

Abstract
Background.
Arterial endothelium is critical in maintaining vessel homeostasis and preventing atherosclerotic cardiovascular disease (CVD). Low-to-moderate alcohol consumption is associated with reduced atherosclerosis and stimulates Notch signaling in endothelial cells. The aim of our study was to determine whether alcohol (EtOH) protects the endothelium against serum amyloid A1 (SAA1)-induced activation/injury, and to determine whether or not this protection is exclusively Notch-dependent.
Methods and Results.
Human coronary artery endothelial cells (HCAEC) were stimulated +/− ‘pro-atherogenic’ SAA1 (1 μM) in the absence or presence of EtOH (25 mM), the Notch ligand DLL4 (3 μg/ml), or the Notch inhibitor DAPT (20 μM). EtOH stimulated Notch signaling in HCAEC as seen by increased Notch receptor and hrt target gene expression. Treatment with EtOH alone, or stimulation of Notch signaling by DLL4, increased eNOS activity and enhanced HCAEC barrier function assessed by trans-endothelial electrical resistance. Moreover, EtOH and DLL4 both inhibited SAA1-induced monolayer leakiness, cell adhesion molecule (ICAM, VCAM) expression, and monocyte adhesion. These effects of EtOH were Notch-dependent as they were blocked with DAPT and by Notch receptor (N1, N4) knockdown. In contrast, EtOH’s inhibition of SAA1-induced inflammatory cytokines (IL-6, IFN-γ) and reactive oxygen species (ROS) were Notch-independent as these effects were unaffected by DAPT or by N1 and/or N4 knockdown.
Conclusions.
Alcohol at moderate levels protects against SAA1-induced endothelial activation via both Notch-dependent and Notch-independent mechanisms. Maintenance of endothelium by alcohol in a non-activated state would be expected to preserve vessel homeostasis and protect against atherosclerosis development.
Keywords: Alcohol, Cardiovascular, Notch, Endothelial, Atherosclerosis, SAA1
INTRODUCTION.
Arteriosclerosis, a hardening and narrowing of the arteries, is the most common pathological condition underlying cardiovascular disease that is the leading cause of death worldwide for both women and men. In the United States alone heart attacks and stroke as a result of arteriosclerosis cause approximately 800,000 deaths annually, with an associated economic cost of >$350 billion (Heart attack and Stroke Statistics- 2020 Update) (Virani et al., 2020).
When it comes to arterial health, the endothelial monolayer covering the vessel lumen is critical in maintaining physiological homeostasis and can be viewed as the gatekeeper (Cahill and Redmond, 2016). Endothelial cell ‘activation’ in response to diverse injurious stimuli (e.g., high LDL cholesterol, smoking, disturbed flow) leading to compromised function has been implicated in thrombosis, hypertension, and diabetes, and is recognized as a key initiating event in arteriosclerosis (Cahill and Redmond, 2016, Sluiter et al., 2021). Compromised endothelium has a pro-atherogenic phenotype typified by a number of changes including reduced nitric oxide (NO) bioavailability and decreased barrier function, together with increased adhesion molecule expression, and greater reactive oxygen species (ROS) and pro-inflammatory cytokine production (Ribeiro et al., 2009). Following endothelial injury, lipids and immune cells can progressively accumulate in the vessel wall and prompt arterial cell populations including medial smooth muscle cells and resident stem cells to differentiate and proliferate, thereby contributing to intimal hyperplasia and vessel wall thickening (Basatemur et al., 2019, Majesky et al., 2011). Maintenance of endothelium in a non-activated state and/or the reversal of endothelial injury are therefore highly desirable therapeutic goals with the promise of ameliorating pathologic vascular remodeling and arteriosclerosis.
Population studies reveal a complex association between alcohol consumption and cardiovascular disease. When compared to abstinence, chronic alcohol abuse and binge drinking are linked with increased cardiovascular-related morbidity and mortality. In contrast, frequent low-moderate consumption (in the range 1–3 drinks/day) seems beneficial (O’Keefe et al., 2014, Rehm and Roerecke, 2017). Various effects on cholesterol levels, blood coagulation, inflammation, and different vascular cells have been investigated, but a complete picture of the precise cell and signaling mechanisms mediating the athero-protective effects of moderate alcohol remains elusive.
We have previously reported that ethanol (EtOH), the main type of alcohol found in beverages, can affect endothelial function, including stimulating their angiogenic activity by activating Notch signaling (Morrow et al., 2008a, Morrow et al., 2014). The Notch pathway is a highly conserved signaling system involved in cell fate control (i.e., dictating whether a cell differentiates, proliferates, undergoes apoptosis etc.) (Morrow et al., 2008b, Shawber and Kitajewski, 2004). Classically known for its role during embyronic development, the importance of Notch signaling in disease processes and to vessel homeostasis in the adult has only begun to be appreciated. There are four types of Notch receptors (N1, 2, 3 and 4), each a single transmembrane protein. N1 and N4 are prevalent in endothelium. Canonical Notch signalling depends on sequential proteolysis by α-secretase and γ-secretase resulting in cleavage of the Notch intracellular domain (NICD) from the receptor, allowing its translocation from the cell membrane to the nucleus where it activates various downstream target genes such as hes and hrt (Artavanis-Tsakonas et al., 1999).
Serum amyloid protein A1 (SAA1), an acute phase protein produced principally by the liver, has several pro-inflammatory properties and has been shown to play a causal role in atherogenesis (Dong et al., 2011, Getz et al., 2016, Shridas and Tannock, 2019). The aim of our study was to determine if moderate alcohol protects the endothelium against SAA1-induced endothelial activation/injury, and to determine whether or not this protection is exclusively Notch-dependent.
MATERIALS AND METHODS
Cell Culture
Human Coronary Artery Endothelial Cells (HCAEC) were obtained from Lonza (Walkersville, MD, USA) and cultured in optimized endothelial cell medium (Cat# CC-3162, CloneticsR, Lonza, Walkersville, MD, USA) along with supplements (FBS, hydrocortisone, hFGF, VEGF, RE-IGF-1, Ascorbic acid, hEGF, GA-1000 & Heparin). Standard humidified water-jacketed CO2 incubators were used to culture cells. HCAEC passage 5–14 were used for all experiments. EtOH (200 proof, ACS/USP Grade, Pharmco Products, Brookfield, CT, USA) was diluted in media to achieve the desired concentration before being added to HCAEC for the time specified. Experimental plates were covered in parafilm to prevent evaporation. We have previously found that under these conditions ethanol concentration (determined by diagnostic kit from Sigma) after 24 hrs of incubation +/− endothelial cells is ~90% of that at the start, indicating no significant metabolism of alcohol by these cells. HCAEC were also treated with the following as indicated; the Notch ligand Delta-like ligand 4 (DLL4, 3 μg/mL; Cat# 1506-D4, R&D Systems, Minneapolis, MN, USA), the Notch signaling inhibitor (a gamma secretase inhibitor, GSI) N-[N-(3,5-Difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl Ester (DAPT, 20 μM; Cat# 2634, Tocris Bioscience, Bristol, United Kingdom), apolipoprotein serum amyloid A-1 protein (SAA 1, 0.5–1.0 μM; Cat# 300-53, PeproTech, Rocky Hill, NJ, United States).
Quantitative real-time reverse transcription PCR (qRT-PCR)
Total RNA was extracted using RNeasy Mini Kit (Cat# 74134, Hilden, Germany) and 1 μg RNA was reverse transcribed to cDNA using iScript cDNA Synthesis kit (Cat# 1708891, Biorad, Hercules, CA, USA). The mRNA expression was determined using SYBR Green master mix (Cat# 4309155, Thermo Fisher Scientific, Waltham, MA, USA) according to manufacturer’s protocol using a QuantStudio 3 (Applied Bosystems, Foster City, CA, USA). The RT-PCR cycling conditions were as follows; DNA polymerase activation at 95°C for 10 min, followed by 40 PCR cycles, each cycle consisting of 95°C for 15 s (denature) and 60°C for 1 min (anneal/extend).
Primer sequences were human Notch 1 forward (5′-CAGGGTGTGCACTGTGAGAT-3′) andreverse (5′-GACAGGCACTCGTTGACATC-3′); human Notch 4 forward (5′-CTAGGGGCTCTTCTCGTCCT-3′) and reverse (5′-CAACTTCTGCCTTTGGCTTC-3′); human HRT 2 forward (5′-GTACCTGAGCTCCGTGGAAG-3′) and reverse (5′-AGTTGTGGAGAGGCGACAAG-3′); human HRT 3 forward (5′-GGTGGGACAGGATTCTTTGA-3′) and reverse (5′-AGCTGTTGAGGTGGGAGAGA-3′); human ICAM-1 forward (5′-AGGATGGCACTTTCCCACTG-3′ and reverse (5′-GGAGAGCACATTCACGGTCA-3′); human VCAM-1 forward (5′-ACTGGCATGGTACGGAGATG-3′) and reverse (5′-CAATGTGACTAAAGGAGGCAGT-3′); human MCP-1 forward (5′-AGCATGAAAGTCTCTGCCGC-3′) and reverse (5′-GGGTACCACGTCTGCTTGGA-3′); human GAPDH forward (5′-CGAGATCCCTCCAAAATCAA-3′) and reverse (5′-TTCACACCCATGGACGAACAT-3′). GAPDH was used as a housekeeping control. The data presented are the average (mean ± SEM). The results were analyzed with Applied Biosystems® qPCR analysis software (ThermoFisher Scientific).
Western blot analysis
HCAEC were rinsed with PBS and lysed with ice-cold 1x RIPA buffer for 5 minutes at 4 °C. Protein concentration was determined using a Bio-Rad protein assay kit II, with bovine serum albumin (BSA) as standard (Cat# 5000002, Bio-Rad, Hercules, CA). Protein concentrations were measured using Bio-Rad reagent. Proteins were separated on 4–20% TGX precast protein gels (Cat# 4561094; Bio-Rad, Hercules, CA, USA) and transferred to nitrocellulose membranes. Membranes were washed twice with 1x TBS (tris-buffered saline), blocked with 5% blocking solution (BSA in PBS) for 1 hour, and incubated with the appropriate primary antibody for 4 hrs at room temperature or overnight at 4° C. Following 3 X TBS washes, the membrane was incubated with secondary antibody conjugated to horseradish peroxidase for 2 h at room temperature. Blots were developed with SuperSignal west Pico Chemiluminescent Substrate (Cat # 34580; Thermo Fisher Scientific, USA) using ChemiDoc XR imaging system (Biorad, Hercules, CA, USA) and the band intensities were quantified using National Institute of Health’s ImageJ software (Bethesda, MD). The following antibodies were used: primary antibody- anti-Notch-1 IC (1:1000 dilution) (Cat # Ab83232); anti-Notch 4 IC (1:1000 dilution) (Cat # Ab199295) and secondary antibody- anti-Rabbit IgG, HRP conjugated (1:50000 dilution) (Cat # Ab97051) from Abcam (Abcam, Cambridge, United Kingdom). Anti-HEY2/HRT2 (1:1000 dilution) (Cat # 501726436, Fisher Scientific, USA); anti-Hey L/HRT3 (1:1000 dilution) (Cat # AB5718, Sigma-Aldrich,USA).
γ-Secretase assay
γ-secretase activity was determined essentially as described previously (Hatch et al., 2015). Integral membrane proteins were separated from HCAEC lysates using ReadyPrep protein extraction kit (Membrane II; BioRad, Cat # 1632084). Membrane protein pellets were resuspended with 1 mL of PBS + CHAPSO (0.25% wt/v) and protein concentration measured by Bradford assay. 40 μg protein was treated with experimental agents for 1 hr at 37° C before γ-secretase fluorogenic substrate (8 μM, Cat # 565764, Sigma-Aldrich) was added and incubated at 37°C overnight. 100 μl samples were then placed in 96 wells and the fluorescence measured using SpectraMax 340PC spectrophotometer at 355 nm excitation/ 440 nm emission wavelength (Molecular Devices, CA, USA). ‘Substrate alone’ sample served as background which was subtracted from all readings.
NOS activity
Endothelial nitric oxide synthase (eNOS) activity was measured in HCAEC lysates using a colorimetric Nitric Oxide Synthase Activity Assay Kit based on the Greiss reaction (Catalog # K205-100, BioVision Inc). L-NG-Nitro-arginine methyl ester (L-NAME) was used as a control NOS inhibitor.
Trans-endothelial electrical resistance (TEER)
TEER was measured according to Ghatak (Ghatak et al., 2016). In brief, HCAEC (P4 – P8) were grown in 12 well Transwells (0.4 μm polyester membrane, polystyrene plates) until ~90% confluency and treated with or without experimental agents as indicated for 1 h at 37°C. Membrane resistance was then measured using chopstick electrodes attached to an Epithelial Voltohmmeter (EVOM2, World Precision Instruments, Inc., Sarasota, FL). The electrode was completely immersed in the media but not touching to the base of the wells. Readings were taken at three different positions (12, 4, 8 o’clock) of each Transwell insert.
siRNA Transfection
Silencer Notch1 & Notch 4 siRNA (Cat# AM16708) and non-specific negative control siRNA (Cat# AM4641) were purchased from Invitrogen; Thermo Fisher Scientific, Waltham, MA, USA. All siRNA transfections were performed using Lipofectamine ™ 3000 Transfection Reagent, according to the manufacturer’s protocol (Cat# L3000015; Invitrogen; Thermo Fisher Scientific, Waltham, MA, USA). In brief, HCAECs (1.25 ×105) in 1 mL of Opti-MEM/reduced serum medium (Cat# 11058022, Thermo Fisher Scientific, Waltham, MA, USA) were seeded on a fibronectin coated 24-well plate. After 24 h at 37°C, the cells were transfected with 200 nM siRNA targeting Notch 1 or Notch 4, or scrambled negative control in 1.5 mL Lipofectamine 3000 reagent. Following transfection for 48 h, cells were treated with or ethanol (25 mM), DLL4 (3 μg/mL), DAPT (10 μM) and/or recombinant human serum amyloid A1 (SAA1, 1μM) for 24 h at 37° C or as indicated. The Notch 1 and Notch 4 knockdown efficiency was confirmed by Western blotting and RT-PCR analysis.
ICAM-1 detection by Immunofluorescence
HCAECs were seeded onto sterilized fibronectin coated cover slips in a 6-well plate. Following treatment, the cells were fixed with 4% paraformaldehyde (PFA) for 15 min at room temperature, then permeabilized (0.1% Triton X-100, 15 min). Cells were blocked with 3% BSA (bovine serum albumin) for 30 min and probed with anti-ICAM-1 antibody (1:100), (Cat# MA5-13021, ThermoFisher Scientific, Waltham, MA, USA) overnight at 4 °C. After washing, FITC-conjugated anti-rabbit IgG antibody (Cat# 62-6511, Thermo Fisher Scientific) was added and incubated for 2 h at 37°C. Cells were mounted with Sigma Fluoroshield with DAPI. Images were captured using a fluorescence microscope (Palmbeam LCM, Zeiss).
Cytokines: Enzyme-Linked Immunosorbant Assay (ELISA)
Supernatants of HCAEC cultures were collected following treatment and stored at −80° C until analysis. Interferon-γ (IFN-γ, Cat# DIF50) and Interleukin-6 (IL-6, Cat# D6060) levels were determined using commercially available ELISA kits (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions.
THP-1 Monocyte Adhesion Assay
HCAEC were grown to 80% confluence in 96-well plates and treated with experimental agents as indicated for 24 h at 37°C. Human monocytic cell line (THP-1) cells were obtained from American Type Culture Collection (Cat# TIB-202, ATCC®, Manassas, VA, USA) and labelled with 2’,7’-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein, acetoxymethyl ester (10 μg/mL) (BCECF-AM; Invitrogen, Carlsbad, CA, USA) for 30 min in RPMI-1640 medium and washed twice with HBSS. THP-1 cells (1 ×103) were then added to HCAEC monolayers, and cultures were incubated at 37°C in a CO2 incubator for 3hr. After incubation, medium was removed and HCAEC were washed twice with PBS to remove unbound THP-1 cells. The fluorescence intensity of remaining THP-1 cells was measured at 480nm Excitation/520nm Emission wavelength range using a SpectraMax 340PC spectrophotometer (Molecular Devices, CA, USA). The number of adherent cells was expressed as fluorescence intensity.
Human Monocyte isolation.
Human white blood cells were collected from leukoreduction filters (Sepacell RS-2000; Fenwal Inc.) after processing of healthy volunteer donor whole blood (American Red Cross) after rinsing with 50 mL of selection buffer (2 mmol/L ethylenediaminetetraacetic acid (EDTA; J.T. Baker Inc.), 2% heat-inactivated fetal bovine serum (Sigma-Aldrich) in Dulbecco’s phosphate- buffered saline (Corning Cellgro)). The filtrate was layered onto Ficoll-Paque PLUS (GE Healthcare) to isolate mononuclear cells by density gradient centrifugation. Mononuclear cells treated with CD14-positive selection beads (BD Biosciences) in selection buffer and subsequent EasySep magnetic purification procedure (Stemcell Technologies) resulted in purified CD14+ monocytes (mean purity 95%), which were cryopreserved in Cryostor CS5 (Biolife Solutions). These primary human monocytes were labelled with BCECF-AM and used in adhesion assays as described above for THP-1 cells.
Reactive Oxygen Species Assay
The production of intracellular reactive oxygen species (ROS) was measured by incubating HCAEC with 10 μM 5-(and-6)-carboxy-21, 71-dichlorodihydrofluorescein diacetate (carboxy-H2 DCFDA, 30 min) (Cat# C400, Invitrogen, Carlsbad, CA, USA), a cell permeable indicator for ROS generation. Cells were then treated with experimental agents for 3 hr, before cells were counterstained with DAPI (1 μg/ml, 10 min) and ROS generation (red fluorescence) detected by microplate reader at 480nm excitation/520nm emission. Images were captured using fluorescence microscope (Palmbeam LCM, Zeiss).
Data analysis
Data are expressed as mean ± SEM. For all experiments, experimental points were performed at least in duplicate, with a minimum of 3 independent experiments performed on different days. An unpaired Student’s t-test was performed to determine differences between 2 groups, while multiple data sets were analysed by ordinary one-way ANOVA followed by a Tukey’s or Dunnett’s multiple comparisons test (GraphPad Prism software V 9.1.0). A probability (p) value of 0.05 was considered significant.
RESULTS
Ethanol stimulates Notch signalling in human coronary artery endothelial cells.
Notch receptor and target gene expression were assessed by qRT-PCR and Western blot in human coronary artery endothelial cells (HCAEC) treated with/without ethanol (EtOH, 25 mM), the Notch ligand Delta-like ligand 4 (DLL4, 3 μg/ml), or serum amyloid A1 (SAA1, 1 μM). Ethanol and DLL4 significantly increased, to similar degrees, Notch receptor 1 and 4 (i.e., the receptor types prevalent in endothelial cells) and target gene hrt2 and hrt3 mRNA and protein expression (Fig 1a, b). Serum amyloid A1 treatment had no effect on Notch receptor or target gene mRNA expression (Fig 1a).
Figure 1.
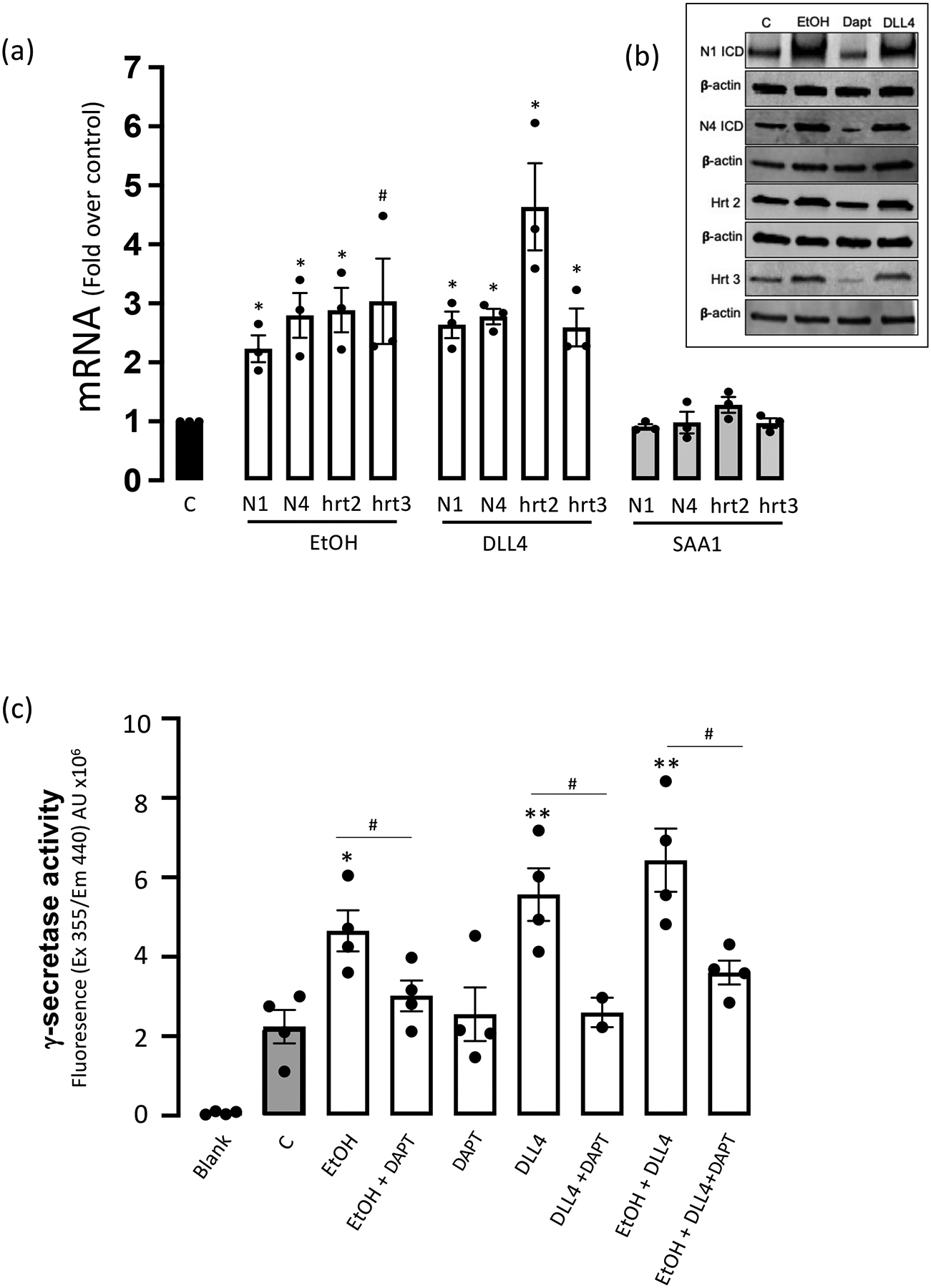
Ethanol stimulates Notch signalling in human coronary artery endothelial cells (HCAEC). (a) Notch receptors 1 and 4 (N1, N4), and target gene hrt2 and hrt3 mRNA levels determined by qRT-PCR in HCAEC treated (24 hr) with or without ethanol (EtOH, 25 mM), Delta like ligand 4 (DLL4, 3 μg/ml), or Serum amyloid protein A1 (SAA1, 1 μM). Data are mean ± SEM, n=3–4, #p<0.05, *p<0.01 vs control. (b) Representative Western Blots for N1ICD, N4ICD, hrt2 and hrt3 in HCAEC treated as indicated. β-actin was used as a loading control. (c) Ethanol stimulates γ-secretase activity in HCAEC. Gamma-secretase activity in solubilized HCAEC membranes was measured using a fluorogenic peptide substrate (C-terminal fragment of β-APP). DAPT was used as a control γ-secretase inhibitor (GSI). Data are mean ± SEM, n=4. *P<0.05, **P<0.01 vs C; #P<0.05 +/− DAPT.
Ethanol stimulates γ-secretase activity in HCAEC.
The effect of EtOH on γ-secretase proteolytic activity necessary for canonical Notch signalling was determined using a fluorogenic peptide substrate, with increased fluorescence indicative of γ-secretase-mediated cleavage. Similar to the Notch ligand DLL4, EtOH treatment significantly increased γ-secretase activity in HCAEC membranes that was DAPT inhibitable (Fig 1c).
Ethanol-stimulated eNOS activity is Notch-dependent.
Nitric oxide (NO), a key vasodilator, is synthesized by the heme-containing calcium and calmodulin-dependent enzyme nitric oxide synthase in endothelial cells (eNOS) from L-arginine in a reaction that produces stoichiometric amounts of L-citrulline. Here, NOS activity was measured in HCAEC lysates by colorimetric assay kit. Ethanol treatment (25 mM, 2 hr) increased eNOS activity (225%) compared to control, an effect that was inhibited by the NOS inhibitor L-NAME (Fig 2a). The ethanol-induced increase in eNOS activity was also inhibited by the Notch signaling inhibitor (i.e., γ-secretase inhibitor, GSI) DAPT (Fig 2a). Similarly, treatment with the Notch ligand DLL4 stimulated HCAEC eNOS activity that was inhibited by L-NAME, and by DAPT (Fig 2a). These data indicate that Ethanol stimulates eNOS activity in HCAEC via a Notch-dependent mechanism.
Figure 2.

(a) Ethanol stimulated eNOS activity is Notch-dependent. Nitric oxide synthase (NOS) activity in human coronary artery endothelial cell (HCAEC) lysates following treatment (2 hr) with or without ethanol (EtOH, 25 mM) or DLL4 (1 μg/ml) in the absence or presence of the NOS inhibitor L-Name (LN, 1 mM), or the γ-secretase/Notch signalling inhibitor DAPT (10 μM). Data are mean ± SEM, n=3. *p<0.01, ** P<0.001.
(b) Ethanol and DLL4 increase HCAEC barrier integrity, and prevent SAA1-induced monolayer leakiness in a Notch-dependent manner. Trans-endothelial electrical resistance (TEER) of HCAEC monolayers in Transwell inserts treated (2 hr) with/without EtOH (25 mM), DAPT (10 μM), DLL4 (3 μg/ml), SAA1 (1 μM), or combinations as indicated. Data are mean ± SEM, n=3. Dotted line denotes control monolayer TEER. Decreased TEER (red) is indicative of reduced barrier function/increased leakiness; whereas increased TEER (green) is indicative of a tighter barrier.
Ethanol improves endothelial barrier integrity, and blocks SAA1-induced monolayer leakiness in a Notch-dependent manner.
The role of the endothelium as a semipermeable barrier regulating the movement of molecules between the blood and the vessel wall is one of its most basic functions (Cahill and Redmond, 2016). Here barrier function of HCAEC grown in Transwell plates was assessed by measuring trans-endothelial electrical resistance (TEER) using chopstick electrodes and a Voltohmmeter as described in Methods. Following ethanol or DLL4 treatment (2 hr), electrical resistance across the HCAEC monolayer was increased compared to control; [181±15 Ω.cm2 (EtOH), 199±10 Ω.cm2 (DLL4) vs 159±10 Ω.cm2 (Control)], while treatment with the Notch inhibitor (DAPT) alone decreased it (Fig 2b). Exposure to SAA1 decreased HCAEC barrier integrity as demonstrated by reduced TEER (117±15 Ω.cm2), an effect prevented by co-treatment with either ethanol or with DLL4 (Fig 2b). The inhibitory effects of both ethanol and DLL4 on SAA1-induced HCAEC leakiness were blocked by DAPT (Fig 2b), indicating the Notch signalling-dependency of these effects
Ethanol inhibits SAA1-induced cell adhesion molecule (CAM) expression in a Notch-dependent manner.
Treatment of control HCAEC with the Notch signalling inhibitor DAPT significantly increased intracellular cell adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule (VCAM) mRNA expression as determined by qRT-PCR (Fig 3). Neither Ethanol nor Dll4 had any significant effect on basal ICAM-1 or VCAM expression (Fig 3e). SAA1 (1 μM, 24 hr) significantly increased (~3–4 fold) ICAM-1 and VCAM mRNA expression in HCAEC (Fig 3c, e). The SAA1-induced increase in ICAM-1 and VCAM mRNA was markedly inhibited by co-treatment with EtOH (25 mM), and with DLL4 (Fig 3c, e). The inhibitory effects of both EtOH and DLL4 on SAA1-induced cell adhesion molecules were Notch-dependent as they were blocked by DAPT (Fig 3c, e). Moreover, Ethanol’s inhibition of SAA1-induced ICAM-1 and VCAM expression was negated when either the N1 or N4 receptor, or both together, were knocked down using specific siRNAs (Fig 3d, f).
Figure 3.
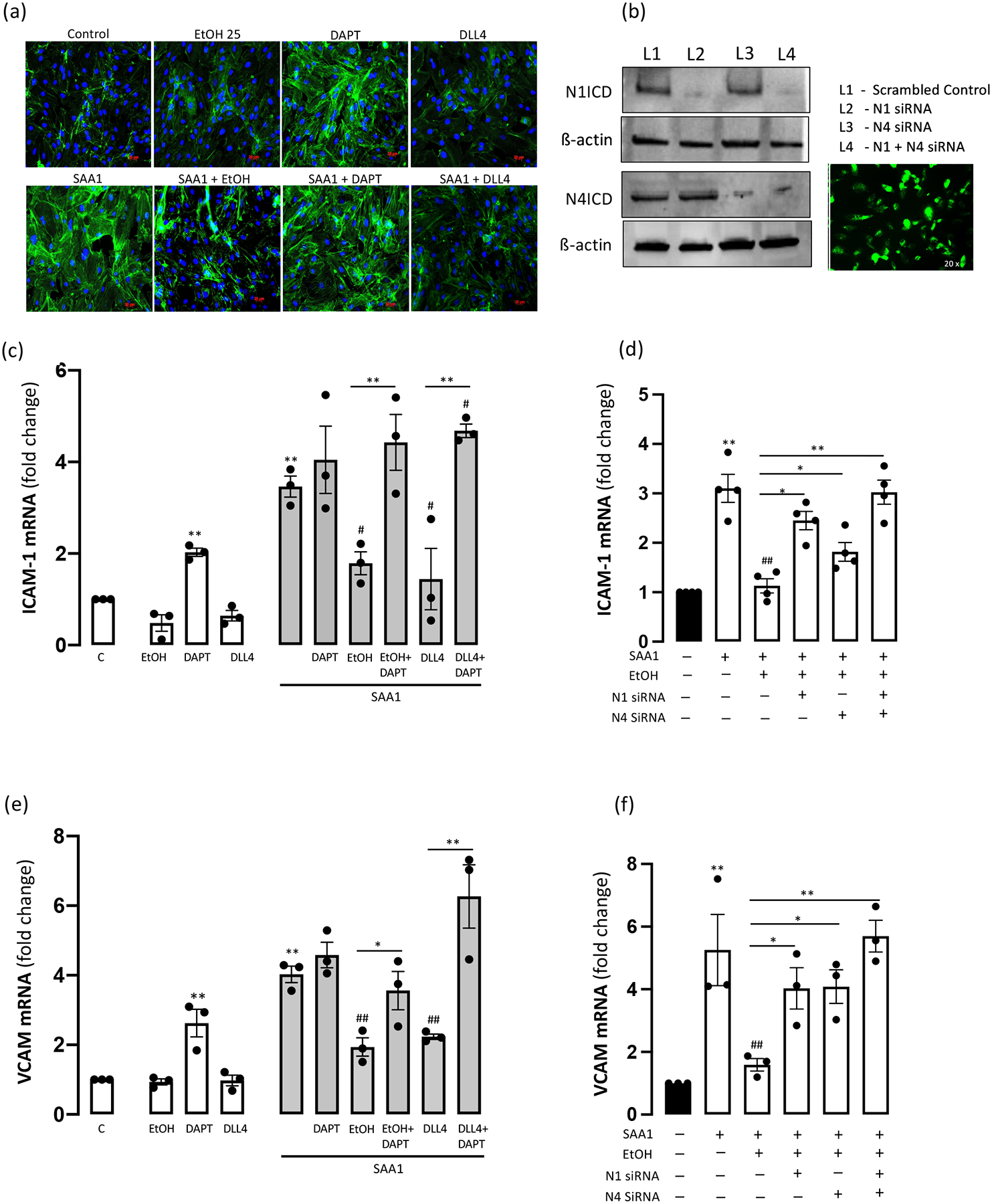
Ethanol inhibition of SAA1-induced cell adhesion molecule expression is Notch-dependent. (a) ICAM-1 expression in HCAEC; representative immunofluorescence images shown. (b) Representative Western blots showing N1 and N4 receptor expression in HCAEC following receptor knockdown as indicated using specific siRNA. β-actin was used as a loading control. Picture shows HCAEC transfected (60–80% efficiency) with GFP using Lipofectamine (x20 magnification). (c) ICAM-1 and (e) VCAM mRNA expression in HCAEC determined by qRT-PCR following treatments (24 hr) as indicated; EtOH (25 mM), DAPT (10 μM), DLL4 (1 μg/ml), SAA1 (1 μM). Cell adhesion molecule mRNA expression (ICAM-1 (d) and VCAM (f)) following SAA1 treatment +/− EtOH in HCAEC with/without N1 and/or N4 knockdown. Data are mean ± SEM, n=3. *P< 0.05, **P< 0.01 vs Control or as indicated; #P<0.05, ##P<0.01 vs SAA1.
SAA1-induced MCP-1 mRNA levels and increased monocyte adhesion are inhibited by ethanol in a Notch-dependent manner.
Monocyte chemoattractant protein-1 (MCP-1) plays a key role in monocyte recruitment and adhesion to the endothelium that is an important early step in atherogenesis (Cahill and Redmond, 2016, Sluiter et al., 2021). SAA1 exposure increased (~6.5 fold) MCP-1 mRNA levels in HCAEC (Fig 4a). While Ethanol had no effect on basal MCP-1 mRNA expression, it strongly inhibited the SAA1-induced MCP-1 response (Fig 4a). Similarly, treatment with DLL4 had no effect on basal MCP-1 but completely inhibited the SAA1-induced MCP-1 response (Fig 4a). These effects of ethanol and DLL4 were partly reversed by DAPT (Fig 4a). Moreover, ethanol’s inhibition of SAA1-induced MCP-1 was prevented when either the N1 or N4 receptor, or both together, were knocked down using specific siRNAs (Fig 4b).
Figure 4.
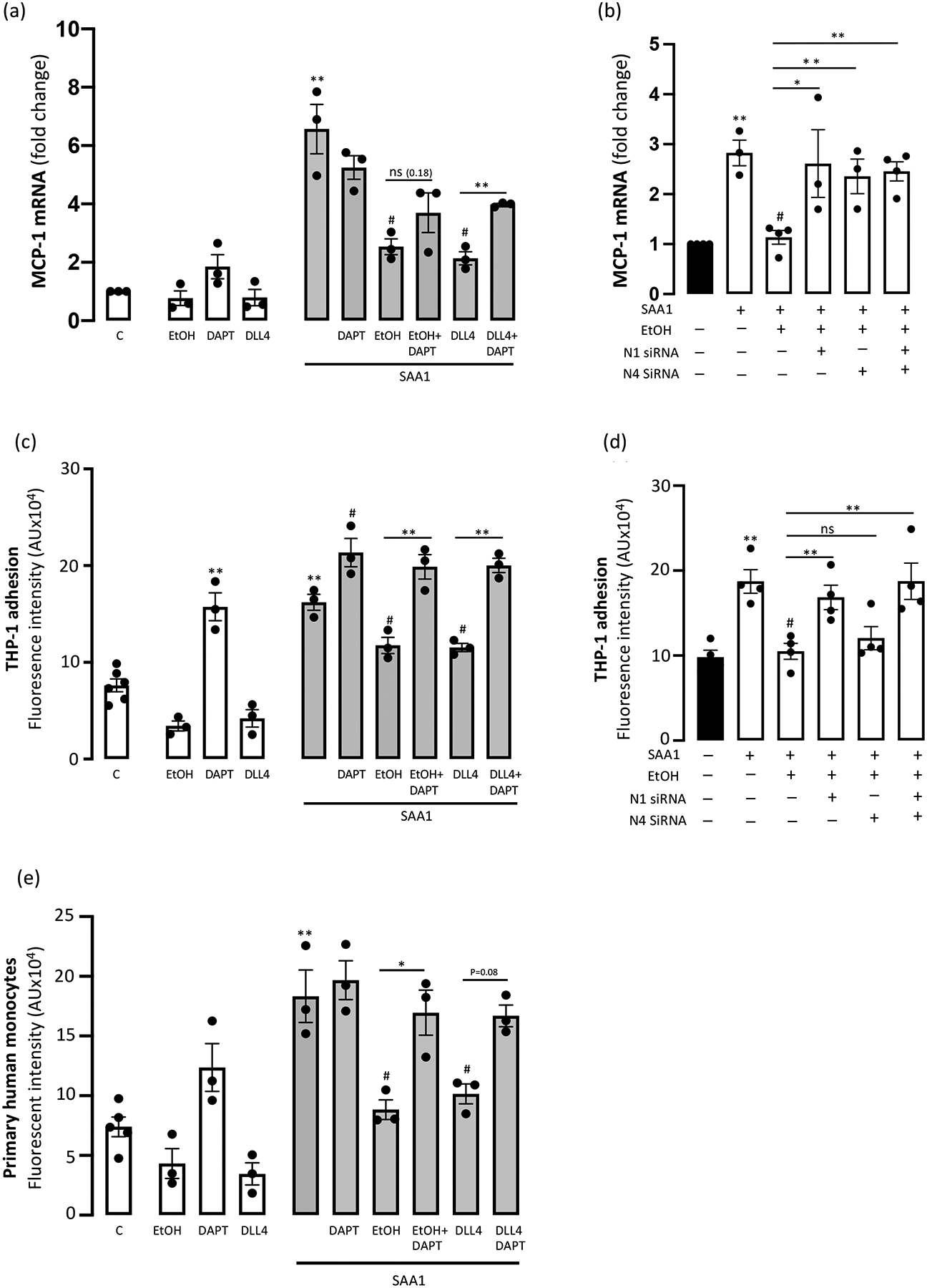
(a) Ethanol inhibition of SAA1-induced MCP-1 expression is Notch-dependent. Monocyte chemoattractant protein-1 (MCP-1) mRNA levels in HCAEC following treatments (24 hr) as indicated; EtOH (25 mM), DAPT (10 μM), DLL4 (1 μg/ml), SAA1 (1 μM). (b) MCP-1 mRNA expression following SAA1 treatment +/− EtOH in HCAEC with/without N1 and/or N4 knockdown.
(c) Ethanol inhibition of SAA1-induced monocyte adhesion is Notch-dependent. THP-1 monocyte adhesion to HCAEC following treatments as above. Data are mean ± SEM, n=3 *P< 0.05, **P< 0.01 vs Control or as indicated, # P<0.05 vs SAA1. (d) THP-1 adhesion following SAA1 treatment +/− EtOH in HCAEC with/without N1 and/or N4 knockdown. (e) Adhesion of primary human monocytes to HCAEC following treatments as above. Data are mean ± SEM, n=3–4. *P< 0.05, **P< 0.01 vs Control or as indicated; #P<0.01 vs SAA1 alone.
Neither Ethanol (25 mM, 24 hr) nor DLL4 had any significant effect on ‘baseline’ THP-1 monocyte adhesion to HCAEC, whereas repression of Notch signalling with DAPT increased THP-1 adhesion (Fig 4c). Serum amyloid protein A1 (SAA1) significantly increased (~200%) THP-1 monocyte adhesion to HCAEC when compared to control, untreated cells (Fig 4c). Co-treatment with either Ethanol or DLL4 inhibited the SAA1-induced increase in THP-1 cell adhesion, effects that were prevented by the Notch inhibitor DAPT (Fig 4c). Similar results were obtained using primary human monocytes (Fig 4e). Knockdown by specific siRNA of the N1 receptor, but not knockdown of N4, reversed the ethanol inhibition of SAA1-stimulated THP-1 monocyte adhesion (Fig 4d). Taken together, these data suggest that ethanol ameliorates SAA1-induced MCP-1 and monocyte adhesion responses via a Notch-dependent mechanism, with the latter effect mediated via the N1 receptor exclusively.
Ethanol’s inhibition of SAA1-induced inflammatory cytokines is Notch-independent.
Cytokine levels in HCAEC supernatants were determined by ELISA. Baseline levels of interleukin-6 (IL-6) and interferon-γ (IFN-γ) were reduced following ethanol or DLL4-exposure, whereas both were significantly increased following Notch signalling repression with DAPT (Fig 5a,c). SAA1 stimulated both IL-6 and IFN-γ levels, compared to controls, responses that were attenuated by co-treatment with ethanol and with DLL4 (Fig 5a, c). DLL4’s inhibition of SAA1-induced IL-6 and IFN-γ was prevented by DAPT. However, Ethanol’s inhibition of SAA1-induced IL-6 and IFN-γ was unaffected in the presence of DAPT and, moreover, was unaffected following N1 and/or N4 receptor knockdown (Fig 5b, d). These data suggest that ethanol’s inhibition of SAA1-induced inflammatory cytokines is Notch-independent.
Figure 5.
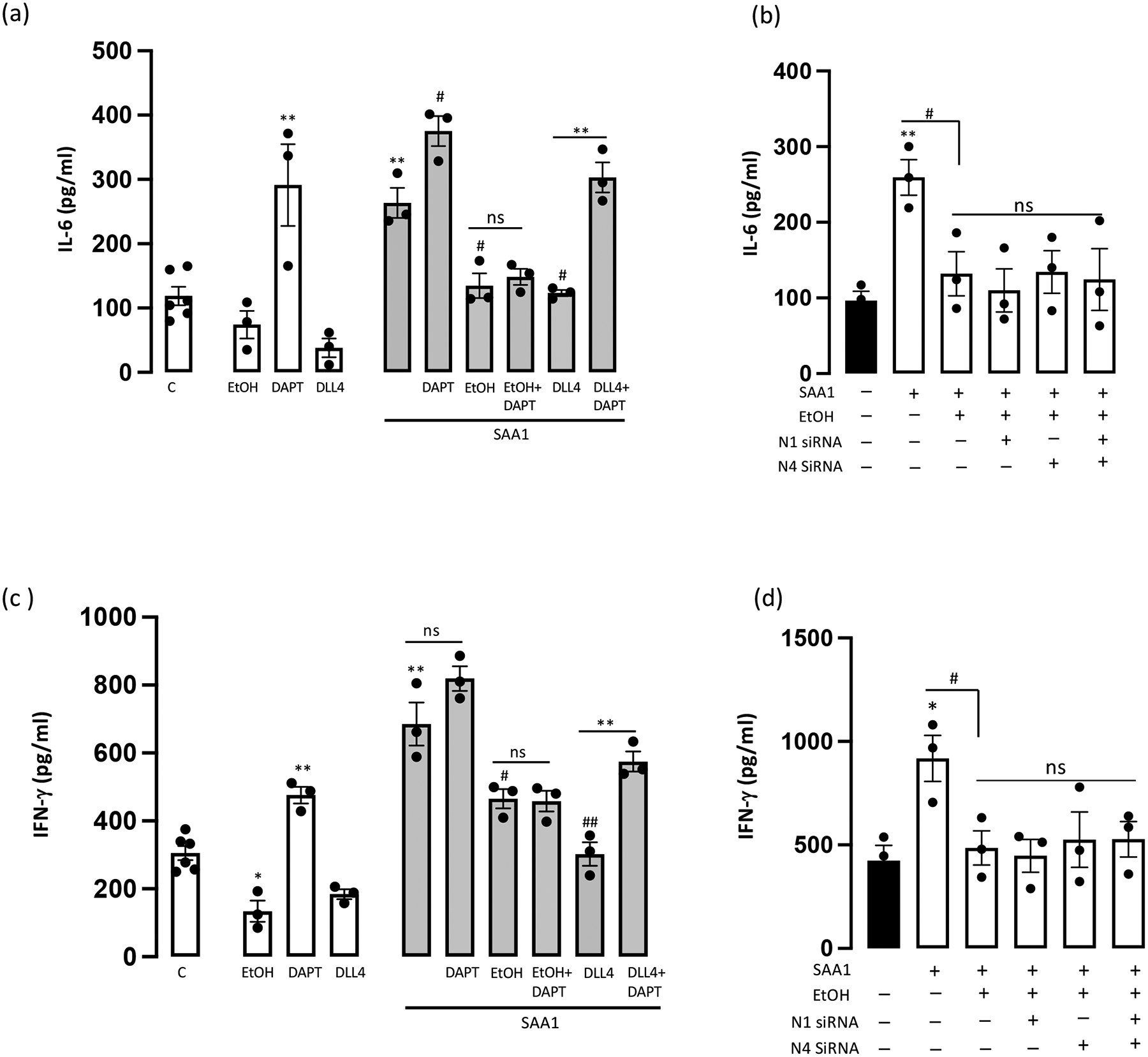
Ethanol inhibition of SAA1-induced inflammatory cytokines is Notch-independent. (a) Interleukin-6 (IL-6) levels in HCAEC supernatants following treatments (24 hr) as indicated; EtOH (25 mM), DAPT (10 μM), DLL4 (1 μg/ml), SAA1 (1 μM). (b) Effect of Ethanol on SAA1-induced IL-6 in HCAEC with/without N1 and/or N4 knockdown. (c) Interferon-gamma (IFN-γ) levels in HCAEC supernatants following treatments (24 hr) as above. (d) Effect of Ethanol on SAA1-induced IFN-γ in HCAEC with/without N1 and/or N4 knockdown. Neither DAPT treatment nor Notch receptor knockdown prevented EtOH’s inhibition of the SAA1-induced IL-6 and IFN-γ response. Data are mean ± SEM, n=3. *P<0.05, **P<0.01 vs C, #P<0.05, ##P<0.01 vs SAA1.
Ethanol inhibits SAA1-stimulated ROS via a Notch-independent mechanism.
Intracellular reactive oxygen species (ROS) generation was measured using a cell-permeable fluorescent probe (Fig 6a). There was no significant effect on basal ROS generation following either ethanol or DLL4 exposure (Fig 6b), while treatment with DAPT alone substantially increased ROS (Fig 6b). Exposure to ‘atherogenic’ SAA1 elicited a robust increase in ROS generation by HCAEC (Fig 6b), a response that was significantly reduced by co-treatment with either ethanol or DLL4 (Fig 6b). The ethanol inhibitory effect on SAA1-induced ROS generation was not affected by the γ-secretase inhibitor DAPT (Fig 6b), and was still present in HCAEC in which N1 and/or N4 receptors were knocked down using specific siRNAs (Fig 6c), suggesting it is a Notch-independent effect.
Figure 6.
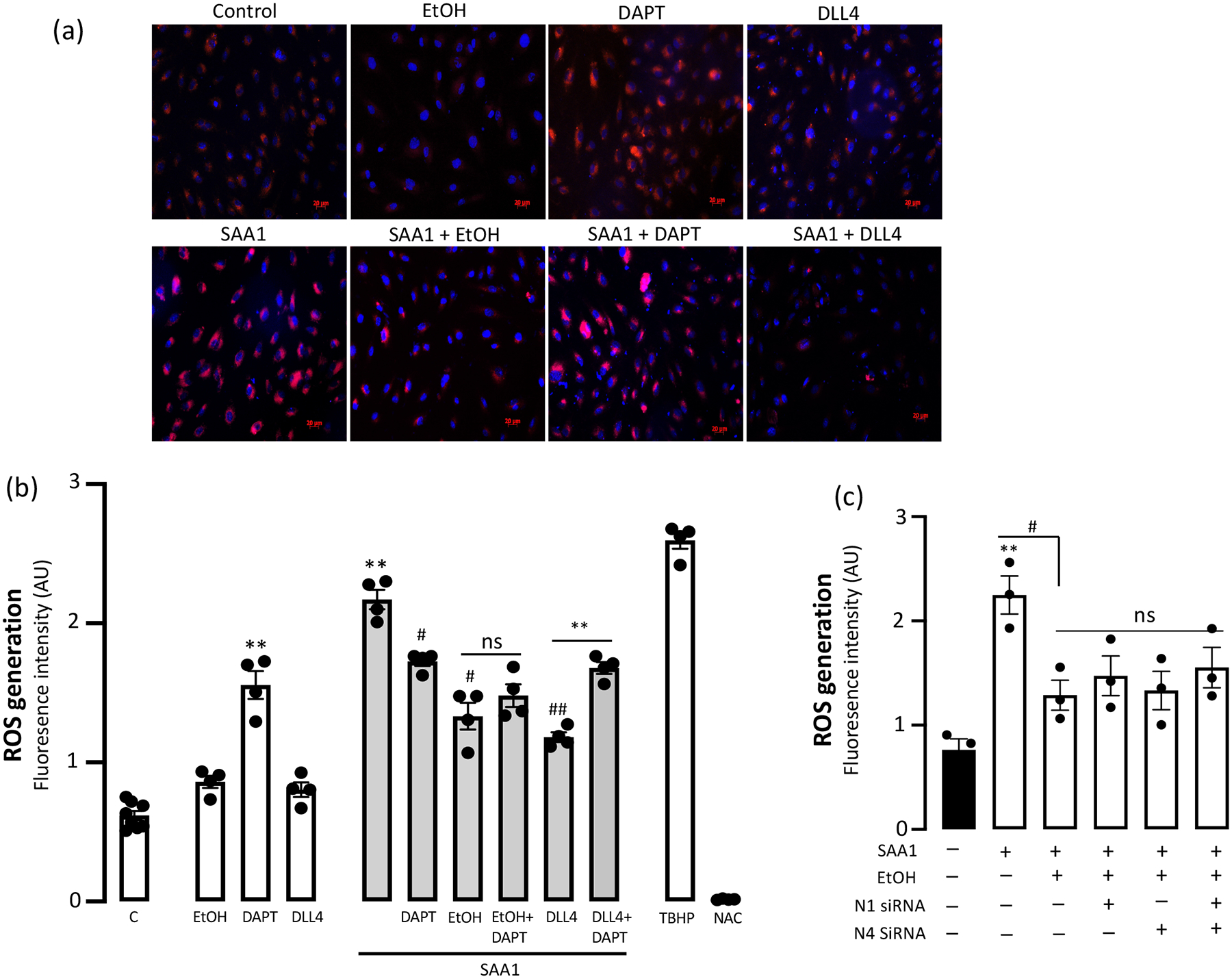
Ethanol inhibition of SAA1-induced Reactive Oxygen Species (ROS) is Notch-independent. (a) (b) Following treatment as specified {EtOH (25 mM), DAPT (10 μM), DLL4 (1 μg/ml), SAA1 (1 μM)} intracellular ROS generation in HCAEC was quantified by microplate reader. TBHP (tert-Butyl hydroperoxide) is a positive control, NAC (N-acetyl Cysteine) is a negative control. Representative images (a) showing ROS (red immunofluoresence), cumulative quantitative data in (b). (c) Effect of Ethanol on SAA1-stimulated ROS generation in HCAEC with/without N1 and/or N4 knockdown. Data are mean ± SEM, n=3. *P< 0.05, **P<0.01 vs C; #P<0.01 vs SAA1, ## P<0.001 vs SAA1. Neither DAPT treatment nor Notch receptor knockdown prevented EtOH’s inhibition of SAA1-induced ROS.
DISCUSSION.
With the ultimate goal of reducing the health burden associated with atherosclerotic cardiovascular disease (CVD), much research is focused on maintaining a healthy endothelium given its importance in vascular homeostasis and disease abatement (Cahill and Redmond, 2016). Compromised endothelial function is linked with risk factors, correlates with disease progression, and is predictive of cardiovascular events. This study investigated the anti-atherogenic potential of moderate levels of alcohol in endothelial cells exposed to a pro-inflammatory serum protein (i.e., SAA1) implicated in the progression of atherosclerosis. We report that alcohol prevents SAA1-induced endothelial activation via both Notch-dependent and -independent mechanisms. Notch-dependent ‘endothelial-protective’ effects of alcohol included those on eNOS activity, endothelial barrier integrity, cell adhesion molecule expression, and monocyte adhesion whereas Notch-independent effects were those on inflammatory cytokines and reactive oxygen species (Fig 7). These data may underpin mechanisms mediating the cardiovascular protective effects associated with moderate alcohol consumption.
Figure 7.
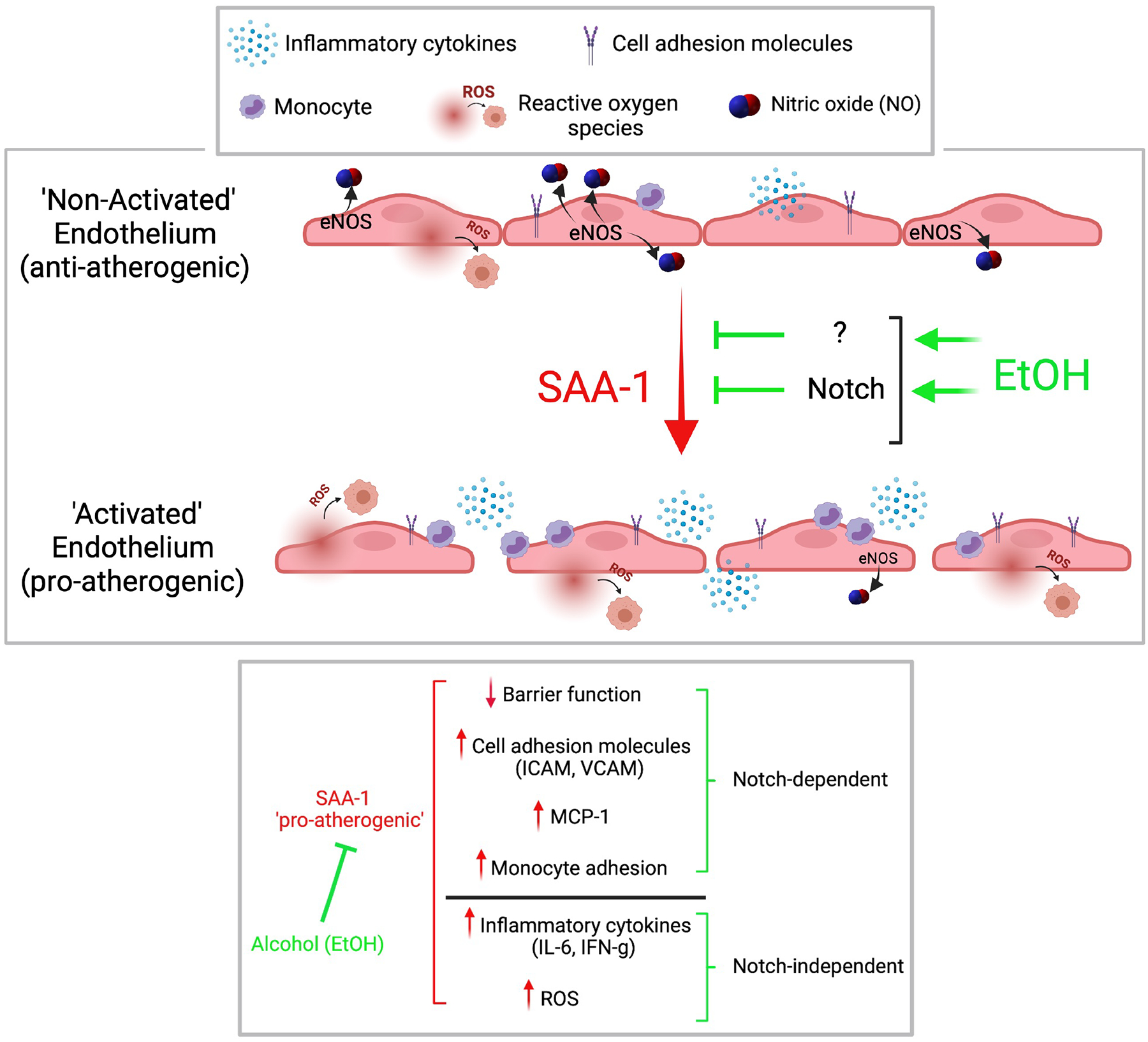
Alcohol at moderate levels inhibits SAA1-induced endothelial activation via both Notch-dependent and Notch-independent mechanisms. Maintenance of endothelium by alcohol in a non-activated state would be expected to preserve vessel homeostasis and protect against atherosclerosis development.
Alcohol consumption is a modifiable behaviour of considerable note in relation to cardiovascular health. Drinking may have either positive or negative effects, largely dependent not only on the amount but also the pattern of consumption (Liu et al., 2011). Meta-analyses of population studies reveal that chronic alcohol abuse and binge drinking is associated with increased cardiovascular-related morbidity and mortality (Rehm and Roerecke, 2017). On the other hand, compared with abstinence, regular alcohol consumption at low to moderate levels is associated with the lowest risk for atherosclerosis-related mortality from myocardial infarction and ischemic stroke (Colpani et al., 2018, Huang et al., 2017). ‘Low-moderate’ alcohol consumption is considered to be in the range 1–3 drinks/day giving rise to blood alcohol levels of 5–25 mM. Elucidating the cell and signaling mechanisms mediating the apparent beneficial effects of low-moderate alcohol is of great interest.
We have previously reported differential effects of ethanol on canonical Notch signalling in vascular cells; inhibitory in smooth muscle (human coronary artery) (Morrow et al., 2010), and stimulatory in endothelial cells (human umbilical vein) (Morrow et al., 2008a, Morrow et al., 2014). Here, the stimulatory effect of ethanol on endothelial Notch signalling was confirmed in human coronary artery endothelial cells. Furthermore, we report for the first time that this response is likely mediated by stimulation of γ-secretase proteolytic activity in these cells by ethanol. γ-secretase is a multi-subunit protease complex composed of presenilin, nicastrin, Aph-1, and Pen-2 (Shih Ie and Wang, 2007). Cleavage mediated by γ-secretase releases the active intracellular domain of the Notch receptor (NICD), a step necessary for signalling to proceed. Taken together with our previously published report (Hatch et al., 2015), these data highlight a novel cell-specific regulation of γ-secretase by alcohol; inhibitory in smooth muscle cells, but stimulatory in endothelium.
While Notch is essential during embryological vascular development (Artavanis-Tsakonas et al., 1999), its recapitulation and influence in adult vasculature is less understood. Endothelial Notch acts as a mechanosensor, as well as a metabolic sensor (Briot et al., 2016, Miloudi et al., 2019). Moreover, a growing body of evidence suggests that Notch pathway dysregulation is involved in the pathophysiology of atherosclerosis, aortic aneurysms, and heart failure (Aquila et al., 2019). Dialling down Notch signalling in arterial endothelial cells, at least under certain conditions, is thought to unlock pro-inflammatory and pro-atherogenic signals, giving rise to the idea that this pathway helps preserve a quiescent/stable endothelial phenotype (Briot et al., 2016, Mack and Iruela-Arispe, 2018). Our findings bolster this concept. Repression of Notch signalling in coronary artery endothelial cells resulted in compromised barrier function, together with increased cell adhesion molecule expression, monocyte adhesion, inflammatory cytokine and ROS production - all of which are generally considered pro-atherogenic changes. Conversely, activation of Notch signalling in these cells stimulated eNOS activity and increased monolayer barrier integrity, while reducing cell adhesion molecule expression, monocyte adhesion, and inflammatory cytokine production - all considered anti-atherogenic changes. These data strongly indicate that Notch signalling acts to maintain endothelium in a non-activated, anti-atherogenic state.
In general agreement with human population data, previous studies in mice describe a protective effect of moderate alcohol consumption on pathologic arterial remodelling (Emeson et al., 2000, Morrow et al., 2010) and, further, highlight medial smooth muscle and resident vascular stem cells as targets for alcohol (Morrow et al., 2010) (Fitzpatrick et al., 2017, Liu et al., 2020). Moreover, various effects of ethanol on endothelial cell function have previously been reported by us and others, both in vitro and in vivo, including those on nitric oxide, endothelin, MCP-1, and monocyte adhesion (Chiva-Blanch et al., 2013, Hendrickson et al., 1999, Morrow et al., 2008a). Here, in addition to determining baseline effects, we investigated the ability of ethanol to protect the endothelium against an atherogenic stimulus, in this instance SAA1, and determined the Notch-dependency of these effects.
Treatment of arterial endothelial cells under basal conditions with Ethanol, at a concentration achievable by moderate consumption, increased eNOS activity in a Notch-dependent manner. Furthermore, stimulation of Notch signalling with DLL4 increased eNOS activity in these cells. NOS activation results in production of the vasodilator nitric oxide (NO), an important protective molecule in the vasculature (Gkaliagkousi and Ferro, 2011). Impaired NO-mediated (i.e., ‘endothelium-dependent’) vasodilation is defined as a hallmark of endothelial dysfunction (Davignon and Ganz, 2004). In addition to its role as a vasodilator, NO is a potent inhibitor of platelet aggregation and adhesion. It also can regulate vascular permeability in a context -dependent manner. While we and others have previously reported a stimulatory effect of ethanol on eNOS activity, our data here are the first to show that this is a Notch-dependent effect. In addition to eNOS stimulation, Ethanol treatment of coronary artery endothelial cells improved their barrier function, decreased ICAM-1 expression, reduced monocyte adhesion to the endothelial monolayer, and reduced IFN-γ cytokine production.
The evolutionarily conserved Serum Amyloid A (SAA) family of proteins are found as normal constituents in blood serum. SAA1 and SAA2 are two representatives that, along with C-reactive protein, are the most prominent members of the acute phase response during which their serum levels rise dramatically after trauma, infection and other stimuli (Sack, 2020). In addition to being plasma biomarkers for cardiovascular events, they have a pathogenic role in the development of atherosclerosis (Getz et al., 2016, Shridas and Tannock, 2019). Indeed, in our study, arterial endothelial cells exposed to SAA1 displayed several robust pro-atherogenic responses. Specifically, SAA1 (which itself had no effect on Notch signalling) increased endothelial monolayer leakiness, increased cell adhesion molecule (ICAM-1 and VCAM) and MCP-1 mRNA expression, and stimulated monocyte adhesion, inflammatory cytokine (IL-6, IFN-γ) production and ROS generation. All of these SAA1-induced ‘pro-atherogenic’ effects were markedly diminished (in most cases completely blocked) by Ethanol co-treatment, or by stimulation of Notch signalling with DLL4. Of the SAA1-responses prevented by Ethanol the TEER, cell adhesion molecule, MCP-1 and THP-1 monocyte adhesion were Notch-dependent as they were abolished by either γ-secretase inhibition or by Notch receptor knockdown.
In contrast, the ethanol-inhibitory effects on SAA1-induced inflammatory cytokines and on ROS generation were Notch-independent as they remained even when Notch signalling was inhibited by a GSI or when Notch receptors were knocked down. A recent study by Mors et al., reported that acute ethanol treatment at high levels (170 mM, 1hr) decreased inflammatory cytokines and neutrophil adhesion in interleukin-1β-stimulated human lung epithelial cells by inhibiting canonical NF-κB signalling (Mors et al., 2017). NF-κB transcription factors (canonical and non-canonical) are particularly important throughout the immune system having both anti-oxidant and pro-oxidant functions, and their upregulation can result in excessive generation of both reactive oxygen species and inflammatory cytokines (Hayden and Ghosh, 2011). It will be of interest in future studies to uncover the precise mechanisms mediating the Notch-independent arterial endothelial effects of moderate alcohol reported here and to investigate any involvement of NF-kB signalling components in this context.
Overall, these data indicate that ethanol, at moderate levels, acts to maintain stimulated endothelium in an non-activated state by both Notch-dependent and -independent mechanisms. The general consequence of such ethanol effects would be expected to preserve vessel homeostasis, despite a pro-atherogenic milieu, and protect against atherosclerosis. These findings might explain the reduced vessel disease and sequalae reported in alcohol consumers.
Acknowledgements.
The authors wish to thank Dr Clive S. Kent and Karl R. Vandermeid for supplying freshly isolated human monocytes. This work was supported in part by grants from the National Institutes of Health RO1AA024082 and R21AA023213 to EMR, and by Science Foundation Ireland grant SFI-11/PI/1128, Health Research Board of Ireland (HRB) grant HRA-POR-2015-1315, and the European Union’s INTERREG VA Programme, managed by the Special EU Programmes Body (SEUPB) to PAC.
Footnotes
Conflict of interest: None declared
REFERENCES
- Aquila G, Kostina A, Vieceli Dalla Sega F, Shlyakhto E, Kostareva A, Marracino L, Ferrari R, Rizzo P, Malaschicheva A (2019) The Notch pathway: a novel therapeutic target for cardiovascular diseases? Expert Opin Ther Targets 23:695–710. [DOI] [PubMed] [Google Scholar]
- Artavanis-Tsakonas S, Rand MD, Lake RJ (1999) Notch signaling: cell fate control and signal integration in development. Science 284:770–776. [DOI] [PubMed] [Google Scholar]
- Basatemur GL, Jorgensen HF, Clarke MCH, Bennett MR, Mallat Z (2019) Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol. [DOI] [PubMed] [Google Scholar]
- Briot A, Bouloumie A, Iruela-Arispe ML (2016) Notch, lipids, and endothelial cells. Curr Opin Lipidol 27:513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill PA, Redmond EM (2016) Vascular endothelium - Gatekeeper of vessel health. Atherosclerosis 248:97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiva-Blanch G, Arranz S, Lamuela-Raventos RM, Estruch R (2013) Effects of wine, alcohol and polyphenols on cardiovascular disease risk factors: evidences from human studies. Alcohol and alcoholism (Oxford, Oxfordshire) 48:270–277. [DOI] [PubMed] [Google Scholar]
- Colpani V, Baena CP, Jaspers L, van Dijk GM, Farajzadegan Z, Dhana K, Tielemans MJ, Voortman T, Freak-Poli R, Veloso GGV, Chowdhury R, Kavousi M, Muka T, Franco OH (2018) Lifestyle factors, cardiovascular disease and all-cause mortality in middle-aged and elderly women: a systematic review and meta-analysis. Eur J Epidemiol 33:831–845. [DOI] [PubMed] [Google Scholar]
- Davignon J, Ganz P (2004) Role of endothelial dysfunction in atherosclerosis. Circulation 109:III27–32. [DOI] [PubMed] [Google Scholar]
- Dong Z, Wu T, Qin W, An C, Wang Z, Zhang M, Zhang Y, Zhang C, An F (2011) Serum amyloid A directly accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Mol Med 17:1357–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emeson EE, Manaves V, Emeson BS, Chen L, Jovanovic I (2000) Alcohol inhibits the progression as well as the initiation of atherosclerotic lesions in C57Bl/6 hyperlipidemic mice. Alcoholism, clinical and experimental research 24:1456–1466. [PubMed] [Google Scholar]
- Fitzpatrick E, Han X, Liu W, Corcoran E, Burtenshaw D, Morrow D, Helt JC, Cahill PA, Redmond EM (2017) Alcohol Reduces Arterial Remodeling by Inhibiting Sonic Hedgehog-Stimulated Stem Cell Antigen-1 Positive Progenitor Stem Cell Expansion. Alcoholism, clinical and experimental research 41:2051–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getz GS, Krishack PA, Reardon CA (2016) Serum amyloid A and atherosclerosis. Curr Opin Lipidol 27:531–535. [DOI] [PubMed] [Google Scholar]
- Ghatak S, Reveiller M, Toia L, Ivanov AI, Zhou Z, Redmond EM, Godfrey TE, Peters JH (2016) Bile Salts at Low pH Cause Dilation of Intercellular Spaces in In Vitro Stratified Primary Esophageal Cells, Possibly by Modulating Wnt Signaling. J Gastrointest Surg 20:500–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkaliagkousi E, Ferro A (2011) Nitric oxide signalling in the regulation of cardiovascular and platelet function. Front Biosci 16:1873–1897. [DOI] [PubMed] [Google Scholar]
- Hatch E, Morrow D, Liu W, Cahill PA, Redmond EM (2015) Ethanol inhibits gamma-secretase proteolytic activity in vascular smooth muscle cells. Alcoholism, clinical and experimental research 39:2115–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S (2011) NF-kappaB in immunobiology. Cell Res 21:223–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson RJ, Cahill PA, Sitzmann JV, Redmond EM (1999) Ethanol enhances basal and flow-stimulated nitric oxide synthase activity in vitro by activating an inhibitory guanine nucleotide binding protein. The Journal of pharmacology and experimental therapeutics 289:1293–1300. [PubMed] [Google Scholar]
- Huang Y, Li Y, Zheng S, Yang X, Wang T, Zeng J (2017) Moderate alcohol consumption and atherosclerosis : Meta-analysis of effects on lipids and inflammation. Wien Klin Wochenschr 129:835–843. [DOI] [PubMed] [Google Scholar]
- Liu W, Harman S, DiLuca M, Burtenshaw D, Corcoran E, Cahill PA, Redmond EM (2020) Moderate Alcohol Consumption Targets S100beta(+) Vascular Stem Cells and Attenuates Injury-Induced Neointimal Hyperplasia. Alcoholism, clinical and experimental research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Redmond EM, Morrow D, Cullen JP (2011) Differential effects of daily-moderate versus weekend binge alcohol consumption on atherosclerotic plaque development in mice. Atherosclerosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack JJ, Iruela-Arispe ML (2018) NOTCH regulation of the endothelial cell phenotype. Current opinion in hematology 25:212–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majesky MW, Dong XR, Regan JN, Hoglund VJ (2011) Vascular smooth muscle progenitor cells: building and repairing blood vessels. Circulation research 108:365–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miloudi K, Oubaha M, Menard C, Dejda A, Guber V, Cagnone G, Wilson AM, Tetreault N, Mawambo G, Binet F, Chidiac R, Delisle C, Buscarlet M, Cerani A, Crespo-Garcia S, Bentley K, Rezende F, Joyal JS, Mallette FA, Gratton JP, Larrivee B, Sapieha P (2019) NOTCH1 signaling induces pathological vascular permeability in diabetic retinopathy. Proceedings of the National Academy of Sciences of the United States of America 116:4538–4547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow D, Cullen JP, Cahill PA, Redmond EM (2008a) Ethanol stimulates endothelial cell angiogenic activity via a Notch- and angiopoietin-1-dependent pathway. Cardiovascular research 79:313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow D, Cullen JP, Liu W, Cahill PA, Redmond EM (2010) Alcohol inhibits smooth muscle cell proliferation via regulation of the Notch signaling pathway. Arteriosclerosis, thrombosis, and vascular biology 30:2597–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow D, Guha S, Sweeney C, Birney Y, Walshe T, O’Brien C, Walls D, Redmond EM, Cahill PA (2008b) Notch and vascular smooth muscle cell phenotype. Circulation research 103:1370–1382. [DOI] [PubMed] [Google Scholar]
- Morrow D, Hatch E, Hamm K, Cahill PA, Redmond EM (2014) Flk-1/KDR mediates ethanol-stimulated endothelial cell Notch signaling and angiogenic activity. Journal of vascular research 51:315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mors K, Horauf JA, Kany S, Wagner N, Sturm R, Woschek M, Perl M, Marzi I, Relja B (2017) Ethanol Decreases Inflammatory Response in Human Lung Epithelial Cells by Inhibiting the Canonical NF-kB-Pathway. Cell Physiol Biochem 43:17–30. [DOI] [PubMed] [Google Scholar]
- O’Keefe JH, Bhatti SK, Bajwa A, DiNicolantonio JJ, Lavie CJ (2014) Alcohol and cardiovascular health: the dose makes the poison…or the remedy. Mayo Clin Proc 89:382–393. [DOI] [PubMed] [Google Scholar]
- Rehm J, Roerecke M (2017) Cardiovascular effects of alcohol consumption. Trends Cardiovasc Med 27:534–538. [DOI] [PubMed] [Google Scholar]
- Ribeiro F, Alves AJ, Teixeira M, Ribeiro V, Duarte JA, Oliveira J (2009) Endothelial function and atherosclerosis: circulatory markers with clinical usefulness. Rev Port Cardiol 28:1121–1151. [PubMed] [Google Scholar]
- Sack GH Jr. (2020) Serum Amyloid A (SAA) Proteins. Subcell Biochem 94:421–436. [DOI] [PubMed] [Google Scholar]
- Shawber CJ, Kitajewski J (2004) Notch function in the vasculature: insights from zebrafish, mouse and man. Bioessays 26:225–234. [DOI] [PubMed] [Google Scholar]
- Shih Ie M, Wang TL (2007) Notch signaling, gamma-secretase inhibitors, and cancer therapy. Cancer research 67:1879–1882. [DOI] [PubMed] [Google Scholar]
- Shridas P, Tannock LR (2019) Role of serum amyloid A in atherosclerosis. Curr Opin Lipidol 30:320–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluiter TJ, van Buul JD, Huveneers S, Quax PHA, de Vries MR (2021) Endothelial Barrier Function and Leukocyte Transmigration in Atherosclerosis. Biomedicines 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Delling FN, Djousse L, Elkind MSV, Ferguson JF, Fornage M, Khan SS, Kissela BM, Knutson KL, Kwan TW, Lackland DT, Lewis TT, Lichtman JH, Longenecker CT, Loop MS, Lutsey PL, Martin SS, Matsushita K, Moran AE, Mussolino ME, Perak AM, Rosamond WD, Roth GA, Sampson UKA, Satou GM, Schroeder EB, Shah SH, Shay CM, Spartano NL, Stokes A, Tirschwell DL, VanWagner LB, Tsao CW (2020) Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 141:e139–e596. [DOI] [PubMed] [Google Scholar]