

Summary
Facilitated dissociation provides a mechanism by which high-affinity complexes can be rapidly disassembled. The negative feedback regulator CITED2 efficiently downregulates the hypoxic response by displacing the hypoxia inducible transcription factor HIF-1α from the TAZ1 domain of the transcriptional coactivators CBP and p300. Displacement occurs by a facilitated dissociation mechanism involving a transient ternary intermediate formed by binding of the intrinsically disordered CITED2 activation domain to the TAZ1:HIF-1α complex. The short lifetime of the intermediate precludes straightforward structural investigations. To obtain insights into the molecular determinants of facilitated dissociation, we model the ternary intermediate by generating a fusion peptide composed of the primary CITED2 and HIF-1α binding motifs. X-ray crystallographic and NMR studies of the fusion peptide complex reveal TAZ1-mediated negative cooperativity that results in nearly mutually exclusive binding of specific CITED2 and HIF-1α interaction motifs, providing molecular level insights into the allosteric switch that terminates the hypoxic response.
Keywords: hypoxic response, intrinsically disordered protein, transcriptional coactivator, competitive binding, allosteric switch, protein-protein interaction
eTOC Blurb
The transcriptional response to hypoxia is regulated by a pair of disordered proteins, HIF-1α and CITED2. CITED2 downregulates HIF-1α-mediated transcription by competing for a shared molecular target by facilitated dissociation. Appling et al. model the intermediate of this process using a fusion peptide comprised of HIF-1α and CITED2 binding motifs.
Graphical Abstract
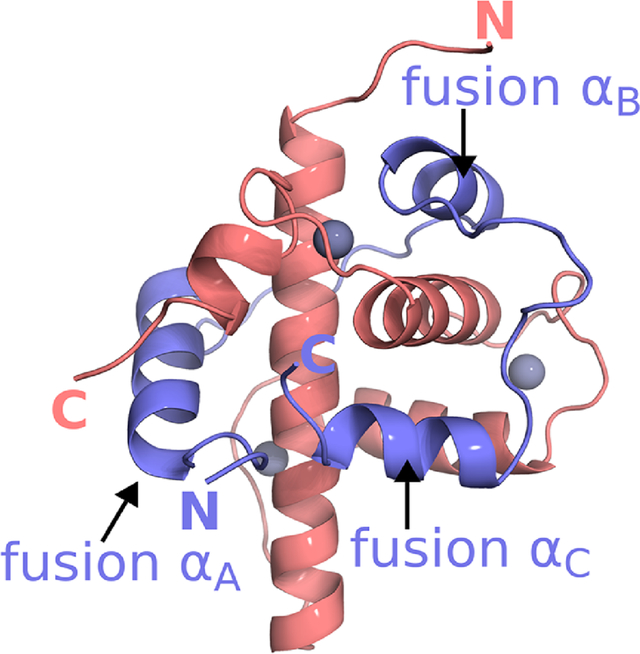
Introduction
Dynamic protein-protein and protein-DNA interactions are central to the regulatory pathways of the cell. Activation and termination of cellular signaling processes is largely governed by the assembly and disassembly of regulatory complexes. Spontaneous, unimolecular dissociation of long-lifetime, high affinity regulatory complexes may be too slow to allow the cell to respond rapidly to changing conditions, and mechanisms have evolved to accelerate the process. Thus, many transcription factors can be actively removed from their DNA target sites by a “facilitated dissociation” or “molecular stripping” mechanism that decreases the lifetime of the DNA complex by enhancing the dissociation rate (Chen et al., 2018; Chen et al., 2015; Graham et al., 2010; Joshi et al., 2012; Kamar et al., 2017; Potoyan et al., 2017). Mechanistically, facilitated dissociation from DNA is aided by multivalent protein-DNA interactions that favor formation of transient ternary intermediates (Chen et al., 2018; Tsai et al., 2016). Facilitated dissociation of protein-protein complexes has also been reported, including accelerated displacement of an antigenic peptide from a Class II MHC complex and stimulated release of an IgE antibody from the high affinity IgE Fc receptor in the presence of an engineered inhibitor (Kim et al., 2012; Schmitt et al., 1999). More recently, facilitated dissociation has been found to play a central role in termination of the cellular response to hypoxia (Berlow et al., 2017). In the present work we apply X-ray crystallography and NMR to elucidate the molecular mechanism of the allosteric switch that terminates the hypoxic response.
Evolution in an oxidizing environment has resulted in the development of regulatory networks that respond to O2 levels and tune cellular metabolism to maintain homeostasis. At the center of the eukaryotic regulatory network is the heterodimeric transcription factor, hypoxia-inducible factor 1 (HIF-1) (Semenza, 2014). Under normoxic conditions, the α-subunit of HIF-1 (HIF-1α) is targeted for proteasomal degradation. In response to hypoxia, HIF-1α is stabilized and cellular HIF-1 levels rise rapidly to activate transcription of stress response genes (Huang et al., 1998; Ivan et al., 2001; Jaakkola et al., 2001; Semenza, 2014; Wang et al., 1995). Interactions between the intrinsically disordered C-terminal transactivation domain of HIF-1α and the TAZ1 domain of the general transcriptional coactivators CREB-binding protein (CBP) or its paralog p300 are essential for activation of HIF-1-mediated transcription (Arany et al., 1996; Ebert and Bunn, 1998). HIF-1 transcriptional activity is regulated through a negative feedback loop driven by the protein CITED2 (Bhattacharya et al., 1999; Henze and Acker, 2010). The gene encoding CITED2 is under the direct transcriptional control of HIF-1 and is strongly induced in response to hypoxia (Bhattacharya et al., 1999). The intrinsically disordered C-terminal activation domain of CITED2 competes with the C-terminal activation domain of HIF-1α for binding to TAZ1 to down-regulate HIF-1-mediated transcription.
The activation domain of HIF-1α folds upon binding to form three α-helices (HIF-1α-αA, αB, αC) that partially encircle TAZ1 (Figure 1B) (Dames et al., 2002; Freedman et al., 2002). In contrast, CITED2 interacts with TAZ1 through a single N-terminal α-helix (CITED2-αA) and an extended region that makes hydrophobic contacts with the TAZ1 α1 and α3 helices (Figure 1A) (De Guzman et al., 2004; Freedman et al., 2003). The HIF-1α and CITED2 activation domains occupy partially overlapping sites on TAZ1 and elicit TAZ1 conformations that differ in the C-terminal region of helix α1 and in the length and orientation of helix α4 (Figure 1). Although the HIF-1α and CITED2 activation domains exhibit little sequence homology, each possesses a 4-amino acid sequence, LP(Q/E)L, that makes similar interactions with TAZ1 in both complexes (Figures 1A, B). We have recently shown that HIF-1α and CITED2, through their competing interactions with TAZ1, function synergistically to form a hypersensitive, unidirectional regulatory switch that efficiently shuts off the hypoxic response (Berlow et al., 2017). The activation domain of CITED2 efficiently displaces the HIF-1α activation domain from the TAZ1:HIF-1α complex by an allosteric mechanism that greatly enhances the rate of HIF-1α dissociation. Remarkably, HIF-1α is ineffective at displacing CITED2 from its complex with TAZ1, even though the binary HIF-1α and CITED2 complexes with TAZ1 have identical affinities (Kd = 10 nM). Kinetic and NMR measurements showed that CITED2 facilitates dissociation of HIF-1α by formation of a transient ternary complex, in which HIF-1α and CITED2 bind simultaneously to TAZ1 and compete for binding of the conserved LP(Q/E)L motif (Berlow et al., 2017). The data suggested a model for the ternary complex in which CITED2 binds to TAZ1 through its αA helix, displacing the weakly bound αA region of HIF-1α, while HIF-1α remains bound to TAZ1 through its αB and αC helices (Berlow et al., 2017; Berlow et al., 2019). However, since the CITED2 αA and the HIF-1α αB and αC helices occupy non-overlapping surfaces of TAZ1, it is not readily apparent why the ternary complex is unstable and is formed only transiently. To reconcile these observations, a mechanism was proposed in which TAZ1-mediated allosteric interactions destabilize the ternary complex and facilitate dissociation of HIF-1α (Berlow et al., 2017).
Figure 1. TAZ1:CITED2 and TAZ1:HIF-1α structures and the design of a CITED2-HIF-1α fusion.

(A) NMR structure of the TAZ1:CITED2 complex (PDB 1R8U; (De Guzman et al., 2004)). TAZ1(CBP 340–439) is shown in cyan and CITED2(220–269) is shown in green. Zinc atoms are shown as dark-gray spheres. Side chains of the CITED2 LPEL motif are shown as light-gray spheres with N and O atoms colored blue and red respectively. Black labels are superimposed on the LPEL side chains. TAZ1 and CITED2 helices and N- and C-termini are labeled for reference. Residues 262–269 of CITED2 are omitted for clarity. (B) NMR structure of the TAZ1:HIF-1α complex (PDB 1L8C; (Dames et al., 2002)). TAZ1(CBP 345–439) is shown in tan and HIF1α(776–826) is shown in red. Zinc atoms are shown as dark-gray spheres. Side chains of the HIF-1α LPQL motif are shown as light-gray spheres with N and O atoms colored blue and red respectively. Black labels are superimposed on the LPQL side chains. TAZ1 and HIF-1α helices and N- and C-termini are labeled for reference. (C) Amino acid sequence of the fusion peptide. Sequences from CITED2 (residues 216–246) and HIF-1α (residues 796–826) are colored green and red respectively and correspond to residues 6–36 and 37–67 of the fusion peptide. The LPEL motif of the CITED2 sequence is shown in black. The N-terminal GSHMS sequence is a cloning artifact (see methods). The block diagram below the sequence denotes the positions of the CITED2 and HIF-1α helical binding motifs.
Formation of the ternary complex is central to the displacement mechanism, but its transient nature has thus far precluded direct characterization of this intermediate in solution and has limited our ability to investigate the structural and dynamic properties that lead to the observed negative cooperativity of CITED2 and HIF-1α binding to TAZ1. To circumvent this problem, we generated a fusion peptide composed of the primary binding motifs of CITED2 and HIF-1α. In the complex between TAZ1 and the fusion peptide, the high local concentration of the CITED2 and HIF-1α binding motifs drives their association with TAZ1, enabling determination of the molecular basis of their competition. Using X-ray crystallography and solution NMR spectroscopy, we have characterized this model of the ternary intermediate to identify a critical component of the negative binding cooperativity between CITED2 and HIF-1α and to determine regions of TAZ1 that mediate the allosteric interaction. Our results provide detailed molecular insights into the mechanism of facilitated dissociation and further contribute to our understanding of how intrinsically disordered proteins compete for shared binding partners.
Results
Designing a CITED2-HIF-1α Fusion Peptide
To gain insights into the molecular mechanism by which CITED2 facilitates dissociation of HIF-1α from its complex with TAZ1, we generated a fusion peptide (Figure 1C) consisting of CITED2 αA and LPEL (CITED2 residues 216–246; residues 6–36 in the fusion peptide) followed immediately by HIF-1α αB and αC (HIF-1α residues 796–826; residues 37–67 in the fusion peptide). We hypothesized that the fusion peptide would bind TAZ1 in a manner representative of the ternary complex, enabling us to define the structure and dynamics of the transient intermediate that govern the displacement process.
Crystal Structure of the TAZ1:fusion Peptide Complex
A crystal structure of the TAZ1:fusion peptide complex was determined to 2 Å resolution (Figure 2A). Data collection and refinement statistics are presented in Table 1. The fusion peptide wraps around TAZ1, with the CITED2 and HIF-1α motifs occupying the same binding sites on TAZ1 as in the parent binary complexes (Dames et al., 2002; De Guzman et al., 2004). A detailed comparison of the TAZ1:fusion peptide crystal structure with the NMR ensembles of the TAZ1:CITED2 and TAZ1:HIF-1α complexes (Dames et al., 2002; De Guzman et al., 2004) is shown in Figure S1. The structures of TAZ1 and the CITED2 and HIF-1α binding motifs are highly conserved in the fusion peptide complex. Although the overall structure of TAZ1 is very similar in all three complexes, local differences are observed at the C-terminal end of helix α1, and in the length and orientation of the α4 helix. The α1 helix is slightly bent in the TAZ1:HIF-1α and TAZ1:CITED2 complexes, but is visibly straightened in the TAZ1:fusion peptide complex. In the fusion peptide complex, the α4 helix is of similar length to that in the TAZ1:HIF-1α complex but assumes the orientation observed in the TAZ1:CITED2 complex (Figure 2B). In addition, there are small shifts in the positions of the αA and αC helices of the fusion peptide relative to the binary CITED2 and HIF-1α complexes and pronounced structural differences in the C-terminal region of the αC helix (Figure S1). In the binary TAZ1:HIF-1α complex, the HIF-1α C-terminal residues follow the trajectory of the αC helix to place the free carboxyl terminus in an electropositive groove formed by the side chains of K365 and R368 on helix α1. In contrast, residues F12 and I13 of the fusion peptide, N-terminal to the αA helix, occlude this site in the crystal structure of the TAZ1:fusion peptide complex. The αC helix of the fusion peptide is disrupted at D64 (equivalent to D823 in HIF-1α) and the last 3 residues project orthogonally towards the C-terminal end of the α3 helix.
Figure 2. Crystal structure of the TAZ1:fusion peptide complex and conformation of the TAZ1 α4 helix.

(A) Crystal structure of the TAZ1:fusion peptide complex. TAZ1 is shown in salmon and the fusion peptide is shown in slate. Zinc atoms are shown as gray spheres. TAZ1 and fusion peptide helices and N- and C-termini are labeled for reference. (B) Superpositions of the TAZ1 α4 helix from the TAZ1:CITED2 (cyan), TAZ1:HIF-1α (tan), and TAZ1:fusion peptide (salmon) structures. Structures were aligned based on residues from the TAZ1 α1 (residues 350–363), α2 (residues 383–397), and α3 (residues 407–420) helices. Residues 426–437, encompassing the TAZ1 α4 helix, are shown.
Table 1.
X-ray diffraction data collection and refinement statistics for the TAZ1:fusion peptide complex
| Data collection a | |
| Beamline | ALS 5.0.3 |
| Wavelength (Å) | 0.97648 |
| Resolution range (Å) | 40.7–2.0 |
| Space Group | P21 |
| Unit cell dimensions (Å, °) | 32.06,50.11,41.26; β=99.19 |
| Total reflections | 60265(3547) |
| Unique reflections | 8465(562) |
| Rmerge (%)b | 11.1(145.5) |
| Rmeas (%)c | 12.0(158.6) |
| Rpim (%)d | 4.5(62.4) |
| CC1/2 (%)e | 87.8(91.0) |
| Completeness (%) | 100.0(99.8) |
| Redundancy | 7.1(6.4) |
| <I>/<σ(I)> | 17.9(1.2) |
| Refinement | |
| Reflections used for Rwork(Rfree) | 7543(882) |
| Number of non-H protein atoms | 1214 |
| Number of water molecules | 36 |
| Number of Zn atoms | 3 |
| Rwork | 0.2107 |
| Rfree | 0.2406 |
| RMS (bond length) | 0.003 |
| RMS (bond angle) | 0.645 |
| Ramachandran: favored, outliers (%) | 95.36,0.66 |
| Clashscore | 5.78 |
| Wilson B-factor (Å2) | 38.4 |
| Average B-factor (Å2) | 65 |
Values in parentheses are for highest resolution shell
Rmerge = ΣhklΣi=1,n|Ii(hkl)-<I(hkl)>|/ΣhklΣi=1,nIi(hkl)
Rmease = Σhkl√(n/(n-1))Σi=1,n|Ii(hkl)-<(I(hkl)>|/Σhkl|Σi=1,nIi(hkl)
Rpim = Σhkl√(1/(n-1))Σi=1,n|Ii(hkl)-<I(hkl)->|/Σ(hkl)Σi=1,nIi=(hkl)
CC1/2 = Pearson Correlation Coefficient between two random half datasets
Number of unfavorable all-atom steric overlaps ≥ 0.4 Å per 1000 atoms
TAZ1 Conformational Exchange in the TAZ1:fusion Peptide Complex
High resolution NMR was used to probe the interactions between TAZ1 and the fusion peptide in solution. Two regions of the 1H-15N HSQC spectra of 15N-labeled TAZ1 in complex with unlabeled CITED2, HIF-1α, or the fusion peptide are shown in Figure 3A and the full HSQC spectra are shown in Figure S2A. There are substantial differences between the spectra, reflecting differences both in the intermolecular interactions of the ligands and in the TAZ1 conformation in the various complexes. Approximately 80% of non-proline backbone amides of 13C,15N-labeled TAZ1 in complex with unlabeled fusion peptide were assigned from triple resonance spectra. In contrast, about 90% of TAZ1 backbone amides were assigned previously for the parent TAZ1:CITED2 and TAZ1:HIF-1α complexes (Dames et al., 2002; De Guzman et al., 2004).
Figure 3. Comparison of 1H-15N HSQC spectra of 15N-labeled TAZ1 in complex with CITED2, HIF-1α, and the fusion peptide.

(A) Regions of superimposed 1H-15N HSQC spectra (900 MHz 1H frequency) of 15N-labeled TAZ1 bound to unlabeled CITED2 (green), HIF-1α (red), or fusion peptide (blue). Backbone amide cross peaks corresponding to TAZ1 α4 helix residues A435 (left panel) and S436 (right panel) are labeled. (B) Weighted average 1H,15N chemical shift differences for 15N-TAZ1 (Δδave=[(ΔδHN)2+(ΔδN/5)2]½) between the CITED2 and fusion peptide complexes (green) and between the HIF-1α and fusion peptide complexes (red). Circles denote residues for which backbone amide resonance assignments are missing for the CITED2 (green), HIF-1α (red), and fusion peptide (blue) complexes. Residues for which there are no circles and no data are prolines. Boxes above the plot indicate the positions of the TAZ1 α1 − α4 helices in the TAZ1:fusion peptide crystal structure; the broken lines indicate the length of the α4 helix in the TAZ1:CITED2 structure. The broken green and red lines inside the graph denote one standard deviation above the 10 % trimmed mean for the TAZ1:CITED2 Vs. TAZ1:fusion peptide and TAZ1:HIF-1α Vs. TAZ1:fusion peptide data sets, respectively. (C) Structure of the TAZ1:fusion peptide complex, with TAZ1 shown in salmon and the fusion peptide in slate. TAZ1 α1 residues for which backbone amide 1H-15N HSQC cross peak intensity is reduced by greater than 60 % in the fusion peptide complex compared to the CITED2 complex are shown as red spheres. Fusion peptide αA and αC residues for which backbone amide resonances are broadened beyond detection or have normalized cross peak intensity less than 0.2 (see Figure 4B) are shown as cyan spheres. Zn atoms are omitted for clarity.
Weighted average TAZ1 1H,15N chemical shift differences between the TAZ1:fusion peptide complex and the TAZ1:CITED2 and TAZ1:HIF-1α complexes are plotted in Figure 3B. The chemical shift differences between the fusion and the CITED2 complexes are in general larger than those between the fusion peptide and HIF-1α complexes. Based on the structures in Figures 1A and 2, we would expect that regions of TAZ1 that contact αA of CITED2 (TAZ1 helices α1 and α4) should show only small chemical shift differences between the CITED2 and fusion peptide complexes, whereas the regions that contact αB and αC of HIF-1α (N-terminus of α1, C-terminus of α2, and α3 helix of TAZ1) should show only small chemical shift differences between the HIF-1α and fusion peptide complexes. Indeed, small chemical shift differences between the HIF-1α and fusion peptide complexes are observed for residues in the α2 and α3 helices (red bars in Figure 3B), confirming that these regions of TAZ1 have similar structure and similar intermolecular contacts in the HIF-1α and fusion peptide complexes, while larger differences are observed for residues in TAZ1 α1 and α4, as expected. The chemical shift differences between the CITED2 and fusion complexes (green bars in Figure 3B) differ greatly for residues in the α2 and α3 regions of TAZ1, also as expected from the structural data. However, the chemical shift differences of residues in α1 are much larger than would be expected on the basis of the crystal structure, suggesting that in solution the interactions between TAZ1 and the fusion peptide αA region differ from those in the TAZ1:CITED2 complex. In addition, there is a greater than 60% reduction in the heights of the Q355, V358, H362, H364, E370, and A372 amide cross peaks in the HSQC spectrum of the fusion complex compared to that of the TAZ1:CITED2 complex (Figure S2B). These residues lie on the face of the TAZ1 α1 helix that contacts the αA region of the fusion peptide (Figure 3C), suggesting the possibility of exchange broadening due to conformational fluctuations in the molecular interface (see following section).
In the crystal structure of the fusion peptide complex, the TAZ1 α4 helix assumes the length observed in the HIF-1α complex and the orientation observed in the CITED2 complex (Figure 2B). Consistent with this observation, chemical shift differences are observed between the fusion peptide complex and both parent complexes for residues in the TAZ1 α4 helix (Figure 3B). The relative magnitudes of the α4 chemical shift differences indicate that the conformation of the TAZ1 α4 helix in the fusion peptide complex differs from that in the HIF-1α complex but does not fully achieve the length and/or conformation observed in the CITED2 complex. These observations are directly reflected in the HSQC spectra (Figure 3A), where cross peaks corresponding to TAZ1 α4 helix residues A435 (left panel) and S436 (right panel) are shown. In the spectrum of the TAZ1:fusion peptide complex (blue), these cross peaks are shifted away from their positions in the TAZ1:HIF-1α spectrum (red) towards their positions in the TAZ1:CITED2 spectrum (green). The A435 and S436 cross peaks in the three spectra (TAZ1:CITED2, TAZ1:HIF-1α, and TAZ1:fusion peptide) fall roughly on a line, suggesting that TAZ1 in the fusion peptide complex likely undergoes a fast exchange process in which the α4 helix interconverts between its conformation in the HIF-1α complex and its conformation in the CITED2 complex.
Fusion Peptide Conformational Exchange in the TAZ1 Complex
The exchange process in the TAZ1:fusion peptide complex was further probed by solution NMR investigation of the isotopically-labeled fusion peptide in complex with unlabeled TAZ1. An overlay of the amide region of 1H-15N HSQC spectra of free and TAZ1-bound 15N-labeled fusion peptide is shown in Figure 4A. The limited 1H chemical shift dispersion of the free fusion peptide spectrum shows that it, like the parent CITED2 and HIF-1α activation domains, is intrinsically disordered. The chemical shift dispersion is increased upon binding to TAZ1, indicating folding of the fusion peptide. However, the amide cross peaks of the bound fusion peptide exhibit heterogenous intensities (Figure 4B) and only 49 of the expected 64 backbone amide cross peaks are observed in spectra of 15N-labeled fusion peptide in complex with unlabeled TAZ1.
Figure 4. Solution NMR characterization of the fusion peptide.

(A) Superimposed 900 MHz 1H-15N HSQC spectra of 15N-labeled fusion peptide bound to unlabeled TAZ1 (blue) and free in solution (coral). Representative amide cross peaks that undergo large shifts in position upon complex formation are labeled. (B) Intensities of backbone amide cross peaks from 1H-15N HSQC spectra of 15N-labeled fusion peptide in complex with unlabeled TAZ1. Intensities are normalized to the intensity of the backbone amide cross peak of the C-terminal fusion peptide residue (N67). Boxes above the plot indicate the positions of the CITED2 αA and HIF1α αB and αC binding motifs. Residues for which backbone amide cross peaks are broadened beyond detection are denoted by blue circles.
The heterogeneity of the cross-peak intensities suggests that exchange broadening is non-uniform across the fusion peptide and is particularly severe for residues in the αA region, where cross peaks are broadened beyond detection. The amide cross peaks of residues 13–18, 20 and 21 in the αA region of TAZ1-bound CITED2 (using the numbering of the fusion peptide from Figure 1) are intense and well-resolved (Figure S3). However, the corresponding cross peaks are entirely missing from spectra of the fusion peptide complex (Figures 5A and S3A), consistent with exchange broadening due to conformational fluctuations in the N-terminal region of αA, where it contacts the TAZ1 α1 helix (Figure 3C). The amide cross peaks associated with residues 22–31 of the bound fusion peptide are generally weak but are observed at chemical shifts that are similar to those of the corresponding residues of the CITED2 binary complex (Figure S3A). Residues L21 – M25 adopt helical structure in both the fusion peptide and CITED2 complexes, based on their similar 13Cα chemical shifts. The chemical shifts of residues 38–49, 52–59, 61, 64, and 67 of the bound fusion peptide are very close to those of HIF resonances in the binary TAZ1:HIF complex (Figure S3A). However, the resonances of several αC residues (60, 63, 65, 66) also exhibit exchange broadening, resulting in low signal intensity (Figure 4B). The differential broadening observed for both fusion peptide and TAZ1 resonances (Figure 3C) provides strong evidence for enhanced conformational exchange relative to the binary complexes in the αA and αC regions of the TAZ1-bound fusion peptide and in the regions of TAZ1 (α1 and α4 helices) that they contact, suggesting that co-occupation of TAZ1 by the αA helix of CITED2 and the αC helix of HIF-1α is unfavorable, i.e. that binding is mutually antagonistic.
Figure 5. Comparison of 1H-15N HSQC spectra of 15N-labeled CITED2, HIF-1α, and fusion peptides in complex with TAZ1.

(A) Overlay of a region of the spectra of 15N-labeled fusion peptide (blue), 15N-labeled CITED2 (green), and 15N-labeled HIF-1α (red) bound to unlabeled TAZ1. (B) Overlay of a region of the spectra of 15N-labeled L63A fusion peptide(black) 15N-labeled CITED2 (green), and 15N-labeled HIF-1α (red) bound to unlabeled TAZ1. Cross peaks from the CITED2 and HIF-1α complexes are shown with only the outer contours for clarity and, in panel A, are connected by lines to the corresponding cross peaks of the fusion peptide complex. Cross peaks from the N-terminus of the HIF-1α complex and the C-terminus of the CITED2 complex have no corresponding cross peak in the fusion peptide complex spectrum and are labeled in smaller font in parentheses. The cross peaks of an amide side chain are denoted sc.
Negative Allosteric Modulation of αA and αC Binding
To test the hypothesized antagonistic binding of the αA and αC regions of the fusion peptide, we performed site-directed mutagenesis to introduce Leu to Ala substitutions designed to impair binding of the αA (L21A) or αC (L63A) helices by disrupting hydrophobic interactions with TAZ1. L21 packs into a hydrophobic pocket formed by V358 in the TAZ1 α1 helix and L432 and A435 in the TAZ1 α4 helix, while L63 makes hydrophobic contacts with L360 and L361 in the TAZ1 α1 helix and I415 in the TAZ1 α3 helix (Figure S4A). 1H-15N HSQC spectra of 15N labeled L21A and L63A fusion peptides free in solution and bound to unlabeled TAZ1 are shown in Figure S4B,C. Overlays of the 1H-15N HSQC spectra of the TAZ1 complexes of HIF-1α, CITED2 and the mutant peptides are shown in Figures 5B, S3B,C. Weighted average backbone 1H,15N chemical shift differences between the original and the mutant fusion peptides in complex with TAZ1 are plotted in Figure 6A. The differences between the two sets of chemical shift differences are striking. The L21A substitution results in only very small chemical shift changes relative to the original fusion peptide complex, whereas dramatic chemical shift changes are observed for residues in the N- and C-terminal regions of the fusion peptide in response to the L63A substitution (Figure 6A). In addition, the L63A substitution results in a decrease in the exchange broadening observed in spectra of the original fusion peptide in complex with TAZ1, enabling observation of backbone amide resonances corresponding to the αA region and assignment using standard triple resonance experiments.
Figure 6. Site-directed mutagenesis to probe TAZ1-mediated allosteric interactions between the fusion peptide αA and αC helices.

(A) Weighted average 1H,15N chemical shift differences between the original 15N-labeled fusion peptide in complex with unlabeled TAZ1 and 15N-labeled L21A (orange) or 15N-labeled L63A (black) fusion peptide in complex with unlabeled TAZ1. Green arrows indicate the sites of mutation. Circles denote residues for which backbone amide cross peaks are broadened beyond detection for the TAZ1 complexes of the original fusion peptide (blue), L21A fusion peptide (orange), and L63A fusion peptide (black). Residues for which there are no circles and no data are prolines. The broken horizontal lines (and horizontal orange arrow) denote one standard deviation above the 10 % trimmed mean for the TAZ1:fusion vs. TAZ1:fusion (L21A) (orange) and TAZ1:fusion vs. TAZ1:fusion (L63A) (black) data sets. Inset: structure of the fusion peptide complex, with TAZ1 shown in gray and the fusion peptide shown in tan. Residues L21 and L63 are labeled and colored green. Zn atoms are shown as gray spheres. (B) Weighted average 1H,15N chemical shift differences for the TAZ1-bound L63A fusion peptide relative to the unbound state (black), to residues 220–245 of CITED in complex with TAZ1 (green, corresponding to residues 10–35 of the fusion peptide), and to residues 795–826 of HIF-1α in complex with TAZ1 (red, corresponding to residues 36–67 of the fusion peptide).The green arrow indicates the site of mutation. The backbone amide resonance corresponding to E32 is broadened beyond detection and is marked with a black circle. The broken black line denotes one standard deviation above the 10 % trimmed mean of the TAZ1:fusion (L63A) vs. free L63A fusion peptide shift differences. The boxes between panels A and B denote the positions of the CITED2 αA and HIF-1α αB and αC binding motifs.
Upon binding to TAZ1, residues in the αA and αB regions of the L63A peptide experience large changes in backbone amide chemical shifts (black bars in Figure 6B), indicating interactions with TAZ1 and folding of the peptide. In contrast, amide cross peaks of residues in the C-terminal region (G49 – N67) are largely unperturbed by binding, showing that this region does not interact with TAZ1 and that the αC helix remains unfolded and disordered. Comparison of the 1H-15N HSQC spectrum of the L63A fusion peptide complex with the spectra of the TAZ1:CITED2 and TAZ1:HIF-1α complexes provides additional insights. The green bars in Figure 6B show that the chemical shifts of backbone amides of residues 9–35 of the L63A fusion peptide bound to TAZ1 are very similar to those of the corresponding residues of the CITED2-TAZ1 complex (residues 219–245, see Figures 5B and S3C), confirming that the αA helix of the L63A peptide folds and makes intimate contacts with TAZ1 in a similar manner to the αA helix of CITED2. Additionally, the chemical shifts of backbone amides of residues 40–45 of the TAZ1-bound L63A fusion peptide are very similar to those of the corresponding residues (HIF-1α 795–804, Figure S3C) in the HIF-1α complex (red bars in Figure 6B), showing that the αB region adopts the same structure and makes similar TAZ1 contacts in both the L63A fusion peptide and HIF-1α complexes. However, residues 50–67 of the TAZ1-bound L63A fusion peptide show large backbone amide chemical shift differences from the corresponding residues (HIF-1α 809–826) of the HIF-1α complex. The backbone amide cross peaks of residues 50–67 have nearly identical 1H and 15N chemical shifts to those of the free L63A peptide (Δδave < 0.07 ppm), indicating dissociation of αC and the αB−αC loop from the surface of TAZ1. From these observations we conclude that the L63A substitution largely abrogates the folding of αC and its interaction with TAZ1, while simultaneously enhancing binding of αA to stabilize the helical structure observed in the TAZ1:CITED2 complex in solution and in the TAZ1:fusion peptide complex in the crystal (Figures 1A, 2A).
In contrast to the large differences observed between the L63A fusion complex and the TAZ1:HIF-1α complex, comparison of the backbone amide chemical shifts for the L21A fusion complex with those of the TAZ1:HIF-1α complex (Figure S5) shows that the αB and αC regions of the L21A peptide are stably bound and adopt similar conformations in the two complexes. In common with the unmodified fusion peptide, backbone amide cross peaks of αA residues are exchange-broadened in the spectrum of the L21A peptide complex. Thus, the mutagenesis experiments clearly show that, in solution, binding of the αA and αC helices is mutually exclusive; the two helices in the fusion peptide cannot bind simultaneously while making optimal contacts with TAZ1. When the αC helix is bound, interactions with the αA region are weak and transient and lead to exchange broadening in the spectrum of the fusion peptide complex. Conversely, impairment of the capacity for αC helix binding, as observed in the L63A fusion peptide complex, allows folding and binding of the αA helix.
The αA helix is stabilized by lattice contacts in the crystal
Given the antagonism observed in binding of the αA and αC helices to TAZ1 in solution, it is at first surprising that both helices are fully folded and docked to TAZ1 in the crystal structure of the fusion peptide complex (Figure 2A). However, examination of the packing in the crystal lattice reveals that the αA helix is sandwiched between its binding site on TAZ1 and the α2 helix of a neighboring TAZ1 molecule (Figure S6). This configuration in the crystal promotes docking of the αA helix to its binding site and enables observation of a complex in which both the αA and αC helices are docked.
Mechanism of TAZ1-Mediated Allostery
To identify the regions of TAZ1 that mediate the negative allostery observed in binding of the αA and αC helices of the fusion peptide, we used NMR to characterize the complexes of 15N-labeled TAZ1 with the L21A and L63A fusion peptides. Comparison of the 1H-15N HSQC spectra of the original and L63A fusion peptide complexes (shown in Figure S7) reveals larger than average chemical shift differences for residues located in the TAZ1 α4 helix and in the αC binding site, at the interface of the α1, α2, and α3 helices of TAZ1 (Figure 7A). The observed chemical shift changes provide further evidence that loss of hydrophobic contacts due to the L63A substitution weakens the interactions between TAZ1 and αC and promotes dissociation of the αC helix. In contrast, the L21A mutation causes little change in the TAZ1 backbone amide chemical shifts except at the C-terminal end of the α4 helix (Figure 7A).
Figure 7. Effects of fusion peptide L21A and L63A mutations on TAZ1 resonances.

(A) Weighted average 1H,15N chemical shift differences for TAZ1 amide cross peaks in complex with unlabeled fusion peptide relative to the L21A (orange) or L63A (black) fusion peptide complexes. The broken orange and black lines and the horizontal orange arrow denote one standard deviation above the 10 % trimmed mean of the L21A and L63A fusion peptide histograms, respectively. The circles indicate residues for which TAZ1 backbone amide cross peaks are weak or missing in spectra of the original fusion peptide (blue) and the L63A fusion peptide (black) complexes. Backbone amide cross peaks of 15N-labeled TAZ1 in complex with unlabeled L21A fusion peptide were assigned based on the similarity of chemical shifts in spectra of the original fusion peptide complex. Missing assignments are mostly associated with cross peaks in highly overlapped regions of the spectrum. Inset: TAZ1:fusion peptide structure, with TAZ1 shown as a gray surface and the bound fusion peptide as a tan cartoon with L63 highlighted in green. Residues for which the weighted average TAZ1 chemical shift differences between the original fusion peptide and L63A fusion peptide complexes (panel A) are greater than one standard deviation above the 10 % trimmed mean are colored red. Boxes above the plot denote positions of the TAZ1 α1 – α4 helices in the TAZ1:fusion peptide crystal structure, with dotted lines indicating the extended α4 helix in the TAZ1:CITED2 structure. (B) Overlays of 1H-15N HSQC spectra (900 MHz 1H frequency) of 15N-labeled TAZ1 in complex with unlabeled CITED2 (green), HIF-1α (red), fusion peptide (blue), L21A fusion peptide (orange), or L63A fusion peptide (black). Cross peaks corresponding to the backbone amides of A435 and S436 are labeled in the left and right panels respectively. (C) Left: Temperature coefficients of backbone amide proton resonances of residues A378, S411, A435, and S436 of TAZ1 in complex with the original (blue), L21A (orange), or L63A (black) fusion peptide. Error bars represent the 68 % confidence interval of the fitted slope. Right: Structure of the TAZ1:fusion peptide complex showing the locations of A378, S411, A435, and S436 as red spheres. Zn atoms are omitted for clarity.
The cross peaks of A435 and S436 in the spectrum of the fusion peptide complex report on the state of TAZ1 α4. In the fusion peptide complex, these cross peaks are located at chemical shifts intermediate between their positions in the spectra of the TAZ1:HIF-1α and TAZ1:CITED2 complexes (Figure 3A). The L21A and L63A substitutions have opposite effects on these cross peaks: the L21A substitution shifts both cross peaks towards their positions in the TAZ1:HIF-1α spectrum whereas the L63A substitution shifts them to positions that are nearly coincident with the corresponding cross peaks in the TAZ1:CITED2 spectrum (Figure 7B).
The orientation and length of the TAZ1 α4 helix is sensitive to the identity of the bound ligand and is shorter in the TAZ1:HIF-1α and TAZ1:fusion peptide complexes than in the TAZ1:CITED2 complex (Figure 2B). To determine whether the L21A and L63A mutations affect the hydrogen-bonded helical structure of α4, we measured backbone amide proton chemical shift temperature coefficients to probe the propensity for formation of intramolecular hydrogen bonds (Baxter and Williamson, 1997). The temperature coefficients of the amide proton chemical shifts of A378 and S411, representing solvent exposed and hydrogen bonded controls, respectively, and two residues in α4, A435 and S436, are shown in Figure 6C and the fitted data are shown in Figure S8. The temperature coefficient of the A378 amide proton resonance (≈ −7 ppb K−1 ) is consistent with its solvent-exposed position while that of the S411 amide (≈ 0 ppb K−1 ) is characteristic of an amide proton involved in an intramolecular hydrogen bond. The temperature coefficients of A378 and S411 in the fusion peptide complex are unaffected by the L21A and L63A mutations. In contrast, the temperature coefficients of A435 and S436 are more negative than −5 ppb K−1 in both the fusion peptide and L21A peptide complexes but become more positive (≈ −3.5 and ≈ −1.5 ppb K−1 for A435 and S436, respectively) in the L63A peptide complex. These data are consistent with A435 and S436 being largely solvent exposed (ie, α4 unfolded) in the original and L21A fusion peptide complexes but engaged in intramolecular hydrogen bonds when TAZ1 is bound to the L63A fusion peptide. Figures 7B and C provide strong evidence that binding and folding of the fusion peptide αA and αC helices are both linked to the TAZ1 α4 helix folding equilibrium. Binding of the CITED2 αA helix extends and stabilizes the TAZ1 α4 helix whereas binding of the HIF-1α αC helix destabilizes α4, suggesting that the α4 region plays a central role in mediating the negative allostery observed in binding of the αA and αC helices of the fusion peptide.
Discussion
Previous studies have shown that the negative feedback regulator CITED2 efficiently displaces HIF-1α from its complex with the TAZ1 domain of CBP/p300 by an associative mechanism involving formation of a transient ternary complex (Berlow et al., 2017). The present work provides insights into the nature of the ternary complex and reveals the structural basis for the negative allosteric coupling between the CITED2 αA and HIF-1α αC binding sites that promotes HIF-1α dissociation.
The ternary complex was modeled using an engineered fusion peptide consisting of the CITED2 αA-LPEL and HIF-1α αB and αC motifs. In contrast to the crystal structure of the TAZ1:fusion peptide complex, where crystal packing stabilizes the interactions between TAZ1 and the CITED2 αA helix, NMR measurements show that contacts between the αA region and TAZ1 are weak and highly dynamic in solution. The C-terminal end of αC also shows exchange broadening in the fusion complex (Figures 4B, 5A, S3A), even though the HIF-1α αB and αC helices dominate the interaction between the fusion peptide and TAZ1. This observation suggests that the αC interactions are weakened compared to those in the parent TAZ1:HIF-1α complex (Berlow et al., 2017; Berlow et al., 2019; Dames et al., 2002). The NMR data for the complexes formed by the original fusion peptide and the L21A and L63A mutants show that binding of the HIF-1α αC helix antagonizes interactions with the CITED2 αA helix and vice versa. This negative allostery appears to be mediated by the α4 helix of TAZ1, which differs in length and orientation depending upon whether CITED2 or HIF-1α is bound. The α4 folding equilibrium is driven in opposite directions: binding of CITED2 αA to TAZ1 stabilizes an extended α4 helix, whereas binding of HIF-1α αC to TAZ1 promotes unfolding of α4. The opposing effects of CITED2 αA and HIF-1α αC binding on the TAZ1 α4 folding equilibrium are a likely source of the observed negative allostery between CITED2 and HIF-1α binding.
The conformational plasticity of TAZ1 plays a central role in the competition between the CITED2 αA and HIF-1α αC helices for binding to TAZ1 (Berlow et al., 2017; Berlow et al., 2019; Ruiz-Ortiz and De Sancho, 2020). NMR relaxation measurements and molecular dynamics simulations reveal fast timescale dynamics in the α4 region of free TAZ1 and the TAZ1:HIF-1α complex that are damped in the binary CITED2 complex (Berlow et al., 2019; Ruiz-Ortiz and De Sancho, 2020), where interactions with the αA helix stabilize extension of helical structure in α4. We propose that allostery arises through thermodynamic coupling that modulates the population of binding-competent states. CITED2 αA binding to TAZ1 increases the population of fully extended α4 helix, which decreases the affinity of the TAZ1–HIF-1α αC interaction. On the other hand, HIF1α αC binding to TAZ1 decreases the population of the extended TAZ1 α4 helix, thereby impairing the TAZ1-CITED2 αA interaction. Therefore, the equilibria of CITED2 αA-TAZ1 and HIF-1α αc-TAZ1 binding interactions are coupled to one another through their individual linkages to the TAZ1 α4 folding equilibrium. Importantly, while the discussion here has focused on the linkage between αA/αC binding and TAZ1 α4 extension, it is important to remember that CITED2 αA and HIF-1α αC are both disordered in solution and must fold upon binding to TAZ1. The CITED2 αA and HIF-1α αC folding equilibria are thus integral components of the network of linked equilibria that determine the outcome of allosteric interactions between αA and αC on the surface of TAZ1.
A number of studies using coarse-grained molecular dynamics simulations to probe the mechanism by which CITED2 drives displacement of HIF-1α from TAZ1 have recently been published (Chu et al., 2020; Ruiz-Ortiz and De Sancho, 2020; Wang and Brooks, 2020). In each of these simulations, the displacement reaction was observed to proceed through an intermediate in which CITED2 is essentially fully bound to TAZ1 while HIF-1α is bound only through αC, with the αA and αB regions fully dissociated. Due to the design of the fusion peptide, the αB and αC helices of HIF-1α both remain bound to TAZ1 in our current model of the ternary complex, which thus represents an earlier step on the displacement pathway than the intermediate observed in the molecular dynamics simulations. It is probable that the hydrophobic residues immediately C-terminal to the LPEL motif of CITED2, which bind in the αB site but are absent from the fusion peptide, play an important role in HIF-1α displacement by competing with the αB helix for binding to TAZ1. These additional hydrophobic contacts would be expected to strengthen the TAZ1:CITED2 interactions, weaken HIF-1α binding by displacing αB, and shift the TAZ1 α4 folding equilibrium towards the CITED2-bound state. Given the negative allostery between the αA and αC binding sites revealed by our current experiments, the enhanced interactions with the CITED2 αA helix would destabilize binding of the HIF-1α αC helix and facilitate dissociation of HIF-1α.
Intrinsically disordered proteins and protein regions constitute a large fraction of the human proteome and are overrepresented in eukaryotic transcriptional regulation and cell signaling pathways (Iakoucheva et al., 2002; Liu et al., 2006; Wright and Dyson, 2015). Given the ubiquity of IDPs, it is unlikely that the facilitated dissociation mechanism regulating the interactions between TAZ1, CITED2, and HIF-1α is unique. Indeed, facilitated dissociation of the disordered histone H1 – prothymosin α complex involving transient ternary complex formation has recently been reported (Borgia et al., 2018; Sottini et al., 2020). Many IDPs contain multiple binding motifs that engage their targets with varying strengths of interaction, enabling the formation of multivalent complexes with non-uniformly distributed binding energy (Berlow et al., 2015; Wright and Dyson, 2015). It is clear that the machinery required for facilitated dissociation is a hallmark of IDPs and it therefore seems likely that many signaling hubs involving IDPs engage in facilitated dissociation. Facilitated dissociation provides a mechanism by which high affinity, long lifetime complexes can be rapidly disassembled. Importantly, this allows for stable “on” signals and fast “off” signals. This is precisely the type of switch-like behavior observed in the TAZ1 / CITED2 / HIF-1α system (Berlow et al., 2017). Both CITED2 and HIF-1α bind to TAZ1 with nanomolar affinity and display slow intrinsic dissociation kinetics (Berlow et al., 2017; Lindström et al., 2018). Without accelerated displacement of HIF-1α by CITED2, the rapid on/off switching characteristic of the hypoxic response would not be possible.
While the hypoxic response is an essential component of human physiology, it is often co-opted by cancer cells, which are generally hypoxic, to promote tumor growth and metastasis (Bertout et al., 2008; Nurwidya et al., 2012; Semenza, 2014). Knowledge of the molecular details of the hypoxic response could be fruitful for the design of therapeutics: the crystal structure of the model ternary intermediate in CITED2-driven displacement of HIF-1α presented in this work should prove useful to the design of small molecule modulators of the displacement reaction. Importantly, we have defined specific interactions within the complex that strengthen or weaken the interaction between TAZ1 and either of its binding partners, providing the opportunity to steer the reaction and the regulatory pathway. Therefore, our findings not only represent a detailed molecular-level dissection of an allosteric network involving IDPs but also provide an avenue for highly targeted therapeutic design.
STAR Methods Text
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Peter E. Wright wright@scripps.edu
Materials Availability
Plasmids generated in this study have been deposited to Addgene. Catalog numbers are given in the key resources table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bacterial and Virus Strains | ||
| E. coli BL21 (DE3) | Agilent | F– ompT hsdSB (rB – mB – ) gal dcm (DE3) |
| E. coli XL-1 Blue competent cells | Agilent | recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac [F ´ proABlacIqZ∆M15 Tn10 (Tetr)] |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| TAZ1 domain of M. musculus CREB-binding protein | This paper | N/A |
| H. sapiens Hypoxia inducible factor 1α C-terminal activation domain | This paper | N/A |
| H. sapiens CITED2 C-terminal activation domain | This paper | N/A |
| H. sapiens CITED2 C-terminal activation domain - H. sapiens Hypoxia inducible factor 1α C-terminal activation domain fusion peptide | This paper | N/A |
| H. sapiens CITED2 C-terminal activation domain - H. sapiens Hypoxia inducible factor 1α C-terminal activation domain fusion peptide L21A-mutant | This paper | N/A |
| H. sapiens CITED2 C-terminal activation domain - H. sapiens Hypoxia inducible factor 1α C-terminal activation domain fusion peptide L63A-mutant | This paper | N/A |
| Critical Commercial Assays | ||
| Deposited Data | ||
| Crystal structure of the CREB-binding protein TAZ1 domain in complex with a CITED2 C-terminal activation domain - Hypoxia inducible factor 1α C-terminal activation domain fusion peptide | This paper | PDB 7LVS |
| NMR solution structure of the CREB-binding protein TAZ1 domain in complex with a CITED2 C-terminal activation domain | De Guzman et al., 2004 | PDB 1R8U |
| NMR solution structure of the CREB-binding protein TAZ1 domain in complex with a Hypoxia inducible factor 1α C-terminal activation domain | Dames et al., 2002 | PDB 1L8C |
| NMR solution structure of TAZ1 | De Guzman et al., 2005 | PDB 1U2N |
| Backbone chemical shift assignments: CITED2 C-terminal activation domain - Hypoxia inducible factor 1α C-terminal activation domain fusion peptide | This paper | BMRB 50866 |
| Backbone chemical shift assignments: CITED2 C-terminal activation domain - Hypoxia inducible factor 1α C-terminal activation domain fusion peptide in complex with the CREB-binding protein TAZ1 domain: TAZ1 | This paper | BMRB 50865 |
| Backbone chemical shift assignments: CITED2 C-terminal activation domain - Hypoxia inducible factor 1α C-terminal activation domain fusion peptide in complex with the CREB-binding protein TAZ1 domain: fusion peptide | This paper | BMRB 50865 |
| Backbone chemical shift assignments: CITED2 C-terminal activation domain - Hypoxia inducible factor 1α C-terminal activation domain fusion peptide L63A mutant in complex with the CREB-binding protein TAZ1 domain: TAZ1 | This paper | BMRB 50867 |
| Backbone chemical shift assignments: CITED2 C-terminal activation domain - Hypoxia inducible factor 1α C-terminal activation domain fusion peptide L63A mutant in complex with the CREB-binding protein TAZ1 domain: fusion peptide | This paper | BMRB 50867 |
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| PCR Primer: 5’-gtccgcggtgatagaaatgggtttggaccgcatcaaggag (fusion peptide L21A mutation) | This paper; Integrated DNA Technologies | N/A |
| PCR Primer: 5’-caaacccatttctatcaccgcggacataagaacttcctcgtcgatgaaatcagtg (fusion peptide L21A mutation) | This paper; Integrated DNA Technologies | N/A |
| PCR Primer: 5’-gctgcggatcaagttaactgatagggatcccctctagaaa (fusion peptide L63A mutation | This paper; Integrated DNA Technologies | N/A |
| PCR Primer: 5’-aacttgatccgcagctctgagtaattcttcaccctgcag (fusion peptide L63A mutation) | This paper; Integrated DNA Technologies | N/A |
| Recombinant DNA | ||
| pET21d encoding M. musculus CREB-binding protein TAZ1 domain | De Guzman et al., 2005 | Addgene #173760 |
| dnaY | Love et al., 2004, Brinkmann et al., 1989 | N/A |
| pET22b encoding M. musculus CREB-binding protein TAZ1 domain and the His6-tagged B1 domain of streptococcal protein G fused to the C-terminal activation domain of H. sapiens hypoxia-inducible factor 1α | Sugase et al., 2008 | Addgene #99343 |
| pET22b encoding M. musculus CREB-binding protein TAZ1 domain and the His6-tagged B1 domain of streptococcal protein G fused to the C-terminal activation domain of H. sapiens CITED2 | Berlow et al., 2017 | Addgene #173761 |
| pET22b encoding M. musculus CREB-binding protein TAZ1 domain and the His6-tagged B1 domain of streptococcal protein G fused to a H. sapiens CITED2 C-terminal activation domain – H. sapiens hypoxia-inducible factor 1α C-terminal activation domain fusion | This manuscript | Addgene #173762 |
| pET22b encoding M. musculus CREB-binding protein TAZ1 domain and the His6-tagged B1 domain of streptococcal protein G fused to a H. sapiens CITED2 C-terminal activation domain – H. sapiens hypoxia-inducible factor 1α C-terminal activation domain fusion harboring the L21A mutation | This manuscript | Addgene #173763 |
| pET22b encoding M. musculus CREB-binding protein TAZ1 domain and the His6-tagged B1 domain of streptococcal protein G fused to a H. sapiens CITED2 C-terminal activation domain – H. sapiens hypoxia-inducible factor 1α C-terminal activation domain fusion harboring the L63A mutation | This manuscript | Addgene #173764 |
| Software and Algorithms | ||
| NmrPipe | Delaglio et al., 1995 | https://www.ibbr.umd.edu/nmrpipe/install.html |
| NMRbox | Maciejewski et al., 2017 | https://nmrbox.org |
| NMRFAM-SPARKY | Lee et al., 2015 | https://nmrfam.wisc.edu/nmrfam-sparky-distribution |
| Bruker Topspin 3.2 | www.Bruker.com | https://www.bruker.com/en/products-and-solutions/mr/nmr-software/topspin.html |
| Bruker XWIN-NMR 3.1 | www.Bruker.com | www.Bruker.com |
| Phenix | Liebschner et al., 2019 | https://www.phenix-online.org |
| Coot | Emsley et al., 2010 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot |
| HKL-2000 | Otwinowski and Minor, 1997 | https://www.hkl-xray.com |
| Arcimboldo Lite | Sammito et al., 2015 | chango.ibmb.csic.es/lite |
| Pymol | https://pymol.org/2/ | https://pymol.org/2/ |
| gnuplot | www.gnuplot.info | www.gnuplot.info |
| GNU Octave | Eaton et al., 2021 | https://octave.org/doc/v6.2.0/ |
| Other | ||
Data and Code Availability
Chemical shift assignment data have been deposited at the BioMagResBank, and coordinates and structure factors have been deposited at the PDB, and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental Model and Subject Details
Source Organism.
Escherichia coli BL21(DE3) (DNAY); Escherichia coli XL-1 Blue competent cells. Culture conditions include LB medium and minimal (M9) medium supplemented with stable isotope labeled precursors as described in detail below.
Method Details
Sample Preparation.
The CITED2-HIF-1α fusion peptide was generated by cloning the sequence coding for human CITED2 residues 216–246 followed immediately by the sequence coding for human HIF-1α residues 796–826 into a co-expression vector with TAZ1 (mouse CBP residues 340–439). The fusion peptide sequence was cloned into the vector immediately downstream of the sequence coding for N-terminally His6-tagged GB1 (Sugase et al., 2008). Site-directed mutagenesis to generate the L21A and L63A fusion peptide constructs was accomplished using standard QuikChange PCR protocols.
The CITED2 C-terminal transactivation domain (residues 216–269 of human CITED2), the HIF-1α C-terminal transactivation domain (residues 776–826 of human HIF-1α), and the CITED2-HIF-1α fusion peptide constructs were expressed in E. coli BL21 (DE3) [DNAY] as His6-tagged GB1 fusion proteins in a coexpression vector with TAZ1 (residues 340–439 of mouse CBP). Cells were grown in M9 minimal media. Cells were cultured at 37 °C to OD600 ~ 0.8 at which time protein expression was induced by the addition of ~ 1 mM IPTG and ~ 100 μM ZnSO4 and growth was continued at 18 °C for an additional 18–21 hours. Cells were harvested by centrifugation at 4 °C and cell pellets were stored at −20 °C prior to protein purification. Cell pellets were resuspended in 40 mL of buffer containing 25 mM Tris pH 8.0, 200 mM NaCl, 8 M urea, and 20 mM imidazole per liter of culture and lysed by sonication at 4 °C. The soluble fraction was isolated by centrifugation at 4 °C. The supernatant was purified at room temperature by nickel affinity chromatography using NiNTA resin and the HIF-1α and CITED2 peptides were separated from the His6-GB1 tag by thrombin cleavage on the resin. The cleaved HIF-1α, CITED2 and fusion peptides were further purified by reversed phase HPLC, using a C4 cartridge (Waters) in acetonitrile/0.1% TFA mobile phase. Pure peptides were lyophilized and stored at −20 °C. Lyophilized peptides were dissolved in 50 – 100 mM Tris buffer pH 8 ~ 10 mM DTT and dialyzed overnight against buffer containing 20 mM Tris pH 6.8, 50 mM NaCl, and 2mM DTT prior to use (Berlow et al., 2017). . After removal of the the His6-GB1 tag by thrombin cleavage, the sequence GSHMS remained at the N-terminus of the CITED2 and fusion peptides and the sequence GSHM at the N-terminus of HIF-1α. TAZ1 was expressed and purified under native conditions. E. coli BL21(DE3) [DNAY] were grown in M9 minimal medium at 37°C to OD600 ~ 0.8. Protein expression was induced by addition of 1 mM IPTG and ~ 100 μM ZnSO4 and growth was continued at 18°C for an additional 18–21 hours. Cells were harvested by centrifugation at 4 °C and cell pellets were stored at −20 °C prior to protein purification. Cells were resuspended in ice-cold buffer (40 mL per liter of cell culture) containing 20 mM Tris pH 8, 40 mM NaCl, 10 μM ZnSO4, 10 mM DTT, and Pierce EDTA-free protease inhibitor cocktail (1 tablet / 150 mL buffer) and lysed by sonication. The soluble lysate fraction was loaded onto HiTrapQ and HiTrapSP columns (GE Healthcare) connected in series that were pre-equilibrated in 20 mM Tris pH 8, 40 mM NaCl, 10 μM ZnSO4, and 2mM DTT. The columns were washed until the UV absorbance at 280 nm returned to baseline. The HiTrapQ column was then removed and bound TAZ1 was subsequently eluted from the HiTrapSP column using a linear gradient to 600mM NaCl in 20 mM Tris pH 8. Fractions containing nearly pure TAZ1 were then further fractionated by size exclusion chromatography on a Superdex75 column (Amersham Biosciences) in 20 mM Tris pH 8, 200 mM NaCl, and 2 mM DTT. Aliquots of pure TAZ1 were made ~ 10 mM in DTT using 1 M DTT stocks, flash frozen with liquid nitrogen, and stored at −80 °C.
TAZ1 and fusion peptides were uniformly labeled with 15N or 15N,13C by expression in M9 minimal media containing 15NH4Cl (1 g L−1) or 15NH4Cl (0.5 g L−1) /15N H4SO4 (0.5 g L−1) as the sole nitrogen sources and 13C glucose (2.5 g L−1) as the sole carbon source.
Protein concentrations were determined by measuring absorbance at 280 nm and using extinction coefficients of 1490 M−1cm−1 for HIF-1α and fusion peptide constructs and 5500 M−1cm−1 for TAZ1 and CITED2. Extinction coefficients were calculated using the ExPASy ProtParam web server.
Samples for NMR and crystallography were prepared by dialysis at 4 °C into NMR/crystallography buffer: 20 mM Tris-Cl pH 6.8, 50 mM NaCl, 2 mM DTT. D2O was added to NMR samples to achieve 5%.
Crystallization and Structure Analysis.
The TAZ1:fusion peptide complex was prepared at a 1:1 TAZ1:peptide molar ratio at 1.2 mM in NMR/crystallography buffer and was crystallized by sitting drop vapor diffusion at 20 °C with a precipitant of 0.1 M HEPES pH 6.87, 28% (w/v) PEG 4000. Crystals were cryoprotected in 30% ethylene glycol / 70% well solution before flash-cooling in liquid nitrogen. Crystallographic data (2.0 Å resolution) were collected at 100 K at the Advanced Light Source at Lawrence Berkeley National Laboratory on Beamline 5.0.3. Data were processed and scaled with the HKL-2000 software suite (Otwinowski and Minor, 1997) in space group P21 with unit cell dimensions 32.06, 50.11, 41.26 and β=99.19. An incomplete polyalanine model consisting of 79 residues (395 atoms) in 6 chains with a CC of 33.5% was generated using Arcimboldo Lite (Rodriguez et al., 2009). The lowest energy NMR structure of TAZ1 (1U2N (De Guzman et al., 2005)) was aligned to the polyalanine model and used as a starting model for refinement. The model was iteratively refined and re-built using Phenix (Liebschner et al., 2019) and Coot (Emsley et al., 2010). The final model contains 155 residues (1213 protein atoms) in 2 chains with 38 modeled waters and 3 Zn atoms. Validation was carried out with Molprobity (Williams et al., 2018) with 94.7%/0.66% of residues in favored/outlier regions of the Ramachandran plot. Structural statistics are listed in Table 1.
NMR Spectroscopy.
NMR spectra were acquired at 298 K, unless otherwise noted, on a Bruker Avance 900 MHz spectrometer with a room temperature triple resonance probe and Bruker Avance 500 and 700 MHz spectrometers equipped with triple resonance cryoprobes. Complexes were prepared by combining isotope labeled TAZ1 or peptide in NMR/crystallography buffer with a 1.2−1.9 fold excess of unlabeled peptide or TAZ1 in the same buffer. Spectra for determining resonance assignments for TAZ1:fusion peptide complexes were acquired at 300 μM – 400 μM concentration of isotope-labeled component. Spectra for determining resonance assignments for the free fusion peptide were obtained at 100 μM concentration. Resonance assignments for TAZ1:CITED2 and TAZ1:HIF-α complexes have been reported previously (Dames et al., 2002; De Guzman et al., 2004). All other NMR experiments utilized 100 μM labeled component.
Backbone resonance assignments for the free and bound fusion peptide, the bound L63A fusion peptide, and TAZ1 bound to the original and L63A fusion peptides were obtained from HNCA, HN(CO)CA, HNCO, HN(CA)CO, HNCACB, and HN(CO)CACB triple resonance spectra (Sattler et al., 1999) as implemented in the TopSpin 3.2 pulse program library. Assignments for the free L63A fusion peptide were made by comparing the 1H-15N HSQC spectra of the L63A and original fusion peptides. Based on the similarity of the spectra, assignments for resolved cross peaks of 15N-labeled TAZ1 in complex with unlabeled L21A fusion peptide were transferred from the 1H-15N HSQC spectrum of the original fusion peptide complex. Similarly, assignments of the bound L21A peptide were transferred from those of the unmodified fusion peptide complex. Triple resonance spectra of free isotopically labeled original fusion peptide as well as isotopically labeled TAZ1 bound to unlabeled original fusion peptide were collected using band-selective amide proton pulses (Lescop et al., 2007; Schanda et al., 2006). The indirect dimensions were uniformly sampled and doubled in size by zero filling. All triple resonance spectra were reconstructed using iterative soft thresholding (Drori, 2007; Stern et al., 2007). NMR data were processed using NMRPipe (Delaglio et al., 1995) and spectra were visualized using SPARKY (Lee et al., 2015) on the NMRbox platform (Maciejewski et al., 2017).
Quantification and Statistical Analysis
Weighted average 1H,15N chemical shift differences were calculated as described in the Figure 3 legend. The 10 % trimmed means and standard deviations of chemical shift difference data sets were determined by calculating the 0.9 quantile of a given data set using GNU Octave, generating a list of chemical shift differences less than the 0.9 quantile, and calculating the mean and standard deviation of this trimmed data set. Where indicated in the figure legends, broken lines inside graphs of chemical shift differences report the sum of the 10 % trimmed mean and standard deviation.
Intensities of cross peaks from 1H-15N-HSQC spectra were obtained using Sparky and were normalized as described in the Figure S2 legend.
1H chemical shift temperature coefficients were calculated by determining the slopes of 1H chemical shift Vs. temperature data using linear regression scripts written in GNU Octave ( where β0 and β1 denote the intercept and 1H chemical shift temperature coefficient respectively, Ti and CSi denote the ith temperature and chemical shift respectively, CI68 denotes the 68 % confidence interval of the 1H chemical shift temperature coefficient, t68 denotes the 68 % single-tailed critical value of the t-distribution, MSR denotes the sum of squared residuals divided by the degrees of freedom, and overbars denote averages).
Additional Resources
See Key Resources Table.
Supplementary Material
Highlights:
Mechanistic insights into regulation of the hypoxic response
Molecular determinants for displacement of HIF-1α by facilitated dissociation
Fusion protein strategy to characterize a model of a transient ternary complex
Structural analysis of a negative allosteric network involving disordered proteins
Acknowledgments
We thank Gerard Kroon for assistance with NMR experiments, Euvel Manlapaz for technical assistance, Mary Lonsdale for assistance in collecting preliminary data, Maria Martinez-Yamout for assistance with HPLC, Henry Tien for assistance with robotic crystallization, and Wright/Dyson lab members for insightful discussions. This work was supported by Grants CA096865 and CA229652 (PEW) and GM131693 (HJD) from the National Institutes of Health and the Skaggs Institute for Chemical Biology. The Berkeley Center for Structural Biology is supported in part by the Howard Hughes Medical Institute. The Advanced Light Source is a Department of Energy Office of Science User Facility under Contract No. DE-AC02-05CH11231.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arany Z, Huang LE, Eckner R, Bhattacharya S, Jiang C, Goldberg MA, Bunn HF, and Livingston DM (1996). An essential role for p300/CBP in the cellular response to hypoxia. Proc. Natl. Acad. Sci. U.S.A 93, 12969–12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter NJ, and Williamson MP (1997). Temperature dependence of 1H chemical shifts in proteins. J. Biomol. NMR 9, 359–369. [DOI] [PubMed] [Google Scholar]
- Berlow RB, Dyson HJ, and Wright PE (2015). Functional advantages of dynamic protein disorder. FEBS Lett 589, 2433–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlow RB, Dyson HJ, and Wright PE (2017). Hypersensitive termination of the hypoxic response by a disordered protein switch. Nature 543, 447–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlow RB, Martinez-Yamout MA, Dyson HJ, and Wright PE (2019). Role of backbone dynamics in modulating the interactions of disordered ligands with the TAZ1 domain of the CREB-binding protein. Biochemistry 58, 1354–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertout JA, Patel SA, and Simon MC (2008). The impact of O2 availability on human cancer. Nat. Rev. Cancer 8, 967–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S, Michels CL, Leung MK, Arany ZP, Kung AL, and Livingston DM (1999). Functional role of p35srj, a novel p300/CBP binding protein, during transactivation by HIF-1. Genes Devel 13, 64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgia A, Borgia MB, Bugge K, Kissling VM, Heidarsson PO, Fernandes CB, Sottini A, Soranno A, Buholzer KJ, Nettels D, et al. (2018). Extreme disorder in an ultrahigh-affinity protein complex. Nature 555, 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann U, Mattes RE, and Buckel P (1989). High-level expression of recombinant genes in Escherichia coli is dependent on the availability of the dnaY gene product. Gene 85, 109–114. [DOI] [PubMed] [Google Scholar]
- Chen T-Y, Cheng Y-S, Huang P-S, and Chen P (2018). Facilitated unbinding via multivalency-enabled ternary complexes: new paradigm for protein–DNA interactions. Acc. Chem. Res 51, 860–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T-Y, Santiago AG, Jung W, Krzemiński Ł, Yang F, Martell DJ, Helmann JD, and Chen P (2015). Concentration- and chromosome-organization-dependent regulator unbinding from DNA for transcription regulation in living cells. Nat. Commun 6, 7445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu W-T, Chu X, and Wang J (2020). Investigations of the underlying mechanisms of HIF-1α and CITED2 binding to TAZ1. Proc. Natl. Acad. Sci. U.S.A 117, 5595–5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dames SA, Martinez-Yamout M, De Guzman RN, Dyson HJ, and Wright PE (2002). Structural basis for Hif-1a/CBP recognition in the cellular hypoxic response. Proc. Natl. Acad. Sci. U.S.A 99, 5271–5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Guzman RN, Martinez-Yamout M, Dyson HJ, and Wright PE (2004). Interaction of the TAZ1 domain of CREB-binding protein with the activation domain of CITED2: regulation by competition between intrinsically unstructured ligands for non-identical binding sites. J. Biol. Chem 279, 3042–3049. [DOI] [PubMed] [Google Scholar]
- De Guzman RN, Wojciak JM, Martinez-Yamout MA, Dyson HJ, and Wright PE (2005). CBP/p300 TAZ1 domain forms a structured scaffold for ligand binding. Biochemistry 44, 490–497. [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Guang Z, Pfeifer J, and Bax A (1995). NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Drori I (2007). Fast l1 minimization by iterative thresholding for multidimensional NMR spectroscopy. EURASIP Journal on Advances in Signal Processing 2007, 020248. [Google Scholar]
- Eaton JW, Bateman D, Hauberg S, and Wehbring R (2021). GNU Octave version 6.2.0 manual: a high-level interactive language for numerical computations.
- Ebert BL, and Bunn HF (1998). Regulation of transcription by hypoxia requires a multiprotein complex that includes hypoxia-inducible factor 1, an adjacent transcription factor, and p300/CREB binding protein. Mol. Cell. Biol 18, 4089–4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Cryst. D 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman SJ, Sun ZY, Kung AL, France DS, Wagner G, and Eck MJ (2003). Structural basis for negative regulation of hypoxia-inducible factor-1a by CITED2. Nat. Struct. Biol 10, 504–512. [DOI] [PubMed] [Google Scholar]
- Freedman SJ, Sun ZY, Poy F, Kung AL, Livingston DM, Wagner G, and Eck MJ (2002). Structural basis for recruitment of CBP/p300 by hypoxia-inducible factor-1a. Proc. Natl. Acad. Sci. U.S.A 99, 5367–5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham JS, Johnson RC, and Marko JF (2010). Concentration-dependent exchange accelerates turnover of proteins bound to double-stranded DNA. Nucl. Acids Res 39, 2249–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henze A-T, and Acker T (2010). Feedback regulators of hypoxia-inducible factors and their role in cancer biology. Cell Cycle 9, 2821–2835. [DOI] [PubMed] [Google Scholar]
- Huang LE, Gu J, Schau M, and Bunn HF (1998). Regulation of hypoxia-inducible factor 1a is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. U.S.A 95, 7987–7992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iakoucheva LM, Brown CJ, Lawson JD, Obradovic Z, and Dunker AK (2002). Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol 323, 573–584. [DOI] [PubMed] [Google Scholar]
- Ivan M, Kondo K, Yang HF, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, and Kaelin WG Jr. (2001). HIFa targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 292, 464–468. [DOI] [PubMed] [Google Scholar]
- Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, et al. (2001). Targeting of HIF-a to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292, 468–472. [DOI] [PubMed] [Google Scholar]
- Joshi CP, Panda D, Martell DJ, Andoy NM, Chen T-Y, Gaballa A, Helmann JD, and Chen P (2012). Direct substitution and assisted dissociation pathways for turning off transcription by a MerR-family metalloregulator. Proc. Natl. Acad. Sci. U.S.A 109, 15121–15126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamar RI, Banigan EJ, Erbas A, Giuntoli RD, Olvera de la Cruz, M., Johnson, R.C., and Marko, J.F. (2017). Facilitated dissociation of transcription factors from single DNA binding sites. Proc. Natl. Acad. Sci. U.S.A 114, E3251–E3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B, Eggel A, Tarchevskaya SS, Vogel M, Prinz H, and Jardetzky TS (2012). Accelerated disassembly of IgE–receptor complexes by a disruptive macromolecular inhibitor. Nature 491, 613–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Tonelli M, and Markley JL (2015). NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31, 1325–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lescop E, Schanda P, and Brutscher B (2007). A set of BEST triple-resonance experiments for time-optimized protein resonance assignment. J. Magn. Reson 187, 163–169. [DOI] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Baker ML, Bunkoczi G, Chen VB, Croll TI, Hintze B, Hung L-W, Jain S, McCoy AJ, et al. (2019). Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Cryst. D 75, 861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindström I, Andersson E, and Dogan J (2018). The transition state structure for binding between TAZ1 of CBP and the disordered Hif-1α CAD. Sci. Rep 8, 7872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Perumal NB, Oldfield CJ, Su EW, Uversky VN, and Dunker AK (2006). Intrinsic disorder in transcription factors. Biochemistry 45, 6873–6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love JJ, Li X, Chung J, Dyson HJ, and Wright PE (2004). The LEF-1 HMG domain undergoes a disorder-to-order transition upon complex formation with cognate DNA. Biochemistry 43, 8725–8734. [DOI] [PubMed] [Google Scholar]
- Maciejewski MW, Schuyler AD, Gryk MR, Moraru II, Romero PR, Ulrich EL, Eghbalnia HR, Livny M, Delaglio F, and Hoch JC (2017). NMRbox: A Resource for Biomolecular NMR Computation. Biophys. J 112, 1529–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurwidya F, Takahashi F, Minakata K, Murakami A, and Takahashi K (2012). From tumor hypoxia to cancer progression: the implications of hypoxia-inducible factor-1 expression in cancers. Anat. Cell Biol 45, 73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, and Minor W (1997). Processing of x-ray diffraction data collected in oscillation mode. In Methods Enzymol Macromol. Crystall. A, Carter CW Jr., and Sweet RM, eds. (New York: Academic Press; ), pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Potoyan DA, Bueno C, Zheng W, Komives EA, and Wolynes PG (2017). Resolving the NFkappaB Heterodimer Binding Paradox: Strain and Frustration Guide the Binding of Dimeric Transcription Factors. J. Am. Chem. Soc 139, 18558–18566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez DD, Grosse C, Himmel S, Gonzalez C, de Ilarduya IM, Becker S, Sheldrick GM, and Uson I (2009). Crystallographic ab initio protein structure solution below atomic resolution. Nat. Methods 6, 651–653. [DOI] [PubMed] [Google Scholar]
- Ruiz-Ortiz I, and De Sancho D (2020). Competitive binding of HIF-1α and CITED2 to the TAZ1 domain of CBP from molecular simulations. Phys. Chem. Chem. Phys 22, 8118–8127. [DOI] [PubMed] [Google Scholar]
- Sammito M, Millán C, Frieske D, Rodríguez-Freire E, Borges RJ, and Usón I (2015). ARCIMBOLDO_LITE: single-workstation implementation and use. Acta Crystallographica D 71, 1921–1930. [DOI] [PubMed] [Google Scholar]
- Sattler M, Schleucher J, and Griesinger C (1999). Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog. NMR Spect 34, 93–158. [Google Scholar]
- Schanda P, VanMelckebeke H, and Brutscher B (2006). Speeding Up Three-Dimensional Protein NMR Experiments to a Few Minutes. J. Am. Chem. Soc 128, 9042–9043. [DOI] [PubMed] [Google Scholar]
- Schmitt L, Kratz JR, Davis MM, and McConnell HM (1999). Catalysis of peptide dissociation from class II MHC-peptide complexes. Proc. Natl. Acad. Sci. U.S.A 96, 6581–6586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL (2014). Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. Mech. Dis 9, 47–71. [DOI] [PubMed] [Google Scholar]
- Sottini A, Borgia A, Borgia MB, Bugge K, Nettels D, Chowdhury A, Heidarsson PO, Zosel F, Best RB, Kragelund BB, et al. (2020). Polyelectrolyte interactions enable rapid association and dissociation in high-affinity disordered protein complexes. Nat. Commun 11, 5736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern AS, Donoho DL, and Hoch JC (2007). NMR data processing using iterative thresholding and minimum l1-norm reconstruction. J. Magn. Reson 188, 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugase K, Landes MA, Wright PE, and Martinez-Yamout M (2008). Overexpression of post-translationally modified peptides in Escherichia coli by co-expression with modifying enzymes. Prot. Express. Purif 57, 108–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai M-Y, Zhang B, Zheng W, and Wolynes PG (2016). Molecular Mechanism of Facilitated Dissociation of Fis Protein from DNA. J. Am. Chem. Soc 138, 13497–13500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GL, Jiang B, Rue EA, and Semenza GL (1995). Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. U.S.A 92, 5510–5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, and Brooks CL (2020). Electrostatic forces control the negative allosteric regulation in a disordered protein switch. J. Phys. Chem. Lett 11, 864–868. [DOI] [PubMed] [Google Scholar]
- Williams CJ, Headd JJ, Moriarty NW, Prisant MG, Videau LL, Deis LN, Verma V, Keedy DA, Hintze BJ, Chen VB, Jain S, Lewis SM, Arendall WB 3rd, Snoeyink J, Adams PD, Lovell SC, Richardson JS, Richardson DC (2018). MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci 27, 293–315/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright PE, and Dyson HJ (2015). Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol 16, 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Chemical shift assignment data have been deposited at the BioMagResBank, and coordinates and structure factors have been deposited at the PDB, and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.