

Abstract
Background and Objectives
There are limited population-based data on small fiber neuropathy (SFN). We wished to determine SFN incidence, prevalence, comorbid conditions, longitudinal impairments, and disabilities.
Methods
Test-confirmed patients with SFN in Olmsted, Minnesota, and adjacent counties were compared 3:1 to matched controls (January 1, 1998–December 31, 2017).
Results
Ninety-four patients with SFN were identified, with an incidence of 1.3/100,000/y that increased over the study period and a prevalence of 13.3 per 100,000. Average follow-up was 6.1 years (0.7–43 years), and mean onset age was 54 years (range 14–83 years). Female sex (67%), obesity (body mass index mean 30.4 vs 28.5 kg/m2), insomnia (86% vs 54%), analgesic-opioid prescriptions (72% vs 46%), hypertriglyceridemia (180 mg/dL mean vs 147 mg/dL), and diabetes (51% vs 22%, p < 0.001) were more common (odds ratio 3.8–9.0, all p < 0.03). Patients with SFN did not self-identify as disabled with a median modified Rankin Scale score of 1.0 (range 0–6) vs 0.0 (0–6) for controls (p = 0.04). Higher Charlson comorbid conditions (median 6, range 3–9) occurred vs controls (median 3, range 1–9, p < 0.001). Myocardial infarctions occurred in 46% vs 27% of controls (p < 0.0001). Classifications included idiopathic (70%); diabetes (15%); Sjögren disease (2%); AL-amyloid (1%); transthyretin-amyloid (1%); Fabry disease (1%); lupus (1%); postviral (1%); Lewy body (1%), and multifactorial (5%). Foot ulcers occurred in 17, with 71% having diabetes. Large fiber neuropathy developed in 36%, on average 5.3 years (range 0.2–14.3 years) from SFN onset. Median onset Composite Autonomic Severity Score (CASS) was 3 (change per year 0.08, range 0–2.0). Median Neuropathy Impairment Scale (NIS) score was 2 at onset (range 0–8, change per year 1.0, range −7.9 to +23.3). NIS score and CASS change >1 point per year occurred in only AL-amyloid, hereditary transthyretin-amyloid, Fabry, uncontrolled diabetes, and Lewy body. Death after symptom onset was higher in patients with SFN (19%) vs controls (12%, p < 0.001), 50% secondary to diabetes complications.
Discussion
Isolated SFN is uncommon but increasing in incidence. Most patients do not develop major neurologic impairments and disability but have multiple comorbid conditions, including cardiovascular ischemic events, and increased mortality from SFN onsets. Development of large fiber involvements and diabetes are common over time. Targeted testing facilitates interventional therapies for diabetes but also rheumatologic and rare genetic forms.
Small fiber neuropathy (SFN) is reported to be a common disorder with population prevalence in a large study being 53 per 100,000.1 The typically painful nature of symptoms, autonomic involvement, and lack of data surrounding clinical features and outcomes can be distressing for both patients and providers, reducing quality of life.2 A specific SFN ICD-10 diagnostic code (G628) has been provided only recently in the United States (October 2015). Consensus diagnostic criteria have emerged and will aid anticipatory guidance, population classification, and study design.3,4 The current diagnostic criteria emphasize the value of quantitative testing, aiding diagnostic precision. Different SFN quantitative approaches such as intraepidermal nerve fiber (IENF) density, thermoregulatory sweat test (TST), quantitative sensory testing (QST) (heat-pain, cold sensation), and autonomic reflex screen (ARS), including Quantitative Sudomotor Autonomic Reflex Test (QSART), enhance localization and objective measurement.5
Much of the earlier research on SFN individually or in combination has not included longitudinal outcomes or detailed neurologic examinations or excluded patients with the development of large fiber involvement. This is important because small fiber dysfunction and large fiber dysfunction often coexist, but painful SFN symptoms are often the presenting symptom and the predominant patient concern.4,6,7 One study examining quality of life with the Short Form-36 questionnaire found significantly worse physical functioning scores at the time of assessment.2 Progressive IENF loss has been reported but without correlation to clinical disability.8 Two studies have documented clinical and neurophysiologic stability over time in a portion of patients with SFN: 46% (n = 21) over 22 months7 and 75% (n = 16) over a mean of 5.3 years.6
The aims of this study were to evaluate patients with SFN without large fiber involvement (pure) at onset for incidence and prevalence and to longitudinally evaluate patients for comorbid conditions, cumulative neurologic impairments (somatic and autonomic), disabilities, and mortality from a population perspective. Idiopathic vs presumed secondary (causal) forms were considered separately for impairments and disability. Outcomes of patients receiving immunotherapy were also reviewed. A geographic, population-based approach was used to allow matched controlled longitudinal follow-up including comparison to patients without neuropathy and our earlier generalized neuropathy cohort.9
Methods
Standard Protocol Approvals, Registrations, and Patient Consents
The study was performed with patient written consent and institutional research board approval.
Cohort Identification
Institutionally available electronic software was used to retrieve patients diagnosed with SFN who presented from January 1, 1998, to December 31, 2017, having had a neurology evaluation who were living in Olmsted or adjacent counties (Dodge, Goodhue, Wabasha, Winona, Fillmore, Mower). Institutional software able to retrieve any chart with specified text was used to retrieve all people with SFN or painful neuropathy, with additional screening using the ICD-10 code for SFN once available (October 2015). All electronically identified charts were reviewed to confirm diagnosis, date of symptom onset (pain, burning, tingling, loss of feeling, autonomic symptoms), and all demographic data. Pain and immunotherapy prescriptions were also queried electronically. Our institution is the only facility offering both EMG and small fiber confirmatory testing in southeastern Minnesota, so all patients meeting inclusion diagnostic testing criteria should be captured. Inclusion required patients with SFN to have had nerve conductions, EMG, and neurology consultation with standardized neuromuscular neurologic examinations scored for motor, reflex, and sensory changes (vibration, proprioception, light touch, pinprick). Patients had to have symptoms of SFN and bedside testing abnormality of small fiber function either pinprick, heat pain, or cold sensation to be included. Onset exclusionary criteria included any weakness attributed to neuropathy at onset, nonsmall fiber sensory loss (proprioception and vibratory loss), absent ankle reflexes in those <65 years of age or those without musculoskeletal or spine explanation, and nerve large fiber peripheral involvement by nerve conductions and EMG. An SFN diagnosis had to be confirmed by TST (anhidrosis at affected site), QSART (below fifth percentile of normal), QST-CASE IV (>97th percentile of normal heat pain or cold sensation), or IENF biopsy (below first percentile of normal).5,10-12 For patients undergoing QST, vibration-detection abnormality (>97th percentile of normal) was exclusionary. These quantitative measures of small fiber function have been available for diagnosis throughout the entire study period excluding IENF density, which began in October 2012.10
Causal Investigations
For idiopathic vs causal designation, all patients had measures for serum glucose (including fasting glucose, glucose challenge, or hemoglobin A1c), vitamin B12, thyroid-stimulating hormone, monoclonal protein testing by immunofixation, autoimmune cascade testing (antinuclear, extractable nuclear antigen, Sjögren SSA and SSB antibodies), and review for excess alcohol or chemotherapeutic exposure. Family history of neuropathy and a complete medical history were reviewed in all patients. Causal association was made for SFN on the basis of known reported causes and temporal association with those disorders and disease onsets.13-15 For patients with SFN in whom >1 temporally associated cause was identified, multifactorial cause was listed. To be considered inherited, a known pathogenic genetic mutation16 needed to be found, and family history alone was not adequate to be considered genetic. American Diabetes Association definitions of diabetes were used (hemoglobin A1c ≥6.5%, fasting blood glucose on 2 separate occasions ≥126 mg/dL, or glucose challenge at 2 hours ≥200 mg/dL). We included patients with diabetic treatment–induced SFN as causal with defined rapid correction of hemoglobin A1c.17 However, for glucose impairment (hemoglobin A1c 5.7%–6.4%, fasting glucose 100–125 mg/dL, glucose challenge 2 hours 140–199 mg/dL), given the debate of its causal potential,18,19 those patients were designated as idiopathic. Expanded testing was also reviewed for those undergoing testing for paraneoplastic or voltage gated potassium channel VGKC immunoglobulin G (IgG) complex disease (LGI1-IgG and CASPR2-IgG subtypes), Lyme disease, α-galactosidase enzymatic and gene sequencing assays, HIV, hepatitis C, cryoglobulins, proteinase-3, celiac disease, angiotensin-converting enzyme, transthyretin-amyloidosis, and SCN9A gene sequencing, among others.
Matched Control Subset
The Rochester Epidemiology Project database was used to match patients with SFN 3:1 (3 controls per 1 case) for sex and age with randomly selected persons in the geographic region, excluding all neuropathy ICD-9 and ICD-10 codes. Control demographic variables, diagnostic codes, and medications were queried with institutional software. Charlson Comorbidity Indices (CCIs) in this SFN cohort were compared to those in our earlier generalized neuropathy publication9 (January 1, 2006–December 31, 2010) generated from ICD-9 codes.
Assessment Scales
Disability was evaluated with serial standardized questionnaires provided at 6-month intervals for all patients returning for care. Surrogate markers included ability to perform activities of daily living; limb weakness; loss of feeling; fall tendency; pain; stair-climbing difficulty; reliance on assistance from gait aid or others; employment status; and living environment. These questions were used in our earlier review of generalized neuropathy.9 Neurologic large fiber neuropathy and SFN impairments were assessed by institutional standardized neurologic examination calculating Neuropathy Impairment Scale (NIS)20 score (0 being normal and 244 being unable to feel all sensory modalities in hands and feet [32 points], having no reflexes, and complete paralysis of all muscles in the body). Because the NIS is predominately a large fiber and motor measurement, a maximal deficit for SFN would be limited to pinprick sensation with loss in hands and feet equating to 8 points. Autonomic impairment was evaluated with the Composite Autonomic Severity Score (CASS) when autonomic testing was available.21 The score is graded from 0 to 10, with 4 points for adrenergic dysfunction and 3 points each for sudomotor and cardiovagal failure. Scores of ≤3 indicate mild impairment, and scores of ≥7 indicate severe autonomic impairment. To assess for progression of neurologic impairments, the NIS and CASS scores were calculated for change over time for patients with SFN when repeat examinations were available.
Disability was scored by modified Rankin Scale (mRS) score (0 = no symptoms; 1 = nondisabling symptoms that do not interfere with daily activities; 2 = no significant disability despite symptoms, able to carry out all usual duties and activities; 3 = moderate disability, requiring some help, but able to walk or dress oneself and eat without assistance; 4 = moderately severe disability, unable to walk, dress self without assistance, and attend to own bodily needs without assistance; 5 = severe disability, bedridden, and requiring constant care and attention; 6 = dead).22 The breakdown of the scoring of mRS based on self-reported surrogate markers is identical to the approach we previously reported in generalized neuropathy.9 Common symptoms reported in SFN with and without autonomic dysfunction were recorded as outlined in Composite Autonomic Symptom Scale 31.23,24
Mortality data were obtained beyond the case ascertainment study period through May 30, 2020. CCIs using ICD-9 and ICD-1025 coding were calculated from chart-extracted diagnoses at the study end date to calculate the scores and comorbid conditions. Causes of death were reviewed in patients with SFN against comorbid conditions.
Statistical Analysis
All statistical analysis was performed on JMP 14.1.0 (SAS Institute Inc, Cary, NC) software, with significance recorded at p < 0.05. Categorical data were analyzed with 2 × 2 testing. Continuous variables were assessed with the Wilcoxon 1-way analysis of variance. Surrogate markers of disability and CCI were compared between patients with SFN and controls. Kaplan-Meier survival curves were plotted for those with and without SFN. Cox proportional hazards regression was used to estimate hazard ratios for survival. The annual incidence and prevalence were calculated from the summed county US Census Bureau decennial censuses and intercensal estimates for 1998 to 2017. The population was stable over time, increasing on average 0.8%/y during the study period. From these results, the means were generated; disease onset was defined as the year of symptom onset; and patients who died were not included in prevalence calculations. Linear slope R2 value correlation was used to assess the incidence trend over the study period.
Data Availability
Deidentified data are available on reasonable request.
Results
Quantitative Testing Confirmed SFN Incidence and Prevalence
Over the study period, 183 patients had SFN mentioned in the diagnosis or assessment fields of the electronic medical record, with 94 meeting all inclusion criteria. Of the 89 patients in whom SFN was excluded, 45% (40 of 89) had onset generalized (large and small fiber involvement by neurologic examination, nerve conduction study/EMG abnormality, or QST abnormality); 24% (21 of 89) had myofascial pain, including fasciitis and tendonitis; 22% (20 of 89) had gout; 15% (13 of 89) had fibromyalgia; and 4% (4 of 89) had other diagnoses. The quantitative testing–confirmed SFN incidence (95% confidence interval [CI]) over the entire study period was 1.3/100,000 inhabitants/year (95% CI 0.9–1.6) with quantitative testing–confirmed SFN prevalence of 13.3/100,000 inhabitants (95% CI 9.9–16.6). There was a significant upward trend in incidence over the study period with a slope of 1.66 (R2 = 0.40, p = 0.02, Figure 1). Increased SFN incidence over time could not be attributed to availability of IENF density testing because 5 of 7 patients with confirmed IENF density reductions with SFN also had multiple positive confirmatory tests (TST, QST, QSART).
Figure 1. Incidence of SFN per 100,000 Inhabitants per Year.
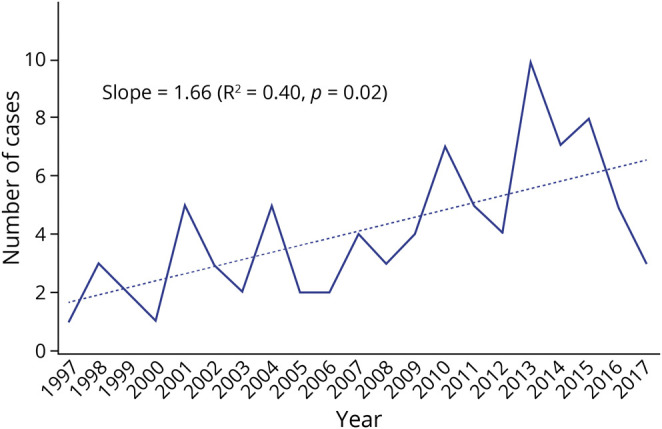
Incidence of small fiber neuropathy (SFN) in Olmsted County, Minnesota, and adjoining counties from January 1, 1998, through December 31, 2017. There was a significant positive upward trend in incidence over the study period.
Demographics
Demographic data are summarized in Table 1. The median age at onset was 54 years (range 14–83 years); average follow-up was 6.1 years (0.7–43 years). There was female predominance (67%). Patients with SFN were significantly more likely than controls to be obese (body mass index [BMI] 30.4 vs 28.5 kg/m2; healthy 18.5–24.9 kg/m2, obese ≥30.0 kg/m2), have higher triglyceride levels (180 mg/dL mean vs 147 mg/dL: normal <150 mg/dL), have sleep difficulties (86% vs 54%), use nonopioid pain prescriptions (67% vs 18%), use opioid analgesics (72% vs 46%), and have higher education (81% vs 66%). Patients with SFN trialed a higher number of different prescription analgesic medications (opioids 2.7 vs 1.8; nonopioids 2.4 vs 1.8). There was no significant difference in reported substance abuse or psychiatric illness compared to controls.
Table 1.
Demographic Characteristics of SFN Cohort

Painful cutaneous symptoms at onset were reported most commonly in the lower extremities (77%, 72 of 94), while 12% (11 of 94) had upper and lower extremity involvement, 5% (5 of 94) had upper extremity involvement only, 3% (3 of 94) had truncal involvement only, and 3% (3 of 94) had truncal and extremity involvement. Chronic onset was typical (80%, 75 of 94), excluding those with truncal only onset (n = 6), of whom 4 presented subacutely. Nociceptive painful symptoms (burning 25%, paresthesia 30%, shock-like pain 29%) often overlapped and occurred in 95% (89 of 94), while complaint of loss of feeling occurred in only 32% (30 of 94). Over the study period, 17 patients with SFN developed foot ulcers (1 requiring toe amputation), and all but 2 of these patients had developed large fiber neuropathy at time of ulcers. Of those, 71% (12 of 17) had diabetes, 18% (3 of 17) had dysproteinemia (2 AL-amyloid, 1 lymphoplasmacytic lymphoma), and 12% (2 of 17) were idiopathic.
Autonomic symptoms were common and generally mild, affecting 85% of patients with SFN. Included among the symptoms were male erectile dysfunction in 58% (18 of 31); constipation in 36% (33 of 94); lightheadedness and palpitations in 36% (33 of 94); urinary symptoms in 33% (31 of 94); diarrhea in 22% (20 of 94); dry eyes and mouth in 22% (20 of 94); sweat abnormalities in 19% (18 of 94); gastroparesis in 6% (6 of 94); and multiple autonomic symptoms in 65% (61 of 94). Quantitative measures of small fiber dysfunction most commonly were found by TST but typically by multiple tests. More than half of the cohort (52%, 49 of 94) had positive SFN-quantified test abnormalities by multiple modalities: TST 75% (70 of 94), ARS 64% (60 of 94), QST 25% (24 of 94), and IENF density 8% (7 of 94). On the ARS, the QSART was the most common abnormality (77%, 46 of 60), with mild often asymptomatic abnormalities of either Valsalva ratio (52%) or head-up tilt (28%).
Polyneuropathy on nerve conductions and EMG evaluations at the first visit was absent (by inclusion), with a median sural sensory amplitude being 8 µV (range 0–26 µV, normal ≥6 µV if age <60 years and ≥0 µV if age ≥60 years). Follow-up EMGs at a median time interval of 18 months (range 6–415 months) were available in 61 patients with SFN. Of those, 36% (22 of 61) evolved to a large fiber length–dependent axonal sensory and motor polyneuropathy consistent with their large fiber sensory and motor and reflex abnormalities on clinical examination.
Causality
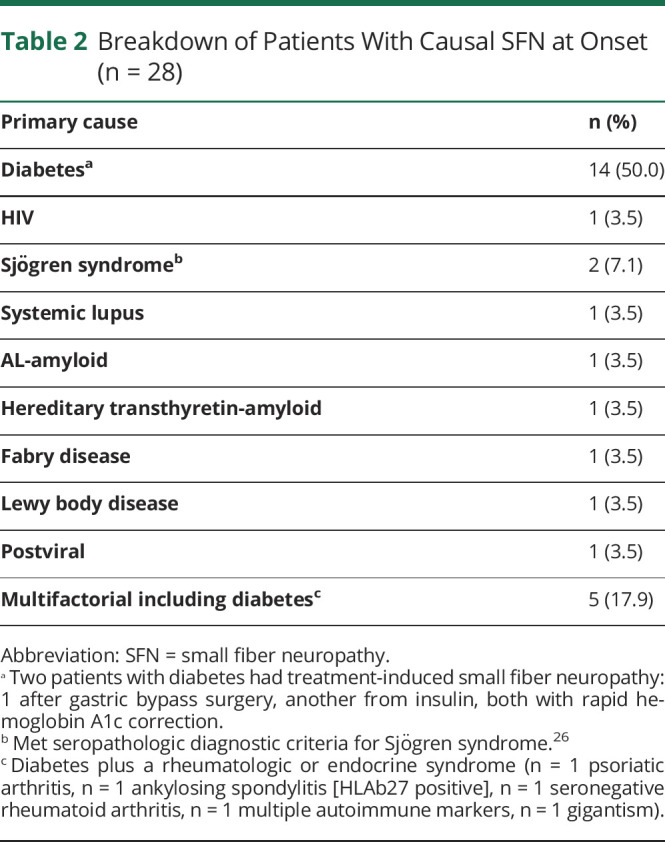
Idiopathic SFN was the most common diagnosis (70%), followed by diabetes (15%). A summary of causal forms is given in Table 2. Glucose impairment or impaired glucose tolerance occurred in 15% at first SFN evaluation. It is important to note that 51% (48 of 94) of the cohort would eventually develop diabetes compared to 22% of controls throughout the study period (p < 0.001). All 14 with glucose impairment and an additional 20 initially normal developed diabetes. Of these diabetics 63% (30 of 48) would develop other end-organ complications of diabetes (retinopathy, nephropathy). Two patients with SFN had treatment-induced diabetic SFN, 1 with a drop in hemoglobin A1c from 9.1% to 6.6% over 2 months after initiation of insulin and another having a drop in hemoglobin A1c from 19% to 11% over 5 months after gastric bypass surgery with normal vitamin B12 and thiamine levels. Five patients with SFN labeled multifactorial had both diabetes and rheumatologic or endocrine disorders (psoriatic arthritis n = 1, ankylosing spondylitis [HLAb27 positive] n = 1, seronegative rheumatoid arthritis n = 1, multiple autoimmune markers n = 1, gigantism n = 1). For most lone rheumatologic disorders, causality was not assigned due to lack of definitive literature establishing causality. Most were instead listed as comorbid conditions within idiopathic classification. Serologic testing for paraneoplastic including VGKC-associated SFN was performed on 36% (34 of 94) with 1 VGKC positive at 0.15 nmol/L (normal <0.03 nmol/L) but negative for the specific LGI1-IgG and CASPR2-IgG targets. Genetic cause was found in only 2 patients with SFN. One had hereditary transthyretin-amyloidosis p.A36P (Ala36Pro) c.166 G>C (GCT>CCT) with strong family history. Her initial symptoms were of burning feet and loss of pinprick sensation for several years before reflex, motor, and autonomic symptoms and signs. One male patient had Fabry disease with chronic intermittent foot pain and irritable bowel diagnosed with SFN at 30 years of age with renal insufficiency. He had no family history but reduced α-galactosidase enzymatic testing and confirmatory Fabry genetic mutation testing.
Table 2.
Breakdown of Patients With Causal SFN at Onset (n = 28)
Comorbid Conditions
Comorbid conditions by CCI are shown in Table 3 for patients with SFN and controls without neuropathy compared to CCI data from our historical generalized neuropathy cohort.9 Ten patients with SFN who developed large fiber involvements were included in the earlier study. The median CCI in the controls was 3 (range 1–9) and significantly less (p < 0.001) than the median CCI of 6 (range 3–9) for patients with SFN but similar to our historical general polyneuropathy cohort. In all CCI categories, patients with SFN had greater prevalence of comorbid conditions compared to controls with no neuropathy. However, compared to the earlier generalized neuropathy cohort, more prevalent comorbid conditions were limited largely to myocardial infarction (23% more prevalent) and rheumatologic disease (18% more prevalent) despite less peripheral vascular disease (p < 0.001). However, among patients with myocardial infarction, peripheral vascular disease was significantly more common (35% vs 9%; p < 0.001) and rheumatologic disease was even more strongly associated (35% vs 6%; p < 0.0003).
Table 3.
Comparison of SFN Comorbidity Prevalence Among Cohorts

Rheumatologic or inflammatory immune-mediated disorders occurred in 28 patients with SFN: rheumatoid arthritis (n = 6); discoid and systemic lupus (n = 3); polymyalgia rheumatica (n = 2); Sjögren disease (n = 2) seropathologically diagnosed26; sicca (n = 5); inflammatory arthritis not otherwise specified (n = 7); Crohn disease (n = 2); and primary biliary cirrhosis (n = 1). In all but 2 patients with Sjögren disease, the rheumatologic inflammatory immune-mediated diagnosis occurred years before the development of SFN while on stable long-term immunotherapies (azathioprine [n = 7]; hydroxychloroquine [n = 12]; infliximab [n = 4]; mycophenolate [n = 1]; oral steroids [n = 20]; methotrexate [n = 6]; and IV immunoglobulin [n = 4]). The 2 patients with Sjögren disease who presented with SFN (designated causal) at the time of their rheumatologic diagnosis reported improvement of neuropathic pain with methotrexate and hydroxychloroquine.
Impairments and Disability
The mean NIS score at the first visit was 2 (range 0–8) and at the last visit was 6 (range 0–65). The average time to develop large fiber involvement was 5.3 years (range 0.2–14.3 years). Among the 18 patients with idiopathic SFN having follow-up neurologic examinations ≥12 months (mean 73 months, range 12–476 months), the mean NIS score worsened by 1.7 points per year (0–7 points). In contrast, among 11 patients with SFN with a known cause having neurologic examination follow-up ≥12 months (mean 50 months, range 12–180 months), the mean NIS score increase was 9.7 points per year (−8 to 32). Of patients with SFN with known SFN cause, only 4 had NIS score worsening >8 points per year. One patient had hereditary transthyretin amyloidosis; 1 had gigantism from endocrine tumor and poorly controlled diabetes; and 2 other isolated poorly controlled diabetes. Only 1 patient had significant improvement in NIS score. That patient with systemic lupus (designated causal) had been changed on immunotherapy (azathioprine to hydroxychloroquine), improving −8 points on NIS. She also reported improvement in pain. No patient with diabetes had clear improvements in their NIS scores, but all patients with diabetes with a hemoglobin A1c ≤7.0% had an NIS score change per year <8 (n = 6). The median CASS of 60 patients with SFN undergoing evaluations at time of diagnosis was 3 (range 0–10) at the first visit, and among 12 patients with SFN with follow-up studies, the mean increase in CASS was only 0.08 per year (range 0.0–2.0) with a median follow-up of 36 months (range 2–144 months).
Review of 67,940 patient-years of standardized questionnaire responses among patients with SFN and controls assessing disability across 12 different surrogate markers revealed 7 having significant difference compared to controls with no neuropathy (Table 4). Despite greater progression of NIS scores in patients with causal SFN vs idiopathic SFN, as a group, statistical differences in surrogate markers of disability were not found except for difficulty taking medications. For the question “Are you disabled?” there was no significant difference between patients with SFN and controls. However, as a group, the SFN cohort at that last visit was more likely than controls to have a higher calculated mRS score (median 1.0, range 0–6 vs 0.0, range 0–6 for controls, p = 0.04).
Table 4.
Serial Standardized Questionnaire Surrogate Markers of Disability

Mortality
Although mean age at death compared to controls was not significantly different at 70 years for patients (interquartile range 57–79 years) vs 73 years (interquartile range 67–82 years, p > 0.05) for controls, there was a significantly higher number of deaths (19%, 18 of 94) in patients with SFN vs controls (12%, 35 of 282, p < 0.0001) from time of symptom onset. In addition, subset analysis of idiopathic vs causal demonstrated greater number of deaths from time of SFN diagnosis in patients with SFN with a known cause vs idiopathic vs controls (Figure 2). Table 5 lists causes of death related to coexisting comorbid conditions, with 50% (9 of 18) associated with diabetic complications.
Figure 2. Survival Comparison From SFN Symptom Onset.

Kaplan-Meier plot showing all-cause mortality across the study population. Curves are adjusted for time of small fiber neuropathy (SFN) symptom onset matching that date in age- and sex-matched controls. Higher frequency of death events occurred only from time of symptom onsets in patients with SFN vs controls with mean age at death not significantly different. Causal vs idiopathic patients had greater death events. Informative risk table set (bottom) displays the number of patients with SFN who were under observation in the specific period.
Table 5.
Deaths Among Patients With SFN and Comorbid Conditions

Discussion
In this longitudinal population-based study with quantitative testing–confirmed SFN, the mean annual incidence of SFN was 1.3 per 100,000 inhabitants with a prevalence of 13.3 per 100,000 (0.013 percentage point prevalence). Incidence increased over the study period. This may relate to increased awareness because it was not explained by test availability. Another possibility is that the increasing BMI in our geographic region,27 a known risk for type 2 diabetes occurrence and hypertriglyceridemia, could be increasing the rates of SFN. Incidence of SFN was much lower compared to the earlier report from the Netherlands,1 but different methodologies were used. In contrast, we did not exclude individuals <20 years of age, we examined a 20-year period (SFN quantitative testing readily available at our institution throughout this time), and we excluded patients with SFN who died in our prevalence calculations. Other differences in patient behavior and health care access, population data, cost, and delivery may also play a role in differing incidence, but these factors are harder to determine. Nevertheless, in our geographic region, pure SFN onset is far less common than generalized neuropathy, for which we found 1.66 percentage point prevalence.9
Patients with SFN in this cohort most often had length-dependent neuropathic pain symptoms that developed insidiously at older age, had frequent autonomic symptoms, and commonly developed large fiber involvement and diabetes over time. Autonomic function testing demonstrates that although autonomic involvement is typical, involvement is generally not severe, and progression is only modest, which aligns with the observation that most have cutaneous rather than autonomic symptoms as their primary difficulty.4 We found higher frequencies of opioid and nonopioid pain medication use, more common than in our earlier generalized neuropathy cohort.9 Sleep difficulties were also more common, consistent with other pain disorders.28 Although pain was a major problem, foot ulcers with insensate injury commonly occurred generally after development of large fiber involvement with diabetes. Patients with SFN were significantly more likely to be female and obese and have elevated triglycerides, consistent with features of the metabolic syndrome.29-31 Metabolic syndrome is reported to be more common in patients with diabetes with neuropathy than in those without neuropathy.30,31 Our findings might also support an association between metabolic syndrome and SFN. Whether improvement of triglycerides or BMI could aid neuropathy severity and pain symptoms will require further prospective investigation, but earlier work has suggested that this may be beneficial.32
Our comorbidity findings demonstrate that patients with SFN are on average sicker than controls without neuropathy have comorbid conditions similar to large fiber polyneuropathy.9 Rheumatologic disease and myocardial infarction are more common in SFN compared to generalized neuropathy. Our finding that the majority of patients with SFN who developed SFN while on immunotherapy for rheumatologic or inflammatory immune-mediated conditions would support a recent double-blind placebo-controlled trial showing that IV immunoglobulin is ineffective in idiopathic SFN.33,34 However, our study also provides anecdotal evidence that rare patients with SFN with contemporaneous onset inflammatory immune disorders can benefit from immunotherapy. Blinded prospective trials of SFN in persons with rheumatologic or inflammatory immune-mediated disorders are needed to clarify the best treatment approaches.
A potentially important observation from our cohort is that myocardial infarction occurred in nearly half of all patients with SFN, much more commonly than in our generalized neuropathy cohort and no neuropathy controls. This may relate to their higher cardiovascular risk factors but also to other unclear polygenic risks, including those described in metabolic syndrome and rheumatologic disorders.35,36 In addition, earlier studies have suggested an association with cardiac disease and autonomic involvements in diabetes.37,38 A lower threshold for cardiovascular screening in patients with SFN is suggested to be helpful. In addition, our data and earlier study39 strongly link SFN with the development of diabetes, including commonly with end-organ damage. More intense longitudinal glucose monitoring, especially among idiopathic forms, is needed because nearly half would eventually develop diabetes.
Except for rare patients with causal SFN (AL-amyloid, familial transthyretin amyloid, Fabry disease, uncontrolled diabetes, and Lewy body disease), NIS and CASS score worsening was nominal. Most patients with idiopathic and causal SFN did not develop major impairments; most had nondisabling symptoms not interfering with daily activities (mRS score 1). When considering mortality, overall survival was comparable to that of matched controls, but when looking at survival from time of SFN onset, mortality was greater in both causal and idiopathic varieties. Death was linked with complications of diabetes in half of patients with SFN.
Previous consensus guidelines for testing in polyneuropathy have included tests for the detection of diabetes, monoclonal proteins, and genetic forms.40 Transthyretin and α-galactosidase gene sequencing was helpful to confirm one patient with hereditary transthyretin neuropathy and another with Fabry disease. Such testing is likely more important now than ever given the increasing availability of Food and Drug Administration–approved therapeutic options for both of those disorders.41,42 While we did not find any patients with paraneoplastic SFN, various IgG autoantibodies (CASPR2, LGI1, ANNA1, CRMP5, Amphiphysin)43-46 can be associated with painful SFN and can lead to a cancer diagnosis. Most patients with a paraneoplastic etiology having SFN also have asymmetric polyradiculoneuropathy with subacute onset, whereas insidious chronic symmetric course is most typical in idiopathic SFN. Thus, if a patient with negative standard evaluation develops features of an immune inflammatory paraneoplastic neuropathy,47 autoantibody testing should be considered.
The primary limitation of this study is its retrospective nature. Prospective studies examining all patients presenting with symptoms of SFN using detailed histories, examinations, and a battery of quantitative measures of small fiber dysfunction are needed to confirm our findings. While we would have liked to include analog pain scales, these were not uniformly collected until after 2017, so pain medication prescribing habits were used as a surrogate, similar to an earlier report.48 Some patients with SFN without quantitative measures of small fiber dysfunction could have been excluded; however, we felt that objective measures were necessary given the broad differential for conditions mimicking SFN.3,4 The small number of patients found may interfere with our ability to accurately capture treatment and causal mechanisms. However, population-based studies are not needed to make those observations.
This longitudinal, population-based, case-controlled cohort study of clinically and objectively defined SFN, not excluding persons developing large fiber dysfunction, provides insights into SFN demographics and disease progression. The disorder appears to be increasing in frequency with greater trends toward obesity. Most patients with SFN do not develop major disability, with neurologic impairments more severe in causal and less severe in idiopathic forms. Cardiovascular ischemic events, development of diabetes, and coexisting rheumatologic comorbid conditions are common, necessitating a multidisciplinary approach to patient care.
Glossary
- ARS
autonomic reflex screen
- BMI
body mass index
- CASS
Composite Autonomic Severity Score
- CCI
Charlson Comorbidity Index
- CI
confidence interval
- ICD-10
International Classification of Diseases, 10th revision
- IENF
intraepidermal nerve fiber
- IgG
immunoglobulin G
- mRS
modified Rankin Scale
- NIS
Neuropathy Impairment Scale
- QSART
quantitative sudomotor autonomic reflex test
- QST
quantitative sensory testing
- SFN
small fiber neuropathy
- TST
thermoregulatory sweat test
- VGKC
voltage gated potassium channel
Appendix. Authors
Footnotes
Study Funding
Funding provided by the Mayo Clinic Foundation, Mayo Clinic Center of Individualized Medicine, Mayo Clinic Center of MS, and Autoimmune Neurology.
Disclosure
Dr. Johnson, Dr. Shouman, Dr. Shelly, Dr. Sandroni, and Dr. Berini report no disclosures relevant to the manuscript. Dr. Dyck reports honorarium from Akcea for talks on TTR amyloid. Dr. Hoffman, Dr. Mandrekar, Dr. Niu, Dr. Lamb, and Dr. Low report no disclosures relevant to the manuscript. Dr. Singer reports serving on the scientific advisory board for Lundbeck and Catalyst Pharmaceuticals and has consulted for Biohaven and Astellas pharmaceuticals. He has received grant support from the NIH (U54NS65736, R01NS092625), Food and Drug Administration (R01FD4789), Boston Scientific, and the American Dysautonomia Institute. He holds a patent on the use of 3,4 diaminopyridine in the treatment of orthostatic hypotension and postural tachycardia syndrome. He is associate editor for Clinical Autonomic Research and serves on the editorial board of Autonomic Neuroscience—Basic and Clinical. Dr. Mauermann reports research support from IONIS, Alnylam, and EIDOS Pharmaceutical. She has received royalties from Oxford Publishing. She serves on the Editorial Board for Mayo Clinic Proceedings. Dr. Mills holds patents on the use of mass spectrometry to measure monoclonal immunoglobulins and has received royalties related to these patents from The Binding Site. Dr. Dubey reports consultation for UCB, Alexion, and Immunovant pharmaceuticals. All compensation for consulting activities is paid directly to Mayo Clinic. He has patents pending for KLHL11 and LUZP4 as biomarkers of neurologic autoimmunity and germ cell tumor. Dr. Staff reports research funding from the NIH (R01CA211887), ALS Association, Target ALS, and Regenerative Medicine Minnesota. He has served as investigator on clinical trials sponsored by Brainstorm Therapeutics, Orion Pharmaceuticals, Biogen, and Medicinova but received no personal compensation for this. He serves as an associate editor for Stem Cell Research & Therapy. Dr. Klein has received teaching honorarium from Akcea pharmaceuticals for lectures on hereditary transthyretin amyloidosis and Fabry disease. He has consulted for Pfizer regarding tafamidis (all compensation for consulting activities is paid directly to Mayo Clinic) and participated in the clinical trials for inotersen and patisiran but received no personal compensation for his participation. Go to Neurology.org/N for full disclosures.
References
- 1.Peters MJ, Bakkers M, Merkies IS, Hoeijmakers JG, van Raak EP, Faber CG. Incidence and prevalence of small-fiber neuropathy: a survey in the Netherlands. Neurology. 2013;81(15):1356-1360. [DOI] [PubMed] [Google Scholar]
- 2.Bakkers M, Faber CG, Hoeijmakers JG, Lauria G, Merkies IS. Small fibers, large impact: quality of life in small-fiber neuropathy. Muscle Nerve. 2014;49(3):329-336. [DOI] [PubMed] [Google Scholar]
- 3.Freeman R, Gewandter JS, Faber CG, et al. Idiopathic distal sensory polyneuropathy: ACTTION diagnostic criteria. Neurology. 2020;95(22):1005-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Devigili G, Rinaldo S, Lombardi R, et al. Diagnostic criteria for small fibre neuropathy in clinical practice and research. Brain. 2019;142(12):3728-3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singer W, Spies JM, McArthur J, et al. Prospective evaluation of somatic and autonomic small fibers in selected autonomic neuropathies. Neurology. 2004;62(4):612-618. [DOI] [PubMed] [Google Scholar]
- 6.Flossdorf P, Haupt WF, Brunn A, et al. Long-time course of idiopathic small fiber neuropathy. Eur Neurol. 2018;79:161-165. [DOI] [PubMed] [Google Scholar]
- 7.MacDonald S, Sharma TL, Li J, Polston D, Li Y. Longitudinal follow-up of biopsy-proven small fiber neuropathy. Muscle Nerve. 2019;60(4):376-381. [DOI] [PubMed] [Google Scholar]
- 8.Khoshnoodi MA, Truelove S, Burakgazi A, Hoke A, Mammen AL, Polydefkis M. Longitudinal assessment of small fiber neuropathy: evidence of a non-length-dependent distal axonopathy. JAMA Neurol. 2016;73(6):684-690. [DOI] [PubMed] [Google Scholar]
- 9.Hoffman EM, Staff NP, Robb JM, St Sauver JL, Dyck PJ, Klein CJ. Impairments and comorbidities of polyneuropathy revealed by population-based analyses. Neurology. 2015;84(16):1644-1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Engelstad JK, Taylor SW, Witt LV, et al. Epidermal nerve fibers: confidence intervals and continuous measures with nerve conduction. Neurology. 2012;79(22):2187-2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Low VA, Sandroni P, Fealey RD, Low PA. Detection of small-fiber neuropathy by sudomotor testing. Muscle Nerve. 2006;34(1):57-61. [DOI] [PubMed] [Google Scholar]
- 12.Dyck PJ, Zimmerman I, Gillen DA, Johnson D, Karnes JL, O'Brien PC. Cool, warm, and heat-pain detection thresholds: testing methods and inferences about anatomic distribution of receptors. Neurology. 1993;43(8):1500-1508. [DOI] [PubMed] [Google Scholar]
- 13.Mendell JR, Sahenk Z. Clinical practice. Painful sensory neuropathy. N Engl J Med. 2003;348(13):1243-1255. [DOI] [PubMed] [Google Scholar]
- 14.Lacomis D. Small-fiber neuropathy. Muscle Nerve. 2002;26(2):173-188. [DOI] [PubMed] [Google Scholar]
- 15.Gibbons CH. Small fiber neuropathies. Continuum. 2014;20(5 Peripheral Nervous System Disorders):1398-1412. [DOI] [PubMed] [Google Scholar]
- 16.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gibbons CH, Freeman R. Treatment-induced neuropathy of diabetes: an acute, iatrogenic complication of diabetes. Brain. 2015;138(pt 1):43-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sumner CJ, Sheth S, Griffin JW, Cornblath DR, Polydefkis M. The spectrum of neuropathy in diabetes and impaired glucose tolerance. Neurology. 2003;60(1):108-111. [DOI] [PubMed] [Google Scholar]
- 19.Dyck PJ, Clark VM, Overland CJ, et al. Impaired glycemia and diabetic polyneuropathy: the OC IG Survey. Diabetes Care. 2012;35(3):584-591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dyck PJ, Sherman WR, Hallcher LM, et al. Human diabetic endoneurial sorbitol, fructose, and myo-inositol related to sural nerve morphometry. Ann Neurol. 1980;8(6):590-596. [DOI] [PubMed] [Google Scholar]
- 21.Low PA. Composite autonomic scoring scale for laboratory quantification of generalized autonomic failure. Mayo Clin Proc. 1993;68(8):748-752. [DOI] [PubMed] [Google Scholar]
- 22.van Swieten JC, Koudstaal PJ, Visser MC, Schouten HJ, van Gijn J. Interobserver agreement for the assessment of handicap in stroke patients. Stroke. 1988;19(5):604-607. [DOI] [PubMed] [Google Scholar]
- 23.Sletten DM, Suarez GA, Low PA, Mandrekar J, Singer W. COMPASS 31: a refined and abbreviated composite autonomic symptom score. Mayo Clin Proc. 2012;87(12):1196-1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Treister R, O'Neil K, Downs HM, Oaklander AL. Validation of the Composite Autonomic Symptom Scale 31 (COMPASS-31) in patients with and without small fiber polyneuropathy. Eur J Neurol. 2015;22(7):1124-1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quan H, Sundararajan V, Halfon P, et al. Coding algorithms for defining comorbidities in ICD-9-CM and ICD-10 administrative data. Med Care. 2005;43(11):1130-1139. [DOI] [PubMed] [Google Scholar]
- 26.Shiboski CH, Shiboski SC, Seror R, et al. 2016 American College of Rheumatology/European League Against Rheumatism classification criteria for primary Sjogren's syndrome: a consensus and data-driven methodology involving three international patient cohorts. Arthritis Rheumatol. 2017;69(1):35-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee TH, Setty PT, Parthasarathy G, et al. Aging, obesity, and the incidence of diverticulitis: a population-based study. Mayo Clin Proc. 2018;93(9):1256-1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Finan PH, Goodin BR, Smith MT. The association of sleep and pain: an update and a path forward. J Pain. 2013;14(12):1539-1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kassi E, Pervanidou P, Kaltsas G, Chrousos G. Metabolic syndrome: definitions and controversies. BMC Med. 2011;9:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Callaghan B, Feldman E. The metabolic syndrome and neuropathy: therapeutic challenges and opportunities. Ann Neurol. 2013;74(3):397-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Callaghan BC, Xia R, Banerjee M, et al. Metabolic syndrome components are associated with symptomatic polyneuropathy independent of glycemic status. Diabetes Care. 2016;39(10):801-807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singleton JR, Marcus RL, Jackson JE, K Lessard M, Graham TE, Smith AG. Exercise increases cutaneous nerve density in diabetic patients without neuropathy. Ann Clin Transl Neurol. 2014;1(15):844-849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Geerts M, de Greef BTA, Sopacua M, et al. Intravenous immunoglobulin therapy in patients with painful idiopathic small fiber neuropathy. Neurology. 2021;96(20):e2534-e2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gibbons CH, Klein MC. IVIG and small fiber neuropathy: the ongoing search for evidence. Neurology. 2021;96(20):929-930. [DOI] [PubMed] [Google Scholar]
- 35.Prasad M, Hermann J, Gabriel SE, et al. Cardiorheumatology: cardiac involvement in systemic rheumatic disease. Nat Rev Cardiol. 2015;12(3):168-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ninomiya JK, L'Italien G, Criqui MH, Whyte JL, Gamst A, Chen RS. Association of the metabolic syndrome with history of myocardial infarction and stroke in the third National Health and Nutrition Examination Survey. Circulation. 2004;109(1):42-46. [DOI] [PubMed] [Google Scholar]
- 37.Maser RE, Mitchell BD, Vinik AI, Freeman R. The association between cardiovascular autonomic neuropathy and mortality in individuals with diabetes: a meta-analysis. Diabetes Care. 2003;26(6):1895-1901. [DOI] [PubMed] [Google Scholar]
- 38.Liu L, Wu Q, Yan H, Chen B, Zheng X, Zhou Q. Association between cardiac autonomic neuropathy and coronary artery lesions in patients with type 2 diabetes. Dis Markers. 2020;2020:6659166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Novella SP, Inzucchi SE, Goldstein JM. The frequency of undiagnosed diabetes and impaired glucose tolerance in patients with idiopathic sensory neuropathy. Muscle Nerve. 2001;24(9):1229-1231. [DOI] [PubMed] [Google Scholar]
- 40.England JD, Gronseth GS, Franklin G, et al. Practice parameter: evaluation of distal symmetric polyneuropathy: role of laboratory and genetic testing (an evidence-based review): report of the American Academy of Neurology, American Association of Neuromuscular and Electrodiagnostic Medicine, and American Academy of Physical Medicine and Rehabilitation. Neurology. 2009;72(1):185-192. [DOI] [PubMed] [Google Scholar]
- 41.Buxbaum JN. Oligonucleotide drugs for transthyretin amyloidosis. N Engl J Med. 2018;379(21):82-85. [DOI] [PubMed] [Google Scholar]
- 42.Azevedo O, Gago MF, Miltenberger-Miltenyi G, Sousa N, Cunha D. Fabry disease therapy: state-of-the-art and current challenges. Int J Mol Sci. 2020;22(1):206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dubey D, Lennon VA, Gadoth A, et al. Autoimmune CRMP5 neuropathy phenotype and outcome defined from 105 cases. Neurology. 2018;90(2):e103-e110. [DOI] [PubMed] [Google Scholar]
- 44.Dubey D, Jitprapaikulsan J, Bi H, et al. Amphiphysin-IgG autoimmune neuropathy: a recognizable clinicopathologic syndrome. Neurology. 2019;93(20):e1873-e1880. [DOI] [PubMed] [Google Scholar]
- 45.Gadoth A, Zekeridou A, Klein CJ, et al. Elevated LGI1-IgG CSF index predicts worse neurological outcome. Ann Clin Transl Neurol. 2018;5(5):646-650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lucchinetti CF, Kimmel DW, Lennon VA. Paraneoplastic and oncologic profiles of patients seropositive for type 1 antineuronal nuclear autoantibodies. Neurology. 1998;50(3):652-657. [DOI] [PubMed] [Google Scholar]
- 47.Klein CJ. Autoimmune-mediated peripheral neuropathies and autoimmune pain. Handb Clin Neurol. 2016;133:417-446. [DOI] [PubMed] [Google Scholar]
- 48.Hoffman EM, Watson JC, St Sauver J, Staff NP, Klein CJ. Association of long-term opioid therapy with functional status, adverse outcomes, and mortality among patients with polyneuropathy. JAMA Neurol. 2017;74(7):773-779. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Deidentified data are available on reasonable request.