

Summary
Select subsets of immune effector cells have the highest propensity to mediate antitumor responses. However, procuring these subsets is challenging, and cell-based immunotherapy is hampered by limited effector cell persistence and lack of on-demand availability. To address these limitations, we generated a triple gene-edited induced pluripotent stem cell (iPSC). The clonal iPSC line was engineered to express a high affinity, non-cleavable version of the Fc receptor CD16a and a membrane-bound IL-15/IL-15R fusion protein. The third edit was knockout of the ecto-enzyme CD38 that hydrolyzes NAD+. NK cells derived from these uniformly engineered iPSCs, termed iADAPT, displayed metabolic features and gene expression profiles mirroring those of cytomegalovirus-induced adaptive NK cells. iADAPT NK cells persisted in vivo in the absence of exogenous cytokine and elicited superior antitumor activity. Our findings suggest that unique subsets of the immune system can be modeled through iPSC technology for effective treatment of patients with advanced cancer.
Keywords: Natural killer (NK) cell, induced pluripotent stem cell (iPSC), immunotherapy, adaptive, multiple myeloma, acute myeloid leukemia, off-the-shelf
Graphical Abstract
eTOC statement
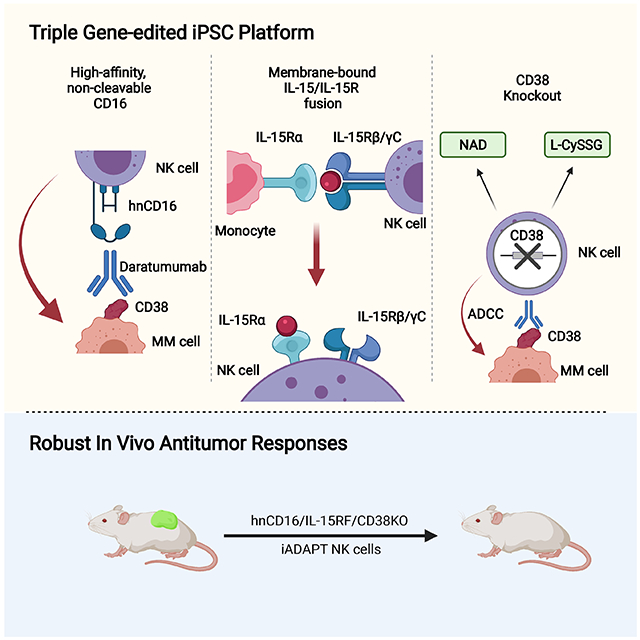
Optimized iPSC-derived NK (iNK) cells have been developed for on demand cancer immunotherapy. Here, Cichocki and colleagues describe a triple gene-edited iNK cell product termed iADAPT NK that persists and functions in vivo in the absence of exogenous cytokines and can be combined with therapeutic antibodies for enhanced tumor targeting.
Introduction
CD8+ T cells and natural killer (NK) cells are critical mediators of antitumor immunity. There have been many efforts to exploit these potent effector cells by either endogenous activation or adoptive transfer. While chimeric antigen receptor (CAR) T cells demonstrated initial success in treating acute B lymphoblastic leukemia (Brentjens et al., 2011; Maude et al., 2014), CAR-T cell efficacy in other malignancies has been less impressive (Hirayama et al., 2019; Schuster et al., 2019; Zou et al., 2018). Moreover, there are limitations to CAR-T cell adoptive therapy including cytokine release syndrome, neurotoxicity, and graft versus host disease (GvHD) in allogeneic settings. NK cells have several advantages over T cells as an adoptive cell therapy product. First, NK cells are safe in the allogeneic setting and do not mediate GvHD, making them amenable for off-the-shelf administration. The toxicity profile for allogeneic NK cell adoptive transfer is also favorable. (Miller et al., 2005; Romee et al., 2016). Allogeneic NK cell adoptive transfer has been tested clinically for the treatment of relapsed, refractory acute myeloid leukemia (AML). However, a minority of patients achieved a complete remission and rarely were responses durable, necessitating additional strategies to improve efficacy (Miller et al., 2005).
We developed a platform for the differentiation and expansion of induced pluripotent stem cell (iPSC)-derived NK (iNK) cells that resemble peripheral blood NK cells and have desirable properties over traditional apheresis products (Valamehr et al., 2014; Cichocki et al., 2020). These cells are cytotoxic against an array of tumor types, maintain robust antitumor function after cryopreservation, and efficiently recruit T cells in vivo (Cichocki et al., 2020). In addition, genetic modifications designed to augment antitumor activity can be introduced with ease. We demonstrated this previously with the generation of iNK cells expressing a modified version of CD16a, which is a low-affinity Fc receptor used by NK cells to mediate antibody-dependent cellular cytotoxicity (ADCC). Upon NK cell activation, CD16a is subject to cleavage by the metalloprotease ADAM17 leading to reduced ADCC (Romee et al., 2013). We previously identified the ADAM17 cleavage site within CD16 and created a high-affinity, non-cleavable version of CD16a (hnCD16) (Jing et al., 2015). Using our single-cell iPSC engineering platform, hnCD16a was introduced into iPSCs. Through clone selection, the ideal engineered iPSC was banked to create the starting material for derivation of iNK cells with uniform expression of hnCD16. Adoptive transfer of hnCD16 iNK cells in combination with rituximab yielded potent antitumor responses and long-term survival in a mouse xenograft model of lymphoma (Zhu et al., 2020).
Having established hnCD16 iNK cells, we sought to introduce additional synthetic modifications to enhance the efficacy and mimic the adaptive NK cell phenotype. First described as an expansion of a unique subset of NK cells in cytomegalovirus (CMV) seropositive individuals (Gumá et al., 2004), adaptive NK cells persist long-term, have enhanced metabolic fitness, exhibit strong ADCC, and likely mediate potent graft-versus-leukemia effects (Lee et al., 2015; Schlums et al., 2015; Schlums et al., 2017; Cichocki et al., 2018, Cichocki et al., 2016; Cichocki et al., 2019). To replicate the adaptive NK cell phenotype, we engineered two additional attributes: knockout of CD38 and the introduction of a membrane-bound IL-15/IL-15 receptor fusion (IL-15RF) molecule. Here, we show that knockout of CD38 and overexpression of both IL-15RF and hnCD16 transgenes imbues iNK cells with augmented innate immune function, ADCC, persistence, and metabolic and transcriptional characteristics associated with adaptive NK cells. We refer to our triple gene-modified iNK cells as iADAPT NK cells. Furthermore, we demonstrate that iADAPT NK cells persist in vivo in the absence of exogenous cytokines, exhibit profound antitumor function, and can be combined with daratumumab for efficient killing of multiple myeloma (MM) and AML cells in both in vitro and in vivo settings.
Results
Adaptive NK cells from CMV seropositive individuals express low levels of CD38 and are more resistant to oxidative stress-induced cell death
CD38 is an ectoenzyme with NAD+ glycohydrolase and ADP-ribosyl cyclase activity (Graeff et al., 2006). It was originally identified on activated T cells but is now known to be more broadly expressed, particularly during inflammation (Reinherz et al., 1980; Shubinsky and Schlesinger, 1997). We assessed surface CD38 levels on major immune subsets present within freshly isolated peripheral blood using a flow cytometry gating strategy (Figure 1A). CD38 levels were high on monocytes, CD56bright NK cells, and CD56dim NK cells, whereas CD38 levels were intermediate on B cells and low on both CD4+ and CD8+ T cells (Figure 1B). With respect to NAD+ metabolism, CD38 overexpression is associated with CD8+ T cell exhaustion, NAD+ depletion, and dysfunction in programmed cell death 1 (PD-1) blockade-resistant cancers (Verma et al., 2019; Chen et al., 2018). An inverse correlation between CD38 and NAD+ dictates long-term antitumor responses by T cells (Chatterjee et al., 2018). Given that adaptive NK cells have genome-wide epigenetic and transcriptional profiles that skew towards those observed for CD8+ T cells (Schlums et al., 2015; Lau et al., 2018), we reasoned that adaptive NK cells, like CD8+ T cells, may express low levels of CD38 at homeostasis. To address this, we examined previously published RNA-seq data comparing canonical (CD56dimCD57−NKG2C− and CD56dimCD57+NKG2C−) and adaptive (CD56dimCD57−NKG2C+ and CD56dimCD57+NKG2C+) NK cell subsets sorted from the peripheral blood of healthy CMV seropositive donors (Cichocki et al., 2018). We observed significantly lower CD38 expression in adaptive NK cell subsets relative to canonical subsets (Figure 1C). This finding was confirmed at the protein level by flow cytometry. NK cells from CMV seronegative donors displayed high surface levels of CD38 regardless of NKG2C and CD57 expression. In contrast, CD38 levels were markedly reduced on adaptive NK cell subsets from CMV seropositive individuals (Figure 1D). To determine whether adaptive NK cells have higher concentrations of intracellular NAD+, we separated NKG2C− and NKG2C+ NK cells freshly isolated from CMV seropositive individuals by magnetic selection and quantified NAD+. As anticipated, we observed elevated levels of NAD+ in NKG2C+ NK cells (Figure 1E).
Figure 1. CMV-induced adaptive NK cells downregulate CD38 and are more resistant to oxidative stress-induced death.

Peripheral blood mononuclear cells were isolated from healthy donors for examination of CD38 surface levels on major immune subsets. (A) Flow cytometry gating strategy used to identify immune subsets. (B) Representative histogram plots of CD38 surface expression (left) and cumulative data from 4 donors (right). (C) Heat map of CD38 mRNA transcript fold expression values assessed by RNA-seq in the indicated NK cell subsets sorted from the peripheral blood of 4 healthy CMV seropositive donors. Values are normalized to the CD56dimCD57−NKG2C− NK cell population. (D) Representative FACS plots of CD57 and NKG2C on gated CD3𢈒CD56dim NK cells from CMV seronegative and seropositive donors and histograms of surface CD38 expression on the indicated NK cell subsets (left). Cumulative data of CD38 mean fluorescence intensity (MFI) on the indicated NK cell subsets from 4 CMV seronegative and 4 CMV seropositive donors (right). Statistical significance was determined by one-way ANOVA with multiple comparisons. *p < 0.05 (E) NKG2C𢈒 and NKG2C+ NK cells were isolated by magnetic selection from 3 CMV seropositive donors and assayed for intracellular concentrations of NAD+. Statistical significance was determined by paired Student’s t-test. *p < 0.05 (F) Peripheral blood NK cells from 4 CMV seropositive were isolated by negative selection and cultured in the presence or absence of 50 μM H2O2 for overnight followed by analysis of apoptosis and cell death by flow cytometry. Show are representative FACS plots of dead cell dye and Annexin V staining for the indicated NK cell subsets (left) and cumulative data plotting the percentages of viable cells after H2O2 treatment (right). Statistical significance was determined by one-way ANOVA with multiple comparisons, and results are from 2 independent experiments. Data are represented as mean ± SEM. *p < 0.05, **p < 0.01
Previous studies have shown that CD38 knockout cells are resistant to oxidative stress through reductions of reactive oxygen species (ROS) levels. (Ge et al., 2010). To determine whether CD38 levels correlate with resistance to oxidative stress-induced NK cell death, we cultured NK cells from CMV seropositive donors with or without hydrogen peroxide (H2O2) and assessed apoptosis and cell death by flow cytometry. Adaptive NK cells were more resistant to cell death induced by oxidative stress as indicated by increased frequencies of viable cells among these subsets (Figure 1F). Together, our data demonstrates associations between reduced CD38 expression, elevated NAD+, and protection against oxidative stress in adaptive NK cells.
CD38KO iNK cells exhibit metabolic traits observed for adaptive NK cells and are resistant to daratumumab-induced fratricide
The association between low CD38 expression on adaptive NK cells and resistance to oxidative stress led us to reason that we could engineer iNK cells for metabolic benefit through knockout of CD38 while simultaneously leveraging this genetic modification for avoidance of NK cell fratricide mediated by the therapeutic anti-CD38 antibody daratumumab. To this end, we generated a bi-allelic CD38 knockout in iPSCs that had been transduced with hnCD16 (Figure 2A). iPSCs were cultured in a cocktail of small molecules and cytokines to induce differentiation into CD34+ hematopoietic progenitor cells (HPCs). Enriched CD34+ HPCs were then cultured under conditions that support NK cell differentiation and expansion using previously published methods (Zhu et al., 2020; Cichocki et al., 2020). The absence of CD38 expression in iNK cells was confirmed by Western blot (Figure 2B) and flow cytometry (Figure 2C). In agreement with our analysis of adaptive NK cells, we found that NAD+, NADH, and ATP levels were significantly higher in hnCD16/CD38KO iNK cells relative to hnCD16 iNK cells (Figure 2D).
Figure 2. hnCD16/CD38KO iNK cells exhibit enhanced metabolic fitness and avoid daratumumab-mediate fratricide.

CRISPR-Cas9 was used to knock out CD38 in iPSCs with transgenic expression of hnCD16. Knockout efficiency was assessed using (A) a T7E1 nuclease assay, (B) Western blot, and (C) flow cytometry. (D) The concentrations of NAD+ and NADH were determined in hnCD16 iNK cells and hnCD16/CD38KO iNK cells using a calorimetric cyclase assay, and ATP concentrations were determined by bioluminescence. Statistical significance was determined by paired Student’s t-test, and results are from 2 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 (E) Real time metabolic profiling of hnCD16 iNK cells and hnCD16/CD38KO iNK cells was performed by Seahorse analysis. Shown are the oxygen consumption rates (OCR) and extracellular acidification rates (ECAR) for 1 of 4 experiments performed. (F) hnCD16 iNK cells and hnCD16/CD38KO iNK cells were cultured in media alone or the indicated concentrations of H2O2 for 1 hour. Levels of mitochondrial superoxide were assessed by MitoSox dye fluorescence. Shown are FACS plots for a representative experiment (left) and cumulative data from 3 independent experiments. Statistical significance was determined by paired Student’s t-test. *p < 0.05 (G) To evaluate daratumumab-induced fratricide, iNK cells and peripheral blood NK cells were cultured with increasing concentrations of daratumumab for 3 hours, and viability was assessed by flow cytometry with staining for 7-AAD and a fixable viability dye. Data is graphed as specific cytotoxicity (% specific death - % spontaneous death) / (1 - % spontaneous death) x 100. Results were compiled from 2 independent experiments. (H) In 3D tumor killing assays, RPMI-8826 cells were cultured in low binding plates for 2 days to form tumor spheroids. The indicated iNK cells were then added at a 2:1 ratio with or without daratumumab. After 5 days, cultures were disrupted, and the remaining viable tumor and iNK cell numbers were quantified by flow cytometry. Results are representative of 2 independent experiments. Data are represented as mean ± SEM.
To determine whether elevated levels of these metabolites were associated with increased metabolic activity, we performed metabolic flux assays to measure mitochondrial oxidative phosphorylation and glycolysis. We observed higher oxygen consumption rates (OCR) and extracellular acidification rates (ECAR) for hnCD16/CD38KO iNK cells relative to hnCD16 iNK cells (Figure 2E), indicative of enhanced metabolic activity. Basal respiration, ATP-linked respiration, and maximum respiration were all higher for hnCD16/CD38KO iNK cells, and a statistical trend towards higher spare respiratory capacity was also observed. Additionally, hnCD16/CD38KO iNK cells exhibited higher rates of glycolysis and a trend toward higher glycolytic reserve (Figure S1). Of note, we previously reported elevated OCR and ECAR for adaptive NK cells relative to canonical NK cells (Cichocki et al., 2018). To determine whether CD38 knockout impacted oxidative stress in iNK cells, hnCD16 iNK cells and hnCD16/CD38KO iNK cells were cultured in media alone, 50 μM H2O2, or 100 μM H2O2. Cells were then analyzed by flow cytometry using MitoSox dye, which emits fluorescence upon oxidation by mitochondrial superoxide. Compared to hnCD16 iNK cells, hnCD16/CD38KO iNK cells exhibited markedly lower mitochondrial superoxide as evidenced by lower frequencies of MitoSox+ cells at both hydrogen peroxide concentrations tested (Figure 2F).
Previous studies suggested a role for CD38 in supporting NK cell degranulation and cytokine release through calcium mobilization (Deaglio et al., 2002; Sconocchia et al., 1999). However, we found no impact of CD38 knockout in calcium flux assays testing iNK cell responses to ionomycin or CD16 crosslinking (Figure S2). To investigate the effect of CD38 knockout in preventing daratumumab-mediated iNK cell fratricide, we performed specific cytotoxicity assays where non-transduced iNK cells, hnCD16 iNK cells, hnCD16/CD38KO iNK cells, and primary NK cells were cultured with daratumumab at a range of concentrations between 0 and 30 μg/ml. The addition of daratumumab to primary NK cells and hnCD16 iNK cells resulted in specific cytotoxicity in a dose-dependent manner. Daratumumab had a minimal effect on non-transduced iNK cells that express CD16 at lower frequencies. As anticipated, no daratumumab-mediated cytotoxicity was observed for hnCD16/CD38KO iNK cells (Figure 2G).
To assess how fratricide impacts MM cell killing, we employed co-culture assays using RPMI-8226 spheroids. RPMI-8226 myeloma cells were added to wells in tissue culture plates and left to form spheroids. Non-transduced iNK cells, hnCD16 iNK cells, and hnCD16/CD38KO iNK cells were then added with or without daratumumab. Five days later, wells were harvested, and the absolute numbers of iNK cells and RPMI-8226 myeloma cells were determined by flow cytometry. Daratumumab-mediated fratricide was evident in wells containing hnCD16 iNK cells, while no fratricide and superior on-target ADCC was observed in wells containing hnCD16/CD38KO iNK cells (Figure 2H). Together, these data show that knockout of CD38 in iNK cells results in favorable metabolic alterations mirroring those observed in adaptive NK cells and protects against daratumumab-mediated fratricide without compromising ADCC.
CD38 knockout results in elevated levels of metabolites associated with glycolysis and cysteine metabolism
To gain a deeper understanding of the metabolic effects associated with CD38 knockout in iNK cells, we performed a comprehensive evaluation of principle metabolites from expanded peripheral blood NK cells, hnCD16 iNK cells, and hnCD16/CD38 KO iNK cells using mass spectrometry. Corroborating our colorimetric assay results, we observed significantly higher NAD+ levels in hnCD16/CD38KO iNK cells. Two intermediates of the glycolysis pathway, 3-phosphoglycerate and phosphoenolpyruvate were markedly higher in hnCD16/CD38KO iNK cells (Figure 3A), consistent with the increased rates of glycolysis observed in metabolic flux assays (Figure 2E). Metabolites selectively enriched in hnCD16/CD38KO iNK cells also included the amino acids cysteine and homocysteine (Figure 3A), which are components of a metabolic pathway leading to cysteine-glutathione disulfide (L-CySSG) formation.
Figure 3. hnCD16/CD38KO iNK cells exhibit elevated concentrations of glycolytic and antioxidant metabolites.

Expanded primary peripheral blood NK cells (n = 7), hnCD16 iNK cells (n = 3) and hnCD16/CD38KO iNK cells (n = 3) were analyzed by mass spectrometry to assess concentrations of key metabolites. Select data are represented in heat map (left) and column form (right). (B) NK cells isolated from peripheral blood by negative selection and (C) non-transduced iNK cells were cultured overnight in media alone or in media containing 50 μM L-CySSG. Cells were then cultured with or without 50 μM H2O2 for 1 hour. Shown are FACS plots of MitoSox dye fluorescence from a representative experiment (left) and cumulative data from 2 independent experiments (right). Statistical significance was determined by paired Student’s t-tests. Data are represented as mean ± SEM. *p < 0.05, **p < 0.01
L-CySSG is a prodrug of glutathione that maintains redox homeostasis and cellular antioxidant levels (Berkeley et al., 2003; Wang and Cynader, 2002). Very high levels of L-CySSG were observed in hnCD16/CD38KO iNK cells relative to hnCD16 iNK cells and expanded peripheral blood NK cells (Figure 3A). To determine whether L-CySSG could protect NK cells against hydrogen peroxide-induced oxidative stress, we isolated peripheral blood NK cells and cultured them overnight in media alone or media containing L-CySSG. Cells were then cultured with or without H2O2 for 1 hour. NK cells pre-incubated with L-CySSG exhibited an ~30% decrease in mitochondrial superoxide frequencies (Figure 3B). Similar results were observed when these assays were performed with non-transduced iNK cells (Figure 3C). Together, these data show that CD38 knockout influences glycolysis and cysteine metabolism. Furthermore, NK cells lacking CD38 have elevated levels of L-CySSG, which may contribute to their relative resistance to oxidative stress.
Expression of a membrane-bound IL-15/IL-15R fusion protein in iNK Cells leads to genome-wide transcriptional alterations that mirror adaptive NK cells
Interleukin (IL)-15 is required for NK cell survival (Cooper et al., 2002; Ranson et al., 2003). We reasoned that transgenic expression of an IL-15/IL-15R fusion protein could provide signals to enhance iNK cell function and persistence. To this end, we transduced hnCD16/CD38KO iPSCs with a membrane-bound IL-15/IL-15R fusion construct (IL-15RF) and differentiated these cells along the NK cell lineage to generate hnCD16/CD38KO/IL-15RF iNK cells. We first performed single cell RNA-seq (scRNA-seq) to compare global gene expression between non-transduced, hnCD16/CD38KO, and hnCD16/CD38KO/IL-15RF iNK cells. As anticipated, given the clonal nature of iNK cells, all three iNK cell lines clustered tightly with no distinct transcript clusters observed for any line (Figure S3). However, differential gene expression analyses showed that several transcripts encoding cytotoxic granule components and signaling molecules were elevated in hnCD16/CD38KO iNK cells and further elevated in hnCD16/CD38KO/IL-15RF iNK cells relative to non-transduced iNK cells (Figure S4).
We also performed Gene Ontology (GO) analyses and found that regulation of immune response, response to virus, and type 1 interferon signaling pathway were among the top enriched pathways in both hnCD16/CD38KO and hnCD16/CD38KO/IL-15RF iNK cells. Additionally, the T cell receptor signaling pathway, adaptive immune response, and T cell activation pathways were enriched in hnCD16/CD38KO/IL-15RF iNK cells (Figure S4). Motivated by our observation that adaptive immune response genes were enriched in hnCD16/CD38KO/IL-15RF iNK cells, we reasoned that these cells may have a broader transcriptional program shared with adaptive NK cells from CMV seropositive individuals. To explore this further, we generated violin plots of top differentially expressed genes in hnCD16/CD38KO and hnCD16/CD38KO/IL-15RF iNK cells relative to non-transduced iNK cells (Figure 4A). The differences between expression levels of all genes shown were also statistically significant when comparing hnCD16/CD38KO iNK cells to hnCD16/CD38KO/IL-15RF iNK cells. scRNA-seq results were validated by both flow cytometry and quantitative RT-PCR (Figure S5). We then analyzed our previously published RNA-seq data comparing canonical and adaptive NK cell subsets sorted from the peripheral blood of CMV seropositive donors (Cichocki et al., 2018) to determine whether there were matching trends (Figure 4B). We found that many genes encoding proteins that dictate NK cell activation and effector function were highly induced in both hnCD16/CD38KO/IL-15RF iNK cells and adaptive NK cells. Genes that were both upregulated and downregulated in hnCD16/CD38KO/IL-15RF iNK cells followed similar patterns in adaptive NK cells (Figure S6). Given the metabolic and transcriptional similarities between hnCD16/CD38KO/IL-15RF iNK cells and adaptive NK cells, we’ve termed these iNK cells “iADAPT” NK cells.
Figure 4. hnCD16/CD38KO/IL-15RF iNK cells and CMV-induced adaptive NK cells share a set of highly upregulated transcripts.

(A) Violin plots of genes that were differentially expressed with statistical significance between hnCD16/CD38KO/IL-15RF iNK cells, hnCD16/CD38KO iNK cells, and non-transduced iNK cells in scRNA-seq analyses. (B) Analysis of the same genes in sorted canonical (CD3−CD56dimNKG2C−) and adaptive (CD3−CD56dimNKG2C+) NK cells from 4 donors analyzed by RNA-seq. Statistical significance was determined by paired Student’s t-test. *p < 0.05, **p < 0.01
iADAPT NK cells exhibit robust serial killing
To assess iADAPT NK cell serial target killing in the absence of exogenous cytokine, we performed sequential killing assays using expanded peripheral blood NK cells from 3 donors and iADAPT NK cells that were thawed from cryopreservation and co-cultured with MM.1R myeloma cells at various E:T ratios in the presence or absence of daratumumab. After 48 hours of live cell imaging, non-adherent cells were collected and transferred into wells containing fresh MM.1R targets for the next 48 hours of live cell imaging. After another 48 hours of live cell imaging, non-adherent cells were again collected and transferred into wells containing fresh MM.1R targets. Thus, effector cells were tested in 3 rounds of target cell killing. During round 1 of the assay, both peripheral blood NK cells and iADAPT NK cells exhibited comparable natural cytotoxicity and daratumumab-mediated ADCC in a dose dependent manner (Figure 5A). However, in round 2, iADAPT NK cells increased their natural cytotoxicity and ADCC, even at low E:T ratios, while peripheral blood NK cells from all three donors lost their ability to kill targets. These trends continued in round 3 of the assay, where only iADAPT iNK cells increased their cytotoxic function after multiple rounds of killing (Figure 5A). Importantly, iADAPT NK cells also raised their cytotoxicity index in round 2 and 3 in the absence of daratumumab (Figure 5B). Similar results were observed in serial killing assays comparing peripheral blood NK cells, non-transduced iNK cells, and iADAPT NK cells (Figure S7). Together, these data show that iADAPT NK cells mediate efficient innate cytotoxicity and serial killing.
Figure 5. iADAPT NK cells sustain natural cytotoxicity and ADCC after multiple rounds of killing.

Expanded peripheral blood NK cells from 3 donors and iADAPT NK cells were thawed and co-cultured with MM.1R myeloma cells transduced with NucLight Red at the indicated E:T ratios in the presence or absence of daratumumab. (A) Target cell killing was assessed over 48 hours by live cell imaging. The remaining effector cells in all wells were collected and transferred to wells containing fresh MM.1R cells. Cytotoxicity was assessed for another 48 hours (round 2). The remaining effector cells in all wells were harvested for a second time and transferred to wells containing fresh MM.1R cells (round 3). Cytotoxicity was assessed by live imaging for another 48 hours. (B) Calculated cytotoxicity index values for each assay condition in IncuCyte assays. All data are normalized to MM.1R myeloma cells alone. Results are representative of 2 independent experiments.
iADAPT NK cells mediate cellular cytotoxicity against primary MM and AML cells when combined with daratumumab
CD38 is highly and uniformly expressed on nearly all MM cells (de Weers et al., 2011), and the first clinical trials conducted with daratumumab as a single agent demonstrated that about 30-50% of patients with relapsed refractory myeloma respond to daratumumab (Usmani et al., 2016). To assess iADAPT NK cell killing of primary MM cells, a bone marrow aspirate was collected from a relapse, refractory MM patient. Cells in the aspirate were labeled with CellTrace dye and co-cultured overnight with expanded peripheral blood NK cells, non-transduced iNK cells, and iADAPT NK cells immediately thawed from cryopreservation at a 2:1 E:T ratio in the presence or absence of daratumumab. Cytotoxicity against primary MM cells was assessed by flow cytometry and analysis of the percentages of CellTrace+ cells expressing CD138, a large glycoprotein that is highly expressed on MM cells (Akhmetzyanova et al., 2020). Of the conditions tested, we found that iADAPT NK cells in combination with daratumumab led to the greatest reduction in the CD138+ MM population (Figure 6A).
Figure 6. iADAPT NK cells mediate ADCC and produce IFN-γ when challenged with primary MM and AML cells.

(A) CD138+ myeloma cells were isolated from a bone marrow aspiration of a patient with relapsed MM and labeled with CellTrace dye and co-cultured overnight at a 2:1 E:T ratio with expanded peripheral blood NK cells or iADAPT NK cells that were thawed from cryopreservation. The percentages of live MM cells, defined as CellTrace+ and CD138+ were assessed by flow cytometry. (B) Primary AML cells from two patients with > 90% blasts were thawed, labeled with CellTrace dye, and cultured overnight with or without ATRA. FACS histogram plots showing surface CD38 expression on HL-60 AML cells and primary, patient-derived AML cells after overnight culture with or without ATRA are shown. Tumor cells were co-cultured at a 2:1 E:T ratio overnight with expanded peripheral blood NK cells or iADAPT NK cells thawed from cryopreservation. The percentages of live (C) HL-60 AML cells and (D) primary, patient-derived AML cells remaining at the end of the co-culture period was determined by flow cytometry and gating on the viable, CellTrace+ population. Intracellular IFN-γ frequencies in expanded NK cells and iADAPT NK cells were also assessed by flow cytometry. Cumulative data for % specific killing and IFN-γ production is shown from primary AML experiments performed with 2 peripheral blood NK cell products and 2 iADAPT iNK cell products in 2 independent experiments. Statistical significance was determined by one-way ANOVA with multiple comparisons. Data are represented as mean ± SEM. *p < 0.05, **p < 0.01
CD38 is expressed on AML cells from ~75% of patients, but there is high variability in expression levels between patients (Williams et al., 2019). One strategy for increasing CD38 levels on AML cells to enhance antibody targeting is treatment with all-trans-retinoic acid (ATRA), which induces CD38 expression through activation of retinoic acid-alpha receptor (Drach et al., 1994). To determine whether iADAPT NK cell cytotoxicity against AML could be enhanced by combinatorial treatment with daratumumab and ATRA, we tested AML cell lines and primary AML cells. Culture overnight with ATRA resulted in a dramatic increase in CD38 expression on HL-60 AML cells and a more moderate increase in CD38 expression on primary AML cells (Figure 6B). HL-60 cells and AML blasts were labeled with CellTrace dye and used as targets in co-culture assays with expanded peripheral blood NK cells and iADAPT iNK cells thawed from cryopreservation. The addition of daratumumab alone or in combination with ATRA did not significantly impact peripheral blood NK cell cytotoxicity against AML cells from either patient. Additionally, peripheral blood NK cells did not produce interferon (IFN)-γ in response to AML targets, likely due to a failure of these cells to recover inflammatory cytokine production after the freeze/thaw cycle. In contrast, iADAPT NK cells co-cultured with HL-60 cells (Figure 6C) and primary AML cells (Figure 6D) exhibited increased cytotoxicity and IFN-γ production, which was further enhanced with the addition of daratumumab. The addition of ATRA most notably improved killing of HL-60 cells where it induced the most profound increase in CD38 surface expression resulting in effective elimination by iADAPT NK cells combined with daratumumab (Figure 6B and 6C). Similar results were observed using the AML cell line THP-1 (Figure S7). Thus, these data demonstrate the potential of using iADAPT NK cells alone or in combination with daratumumab and ATRA for the treatment of AML.
iADAPT NK cells persist in vivo and display robust in vivo antitumor responses against MM and AML
Next, we sought to assess the persistence of iADAPT NK cells in vivo. To this end, we intravenously (i.v.) injected NOD.Cg-PrkdcscidIl2rgtm1Wj1/SzJ (NSG) mice with 1.2x107 expanded peripheral blood NK cells or iADAPT NK cells thawed from cryopreservation on days 1, 8, and 15. The cell dose and dosing schedule were chosen to mimic a proposed clinical trial to test the safety and efficacy of iADAPT NK cells in patients with advanced cancer. Blood draws were performed weekly, and human CD45+CD56+CD16+ NK cell numbers were quantified by flow cytometry. Peripheral blood NK cell numbers peaked at approximately 1-3 cells per μl and less than 0.02 percent of the blood population after the final dose and then quickly declined, which was expected given the lack of cytokine support (Figure 7A, B). In contrast, iADAPT NK cells peaked at nearly 100 cells per μl and greater than 20 percent of the blood population and persisted at markedly higher levels for 4 weeks after the last injection before declining to low, but still detectable levels at day 43 (Figure 7A, B).
Figure 7. iADAPT NK cells mediate robust antitumor function in vivo.

(A) To assess persistence, NSG mice were injected with 1.2x107 expanded peripheral blood NK cells or iADAPT NK cells thawed from cryopreservation on days 1, 8, and 15. Mice were bled on days 8 (before second NK injection), 15 (before 3rd NK injection), 16, 22, 29, 36, and 43. (B) peripheral blood NK cells and iADAPT NK cells were identified by flow cytometry as hCD45+hCD56+hCD16+mCD45− cells. To assess antitumor function, NSG mice were engrafted with 1.5x106 HL-60 cells transduced with the firefly luciferase gene. After 4 days, groups of mice (5 mice per group) received no treatment (HL-60 alone), 3 infusions of thawed expanded peripheral blood NK cells, hnCD16/CD38KO iNK cells, hnCD16/IL-15RF iNK cells, iADAPT NK cells. Each dose consisted of 1x107 cells, and mice were treated weekly for 3 weeks. Bioluminescent imaging (BLI) was performed weekly to track tumor burden. Shown are (C) raw BLIs and (D) quantification of BLI data through day 28. (E) NSG mice were engrafted with 5x105 luciferase-expressing MM.1S cells. After 3 days, groups of mice (4 mice per group) received no treatment (MM.1S alone), 3 infusions of daratumumab alone, 3 infusions of iADAPT NK cells alone, or 3 infusions of iADAPT NK cells in combination with daratumumab. Each dose consisted of 1x107 cells, and mice were treated weekly for 3 weeks. Bioluminescent imaging (BLI) was performed weekly to track tumor burden. (F) Quantification of BLI data through day 35. (G) Area under the curve (AUC) data quantified from BLI imaging. AUC values were statistically significant for the daratumumab alone and hnCD16/CD38KO/IL-15R fusion iNK cells + daratumumab groups relative to the MM.1S alone group. Data is representative of 3 independent experiments. P values were determined by one-way ANOVA. Data are represented as mean ± SD. *p < 0.05, **p < 0.01, ****p < 0.0001
To compare the in vivo antitumor activity of iADAPT NK cells to iNK cells without the full complement of genetic modifications present in iADAPT NK cells, we set up a disseminated xenogeneic AML model. Human HL-60 cells transduced with the gene encoding firefly luciferase were injected i.v. into NSG mice that did not receive any exogenous cytokine support traditionally provided in NK cell xenograft models. Groups of mice then received hnCD16/IL-15RF iNK cells, hnCD16/CD38KO iNK cells or iADAPT NK cells (3 doses of cells immediately thawed from cryopreservation). Tumor bioluminescence (BLI) was measured weekly for 4 weeks. While hnCD16/IL-15RF iNK cells and hnCD16/CD38KO iNK cells only displayed modest control of tumor growth, iADAPT iNK cells mediated strong antitumor effects without the need for cytokine support (Figure 7C, D).
To evaluate iADAPT NK cell in vivo antitumor function in combination with daratumumab, we employed a more aggressive xenogeneic MM model. Human MM.1S cells expressing firefly luciferase were injected i.v. into NSG mice. After allowing the tumor to establish, groups of mice received daratumumab alone, iADAPT NK cells alone (3 doses of cells immediately thawed from cryopreservation), or daratumumab plus iADAPT NK cells. Tumor BLI was assessed weekly for 5 weeks (Figure 7E). Consistent with previous studies, daratumumab alone demonstrated single agent activity in immunodeficient NSG mice (De Weers et al., 2011). As expected, the administration of iADAPT NK cells alone did not impact tumor progression in this xenogeneic model. However, the combination of iADAPT NK cells and daratumumab had a potent antitumor effect, with a 99% reduction in the area under the curve (AUC) calculated from BLI measurements compared to untreated tumor-bearing mice (3.9x107 vs. 8x1010 photons/sec.) and an 89% reduction in AUC compared to daratumumab alone (3.9x107 vs. 3.5x108 photons/sec.) (Figure 7F, G). Together, these data show that iADAPT NK cells mediate robust, durable in vivo antitumor activity without the need for exogenous cytokine support. These cells represent an effective treatment strategy alone or in combination with daratumumab.
Discussion
We use the term ‘adaptive’ in reference to the unique subsets of NK cells that arise in response to CMV. Adaptive NK cells are phenotypically diverse (Schlums et al., 2015; Lee et al., 2015), have a genome-wide epigenetic profile that parallels cytotoxic effector CD8+ T cells (Schlums et al., 2015), and are long-lived (Schlums et al., 2017). Functionally, adaptive NK cells excel at ADCC and IFN-γ production (Schlums et al., 2015; Lee et al., 2015; Luetke-Eversloh et al., 2014). Metabolically, adaptive NK cells exhibit elevated mitochondrial oxidative phosphorylation and glycolysis, as well as increased levels of ATP (Cichocki et al., 2018). Given these unique characteristics, there is interest in the question of whether adaptive NK cells could be exploited for cancer immunotherapy. Support for this idea comes from studies of immune reconstitution after allogeneic hematopoietic cell transplant (HCT) where there is an association between adaptive NK cell expansion in response to CMV reactivation and relapse protection (Cichocki et al., 2016; Cichocki et al., 2019).
Here, we describe an iPSC-derived NK cell product termed iADAPT NK with multiple customized attributes that can be produced in compliance with good manufacturing practices for off-the-shelf immunotherapy. We show that iADAPT NK cells share metabolic and transcriptional features with adaptive NK cells. These iNK cells exhibited enhanced serial killing and in vivo persistence in the absence of exogenous cytokines. The persistence of these iADAPT NK cells at high levels in peripheral blood for several weeks after adoptive transfer in the absence of exogenous cytokine is of considerable importance to the use of these cells for immunotherapy. Traditionally, infusions of either IL-2 or IL-15 are required to support the expansion and persistence of adoptively transferred peripheral blood NK cells (Miller et al., 2005; Cooley et al., 2019). However, there can be counterproductive consequences related to the infusion of these cytokines. IL-2 is a strong mitogen for T regulatory cells that can inhibit NK cell antitumor efficacy (Bachanova et al., 2014). IL-15 may be preferable given that it does not expand T regulatory cells, but it activates and expands cytotoxic CD8+ T cells that can mediate rejection of the NK cell graft (Cooley et al., 2019). The autonomous persistence of hnCD16/CD38KO/IL-15RF iNK cells in vivo obviates the need for cytokine dosing, which reduces treatment cost and avoids unwanted immunoregulatory responses.
iADAPT NK cells can be combined with daratumumab to trigger ADCC and IFN-γ production in response to cancer cells lines and patient-derived MM and AML cells. Moreover, induction of CD38 expression by ATRA could further enhance iADAPT NK cell ADCC against AML cells. Importantly, our xenogeneic adoptive transfer experiments show that iADAPT NK cells mediate superior tumor control as a monotherapy relative to iNK cells that do not have the full complement of genetic modifications. In adoptive transfer experiments with engrafted MM.1S cells, which are resistant to NK cell natural cytotoxicity, iADAPT NK cells mediated strong in vivo antitumor function when combined with daratumumab. iADAPT NK cells represent an attractive off-the-shelf source of cells for multiple cancer immunotherapy indications. Taken together, the preclinical data presented here supports further testing of iADAPT NK cells in FDA-approved phase I clinical trials to treat patients with advanced cancer (NCT04614636).
Limitations of study
While we were able to clearly demonstrate the negative impact of daratumumab-mediated NK cell fratricide on cytotoxic function and the mitigation of this effect by CD38 knockout in vitro, we were unable to definitively confirm the antitumor benefit of eliminating NK cell fratricide through CD38 knockout in vivo. This may be due to the effectiveness of daratumumab despite its NK cell depleting activity, which has been reported clinically (Casneuf et al., 2017). Alternatively, it may be due to limitations of the xenogeneic model. Additionally, high doses of cryopreserved iNK cells were used in this study to achieve in vivo antitumor efficacy relative to typical CAR T cell doses. This may be due to the expression of inhibitory receptors on iNK cells. While this is a potential drawback, we have previously shown that iNK cells exhibit a high level of proliferation during the differentiation and expansion process resulting in 1x106-fold expansions (Cichocki et al., 2020). Our scaled-up manufacturing process can generate billions of cells from a single production run, accommodating multiple dosing for patients enrolled in trials. The phase I clinical trial will monitor for any potential adverse effects associated with dose escalation of iNK cells.
STAR Methods
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Frank Cichocki (cich0040@umn.edu).
Materials availability
The study did not generate new unique reagents.
Data and code availability
Raw and processed single cell RNA-seq datasets generated in this study are available through the Gene Expression Omnibus repository hosted by the National Center for Biotechnology Information under the accession number GSE168936.
Experimental model and subject details
Animals
NOD.Cg-PrkdcscidIl2rgtm1Wj1/SzJ mice (Jackson Laboratories) were housed in the institution’s Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)-accredited animal care facility, which is in a dedicated building on the University of Minnesota campus. Autoclaved specific pathogen-free cages were in one of two 300 square foot rooms. In addition to oversight by a board-certified laboratory animal veterinarian, all animal technicians are rigorously trained and certified. Microbiological, clinical pathological, and necropsy diagnostic facilities are available on site. Experiments were balanced by sex. All experiments were reviewed and approved by the University of Minnesota Institutional Animal Care Committee (IACUC) under the protocol 1907-37257A.
Cell lines
MM.1S, MM.1R, RPMI-8226, HL-60, THP-1, K562, and OP9 cell lines were obtained from the American Tissue Culture Collection (ATCC). All cell lines were cultured in RPMI 1640 (Corning) supplemented with 10% fetal bovine serum (Hyclone) and penicillin/streptomycin. Cells were kept at low passage and maintained at 37°C and 5% CO2 and tested for mycoplasma by PCR monthly.
Method details
iNK cell culture and phenotyping
Human iPSC differentiation to iCD34+ cells, differentiation along the NK cell lineage, and expansion of the NK cell population were performed as previously described (Cichocki et al., 2020, Valamehr et al., 2014; Valamehr et al., 2012; Tsutsui et al., 2011). Briefly, in most cases iPSCs were differentiated towards the mesoderm and CD34+ hematopoietic progenitor stages in StemPro34 media (Thermo Fisher) supplemented with BMP4 (Life Technologies), bFGF (Life Technologies). CD34+ cells were subsequently enriched prior to differentiation into iNK cells. At the beginning of the iNK cell differentiation culture, iCD34+ cells were plated on OP9 cells in B0 media (Cichocki et al., 2010) to support NK cell differentiation from hematopoietic progenitors. After 20-30 days of culture, iNK cells were harvested and co-cultured with irradiated K562 cells transduced with membrane-bound IL-22 and 4-1BB ligand (4-1BBL) constructs. In supplemented B0 media for 2-to-4 weeks.
iADAPT iPSCs were created by CRISPR-mediated targeted integration. The donor plasmid contained IL-15RF and hnCD16 in a 2A peptide-connected bi-cistronic expression cassette flanked by two homology arms to facilitate the targeted integration at the CD38 locus. IL-15RF was constructed by combining IL-15 (GenBank accession # NM_000585) and IL-15 receptor alpha (GenBank accession # 002189), and hnCD16 was constructed as described previously (Jing et al., 2015). To generate iADAPT NK cells, AsCpf1 nuclease (Aldevrong), a CD38-targeting gRNA (target sequence: TCCCCGGACACCGGGCTGAAC), and the donor plasmid were combined with iPSCs and transfected using a Neon electroporation device (Thermo Fisher) following the manufacturer’s protocol. Once electroporated, cells were plated and clones were screened by sequencing. Individual clones were selected as iADAPT iPSCs. To generate hnCD16/CD38KO iPSCs, iPSCs were first transfected with the CD38-targeting gRNA described above. iPSC clones were screened by sequencing, and the selected CD38KO clone was transduced with lentivirus containing hnCD16. To generate hnCD16/IL-15RF iPSCs, iPSCs were engineered by sequential transduction of lentivirus containing IL-15RF and lentivirus containing hnCD16, respectively. All engineered iPSCs were maintained in media consisting of DMEM/F12 (Mediatech), 20% v/v knockout serum replacement (Invitrogen), 1% v/v non-essential amino acids (Mediatech), 2 mM L-glutamine (Mediatech), 100 mM β-mercaptoethanol (Invitrogen), 10 ng/mL bFGF (Invitrogen), and the small molecules PD0325901, CHIR99021, Thiazovivin, and SB431542 (all Biovision). For single-cell dissociation, iPSCs were washed once with phosphate buffered saline (Mediatech) and treated with Accutase (Millipore) at 37°C followed by pipetting to ensure single cell dissociation. The single-cell suspension was then processed for continued culture on Matrigel or induced to differentiation into iNK cells. The efficacy of CD38 knockout was determined using a standard T7 endonuclease assay (New England Biolabs). The following fluorescently conjugated antibodies were used for phenotypic analysis of iNK cells by flow cytometry: anti-CD56 (HCD56), anti-CD38 (HIT2), and anti-CD16 (3G8). All antibodies were purchased from BioLegend. Flow cytometry was performed on an LSR II instrument (BD Biosciences). Flow cytometry data was analyzed with FlowJo software (v10.7.1) (BD).
Western blot
iNK cells were lysed with radioimmunoprecipitation assay (RIPA) buffer containing a protease inhibitor cocktail (Sigma Aldrich). 10 μg of total protein was migrated through a 7.5% bis/acrylamide gel. Protein was then transferred to PVDF membranes using the iBlot system (Invitrogen). Membranes were blocked with 5% albumin or 5% milk and incubated overnight at 4°C with antibodies specific for CD38 (14637S; Cell Signaling) and β-actin (sc-47778; Santa Cruz) followed by incubation with a horseradish peroxidase-conjugated secondary antibody (Cell Signaling). Chemiluminescence was detected with Pico and Femto substrate (Thermo Fisher), and images were acquired with a BioLite Xe (UVP).
Calcium flux analysis
iNK cells were stained with Indo-1 AM (Thermo Fisher) per the manufacturer’s protocol and co-stained with CD56 and a fixable live/dead indicator dye (Invitrogen). For stimulation through CD16, cells were pre-coated with an anti-CD16 antibody (3G8; BioLegend). Cells were run for 30 seconds on a FACS Fortessa X-30 (BD Biosciences) to obtain baseline measurements of free and bound calcium by measuring the shift in emission spectra. Then, goat anti-mouse IgG f(ab’)2 (Jackson ImmunoResearch Laboratories) was added as a crosslinking agent, and cells were immediately returned to the flow cytometer. Data was collected for an additional 4 minutes. For inonomycin stimulation, non-antibody coated iNK cells were first run unstimulated for 30 seconds followed by addition of ionomycin to achieve a final concentration of 1 μM, then run for an additional 4 minutes. Calcium flux was calculated based on the ratio of free and bound calcium over time.
Metabolic assays
Seahorse assays were performed according to the manufacturer’s instructions with modifications to simultaneously analyze glycolysis and oxidative mitochondrial metabolism using the Seahorse XF Glycolysis Stress Test Kit and the Seahorse XF Cell Mito Stress Kit (Agilent Technologies). Briefly, iNK cells were washed and resuspended in glucose-free media (Gibco). 1.5x105 cells were plated per well in triplicate and analyzed with a Seahorse Xfe96 Analyzer (Agilent Technologies). Glucose, oligomycin, FCCP, sodium pyruvate, rotenone, and antimycin A were serially injected to measure metabolic function. SRC measurements were calculated as average maximal OCR values minus average basal OCR values. ATP-linked respiration was calculated as average basal OCR values minus average post-oligomycin values. Glycolysis was calculated as average post-glucose ECAR values minus average basal ECAR values. Glycolytic reserve was calculated as average maximal ECAR values minus post-glucose ECAR values. For ATP quantification assays, 1x105 iNK cells per well were analyzed using the ATP Bioluminescence Assay Kit HS II (Sigma Aldrich) and analyzed with an Infinite M200 PRO Luminometer (Tecan). NAD+ and NADH concentrations were quantified using the NAD/NADH Cell-Based Assay Kit (Cayman Chemical) as per the manufacturer’s instructions and analyzed with an Infinite M200 PRO Luminometer. For analyses of oxidative stress, iNK cells were cultured with hydrogen peroxide (Sigma Aldrich) for 1 hour. Following treatment, cells were cultured in serum-free media containing 5 mM MitoSox Indicator Dye (Thermo Fisher). Cells were then washed and counter stained with anti-CD56 antibody and fixable viability dye for flow cytometry analysis. For mass spectrometry analysis of metabolites, iNK cells and expanded peripheral blood NK cells were snap frozen in liquid nitrogen and sent to Metabolon (Durham, NC).
iNK cell fratricide assays
Peripheral blood NK cells and iNK cells were cultured for 3 hours in the presence of daratumumab (Janssen) at concentrations ranging from 0-30 μg/ml. Cells were then stained with a fluorescently conjugated anti-CD56 antibody, fixable viability dye, and 7-AAD (Thermo Fisher) for flow cytometry analysis. NK cells were gated based on CD56 expression, and viable cell percentages were determined based on exclusion of the dead cell dye and 7-AAD staining. Specific cytotoxicity was calculated as (% specific death - % spontaneous death) / (1- % spontaneous death) x 100.
3-dimensional tumor spheroid cytotoxicity assays
1x104 RPMI-8226 myeloma cells transduced with NucLight Red (Sartorius) were seeded into 96-well ultra-low binding plates (Corning). Cells were cultured for 48-72 hours to allow for spheroid formation. Next, 4x104 iNK cells were gently added to each well with or without daratumumab at a final concentration of 10 μg/ml, and cells were co-cultured for 5 days. At the end of the culture, cells in each well were disrupted into a single cell suspension and stained with a fluorescently conjugated CD56 antibody and fixable viability dye for flow cytometry analysis. Tumor cells were quantified based on NucLight Red, and iNK cells were quantified based on CD56 expression.
RNA-seq and single cell RNA-seq analyses
RNA-seq on subsets of canonical and adaptive NK cells from CMV seropositive donors was performed as previously described (Cichocki et al., 2018). The RNA-seq data can be found under the GEO accession number GSE117614. For scRNA-seq experiments, iNK cells were washed in DPBS + 0.04% BSA and submitted to the University of Minnesota Genomics Center (UMGC) for sample preparation and sequencing. Briefly, we used the Chromium Single Cell 3’ Reagent Kit (v3 Chemistry) for library preparation (10X Genomics). Libraries were sequenced on a NovaSeq 6000 (Illumina) with a sequencing depth of at least 5x104 reads per cell. Raw FASTQ files were processed with CellRanger software v4.0.0 (10X Genomics) for read alignment, filtering, barcode counting, and unique molecular identifier (UMI) counting. Reads were simultaneously aligned and mapped to the GRch38 human genome sequence. Low quality cells were excluded during the initial quality control step by removing cells with fewer than 1,500 gene expressed and cells expressing more than 7,500 genes. Cells with more than 20% mitochondria-associated genes were also removed. Log-normalization was performed on gene expression values for each barcode by scaling the total number of transcripts and multiplying by 1x103. Values were log-transformed for further downstream analysis. scRNA-seq data can be found under the GEO accession number GSE168936.
Isolation and expansion of peripheral blood NK cells
De-identified Trima cones were obtained from the Memorial Blood Center (Saint Paul, MN). Mononuclear cells were isolated by density gradient centrifugation using Ficoll-HiPaq (GE Healthcare). NK cells were then enriched using EasySep Human NK Cell Enrichment Kits (StemCell Technologies) per the manufacturer’s instructions. For ex vivo expansion, peripheral blood NK cells were co-cultured with irradiated, gene-modified K562 cells for 14 days in B0 media with 250 U/ml IL-2.
In vitro assays to assess cytotoxicity against AML cell lines, primary AML cells, and primary MM cells
Primary AML cells, HL-60 cells, and THP-1 cells were cultured in the presence or absence of 1 μM all-trans retinoic acid (Sigma Aldrich) overnight. The following day, targets were removed from culture, counted, and labeled with CellTrace Violet (Thermo Fisher) per the manufacturer’s instructions. Labeled targets were then co-cultured with expanded peripheral blood NK cells or iADAPT NK cells thawed immediately from cryopreservation at a 2:1 E:T ratio in the presence or absence of daratumumab (10 μg/ml) for 5 hours. Cells were then stained for Live/Dead Near IR (Thermo Fisher) and analyzed by flow cytometry. A fresh bone marrow aspirate was obtained from a relapsed MM patient, and cells were stained with CellTrace Violet and a fluorescently conjugated antibody against CD138 (281-2; Biolegend). Bone marrow aspirate cells were then co-cultured with expanded peripheral blood NK cells, non-transduced iNK cells, or iADAPT iNK cells in the presence or absence of daratumumab (10 μg/ml) overnight. The percentages of live CD138+ MM cells remaining in each culture was determined by flow cytometry. Percent specific killing was calculated with the formula (1-[% live tumor cells in the NK cell co-culture condition/% live tumor cells in the tumor alone condition]) x 100.
In vivo xenogeneic adoptive transfer experiments
For the HL-60 model, 25 six-to-eight-week-old NSG mice were injected i.v. with 1.5x106 luciferase-expressing HL-60 cells. After allowing the tumor to engraft for 4 days, bioluminescence imaging was performed to quantify tumor burden and balance mice evenly into 5 groups. Groups of mice received either no treatment or 3 i.v. doses (once per week for 3 weeks) of 1x107 expanded peripheral blood NK cells, hnCD16/CD38KO iNK cells, hnCD16/IL-15RF iNK cells or iADAPT NK cells immediately thawed from cryopreservation. Bioluminescence imaging was performed weekly to monitor tumor progression, and mice were sacrificed when they showed signs of morbidity. For in vivo experiments with MM.1S cells, six-to-eight-week-old NSG mice were injected i.v. with 5x105 luciferase-expressing MM.1S cells. After allowing the tumor to engraft for 3 days, bioluminescence imaging was performed to quantify tumor burden and balance mice evenly into 4 groups. Groups of mice received daratumumab alone (8 mg/kg), 3 i.v. doses (once per week for 3 weeks) of 1x107 iADAPT NK cells immediately thawed from cryopreservation, or 3 i.v. doses of iADAPT NK cells in combination with daratumumab. Bioluminescence imaging was performed weekly to monitor tumor progression. Imaging was conducted using an IVIS Spectrum, and images were analyzed using Live Imaging Software (Perkin Elmer). All studies were performed under an approved protocol by the Institutional Animal Care and Use committee of the University of Minnesota.
Quantification and statistical analysis
Statistics for differences in CD38 levels on primary NK cell subsets and the percentages of viable NK cells after H2O2 challenge were determined by one-way ANOVA with multiple comparisons. Statistics for differences in NAD+, NADH, and ATP concentrations in NK cell subsets and iNK cells were determined by paired Student’s t-test. Statistics for MitoSox dye fluorescence in NK cells challenged with H2O2 and differences between metabolite levels between iNK cell lines were determined by paired Student’s t-test. One-way ANOVA with multiple comparisons was used to determine statistical significance in assays testing iNK cell and expanded primary NK cell function against tumor cell lines and primary AML cells. Statistical significance for xenogeneic adoptive transfer experiments were determined by one-way ANOVA with multiple comparisons. All relevant quantifications were analyzed by GraphPad Prism, and error bars indicate mean ± SEM. Details of “n” values describing the number of experimental repeats or mice are provided in the figure legends.
Supplementary Material
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Daratumumab | Janssen | N/A |
| Rabbit polyclonal anti-CD38 | Cell Signaling Technology | Cat # 14637S; RRID: AB_2798550 |
| Mouse monoclonal anti-β-Actin | Santa Cruz Biotechnology | Cat # sc-47778; RRID: AB_2714189 |
| Horse monoclonal anti-mouse IgG, HRP-linked | Cell Signaling Technology | Cat # 7076; RRID: AB_330924 |
| Goat monoclonal anti-rabbit IgG, HRP-linked | Cell Signaling Technology | Cat # 7074; RRID: AB_2099233 |
| Goat anti-mouse AffiniPure F(ab’)2 Fragment | Jackson ImmunoResearch Laboratories | Cat # 115-006-003; RRID: AB_2338466 |
| Mouse monoclonal anti-CD45 | BioLegend | Cat # 304008; RRID: AB_314396 |
| Mouse monoclonal anti-CD3 | BioLegend | Cat # 317330; RRID: AB_11219196 |
| Mouse monoclonal anti-CD19 | BioLegend | Cat # 363016; RRID: AB_2564207 |
| Rat monoclonal anti-CD4 | BioLegend | Cat # 357414; RRID: AB_2565666 |
| Mouse monoclonal anti-CD8 | BioLegend | Cat # 344704; RRID: AB_1877178 |
| Mouse monoclonal anti-CD14 | BioLegend | Cat # 325606; RRID: AB_830679 |
| Mouse monoclonal anti-CD56 | BioLegend | Part: 92189 (custom order) |
| Mouse monoclonal anti-NKG2C | R&D Systems | Cat # FAB138P; RRID: AB_2132983 |
| Mouse monoclonal anti-CD16 | BioLegend | Cat # 302006; RRID: AB_314206 |
| Mouse monoclonal anti-IFN-γ | BioLegend | Part: 93705 (custom order) |
| Mouse monoclonal anti-Perforin | BioLegend | Cat # 308106; RRID: AB_314704 |
| Mouse monoclonal anti-Granzyme B | BioLegend | Cat # 372206; RRID: AB_2687030 |
| Mouse monoclonal anti-Granzyme K | BioLegend | Cat # 370507; RRID: AB_2632845 |
| Mouse monoclonal anti-CD247 | BioLegend | Cat # 644105; RRID: AB_2565720 |
| Bacterial and virus strains | ||
| Incucyte NucLight Red Lentivirus Reagent (EF1α, Puro) | Sartorius | Cat # 4625 |
| Biological samples | ||
| Patient AML cells | Translational Therapy Laboratory – University of Minnesota | http://cancer.umn.edu/translational-therapy-shared-resource |
| Patient multiple myeloma cells | Clinics and Surgery Center (CSC) – University of Minnesota | http://ctsi.umn.edu/about/programs-initiatives/research-services-ctrs/clinics-and-surgery-center-csc |
| Trima cones (human donor peripheral blood) | Memorial Blood Center – Saint Paul, MN | http://mbc.org |
| Chemicals, peptides, and recombinant proteins | ||
| Retinoic acid | Sigma Aldrich | Cat # 302-79-4 |
| Hydrogen peroxide | Sigma Aldrich | Cat # 7722-84-1 |
| Indo-1, AM, cell permeant | Thermo Fisher | Cat # I1223 |
| Protease Inhibitor Cocktail | Sigma Aldrich | Cat # P8340-1ML |
| SuperSignal West Pico PLUS Chemiluminescent Substrate | Thermo Fisher | Cat # PI34577 |
| SuperSignal West Femto Maximum Sensitivity Substrate | Thermo Fisher | Cat # PI34096 |
| T7 Endonucleaase I | New England Biolabs | Cat # M0302 |
| MitoSOX Red Mitochondrial Superoxide Indicator | Thermo Fisher | Cat # M36008 |
| 7-AAD (7-Aminoactinomycin D) | Thermo Fisher | Cat # A1310 |
| Annexin V | Thermo Fisher | Cat # 556419 |
| LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit | Thermo Fisher | Cat # L34976 |
| CellTrace Violet Cell Proliferation Kit | Thermo Fisher | Cat # C34557 |
| L-Cysteine-glutathione disulfide | Cayman Chemical | Cat # 17582 |
| Critical commercial assays | ||
| Power SYBR Green Master Mix | Thermo Fisher | Cat # 4367659 |
| Chromium Single Cell 3’ Reagent Kit (v3 Chemistry) | 10X Genomics | Cat # PN-1000075 |
| EasySep NK Cell Enrichment Kit | StemCell Technologies | Cat # 19055 |
| ATP Bioluminescence Assay Kit HS II | Sigma Aldrich | Cat # 11699709001 |
| NAD/NADH Cell-Based Assay Kit | Cayman Chemical | Cat # 600480 |
| Seahorse XFp Cell Mito Stress Test Kit | Agilent | Cat # 103015-100 |
| Seahorse XFp Glycolysis Stress Test Kit | Agilent | Cat # 103020-100 |
| Anti-PE-MicroBeads | Miltenyi Biotech | Cat # 130-048-801 |
| Deposited data | ||
| Raw and analyzed single cell RNA-seq data | This paper | GSE168936 |
| Experimental models: Cell lines | ||
| Engineered K562 cells (mbIL-21 and 41BBL) | Fate Therapeutics | N/A |
| RPMI-8226 | ATCC | CRM-CLL-155 |
| MM.1S | ATCC | CRL-2974 |
| MM.1R | ATCC | CRL-2975 |
| HL-60 | ATCC | CCL-240 |
| THP-1 | ATCC | TIB-202 |
| Experimental models: Organisms/strains | ||
| Mouse: NOD.Cg-PrkdcscidIL2rgtm1Wjl/SzJ | Jackson Laboratory | JAX: 005557 |
| Oligonucleotides | ||
| CD3E forward primer: TACCTGAGGGCAAGAGTGTGT | Integrated DNA Technologies | Custom order |
| CD3E reverse primer: CCCAGTGATGCAGATGTCCAC | Integrated DNA Technologies | Custom order |
| IL32 forward primer: GTGGGGAGTTGGGGTCC | Integrated DNA Technologies | Custom order |
| IL32 reverse primer: TCAGAGAGGACCTTCGGGA | Integrated DNA Technologies | Custom order |
| GZMH forward primer: GCTGACAGTGCAGAAGGACT | Integrated DNA Technologies | Custom order |
| GZMH reverse primer: CCGGAGTCCCCCTTGAAAC | Integrated DNA Technologies | Custom order |
| ACTB forward primer: CGCGAGAAGATGACCCAGATC | Integrated DNA Technologies | Custom order |
| ACTB reverse primer: TTGCTGATCCACATCTGCTGG | Integrated DNA Technologies | Custom order |
| KLRC2 primers | Thermo Scientific | Hs02379574_g1 |
| Recombinant DNA | ||
| pKT2/PGK-Bsd:GFP CLP-Luc | This paper | N/A |
| Software and algorithms | ||
| FlowJo v.10.7.1 | Becton Dickinson | http://flowjo.com |
| CellRanger | 10X Genomics | http://10xgenomics.com |
| Seurat R package, v.3.2.0 | Stuart et al., 2019 | http://github.com/satijalab/seurat/ |
| ggplot 2 R package v.3.3.2 | http://ggplot2.tidyverse.org | |
| GOplot R package v.1.0.2 | Walter et al., 2015 | http://cran.r-project.org/web/packages/GOplot |
| Incucyte Base Software | Sartorius | http://essenbioscience.com/en/products/software/incucyte-base-software |
| Incucyte Spheroid Analysis Software Module | Sartorius | Cat #: 9600-0019 |
| GraphPad Prism | GraphPad | https://graphpad.com |
Highlights.
iADAPT NK cells and adaptive NK cells share metabolic and transcriptional features.
iADAPT NK cells are cytokine autonomous
iADAPT NK cells display enhanced innate immune function
Acknowledgements:
This work was supported by NIH R00 HL123638 (F.C.), NIH P01 CA111412 (J.S.M), NIH P01 CA65493 (J.S.M., B.R.B.), NIH R35 CA197292 (J.S.M), NIH R01 CA203348 (B.W.), NIH R37 AI34495 (B.R.B.) and research funds provided by Fate Therapeutics (J.S.M.).
Footnotes
Declaration of Interests: F.C. and J.S.M. are paid consultants for Fate Therapeutics and they receive research funds from this relationship. J.S.M. serves on the Scientific Advisory Board of OnkImmune, Nektar, Magenta and is a paid consultant consult for GT BioPharma and Vycellix (all unrelated to the content of this manuscript). He receives research funds from these relationships. B.R.B is a paid consultant/advisor for Regeneron Pharmaceuticals, Inc, Incyte Corp., Obsidian Therapeutics, Bristol-Myers Squibb and Bluerock. He receives research support from Tmunity, Bluerock, Kadmon, and Rheos and is co-founder of Tmunity. None of these relationships conflict with the content of the published research. He also receives research support from Fate Therapeutics for research unrelated to the content of this report. R.B., S.G., S.M., R.A., P.R., M.Q. G., G.B., M.M., J.H., T.D., T.T.L., and B.V. are employees of Fate Therapeutics. Fate Therapeutics owns patents (METHODS AND COMPOSITIONS FOR INDUCING HEMATOPOIETIC CELL DIFFERENTIATION; Patent No. 10,626,372) covering the iPSC derived NK cells.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akhmetzyanova I, McCarron MJ, Parekh S, Chesi M, Bergsagel PL, and Fooksman DR (2020). Dynamic CD138 surface expression regulates switch between myeloma growth and dissemination. Leukemia. 34, 245–256. 10.1038/s41375-019-0519-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachanova V, Cooley S, Defor TE, Verneris MR, Zhang B, McKenna DH, Curtsinger J Panoskaltsis-Mortari A, Lewis D, Hippen K, et al. (2014). Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood. 123, 3855–3862. 10.1182/blood-2013-10-532531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkeley LI, Cohen JF, Crankshaw DL, Shirota FN, and Nagasawa HT (2003). Hepatoprotection by L-cysteine-glutathione mixed disulfide, a sulfhydryl-modified prodrug of glutathione. J. Biochem. Mol. Toxicol 17, 95–97. 10.1002/jbt.10069. [DOI] [PubMed] [Google Scholar]
- Brentjens RJ, Rivière I, Park JH, Davila MJ, Wang X, Stefanski J, Taylor C, Yeh R, Bartido S, Borquez-Ojeda O, et al. (2011). Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 118, 4817–4828. 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casneuf T, Xu XS, Adams HC 3rd, Axel AE, Chiu C, Khan I, Ahmadi T, Yan X, Lonial S, Plesner T, et al. (2017). Effects of daratumumab on natural killer cells and impact on clinical outcomes in relapsed or refractory multiple myeloma. Blood Adv. 1, 2105–2114. 10.1182/bloodadvances.2017006866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Diao L, Yang Y, Yi X, Rodriguez BL, Li Y, Villalobos PA, Cascone T, Liu X, Tan L, et al. (2018). CD38-Mediated Immunosuppression as a Mechanism of Tumor Cell Escape from PD-1/PD-L1 Blockade. Cancer Discov. 8, 1156–1175. 10.1158/2159-8290.CD-17-1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichocki F, and Miller JS (2010). In vitro development of human Killer-Immunoglobulin Receptor-positive NK cells. Methods Mol. Biol 612, 15–26. 10.1007/978-1-60761-362-6_2. [DOI] [PubMed] [Google Scholar]
- Cichocki F, Cooley S, Davis Z DeFor TE Schlums H, Zhang B, Brunstein CG, Blazar BR, Wagner J, Diamond DJ, et al. (2016). CD56dimCD57+NKG2C+ NK cell expansion is associated with reduced leukemia relapse after reduced intensity HCT. Leukemia. 30, 456–463. 10.1038/leu.2015.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichocki F, Wu C-Y, Zhang B, Felices M, Tesi B, Tuininga K, Dougherty P, Taras E, Hinderlie P, Blazar BR, Bryceson YT, et al. (2018). ARID5B regulates metabolic programming in human adaptive NK cells. J. Exp. Med 215, 2379–2395. 10.1084/jem.20172168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichocki F, Taras E, Chiuppesi F, Wagner JE, Blazar BR, Brunstein C, Luo X, Diamond DJ, Cooley S, Weisdorf DJ, et al. (2019). Adaptive NK cell reconstitution is associated with better clinical outcomes. JCI Insight. 4, e125553. 10.1172/jci.insight.125553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichocki F, Bjordahl R, Gaidarova S, Mahmood S, Abujarour R, Wang H, Tuininga K, Felices M, Davis ZB, Bendzick L, et al. (2020). iPSC-derived NK cells maintain high cytotoxicity and enhance in vivo tumor control in concert with T cells and anti-PD-1 therapy. Sci. Transl. Med 12, eaaz5618. 10.1126/scitranslmed.aaz5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooley S, He F, Bachanova V, Vercellotti GM, DeFor TE, Curtsinger JM, Robertson P, Grzywacz B, Conlon KC, Waldmann TA, et al. (2019). First-in-human trial of rhIL-15 and haploidentical natural killer cell therapy for advanced acute myeloid leukemia. Blood Adv. 3, 1970–1980. 10.1182/bloodadvances.2018028332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper MA, Bush JE, Fehnigher TA, VanDeusen JB, Waite RE, Liu Y, Aguila HL, and Caligiuri MA (2002). In vivo evidence for a dependence on interleukin 15 for survival of natural killer cells. Blood. 100, 3633–3638. 10.1182/blood-2001-12-0293. [DOI] [PubMed] [Google Scholar]
- de Weers M, Tai Y-T, van der Veer MS, Bakker JM, Vink T, Jacobs DCH, Oomen LA, Peipp M, Valerius T, Slootstra JW, et al. (2011). Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J. Immunol 186, 1840–1848. 10.4049/jimmunol.1003032. [DOI] [PubMed] [Google Scholar]
- Deaglio S, Zubiaur M, Gregorini A, Bottarel F, Ausiello CM, Dianzani U, Sancho J, and Malavasi F (2002). Human CD38 and CD16 are functionally dependent and physically associated in natural killer cells. Blood. 99, 2490–2498. 10.1182/blood.v99.7.2490. [DOI] [PubMed] [Google Scholar]
- Drach J, McQueen T, Engel H, Andreeff M, Roberston KA, Collins SJ, Malavasi F, and Mehta K (1994). Retinoic acid-induced expression of CD38 antigen in myeloid cells is mediated through retinoic acid receptor-alpha. Cancer Res. 54, 1746–1752. [PubMed] [Google Scholar]
- Ge Y, Jiang W, Gan L, Wang L, Sun C, Ni P, Liu Y, Wu S, Gu L, Zheng W, et al. (2010). Mouse embryonic fibroblasts from CD38 knockout mice are resistant to oxidative stresses through inhibition of reactive oxygen species production and Ca(2+) overload. Biochem. Biophys. Res. Commun 399, 167–172. 10.1016/j.bbrc.2010.07.040. [DOI] [PubMed] [Google Scholar]
- Graeff R, Liu Q, Kriksunov IA, Hao Q, and Lee HC (2006). Acidic residues at the active sites of CD38 and ADP-ribosyl cyclase determine nicotinic acid adenine dinucleotide phosphate (NAADP) synthesis and hydrolysis activities. J. Biol. Chem 281, 28951–28957. 10.1074/jbc.M604370200. [DOI] [PubMed] [Google Scholar]
- Gumá M, Angulo A, Vilches C, Gómez-Lozano N, Malats N, and López-Botet M (2004). Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood. 104, 3664–3671. 10.1182/blood-2004-05-2058. [DOI] [PubMed] [Google Scholar]
- Hirayama AV, Gauthier J, Hay KA, Voutsinas JM, Wu Q, Pender BS, Hawkins RM, Vakil A, Steinmetz RN, Riddell SR, et al. (2019). High rate of durable complete remission in follicular lymphoma after CD19 CAR-T cell immunotherapy. Blood. 134, 636–640. 10.1182/blood.2019000905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing Y, Ni Z, Wu J, Higgins L, Markowski TW, Kaufman DS, and Walcheck B (2015). Identification of an ADAM17 cleavage region in human CD16 (FcγRIII) and the engineering of a non-cleavable version of the receptor in NK cells. PLoS One. 10, e0121788. 10.1371/journal.pone.0121788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau CM, Adams NM, Geary CD, Weizman O-E, Rapp M, Pritykin Y, Leslie CS, and Sun JC (2018). Epigenetic control of innate and adaptive immune memory. Nat. Immunol 19, 963–972. 10.1038/s41590-018-0176-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Zhang T, Hwang I, Kim A, Nitschke L, Kim M, Scott JM, Kamimura Y, Lanier LL, and Kim S (2015). Epigenetic modification and antibody-dependent expansion of memory-like NK cells in human cytomegalovirus-infected individuals. Immunity. 42, 431–432. 10.1016/j.immuni.2015.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luetke-Eversloh M, Hammer Q, Durek P, Nordström K, Gasparoni G, Pink M, Hamann A, Walter J, Chang H-D Dong J, et al. (2014). Human cytomegalovirus drives epigenetic imprinting of the IFNG locus in NKG2Chi natural killer cells. PLoS Pathog. 10, e1004441. 10.1371/journal.ppat.1004441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, et al. (2014). Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med 371, 1507–1517. 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, McKenna D, Le C, Defor TE, Burns LJ, et al. (2005). Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 105, 3051–3057. 10.1182/blood-2004-07-2974. [DOI] [PubMed] [Google Scholar]
- Ranson T, Vosshenrich CA, Corcuff E, Richard O, Müller W, and Di Santo JP (2003). IL-15 is an essential mediator of peripheral NK-cell homeostasis. Blood. 101, 4887–4893. 10.1182/blood-2002-11-3392. [DOI] [PubMed] [Google Scholar]
- Reinherz EL, Kung PC, Goldstein G, Levey RH, and Schlossman SF (1980). Discrete stages of human intrathymic differentiation: analysis of normal thymocytes and leukemic lymphoblasts of T-cell lineage. Proc. Natl. Acad. Sci. U S A 77, 1588–1592. 10.1073/pnas.77.3.1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romee R, Foley B, Lenvik T, Wang Y, Zhang B, Ankarlo D, Luo X, Cooley S, Verneris MR, Walcheck B, et al. (2013). NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood. 121, 3599–3608. 10.1182/blood-2012-04-425397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, Leong JW, Abdel-Latif S, Schneider SE, Willey S, et al. (2016). Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci. Transl. Med 8, 357ra123. 10.1126/scitranslmed.aaf2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlums H, Cichocki F, Tesi B, Theorell J, Béziat V, Holmes TD, Han H, Chiang SCC, Foley B, Mattsson K, et al. (2015). Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity. 42, 443–456. 10.1016/j.immuni.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlums H, Jung M, Han H, Theorell J, Bigley V, Chiang SCC, Allan DS, Davidson-Moncada JK, Dickinson RE, Holmes TD, et al. (2017). Adaptive NK cells can persist in patients with GATA2 mutation depleted of stem and progenitor cells. Blood. 129, 1927–1939. 10.1182/blood-2016-08-734236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, Jäger U, Jaglowski S, Andreadis C, Westin JR, et al. (2019). Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Eng. J. Med 380, 45–56. [DOI] [PubMed] [Google Scholar]
- Sconocchia G, Titus JA, Mazzoni A, Visintin A, Pericle F, Hicks SW, Malavasi F, and Segal DM (1999). CD38 Triggers Cytotoxic Responses in Activated Human Natural Killer Cells. Blood. 94, 3864–3871. [PubMed] [Google Scholar]
- Shubinsky G, and Schlesinger M (1997). The CD38 lymphocyte differentiation marker: new insight into its ectoenzymatic activity and its role as a signal transducer. Immunity. 7, 315–324. 10.1016/s1074-7613(00)80353-2. [DOI] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell. 177, 1888–1902. 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui H, Valamehr B, Hindoyan A, Qiao R, Ding X, Guo S, Witte ON, Liu X, Ho C-M, and Wu H (2011). An optimized small molecule inhibitor cocktail supports long-term maintenance of human embryonic stem cells. Nat. Commun 2, 167. 10.1038/ncomms1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usmani SZ, Weiss BM, Plesner T, Bahlis NJ, Belch A, Lonial S, Lokhorst HM, Voorhees PM, Richardson PG, Chari A, et al. (2016). Clinical efficacy of daratumumab monotherapy in patients with heavily pretreated relapsed or refractory multiple myeloma. Blood. 128, 37–44. 10.1182/blood-2016-03-705210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valamehr B, Abujarour R, Robinson M, Le T, Robbins D, Shoemaker D, and Flynn P (2012). A novel platform to enable the high-throughput derivation and characterization of feeder-free human iPSCs. Sci. Rep 2, 213. 10.1038/srep00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valamehr B, Robinson M, Abujarour R, Rezner B, Vranceanu F, Le T, Medcalf A, Lee TT, Fitch M, Robbins D, et al. (2014). Platform for induction and maintenance of transgene-free hiPSCs resembling ground state pluripotent stem cells. Stem Cell Reports. 2, 366–381. 10.1016/j.stemcr.2014.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma V, Shrimali RK, Ahmad S, Dai W, Wang H, Lu S, Nandre R, Gaur P, Lopez J, Sade-Feldman M, et al. (2019). PD-1 blockade in subprimed CD8 cells induces dysfunctional PD-1+CD38hi cells and anti-PD-1 resistance. Nat. Immunol 20, 1231–1243. 10.1038/s41590-019-0441-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter W, Sánchez-Cabo F, and Ricote M (2015). GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics. 37, 2912–2914. 10.1093/bioinformatics/btv300. [DOI] [PubMed] [Google Scholar]
- Wang XF, and Cynader MS (2002). Astrocytes Provide Cysteine to Neurons by Releasing Glutathione. J. Neurochem 74, 1434–1442. 10.1046/j.1471-4159.2000.0741434.x. [DOI] [PubMed] [Google Scholar]
- Williams BA, Law A, Hunyadkurti J, Desilets S, Leyton JV, and Keating A (2019). Antibody Therapies for Acute Myeloid Leukemia: Unconjugated, Toxin-Conjugated, Radio-Conjugated and Multivalent Formats. J. Clin. Med 8, 1261. 10.3390/jcm8081261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Xu W, and Li J (2018). Chimeric antigen receptor-modified T cell therapy in chronic lymphocytic leukemia. J. Hematol. Oncol 11, 130. 10.1186/s13045-018-0676-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Blum RH, Bjordahl R, Gaidarova S, Rogers P, Lee TT, Abujarour R, Bonello GB, Wu J, Tsai PF, et al. (2020). Pluripotent stem cell-derived NK cells with high-affinity noncleavable CD16a mediate improved antitumor activity. Blood. 135, 399–410 10.1182/blood.2019000621. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and processed single cell RNA-seq datasets generated in this study are available through the Gene Expression Omnibus repository hosted by the National Center for Biotechnology Information under the accession number GSE168936.