

Summary
Maternal stress can have long-lasting epigenetic effects on offspring. To examine how epigenetic changes are triggered by stress, we examined the effects of activating the universal stress-responsive heat-shock transcription factor HSF-1 in the germline of Caenorhabditis elegans. We show that when activated in germ cells HSF-1 recruits MET-2, the putative histone 3 lysine 9 (H3K9) methyltransferase responsible for repressive H3K9me2 marks in chromatin, and negatively bookmarks the insulin-receptor daf-2 and other HSF-1-target genes. Increased H3K9me2 at these genes persists in adult progeny and shifts their stress response strategy away from inducible chaperone expression as a mechanism to survive stress, to rely instead on decreased Insulin/ IGF-1-like signaling (IIS). Depending on the duration of maternal heat-stress exposure, this epigenetic memory is inherited by the next generation. Thus, paradoxically, HSF-1 recruits the germline machinery normally responsible for erasing transcriptional memory, but instead, establishes a heritable epigenetic memory of prior stress exposure.
Keywords: Heat shock, HSF1, C.elegans, H3K9 methyltransferase, MET-2, epigenetic, daf-2, stress, transgenerational, memory, transcriptional plasticity
Graphical Abstract
eTOC Blurb
Das et al. show that the transcription factor HSF-1, activated in the C. elegans germline by heat-shock, bookmarks target genes by directly recruiting the putative H3K9methyltransferase MET-2. This causes a transcriptional switch in offspring away from inducible chaperone expression as a stress survival mechanism towards decreased Insulin/IGF-1-like signaling and DAF-16.
Introduction
Many organisms display transgenerational plasticity whereby the memory of parental exposure to stress alters later life cellular responses and survival mechanisms of offspring (Brunet and Berger, 2014; D'Urso and Brickner, 2017; Leimar and McNamara, 2015; Sen et al., 2016). How epigenetic changes that occur during perinatal life are maintained to affect later-life physiology is unclear. This question is underscored because during normal development, in all animals, chromatin modifications acquired during germline transcriptional activity are removed at fertilization to epigenetically reprogram the embryo (D'Urso and Brickner, 2017; Guo et al., 2017; Kaneshiro et al., 2019). These reprograming mechanisms remove marks of active gene expression that are added during normal germline transcription, and in some cases increase repressive marks at germline active genes, to prevent their ectopic expression in subsequent generations (Andersen and Horvitz, 2007; Bessler et al., 2010; Carpenter et al., 2021; Rechtsteiner et al., 2019). Nevertheless, in many organisms the memory of early-life stress exposure persists through meiosis and cycles of mitosis to alter later-life cellular programs through as yet poorly understood mechanisms (Brunet and Berger, 2014; D'Urso and Brickner, 2017; Leimar and McNamara, 2015; Sen et al., 2016). This early life programming, or maternal effect, can be beneficial for progeny, altering their phenotypes to decrease predation risk (Mommer and Bell, 2014); alternatively, it can be detrimental as has been observed in humans where maternal stress increases the later-life susceptibility of offspring to metabolic and neuropsychiatric disease (Brown et al., 2019; Tartaglione et al., 2016; Tobi et al., 2014; Zimmet, 2017).
As in many organisms, in the nematode Caenorhabditis elegans environmental stressors that impact the parent also affect the unfertilized gametes. In C. elegans, epigenetic mechanisms that affect chromatin in one generation have been found to alter offspring lifespan and behavior in subsequent generations (Baugh and Day, 2020; Baugh and Hu, 2020; Camacho et al., 2018; Carpenter et al., 2021; Furuhashi et al., 2010; Greer et al., 2014; Greer et al., 2011; Houri-Ze'evi et al., 2016; Jobson et al., 2015; Kaneshiro et al., 2019; Katz et al., 2009; Kerr et al., 2014; Kreher et al., 2018; Lev et al., 2017; Lim and Brunet, 2013; Minkina and Hunter, 2018; Moore et al., 2019; Posner et al., 2019; Rechavi et al., 2014; Tabuchi et al., 2018; Wan et al., 2021). In the parent, stress activates the heat-shock transcription factor, HSF1 (HSF-1 in C. elegans), which increases the expression of protective heat-shock protein (hsp) genes to counteract stress-induced damage (Gomez-Pastor et al., 2018; Vihervaara et al., 2018). We previously found that even a brief and transient exposure to increased temperatures also activates HSF-1 in the germline of C. elegans (Das et al., 2020). Therefore, to understand how early-life stress exposure alters later-life physiology, we examined the later-life survival mechanisms and proteostasis of animals that had activated HSF-1, early-in-life, as germ cells.
Results
HSF-1 germline activity alters the later-life stress-resilience and proteostasis in animals that develop from these germline cells.
We first established an experimental protocol to activate HSF-1 in germ cells before their fertilization and identify offspring generated from these germ cells (Figure 1A). To do this, we exposed one-day-old, gravid mothers to 34°C heat-shock. During this stress exposure, oocytes, and sperm resident in the animal’s germline as part of the germline syncytium also experience heat-shock. Following heat-shock, animals were moved to normal growth temperatures of 20°C where the heat-shocked oocytes completed maturation, were fertilized by the heat-shocked sperm, and developed into adults. To activate HSF-1 in the germline, we used different regimens consisting of both long heat exposures (30-minutes or 60-minutes at 34°C) typically used to activate HSF-1 throughout the animal (Das et al., 2020; Kumsta et al., 2017; Labbadia and Morimoto, 2015; Li et al., 2016) and a shorter, 5-minute heat-shock at 34°C, which induces HSF-1-dependent expression of its main target hsp genes, hsp-70 (C12C8.1) and the hsp-70-like gene, F44E5.4/.5, almost exclusively in the germline (Das et al., 2020). We confirmed that heat-shock triggered HSF-1 binding at germline chromatin by using ChIP-PCR (Chromatin immunoprecipitation followed by PCR) to measure HSF-1 occupancy at the promoter regions of hsp genes in chromatin extracted from whole animals after hsf-1 was knocked down only in the germline using RNA interference (RNAi) (Figures S1A-S1C; see Figure S1D for antibody and RNAi validation). For these experiments, animals that expressed endogenous HSF-1 tagged with 3X FLAG (Das et al., 2020) were used and HSF-1 occupancy at the syp-1 promoter served as a control (Figure S1C). Germline knockdown of hsf-1 abolished HSF-1 binding to target hsp promoter regions upon a 5-minute heat-shock, and this remained reduced upon longer heat exposure (Figures S1A and S1B). This confirmed that HSF-1 is indeed activated in germline cells upon heat-shock, and during the first 5-minutes of heat exposure, HSF-1 binding to target hsp genes occurs almost exclusively in germline cells.
Figure 1: HSF-1 germline activity alters the stress resilience and proteostasis mechanisms of progeny that develop from stressed germ cells.

(A) Experimental procedure (also see STAR Methods).
(B) Stress resilience of F1 progeny of C. elegans mothers subjected to a short (5-minute) or long (30-minute or 60-minute) heat-shock (HS).. X-axis: the time interval at 20°C after the maternal heat-shock at which progeny were collected. Non-heat shocked control: NHS(Ctrl). Y axis: Percent F1 progeny surviving severe heat-stress. Legend: duration of maternal heat-shock. n=3-6 experiments/time point. Each experiment represents 3-20 P0 mothers/condition/time interval and an average of 24.3±2.3 F1 progeny/time interval/experiment for the short heat-shock experiments and 50.4±7.8 F1 progeny/time interval/experiment for long heat-shock experiments.
(C) Stress resilience of F1 progeny of NHS(Ctrl), and 5-minute heat-shocked P0 mothers, following hsf-1 germline RNAi. Control (Ctrl) RNAi: L4440 empty vector. n=4-8 experiments, 27.4±0.8 and 23.4±2.7 F1 progeny/experiment on L4440 and hsf-1 RNAi respectively.
(D) Stress resilience of F2 and F3 progeny of NHS(Ctrl) and heat-shocked P0 mothers. Heat-shock: 5-minute, 30-minute and 60-minute. Y axis: Percent NHS(Ctrl), F2 and F3 progeny that surive severe heat-stress. Legend indicates duration of maternal heat-shock. Each experiment represents 3-20 P0 mothers/condition and F2 and F3s collected from 5-40 F1 or F2 progeny/condition.
(E) Number of polyglutamine aggregates in day-one to day-four adult F1 progeny of NHS(Ctrl) and heat-shocked P0 mothers. Heat-shock: 5-minute, 30-minute or 60-minute. n=3 experiments, 25 F1 progeny/experiment from 3-20 P0 mothers/condition.
B-E: Data show Mean ± Standard Error of the Mean. *: p<0.05, **: p<0.01, ***: p<0.001, ns: non-significant; Unpaired Student’s t-test.
See also Figures S1 and S2
By determining the number of fertilized eggs in the uterus and unfertilized oocytes in the germline after stress exposure (8.6 ± 0.5/animal), and the egg laying rates of mothers (Figures S1E and S1F), we established that embryos collected 2 hours after a 5-minute maternal heat-shock, 4 hours after a 30-minute heat-shock, or 8 hours after a 60-minute heat-shock were embryos that were reliably generated by oocytes in diakinesis that were resident in the maternal germline during heat exposure, but fertilized by heat-shocked sperm at 20°C during maternal recovery (Figures S1E and S1F). Therefore, embryos laid 2-4 hours, 4-8 hours or 8-12 hours following a 5-minute, 30-minute or 60-minute maternal heat-shock respectively, were used for all experiments (Figure 1A). These embryos were allowed to develop into one-day-old adults at 20°C, subjected to a severe heat-shock at 37°C, and scored for survival to determine whether stress exposure in utero had altered their later-life stress resilience. We found that progeny that developed from germ cells that had been exposed to heat-shock while in the maternal germline were more resilient to severe heat stress and significantly more survived compared to those from unstressed germ cells (Figure 1B). Moreover, while embryos collected between 2-4 hours post 5-minute maternal heat-shock were more stress resistant, oocytes fertilized and laid later after maternal stress (collected 8-12 hours post 5-minute maternal heat-shock), did not display this heightened thermotolerance (Figure 1B), suggesting that the early life programming of stress resilience was occuring through oocytes, and not sperm, since the same heat-shocked sperm also fertilize these later-laid embryos.
To more directly assess whether stress resilience was transmitted through sperm or oocytes, adult C. elegans males were heat-shocked for 60 minutes, allowed to mate with non-heat shocked hermaphrodites, and stress resilience of the F1 hermaphrodite cross-progeny generated from their heat-shocked sperm was measured (Figure S2A). Since the numbers of males and hermaphrodite progeny on the plate following mating were roughly equal, this assured us that we were examining cross-progeny and not self-progeny (see Star Methods). F1 hermaphrodite progeny generated by the fertilization of heat-shocked male sperm with non-heat shocked oocytes did not show increased stress resilience and were as stress tolerant as F1 hermaphrodite cross-progeny generated from non-heat shocked male sperm, or control progeny derived from unmated, non-heat shocked mothers (Figure S2B). While a contribution from sperm could not be completely excluded given the physiological differences between males and hermaphrodites, these data strongly suggested that stress resilience was being transmitted through the oocyte.
Downregulation of hsf-1 only in the maternal germline using RNAi was sufficient to abolish the stress tolerance in F1 offspring (Figure 1C; see Figure S1D for validation of RNAi). To test whether this reflected the requirement for HSF-1 stress-induced activity, or basal activity, we measured the stress resilience of animals harboring a mutation in hsf-1, hsf-1 (sy441) (Hajdu-Cronin et al., 2004). These animals express a truncated HSF-1 protein lacking the transactivation domain and HSF-1 binds constitutively to the promoter regions of its target genes in the absence of heat-shock, but is deficient in heat-shock induced binding (Figures S2C and S2D) and transcription (Das et al., 2020). Progeny that developed from heat-shocked germ cells of hsf-1 (sy441) animals also did not display enhanced stress resilience (Figure S2E). Together, these data showed that stress induced activity of germline HSF-1 was altering the stress resilience of progeny that developed from these stressed germ cells.
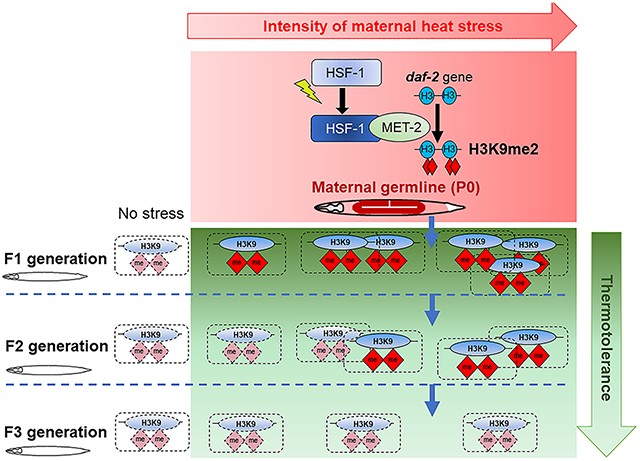
We next asked whether the increased stress-resilience seen in F1 progeny of heat-shocked mothers could be inherited by subsequent generations. To test this, we subjected C. elegans mothers to a 5-minute, 30-minute or 60-minute heat-shock, collected F1 progeny at 2-4 hours, 4-8 hours or 8-12 hours respectively following maternal heat-shock, allowed them to lay eggs at control temperatures of 20°C, and tested the stress resilience of the F2 and F3 generations after they developed into adults at 20°C (Figure 1D; Figure S2F). Remarkably, enhanced heat resilience was inherited by the F2 generation of P0 mothers subjected to the 60-minute heat-shock exposure but did not extend beyond the F1 generation when the P0 mothers were subjected to only a short, 5-minute heat-shock (Figure 1D). Fewer F2 progeny were stress-resilient compared to F1 progeny, but nevertheless the effects were consistent and significant (compare Figures 1D and 1B). More F2 progeny of P0 mothers subjected to a 30-minute heat-shock also survived severe heat-stress compared to progeny of control non-heat shocked P0 mothers, but the effects varied and were not significant (Figure 1D). None of the F3 generations of heat-shocked P0 mothers displayed enhanced heat-resilience (Figure 1D). Thus, it appeared that maternal stress exposure conferred long-lasting, and in some cases heritable inter-generational changes on germ cells resident within the mother during stress exposure. Moreover, there seemed to be a ‘maternal dose-effect’ on progeny stress resilience, with longer maternal stress exposure conferring protection to more offspring and persisting into the next generation.
Resilience to heat stress depends on molecular mechanisms that modulate protein homeostasis. Consistent with their ability to better survive severe temperatures, progeny of mothers exposed to heat-shock also partially suppressed the accumulation of misfolded and aggregated polyglutamine (polyQ) expansion proteins that begin to aggregate later in life (Figures 1E and S2G) (Morley et al., 2002; Prahlad and Morimoto, 2011). This occurred even though steady-state expression levels of polyQ expansion proteins in progeny of stressed mothers were similar to that in offspring of non-stressed mothers (Figure S2H). Thus, in addition to increasing the ability of progeny to survive heat-stress, early life stress exposure also altered the cellular mechanisms of protein homeostasis in offspring.
HSF-1 recruits MET-2 to increase H3K9me2 levels at HSF-1 target genes in germ cells.
In C. elegans, the epigenetic reprogramming of embryos occurs through the activities of the H3K4 demethylases SPR-5 and RBR-2 which remove H3K4me3 marks added at spermatogenesis genes during normal germline transcription (Carpenter et al., 2021; Christensen et al., 2007; Greer et al., 2014; Katz et al., 2009; Kerr et al., 2014; Nottke et al., 2011). These demethylases synergize with the putative methyltransferase MET-2 responsible for the addition of repressive H3K9me2 modifications, and with other repressive methyltransferases, together erase the memory of germline transcription to prevent ectopic gene expression in the next generation (Andersen and Horvitz, 2007; Bessler et al., 2010; Carpenter et al., 2021; Greer et al., 2014; Katz et al., 2009; Kerr et al., 2014; Rechtsteiner et al., 2019). Since adult offspring of stressed mothers were more stress resilient, we first considered the possibility that HSF-1-dependent transcription that occurred in heat-shocked mothers’ germlines had somehow escaped germline reprogramming, allowing HSF-1 to preserve the memory of prior transcription and more readily increase the expression of stress-protective hsp genes in the next generation. To test this, we investigated whether F1 progeny that developed from oocytes of mothers subjected to a 5-minute heat-shock had enhanced hsp expression upon heat-shock. This was not the case; instead of being enhanced, heat-shock induced hsp expression in these F1 progeny was attenuated. This was evident from the lower levels of hsp-70 and F44E5.4/.5 mRNA (Figures S3A and S3B), as well as from the decreased binding of HSF-1 to the promoter regions of hsp genes (Figures S3C and S3D) in the offspring of heat-shocked mothers, when compared to offspring of unstressed mothers. We asked whether offspring of heat-shocked mothers expressed higher basal levels of the molecular chaperone proteins HSP70 (HSP-1 in C. elegans) and HSP90 (DAF-21 in C. elegans) which inhibit HSF-1 activation but can increase thermotolerance, as this might explain the apparently contradictory results of increased stress relilience and decreased HSF-1 stress-inducibility. However, this was also not the case: Western blot analysis using antibodies to endogenous HA tagged HSP-1 (Figure S3E) and DAF-21 [Figure S3F;also see (Blanchette et al., 2015; Desse et al., 2021)] showed no differences in these protein levels between offspring of stressed mothers and non-stressed mothers. Inducible HSP-70 protein levels were also not higher in animals that had experienced early life stress exposure (Figure S3G). These data indicated that the increased later-life stress resilience of animals that had experienced in utero stress could not be attributed to mechanisms that maintained HSF-1 regulatory regions in a permissive state, nor was it due to the persistence of gene products that resulted from prior HSF-1 activation.
However, these data also suggested that MET-2 activity that occurs during normal germline reprogramming might be increasing H3K9me2 levels at the transcription start sites or promoter proximal regions of genes induced by HSF-1 in the germline, since the persistence of such marks could account for the muted expression of hsp genes in these progeny later in life (Barski et al., 2007; Rechtsteiner et al., 2019). Supporting this, knockdown of met-2 in the maternal germline restored the inducible levels of hsp-70 in adults that developed from heat-shocked mothers (Figure S4A; validation of RNAi, Figure S4B). Therefore, we used ChIP-PCR to directly measure H3K9me2 levels at the 5’-UTR regions of HSF-1 target hsp genes following HSF-1 germline activation (Figure 2A). For this, we exposed mothers to a 5-minute heat-shock, which induces HSF-1 binding to these hsp promoters predominantly in the germline (see Figures S2C and S2D). We found that within 2 hours following maternal heat-shock during which time oocytes progress through the maternal germline, H3K9me2 abundance at the promoter proximal 5’-UTR regions of hsp-70 and F44E5.4/.5 increased (Figures 2A and S4C). The small heat-shock protein gene, hsp-16.11, another germline target of HSF-1 (Das et al., 2020) also accrued H3K9me2 at its 5’-UTR region (Figure 2A and S4C). This increase in H3K9me2 required MET-2, as RNAi-induced knockdown of met-2 only in the maternal germline suppressed the H3K9me2 increase (Figure 2B, also see Figure S4D). The heat-shock induced increase in H3K9me2 levels also required the stress-induced activity of HSF-1 and was not observed in hsf-1 (sy441) animals, although, consistent with the constitutive DNA-binding activity of this mutant HSF-1, hsf-1 (sy441) mutant animals had slightly elevated levels of basal H3K9me2 at some genes (Figures 2A and S4C).
Figure 2: HSF-1 recruits MET-2 to HSF-1 target genes upon heat-shock and increases H3K9me2 at their 5’-UTR regions.

(A) H3K9me2 occupancy [relative to non-heat shocked control values [NHS(Ctrl)] of each respective strain; for data normalized to wild-type NHS(Ctrl), see Figure S4C] at the promoter proximal 5’-UTR regions of hsp-70(C12C8.1) I, F44E5.4/.5 II, hsp-16.11 V, hsp-16.2 V, hsp-16.41 V, unc-23 V, nurf-1 II, K10D3.6 I, C32H11.4 IV and pha-4 V in P0 wild-type and hsf-1 (sy441) I under NHS(Ctrl) conditions, immediately upon heat-shock (HS), and 2 hours after heat-shock (HS+rec). Heat-shock: 5-minutes. n=4-7 experiments. See STAR Methods for 5’-UTR regions assayed.
(B) H3K9me2 occupancy [relative to respective RNAi NHS(Ctrl); also see Figure S4D] at 5’-UTR of hsp-70(C12C8.1) I, F44E5.4/.5 II and hsp-16.11 V. H3K9me2 occupancy was assessed in P0 animals subjected to control and met-2 germline RNAi, under NHS(Ctrl) conditions and at 2 hours after a 5-minute heat-shock (HS+rec). n=4-8 experiments. Control RNAi: L4440.
(C) Representative Western blot from co-immunoprecipitation experiment using anti-HA antibody and lysates from NHS(Ctrl) and heat-shocked (30-minute), wild-type and hsf-1(sy441)I P0 animals expressing FLAG::HSF-1 and MET-2::HA. Left: antibodies used. 10% of Input is included. n=3 experiments. For antibody validation see Figure S1D and S4B.
(D) Representative micrographs showing projections of confocal z-sections through nuclei of diakinesis oocytes in wild-type and hsf-1(sy441) I animals, in NHS(Ctrl) and upon heat-shock (30-minute; also see Figure S6). Arrows: HSF-1 and MET-2 colocalization in nSBs at condensed chromosomes (DAPI) upon heat-shock. Dotted lines: nuclei. * somatic gonad sheath cell where HSF-1 constitutively forms nSBs colocalized with MET-2. Panels from left to right: overlap of HSF-1 immunostaining (red) with DAPI (blue), MET-2 immunostaining alone (green), and overlap of HSF-1 (red) and MET-2 (green) immunostaining.
A, B: Data: Mean ± Standard Error of the Mean. *: p<0.05, **: p<0.01, ***: p<0.001, ns: non-significant; ANOVA with Tukey’s correction.
D: Scale bar:5μm.
See also Figures S1, S3, S4, S5 and S6
We tested whether H3K9me2 levels increased at other HSF-1-germline target genes by examining H3K9me2 levels at five other heat-inducible genes, hsp16.2, hsp16.41, unc-23, nurf-1, and K10D3.6 (Brunquell et al., 2016; Das et al., 2020; Garrigues et al., 2019; Li et al., 2016) and two genes, pha-4 and C32H11.4 that are not upregulated by heat-shock, but are upregulated upon HSF-1 overexpression and lifespan-extension (Hsu et al., 2003; Sural et al., 2019). We confirmed that all seven genes are direct heat-shock targets of HSF-1 by ChIP-PCR (Figure S4E; HSF-1 occupancy at unc-23 upon heat-shock was higher but varied between repeats), and that the heat-induced genes are germline-expressed by measuring total heat-shock induced mRNA levels by qPCR after knocking down hsf-1 only in the germline (Figure S5A). Indeed, H3K9me2 levels increased at the 5’-UTR regions of all seven genes following heat-shock in a HSF-1-dependent manner (Figures 2A and S4C).
Given these findings, we questioned whether the heat-shock treatment itself triggered a non-specific increase in total H3K9me2. This was not the case and total H3K9me2 levels remained unaltered in wild-type mothers before and after heat-shock exposure as assessed by Western blots (Figure S5B). hsf-1(sy441) animals expressed higher basal levels of total H3K9me2, but also had elevated total H3 levels, neither of which changed upon heat-shock, as determined by normalizing total H3K9me2 and H3 levels to β-tubulin (Figure S5B). Thus, the increase in H3K9me2 observed by ChIP-PCR appeared to be due to the stress-induced activity of HSF-1 at target genes.
We tested whether HSF-1 directly interacted with MET-2 upon heat-shock activation. Using strains where wild type and hsf-1(sy441) mutants were tagged at their endogenous loci with a 3XFLAG tag and met-2 was tagged at its endogenous locus using 3X HA, we found that full-length, wild-type HSF-1 co-immunoprecipitated with MET-2 upon heat-shock but not under non-stressed conditions (Figure 2C, lane 2; compare to the lack of immunoprecipitation with non-specific mouse IgG. Antibody specificity in Figure S4B; MET-2 appears as a doublet). Upon heat-shock, or under control conditions, MET-2 did not associate with the truncated HSF-1 that lacked the transactivation domain in the hsf-1 (sy441) mutants (Figure 2C).
The interaction between HSF-1 and MET-2 was also supported by immunolocalization studies. In the absence of heat-shock, both wild type and mutant HSF-1 localize throughout the nucleus in a diffuse pattern (Figure 2D, top panel). Upon heat-shock or exposure to other stressors, C. elegans HSF-1 forms distinct foci of unknown function within the nucleus known as nuclear stress bodies, nSB., (Deonarine et al., 2021; Morton and Lamitina, 2013; Ooi and Prahlad, 2017; Tatum et al., 2015). MET-2 colocalized with HSF-1 nSBs upon heat-shock (Figures 2D and S6). HSF-1 and MET-2 nSBs were distinctly visible in diakinesis oocytes (Figure 2D), in germ cells in earlier stages of meiosis (pachytene; Figures S6A, S6B, S6C and S6D) and in somatic intestinal cells (Figures S6E and S6F) upon a longer, 30 minutes, heat-shock but only apparent in germline cells upon a short, 5-minute heat-shock (Figure S6A). In hsf-1 (sy441) mutant animals, HSF-1 did not form nSBs in diakinesis oocytes upon heat-shock (Figure 2D), and nSBs formed in pachytene nuclei appeared disorganized (Figures S6B, S6C and S6D). MET-2 failed to form foci upon heat-shock or co-localize with the mutant HSF-1 nSBs in hsf-1(sy441) germlines and remained largely uniform throughout the nuclear periphery in pachytene cells or spread throughout the cytoplasm in diakinesis (Figures 2D, S6B, S6C and S6D).
Thus, together, these studies indicated that HSF-1 directly recruits MET-2 to increase H3K9me2 at HSF-1 germline target genes upon heat-shock, likely through its C-terminal transactivation domain.
H3K9me2 levels are increased in embryonic nuclei and persist at genomic loci later-in life in animals subject to in utero stress exposure.
For the increased H3K9me2 marks accrued in the maternal germline to serve as an epigenetic memory of early life stress exposure, they would have to persist in the adult offspring of heat-shocked parents. We examined if this was the case. Consistent with the stress-induced interaction between HSF-1 and MET-2 in diakinesis oocytes, H3K9me2 levels were increased in the oocytes of heat-shocked mothers and remained elevated for at least up to two hours even after a short, 5-minute heat-shock (Figures 3A and 3B). In contrast, in the oocytes of unstressed C. elegans mothers H3K9me2 was undetectable, or barely detectable, as previously reported [Figures 3A and 3B, (Bessler et al., 2010)].
Figure 3: H3K9me2 levels remain elevated in adult F1 progeny subjected to in utero stress.

(A) Representative micrographs showing H3K9me2 staining in projections of confocal z-sections through the germline of P0 wild-type mothers under non-heat shocked control conditions [NHS(Ctrl)] and upon heat-shock. Heat-shock: 5-minute (5min HS) and 60-minute (60min HS). Insets: magnified images of diakinesis oocytes.
(B) Percent H3K9me2 positive diakinesis oocytes per gonad in P0 NHS(Ctrl) and upon heat-shock. Heat-shock: 5-minute and 60-minute. Percent H3K9me2 positive oocytes present in the P0 germline 2 hours after the 5-minute heat-shock (5min HS+rec) are also shown (corresponds to data in Figure 2A). n=2-6 experiments; oocytes in 3-15 gonads quantified/experiment.
(C) H3K9me2 occupancy [relative to NHS(Ctrl) from each respective condition] at the 5’-UTR regions of hsp-70(C12C8.1) I, F44E5.4/5 II, hsp-16.11 V, C32H11.4 IV and pha-4 V in day-one adult F1 progeny of NHS(Ctrl), and 5-minute heat-shocked, P0 mothers subjected to germline RNAi. RNAi: hsf-1 and control RNAi (L4440 empty vector). n=5 experiments.
(D) Top: Representative Western blot of lysates from day-one-adult F1 progeny of NHS(Ctrl) and heat-shocked P0 mothers. Heat-shock: 5-minutes and 30-minutes. Antibodies: anti-H3K9me2 (above) and anti-Histone H3 (below). Lysate from met-2(n4256) III animals show specificity of antibody. Also see Figure S7A. Bottom: H3K9me2 levels quantified relative to total H3, and normalized to NHS(Ctrl). n=6 experiments.
(E) Representative micrographs showing H3K9me2 staining in F1 embryos from NHS(Ctrl) and heat-shocked P0 mothers. Heat-shock: 5-minutes and 60-minutes.
(F) Fluorescent intensity (arbitrary units) in H3K9me2-stained nuclei of 2-4 cell stage, and <100 cell stage F1 embryos from NHS(Ctrl) and heat-shocked P0 mothers. Heat-shock: 5-minutes and 60-minutes. n=2-4 experiments, 17-47 nuclei/condition for 2-4 cell stage embryos, and 103-105 nuclei/condition for <100 cell stage embryo.
B-D, F: Data show Mean ± Standard Error of the Mean. *: p<0.05, **: p<0.01, ***: p<0.001, ns: non-significant; ANOVA with Tukey’s correction, and Unpaired Student’s t-test.
A: Scale bar:30μm. E: Scale bar: 15μm.
See also Figure S7
The increased abundance of H3K9me2 at the 5’-UTR of HSF-1 target genes also persisted in adult progeny that developed from these oocytes (Figure 3C: five HSF-1 target genes tested by ChIP-PCR: hsp-70, F44E5.4/.5, hsp-16.11, pha-4 and C32H11.4). H3K9me2 levels did not increase or increased only modestly in progeny of mothers subjected to RNAi-induced germline knockdown of hsf-1 prior to heat-shock (Figure 3C). Moreover, while heat-shocked mothers did not show any changes in total H3K9me2 levels (Figure S5B), progeny that developed into adults from heat-shocked oocytes had more total H3K9me2 (Figure 3D). This was readily apparent by Western blot analysis in progeny of mothers subjected to a 5-minute and 30-minute heat-shock (Figure 3D) and was also true for progeny of mothers subjected to the 60-minute heat-shock, although because these latter animals had lesser total H3, the net increase was less obvious (Figure S7A; see also Figures 7B and S7G, showing sterility as a possible explanation for lower H3 levels).
Figure 7: Maternal stress-induced increase in H3K9me2 at daf-2 in reversed over generations.

(A) Top: Number of eggs laid every hour within the time interval on the X-axis, by non-heat shocked control [NHS(Ctrl)], and heat-shocked P0 mothers. Bottom: Percent hatched embryos from NHS(Ctrl) and heat-shocked P0 mothers. Heat-shock: 5-minute and 60-minute.
(B) Representative micrographs showing germline and uterus of F1 progeny from NHS(Ctrl), and heat-shocked P0 mothers. Heat-shock: 5-minute and 60-minute. Note all F1 from NHS(Ctrl) and 5min HS P0 mothers, and some F1 from 60min HS P0 mothers are fertile (marked by dotted lines), but 52.5±8.4% F1 from 60min HS P0 mothers are sterile (right). n=3 experiments, 17-101 F1s scored/experiment
(C) H3K9me2 occupancy at regions a , b, c and d, of the daf-2 locus (top), in day one adult F2 progeny from NHS(Ctrl), and heat-shocked P0 mothers. Heat-shock: 5-minute and 60-minute. n=2-3 experiments. Compare to H3K9me2 levels in Figure 6B.
(D) Model: germline activity of HSF-1 induces a MET-2-dependent increase in H3K9me2 at the daf-2 gene and consequently DAF-16 is activated to confer thermotolerance; this repression of daf-2 is inherited by subsequent generations depending on the severtity of maternal stress.
A, C: Data show Mean ± Standard Error of the Mean. *: p<0.05, **: p<0.01, ***: p<0.001, ns: non-significant; ANOVA with Tukey’s correction, and Unpaired Student’s t-test.
B: Scale bar:50μm.
See also Figure S7
We conducted immunostaining for H3K9me2 on 2-4 cell stage embryos, and embryos in early stages of development dissected from heat-shocked and non-heat shocked mothers soon after fertilization to determine whether the increased H3K9me2 levels persisted unaltered though embryogenesis (Figures 3E and 3F). As previously reported (Mutlu et al., 2019; Mutlu et al., 2018), normally, H3K9me2 is barely detectable in embryos at fertilization and increases through embryogenesis to be visible by gastrulation in all embryonic nuclei (Mutlu et al., 2019; Mutlu et al., 2018). H3K9me2 was still not visible by immunofluorescence microscopy in the 2-4 cell stage embryos generated from heat-shocked oocytes, likely due to the limited sensitivity of the technique, but by gastrulation and after, the nuclei of embryos from heat-shocked oocytes had significantly higher levels of H3K9me2 (Figures 3E and 3F).
These data indicated that the MET-2-dependent increase in H3K9me2 that occurred at HSF-1 targets in the maternal germline following heat-shock, persisted in offspring through some mechanism initiated during the initial heat-shock. Thus, perhaps contrarily, MET-2 activity which increases H3K9me2 modifications at germline active genes to erase germline transcriptional memory, was by this very process, reinforcing the memory of HSF-1 activation by increasing H3K9me2 levels in progeny that had been subjected to stress in utero.
HSF-1-dependent recruitment of MET-2 in germ cells is required for enhanced stress resilience later-in-life.
These data presented a conundrum since they indicated that early life stress exposure as germ cells dampened, in adults that developed from these germ cells, the stress-inducibility of the very hsp genes and other HSF-1-dependent targets that are normally needed for surviving stress, but at the same time, increased their stress resilience. We therefore first asked whether MET-2 activity was required for later life stress resilience or if it was a coincident, unrelated phenomenon. To test this, we used RNAi to knock down met-2 in the maternal germline and assayed the stress resilience of offspring of heat-shocked mothers (Figure 4A). Knocking down maternal germline met-2 abolished their stress resilience (Figure 4A; see Figure S4B for RNAi validation). As controls, we also examined the effects of knocking down other chromatin modifiers that repress transcription (Figure 4A). Knocking down SET-25, the putative H3 lysine 9 (H3K9) methyltransferase responsible for H3K9me3 modification of chromatin (Garrigues et al., 2015; Rechtsteiner et al., 2019) decreased the average stress resilience of progeny but did not have a significant or consistent effect. Knockdown of MES-2 (Holdeman et al., 1998), the C. elegans PRC2 homolog, decreased offspring stress resilience after maternal stress exposure, and the knock-down of another SET domain protein SET-1 (Vielle et al., 2012) did not alter the protective effects of maternal heat-shock. We confirmed that MET-2 was required for the thermotolerance of progeny of heat-shocked mothers by also using the deletion mutant met-2 (n4256) III (Figure 4B). The rescue of polyQ aggregation in progeny of stressed mothers was also dependent on maternal met-2, as RNAi-mediated knockdown of met-2 in the mother prevented the suppression of protein aggregation in offspring (Figure 4C). While complex alterations in stress resilience that resulted from the RNAi-mediated knock down of multiple putative methyltransferases suggested that a coordinated change in chromatin modifications by these enzymes might be required for the persistence of thermotolerance throughout the lifetime of the fertilized oocyte, these experiments nevertheless confirmed that maternal MET-2 was required for progeny stress resilience: decreasing germline met-2 abolished early life programming of stress resilience.
Figure 4: Germline MET-2 is required for enhanced stress resilience in progeny.

(A) Stress resilience of F1 progeny from control non-heat shocked [NHS(Ctrl)] and 5-minute heat-shocked P0 mothers subject to germline RNAi. RNAi indicated on the X-axis. Control RNAi: L4440 empty vector. n=5-14 experiments, 22.7±2.7 to 30.0±2.8 F1/ experiment.
(B) Stress resilience of F1 progeny of wild-type and met-2 (n4256) III animals. F1 progeny are from NHS(Ctrl) and heat-shocked P0 mothers. Heat-shock: 5-minutes and 60-minutes. n=3-9 experiments.
(C) Number of polyglutamine aggregates scored in day-one to day-four adult F1 progeny of NHS(Ctrl) and 5-minute heat-shocked P0 mothers. P0 mothers were subjected to met-2 and control (L4440 empty vector) RNAi. n=3 experiments, 15 F1 animals/experiment obtained from 3-20 P0 mothers/condition/experiment.
A-C: Data show Mean ± Standard Error of the Mean. *: p<0.05, **: p<0.01, ***: p<0.001, ns: non-significant; Unpaired Student’s t-test
HSF-1-dependent MET-2 activity downregulates the Insulin/ IGF-1 like signaling pathway (IIS) to increase stress resilience.
How then were germline HSF-1 and MET-2 increasing the stress resilience of progeny upon in utero stress? One intriguing possibility was that HSF-1-dependent MET-2 recruitment increased repressive H3K9me2 at some other HSF-1 target(s), whose repression activated other stress protection pathways. In C. elegans, stress resilience is largely due to the activities of either HSF-1 or the FOXO transcription factor DAF-16 (Kumsta et al., 2017; McColl et al., 2010). Knocking down hsf-1 only in the progeny of heat-shocked mothers had no effect, consistent with the attenuated induction of hsps in these offspring (Figure 5A). Knocking down daf-16 only in progeny of heat-shocked mothers, on the other hand, suppressed their enhanced stress resilience (Figure 5A; see Figure S7B for RNAi validation). This contrasted with the maternal germline requirements for hsf-1 and daf-16: knocking down germline daf-16 decreased the basal thermotolerance of progeny, but still allowed the acquisition of enhanced stress resistance in the in utero heat-shocked progeny (Figure 5B), while, as shown previously (Figure 1C), hsf-1 germline knock-down abolished enhanced stress resistance. Thus, DAF-16 activity was responsible for the observed thermotolerance, and maternal stress exposure appeared to have shifted the stress resilience strategy of the progeny to become less reliant on HSF-1, and more reliant on the activation of DAF-16. Since the protective activity of DAF-16 can be triggered by attenuated signaling of the sole C. elegans insulin/IGF-1 receptor, DAF-2, we tested (i) whether HSF-1 bound the 5’-UTR region of daf-2 upon heat-shock, (ii) whether HSF-1 and MET-2 activity increased the abundance of H3K9me2 at the daf-2 gene in heat-shocked mothers, (iii) whether this increase persisted in progeny and (iv) whether DAF-16 was indeed activated in progeny to account for their stress resilience.
Figure 5: DAF-16 activity in the F1 progeny of heat-shocked mothers is required for their enhanced stress resilience.

(A) Top: Experimental protocol to test the dependency of stress resilience in F1 progeny. Note:F1 progeny and not the P0 maternal germline were subjected to daf-16 or hsf-1 RNAi. Bottom: Stress resilience of F1 progeny from control non-heat shocked [NHS(Ctrl)], and heat-shocked, P0 mothers. Heat-shock: 5-minutes and 60-minutes. Control RNAi: L4440. n=3-10 experiments, 19.3±4.3 to 28.4±4.8 F1 progeny/experiment/condition.
(B) Stress resilience of F1 progeny from NHS(Ctrl) and heat-shocked P0 mothers. Heat-shock: 5-minute and 60-minute. P0 mothers were subjected to daf-16 germline RNAi. n=5-8 experiments, 24.4±2.7 to 35.2±2.0 F1 progeny/experiment/condition.
(C) Schematic of daf-2 gene regions analyzed for HSF-1 binding (5’ Far; −9071 to −8954), and H3K9me2 occupancy: a (−172 to −70), b (Intron 1: +3179 to +3279), c (Intron 2: +8046 to +8177) and d (Exon 9: +15719 to +15834).
(D) HSF-1 occupancy at the 5’-far region of daf-2 gene in NHS(Ctrl)] and heat-shocked wild-type and hsf-1 (sy441) P0 mothers. Heat-shock: 5-minute and 60-minute. n=3 experiments. Data normalized to NHS(Ctrl) wild-type and hsf-1(sy441) values respectively (also see Figure S7D).
(E) H3K9me2 occupancy at regions a, b, c, and d, depicted in C, in wild-type and hsf-1 (sy441) P0 mothers. Conditions: NHS(Ctrl), 2 hours after a 5-minute heat-shock (5min HS+rec), and immediately after a 60-minute heat-shock. n=3-4 experiments. H3K9me2 occupancy shown relative to NHS(Ctrl) wild-type and NHS(Ctrl) hsf-1 (sy441) values respectively. (also see Figure S7F).
A, B, D, E: Data show Mean ± Standard Error of the Mean. *: p<0.05, **: p<0.01, ***: p<0.001, ns: non-significant; ANOVA with Tukey’s correction, and Unpaired Student’s t-test.
See also Figure S7
To assess whether HSF-1 bound to the 5’-UTR regions of daf-2, we reanalyzed publicly available ChIP-seq data on HSF-1 binding sites in C. elegans [(Li et al., 2016); see Star Methods]. We identified a region that contained a putative heat shock element (HSE) motif in the daf-2 5’-UTR (p<0.01; Figure S7C) and tested whether HSF-1 bound to this region upon heat-shock. As determined by ChIP-PCR, HSF-1 occupancy at the daf-2 5’-UTR region increased with 5-minute heat-shock, just as it does at hsp genes, and remained elevated also in 60-minute heat-shocked animals (5’-far; Figures 5C and 5D). Also, as seen with the other HSF-1 targets, this heat-shock induced increase did not occur in hsf-1 (sy441) mutants (Figure 5D), although again, the mutant HSF- showed constitutive binding at this region (Figures S7D). Interestingly, as with pha-4 and C32H11.4, daf-2 mRNA levels did not change with heat-shock in wild-type animals or hsf-1 (sy441) upon heat-shock (Figure S7E).
We next tested whether H3K9me2 levels increased across the daf-2 gene upon heat-shock. Since multiple isoforms of daf-2 have been described, we assayed H3K9me2 abundance not only at the 5’-UTR regions containing the daf-2 promoter, but also across the daf-2 gene body that has been shown to be amenable to epigenetic modification [(Jin et al., 2011); Figure 5C]. H3K9me2 levels increased across daf-2 in mothers that had been subjected to a 5-minute heat-shock and were more elevated in mothers subjected to the longer (60-minutes) heat-shock (Figures 5C and 5E). This increase in H3K9me2 was HSF-1-dependent, and H3K9me2 levels did not increase in heat-shocked hsf-1(sy441) mutants although, as expected, H3K9me2 was elevated under basal conditions (Figures 5C, 5E and S7F). Moreover, like the increased H3K9me2 marks at other HSF-1 targets, higher levels of H3K9me2 persisted at the daf-2 gene in progeny of stressed mothers, both when the mothers were subjected to a short (5-minute) and long (30-minute) heat-shock (Figures 6A and 6B) and this was dependent on germline HSF-1 and MET-2 activity being suppressed upon hsf-1 or met-2 germline RNAi (Figure 6C).
Figure 6: HSF-1 and MET-2 in the maternal germline heritably silence the daf-2 gene and activate DAF-16 in progeny.

(A) Schematic of daf-2 gene regions assayed for H3K9me2 occupancy (as in Figure 5C).
(B) H3K9me2 occupancy at regions a, b, c and d, depicted in A, in day-one adult F1 progeny from non-heat shocked control [NHS(Ctrl)], and heat-shocked P0 mothers. Heat-shock: 5-minutes and 30-minutes. n=5-7 experiments. H3K9me2 occupancy shown relative to NHS(Ctrl) wild-type values.
(C) H3K9me2 occupancy at regions a , b, c and d in day-one adult F1 progeny from NHS(Ctrl), and heat-shocked P0 mothers subjected to hsf-1 or met-2 germline RNAi. Heat-shock: 5-minute. n=5-7 experiments. H3K9me2 occupancy shown relative to NHS(Ctrl) from each respective treatment. Control RNAi : L4440.
(D) Average daf-2 mRNA levels in F1 progeny of NHS(Ctrl), and heat-shocked P0 mothers. Heat-shock: 5-minutes and 60-minutes. mRNA levels are relative to pmp-3 and normalized to that in F1 progeny from NHS(Ctrl) P0 mothers. n=5-6 experiments.
(E) Representative micrographs showing DAF-16::GFP localization in day-one adult F1 progeny from NHS(Ctrl), and heat-shocked P0 mothers. Heat-shock: 5-minute and 60-minute.
(F) Percent F1 progeny that display DAF-16::GFP nuclear localization as day one adults. n=3-4 experiments, 20-35 progeny/experiment.
(G) Average sod-3, mtl-1 and lys-1 mRNA levels in day one adult F1 progeny from NHS(Ctrl) and 60-minute heat-shocked P0 mothers. mRNA levels are relative to pmp-3, and normalized to that in F1 progeny from NHS(Ctrl) P0 mothers. n=3-5 experiments, 30 F1s from 15 mothers/experiment.
B-D, F, G: Data show Mean ± Standard Error of the Mean. *: p<0.05, **: p<0.01, ***: p<0.001, ns: non-significant; ANOVA with Tukey’s correction, and Unpaired Student’s t-test.
E: Scale bar:30μm.
See also Figure S7
Increased H3K9me2 at daf-2 predicted that progeny of heat-shocked mothers would express less daf-2 mRNA. This was the case (Figure 6D). The decrease in daf-2 expression was functional and DAF-16 was activated in these F1 progeny, even when they became adults (Figure 6E and 6F). This was assayed by the increased nuclear localization of a DAF-16::GFP transgenic protein in F1 progeny of stressed mothers grown under control conditions (Figure 6E and 6F) and confirmed by the increased expression of known DAF-16 target genes (Figure 6G). Thus, maternal stress exposure of C. elegans and germline HSF-1 activity induced a MET-2-dependent increase in H3K9me2 marks at the daf-2 locus, to program the activity of the Insulin-like signaling pathway (ILS) and stress-resiliency in the next generation.
Maternal stress-induced H3K9me2 increase at daf-2 is reversed over generations.
The apparent ease with which HSF-1 germline activity could alter the physiology and stress resilience of offspring presented the question why stress-resilience had not been fixed in C. elegans populations. The continuous repression of daf-2 promotes an alternate, stress-resistant but reproductively delayed larvae stage called dauer (Kenyon et al., 1993; Kimura et al., 1997; Ogg et al., 1997; Tissenbaum and Ruvkun, 1998). Therefore, one answer could be that the conditions that increased stress resilience also caused phenotypic alterations that limited heritable transmission. This was the case and 60-minute heat-shocked mothers showed a marked delay in egg laying (Figure 7A; top) and a significant number of their embryos did not hatch (Figure 7A; bottom). In addition, over half of the embryos that did hatch and develop into adults, were sterile (Figure 7B; 52±8.9%, n=3 experiments, 40-50 F1s scored/experiment). In contrast 0% of the control non-heat shocked mothers laid sterile offspring. However, despite these significant fitness disadvantages, the remaining fertile F1 progeny of 60-minute heat-shocked mothers did produce F2 progeny that were stress-tolerant (Figure 1D). Moreover, progeny of mothers exposed to heat-shock for 5 minutes also displayed enhanced stress tolerance (Figure 1B) and did not display these overt fitness disadvantages: they produced viable fertile embryos (Figure 7A; 0% sterility, n=3 experiments, 40-50 F1 scored/experiment). Therefore, it appeared that other mechanisms prevented stress tolerance from becoming a fixed trait in C. elegans populations. Since the number of generations displaying stress resilience depended on the severity of maternal stress exposure, whereby stress-tolerance was limited to the F1 generation if the mothers were exposed to a 5-minute heat-shock and persisted into the F2 generation when mothers were heat-shocked for 60-minutes, we predicted that elevated H3K9me2 levels at daf-2 were erased over generations, resetting stress-tolerance in the population. To test this, we examined H3K9me2 levels at daf-2 in the F2 generation derived from mothers subjected to a 5-minute and a 60-minute heat-shock (Figure 7C). Indeed, in the F2 generation from 5-minute heat-shocked mothers, H3K9me2 levels at daf-2 were no longer elevated and had returned to wild-type levels (Figure 7C). H3K9me2 levels at daf-2 in the F2 progeny of mothers subjected to a long (60-minute) heat-shock remined elevated but were lower than that in F1 progeny from P0 mothers subjected to a long heat-shock (Figure 7C; compare Figure 7C with Figure 6B). These studies suggested that although HSF-1 activity upon maternal stress exposure increased H3K9me2 levels at daf-2 to increase offspring stress tolerance, and although these enhanced levels could even be heritable and transmitted to the grand-children (F2 generation), there appeared to be a mechanism, likely through demethylase activity, that erased these marks over generations, reverting stress tolerance within populations to pre-existing levels (Figure 7D).
Discussion
Stress and the epigenetic control of the Insulin-like signaling pathway.
These studies demonstrate a mechanism whereby in response to maternal stress exposure, the conserved transcription factor HSF-1 recruits the putative histone H3K9 SET domain methyltransferase MET-2 to HSF-1 target genes in the germline, including to the insulin-like receptor gene daf-2, to increase heterochromatin, repress subsequent expression, establish a memory of prior stress exposure, and alter the animals’ later-life stress-response strategy. HSF-1 mediated increase in H3K9me2 appears to be a consequence of normal germline reprogramming mechanisms that act to erase the memory of germline transcriptional activity in all animals. Given the conservation of these pathways, we propose that HSF-1-mediated heterochromatinization through the recruitment of MET-2/SETDB1 (or other H3K9methyltransferases) could also be activated in humans in response to adverse intrauterine environments such as toxins, pathogens, or starvation and cause similar epigenetic changes during perinatal development. Such a mechanism could be responsible for the effects of in utero nutrient depravation on subsequent generations as occurred during natural historical events such as the Dutch Hunger Winter (Stanner and Yudkin, 2001; Yarde et al., 2013). In addition, given the role of H3K9me1/2/3 heterochromatin in chromosome segregation, genome integrity, and gene expression (Allshire and Madhani, 2018) it is also possible that HSF-1-dependent repressive H3K9me2 bookmarking upon heat-shock serves other functions besides establishing increased stress tolerance. This remains to be explored.
The role of HSF-1-induced increase in H3K9me2 in homeostatic plasticity and hormesis.
There are similarities between the heat-shock induced transcriptional plasticity in our studies and treatments that induce hormesis, a phenomenon where prior exposure to a low dose of a stressor reduces toxicity from a subsequent harmful exposure (Chou et al., 2019; Kumsta et al., 2017; Matai et al., 2019; Schulz et al., 2007; Van Voorhies, 2001). We have yet to investigate whether the interaction between HSF-1 and MET-2 follows the biphasic dose-response relationship characteristic of hormetic responses (Calabrese and Baldwin, 1998, 2001; Calabrese et al., 2020; Giordano et al., 2008). HSF-1 and MET-2 are essential during normal development (Akerfelt et al., 2007; Sridharan et al., 2013), and beneficial for lifespan extension (Lee et al., 2019), but detrimental when upregulated in cancer (Alasady and Mendillo, 2020; Zhang and Adams, 2007), or dysregulated in neurodegenerative diseases (Berson et al., 2018; Neef et al., 2011), suggesting that their interaction is unlikely to be monotonic.
The set-point of stress-responses and choice of stress-response strategies
For poorly understood reasons, stress responses are activated at widely varying thresholds even between genetically identical individuals (de Nadal et al., 2011; Vihervaara et al., 2018). The increase in H3K9me2 at HSF-1 target genes following an initial stress exposure provides one mechanism by which the threshold of stress can be set based on prior experience. In the studies described here, HSF-1-dependent increase in H3K9me2 biases the subsequent stress responsive transcriptional program towards one that relies on the insulin-like signaling pathway (ILS) which is arguably more protective against chronic, rather than an acute stress exposure (Hsu et al., 2003; Murphy et al., 2003; Ogg et al., 1997). This suggests that such a mechanism might have evolved to protect organisms from the continued presence of the stressor, as might be expected to happen in the natural habitat. Thus, it is notable that increases in H3K9me2 at HSF-1 targets appear to be remarkably easy to acquire, even with a short and transient heat-shock, and are lost easily over a few generations. Previous work from our lab have demonstrated that, in addition to being activated in response to protein damage (Gomez-Pastor et al., 2018; Morimoto, 1998; Vihervaara et al., 2018), HSF-1 can also be rapidly activated in anticipation of a stressor (allostasis) through sensory neural networks (Das et al., 2020; Ooi and Prahlad, 2017; Tatum et al., 2015). Thus, epigenetic alterations to germline chromatin could occur and be reversed frequently, during an organism’s reproductive lifespan.
Limitations of the Study
Our studies show that HSF-1 recruits MET-2 to negatively bookmark the insulin-receptor daf-2 and other HSF-1-target genes upon in utero stress. We did not investigate the genome-wide changes in occupancy of HSF-1 and MET-2 upon heat-shock, their relationship to H3K9me2, or whether bookmarking persists over differing durations at different loci. Such studies could be essential to elucidate the global consequences of gene bookmarking by HSF-1. Furthermore, the increase in global levels of H3 and H3K9me2 that occur in hsf-1(sy441) mutants is intriguing and suggests that HSF-1-dependent modulation of chromatin may differ under conditions of acute stress, which drives HSF-1 binding to its target genes, and conditions where HSF-1 remains bound but is not stress-inducible, as may occur upon its dysregulation in disease. This remains to be investigated.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to, and will be fulfilled by, the lead contact, Veena Prahlad (veena-prahlad@uiowa.edu).
Materials availability
All C. elegans strains generated in the laboratory will be deposited with the Caenorhabditis Genetics Center (CGC) and will be available from the lead contact upon request.
Data and code availability
Original western blot images and microscopy data reported in this paper have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the Key Resources Table.
This paper does not report original code, but uses publicly available software to re-analyze publicly available ChIP-seq data. The pipeline scripts for re-analysis has been deposited at Mendeley and are publicly available as of the date of publication. DOIs and GEO accession are listed in the Key Resources Table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies (also see Table S1) | ||
| Mouse monoclonal anti-FLAG | Sigma Aldrich | Cat No. F-3165, RRID:AB_259529 |
| Mouse monoclonal anti-FLAG M2 | Sigma Aldrich | Cat No. F1804, RRID:AB_262044 |
| Rabbit polyclonal anti-HA | Abcam | Cat No. ab9110, RRID:AB_307019 |
| Rabbit polyclonal anti-H3K9me2 | Diagenode | Cat No. C15410060, RRID:AB_2892672 |
| Mouse monoclonal anti-H3K9me2 | Abcam | Cat No. ab1220, RRID:AB_449854 |
| Pierce Anti-HA Magnetic beads | Thermo Fisher Scientific | Cat No. 88837, RRID:AB_2861399 |
| Anti-FLAG M2 magnetic beads | Sigma-Aldrich | Cat No. M-8823, RRID:AB_2637089 |
| Mouse monoclonal anti-polyglutamine | Sigma Aldrich | Cat No. P1874, RRID:AB_532270 |
| Rabbit anti-HSP90 | Cell Signaling Technology | Cat No. 4874, RRID:AB_2121214 |
| Rabbit polyclonal anti-histone H3 | Abcam | Cat No. ab1791, RRID:AB_302613 |
| Mouse monoclonal anti-α-tubulin | Developmental Studies Hybridoma Bank (DSHB) | Cat No. AA4.3, RRID:AB_579793 |
| Normal Anti-mouse IgG | Abcam | Cat No. ab188776 |
| IRDye 800CW conjugatedSheep anti-mouse IgG (H+L) | Rockland Immunochemicals | Cat No. 610-631-002, RRID:AB_220142 |
| Alexa Fluor 680 conjugated goat anti-rabbit IgG (H+L) | Thermo Fisher Scientific | Cat No. A21109, RRID:AB_2535758 |
| Alexa Fluor 647 conjugated goat anti-mouse IgG (H+L) | Thermo Fisher Scientific | Cat No. A-21236, RRID:AB_2535805 |
| Alexa Fluor 488 conjugated goat anti-rabbit IgG (H+L) | Thermo Fisher Scientific | Cat No. A-11008, RRID:AB_143165 |
| Chemicals, peptides, and recombinant proteins | ||
| Vectashield mounting medium with DAPI | Vector Laboratories | Cat No. H-1200, RRID:AB_2336790 |
| 16% paraformaldehyde | Electron Microscopy Science | Cat No. 15710 |
| Formaldehyde | Sigma Aldrich | Cat No. 252549 |
| Halt Protease Inhibitor Cocktail (100X) | Halt | Cat No. 78438 |
| Salmon Sperm DNA | Invitrogen | Cat No. 15632–011 |
| ChIP DNA purification kit | Zymo Research | Cat No. D5205 |
| TURBO DNA-free Kit | Life Technologies | Cat No. AM1907, |
| iScript cDNA Synthesis Kit | Bio-Rad | Cat No. 170–8891 |
| PowerUp SYBR Green Master Mix | Thermo Fisher Scientific | Cat No. A25742 |
| Deposited data | ||
| Raw data (Mendeley data) | This paper | DOI: 10.17632/f38gnsgwvy.1 |
| Reanalyzed ChiP-seq data | Li et al, 2016 | GEO: GSE81523 |
| Experimental models: Organisms/strains (also see Table S2) | ||
| C. elegans: Strain N2 wild-type | Caenorhabditis Genetic Center | N/A |
| C. elegans: Strain PS3551: hsf-1(sy441)I | Caenorhabditis Genetic Center | N/A |
| C. elegans: Strain AM140: rmIs132 [unc-54p::Q35::YFP] | Caenorhabditis Genetic Center | N/A |
| C. elegans: Strain DCL569: mkcSi13 II; rde-1(mkc36) V | Caenorhabditis Genetic Center | N/A |
| C. elegans: Strain MT13293: met-2(n4256) III | Caenorhabditis Genetic Center | N/A |
| C. elegans: Strain: TJ356: zIs356 [daf-16p::daf-16a/b::GFP + rol-6(su1006)] IV | Caenorhabditis Genetic Center | N/A |
| C. elegans: hsf-1::3Xflag(I) | Das et al., 2020 | N/A |
| C. elegans: 3Xflag::hsf-1(I) | Prahlad lab | N/A |
| C. elegans: 3Xflag:: hsf-1(sy441)(I) | Prahlad lab | N/A |
| C. elegans: Strain: PHX2467: 3XHA::hsp-1(IV) | Prahlad lab/Suny Biotech | N/A |
| C. elegans: Strain: PHX2141: hsp-70::3Xflag(I) | Prahlad lab/Suny Biotech | N/A |
| C. elegans: Strain: PHX3574: met-2::3XHA(III) | Prahlad lab/Suny Biotech | N/A |
| C. elegans: met-2::3XHA(III); 3Xflag::hsf-1(I) | Prahlad lab | N/A |
| C. elegans: met-2::3XHA(III); 3Xflag:: hsf-1(sy441)(I) | Prahlad lab | N/A |
| C. elegans: mkcSi13 II; rde-1(mkc36) V; hsf-1::3Xflag(I) | Prahlad lab | N/A |
| Oligonucleotides | ||
| Primers are listed in Table S3 | ||
| Software and algorithms | ||
| LAS X | Leica Microsystems | RRID:SCR_013673 |
| ImageJ (FIJI) | NIH | RRID:SCR_003070 |
| GraphPad Prism (9.0.0) | GraphPad Software | RRID:SCR_002798 |
| Image Studio | LI-COR | RRID:SCR_015795 |
| NCBI SRA Toolkit | SRA Toolkit Development Team | https://github.com/ncbi/sra-tools |
| Snakemake | Mölder, Jablonski et al. 2021 | https://github.com/snakemake/snakemake/blob/26373e66fbaba04fa076aebb24ad869f41f228b9/docs/index.rst |
| Bowtie2 | Langmead, Trapnell et al. 2009 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Sambamba | Tarasov, Vilella et al. 2015 | https://lomereiter.github.io/sambamba/ |
| Bedtools | Quinlan and Hall 2010 | https://github.com/arq5x/bedtools2 |
| modENCODE Blacklist | Amemiya, Kundaje et al. 2019 | https://github.com/Boyle-Lab/Blacklist/ |
| MACS2 | Zhang, Liu et al. 2008 | https://github.com/macs3-project/MACS |
| ucsc-wigcorrelate | UCSC Genome Bioinformatics Group | https://github.com/ucscGenomeBrowser/kent/tree/master/src/utils |
| Rstudio | RStudio Team (2020) | https://www.rstudio.com/ |
| “regioner” R package | Gel, Díez-Villanueva et al. 2015 | https://bioconductor.org/packages/release/bioc/html/regioneR.html |
| MEME Suite v 5.3.2 FIMO |
Bailey, Johnson et al. 2015 Grant, Bailey et al. 2011 |
https://meme-suite.org/meme/meme-software/5.3.2/meme-5.3.2.tar.gz |
| Other | ||
| PierceTM Protein A/G magnetic beads | Thermo Fisher Scientific | Cat No. 88802 |
| Precellys 24 homogenizer | Bertin Corp. | N/A |
| Bioruptor Pico Sonication System | Diagenode | Cat No. B0106001 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
C. elegans strains
C. elegans used in this study are listed in Table S2. Strains were procured from Caenorhabditis Genetics Center (CGC, Twin Cities, MN), generated in the Prahlad laboratory, or generated by SunyBiotech (Suzhou, Jiangsu, China 215028).
Generation of CRISPR strains
CRISPR/Cas9 was used to create following C. elegans strains by adding different tags at endogenous loci:
| Background strain |
Tag | Position of the tag | Gene | Resulting strain |
|---|---|---|---|---|
| N2 | 3X FLAG | N terminus (after the initiation codon) | endogenous hsf-1(I) gene | FLAG::HSF-1 |
| PS3551 | 3X FLAG | N terminus (after the initiation codon) | endogenous hsf-1(sy441)I gene | FLAG::HSF-1(sy441) |
| N2 | 3X FLAG | C terminus (before the termination codon) | endogenous hsf-1(I) gene | HSF1::FLAG |
| N2 | 3X HA | C terminus (before the termination codon) | endogenous met-2 (III) gene | PHX3574 (MET-2::HA) |
| N2 | 3X FLAG | C terminus (before the termination codon) | endogenous hsp-70 (I) (C12C8.1) gene | PHX2141 (C12C8.1::FLAG) |
| N2 | 3X HA | N terminus (after the initiation codon) | endogenous hsp-1 (IV) gene | PHX2467 (HA::HSP-1) |
Generation of (a) met-2::3XHA(III); 3XFLAG::hsf-1(I) (b) met-2::3XHA(III); 3XFLAG::hsf-1(sy441)I (c) mkcSi13 II; rde-1(mkc36) V; hsf-1::FLAG worms
The met-2::3XHA(III) strain was crossed into the hsf-1::3Xflag (I) strain or hsf-1(sy441)::3Xflag (I) strain and verified by PCR. mkcSi13 II; rde-1(mkc36) V (DCL569) strain was crossed into the hsf-1::3Xflag (I) strain and verified by PCR.
Growth conditions of C. elegans strains
All strains were grown and maintained at low densities at 20°C. Animals were grown in 20°C incubators (humidity controlled) on 60mm nematode growth media (NGM) plates by passaging 8–10 L4s onto a plate, and 4 days later, picking L4 animals onto fresh plates for experiments. Animals were fed Escherichia coli OP50 obtained from Caenorhabditis Genetics Center (CGC) that were seeded onto culture plates 2 days before use. The NGM plate thickness was controlled by pouring 8.9 ml of autoclaved liquid NGM per 60 mm plate. Plates used in experiments were an average weight of 13.5 ± 0.2 g. Plates that varied from these measurements were not used for experiments. Plate weights immediately upon pouring, and prior to use in experiments were monitored and served as the readout for changes in humidity that occurred in the laboratory. Laboratory temperature was maintained at 20°C to 22°C and carefully monitored throughout the experimental procedures. All animals included in the experiments, unless otherwise stated, were one-day-old hermaphrodites that were age-matched either by (a) bleaching and starting the experiment after 75–78 hrs. or (b) picking as larval stage 4 (L4) juveniles 24 to 26 hrs. before the start of the experiment.
METHOD DETAILS
Heat shock
The heat shock protocol described in Das et al., 2020 (Das et al., 2020) was used. Briefly, NGM plates (8.9 ml liquid NGM/plate, weight 13.5 ± 0.2 g) were seeded with 250-300 μl OP50 culture in the center and allowed to dry at room temperature for 48 hr. L4 hermaphrodites were passaged onto these plates, or worms were bleach- hatched onto these plates and allowed to grow to Day1 adults. All heat shock experiments were performed with one-day-old gravid animals. To induce the heat shock response, NGM plates containing one-day-old animals were sealed with parafilm and immersed in a water bath (product no. ITEMP 4100 H21P 115V, Fischer Scientific, Pittsburgh, PA) pre-warmed to 34°C, for different durations as mentioned. The rate of temperature increase using this method, at the surface of plates of said thickness has been previously monitored using a thermocouple (Tatum et al., 2015). When required, animals were recovered in the 20°C incubator following heat-shock for different durations. For experiments involving F1 progeny, eggs from non-heat shocked control or heat-shocked mothers (P0) were collected on fresh plates and F1 progeny were allowed to grow to day-one adults in the 20°C incubator. F2 or F3 progeny were collected from F1 or F2 animals respectively, and also grown at 20°C.
Obtaining synchronized embryos by ‘bleach-hatching’.
C. elegans populations containing 250-300 gravid adults were generated by passaging L4s, as described above. Five days later, the plates consisted mainly of gravid day-one adults. These plates were used for obtaining embryos by bleach-induced solubilization of adults. Animals were washed off the plates with 1X PBS and pelleted by centrifuging at 2655 X G for 30 s. The PBS was removed carefully, and worms were gently vortexed in the presence of bleaching solution [250 μl 1N NaOH, 200 μl standard (regular) bleach, 550 μl sterile water] until all the worm bodies had dissolved (approximately 5–6 min), and only eggs were visible. The eggs were pelleted by centrifugation (2655 X G for 45 s) and the bleaching solution was removed. Eggs were washed with sterile water 3-4 times and then counted. The eggs were seeded on fresh OP50 or RNAi plates in required numbers (~200 eggs/plate for chromatin immunoprecipitation) and allowed to grow to day-one-adults under standard condition (20°C). If eggs remained unhatched, these plates were discarded.
Assessing egg laying rates and F1 embryo hatching rates following heat shock
Wild-type (N2) L4s were picked on fresh OP50 plates the day before the experiment. After 24-26hr, one-day-old animals were either heat shocked at 34°C for 5, 30 or 60 min in the water bath as described above or left untreated (non-heat shocked control). Heat-shocked animals were recovered at 20°C following heat shock and five heat-shocked, and non-heat shocked control animals were moved to new OP50 plates at different time intervals (0hr, 2hr, 4hr and 8 hr.) Eggs laid by control non-heat shocked animals, and heat-shocked animals between 2-4hr, 4-8hr and 8-12hr were counted. Cumulative numbers of eggs, and egg laying rates per worm per hour per treatment condition were calculated by counting the number of embryos laid every 2 hours for upto 12 hours. To determine embryo survival and hatching rates, eggs were allowed to hatch at 20°C and the number of live progenies were scored 48 hr. later. As described earlier, eggs are extremely susceptible to heat stress and >50% eggs laid between 0-2 hr. post heat shock didn’t hatch (Das et al., 2020).
Thermotolerance assay
Three control non-heat shocked P0 mothers, three P0 mothers subjected to heat-shock at 34°C for 5 minutes, five P0 mothers subjected to the 30-minute heat shock, and twenty P0 mothers subjected to the 60-minute heat shock were allowed to lay eggs for different time intervals (2–4 hr. or 4—8 hr. or 8—12 hr. post-heat shock as described) and then all P0 mothers were removed from the plates. The differing numbers of P0 mothers were required to generate plates with F1 progenies of comparable densities (30-50 F1 progenies/plate/condition) since P0 animals subjected to longer durations of heat-shock lay fewer eggs all of which do not hatch. Plates were then kept in an incubator at 20°C until the F1 progeny become day-one adults. Thermotolerance of the F1 progenies was determined by subjecting them to a prolonged (2 hr. 15 min) heat exposure of 37°C and then allowing them to recover at 20°C overnight. This severe heat-shock condition was chosen after prior experiments to titrate death of control, non-heat shocked progeny to <~50% to prevent ceiling effects. The percent animals that survived the prolonged heat shock was scored 24 hrs. later. To assess the thermotolerance of F2 and F3 generations we had to prevent that the plates with the 30-50 F1 progeny become depleted of bacterial food due to the numbers of F2 embryos that would be laid by the 30-50 F1s. Therefore 3,3,5 and 40 random F1 progenies from control P0, 5-minute heat shocked P0, 30-minute heat shocked P0 and 60-minute heat-shocked P0 respectively, were selected from the F1 plates and allowed to lay eggs on a new plate for 4 hours to generate the F2 generation. These F2 progeny were tested for thermotolerance. The numbers of F1 were chosen after determining that the F2 progeny would also be ~30-50/plate, retaining plate densities for comparison. More F1 progeny from the 60-minute heat shocked P0 were required due to high rates of sterility and the low egg laying rates of these F1. Thermotolerance of F2 progenies was determined as described above. A similar procedure was used to generate F3 progeny; however, in this case fewer (five) F2 progeny from 60-minute heat shocked mothers were used, as the F2 generation were not sterile or compromised in egg laying/hatching.
Thermotolerance assay with mated progeny
Fifteen wild-type day-one males were either heat-shocked at 34°C for 60 min or not heat-shocked (control) and then transferred onto plates containing three L4 wild-type hermaphrodites and allowed to mate. Mating was ascertained by counting, post-hoc, the numbers of male and hermaphrodite F1 progeny laid by these hermaphrodites (the male: hermaphrodite ratio of F1 progeny was 0.78±0.03 and 0.71±0.03 for wild-type worms mated with control non-heat shocked males and heat-shocked males respectively). About 30-50 L4 hermaphrodites (F1 progeny) were moved onto new plates and 24 hr. later, their thermotolerance as day-one adults was assessed as described above.
RNA interference (RNAi)
RNAi experiments were conducted using the standard feeding RNAi method. Bacterial clones expressing the control (empty vector pL4440) construct and the dsRNA targeting different C. elegans genes were obtained from the Ahringer RNAi library (Lee et al., 2003) now available through Source Bioscience (https://www.sourcebioscience.com/errors?aspxerrorpath=/products/life-science-research/clones/rnai-resources/c-elegans-rnai-collection-ahringer/). All RNAi clones used in experiments were sequenced for verification before use. For RNAi experiments, RNAi bacteria with empty (pL4440 vector as control) or the dsRNA-expressing plasmid were grown overnight in LB liquid culture containing ampicillin (100 μg/ml) and tetracycline (12.5 μg/ml) and then induced with IPTG (1 mM) for 2 hr. before seeding the bacteria on NGM plates supplemented with ampicillin (100 μg/ml), tetracycline (12.5 μg/ml) and IPTG (1 mM). Bacterial lawns were allowed to grow for 48 hrs. before the start of the experiment. RNAi-induced knockdown was conducted by (a) dispersing the bleached eggs onto RNAi plates or (b) feeding L4 animals for 24 hrs. (as they matured from L4s to 1-day-old adults) or (c) feeding animals for over one generation, where second-generation animals were born and raised on RNAi bacterial lawns (for hsf-1 RNAi). mkcSi13 II; rde-1(mkc36) V (DCL569) worms were used for germline-specific RNAi experiments (Zou et al., 2019). For experiments with F1 progeny, mothers (P0) were grown on RNAi plates and then eggs were collected on plates containing OP50 bacteria, so that worms in F1 generation were grown on regular plate until they were day-one-adults. Unless otherwise mentioned, day-one adults were used for experiments.
Scoring and imaging of polyglutamine (polyQ) aggregates
Transgenic animals [rmIs132 (unc-54p::Q35::YFP) expressing polyglutamine (polyQ) expansion proteins fused with fluorescent proteins in their muscle] were used for the experiment. Animals growing on OP50 bacteria or RNAi bacteria (with empty L4440 vector or met-2 RNAi construct) were either left untreated (non-heat shock control) or heat-shocked at 34°C for the indicated times. Eggs were collected from non-heat shocked controls and heat-shocked P0 mothers over the following time intervals: (a) eggs laid between 2-4hr post 5min heat shock, (b) eggs laid between 4-8hr post 30min heat shock and (c) eggs laid between 8-12hr post 60min heat shock. These eggs were allowed to grow to day-one adults (F1 progeny) under standard condition (20°C). Aggregates were scored in one-day-old adults, and then every 24h interval over a period of 4 days using a Leica fluorescent stereomicroscope (MZFLIII) with the EYFP filter set (excitation, 510/20; emission, 560/40). Aggregates were recognized visually, based on experience from FRAP regarding which foci did not recover following photobleaching. Aggregates too close to be resolved under the stereomicroscope, or smaller satellite aggregates scattered near a large aggregate were accounted as one. This system was consistently maintained for all experiments. The number of aggregates in each worm was scored blind, by independent investigators and yielded very similar results. Approximately 100 animals were scored, corresponding to more than three biological samples. Images of the Q35 expressing animals were collected using a Leica TCS SPE Confocal Microscope (Leica) with a 63X oil objective. Z-planes of 0.35 microns were collected through the whole animal with settings optimized at the beginning so that that the intensities of the polyQ aggregates were not saturated. The same settings were maintained to image all animals. Q35 expressing animals were imaged on Day 4. LAS AF software (Leica, RRID:SCR_013673) was used to obtain and process Z-stack.
Sterility Assays
Wild-type day-one worms growing on OP50 bacteria were left untreated (non-heat shocked control) or heat shocked at 34°C for either 5min or 60min. Eggs (F1) were collected at 20°C during different time intervals as described above. When the F1 progenies become day-one adults, they were visualized, and then separated onto new OP50 plate based on the presence or absence of visible embryos in their uterus. Sterility was ascertained by checking for the presence of eggs on plates on the next day. Images of worms were obtained using a Leica TCS SPE Confocal Microscope (Leica). The plates were imaged under a Leica S9i Digital Stereo Microscope (Leica).
Immunostaining of dissected worms
Immunostaining of dissected worms was performed according to previously published methods (Das et al., 2020) described below. Specifically, non-heat shocked control or heat-shocked (34°C for indicated time) animals were transferred to 8 μl 1X PBS (pH 7.4) on a cover glass (catalog no. 3800105, Leica biosystem). Worms were quickly dissected with a blade (catalog no. 4-311, Integra Miltex), and fixed with freshly prepared 4% paraformaldehyde (catalog no. 15710, Electron Microscopy Science) for 6 min. A charged slide (Superfrost Plus, catalog no. 12-550-15, Thermo Fisher Scientific) was then placed over the cover glass, and the preparation immediately placed on a pre-chilled freezing block on dry ice for at least 10 min. The cover glass was quickly removed, and the samples were post-fixed with pre-chilled 100% methanol (−20°C) for 2 min. The slides were washed in 1X PBST (1X PBS with 0.1% Tween-20), and then blocked in 1X PBST with 0.5% BSA for 1 hr. Samples were incubated at 4°C overnight with primary antibody diluted in 1X PBST. The next day, slides were rinsed in 1X PBST, and incubated with secondary antibody for 2 hr. After washing samples in 1x PBST, they were mounted in Vectashield mounting medium with DAPI (catalog no. H-1200, Vector Laboratories). The antibodies used in the experiment are: mouse anti-FLAG M2 antibody (catalog no. F-3165, Sigma Aldrich, RRID:AB_259529); Rabbit anti-HA antibody (catalog no. ab9110, Abcam, RRID:AB_307019); Rabbit anti-H3K9me2 antibody (catalog no. C15410060, Diagenode, RRID:AB_2892672); Alexa Fluor 647 goat anti-mouse IgG (H+L) antibody (catalog no. A-21236, Thermo Fisher Scientific, RRID:AB_2535805); Alexa Fluor 488 goat anti-rabbit IgG (H+L) antibody (catalog no. A-11008, Thermo Fisher Scientific, RRID:AB_143165). Antibody dilutions are mentioned in Table S1. Imaging of slides was performed using a Leica TCS SPE Confocal Microscope (Leica) with a 63X oil objective. LAS AF software (Leica, RRID:SCR_013673) was used to obtain and process the Z-stack. The LAS AF software (Leica) was also used to quantify the number of oocytes labelled by H3K9me2 (Threshold setting was adjusted to yield a similar low reading for the control of each experiment). For analysis of colocalization between HSF-1 and MET-2, ImageJ software (RRID:SCR_003070) was used; a line was drawn on one-section of Z-stack, and then intensities along the line for each channel were measured. Line scan graphs were generated by plotting the immunofluorescence intensity along the freely positioned line at the periphery of the nucleus.
Immunostaining of Embryos
Embryos were obtained from non-heat shocked P0 control mothers and heat-shocked P0 mothers (34°C for 5 and 60min followed by 5 hrs. recovery at 20°C) by dissecting animals in 1X PBS (pH7.4) on a cover glass. Immediately following dissection, a charged slide was placed over the cover glass and samples were frozen on a pre-chilled freezing block in liquid nitrogen for at least 10 min, and then immersed in liquid nitrogen for 2 min. After freezing for an additional 1 hour at −80°C, the cover glass was quickly removed. Embryos were subsequently fixed at −20°C in pre-chilled, 100% methanol and 100% acetone for 30 min each. The slides were then incubated successively, in pre-chilled 70%, 50%, and 30% ethanol (−20°C) for 1 min each and washed twice in 1X PBST. The remaining process (blocking to imaging) was performed as described above. Rabbit anti-H3K9me2 antibody (catalog no. C15410060, Diagenode, RRID:AB_2892672) and Alexa Fluor 488 goat anti-rabbit IgG (H+L) antibody (catalog no. A-11008, Thermo Fisher Scientific, RRID:AB_143165) were used. Antibody dilutions are mentioned in Table S1. All images were quantified using Image J; H3K9me2 intensity for each nucleus was measured as raw integrated density divided by area.
DAF-16::GFP nuclear localization
Transgenic animals expressing DAF-16::GFP (zIs356) were used for the experiment. Non-heat shocked control P0 mothers, or P0 mothers heat-shocked at 34°C for 5 and 60 min were allowed to lay eggs (2-4 hr. eggs post 5min heat shock and 8-12 hr. eggs post 60min heat shock) on fresh NGM plates seeded with OP50 bacteria. The F1 embryos were allowed to grow at 20°C to day-1 adults. Day-one progeny (F1) were mounted on a 2% agar pad containing a drop of 10mM levamisole for immobilization. All worms were imaged within 10 min using a Leica TCS SPE Confocal Microscope (Leica) with a 63X oil objective. LAS AF software (Leica) was used to obtain images, as well as to score the number of worms that showed DAF-16 localization in nuclei.
RNA extraction and real-time quantitative reverse-transcriptase PCR (qRT-PCR)
Unless otherwise mentioned, RNA was extracted from day-one adults. Animals were either passaged as L4s, 24 hrs. before harvesting (for P0 animals) or harvested as day-one adults (for F1 progeny) at densities of 20-30 worms/plate. RNA was extracted as previously described (Chikka et al., 2016). Briefly, RNA samples were harvested in 50 μl of Trizol (catalog no. 400753, Life Technologies) and snap-frozen immediately in liquid nitrogen. Samples were thawed on ice and 200 μl of Trizol was added, followed by brief vortexing at room temperature. Samples were then lysed using a Precellys 24 homogenizer (Bertin Corp.). RNA was purified as detailed with appropriate volumes of reagents modified for the starting volume of 250 μl of Trizol. The RNA pellet was dissolved in 17 μl of RNase-free water. The purified RNA was then treated with deoxyribonuclease using the TURBO DNA-free kit (catalog no. AM1907, Life Technologies) as per the manufacturer’s protocol. cDNA was generated by using the iScript cDNA Synthesis Kit (catalog no. 170–8891, Bio-Rad). qRT-PCR was performed using PowerUp SYBR Green Master Mix (catalog no. A25742, Thermo Fisher Scientific) in QuantStudio 3 Real-Time PCR System (Thermo Fisher Scientific, RRID:SCR_018712) at a 10 μl sample volume, in a 96-well plate (catalog no. 4346907, Thermo Fisher Scientific). The relative amounts of mRNA were determined using the ΔΔCt method for quantitation. We selected pmp-3 as an appropriate internal control for gene expression analysis in C. elegans based on previous experiments (Das et al., 2020).
mRNA values (relative to pmp-3) were normalized to either that of the non-heat shocked control values of wild-type animals, or to the non-heat shocked control values for each genotype or RNAi treatment (specified in figure legends). Each experiment was repeated a minimum of three times. For qPCR reactions, the amplification of a single product with no primer dimers was confirmed by melt-curve analysis performed at the end of the reaction. Reverse transcriptase-minus controls were included to exclude any possible genomic DNA amplification. Primers were designed using Primer3 software (RRID:SCR_003139) and generated by Integrated DNA Technologies. The primers used for the qRT-PCR analysis are listed in Table S3.
Co-immunopreciptation (Co-IP)
Approximately 1,000-1,200 day-one adult animals per condition (non-heat shocked control or heat shock at 34°C for 30 min) were collected in M9 buffer [3 g KH2PO4, 6 g Na2HPO4, 5 g NaCl, 1 ml 1 M MgSO4, water to 1 liter] and snap frozen in liquid nitrogen. Worm pellets were resuspended with equal volume of pre-chilled 2x lysis buffer [60mM HEPES (pH7.4), 100 mM KCl, 4 mM MgCl, 0.1% Triton X-100, 10% glycerol, supplemented with 2 mM DTT and protease inhibitor cocktail (Jedamzik and Eckmann, 2009)]. After lysing with a Precellys 24 homogenizer (Bertin Corp.), samples were centrifuged at 200 XG for 5 min at 4°C to remove worm debris. 10% of worm lysates was set aside and snap frozen to use as input. For immunoprecipitation of HA tagged MET-2, the remaining lysates were incubated at 4°C for 2 hours with mouse anti-HA magnetic bead (catalog no. 88837, Pierce, RRID:AB_2861399). Before incubation, the beads were pre-cleared with pre-cold 1X lysis buffer. Anti-mouse IgG antibody (catalog no. ab188776, Abcam) was used as negative control. Before incubation with samples, anti-mouse IgG antibody was conjugated with Protein A/G magnetic beads (catalog no. 88802, Pierce) at 4°C for 1 hr. Beads were then washed with pre-chilled wash buffer [30 mM HEPES, pH = 7.4, 100 mM KCl, 2mM MgCl, 0.1% Triton X-100, supplemented with 1 mM DTT], and mixed with 4X Laemmli sample buffer (catalog no. 1610737, Bio-Rad) supplemented with 10% β-mercaptoethanol. Immunoprecipitated protein mixture was eluted, denatured by boiling and analyzed by western blotting with mouse anti-FLAG M2 antibody (catalog no. F1804, Sigma Aldrich, RRID:AB_262044) and rabbit anti-HA antibody (catalog no. ab9110, Abcam, RRID:AB_307019). Antibody dilutions are mentioned in Table S1.
Western Blotting
Western blot analysis was performed with adult day-1 animals. For protein analysis, 15–50 worms (depending on the experimental requirement) were harvested in 15 μl of 1X PBS (pH 7.4), and then 4X Laemmli sample buffer (catalog no. 1610737, Bio-Rad) supplemented with 10% β-mercaptoethanol was added to each sample. Samples were then boiled for 30 min. Comparable numbers of animals were loaded/lane. Whole-worm lysates were resolved on SDS-PAGE gels and transferred onto nitrocellulose membrane (catalog no. 1620115, Bio-Rad). Membranes were blocked with Odyssey Blocking Buffer (part no. 927–50000, LI-COR). Immunoblots were imaged using LI-COR Odyssey Infrared Imaging System (LI-COR Biotechnology, Lincoln, NE). Mouse anti-FLAG M2 antibody (catalog no. F1804, Sigma Aldrich, RRID:AB_262044) was used to detect HSF-1::FLAG and C12C1.8::FLAG. Rabbit anti-HA antibody (catalog no. ab9110, Abcam, RRID:AB_307019) was used to detect MET-2::HA and HSP-1::HA. Mouse anti-polyglutamine antibody (catalog no. P1874, Sigma Aldrich, RRID:AB_532270) was used to detect polyQ expression in Q35::YFP animals (Prahlad and Morimoto, 2011). Rabbit anti-HSP90 antibody (catalog no. 4874, Cell Signaling Technology, RRID:AB_2121214) was used to detect DAF-21 level. Mouse anti-H3K9me2 antibody (catalog no. ab1220, Abcam, RRID:AB_449854) was used to detect dimethylated H3K9 and rabbit anti-histone H3 antibody (catalog no. ab1791, Abcam, RRID:AB_302613) was used to detect total histone H3 level. Mouse anti-α-tubulin primary antibody (catalog no. AA4.3, RRID:AB_579793), developed by C. Walsh, was obtained from the Developmental Studies Hybridoma Bank (DSHB), created by the National Institute of Child Health and Human Development (NICHD) of the National Institute of Health (NIH), and maintained at the Department of Biology, University of Iowa. The following secondary antibodies were used: Sheep anti-mouse IgG (H and L) Antibody IRDye 800CW Conjugated (catalog no. 610-631-002, Rockland Immunochemicals, RRID:AB_220142) and Alexa Fluor 680 goat anti-rabbit IgG (H+L) (catalog no. A21109, Thermo Fisher Scientific, RRID:AB_2535758). Antibody dilutions are mentioned in Table S1. Protein expression (relative to α-tubulin levels) in different samples was measured using the LI-COR Image Studio software (RRID:SCR_015795). Fold change of protein levels was calculated relative to non-heat shocked wild-type controls (or RNAi control, L4440). For quantifying polyQ expression in Q35::YFP animals day-one adults were chosen as the majority of polyglutamine is still soluble at that age.
Identification of Heat Shock Elements (HSE) in the 5’-UTR region of daf-2.
Chip-seq reads were downloaded from the NBCI Gene Expression Omnibus GSE81523. ChIP-seq reads were aligned to the WBcel235 /ce11 assembly of the C. elegans genome with bowtie 2 using the standard parameters. Reads were filtered using sambamba view (Tarasov et al., 2015) to remove unaligned reads and then filtered against modENCODE Blacklist (Amemiya et al., 2019). Peaks for the individual replicates were called using MACS2 (Zhang et al., 2008), using the parameters -g ce --bdg --keep-dup=auto -m 5 30 -q 0.01. To combine the replicates, the build signal track was generated using MACS bdgcmp and Pearson correlations between replicates were calculated by wigcorrelate, Peaks were called again on the combined replicates using MACS2. The overlap and the Fisher’s exact test were performed by using Bedtools Intersect, the overlap permutation test was performed using the R package “regioner” with 1000 randomizations. We defined the HSF-1 bound regions, by centralizing and extending a region of 101 bp at the HSF-1 summits called by MACS2 (Zhang et al., 2008). We used the tool FIMO (Grant et al., 2011) in the MEME Suite v 5.3.2 (Bailey et al., 2009) to identify the HSE motif “TTCNNGAA” in the WBcel235(ce11) genome and in the HSF1 bound regions, using a threshold 0.01, ex.: “fimo --bgfile ce11.markov -file --thresh 0.01 hsf1_hse_meme ce11.fa”. The Markov background model was defined using the tool fasta-get-markov (Bailey et al., 2009), with the sequences of the WBcel235(ce11) and the HSF-1 bound regions. The DOIs for the pipeline and Software used are listed in the Key Resource Table.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed according to previously published methods (Das et al., 2020) described below. Specifically, for ChIP in P0 animals (mothers), ~500 animals per condition (non-heat shocked control, or heat shock at 34°C for indicated times) were obtained by washing off two plates of synchronized day-one adult animals obtained by the ‘bleach-hatch’ method. For ChIP in F1 animals (progeny), ~400-500 F1 day-one adult animals per condition (or F2) were generated from P0 animals (non-heat shocked controls, or heat shock at 34°C for indicated time), by collecting the P0 eggs (as described above and in figures/ figure legends) and allowing these eggs to mature into day-one adults. Animals were washed quickly, with 1X PBS (pH 7.4). Cross-linking was performed with freshly prepared 2% formaldehyde (catalog no. 252549, Sigma Aldrich) at room temperature for 10 min. 1 ml of formaldehyde was typically required to ensure consistent results from the harvested samples. Tris (pH 7.4) was added to a final concentration of 250 mM to quench the samples, and the samples were quenched at room temperature for 10 min. Samples were then washed three times in ice-cold 1X PBS supplemented with protease inhibitor cocktail and snap-frozen in liquid nitrogen. The worm pellet was resuspended in FA buffer [50 mM HEPES (pH 7.4), 150 mM NaCl, 50 mM EDTA, 1% Triton-X-100, 0.5% SDS and 0.1% sodium deoxycholate], supplemented with 1 mM DTT and protease inhibitor cocktail. The large excess of EDTA was used after extensive prior optimization experiments, as we discovered that there was an unknown source of DNAse (likely from the bactopeptone in agarose plates or OP50 bacteria) that limited DNA yield. Following the addition of FA buffer, the suspended worm pellet was lysed using a Precellys 24 homogenizer (Bertin Corp.), and then sonicated in a Bioruptor Pico Sonication System (catalog no. B0106001, Diagenode) (15 cycles of 30 s on/off). Samples were centrifuged at 11731 XG for 5 min at 4°C to remove worm debris. For all HSF-1 ChIP experiments, endogenous HSF-1 was immunoprecipitated with anti-FLAG M2 magnetic bead (catalog no. M-8823, Sigma-Aldrich, RRID:AB_2637089). Beads were first pre-cleared with chromatin isolated from wild-type worms not having any FLAG tag and salmon sperm DNA (catalog no. 15632–011, Invitrogen). Worm lysate was incubated at 4°C overnight with the pre-cleared FLAG beads. For all other ChIP experiments, Protein A/G Magnetic Beads (catalog no. 88802, Pierce) pre-cleared with salmon sperm DNA was used. Worm lysates were first pre-cleared with pre-cleared magnetic beads and then the resulting pre-cleared worm lysate was incubated at 4°C overnight with anti-H3K9me2 antibody (Lee et al., 2019) (catalog no. ab1220, Abcam, RRID:AB_449854). Subsequently, the pre-cleared magnetic beads were added, and the mixture incubated for another 3–4 hr. Beads were washed with low salt, high salt and LiCl wash buffers and then eluted in buffer containing EDTA, SDS and sodium bicarbonate [20mM EDTA, 1% SDS, 0.1M sodium bicarbonate]. The elute was incubated with RNase A and then de-crosslinked overnight in presence of Proteinase K and NaCl. The DNA was purified by ChIP DNA purification kit (catalog no. D5205, Zymo Research). qPCR analysis of DNA was performed as described above using primer sets specific for different target genes. Promoter region of syp-1 was amplified for all HSF1-ChIP experiments to quantify non-specific binding of HSF-1. For all ChIP experiments, 10% of total lysate was used as ‘input’ and chromatin immunoprecipitated by different antibodies were expressed as % input values. All relative changes were normalized to either that of the wild-type non-heat shocked control or the non-heat shocked control of each genotype or RNAi treatment (specified in figure legends). Fold changes were calculated by ΔΔCt method. The sequence and position of the primers used for ChIP experiments, and the expected amplicon sizes are listed in Table S3 and depicted in Figure S8.
QUANTIFICATION AND STATISTICAL ANALYSIS
Each ‘experiment’ refers to a biological repeat. No statistical methods were used to predetermine sample size. The experiments were not randomized, but some were blinded. The statistical details of experiments could be found in corresponding figure legends. The data were analyzed by using Student’s t-test and/or one-way ANOVA with Tukey’s correction (GraphPad Prism software) as described in respective figure legends. P values are indicated as follows: *p<0.05; **p< 0.01; ***p<0.001.
Supplementary Material
Table S1: Antibodies used in this study. Related to STAR Methods.
Table S2: Strains used in this study. Related to STAR Methods.
Table S3: Primers used in this study. Related to STAR Methods.
Highlights.
Germline activation of HSF-1 triggers heritable epigenetic changes in C. elegans
HSF-1 recruits the putative H3K9methyltransferase MET-2 to bookmark daf-2
HSF-1 causes transcriptional switching of stress response strategies of offspring
Maternal heat-shock duration sets the heritability of H3K9me2 bookmarking.
Acknowledgements
We thank the V.P. laboratory, Drs. Sarit Smolikove, Josep Comeron, Jian Li, Tali Gidalevitz and Yair Argon for comments, Drs. Rachel Reichman and Smolikove for advice with CRISPR/Cas9, Dr. Cruz-Corchado for data analyses, and Ms. Sermet for assistance with crosses. Nematode strains were provided by the Caenorhabditis Genetics Center (CGC) (funded by the NIH Infrastructure Programs P40 OD010440). This work was supported by NIH R01 AG 050653 (V.P.).
Footnotes
Declaration of Interests
The authors declare no competing interests
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akerfelt M, Trouillet D, Mezger V, and Sistonen L (2007). Heat shock factors at a crossroad between stress and development. Ann N Y Acad Sci 1113, 15–27. [DOI] [PubMed] [Google Scholar]
- Alasady MJ, and Mendillo ML (2020). The Multifaceted Role of HSF1 in Tumorigenesis. Adv Exp Med Biol 1243, 69–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allshire RC, and Madhani HD (2018). Ten principles of heterochromatin formation and function. Nat Rev Mol Cell Biol 19, 229–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amemiya HM, Kundaje A, and Boyle AP (2019). The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Scientific Reports 9, 9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen EC, and Horvitz HR (2007). Two C. elegans histone methyltransferases repress lin-3 EGF transcription to inhibit vulval development. Development 134, 2991–2999. [DOI] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, and Noble WS (2009). MEME Suite: tools for motif discovery and searching. Nucleic Acids Research 37, W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, and Zhao K (2007). High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837. [DOI] [PubMed] [Google Scholar]
- Baugh LR, and Day T (2020). Nongenetic inheritance and multigenerational plasticity in the nematode C. elegans. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baugh LR, and Hu PJ (2020). Starvation Responses Throughout the Caenorhabditis elegans Life Cycle. Genetics 216, 837–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berson A, Nativio R, Berger SL, and Bonini NM (2018). Epigenetic Regulation in Neurodegenerative Diseases. Trends Neurosci 41, 587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessler JB, Andersen EC, and Villeneuve AM (2010). Differential localization and independent acquisition of the H3K9me2 and H3K9me3 chromatin modifications in the Caenorhabditis elegans adult germ line. PLoS Genet 6, e1000830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette CR, Perrat PN, Thackeray A, and Benard CY (2015). Glypican Is a Modulator of Netrin-Mediated Axon Guidance. PLoS Biol 13, e1002183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A, Fiori LM, and Turecki G (2019). Bridging Basic and Clinical Research in Early Life Adversity, DNA Methylation, and Major Depressive Disorder. Front Genet 10, 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, and Berger SL (2014). Epigenetics of aging and aging-related disease. J Gerontol A Biol Sci Med Sci 69 Suppl 1, S17–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunquell J, Morris S, Lu Y, Cheng F, and Westerheide SD (2016). The genome-wide role of HSF-1 in the regulation of gene expression in Caenorhabditis elegans. BMC Genomics 17, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese EJ, and Baldwin LA (1998). Hormesis as a biological hypothesis. Environ Health Perspect 106 Suppl 1, 357–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese EJ, and Baldwin LA (2001). Hormesis: a generalizable and unifying hypothesis. Crit Rev Toxicol 31, 353–424. [DOI] [PubMed] [Google Scholar]
- Calabrese EJ, Mattson MP, Dhawan G, Kapoor R, Calabrese V, and Giordano J (2020). Hormesis: A potential strategic approach to the treatment of neurodegenerative disease. Int Rev Neurobiol 155, 271–301. [DOI] [PubMed] [Google Scholar]
- Camacho J, Truong L, Kurt Z, Chen YW, Morselli M, Gutierrez G, Pellegrini M, Yang X, and Allard P (2018). The Memory of Environmental Chemical Exposure in C. elegans Is Dependent on the Jumonji Demethylases jmjd-2 and jmjd-3/utx-1. Cell Rep 23, 2392–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter BS, Lee TW, Plott CF, Rodriguez JD, Brockett JS, Myrick DA, and Katz DJ (2021). C. elegans establishes germline versus soma by balancing inherited histone methylation. Development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikka MR, Anbalagan C, Dvorak K, Dombeck K, and Prahlad V (2016). The Mitochondria-Regulated Immune Pathway Activated in the C. elegans Intestine Is Neuroprotective. Cell Rep 16, 2399–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou WY, Lin YC, and Lee YH (2019). Short-term starvation stress at young adult stages enhances meiotic activity of germ cells to maintain spermatogenesis in aged male Caenorhabditis elegans. Aging Cell 18, e12930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen J, Agger K, Cloos PA, Pasini D, Rose S, Sennels L, Rappsilber J, Hansen KH, Salcini AE, and Helin K (2007). RBP2 belongs to a family of demethylases, specific for tri-and dimethylated lysine 4 on histone 3. Cell 128, 1063–1076. [DOI] [PubMed] [Google Scholar]
- D'Urso A, and Brickner JH (2017). Epigenetic transcriptional memory. Curr Genet 63, 435–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Ooi FK, Cruz Corchado J, Fuller LC, Weiner JA, and Prahlad V (2020). Serotonin signaling by maternal neurons upon stress ensures progeny survival. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Nadal E, Ammerer G, and Posas F (2011). Controlling gene expression in response to stress. Nat Rev Genet 12, 833–845. [DOI] [PubMed] [Google Scholar]
- Deonarine A, Walker MWG, and Westerheide SD (2021). HSF-1 displays nuclear stress body formation in multiple tissues in Caenorhabditis elegans upon stress and following the transition to adulthood. Cell Stress Chaperones. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desse VE, Blanchette CR, Nadour M, Perrat P, Rivollet L, Khandekar A, and Benard CY (2021). Neuronal post-developmentally acting SAX-7S/L1CAM can function as cleaved fragments to maintain neuronal architecture in C. elegans. Genetics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuhashi H, Takasaki T, Rechtsteiner A, Li T, Kimura H, Checchi PM, Strome S, and Kelly WG (2010). Trans-generational epigenetic regulation of C. elegans primordial germ cells. Epigenetics Chromatin 3, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrigues JM, Sidoli S, Garcia BA, and Strome S (2015). Defining heterochromatin in C. elegans through genome-wide analysis of the heterochromatin protein 1 homolog HPL-2. Genome Res 25, 76–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrigues JM, Tsu BV, Daugherty MD, and Pasquinelli AE (2019). Diversification of the Caenorhabditis heat shock response by Helitron transposable elements. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano J, Ives JA, and Jonas WB (2008). Hormetic responses in neural systems: consideration, contexts, and caveats. Crit Rev Toxicol 38, 623–627. [DOI] [PubMed] [Google Scholar]
- Gomez-Pastor R, Burchfiel ET, and Thiele DJ (2018). Regulation of heat shock transcription factors and their roles in physiology and disease. Nat Rev Mol Cell Biol 19, 4–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Beese-Sims SE, Brookes E, Spadafora R, Zhu Y, Rothbart SB, Aristizabal-Corrales D, Chen S, Badeaux AI, Jin Q, et al. (2014). A histone methylation network regulates transgenerational epigenetic memory in C. elegans. Cell Rep 7, 113–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Maures TJ, Ucar D, Hauswirth AG, Mancini E, Lim JP, Benayoun BA, Shi Y, and Brunet A (2011). Transgenerational epigenetic inheritance of longevity in Caenorhabditis elegans. Nature 479, 365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G, von Meyenn F, Rostovskaya M, Clarke J, Dietmann S, Baker D, Sahakyan A, Myers S, Bertone P, Reik W, et al. (2017). Epigenetic resetting of human pluripotency. Development 144, 2748–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajdu-Cronin YM, Chen WJ, and Sternberg PW (2004). The L-type cyclin CYL-1 and the heat-shock-factor HSF-1 are required for heat-shock-induced protein expression in Caenorhabditis elegans. Genetics 168, 1937–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdeman R, Nehrt S, and Strome S (1998). MES-2, a maternal protein essential for viability of the germline in Caenorhabditis elegans, is homologous to a Drosophila Polycomb group protein. Development 125, 2457–2467. [DOI] [PubMed] [Google Scholar]
- Houri-Ze'evi L, Korem Y, Sheftel H, Faigenbloom L, Toker IA, Dagan Y, Awad L, Degani L, Alon U, and Rechavi O (2016). A Tunable Mechanism Determines the Duration of the Transgenerational Small RNA Inheritance in C. elegans. Cell 165, 88–99. [DOI] [PubMed] [Google Scholar]
- Hsu AL, Murphy CT, and Kenyon C (2003). Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science 300, 1142–1145. [DOI] [PubMed] [Google Scholar]
- Jedamzik B, and Eckmann CR (2009). Analysis of in vivo protein complexes by coimmunoprecipitation from Caenorhabditis elegans. Cold Spring Harb Protoc 2009, pdb prot5299. [DOI] [PubMed] [Google Scholar]
- Jin C, Li J, Green CD, Yu X, Tang X, Han D, Xian B, Wang D, Huang X, Cao X, et al. (2011). Histone demethylase UTX-1 regulates C. elegans life span by targeting the insulin/IGF-1 signaling pathway. Cell Metab 14, 161–172. [DOI] [PubMed] [Google Scholar]
- Jobson MA, Jordan JM, Sandrof MA, Hibshman JD, Lennox AL, and Baugh LR (2015). Transgenerational Effects of Early Life Starvation on Growth, Reproduction, and Stress Resistance in Caenorhabditis elegans. Genetics 201, 201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneshiro KR, Rechtsteiner A, and Strome S (2019). Sperm-inherited H3K27me3 impacts offspring transcription and development in C. elegans. Nat Commun 10, 1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz DJ, Edwards TM, Reinke V, and Kelly WG (2009). A C. elegans LSD1 demethylase contributes to germline immortality by reprogramming epigenetic memory. Cell 137, 308–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon C, Chang J, Gensch E, Rudner A, and Tabtiang R (1993). A C. elegans mutant that lives twice as long as wild type. Nature 366, 461–464. [DOI] [PubMed] [Google Scholar]
- Kerr SC, Ruppersburg CC, Francis JW, and Katz DJ (2014). SPR-5 and MET-2 function cooperatively to reestablish an epigenetic ground state during passage through the germ line. Proc Natl Acad Sci U S A 111, 9509–9514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura KD, Tissenbaum HA, Liu Y, and Ruvkun G (1997). daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science 277, 942–946. [DOI] [PubMed] [Google Scholar]
- Kreher J, Takasaki T, Cockrum C, Sidoli S, Garcia BA, Jensen ON, and Strome S (2018). Distinct Roles of Two Histone Methyltransferases in Transmitting H3K36me3-Based Epigenetic Memory Across Generations in Caenorhabditis elegans. Genetics 210, 969–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumsta C, Chang JT, Schmalz J, and Hansen M (2017). Hormetic heat stress and HSF-1 induce autophagy to improve survival and proteostasis in C. elegans. Nat Commun 8, 14337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbadia J, and Morimoto RI (2015). Repression of the Heat Shock Response Is a Programmed Event at the Onset of Reproduction. Mol Cell 59, 639–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SS, Lee RY, Fraser AG, Kamath RS, Ahringer J, and Ruvkun G (2003). A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat Genet 33, 40–48. [DOI] [PubMed] [Google Scholar]
- Lee TW, David HS, Engstrom AK, Carpenter BS, and Katz DJ (2019). Repressive H3K9me2 protects lifespan against the transgenerational burden of COMPASS activity in C. elegans. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leimar O, and McNamara JM (2015). The evolution of transgenerational integration of information in heterogeneous environments. Am Nat 185, E55–69. [DOI] [PubMed] [Google Scholar]
- Lev I, Seroussi U, Gingold H, Bril R, Anava S, and Rechavi O (2017). MET-2-Dependent H3K9 Methylation Suppresses Transgenerational Small RNA Inheritance. Curr Biol 27, 1138–1147. [DOI] [PubMed] [Google Scholar]
- Li J, Chauve L, Phelps G, Brielmann RM, and Morimoto RI (2016). E2F coregulates an essential HSF developmental program that is distinct from the heat-shock response. Genes Dev 30, 2062–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JP, and Brunet A (2013). Bridging the transgenerational gap with epigenetic memory. Trends Genet 29, 176–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matai L, Sarkar GC, Chamoli M, Malik Y, Kumar SS, Rautela U, Jana NR, Chakraborty K, and Mukhopadhyay A (2019). Dietary restriction improves proteostasis and increases life span through endoplasmic reticulum hormesis. Proc Natl Acad Sci U S A 116, 17383–17392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColl G, Rogers AN, Alavez S, Hubbard AE, Melov S, Link CD, Bush AI, Kapahi P, and Lithgow GJ (2010). Insulin-like signaling determines survival during stress via posttranscriptional mechanisms in C. elegans. Cell Metab 12, 260–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minkina O, and Hunter CP (2018). Intergenerational Transmission of Gene Regulatory Information in Caenorhabditis elegans. Trends Genet 34, 54–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mommer BC, and Bell AM (2014). Maternal experience with predation risk influences genome-wide embryonic gene expression in threespined sticklebacks (Gasterosteus aculeatus). PLoS One 9, e98564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RS, Kaletsky R, and Murphy CT (2019). Piwi/PRG-1 Argonaute and TGF-beta Mediate Transgenerational Learned Pathogenic Avoidance. Cell 177, 1827–1841 e1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto RI (1998). Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev 12, 3788–3796. [DOI] [PubMed] [Google Scholar]
- Morley JF, Brignull HR, Weyers JJ, and Morimoto RI (2002). The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc Natl Acad Sci U S A 99, 10417–10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton EA, and Lamitina T (2013). Caenorhabditis elegans HSF-1 is an essential nuclear protein that forms stress granule-like structures following heat shock. Aging Cell 12, 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, and Kenyon C (2003). Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424, 277–283. [DOI] [PubMed] [Google Scholar]
- Mutlu B, Chen HM, Gutnik S, Hall DH, Keppler-Ross S, and Mango SE (2019). Distinct functions and temporal regulation of methylated histone H3 during early embryogenesis. Development 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutlu B, Chen HM, Moresco JJ, Orelo BD, Yang B, Gaspar JM, Keppler-Ross S, Yates JR 3rd, Hall DH, Maine EM, et al. (2018). Regulated nuclear accumulation of a histone methyltransferase times the onset of heterochromatin formation in C. elegans embryos. Sci Adv 4, eaat6224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neef DW, Jaeger AM, and Thiele DJ (2011). Heat shock transcription factor 1 as a therapeutic target in neurodegenerative diseases. Nat Rev Drug Discov 10, 930–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nottke AC, Beese-Sims SE, Pantalena LF, Reinke V, Shi Y, and Colaiacovo MP (2011). SPR-5 is a histone H3K4 demethylase with a role in meiotic double-strand break repair. Proc Natl Acad Sci U S A 108, 12805–12810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, and Ruvkun G (1997). The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature 389, 994–999. [DOI] [PubMed] [Google Scholar]
- Ooi FK, and Prahlad V (2017). Olfactory experience primes the heat shock transcription factor HSF-1 to enhance the expression of molecular chaperones in C. elegans. Sci Signal 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posner R, Toker IA, Antonova O, Star E, Anava S, Azmon E, Hendricks M, Bracha S, Gingold H, and Rechavi O (2019). Neuronal Small RNAs Control Behavior Transgenerationally. Cell 177, 1814–1826 e1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prahlad V, and Morimoto RI (2011). Neuronal circuitry regulates the response of Caenorhabditis elegans to misfolded proteins. Proc Natl Acad Sci U S A 108, 14204–14209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechavi O, Houri-Ze'evi L, Anava S, Goh WSS, Kerk SY, Hannon GJ, and Hobert O (2014). Starvation-induced transgenerational inheritance of small RNAs in C. elegans. Cell 158, 277–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechtsteiner A, Costello ME, Egelhofer TA, Garrigues JM, Strome S, and Petrella LN (2019). Repression of Germline Genes in Caenorhabditis elegans Somatic Tissues by H3K9 Dimethylation of Their Promoters. Genetics 212, 125–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, and Ristow M (2007). Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab 6, 280–293. [DOI] [PubMed] [Google Scholar]
- Sen P, Shah PP, Nativio R, and Berger SL (2016). Epigenetic Mechanisms of Longevity and Aging. Cell 166, 822–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridharan R, Gonzales-Cope M, Chronis C, Bonora G, McKee R, Huang C, Patel S, Lopez D, Mishra N, Pellegrini M, et al. (2013). Proteomic and genomic approaches reveal critical functions of H3K9 methylation and heterochromatin protein-1gamma in reprogramming to pluripotency. Nat Cell Biol 15, 872–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanner SA, and Yudkin JS (2001). Fetal programming and the Leningrad Siege study. Twin Res 4, 287–292. [DOI] [PubMed] [Google Scholar]
- Sural S, Lu TC, Jung SA, and Hsu AL (2019). HSB-1 Inhibition and HSF-1 Overexpression Trigger Overlapping Transcriptional Changes To Promote Longevity in Caenorhabditis elegans. G3 (Bethesda) 9, 1679–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabuchi TM, Rechtsteiner A, Jeffers TE, Egelhofer TA, Murphy CT, and Strome S (2018). Caenorhabditis elegans sperm carry a histone-based epigenetic memory of both spermatogenesis and oogenesis. Nat Commun 9, 4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, and Prins P (2015). Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglione AM, Venerosi A, and Calamandrei G (2016). Early-Life Toxic Insults and Onset of Sporadic Neurodegenerative Diseases-an Overview of Experimental Studies. Curr Top Behav Neurosci 29, 231–264. [DOI] [PubMed] [Google Scholar]
- Tatum MC, Ooi FK, Chikka MR, Chauve L, Martinez-Velazquez LA, Steinbusch HWM, Morimoto RI, and Prahlad V (2015). Neuronal serotonin release triggers the heat shock response in C. elegans in the absence of temperature increase. Curr Biol 25, 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissenbaum HA, and Ruvkun G (1998). An insulin-like signaling pathway affects both longevity and reproduction in Caenorhabditis elegans. Genetics 148, 703–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobi EW, Goeman JJ, Monajemi R, Gu H, Putter H, Zhang Y, Slieker RC, Stok AP, Thijssen PE, Muller F, et al. (2014). DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat Commun 5, 5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Voorhies WA (2001). Hormesis and aging. Hum Exp Toxicol 20, 315–317; discussion 319-320. [DOI] [PubMed] [Google Scholar]
- Vielle A, Lang J, Dong Y, Ercan S, Kotwaliwale C, Rechtsteiner A, Appert A, Chen QB, Dose A, Egelhofer T, et al. (2012). H4K20me1 contributes to downregulation of X-linked genes for C. elegans dosage compensation. PLoS Genet 8, e1002933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihervaara A, Duarte FM, and Lis JT (2018). Molecular mechanisms driving transcriptional stress responses. Nat Rev Genet 19, 385–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan QL, Meng X, Dai W, Luo Z, Wang C, Fu X, Yang J, Ye Q, and Zhou Q (2021). N(6)-methyldeoxyadenine and histone methylation mediate transgenerational survival advantages induced by hormetic heat stress. Sci Adv 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarde F, Broekmans FJ, van der Pal-de Bruin KM, Schonbeck Y, te Velde ER, Stein AD, and Lumey LH (2013). Prenatal famine, birthweight, reproductive performance and age at menopause: the Dutch hunger winter families study. Hum Reprod 28, 3328–3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, and Adams PD (2007). Heterochromatin and its relationship to cell senescence and cancer therapy. Cell Cycle 6, 784–789. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based Analysis of ChIP-Seq (MACS). Genome Biology 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmet PZ (2017). Diabetes and its drivers: the largest epidemic in human history? Clin Diabetes Endocrinol 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Wu D, Zang X, Wang Z, Wu Z, and Chen D (2019). Construction of a germline-specific RNAi tool in C. elegans. Sci Rep 9, 2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Antibodies used in this study. Related to STAR Methods.
Table S2: Strains used in this study. Related to STAR Methods.
Table S3: Primers used in this study. Related to STAR Methods.
Data Availability Statement
Original western blot images and microscopy data reported in this paper have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the Key Resources Table.
This paper does not report original code, but uses publicly available software to re-analyze publicly available ChIP-seq data. The pipeline scripts for re-analysis has been deposited at Mendeley and are publicly available as of the date of publication. DOIs and GEO accession are listed in the Key Resources Table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies (also see Table S1) | ||
| Mouse monoclonal anti-FLAG | Sigma Aldrich | Cat No. F-3165, RRID:AB_259529 |
| Mouse monoclonal anti-FLAG M2 | Sigma Aldrich | Cat No. F1804, RRID:AB_262044 |
| Rabbit polyclonal anti-HA | Abcam | Cat No. ab9110, RRID:AB_307019 |
| Rabbit polyclonal anti-H3K9me2 | Diagenode | Cat No. C15410060, RRID:AB_2892672 |
| Mouse monoclonal anti-H3K9me2 | Abcam | Cat No. ab1220, RRID:AB_449854 |
| Pierce Anti-HA Magnetic beads | Thermo Fisher Scientific | Cat No. 88837, RRID:AB_2861399 |
| Anti-FLAG M2 magnetic beads | Sigma-Aldrich | Cat No. M-8823, RRID:AB_2637089 |
| Mouse monoclonal anti-polyglutamine | Sigma Aldrich | Cat No. P1874, RRID:AB_532270 |
| Rabbit anti-HSP90 | Cell Signaling Technology | Cat No. 4874, RRID:AB_2121214 |
| Rabbit polyclonal anti-histone H3 | Abcam | Cat No. ab1791, RRID:AB_302613 |
| Mouse monoclonal anti-α-tubulin | Developmental Studies Hybridoma Bank (DSHB) | Cat No. AA4.3, RRID:AB_579793 |
| Normal Anti-mouse IgG | Abcam | Cat No. ab188776 |
| IRDye 800CW conjugatedSheep anti-mouse IgG (H+L) | Rockland Immunochemicals | Cat No. 610-631-002, RRID:AB_220142 |
| Alexa Fluor 680 conjugated goat anti-rabbit IgG (H+L) | Thermo Fisher Scientific | Cat No. A21109, RRID:AB_2535758 |
| Alexa Fluor 647 conjugated goat anti-mouse IgG (H+L) | Thermo Fisher Scientific | Cat No. A-21236, RRID:AB_2535805 |
| Alexa Fluor 488 conjugated goat anti-rabbit IgG (H+L) | Thermo Fisher Scientific | Cat No. A-11008, RRID:AB_143165 |
| Chemicals, peptides, and recombinant proteins | ||
| Vectashield mounting medium with DAPI | Vector Laboratories | Cat No. H-1200, RRID:AB_2336790 |
| 16% paraformaldehyde | Electron Microscopy Science | Cat No. 15710 |
| Formaldehyde | Sigma Aldrich | Cat No. 252549 |
| Halt Protease Inhibitor Cocktail (100X) | Halt | Cat No. 78438 |
| Salmon Sperm DNA | Invitrogen | Cat No. 15632–011 |
| ChIP DNA purification kit | Zymo Research | Cat No. D5205 |
| TURBO DNA-free Kit | Life Technologies | Cat No. AM1907, |
| iScript cDNA Synthesis Kit | Bio-Rad | Cat No. 170–8891 |
| PowerUp SYBR Green Master Mix | Thermo Fisher Scientific | Cat No. A25742 |
| Deposited data | ||
| Raw data (Mendeley data) | This paper | DOI: 10.17632/f38gnsgwvy.1 |
| Reanalyzed ChiP-seq data | Li et al, 2016 | GEO: GSE81523 |
| Experimental models: Organisms/strains (also see Table S2) | ||
| C. elegans: Strain N2 wild-type | Caenorhabditis Genetic Center | N/A |
| C. elegans: Strain PS3551: hsf-1(sy441)I | Caenorhabditis Genetic Center | N/A |
| C. elegans: Strain AM140: rmIs132 [unc-54p::Q35::YFP] | Caenorhabditis Genetic Center | N/A |
| C. elegans: Strain DCL569: mkcSi13 II; rde-1(mkc36) V | Caenorhabditis Genetic Center | N/A |
| C. elegans: Strain MT13293: met-2(n4256) III | Caenorhabditis Genetic Center | N/A |
| C. elegans: Strain: TJ356: zIs356 [daf-16p::daf-16a/b::GFP + rol-6(su1006)] IV | Caenorhabditis Genetic Center | N/A |
| C. elegans: hsf-1::3Xflag(I) | Das et al., 2020 | N/A |
| C. elegans: 3Xflag::hsf-1(I) | Prahlad lab | N/A |
| C. elegans: 3Xflag:: hsf-1(sy441)(I) | Prahlad lab | N/A |
| C. elegans: Strain: PHX2467: 3XHA::hsp-1(IV) | Prahlad lab/Suny Biotech | N/A |
| C. elegans: Strain: PHX2141: hsp-70::3Xflag(I) | Prahlad lab/Suny Biotech | N/A |
| C. elegans: Strain: PHX3574: met-2::3XHA(III) | Prahlad lab/Suny Biotech | N/A |
| C. elegans: met-2::3XHA(III); 3Xflag::hsf-1(I) | Prahlad lab | N/A |
| C. elegans: met-2::3XHA(III); 3Xflag:: hsf-1(sy441)(I) | Prahlad lab | N/A |
| C. elegans: mkcSi13 II; rde-1(mkc36) V; hsf-1::3Xflag(I) | Prahlad lab | N/A |
| Oligonucleotides | ||
| Primers are listed in Table S3 | ||
| Software and algorithms | ||
| LAS X | Leica Microsystems | RRID:SCR_013673 |
| ImageJ (FIJI) | NIH | RRID:SCR_003070 |
| GraphPad Prism (9.0.0) | GraphPad Software | RRID:SCR_002798 |
| Image Studio | LI-COR | RRID:SCR_015795 |
| NCBI SRA Toolkit | SRA Toolkit Development Team | https://github.com/ncbi/sra-tools |
| Snakemake | Mölder, Jablonski et al. 2021 | https://github.com/snakemake/snakemake/blob/26373e66fbaba04fa076aebb24ad869f41f228b9/docs/index.rst |
| Bowtie2 | Langmead, Trapnell et al. 2009 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Sambamba | Tarasov, Vilella et al. 2015 | https://lomereiter.github.io/sambamba/ |
| Bedtools | Quinlan and Hall 2010 | https://github.com/arq5x/bedtools2 |
| modENCODE Blacklist | Amemiya, Kundaje et al. 2019 | https://github.com/Boyle-Lab/Blacklist/ |
| MACS2 | Zhang, Liu et al. 2008 | https://github.com/macs3-project/MACS |
| ucsc-wigcorrelate | UCSC Genome Bioinformatics Group | https://github.com/ucscGenomeBrowser/kent/tree/master/src/utils |
| Rstudio | RStudio Team (2020) | https://www.rstudio.com/ |
| “regioner” R package | Gel, Díez-Villanueva et al. 2015 | https://bioconductor.org/packages/release/bioc/html/regioneR.html |
| MEME Suite v 5.3.2 FIMO |
Bailey, Johnson et al. 2015 Grant, Bailey et al. 2011 |
https://meme-suite.org/meme/meme-software/5.3.2/meme-5.3.2.tar.gz |
| Other | ||
| PierceTM Protein A/G magnetic beads | Thermo Fisher Scientific | Cat No. 88802 |
| Precellys 24 homogenizer | Bertin Corp. | N/A |
| Bioruptor Pico Sonication System | Diagenode | Cat No. B0106001 |