E1B). However, HIV-1–infected subjects had increased CD8+ T cells in the blood and a reduced CD4/CD8 T-cell ratio compared with uninfected subjects (Figures E1B and E1C). Smokers had higher total numbers of BAL fluid cells (Figure 1A), irrespective of HIV-1 status, which was driven by an increase in the number of macrophages and granulocytes (Figure E2). The number and frequency of BAL fluid CD4+ T cells was reduced in smokers compared with nonsmokers, regardless of HIV-1 status (Figure 1B). We also observed an increased frequency and a trend toward increased numbers of BAL fluid CD8+ T cells in HIV-1–infected nonsmokers compared with uninfected nonsmokers. BAL fluid CD8+ T-cell frequency was reduced in smokers, independent of HIV-1 status, and HIV-1–infected smokers had reduced numbers of BAL fluid CD8+ T cells compared with HIV-1–infected nonsmokers (Figure 1C). The BAL fluid CD4/CD8 T-cell ratio was reduced in HIV-1–infected subjects compared with uninfected subjects (Figure E1D), in findings similar to those observed in blood. There were no significant differences in the frequencies of CD4+ or CD8+ T cells expressing markers of anergy or exhaustion, including CD28, PD-1, or BTLA (Figure E3). Thus, HIV-1 is associated with an increase in the BAL fluid CD8+ T-cell frequency in nonsmokers. However, smoking reduces CD4+ and CD8+ T cells in the airspaces independently of HIV-1 status.
Figure 1.
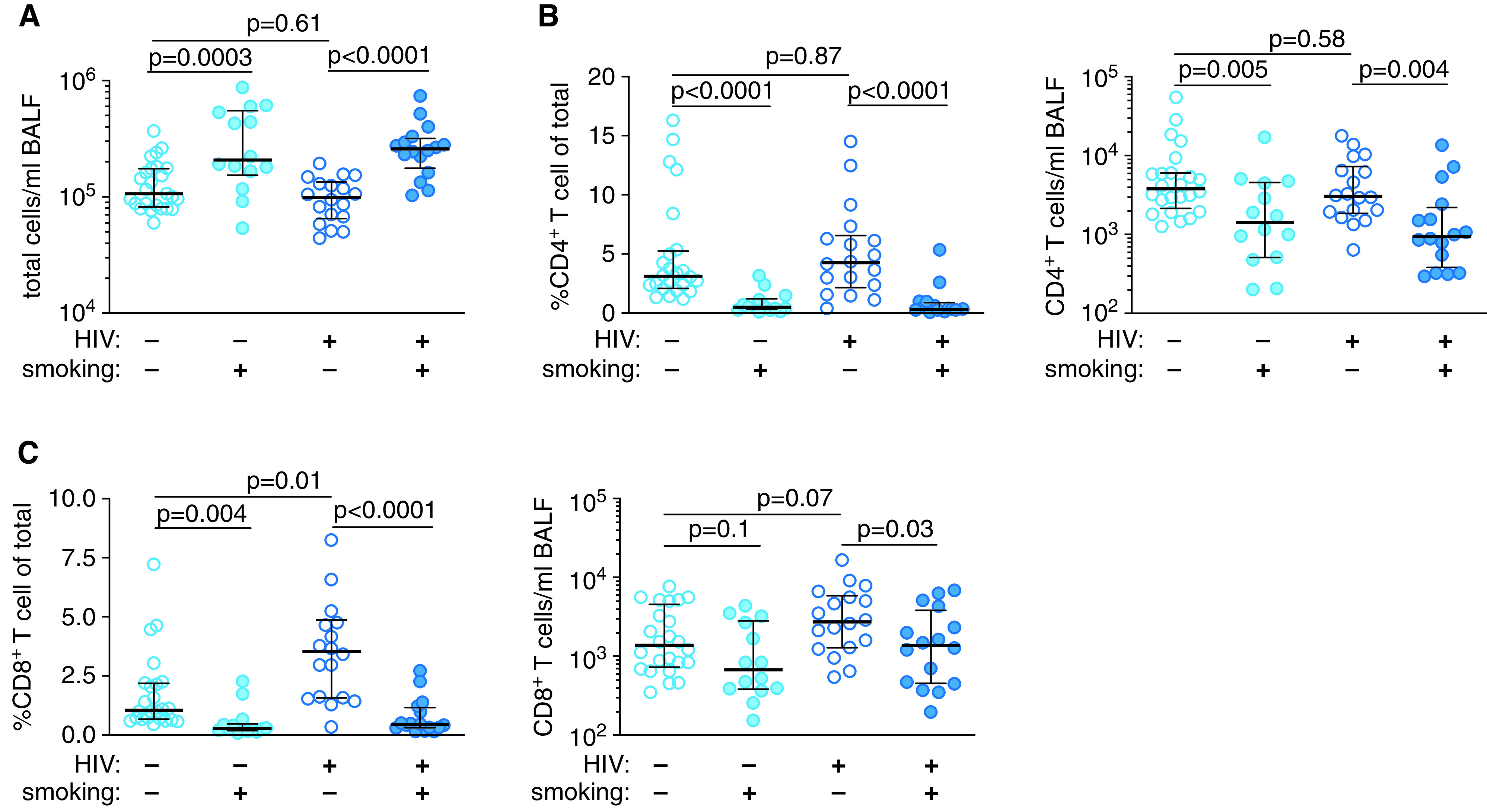
Smokers have reduced BAL fluid (BALF) CD4+ and CD8+ T cells. (A) Total cells per milliliter of BALF. (B) CD4+ T-cell frequency and number per milliliter of BALF. (C) CD8+ T-cell frequency and number per milliliter of BALF. P values were determined by using nonparametric one-way ANOVA with the Dunn’s test for multiple comparisons. Scatter plots are labeled with the median and interquartile range. HIV = human immunodeficiency virus.
Smoking and HIV-1 Increase CCR5+CXCR3+ T Cells
To better understand the smoking-associated decrease in BAL fluid T cells, we assessed T-cell apoptosis and proliferation. There were no differences in the frequency of Annexin V+CD95+ or Ki67+ CD4+ or CD8+ T cells between groups (Figures 2A, 2B, and E4). Thus, the decrease in T cells in smokers was not explained by increased apoptosis or decreased proliferation. Therefore, we examined expression of the lung-homing chemokine receptors CCR5 and CXCR3 (28–30). The frequency of BAL fluid CCR5+CXCR3+ CD4+ and CD8+ T cells was increased in uninfected smokers compared with nonsmokers (Figures 2C and E4) and further increased in HIV-1–infected subjects compared with uninfected subjects. Similar increases in CCR5+CXCR3+ CD4+ and CD8+ T cells from blood were observed with smoking and HIV-1 (Figure E5). Thus, smoking and HIV-1 increase expression of the lung-homing chemokine receptors CCR5 and CXCR3 on T cells.
Figure 2.
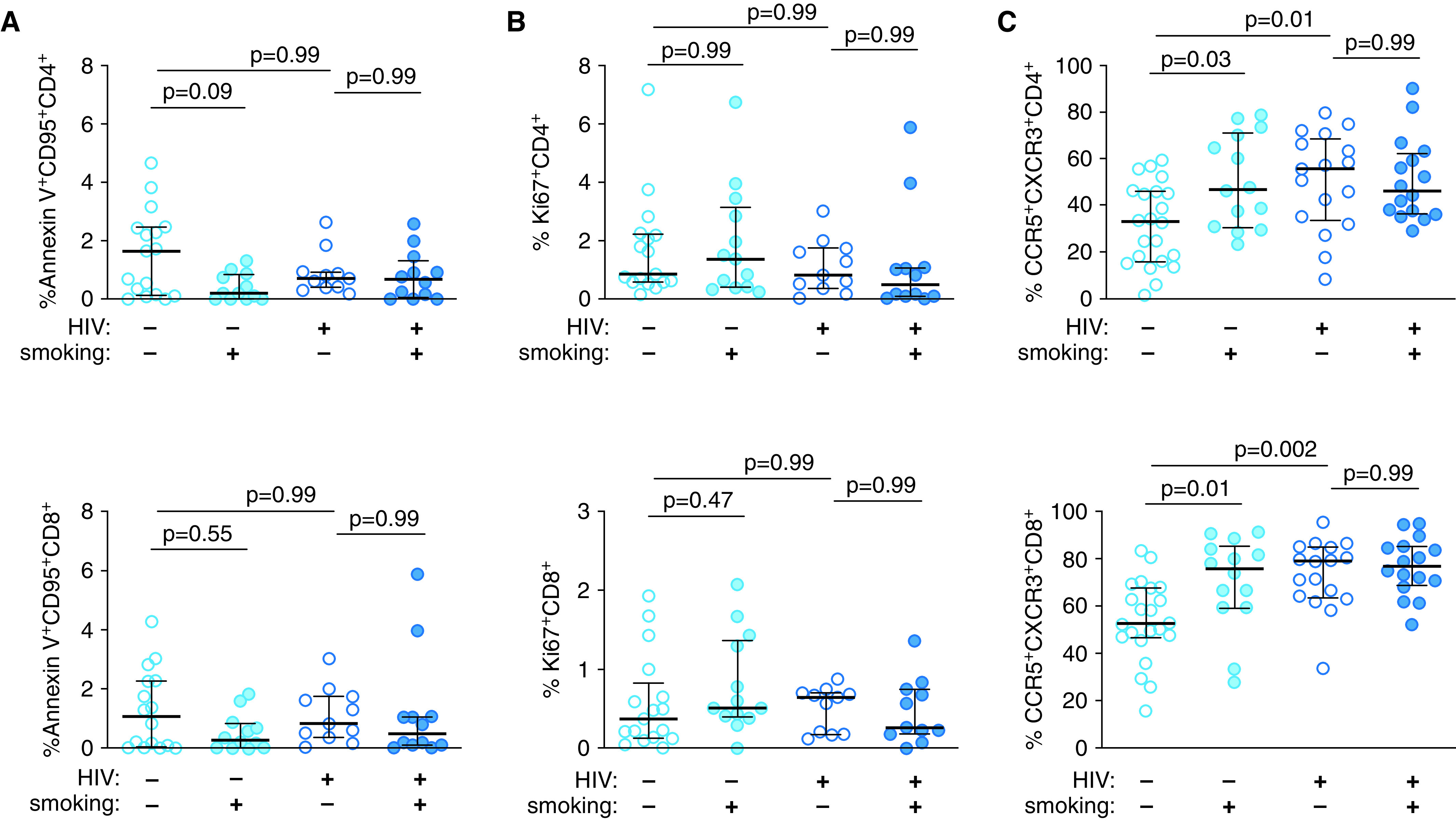
Smoking and HIV-1 increase the frequency of CCR5+CXCR3+ CD4+ and CD8+ T cells in BALF. The frequencies of BALF CD4+ T cells (top) and CD8+ T cells (bottom) identified as (A) being apoptotic by using annexin V staining, (B) proliferating by using intracellular staining for the marker Ki67, and (C) lung-homing by measuring the expression of the chemokine receptors CCR5 and CXCR3 are shown. P values were determined by using nonparametric one-way ANOVA with the Dunn’s test for multiple comparisons. Scatter plots are labeled with the median and interquartile range.
Smoking Reduces CXCL10 and CCL5 in BAL Fluid
Despite an increase of CCR5+CXCR3+ T cells in the blood and BAL fluid, we observed fewer total BAL fluid CD8+ T cells in smokers. Therefore, we measured RNA and protein levels of CXCL10 and CCL5 in bulk BAL fluid cells and cell-free BAL supernatants, respectively. CXCL10 is the primary ligand for CXCR3, and CCL5 is the ligand for CCR5. RNA and protein levels of CXCL10 were reduced in smokers compared with nonsmokers, independent of HIV-1 status (Figure 3A). CCL5 RNA and protein concentrations were reduced only in HIV-1–infected smokers (Figure 3B). We observed the same pattern for the CXCR3-binding chemokines CXCL9 and CXCL11 but not for other CC- or CXC-family chemokines (Figures E6 and E7 and Table E4). In addition, we found a significant correlation between the protein concentrations of CXCL10 and CCL5 in BAL fluid and the BAL fluid CD8+ T-cell numbers (Figure 3C) that was not observed for BAL fluid CD4+ T cells (Figure 3D). These data suggest that smoking leads to specific downregulation of chemokines that mediate CD8+ T-cell recruitment to the airspaces.
Figure 3.
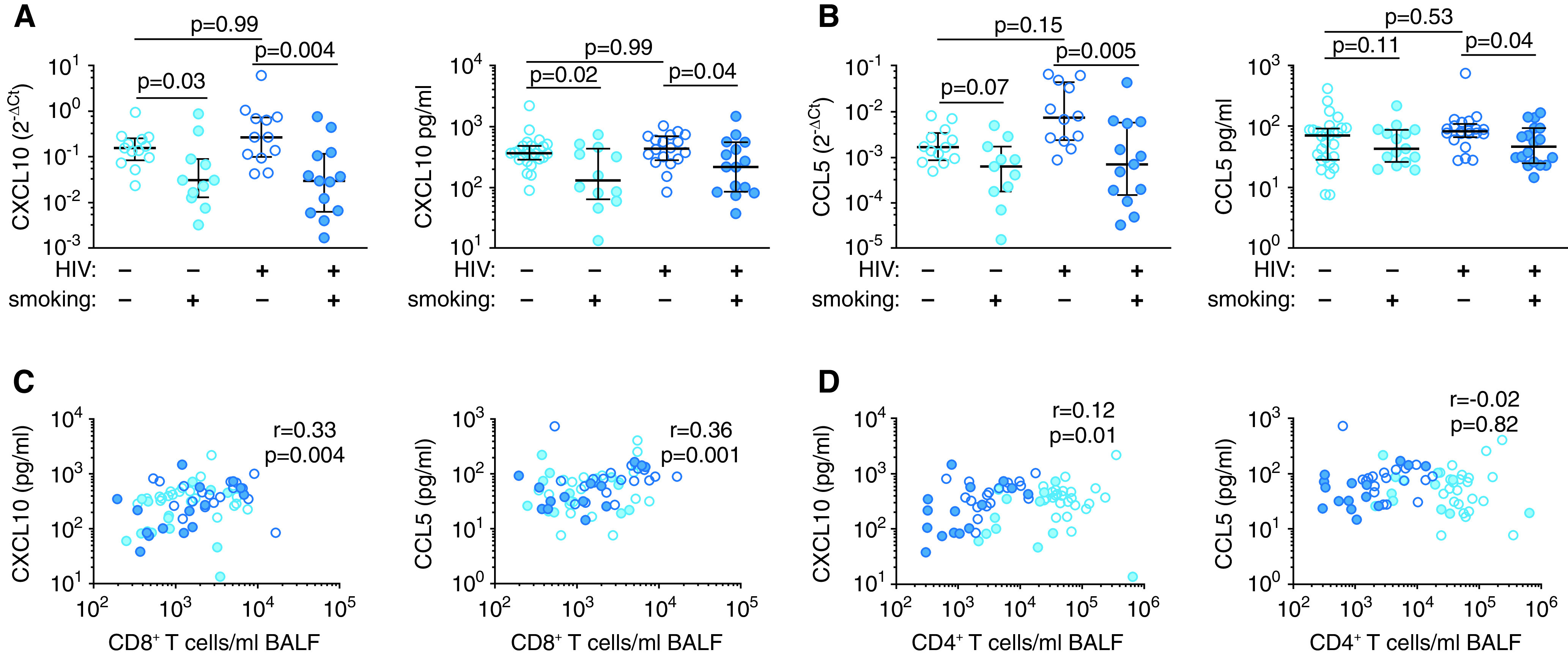
Smoking is associated with decreased CXCL10 and CCL5 in the BALF. Expression of (A) CXCL10 and (B) CCL5 measured by quantitative real-time PCR analysis of bulk BALF cells (left panels) and an ELISA of the BALF (right panels). Correlation of BALF CXCL10 (left panels) and CCL5 (right panels) protein concentrations with BALF (C) CD8+ and (D) CD4+ T-cell numbers. P values were determined by using nonparametric one-way ANOVA with the Dunn’s test for multiple comparisons in A and B and the Spearman’s correlation test in C and D. Scatter plots are labeled with the median and interquartile range. Ct = cycle threshold.
Cigarette Smoke Decreases MDM CXCL10 Production and T-Cell Chemotaxis
Macrophages are the primary cell type in the alveolar space and are a major source of CXCL10 and CCL5 (31, 32). To investigate whether smoking directly affects production of these chemokines, we exposed MDMs to CSE and then quantified T-cell chemotaxis and chemokine production in a Transwell assay (Figure E8). Exposure of MDMs to CSE reduced RNA amounts and protein concentrations of CXCL10 and CCL5 RNA (Figures 4A and E9A). Protein concentrations of CCL5 were only decreased in CSE-exposed MDMs incubated with peripheral blood mononuclear cells from HIV-1–infected donors (Figure 4A), consistent with our observations in BAL fluid (Figure 3B). The CSE-mediated decrease in CXCL10 production was dose dependent and not accompanied by increased cell death (Figure E9B). Exposure of MDMs to CSE reduced CD4+ and CD8+ T-cell migration independently of HIV-1 status (Figure 4B). Similarly, antibody-mediated inhibition of CXCR3 prevented CD8+ T-cell chemotaxis (Figures 4C and E9C). Importantly, recombinant CXCL10 but not CCL5 rescued CSE-mediated impairment of T-cell chemotaxis (Figures 4D and E9D). These results demonstrate that CXCL10 binding of CXCR3 directs T-cell migration and suggest that the reduction of BAL fluid T cells in HIV-1–infected smokers is due to smoking-induced impairment in the production of T cell–recruiting chemokines.
Figure 4.
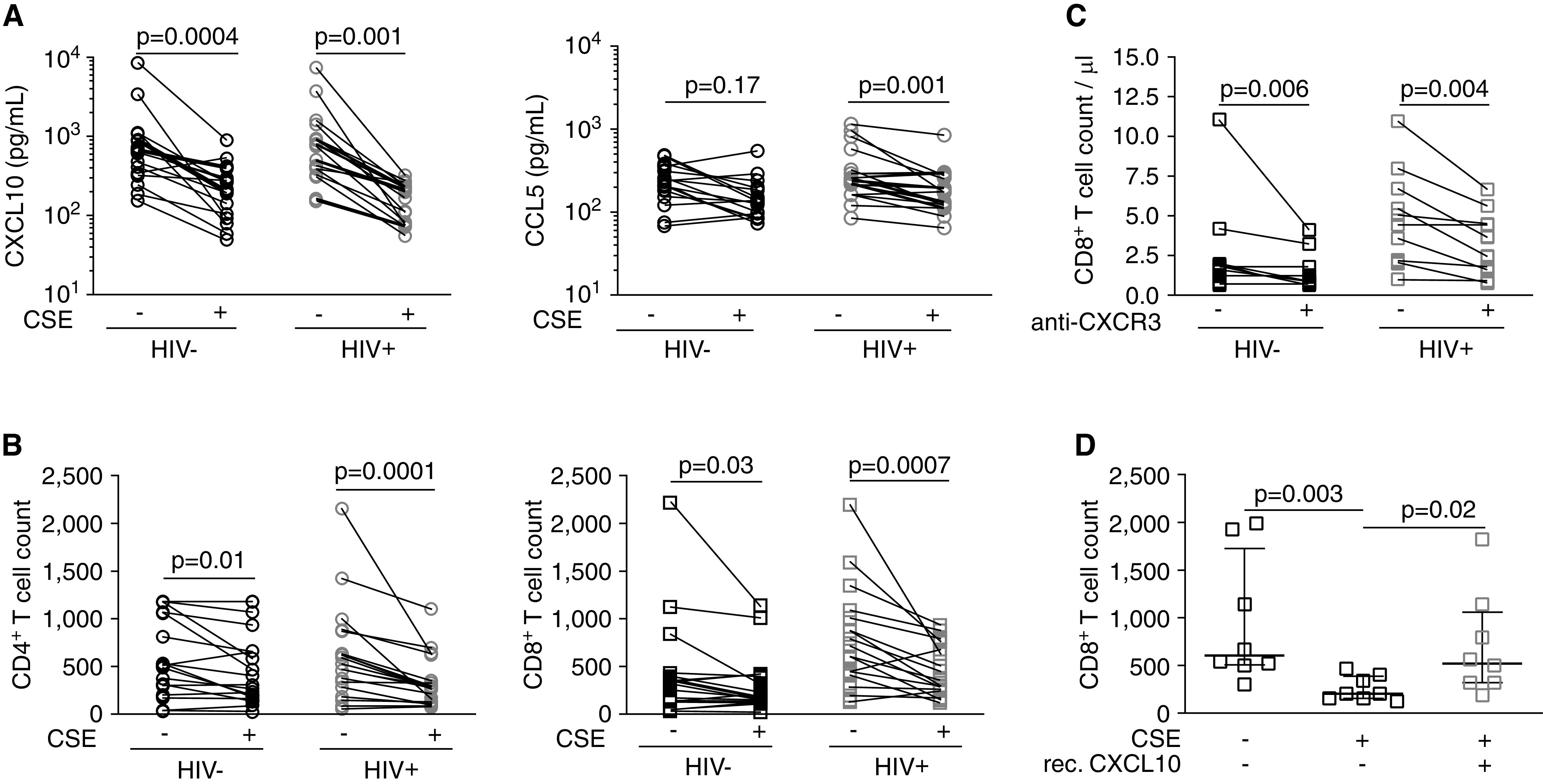
Cigarette smoke extract (CSE) decreases monocyte-derived macrophage (MDM) CXCL10 production and CD8+ T-cell chemotaxis in vitro. (A) CXCL10 (left) and CCL5 (right) protein in CSE-exposed MDM co-culture supernatants measured by using an ELISA. (B) Chemotaxis of CD4+ T cells (left) and CD8+ T cells (right) toward CSE-exposed MDMs. (C) Chemotaxis of CD8+ T cells in the presence of anti-CXCR3 antibody. (D) Chemotaxis of CD8+ T cells from HIV-1–infected subjects toward CSE-exposed MDMs in the presence of rec. CXCL10 (100 ng/ml). P values were determined by using a Wilcoxon signed rank test. rec. = recombinant.
CD8+ T Cells Are Increased in the Airway Mucosa of HIV-1– infected Smokers
Increased numbers of CD8+ T cells in the airways have been associated with the development and severity of COPD. However, we found that HIV-1–infected smokers have a significant reduction in BAL fluid CD8+ T cells compared with HIV-1– infected nonsmokers. BAL is widely used to study immune responses in the lung but provides only an indirect measure of mucosal inflammation. We therefore directly sampled the airway mucosa by using endobronchial brushing and quantified the number of T cells recovered relative to the number of CD326+ epithelial cells. The majority of T cells in the mucosa were CD8+ T cells, and no differences were observed in the number of CD4+ T cells across groups (Figure E10). However, we found an increase in the number of mucosal CD8+ T cells in HIV-1–infected smokers compared with uninfected nonsmokers (Figure 5A). Nearly all of the mucosal CD8+ T cells were CD69+CD103+ tissue-resident memory T (TRM) cells (Figures 5B, 5C, and E11). In contrast, only a minority of BAL fluid CD8+ T cells were TRM cells, and CD8+ TRM cells were not present in the blood. These results suggest that endobronchial brushing samples a distinct immunologic compartment compared with BAL and blood sampling. We therefore compared mucosal CD8+ T cells across groups. Although almost all of the mucosal CD8+ T cells were TRM cells in uninfected nonsmokers, both smoking and HIV-1 increased the number of mucosal CD8+CD62L−CD45RO+ effector memory T (TEM) cells (Figures 5D, 5E, and E11). HIV-1–infected smokers had the highest number of mucosal CD8+ TEM cells, in addition to having an increase in TRM cells. Notably, the mucosal CD8+ TEM cell numbers mirror the profile of circulating CXCR3+CCR5+CD8+ T cells (Figure E5). To determine whether mucosal CD8+ T cells are associated with subclinical changes in the lung structure, we correlated the number of cells with the mean Fgas obtained via chest CT imaging. Fgas measures lung aeration, with higher values indicating a larger component of gas and lower values indicating a higher component of blood or tissue. We found an inverse correlation between the number of mucosal CD8+ T cells and the mean Fgas (Figure 5F). Taken together, these data suggest that circulating CD8+ T cells are recruited into the lungs of HIV-1–infected smokers and then retained in the airway mucosa. Accumulation of mucosal CD8+ T cells correlates with lower lung aeration, suggesting that these cells are associated with inflammation and remodeling.
Figure 5.
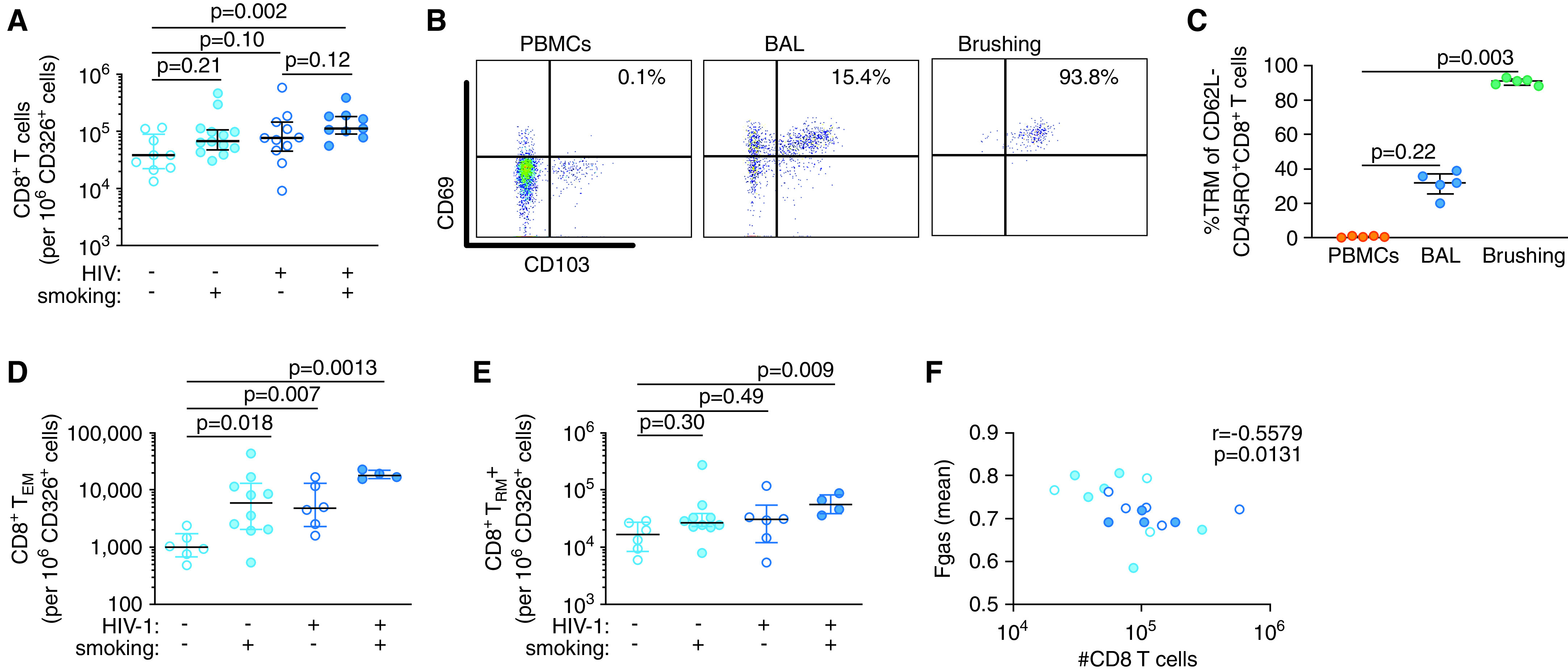
CD8+ T cells are increased in the airway mucosa of HIV-1–infected smokers. (A) The number of mucosal CD8+ T cells per 106 CD326+ epithelial cells in endobronchial brush samples. (B) Representative flow cytometry plots of CD8+ tissue-resident memory T (TRM) cells in blood, BALF, and endobronchial brush samples. (C) Frequency of CD8+ TRM cells in blood, BALF, and endobronchial brush samples. Number of CD8+ (D) TEM cells and (E) TRM cells in endobronchial brush samples per 106 CD326+ epithelial cells. (F) Correlation of mucosal CD8+ T-cell numbers and mean Fgas as a measure of lung aeration. P values were determined by using nonparametric one-way ANOVA with the Dunn’s test for multiple comparisons (in A and C–E) and the Spearman’s correlation test (in F). Scatter plots are labeled with the median and interquartile range. Fgas = fraction of gas content; PBMC = peripheral blood mononuclear cell; TEM = effector memory T cells.
Discussion
PLHIV are at increased risk of developing COPD, and both smoking and HIV-1 have been identified as independent risk factors for COPD (1, 2, 8). In this study, we directly compared the effects of HIV-1 and chronic smoke exposure on T cells in the airspaces and airway mucosa of at-risk individuals before the onset of clinical disease. HIV-1 infection was associated with an increase in BAL fluid CD8+ T cells in nonsmokers. However, smoking decreased the frequency of BAL fluid T cells independently of HIV-1 status and was associated with lower expression of T cell–recruiting chemokines in the BAL fluid. Importantly, the decrease in BAL fluid CD8+ T cells was accompanied by an increase in mucosal CD8+ T cells in HIV-1–infected smokers. Thus, our data suggest that HIV-1 infection and smoking lead to retention of CD8+ T cells within the airway mucosa. Furthermore, we found that the number of mucosal CD8+ T cells inversely correlated with the mean lung aeration as measured by using chest CT (24). In the absence of clinically apparent structural lung disease, lower lung aeration suggests that mucosal CD8+ T cells may be associated with lung inflammation and remodeling.
Our study confirmed that HIV-1–infected nonsmokers have increased CD8+ T cells in the alveolar space (28, 33–35). This parallels the persistent expansion of circulating CD8+ T cells observed in HIV-1–infected individuals despite viral suppression. Interestingly, smoking completely abrogated the HIV-1–induced increase in BAL fluid CD8+ T cells by specifically inhibiting production of the T cell–recruiting chemokine CXCL10. There was a trend toward lower CCL5 in smokers that only reached significance in HIV-1–infected subjects, suggesting an additional suppressive effect of HIV-1 on CCL5 production. Importantly, CXCL10 and CCL5 expression correlated with the number of BAL fluid CD8+ T cells.
Alveolar macrophages are the dominant cell type in the BAL fluid, and in concert with other cells, including T cells, are known to be significant producers of CXCL10 and CCL5 (28, 31, 32). The reduced expression of these chemokines in smokers in vivo was replicated in vitro. Furthermore, expression of other chemokines was not altered, implying that smoking-induced suppression of chemokine production from macrophages is relatively specific. Indeed, smoking-induced suppression of CXCL10 and CCL5 production by alveolar macrophages progresses with the development of COPD (36). Several components of cigarette smoke might contribute to suppression of T cell–recruiting chemokines. For example, nicotine inhibits proinflammatory cytokine production by alveolar macrophages (37), and cigarette smoke contains free radicals and aryl hydrocarbons that induce oxidative stress and apoptosis (38, 39). Further investigation is required to identify the essential components of cigarette smoke that cause downregulation of CXCL10 and CCL5.
In contrast to CD8+ T cells in the airspaces, airway mucosal CD8+ T cells were increased in HIV-1–infected smokers and trended higher in uninfected smokers and HIV-1–infected nonsmokers. This paralleled the frequency of circulating CXCR3+CCR5+CD8+ T cells and suggests that smoking and HIV-1 synergize to promote retention of CD8+ T cells in the mucosa. The increase in mucosal CD8+ T cells was driven by an increase in TEM cells and, in HIV-1–infected smokers, an expansion in the number of TRM cells. The TEM pool is expanded in chronic HIV-1 infection (40), and TRM numbers in the lung are determined by the balance of TRM apoptosis and replenishment by circulating TEM cells (41). Thus, the increase in mucosal TRM cells in HIV-1–infected smokers may be driven by persistent recruitment of TEM cells to the lung, impaired TRM-cell apoptosis, or both.
To our knowledge, endobronchial brushing has not previously been used to characterize CD8+ T-cell responses in the lung. Our data demonstrate that the proportion of CD8+ TRM cells in the mucosa is higher than that in the BAL fluid and indicate that TEM and TRM cells occupy specific niches within the lung. Endobronchial sampling was performed after the airways were lavaged. However, it is not clear whether mucosal CD8+ T cells are localized in the submucosal space or whether they are luminal but tightly associated with the epithelium. Regardless, endobronchial brushing appears to access a unique immune compartment and should prove to be a useful technique for studying CD8+ TRM cells in COPD and other lung diseases.
A substantial body of literature supports a role for CD8+ T cells in COPD. CD8+ T-cell depletion in a mouse model of emphysema attenuates inflammation and protects against alveolar destruction (11, 14). Furthermore, the number of CD8+ T cells in the airways correlates with the severity of airway obstruction and the presence of emphysema in patients with COPD (12, 13). It is not known whether the increase in CD8+ T cells observed in COPD is driven by an expansion of TEM cells, TRM cells, or both, and the specific roles of these subsets in COPD pathogenesis have not been elucidated.
Our study has several limitations. All of the HIV-1–infected subjects in our cohort were virally suppressed with cART. Thus, it is possible that some of the differences observed between HIV-1–uninfected subjects and HIV-1–infected subjects are related to cART rather than to HIV-1 infection itself. In addition, we studied subjects at a single point in time before the onset of structural lung disease. Longitudinal studies are needed to determine whether retention of CD8+ T cells in the airway mucosa leads to clinically apparent COPD over time. Nevertheless, our data reveal a striking increase in airway mucosal CD8+ T cells in HIV-1–infected smokers and suggest that this group of patients may benefit from targeted efforts aimed at smoking cessation.
Taken together, the findings of our study indicate that smoking and HIV-1 promote the migration of CD8+ T cells from the circulation into the lung through upregulation of CXCR3 and CCR5. In HIV-1–infected nonsmokers, CD8+ T cells are recruited from the mucosa into the airspaces by macrophages producing CXCL10 and CCL5. Conversely, suppression of CXCL10 and CCL5 production by chronic tobacco exposure results in accumulation of CD8+ T cells in the airway mucosa of HIV-1–infected smokers. Thus, our data identify a potential mechanism by which HIV-1 and smoking may accelerate the development of COPD.
Acknowledgments
Acknowledgment
The authors thank the Ragon Institute Imaging Core Facility. The authors also thank the staff from the Massachusetts General Hospital Departments of Nuclear Medicine and Nuclear Pharmacy for their assistance with this project, including S. Weise, M. Cournoyer, J. Correia, E. Lee, P. Rice, M. Bruen, E. Beloin, K. Vernon, and D. Yokell. The authors also thank F. Ruzicka, S. Ip, D. Worrall, M. Kone, V. Kelly, and the Pulmonary Special Procedures Unit for their assistance with this study.
Footnotes
Supported by the National Institutes of Health grants 1U01HL121827-01 (B.D.M. and D.S.K.) and R01DA042685 (D.S.K.). The Ragon Institute Imaging Core Facility is supported in part by the Harvard University Center for Acquired Immunodeficiency Syndrome Research, a National Institutes of Health–funded program (5 P30 AI060354010).
Author Contributions: B.C., J.L.C., B.D.M., and D.S.K. conceived of and designed the study. B.C., J.L.C., S.J.G., A.H.L., A.D., A.C.L.-P., A.E.S., P.K., T.W., R.S.H., and B.D.M. acquired samples and experimental data. B.C., J.L.C., M.G., P.K., and T.W. analyzed the data. B.C., J.L.C., P.K., T.W., B.D.M., and D.S.K. interpreted the data. B.C., J.L.C., B.D.M., and D.S.K. drafted and edited the manuscript. B.C., J.L.C., S.J.G., A.H.L., A.D., A.C.L.-P., A.E.S., M.G., P.K., T.W., R.S.H., B.D.M., and D.S.K. approved the final version to be published.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2021-0168OC on June 24, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Crothers K, Butt AA, Gibert CL, Rodriguez-Barradas MC, Crystal S, Justice AC. Veterans Aging Cohort 5 Project Team. Increased COPD among HIV-positive compared to HIV-negative veterans. Chest. 2006;130:1326–1333. doi: 10.1378/chest.130.5.1326. [DOI] [PubMed] [Google Scholar]
- 2. Crothers K, Huang L, Goulet JL, Goetz MB, Brown ST, Rodriguez-Barradas MC, et al. HIV infection and risk for incident pulmonary diseases in the combination antiretroviral therapy era. Am J Respir Crit Care Med. 2011;183:388–395. doi: 10.1164/rccm.201006-0836OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bigna JJ, Kenne AM, Asangbeh SL, Sibetcheu AT. Prevalence of chronic obstructive pulmonary disease in the global population with HIV: a systematic review and meta-analysis. Lancet Glob Health. 2018;6:e193–e202. doi: 10.1016/S2214-109X(17)30451-5. [DOI] [PubMed] [Google Scholar]
- 4. Mokdad AH, Ballestros K, Echko M, Glenn S, Olsen HE, Mullany E, et al. US Burden of Disease Collaborators. The state of US health, 1990-2016: burden of diseases, injuries, and risk factors among US states. JAMA. 2018;319:1444–1472. doi: 10.1001/jama.2018.0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Løkke A, Lange P, Scharling H, Fabricius P, Vestbo J. Developing COPD: a 25 year follow up study of the general population. Thorax. 2006;61:935–939. doi: 10.1136/thx.2006.062802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tager IB, Speizer FE. Risk estimates for chronic bronchitis in smokers: a study of male-female differences. Am Rev Respir Dis. 1976;113:619–625. doi: 10.1164/arrd.1976.113.5.619. [DOI] [PubMed] [Google Scholar]
- 7. Mdodo R, Frazier EL, Dube SR, Mattson CL, Sutton MY, Brooks JT, et al. Cigarette smoking prevalence among adults with HIV compared with the general adult population in the United States: cross-sectional surveys. Ann Intern Med. 2015;162:335–344. doi: 10.7326/M14-0954. [DOI] [PubMed] [Google Scholar]
- 8. Gingo MR, George MP, Kessinger CJ, Lucht L, Rissler B, Weinman R, et al. Pulmonary function abnormalities in HIV-infected patients during the current antiretroviral therapy era. Am J Respir Crit Care Med. 2010;182:790–796. doi: 10.1164/rccm.200912-1858OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Agustí A, Hogg JC. Update on the Pathogenesis of Chronic Obstructive Pulmonary Disease. N Engl J Med. 2019;381:1248–1256. doi: 10.1056/NEJMra1900475. [DOI] [PubMed] [Google Scholar]
- 10. Cosio MG, Saetta M, Agusti A. Immunologic aspects of chronic obstructive pulmonary disease. N Engl J Med. 2009;360:2445–2454. doi: 10.1056/NEJMra0804752. [DOI] [PubMed] [Google Scholar]
- 11. Maeno T, Houghton AM, Quintero PA, Grumelli S, Owen CA, Shapiro SD. CD8+ T cells are required for inflammation and destruction in cigarette smoke-induced emphysema in mice. J Immunol. 2007;178:8090–8096. doi: 10.4049/jimmunol.178.12.8090. [DOI] [PubMed] [Google Scholar]
- 12. O’Shaughnessy TC, Ansari TW, Barnes NC, Jeffery PK. Inflammation in bronchial biopsies of subjects with chronic bronchitis: inverse relationship of CD8+ T lymphocytes with FEV1. Am J Respir Crit Care Med. 1997;155:852–857. doi: 10.1164/ajrccm.155.3.9117016. [DOI] [PubMed] [Google Scholar]
- 13. Peinado VI, Barberá JA, Abate P, Ramírez J, Roca J, Santos S, et al. Inflammatory reaction in pulmonary muscular arteries of patients with mild chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1999;159:1605–1611. doi: 10.1164/ajrccm.159.5.9807059. [DOI] [PubMed] [Google Scholar]
- 14. Podolin PL, Foley JP, Carpenter DC, Bolognese BJ, Logan GA, Long E, III, et al. T cell depletion protects against alveolar destruction due to chronic cigarette smoke exposure in mice. Am J Physiol Lung Cell Mol Physiol. 2013;304:L312–L323. doi: 10.1152/ajplung.00152.2012. [DOI] [PubMed] [Google Scholar]
- 15. Freeman CM, Curtis JL, Chensue SW. CC chemokine receptor 5 and CXC chemokine receptor 6 expression by lung CD8+ cells correlates with chronic obstructive pulmonary disease severity. Am J Pathol. 2007;171:767–776. doi: 10.2353/ajpath.2007.061177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Grumelli S, Corry DB, Song LZ, Song L, Green L, Huh J, et al. An immune basis for lung parenchymal destruction in chronic obstructive pulmonary disease and emphysema. PLoS Med. 2004;1:e8. doi: 10.1371/journal.pmed.0010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 18. Trautmann L, Janbazian L, Chomont N, Said EA, Gimmig S, Bessette B, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med. 2006;12:1198–1202. doi: 10.1038/nm1482. [DOI] [PubMed] [Google Scholar]
- 19. Morris A, George MP, Crothers K, Huang L, Lucht L, Kessinger C, et al. Lung HIV Study. HIV and chronic obstructive pulmonary disease: is it worse and why? Proc Am Thorac Soc. 2011;8:320–325. doi: 10.1513/pats.201006-045WR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Serrano-Villar S, Sainz T, Lee SA, Hunt PW, Sinclair E, Shacklett BL, et al. HIV-infected individuals with low CD4/CD8 ratio despite effective antiretroviral therapy exhibit altered T cell subsets, heightened CD8+ T cell activation, and increased risk of non-AIDS morbidity and mortality. PLoS Pathog. 2014;10:e1004078. doi: 10.1371/journal.ppat.1004078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Triplette M, Attia EF, Akgün KM, Soo Hoo GW, Freiberg MS, Butt AA, et al. A low peripheral blood CD4/CD8 ratio is associated with pulmonary emphysema in HIV. PLoS One. 2017;12:e0170857. doi: 10.1371/journal.pone.0170857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Alford SK, van Beek EJ, McLennan G, Hoffman EA. Heterogeneity of pulmonary perfusion as a mechanistic image-based phenotype in emphysema susceptible smokers. Proc Natl Acad Sci USA. 2010;107:7485–7490. doi: 10.1073/pnas.0913880107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hueper K, Vogel-Claussen J, Parikh MA, Austin JH, Bluemke DA, Carr J, et al. Pulmonary microvascular blood flow in mild chronic obstructive pulmonary disease and emphysema: the MESA COPD study. Am J Respir Crit Care Med. 2015;192:570–580. doi: 10.1164/rccm.201411-2120OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kohli P, Kelly VJ, Hibbert KA, Corleis B, Kone M, Cho JL, et al. PET imaging reveals early pulmonary perfusion abnormalities in HIV infection similar to smoking. J Nucl Med. 2021;62:405–411. doi: 10.2967/jnumed.120.245977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cho JL, Ling MF, Adams DC, Faustino L, Islam SA, Afshar R, et al. Allergic asthma is distinguished by sensitivity of allergen-specific CD4+ T cells and airway structural cells to type 2 inflammation. Sci Transl Med. 2016;8:359ra132. doi: 10.1126/scitranslmed.aag1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bryson BD, Rosebrock TR, Tafesse FG, Itoh CY, Nibasumba A, Babunovic GH, et al. Heterogeneous GM-CSF signaling in macrophages is associated with control of Mycobacterium tuberculosis. Nat Commun. 2019;10:2329. doi: 10.1038/s41467-019-10065-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Corleis B, Lisanti AC, Körner C, Schiff AE, Rosenberg ES, Allen TM, et al. Early type I interferon response induces upregulation of human β-defensin 1 during acute HIV-1 infection. PLoS One. 2017;12:e0173161. doi: 10.1371/journal.pone.0173161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Agostini C, Facco M, Siviero M, Carollo D, Galvan S, Cattelan AM, et al. CXC chemokines IP-10 and mig expression and direct migration of pulmonary CD8+/CXCR3+ T cells in the lungs of patients with HIV infection and T-cell alveolitis. Am J Respir Crit Care Med. 2000;162:1466–1473. doi: 10.1164/ajrccm.162.4.2003130. [DOI] [PubMed] [Google Scholar]
- 29. Kohlmeier JE, Cookenham T, Miller SC, Roberts AD, Christensen JP, Thomsen AR, et al. CXCR3 directs antigen-specific effector CD4+ T cell migration to the lung during parainfluenza virus infection. J Immunol. 2009;183:4378–4384. doi: 10.4049/jimmunol.0902022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kohlmeier JE, Miller SC, Smith J, Lu B, Gerard C, Cookenham T, et al. The chemokine receptor CCR5 plays a key role in the early memory CD8+ T cell response to respiratory virus infections. Immunity. 2008;29:101–113. doi: 10.1016/j.immuni.2008.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Devergne O, Marfaing-Koka A, Schall TJ, Leger-Ravet MB, Sadick M, Peuchmaur M, et al. Production of the RANTES chemokine in delayed-type hypersensitivity reactions: involvement of macrophages and endothelial cells. J Exp Med. 1994;179:1689–1694. doi: 10.1084/jem.179.5.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang J, Nikrad MP, Travanty EA, Zhou B, Phang T, Gao B, et al. Innate immune response of human alveolar macrophages during influenza A infection. PLoS One. 2012;7:e29879. doi: 10.1371/journal.pone.0029879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Neff CP, Chain JL, MaWhinney S, Martin AK, Linderman DJ, Flores SC, et al. Lymphocytic alveolitis is associated with the accumulation of functionally impaired HIV-specific T cells in the lung of antiretroviral therapy-naive subjects. Am J Respir Crit Care Med. 2015;191:464–473. doi: 10.1164/rccm.201408-1521OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Twigg HL, Soliman DM, Day RB, Knox KS, Anderson RJ, Wilkes DS, et al. Lymphocytic alveolitis, bronchoalveolar lavage viral load, and outcome in human immunodeficiency virus infection. Am J Respir Crit Care Med. 1999;159:1439–1444. doi: 10.1164/ajrccm.159.5.9808031. [DOI] [PubMed] [Google Scholar]
- 35. Twigg HL, III, Weiden M, Valentine F, Schnizlein-Bick CT, Bassett R, Zheng L, et al. AIDS Clinical Trials Group Protocol 723 Team. Effect of highly active antiretroviral therapy on viral burden in the lungs of HIV-infected subjects. J Infect Dis. 2008;197:109–116. doi: 10.1086/523766. [DOI] [PubMed] [Google Scholar]
- 36. Shaykhiev R, Krause A, Salit J, Strulovici-Barel Y, Harvey BG, O’Connor TP, et al. Smoking-dependent reprogramming of alveolar macrophage polarization: implication for pathogenesis of chronic obstructive pulmonary disease. J Immunol. 2009;183:2867–2883. doi: 10.4049/jimmunol.0900473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Matsunaga K, Klein TW, Friedman H, Yamamoto Y. Involvement of nicotinic acetylcholine receptors in suppression of antimicrobial activity and cytokine responses of alveolar macrophages to Legionella pneumophila infection by nicotine. J Immunol. 2001;167:6518–6524. doi: 10.4049/jimmunol.167.11.6518. [DOI] [PubMed] [Google Scholar]
- 38. Rao P, Ande A, Sinha N, Kumar A, Kumar S. Effects of cigarette smoke condensate on oxidative stress, apoptotic cell death, and HIV replication in human monocytic cells. PLoS One. 2016;11:e0155791. doi: 10.1371/journal.pone.0155791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Valavanidis A, Vlachogianni T, Fiotakis K. Tobacco smoke: involvement of reactive oxygen species and stable free radicals in mechanisms of oxidative damage, carcinogenesis and synergistic effects with other respirable particles. Int J Environ Res Public Health. 2009;6:445–462. doi: 10.3390/ijerph6020445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Appay V, Papagno L, Spina CA, Hansasuta P, King A, Jones L, et al. Dynamics of T cell responses in HIV infection. J Immunol. 2002;168:3660–3666. doi: 10.4049/jimmunol.168.7.3660. [DOI] [PubMed] [Google Scholar]
- 41. Slütter B, Van Braeckel-Budimir N, Abboud G, Varga SM, Salek-Ardakani S, Harty JT. Dynamics of influenza-induced lung-resident memory T cells underlie waning heterosubtypic immunity. Sci Immunol. 2017;2:eaag2031. doi: 10.1126/sciimmunol.aag2031. [DOI] [PMC free article] [PubMed] [Google Scholar]