

Summary
Biosynthesis scales with cell size such that protein concentrations generally remain constant as cells grow. As an exception, synthesis of the cell-cycle inhibitor Whi5 ‘sub-scales’ with cell size so that its concentration is lower in larger cells to promote cell cycle entry. Here, we find that transcriptional control uncouples Whi5 synthesis from cell size and, screening for similar genes, identify histones as the major class of sub-scaling transcripts besides WHI5. Histone synthesis is thereby matched to genome content rather than cell size. Such sub-scaling proteins are challenged by asymmetric cell division because proteins are typically partitioned in proportion to newborn cell volume. To avoid this fate, Whi5 uses chromatin-binding to partition similar protein amounts to each newborn cell regardless of cell size. Finally, disrupting both Whi5 synthesis and chromatin-based partitioning weakens G1 size control. Thus, specific transcriptional and partitioning mechanisms determine protein sub-scaling to control cell size.
Graphical Abstract
eTOC blurb
Most proteins increase in amount as cell size increases. However, Swaffer et al. show that transcriptional and chromatin-based partitioning mechanisms uncouple Whi5 and histone protein amounts from cell size. This results in Whi5 protein concentration reflecting cell size and delaying cell-cycle entry in smaller cells to control cell size.
A striking feature of cell growth is that total protein and RNA amounts per cell increase approximately in proportion to cell volume (Fig. 1A) (Crissman and Steinkamp, 1973; Fraser and Nurse, 1978, 1979). To achieve this coordinated scaling of macromolecules with cell size, larger cells have higher global transcription and protein synthesis rates (Creanor and Mitchison, 1982; Elliott, 1983; Elliott and McLaughlin, 1979; Elliott et al., 1979; Padovan-Merhar et al., 2015; Sun et al., 2020; Zhurinsky et al., 2010). This size-scaling is of general importance because it ensures macromolecule copy number is proportional to cell volume and therefore concentrations are kept approximately constant as a cell grows (Fig. 1B) (Marguerat and Bahler, 2012; Neurohr et al., 2019). Nuclear volume also scales in proportion to cell volume meaning that nuclear concentrations are also expected to be constant (Jorgensen et al., 2007; Neumann and Nurse, 2007).
Figure 1 ∣. WHI5 mRNA does not scale with cell size.

See also Figures S1&S3.
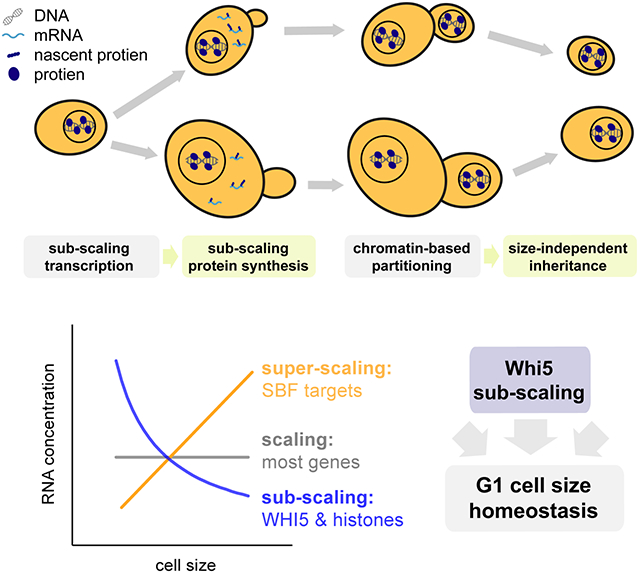
(A-C) Schematics illustrating scaling and sub-scaling gene expression. (A) Total protein and RNA copy numbers per cell generally scale with to cell volume so that concentrations remain constant during growth. (B) However, some proteins sub-scale with size such that protein amounts are constant as a function of size and therefore protein concentrations decrease with cell size, which (C) could result from regulation at any step of gene expression.
(D-F) Cells in S/G2/M were sorted into four bins based on the intensity of total protein dye. See Fig. S1 and STAR Methods for details. (D) Histogram of total protein content per cell in each bin remeasured after sorting. (E) Normalized Transcripts Per Million (TPM / mean TPM) for WHI5 and MDN1 mRNA in cells of different sizes (total protein content). The mean (±range) of two biological replicates is plotted. Changes in TPM are proportional to changes in mRNA concentration. (F) Normalized TPM x total-mRNA for WHI5 and MDN1 mRNA in cells of different sizes (total protein content). Mean (±range) of two biological replicates is plotted. Changes in TPM x total mRNA are proportional to changes in mRNA amount. Relative total mRNA per cell was determined by the number of reads relative to those from a fixed number of S. pombe cells added to the sample.
(G-I) single-molecule Fluorescence In Situ Hybridization (smFISH) analysis of WHI5 and MDN1 mRNA. (G) Representative smFISH images. (H&I) mRNA counts per cell as a function of cell volume for WHI5 and MDN1 determined by smFISH, n=567 cells. Linear regression (solid line) and 95% confidence interval (dashed lines) are shown. Data are pooled from two biological replicates. The same data with replicates plotted independently are shown in Fig. S3A&B.
The importance of biosynthetic size-scaling is underscored by experiments where cells are genetically manipulated to be excessively large. In these cases, protein and RNA synthesis can no longer keep pace with the expanding cell volume and the cytoplasm starts to dilute (Neurohr et al., 2019; Zhurinsky et al., 2010). This results in the failure of many key cellular processes including cell cycle progression and conditional gene expression programs (Neurohr et al., 2019). Importantly, the breakdown in biosynthesis only occurs in extremely large cells. To prevent themselves from becoming excessively large, proliferating cells coordinate cell division with cell growth so that larger cells grow less before entering the cell division cycle and dividing (Johnston et al., 1977; Turner et al., 2012).
The processes of cell size control and of biosynthetic scaling are deeply connected because one mechanism of size control relies on the differential scaling of cell cycle regulators with cell size. While it is generally assumed that most individual proteins exhibit the general size-scaling behavior and therefore remain at constant concentration, one notable exception is the budding yeast cell cycle inhibitor Whi5 (Schmoller et al., 2015). Whi5 synthesis occurs during the S/G2/M stages of the cell cycle before it is translocated into the nucleus at the end of mitosis to inhibit the SBF cell cycle transcription factor in the following G1. Quantification of Whi5 synthesis revealed that the rate of Whi5 synthesis does not increase in proportion to cell size – a behavior defined as protein sub-scaling (Fig. 1B). Whi5 sub-scaling means that an approximately constant amount of Whi5 is made in each cell cycle independent of cell size and results in larger new born cells having a lower Whi5 concentration (Schmoller et al., 2015)(Fig. 1B). Whi5 is then diluted in G1 as cells grow, reducing its concentration and so reducing its ability in larger cells to inhibit the SBF transcription factor that promotes cell-cycle entry. Interestingly, examination of a variety of extracellular growth conditions showed that a similar number of Whi5 molecules are made in all conditions tested, implying Whi5 synthesis is uncoupled from the cellular growth rate as well as cell size (Qu et al., 2019).
This idea of cell cycle regulators differentially scaling during G1 has been expanded on by recent work in budding yeast examining cells arrested in G1 for increasing amounts of time. This revealed that as a G1 arrest is prolonged, a number of cell cycle activators increase in concentration (super-scale) while certain cell cycle inhibitors decreased in concentration (sub-scale) to promote the cell cycle entry of larger cells (Chen et al., 2020). Such a size-dependent super-scaling concentration increase was first observed in the fission yeast S. pombe for the cell cycle activator cdc25 (Keifenheim et al., 2017).
Despite this progress, the underlying molecular mechanisms determining the relationship between cell size and the expression of individual proteins remain largely unknown. It is both unclear what mechanisms scales most biosynthesis with cell size and what additional mechanisms uncouple the synthesis of sub-scaling proteins such as Whi5 from the general trend. It also remains unclear how pervasive sub-scaling behavior is and which other categories of proteins sub-scale to differentially coordinate other aspects of cell biology with cell size.
To address these questions surrounding the size-scaling of gene expression, we have used multiple orthogonal high-throughput and single-cell approaches. We identified a transcriptional control mechanism uncoupling Whi5 synthesis from cell size. Besides WHI5, we identified histones as the major class of sub-scaling gene products by analyzing the transcriptome of differently sized cells progressing through the cell cycle. For stable proteins such as Whi5, we show how budding yeast’s asymmetry in cell division presents a challenge to their sub-scaling expression. This is because the default manner in which proteins are partitioned is in proportion to the volume of the new-born cell which would result in the smaller daughter cell inheriting proportionally less protein – effectively undoing the sub-scaling synthesis of the preceding cell cycle. To avoid this fate, Whi5 uses chromatin-binding to segregate a similar number of protein molecules to each new-born cell regardless of their size. Finally, we disrupted both Whi5 sub-scaling synthesis and its chromatin-based partitioning and show that together these mechanisms are required for proper G1 size control in budding yeast.
WHI5 mRNA does not scale with cell size
First, we set out to determine at what stage of gene expression Whi5 sub-scaling originates. In principle, any step of gene expression could be regulated in a manner that results in sub-scaling protein levels (Fig. 1C). Importantly, for Whi5 sub-scaling is not achieved through negative feedback on protein amounts because multiple copies of the WHI5 gene result in a proportional increase in the number of proteins made per cell cycle (Qu et al., 2019; Schmoller et al., 2015). To determine whether Whi5’s sub-scaling behavior originates at the protein or transcript level we isolated S. cerevisiae cells of different sizes by FACS using total cellular protein content as a proxy for cell size. To do this we stained cells with an amine reactive NHS ester dye which binds bulk protein, sorted cells into four bins based on dye intensity, and performed RNA-seq on each bin (Fig. 1D & S1A). The protein dye intensity in each bin was well correlated with total mRNA content and cell volume, confirming the protein dye is a good proxy for cell size (Fig. S1B-E). Total mRNA was quantified by adding a fixed number of S. pombe cells as a spike-in before RNA extraction and then determining the ratio of S. cerevisiae to S. pombe reads. Cell volume was measured by Coulter counter after sorting. WHI5 mRNA transcripts per million (TPM) decreased as cell size increased, which implies that the WHI5 mRNA concentration is lower in larger cells. In contrast, MDN1 mRNA TPM, as representative of scaling gene expression, was constant (Fig 1E).
To estimate the relative mRNA amount per cell, we then normalized the TPM value to the total mRNA amount per cell determined using the S. pombe spike-in. WHI5 mRNA amount per cell was constant as a function of size while MDN1 mRNA increased linearly (Fig. 1F). To corroborate this finding, we performed single-molecule FISH in individual cells while also measuring the size of each individual cell (Fig. 1G-I). Consistent with the RNA-seq data, the number of WHI5 transcripts per cell did not increase with cell size whereas the number of MDN1 transcripts did (Fig. 1H-I & S3A-B).
Cell cycle analysis of WHI5 sub-scaling
Next, we sought to test if the sub-scaling behavior of WHI5 mRNA we observed is simply a consequence of the cell cycle rather than cell size per se. This is a possibility because WHI5 mRNA is cell cycle regulated and peaks in S phase (Pramila et al., 2006) (Fig. S3D) and cells later in the cell cycle are larger on average. To control for this possibility, we isolated cells in early G1 by centrifugal elutriation and arrested them in G1 for increasing amounts of time to generate populations of cells of increasing sizes. Cells were then released from the G1 arrest resulting in cultures of cells synchronously traversing the entire cell cycle but at different sizes, which we then sampled for RNA-seq analysis (Fig. 2A-B & S2). These differently sized cultures progressed through the cell cycle with similar kinetics as assessed by either DNA-content (Fig. 2A & S2D-E) or the timing in expression of the different classes of cell cycle genes (Fig. S2F-G). Consistent with prior work (Pramila et al., 2006), we observed that WHI5 expression peaks in S phase. We also found that expression peaks at a lower level in larger cells, consistent with WHI5 mRNA sub-scaling with cell size (Fig. 2C). The total amount of WHI5 expression across the entire cell cycle can then be estimated as the area under the curve, which again shows that the concentration of all the WHI5 mRNA made over the cell cycle decreases as cell size increases (Fig. 2D). Consistent with this, if we restrict our smFISH analysis to only those cells in early S/G2/M, when WHI5 expression peaks, the number of WHI5 transcripts is still uncorrelated with cell size (Fig. 2E & S3E-F). Taken together, this group of experiments strongly suggests that WHI5 transcript levels are responsible for the sub-scaling expression of Whi5 protein.
Figure 2 ∣. WHI5 sub-scaling occurs across the cell cycle and is encoded in the WHI5 promoter.

See also Figures S1&S3.
(A-D) G1 cells of different sizes (small, medium, and large) were arrested for increasing amounts of time in G1 using a temperature sensitive cdc28-13 allele at 37°C. Cells were then released from G1 to progress synchronously through a full cell cycle and analyzed by RNA-seq. See Fig. S2 for details. (A) DNA content analysis determined by flow cytometry. (B) Size distributions at point of release from G1 arrest (top panel) and at mid S-phase (bottom panel, corresponds to the 40-minute time point). (C) WHI5 mRNA TPM and (D) the Area Under the Curve (AUC) of mean normalized WHI5 mRNA TPM for small, medium large cells synchronously progressing through the cell cycle. The AUC mean (± range) of two biological replicates is plotted.
(E) mRNA counts per cell for WHI5 as a function of cell size in early S/G2/M cells determined by smFISH; n=156 cells. Early S/G2/M cells were defined as budded cells with a small (≤ 0.2) bud-to-mother volume ratio. Linear regression (solid line) and 95% confidence interval (dashed lines) are shown. Data are pooled from two biological replicates. The same data with replicates plotted independently, including data for MDN1, are shown in Fig. S3E&F.
(F-H) Protein synthesis rates normalized to the mean as a function of cell volume at budding were determined by time-lapse fluorescence microscopy measuring Whi5-mCitrine expressed from (F) the endogenous WHI5 promoter or (H) the ACT1 promoter, and (G) mCitrine expressed alone from the WHI5 promoter. Synthesis rates were determined as in Schmoller et al. (2015) for single cells using linear fits to protein amount traces for the period between bud emergence and cytokinesis (S/G2/M). Data are binned according to cell volume at budding and the mean (±SEM) of each bin is plotted. Un-binned single-cell values from the same data are plotted in Fig. S3G-I.
WHI5 sub-scaling is encoded in its promoter
Having established that WHI5 mRNA subscales with cell size, we sought to test whether this sub-scaling is encoded in its promoter. If this were the case, then the WHI5 promoter should be both necessary and sufficient for sub-scaling protein expression. Previously, we reported that the protein synthesis rate of Whi5-mCitrine sub-scales with cell size, which means that larger cells do not synthesize Whi5-mCitrine proportionally faster than smaller cells (Schmoller et al., 2015). To test if the WHI5 promoter is sufficient for this sub-scaling, we compared the size-dependency of Whi5-mCitrine synthesis rates with that of a reporter mCitrine also expressed from the WHI5 promoter. Whi5-mCitrine and the mCitrine reporter are both synthesized in a sub-scaling manner indicating that the WHI5 promoter is sufficient for sub-scaling synthesis (Fig. 2F-G). In contrast, when we expressed Whi5-mCitrine from a scaling promoter (ACT1pr) its synthesis rate increases with cell size (Fig. 2H). Together, these experiments demonstrate that WHI5 is transcribed in a sub-scaling manner and that the WHI5 promoter is both necessary and sufficient for the sub-scaling synthesis pattern of Whi5.
Histones are a rare class of sub-scaling genes
Having shown that sub-scaling expression of WHI5 is due to a transcriptional mechanism, we sought to determine which other cellular processes are similarly uncoupled from cell size. To do this, we analyzed our RNA-seq experiments of different sized cells. We found 15 transcripts that behaved similarly to WHI5 in both the size-sort and the elutriation arrest-release experiments (Figure S4A). These genes are enriched for GO terms related to chromatin and revealed histones as the major class of sub-scaling genes as 9 of the 15 identified genes encode histones (Fig. 3A). Histone mRNA TPMs are clearly lower in the larger sorted cells than the smaller ones (Fig. 3B & S4B). Similar to WHI5, this sub-scaling is not a consequence of their cell cycle regulated expression because the same trend was observed in the timecourse experiments where cells of different sizes synchronously progress through the cell cycle (Fig. 3C-D & S4C).
Figure 3 ∣. Histones are a rare class of sub-scaling genes.

See also Figures S1-2 & S4-6.
(A) Gene ontology terms enriched in sub-scaling genes. 9 of the 16 sub-scaling genes encode histones and one is WHI5. See Fig. S4A and STAR Methods for classification details.
(B) Normalized TPM (TPM / mean TPM) for sub-scaling histone mRNAs in cells of different sizes (total protein content). The mean (±range) of two biological replicates is plotted. Changes in TPM are proportional to changes in mRNA concentration. See Fig. S1 for experimental details.
(C) HTB2 mRNA TPM for small, medium, and large cells synchronously progressing through the cell cycle as in Fig. 2C-D. See Fig. S2 for experimental details.
(D) The Area Under the Curve (AUC) of mean normalized sub-scaling histone mRNA TPM of small, medium, and large cells synchronously progressing through the cell cycle. The AUC mean (±range) of two biological replicates is plotted.
(E) Pearson correlation coefficient R for the correlation between Histone mRNA levels, relative to wild-type, in 1,484 gene deletion strains (Kemmeren et al., 2014; O'Duibhir et al., 2014) and the cell size of the respective gene deletions for four different data sets of size measurements (Hoose et al., 2012; Jorgensen et al., 2002; Ohya et al., 2005; Soifer and Barkai, 2014). Each point represents an individual mRNA species. Histone mRNAs are shown in blue. The individual regression fits for the histone transcript levels with cell size determined by Jorgensen et al. are shown in Fig. S4D.
(F) Gene ontology terms enriched in super-scaling genes. See Fig. S6A and STAR Methods for classification details.
(G) Normalized TPM (TPM / mean TPM) for example super-scaling mRNAs, specifically those known as targets of the SBF transcription factor, in cells of different sizes (total protein content). The mean (±range) of two biological replicates is plotted. Changes in TPM are proportional to changes in mRNA concentration. See Fig. S1 for experimental details.
(H) Schematics illustrating the scaling, sub-scaling and super-scaling trends of gene expression, representative of most genes, WHI5 and histones, and a subset of SBF targets respectively.
To further test for sub-scaling expression of histone transcripts, we examined microarray data for 1,484 strains each containing a single gene deletion (Kemmeren et al., 2014; O'Duibhir et al., 2014). We compared the level of a given transcript in each deletion strain with the cell size of the same deletion strain and then calculated the Pearson R coefficient for the correlation between transcript levels and cell size across all 1,484 deletion strains. We repeated this using four different cell size datasets acquired as part of independent genome-wide screens utilizing multiple different methodologies for measuring cell size (Hoose et al., 2012; Jorgensen et al., 2002; Ohya et al., 2005; Soifer and Barkai, 2014). This revealed a clear negative correlation between histone mRNA levels and cell size (Fig. 3E & S4D), meaning that histone mRNA concentrations are lower relative to the rest of the transcriptome in deletion strains with a larger cell size. Indeed, histones populate the most extreme negative end of the spectrum of transcripts in all four datasets (Fig. 3E) in contrast to typical transcripts such as those encoding RNA polymerase II subunits (Fig. S5).
We next sought to identify super-scaling genes whose mRNA concentrations increase in larger cells (Fig. S6A). To do this, we again analyzed how gene expression through the cell cycle changes as a function of cell size (Fig. S1 & S2). This identified several super-scaling cell cycle regulated transcripts including SBF regulated genes such as CLN2 (Fig. 3F-H & S6B-C). That these cell cycle regulated genes super-scale, whereas histones and WHI5 sub-scale, despite both sets peaking in expression at a similar time, highlights how these differential scaling properties are unlikely to be due to a conflation of cell cycle progression with cell size. Consistent with this conclusion, many other cell cycle regulated transcripts, including the B-type cyclins, do not sub-scale (Fig. S6D-E).
Histone protein synthesis is uncoupled from cell size
The sub-scaling expression of histone mRNAs suggests that histone protein expression is coordinated with genome content rather than cell size and predicts that histone protein synthesis should also not scale proportionally with cell size. To examine this, we first analyzed two published datasets of flow cytometry measurements across the collection of strains in which each individual open reading frame was fused to GFP (Parts et al., 2014). We compared the relationship between GFP fluorescence (protein amount) and side scatter (SSC-A, cell size) (Fig. 4A). This revealed that histone protein amounts show a weaker dependence on cell size than the average protein in the proteome, i.e., that the slope between cell size (SSC-A) and GFP intensity is smaller (Fig. 4B-C). To confirm that histone protein synthesis does indeed sub-scale, we analyzed the synthesis of the histones Hta2 and Htb2 using time-lapse fluorescence microscopy and compared this to the scaling control of mCitrine expressed from an ACT1pr. We then compared the volume growth during each cell cycle with the amount of new protein synthesized in that cell cycle. As expected, cells that grew more between birth and division expressed correspondingly more ACT1pr-mCitrine. In contrast Hta2 and Htb2 synthesis sub-scales with cell growth in a manner comparable to WHI5pr-WHI5 but not ACT1pr-WHI5 (Fig. 4D-E). Taken together, these experiments identify histones as a rare class of sub-scaling genes whose transcription and protein synthesis are uncoupled from cell size. In this way, histone production can be matched with genome-content rather than cellular growth.
Figure 4 ∣. Histone protein synthesis does not scale with cell size.

(A-C) Analysis of size-dependent expression in the genome-wide collection of GFP fusion strains measured by flow-cytometry (Parts et al., 2014). The slope of the linear fit between cell size (SCC-A) and GFP signal in budded cells was used to estimate the degree of size-dependence for each protein. See STAR Methods for details.
(A) Plot of example protein-GFP levels (intensities normalized to the mean intensity) against cell size. Grey dots denote bin means. Red lines show the linear regression to the un-binned data.
(B) Slope values of 1752 proteins analyzed in two replicates. Slopes closer to 0 correspond to sub-scaling behavior. Histone proteins are shown in blue.
(C) Average slope values for histones (blue) and all other proteins (grey). Four histone were present in the 1752 proteins analyzed. Histone proteins have significantly smaller slopes than the average protein (** p=0.0014; **** p <0.0001).
(D) The amount of mCitrine (expressed from the scaling ACT1 promoter), Hta2-GFP, and Htb2-GFP synthesized (ΔFP normalized to its mean) between birth and division plotted against the amount of cell growth (Δvolume normalized to its mean) determined by single cell time-lapse fluorescence microscopy. Data are binned by Δvolume and the bin means (±SEM) are plotted. Dashed line shows perfect scaling (x=y).
(E) Slope of the robust linear fits to single cell values of ΔFP against Δvolume. Error bars show the standard error of the slope. Slopes for Whi5 scaling are also shows for comparison.
Inheritance of sub-scaling protein levels requires chromatin-based partitioning
Both Whi5 and histones are stable proteins synthesized in a sub-scaling manner during the S/G2/M phases of the cell cycle, meaning that their amounts in G1 are determined by inheritance from previous cell cycles. For typical proteins, which are partitioned along with the cytoplasm or nucleoplasm, concentrations are expected to be similar in the mother and daughter cells at the point of cell division, as is observed for a freely diffusing mCitrine (Fig. 5A&B). Thus, the asymmetric division of budding yeast is a problem for maintaining the protein-level sub-scaling of Whi5 and histones because smaller daughter cells would inherit fewer proteins if they were partitioned in proportion to cell volume. Instead, to maintain size-independent amounts, a mechanism partitioning equal amounts to the daughter and mother cells is required. This is indeed the case for Whi5, which is not partitioned evenly by volume as seen by the increased bud-to-mother concentration ratio at cytokinesis (Fig. 5B). This suggests that differently sized G1 cells could inherit a more similar amount of Whi5 during cell division than would be expected for a typical protein partitioned by volume.
Figure 5 ∣. Maintenance of Whi5 sub-scaling requires chromatin-based partitioning during asymmetric division.

See also Figure S7.
(A) Schematic illustrating the different regimes of protein partitioning at cell division which can be quantified by comparing the mother-to-bud protein concentration ratio at cytokinesis. A ratio ~ 1 is expected for proteins partitioned in proportion to volume. A ratio > 1 is expected for proteins that are partitioned by protein amounts.
(B) The bud-to-mother concentration ratios for Whi5-mCitrine, free mCitrine, and Whi5(WIQ)-mCitrine at cytokinesis. Whi5(WIQ)-mCitrine has reduced recruitment to DNA (Travesa et al., 2013) (Fig. 5C & S7A). **** p < 0.0001..
(C) Anti-Flag ChIP-seq experiments were performed to compare Whi5, Whi5(WIQ) and GFP-NLS. Average RPM metagene plot upstream of all SBF regulated genes (as defined by Ferrezuelo et al., 2010) is shown. ChIP signal around individual SBF binding sites, including additional replicates and controls, is shown in Fig S7A..
(D) Computational simulation of protein amounts at birth as a function of daughter cell volume. Four conditions were simulated where protein expression was either in proportion to cell size (scaling) or independent of cell size (sub-scaling), and protein partitioning is either by amount or in proportion to cell volume. See STAR methods for details. Individual simulated cells (light blue) as well as bin means (dark blue) are plotted. Protein concentrations from the same simulation are shown in Fig. S7B.
(E) Protein amount at birth (normalized to the mean) as a function of daughter cell volume at birth for WHI5pr-WHI5-mCitrine and WHI5pr-WHI5(WIQ)-mCitrine cells. Data are binned according to cell size at birth and the bin means (±SEM) are plotted. Un-binned single-cell values of the same data are plotted in Fig. S7C.
(F) Example time-lapse images of WHI5-mCitirine MYO1-3xmKate2 cells before, during, and after cytokinesis (from left to right), defined as the moment of Myo1 loss from the bud neck.
To quantitatively assess the impact of amount- and volume-based partitioning modalities on the amounts of inherited protein, we employed a full cell-cycle model that simulates growth and division of a population of budding yeast cells. This model was parameterized by single-cell microscopy measurements and therefore accounts for cell-to-cell variability and the size-dependence of cell cycle progression (Chandler-Brown et al., 2017). To this model, we added a protein synthesized either at a rate proportional to cell size (scaling) or independent of cell size (sub-scaling). At division, these proteins were then either partitioned in proportion to cell-volume or with the bud-to-mother concentration ratio of ~1.4 measured for Whi5, which corresponds to approximately 50% of the protein being partitioned by amount. Our simulations show that the amount of Whi5 inherited in G1 should scale significantly more with cell size if it were partitioned according to cell volume rather than, as we estimated, with ~50% partitioned by volume and ~50% by amount (Fig. 5D & S7B). This is in part because bud size varies significantly even for mothers with the same volume. Taken together, this computational analysis shows how both partitioning by amount and sub-scaling synthesis should be required to ensure Whi5 protein sub-scaling in G1.
We hypothesized that the amount-based partitioning of Whi5 could be achieved by utilizing the equal partitioning of the genome during cell division. This possibility was suggested by the fact that Whi5 binds the DNA-bound SBF transcription factor complex. To test this, we analyzed the partitioning of a Whi5 mutant, Whi5(WIQ), that does not bind SBF and is not recruited to the SBF binding sites in the CLN2 or SVS1 promoters (Travesa et al., 2013). First, we confirmed that the WIQ mutation reduces Whi5 binding at SBF-bound DNA elements across the genome by ChIP-seq (Fig. 5C & S7A). Next, we analyzed single cells expressing Whi5(WIQ)-mCitrine, which revealed that Whi5(WIQ) has a lower bud-to-mother concentration ratio at cytokinesis than wild-type Whi5. This supports our model that partitioning by amount is mediated by chromatin binding and is consistent with our estimate of approximately 50% of Whi5 being chromatin-bound at this stage (Fig. 5A).
The chromatin-based partitioning we propose requires that Whi5 has access to bind the genome before cell division. Because Whi5 localization is dynamically regulated during the cell cycle (Costanzo et al., 2004; de Bruin et al., 2004), we examined the timing of Whi5 nuclear import with respect to cytokinesis (Fig. 5F). This clearly shows Whi5 is re-imported into the nucleus before cytokinesis, defined by loss of Myo1 from the bud neck, consistent with previous data (Di Talia et al., 2007).
Finally, to directly test if Whi5 partitioning is important for the size-dependence of Whi5 inheritance as predicted by our model (Fig. 5D & S7B), we examined the relationship between cell size at birth and Whi5 amount for wild-type Whi5 and the Whi5(WIQ) protein variant that cannot bind chromatin (Fig. 5E). This shows that Whi5(WIQ) amounts at birth are higher in larger cells and lower in smaller cells when compared to wild-type Whi5, demonstrating that when chromatin-binding of Whi5 is disrupted, Whi5 sub-scaling in G1 is also disrupted (Fig. 5E). Thus, our data support a model where Whi5 is imported into the nucleus before cell division, which allows ~50% of Whi5 to bind to chromatin and be partitioned with the genome into the mother and bud at division. This ensures that daughter cells inherit more similar amounts of Whi5 despite their differences in cell size at division.
Whi5 sub-scaling contributes to G1 size control
We have previously proposed that Whi5 contributes to G1 size control because: (i) Whi5 concentration is lower in larger cells, and (ii) increasing Whi5 expression increases cell size in a dose-dependent manner (Schmoller et al., 2015). However, it was recently reported that a strain designed to constitutively express WHI5 from a GAL1 promoter, so that its amount scaled with size in G1, still had effective G1 size control as assayed by the single cell correlation between growth in G1 and cell size at birth (Barber et al., 2020). Yet, this same study showed that increasing Whi5 dosage resulted in larger cells on average in the population. Thus, taken at face value, Barber et al.’s result suggests the paradoxical conclusion that Whi5 concentration controls cell size at the population level but is not important in G1 in single cells.
To clarify this issue, we sought to test the function of Whi5 sub-scaling in G1 size control by generating cells with a constant, size-independent Whi5 concentration. To do this, we used the fact that we have now established that Whi5 expression relies on a combination of sub-scaling transcription and chromatin-based partitioning and proceeded to disable both mechanisms (Fig. 6). For these analyses we used the bck2Δ background because BCK2 and WHI5 may be involved in parallel size control pathways (Schmoller et al., 2015) and we are here focusing on the BCK2-independent branch of G1 size control. We have also focused our analysis specifically on daughter cells growing in a poor non-fermentable carbon source (1% ethanol + 2% glycerol) because under these conditions G1 size control is most pronounced, in part due to the smaller and more variable size of new born daughters (Di Talia et al., 2007; Qu et al., 2019). The memory of Whi5 partitioning throughout G1 phase relies on it being a stable protein. We therefore destabilized Whi5 by fusing Whi5-mCitrine with the Cln2 PEST degron (Mateus and Avery, 2000). This allowed us to bypass size-independent partitioning because any Whi5 inherited from the previous cell cycle will be degraded early in G1. We expressed this Whi5-mCitrine-PEST fusion protein from a synthetic TET promoter that is conditionally activated by anhydrotetracycline in a dose-dependent manner, and whose expression scales with cell size (Azizoğlu et al., 2020). We grew cells in anhydrotetracycline at a concentration where the average cell size was similar to that of cells expressing WHI5-mCitrine from its own promoter (WHI5pr-WHI5-mCitrine) (Fig. 6A). Thus, the TETpr-WHI5-mCitrine-PEST strain allows us to generate G1 cells where Whi5 no longer sub-scales with cell size. Instead, Whi5 concentrations in new-born daughter cells start high due to the chromatin-based partitioning, but are then reduced to the steady-state level via protein degradation in the first ~30 minutes of G1 (Fig. 6B-C). This contrasts with WHI5-mCitrine expressed from its own promoter, which is steadily diluted as cells grow during G1 (Fig. 6B).
Figure 6 ∣. Disruption of Whi5 sub-scaling weakens G1 size control.

See also Figure S8.
(A) Median cell volume (average of two independent measurements), measured by Coulter counter of cells expressing WHI5pr-WHI5-mCitrine (grey) or TETpr-WHI5-mCitrine-PEST (blue) in the presence of anhydrotetracycline to induce expression from the size-scaling TET promoter. The fused PEST domain destabilizes Whi5 to eliminate Whi5 synthesized in the preceding cell cycle from new-born G1 cells.
(B-F) Single cell time-lapse microscopy was performed on bck2Δ strains expressing either WHI5pr-WHI5-mCitrine or TETpr-WHI5-mCitrine-PEST. All analysis is restricted to daughter cells. D-F show binned data where no more than 2 cells are outside the bin limits.
(B) Mean Whi5 concentration (±SEM) as a function of time from birth in all daughter cells that completed G1.
(C) Mean Whi5 concentration (±SEM) at 42 minutes after birth as a function of daughter cell volume at birth in all cells that completed G1.
(D) Cell growth in G1 as a function of cell volume at birth for daughter cells that completed G1. Data are binned according to cell volume at birth and the bin means (±SEM) are plotted. Un-binned single-cell values of the same data are plotted in Fig. S8C-D.
(E) Cell volume growth during S/G2/M (i.e. between the first budding event and the subsequent cell division) as a function of cell volume at budding. Data are binned according to cell volume at budding for all cells that completed the cell cycle and the bin means (±SEM) are plotted.
(F) Cell volume growth between birth and cell division as a function of cell volume at birth. Data are binned according to cell volume at birth for cells with a completed cell cycle and the bin means (±SEM) are plotted.
To determine the effect of Whi5 sub-scaling on cell size control, we compared size-dependent cell cycle progression in WHI5pr-WHI5-mCitrine and TETpr-WHI5-mCitrine-PEST cells. When we examined G1 size control in WHI5pr-WHI5-mCitrine cells, a clear anticorrelation between cell volume at birth and volume growth in G1 was observed, as expected and previously reported (Figs. 6D & S8C) (Di Talia et al., 2007). In contrast, this anticorrelation between cell volume at birth and volume growth in G1 was weaker in TETpr-WHI5-mCitrine-PEST cells, showing that Whi5 sub-scaling and dilution are important for cell size control in G1 (Figs. 6D & S8D). We did not observe any significant perturbation to the relationship between size at budding and growth during S/G2/M (Fig. 6E). As previously reported, in wild-type cells the two independent phases of G1 and S/G2/M combine to form an ‘apparent adder’, where an approximately fixed absolute amount of volume growth occurs during each entire cell cycle (Chandler-Brown et al., 2017). In contrast, TETpr-WHI5-mCitrine-PEST cells display a positive correlation between size at birth and total cell cycle volume growth (Fig. 6F). These data show that G1 size control relies on the sub-scaling behavior of Whi5 protein concentration.
While the above results are in support of the Whi5-dilution model for size control, we do not know why Barber et al.’s conceptually similar GAL1pr-WHI5 experiment did not reveal a similar effect. One possibility is that Barber et al. only examined the effect of replacing the promoter and not removing the chromatin-based partitioning that contributes to Whi5 sub-scaling. We sought to test this by expressing stable Whi5 from a weaker promoter (TETpr(weak)-WHI5-mCitrine), which was necessary to titrate in Whi5 to match endogenous levels. This also resulted in a clear reduction in G1 size control (Fig. S8E-F). However, TETpr(weak)-WHI5-mCitrine expression was significantly noisier than TETpr-WHI5-mCitrine-PEST or the wild type WHI5pr-WHI5-mCitrine (Fig. S8G-H). While the increased noise means we cannot make the like-for-like comparisons necessary to isolate the of contribution chromatin partitioning, these data do provide additional evidence that the single-cell Whi5 concentrations in G1 are important in coupling birth size to G1 growth. In contrast, the TETpr-WHI5-mCitrine-PEST has similar expression noise to WHI5pr-WHI5-mCitrine, meaning that a direct comparison is more appropriate (Fig. 6). In this situation (i.e., TETpr-WHI5-mCitrine-PEST), the anticorrelation between birth size and G1 growth is reduced, but not completely lost (Figs. 6D & S8B-D), consistent with the idea that other pathways feed into the G1/S transition to couple growth and cell cycle entry (Chen et al., 2020). Nevertheless, our data clearly indicates that Whi5 dilution and sub-scaling contribute significantly to G1 cell size control in budding yeast.
Discussion
In conclusion, while most genes are expressed proportionally to cell size, a handful are not and instead sub-scale with cell size so that they are diluted in larger cells (Fig. 1A&B). Such sub-scaling is already apparent in the mRNA amounts for both histones and WHI5 across the cell cycle. Our WHI5 promoter-swap experiments indicate that specific promoter elements are at least partly responsible for sub-scaling synthesis. In addition to this transcriptional control, sub-scaling proteins also need a dedicated mechanism to ensure that more equal protein amounts are inherited by differently sized cells during asymmetric cell division. We discovered that Whi5 uses chromatin-binding as a mechanism to segregate approximately equal numbers of molecules to each new-born cell and thereby ensure protein synthesized in the preceding cell cycle is inherited by new-born cells independently of their size (Fig. 7).
Figure 7 ∣. Summary schematic.

A small class of genes including the cell cycle inhibitor WHI5 and histones are transcribed in a sub-scaling manner, resulting in sub-scaling protein synthesis during the cell cycle. Sub-scaling proteins must also be partitioned independent of daughter cell size to retain sub-scaling after cell division, which is achieved through chromatin-based partitioning.
Histone genes dominate the sub-scaling gene class
Through a combination of multiple transcriptomic and proteomic screens we identified histones as the major class of sub-scaling genes in addition to WHI5. Sub-scaling of histone synthesis maintains a stoichiometric relationship between the amount of histones and the genome without engaging wasteful feedback mechanisms, which are known to operate when histone expression is artificially perturbed (Claude et al., 2021; Cross and Smith, 1988; Gunjan and Verreault, 2003; Moran et al., 1990; Norris and Osley, 1987). Our findings, in conjunction with those of Claude et al., (2021), show that the amount of histone synthesis in each cell cycle better reflects the binary increase in genome content rather than variations in cell size. We speculate that this helps prevent unwanted variations in chromatin structure and accessibility in differently sized cells.
It is intriguing that both histones and WHI5 are sub-scaling and are also cell cycle regulated whereas many other cell-cycle regulated genes are not sub-scaling. Indeed, multiple SBF targets have the opposite behavior and are super-scaling (Fig. 3). This raises the question of how sub-scaling transcription arises and whether the mechanisms for Whi5 and histones are related to one another. Because histones are so highly expressed, one possibility is that histone mRNA sub-scaling arises from their promoters or gene bodies becoming saturated with TFs or polymerase. In this way, the peak expression of sub-scaling genes may be limited by the DNA copy number (Claude et al., 2021). In contrast, Whi5 is orders of magnitude less abundant than histones. Thus, it is harder to envisage a situation where the Whi5 gene is saturated and raises the possibility that very different regulatory mechanisms could have arisen to regulate histone and Whi5 sub-scaling.
Whi5 sub-scaling is one of multiple inputs for G1 size control
In the case of WHI5, the function of sub-scaling synthesis is to control cell size. Whi5 functions in early G1 of the cell cycle to inhibit the SBF transcription factor and thereby delay cell cycle progression (Costanzo et al., 2004; de Bruin et al., 2004). Whi5 is then inactivated by phosphorylation by cyclin-Cdk complexes that also drive its exclusion from the nucleus at Start, the point of commitment to cell division (Doncic et al., 2011). Importantly, the exclusion of Whi5 from the nucleus at Start marks the end of the most size-dependent part of the cell division cycle (Di Talia et al., 2007). The sub-scaling of Whi5 allows its concentration to reflect and control cell size. That all cells are born with similar amounts of Whi5 protein, which is then diluted in G1, means that the Whi5 concentration is a readout of current cell size. Since Whi5 is a cell cycle inhibitor, its higher concentration in smaller cells delays their Start transition so that they have more time to grow in G1. Conversely, larger cells have lower concentrations of Whi5 and more rapidly enter the cell division cycle.
While two recent studies presented claims that Whi5 is not diluted in G1 (Dorsey et al., 2018; Litsios et al., 2019), our re-examination of Litsios et al.’s own data clearly shows Whi5 dilution in G1 and that their interpretation was in fact due to a normalization artefact. We have detailed this re-analysis in a recent response to these claims (Schmoller et al., 2020), where we also discuss technical problems apparent in Dorsey et al.’s experiments, rendering their data largely uninterpretable. We also note that at least seven different laboratories have reported Whi5 dilution in the literature, leaving the claims of Dorsey et al. isolated (Barber et al., 2020; Lucena et al., 2018; Qu et al., 2019; Schmoller et al., 2020; Schmoller et al., 2015).
The role of Whi5 sub-scaling for cell size control in G1 is demonstrated by our experiments where we synthetically disable Whi5 sub-scaling and observe a weakened G1 size control (Fig. 6). To remove Whi5 sub-scaling we bypassed both the sub-scaling transcription and the chromatin-based partitioning mechanisms. Our data indicate that while Whi5 sub-scaling and dilution contribute significantly to budding yeast G1 cell size control, they do not remove it completely. This is consistent with the presence of additional size control mechanisms (Chen et al., 2020).
In addition to Whi5, there are several other key regulators of Start, including the SBF transcription factor, that could contribute to the size-dependence of G1 (Andrews and Herskowitz, 1989; Eser et al., 2011; Ferrezuelo et al., 2010; Koch et al., 1993; Nasmyth and Dirick, 1991). Crucially, SBF regulates the transcriptional activation of G1 cyclins that complete the positive feedback loop defining Start as the commitment point to enter the cell cycle (Doncic et al., 2011; Skotheim et al., 2008). SBF was previously identified as a common regulator of certain super-scaling genes in a study examining gene expression in cells of different size in G1 phase (Chen et al., 2020). However, because Whi5 is diluted in larger G1 cells it is possible to interpret the SBF super-scaling as downstream of the size-dependent Whi5 dynamics. Interestingly, we found here that the expression of a number of SBF targets, including the G1 cyclin CLN2, super-scales throughout the cell cycle. This is unlikely to be a downstream effect of Whi5 sub-scaling because by this stage in the cell cycle Whi5 has been phosphorylated, inactivated, and exported from the nucleus. Thus, it appears likely that Whi5 concentration is not the only size-dependent signal regulating SBF activated transcription. We find it intriguing that both the super-scaling and sub-scaling genes we identified here are predominantly cell cycle regulated transcripts whose expression peaks outside of G1.
Comparison with Barber et al., (2020)
The experiments present here where we engineered Whi5 to no longer sub-scale with size (Fig. 6) are related to a previous study, which concluded that constitutive Whi5 expression does not alter G1 size control (Barber et al., 2020). Barber et al. also replicated the prior observations that Whi5 concentration decreases in larger cells and that increasing Whi5 expression results in increases in the average cell size in a dose-dependent manner. Thus, taken together, Barber et al.’s data leads us to the paradoxical interpretation that Whi5 concentrations are important at the population level, but, somehow, are not important in single cells. In contrast, when we eliminated Whi5 sub-scaling we did observe that G1 size control is compromised - i.e., the anti-correlation between size at birth and growth in G1 was weaker (Fig. 6).
A few differences between Barber et al’s and our experiments may help explain this apparent discrepancy. First, Barber et al’s approach was designed to only eliminate the sub-scaling synthesis, whereas our approach has been designed to eliminate sub-scaling synthesis and chromatin-based partitioning, both of which we show contribute to Whi5 sub-scaling in G1 (Fig. 5). Second, even though Whi5 is meant to be constituently expressed in their experiments, it is partially diluted in much of G1 (cf. Fig. S4B from Barber et al.). Thus, one plausible explanation is that Whi5 sub-scaling has not been fully eliminated in Barber et al’s experiment. It is also possible that the effects on size control are more pronounced in the bck2Δ background, in which we have performed our analysis. If this is the case, it would indicate that BCK2 is involved in an additional size control mechanism. Finally our experiments are performed in in a non-fermentable carbon source (ethanol and glycerol) where G1 size control is most pronounced (Di Talia et al., 2007). In contrast, Barber et al. used alternative carbon sources (raffinose and galactose) necessary to express their GALIpr-WHI5 construct. This may also account for some part of this apparent discrepancy as cells in different culturing conditions utilize Whi5 dilution to different extents (Qu et al., 2019).
Finally, we emphasize that we view our interpretations as the most parsimonious because it does not need to evoke any paradoxical effects where Whi5 concentrations can be important at the population level, but not at the single cell level. We also note that our results are consistent with Chen et al. (2020), who expressed WHI5 from a CLN2 promoter and observed reduced G1 size control.
The role of Whi5 dilution in G1
It is important to note that our Whi5 dilution model does not propose to explain exactly why any given cell enters the cell cycle precisely when it does. Instead, it addresses the question of how cells measure their size and then input this information, alongside other size-independent inputs, into the decision to divide. Importantly, the relationship between size at birth and G1 duration exhibits significant cell-to-cell variability (Di Talia et al., 2007; Lord and Wheals, 1981). Our model is therefore not that cells progress through Start at a precise Whi5 concentration threshold. Rather, our model is that the stochastic rate of progression through Start is modulated by the size-dependent Whi5 concentration, as well as additional cell size-dependent and cell size-independent mechanisms. Moreover, we support the recent conclusion that the relative importance of Whi5 sub-scaling and dilution will vary significantly in different growth conditions (Qu et al., 2019). For instance, G1 is longer and cell size control is more pronounced in daughter cells born in poorer carbon sources (e.g., non-fermentable carbons such as ethanol and glycerol) compared to those born in richer carbon sources (e.g., glucose). It is also under poorer carbon sources that Whi5 concentrations are highest (Qu et al., 2019). Thus, daughter cells born in rich carbon conditions will have lower Whi5 concentrations combined with a shorter G1 which results in less Whi5 dilution simply because the extent of dilution is determined by the amount of volume growth in G1. Consistent with this picture, it is precisely in these rich conditions that size control, measured as the degree of inverse correlation between G1 duration and cell size at birth, is weakest (Di Talia et al., 2007).
We also note that the gradual and condition-specific changes that dilution imparts on Whi5 concentration can be readily integrated into the all or nothing decision to enter the cell cycle by the G1/S positive feedback loop (Doncic et al., 2011; Skotheim et al., 2008). As Whi5 concentration is diluted this would promote a gradual increase in the number of active SBF complexes, resulting in the accumulation of low levels of CLN1/2 mRNA molecules. Then, once above some threshold level of CLN1/2 mRNA, the positive feedback switch is flipped, and cells are committed to cell cycle entry (Heldt et al., 2018).
Chromatin binding provides an elegant mechanism for equal protein segregation independent of daughter cell size
When examining the inheritance of Whi5 protein levels during cell division we discovered that the inherent asymmetry at cytokinesis poses a problem for sub-scaling proteins. If a protein were simply partitioned in proportion to the relative volumes of newborn cells, sub-scaling would be lost. Instead, we found cells use chromatin-binding to harness the faithful segregation of sister chromosomes to partition approximately equal protein amounts to newborn cells regardless of their size (Fig. 7). This partitioning mechanism is especially relevant when there is a major size asymmetry at cytokinesis, as is the case for budding yeast and some metazoan cell divisions including D. melanogaster neuroblasts and cells in the early C. elegans embryo (Chia et al., 2008; Sulston et al., 1983). Thus, while it has long been appreciated that big and small cells both have the same amount of DNA, here we identified a set of genes that are similarly sub-scaling. It is both curious and elegant that the sub-scaling gene set includes WHI5, which regulates DNA replication, while the DNA itself is used as a scaffold for the synthesis and maintenance of sub-scaling protein concentrations.
Limitations of the study
While we have established here that a transcriptional and chromatin-partitioning mechanism impart Whi5 sub-scaling, the specific regulatory molecular mechanism(s) that brings about the transcriptional sub-scaling remains unknown. Moreover, it is unclear if the specific sub-scaling mechanism for Whi5 is shared by other sub-scaling genes including histones. Thus, defining the precise promoter elements and regulatory factor(s) responsible for this effect represents an important avenue of future work to further elucidate the basis by which gene expression is differentially regulated with cell size.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jan Skotheim (skotheim@stanford.edu).
Materials availability
All plasmids and strains generated in this study are available from the lead contact upon request without restriction.
Data and code accessibility
RNA-seq and CHIP-seq data associated with this study are available at the GEO repository and assigned accession number GEO: GSE167842.
All original code (i.e.. that used for the cell cycle and protein partitioning model simulations) has been deposited at Zendo and is publicly available as of the date of publication. The DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| M2 anti-FLAG antibody | SIGMA | Cat# F1804 (RRID:AB_262044) |
| SV5-Pk1 anti-V5 antibody | BioRad | Cat# MCA1360G (RRID:AB_1172162) |
| Chemicals, peptides, and recombinant proteins | ||
| Alexa Fluor™ 647 NHS Ester dye | TdermoFisher Scientific | A20006 |
| MDN1 probes coupled to tde fluorescein amidite dye | Biosearch Technologies, Stellaris Custom Probes | NA |
| WHI5 probes coupled to tde Quasar570 dye | Biosearch Technologies, Stellaris Custom Probes | NA |
| Critical commercial assays | ||
| direct-zol RNA microprep kit | Zymo Research | R2061 |
| NEBNext Poly(A) mRNA Magnetic Isolation Module | NEB | E7490 |
| NEBNext Ultra II RNA Library Prep Kit for Illumina | NEB | E7775 |
| NEBNext Ultra II DNA Library Prep kit for Illumina | NEB | E7645 |
| CellASIC ONIX plate for haploid yeast cells | Milipore SIGMA | Y04C |
| Deposited data | ||
| RNA-seq data | This study | GEO: GSE167842 |
| CHIP-seq data | This study | GEO: GSE167842 |
| Experimental models: Organisms/strains | ||
| S. cerevisiae: W303; MATa, ADE2, cdc28-13 | Amon lab | A17896 |
| S. cerevisiae: W303; MATa, ADE2, WHI5-mCitrine::URA3 | This study | DCB99 |
| S. cerevisiae: W303; MATa, bar1Δ::HisG, whi5Δ::LEU2, cln3Δ::hphMX6 | This study | MK551-1 |
| S. cerevisiae: W303; MATalpha, ADE2, HTB2-sfGFP::HisMX6, myoI-3xmKate2::kanMX6 | This study | MS118 |
| S. cerevisiae: W303; MATalpha, ADE2, HTA2-sfGFP::HisMX6, myoI-3xmKate2::kanMX6 | This study | MS120 |
| S. cerevisiae: BY4741; HTB2-GFP | GFP collection (Huh et al., 2003) | HTB2-GFP |
| S. cerevisiae: W303; MATa, ADE2, ura3:: WHI5pr(1kb)-mCitrine-CYC1term | This study | DCB72 |
| S. cerevisiae: W303; ADE2, WHI5-mCitrine-HIS3 | Lab collection | KSY108-1 |
| S. cerevisiae: W303; ADE2, whi5Δ::KanMX6 URA3::WHI5pr(1kb)-WHI5-WIQ-mCitrine- ADH1term | This study | KSY190-2 |
| S. cerevisiae: W303; ADE, URA3::ACT1pr(1kb)-Whi5-mCitrine-CYC1term | This study | KSY160-2 |
| S. cerevisiae: W303; ADE2, URA3:: ACT1pr(1kb)-mCitrine-CYC1term | Lab collection | KSY158-1 |
| S. cerevisiae: W303; bar1Δ::HisG, URA3:: GFP-3xNLS-FLAG | This study | MS534 |
| S. cerevisiae: W303; bar1Δ::HisG, URA3:: LacI-GFP-3xNLS-FLAG | This study | MS535 |
| S. cerevisiae: W303; bar1Δ::HisG, whi5Δ::LEU2, URA3::WHI5pr(1kb)-3xFLAG-WHI5-CYC1term | This study | MS536 |
| S. cerevisiae: W303; bar1Δ::HisG, URA3::WHI5pr(1kb)-3xFLAG-WHI5(WIQ)-CYC1term | This study | MS537 |
| S. cerevisiae: W303; bar1Δ::HisG, SWI4-V5::hphMX6 | This study | MK653-1 |
| S. cerevisiae: W303; bar1Δ::HisG, SWI6-V5::hphMX6 | This study | MK645-1 |
| S. cerevisiae: W303; MATalpha, ADE whi5Δ::cgTRP; bck2Δ::hphNT1, myoI-3xmKate2::kanMX6, leu1::PtetO7.1-WHI5-mCitrine-PEST::LEU1, WTC846(PrtetO-7.1-TetR, PrRNR2-TetR-TUP1)::HIS3 | This study | MS364 |
| S. cerevisiae: W303; MATalpha, ADE, whi5Δ::cgTRP; bck2Δ::hphNT1, myoI-3xmKate2::kanMX6, WHI5pr[1kb]-WHI5-mCitrine::URA3, WTC846(PrtetO-7.1-TetR_PrRNR2-TetR-TUP1)::HIS3 | This study | MS367 |
| S. cerevisiae: W303; MATalpha, ADE, whi5Δ::cgTRP; bck2Δ::hphNT1, myoI-3xmKate2::kanMX6, leu1::PtetO7.1(weak)-WHI5-mCitrine::LEU1, WTC846(PrtetO-7.1-TetR, PrRNR2-TetR-TUP1)::HIS3 | This study | MS391 |
| S. pombe: 972 h- | Lab collection | 972 |
| Software and algoritdms | ||
| Cell cycle and protein partitioning modeling | This study and Chandler-Brown et al., 2017 | https://doi.org/10.5281/zenodo.5549357 |
| YeaZ algoritdm | Dietler et al., 2020 | www.quantsysbio.com |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast genetics
Standard procedures were used for Saccharomyces cerevisiae strain construction. Full genotypes of all strains used in this study are listed in Table S1 and the Key Resource Table. The strain for constitutive expression of WHI5-mCitrine-PEST (MS364) was constructed using the WTC846 tetracycline responsive promoter system (Azizoğlu et al., 2020). The strain for constitutive expression of WHI5-mCitrine (MS391) was constructed using a truncated (565bp) version of the WTC846 tetracycline responsive promoter in combination with a weaker kozak sequence (AAGGGAAAAGGGAAA) as detailed in Azizoğlu et al., 2020.
METHOD DETAILS
Transcriptomic analysis of size-sorted cells
To determine transcript levels in cells of different sizes, S/G2/M cells were sorted according to total protein content by fluorescence activated cell sorting (FACS) before RNA extraction and sequencing. 500 mL S. cerevisiae (HTB2-GFP) was grown (synthetic complete media + 2% glucose at 30 °C), and fixed at O.D. ~ 0.3 by addition of 500ml 80% methanol 20mM TRIS (−20 °C) and then incubated at −20 °C for 30 minutes. Cells were fixed to prevent gene expression changes during the course of the cell sorting, which requires multiple generation equivalents of time to complete. Cells were pelleted (13k rpm, 3 minutes) and washed 3x in PBS, before gentle sonication and then addition of 5 μg/ml total protein dye (Alexa Fluor™ 647 NHS Ester dye; ThermoFisher Scientific; A20006) and incubation (4 °C, 30 minutes). Cells were again pelleted (13k rpm, 3 minutes) and washed 3x in PBS to remove excess dye before again being sonicated. Cells from four different size fractions were sorted on a FACSAria II sorter (BD Biosciences) according to the following strategy. First, singlets were gated based on scatter (FSC and SSC), then S/G2/M cells were identified using Htb2-GFP signal, and then finally, four bins of different total protein content cells were sorted based on the total protein intensity (bin 1 = lowest signal, bin 4 = highest, Fig. S1A). The fidelity of the total protein dye sort was confirmed by re-analyzing 10,000 cells on the same sorter (Fig. 1D & S1A). Furthermore, total protein content was validated as a proxy for size by measuring the cell volume of the different protein dye sorted cells using a Coulter counter (Fig. S1C-E).
Two biological replicates were performed. Within each biological replicate, two technical replicates were performed for each size bin so that four replicates were performed per bin in total. For the different bins within the same replicate set, a constant number of S. pombe cells, fixed as above, were added as a spike-in to measure total RNA content per S. cerevisiae cell. The number of S. cerevisiae cells and S. pombe cells (972 h-) per sample was constant within a set of replicates but varied slightly between each set of replicates (5-10 million S. cerevisiae cells and 5-10 million S. pombe cells). To maximize the number of reads from the experimental S. cerevisiae samples, S. pombe cells were nitrogen starved (grown in EMM before media switch to EMM - NH4Cl for 24 hours at 30 °C) because this reduces their mRNA concentration (Marguerat et al., 2012). Cells were then pelleted (4k rpm, 15 minutes), and their RNA extracted and sequenced as described below. For each set of replicates, the S. pombe spike-in was added independently and RNA was extracted independently.
To estimate the relative amount of mRNA per cell in each size bin, the number of S. cerevisiae reads per S. pombe read was calculated (see RNA-seq data processing below) and then normalized to the mean value within a set of replicates. The normalized total mRNA per sample was then averaged between the four replicates (Fig. S1B).
Transcriptomic analysis of differently sized cells synchronously progressing through the cell cycle
To determine transcript levels during the cell cycle in cells of different sizes, cells were elutriated and arrested in G1 for different amounts of time. Samples were then collected during the synchronous progression from G1 through S, G2 and M phases of the cell cycle for RNA extraction and sequencing (See Fig. S2 for schematic of experimental design). Specifically, 4 L S. cerevisiae (A17896: W303 cdc28-13) were grown in synthetic complete (SC) media with 2% glucose at 25 °C to OD ~0.75 and then collected on a filter membrane and resuspend in ice-cold SC media (no carbon source). Cells were then sonicated (3 x 20 seconds, 3 minutes on ice between sonication cycles) and loaded into a JE 5.0 elutriation rotor fitted for a two-chamber run (Beckman Coulter) in a J6-MI Centrifuge (2.4krpm, 4 °C). The elutriation chambers were pre-equilibrated and run with SC media (4 °C, no carbon source). The pump speed was gradually increased until G1 cells with minimal debris were collected. G1 fractions were then collected on a filter and resuspended in 37 °C conditioned SC media + 2% glucose in a 37 °C shaking water-bath (OD ~ 0.1). The G1 arrest was maintained at 37 °C until cells reached either 36-39 fL (small), 67-69 fL (medium) or 129-131 fL (large) as determined by Coulter counter (Fig. S2B-C). When they reached the target size, cells were released from the G1 arrest. To do this, cells were collected on a filter membrane and resuspended in 25 °C SC media + 2% glucose (OD ~0.35). Samples for size measurement by Coulter counter, DNA-content analysis, and RNA-extraction were taken at 10-minute intervals after release with the 0-minute time point being designated as the time point 30 minutes before the onset of DNA replication (Fig. S2D-E). For small cells, the 0-minute time point was collected 40-50 minutes after the shift to the permissive temperature, for medium cells the 0-minute time point was collected 10 minutes after the shift to the permissive temperature and for large cells the 0-minute time point was collected 0 minutes after shift to the permissive temperature. Two biological replicates were performed.
For DNA-content analysis, 0.4 mL culture was added to 1 mL 100% 4 °C ethanol and stored at 4 °C. Cells were pelleted (13 krpm, 2 minutes), washed, and resuspended in 50 mM Sodium Citrate (pH = 7.2), incubated with 0.2 mg/mL RNAse A (overnight, 37 °C) and then 0.4 mg/mL proteinase K (1 hour, 50 °C) before addition of 25 μM Sytox Green (ThermoFisher Scientific). Cells were then sonicated and DNA-content was analyzed for >10000 events on a FACScan Analyzer (BD Biosciences). For RNA-extraction 1.5 mL cells were pelleted (13 krpm, 30 seconds) and snap frozen in liquid N2. Samples were then thawed in TRI Reagent (Zymo Research) and RNA was extracted as described below (RNA extraction and sequencing).
RNA extraction and sequencing
To extract RNA, cell pellets were lysed in 300 μL TRI Reagent (Zymo Research) by bead beating using a Fastprep 24 (4 °C, settings: 5.0 m/s, 1 x 30 seconds). Cell debris was pelleted (13 krpm, 5 minutes) and the supernatant recovered. RNA was then extracted using the direct-zol RNA microprep kit (Zymo Research). mRNA was enriched using the NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB, E7490) and NEBNext Ultra II RNA Library Prep Kit for Illumina® (NEB, #E7775) was then used to prepare libraries for paired-end (2x150 bp) Illumina sequencing (Novogene). More than 20 million reads were sequenced per sample.
Single-molecule fluorescence in situ hybridization (smFISH)
smFISH was used to image WHI5 and MDN1 mRNAs in single cells. A Whi5-mCitrine tagged strain (DCB099) was used to discriminate pre- and post-Start G1 based on Whi5-mCitrine nuclear localization (Doncic et al., 2011). Early S/G2/M cells were defined as budded cells with a small (≤ 0.2) bud-to-mother volume ratio. Cells were grown at 30 °C in synthetic complete (SC) media + 2% glycerol + 1% ethanol. Two biological replicates were performed. Each biological replicate contained two technical replicates (i.e., two independent hybridizations to cells from the same culture). In addition, two negative controls were performed regularly where (i) FISH probes were omitted and (ii) a whi5Δ (MK551-1) strain was analyzed.
The smFISH protocol (detailed below) was optimized based on protocols from multiple prior studies (Raj and Tyagi, 2010; Trcek et al., 2012; Tutucci et al., 2018; Youk et al., 2010; Zenklusen et al., 2008). 45 mL cells (OD600 ~0.2) were fixed with 5 mL 37% formaldehyde and incubated (45 minutes, room temperature, rotating). Cells were then pelleted (1600 g, 5 minutes) and washed twice in 1 mL of ice-cold fixation buffer (pH 7.5, 218 mg/mL sorbitol, 84 mM potassium phosphate dibasic, 16 mM potassium phosphate monobasic, dissolved in water for RNA work (Thermo Fisher Scientific, BP561-1)). Cells were again pelleted and resuspended in 900 μL fixation buffer and gently sonicated before 100 μL of 200 mM RNase inhibitor vanadyl ribonucleoside complex was added (New England BioLabs, S1402S). Cells were then digested by adding 3.5-5 μL zymolyase stock (5 mg/mL 100T, MP Biomedicals, 0832093) and incubated (70-80 minutes, 30 °C, rotating). Cells were then pelleted (400 g, 6 minutes) and washed twice in 1 mL of ice-cold fixation buffer to stop digestion and finally permeabilized by resuspension in 1ml 70% ethanol. Permeabilized cells were kept at 4 °C for 1 to 3 days. 300 μL of the permeabilized cells per hybridization sample were then pelleted (400 g, 7 minutes) and washed in 500 μL of wash buffer A (Biosearch Technologies, SMF-WA1-60: prepared fresh on the day of use according to manufacturer’s instructions always using a fresh aliquot of deionised formamide (EMD Millipore, S4117, stored at −20 °C)). Permeabilized cells were then resuspended in 100 μL hybridisation solution (Biosearch Technologies, SMF-HB1-10) containing 1-3x of standard probe concentrations for WHI5 and MDN1 probes, 10 mM VRC and 0.5 mg/mL smFISH probe competitor E. coli tRNA (Roche, TRNAMRE- RO). Note VRC and probe competitor were omitted for half the cells analyzed in replicate 1. For MDN1 mRNAs two probe sets, totaling 86 probes (38 + 48), coupled to the FAM (fluorescein amidite) dye (Biosearch Technologies, Stellaris Custom Probes) were used. The sequences of these probes were taken from Tutucci et al., 2018 (MDN1-3’ORF and MDN1-ORF). For WHI5, a set of 46 probes coupled to the Quasar570 dye (Biosearch Technologies, Stellaris Custom Probes) were used. WHI5-mCitrine Probe sequences were designed using Stellaris Probe Designer applied to the WHI5-mCitrine mRNA sequence. Probes were hybridized in the dark (30 °C, overnight, with end-over-end rotation). 100 μL of wash buffer A was then added before cells were pelleted (400 g, 8 minutes) and supernatant was aspirated. Cells were resuspended in 1 mL wash buffer A, incubated in the dark (30° C, 30 minutes), pelleted (400 g, 6 minutes), resuspended in 1 mL wash buffer A +350 μg/mL calcofluor white (Sigma, F3543), and again incubated in the dark (30 °C, 30 minutes). Cells were then resuspended in 1 mL wash buffer B (Biosearch Technologies, SMF-WB1-20), incubated for 2- 5 minutes at room temperature, pelleted (400 g, 6 minutes) and resuspended in 2 to 3 drops (~75 μL) of Vectashield Antifade Mounting Medium (Vector Laboratories, H-1000). This suspension was then mixed thoroughly by pipetting to separate clumped cells. 1.5 μL of this solution was mounted on an acid washed slide and imaged on a wide-field epifluorescence Zeiss Observer Z1 microscope (63X/1.4NA oil immersion objective and a Colibri LED module). 30-step z-stacks (step size = 200 nm) were imaged. Cell outlines were identified using phase contrast images. Quasar570 probes (WHI5) were imaged in the orange channel (white LED module for 555 nm wavelength, 100% light power, 5 sec exposure per stack image). Whi5-mCitrine protein and FAM probes (MDN1) were imaged in the yellow channel (505 nm LED module, 100% light power, 3.5 seconds exposure time). FAM FISH probes alone were imaged in the green channel (470 nm LED module, 75% light power, 5 seconds exposure time). Calcofluor white stain was imaged in the blue channel (365 nm LED module, 25% light power, 20 ms exposure). Under these conditions, no significant photobleaching was observed after taking multiple images of the same cells.
smFISH Image analysis was performed manually using ImageJ (version 2.0.0). Single cells were manually selected in each image. A cell was only selected if its morphology was sufficiently intact (following zymolyase treatment) and if the absence/presence of a bud and the nuclear/cytoplasmic localization of Whi5-mCitrine protein could be assigned. Cell size was measured by drawing cell outlines in the phase z-plane with the largest cell area, fitting a two-dimensional ellipse, and then rotating the ellipse along its major axis to obtain a volume estimate. Separately calculated volumes for mothers and buds were added together. Absolute counts of WHI5-mCitrine and MDN1 mRNAs in single cells were obtained by manual counting and single dots were counted as one mRNA (i.e., we did not quantify single dot intensities to try to discern multiple overlapping mRNAs).
Live cell microscopy
Cells were grown to early log phase in synthetic complete (SC) media + 2% glycerol + 1% ethanol and gently sonicated before being loaded into a CellASIC Y04C microfluidics plate (Milipore SIGMA) under continuous media flow at 2 psi. Imaging, image segmentation, and pedigree tracking was performed as previously described (Doncic et al., 2013; Schmoller et al., 2015) with the exception of the experiments in Fig. 4D, Fig. 6 and Fig. S8 which were segmented and tracked using the convolutional neural network YeaZ algorithm (Dietler et al., 2020). For experiments in Fig. 4D, Fig. 6 and Fig. S8, Myo1-3xmKate2 signal at the bud neck was also used aid the determination of mother-bud pairs and cytokinesis timing. Cells expressing GFP proteins were exposed for 15 milliseconds (505nm Colibri LED, 25% power), cells expressing mCitrine were exposed for 400 ms (505nm Colibri LED, 25% power) and cells expressing mKate2 were exposed for 1 second (555nm Colibri LED, 25% power).
Background subtraction for variation in background fluorescence in each frame of the movie was performed as previously described (Chandler-Brown et al., 2017). Briefly, in each frame cell and non-cell area was defined. A 4-pixel average filter was then applied, and the background was taken to be the median filtered pixel value of the non-cell area, except for the experiments in Fig. 4D, Fig. 6 and Fig. S8, where background fluorescence was measured using median pixel values from a collection of flatfield images. After subtraction of non-cell background fluorescence, differences in cell volume dependent autofluorescence were accounted for as previously described (Schmoller et al., 2015). Briefly, cell volume dependent autofluorescence was estimated by imaging an untagged strain, quantifying cell volumes and autofluorescence for all cell bodies and then fitting a linear regression to volume vs total fluorescence signal. The linear regression was then used to interpolate the cellular autofluorescence of all other cells based on their cell volume, which was then subtracted from the quantified signal. Note that the analysis of WHI5pr-WHI5-mCitrine (Fig. 2F, S3G & 5B&E) includes cells previously imaged for analysis reported in Schmoller et al., 2015.
Synthesis rates (shown in Fig. 2F-H and S3G-I) were estimated as previously described (Schmoller et al., 2015). Briefly, protein synthesis rates were calculated by fitting a linear regression to the quantified protein amounts against time, for the period between budding and cell division. This was done for all individual cells that completed the cell cycle. The slope of the linear fit was then used as the synthesis rate. The Δprotein and Δvolume calculations (shown in Fig. 4D-E) were calculated by taking the differences between the median of the last three frames and the median of the first three frames for all cell that completed the cell cycle.
ChIP-seq experiments
Cells expressing Swi4-V5, Swi6-V5, 3xFLAG-WHI5, 3xFLAG-WHI5, GFP-NLS-5xFLAG or LacI-GFP-NLS-5xFLAG were grown in SC media with 2% glycerol 1% ethanol. 500 mL of cells at OD ~0.5 were fixed with 1% formaldehyde (30 minutes) and quenched with 0.125 M glycine (5 minutes). Fixed cells were washed twice in cold PBS, pelleted, snap-frozen and stored at −80 °C. Cell lysis and ChIP reactions were performed as previously described (Hu et al., 2015) with minor modifications. Pellets were lysed in 300 μL FA lysis buffer (50 mM HEPES–KOH pH 8.0, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 1 mM PMSF, Roche protease inhibitor) with ~1 mL ceramic beads using a Fastprep-24 (MP Biomedicals). The entire lysate was then collected and adjusted to 1 mL before sonication to ~200bp fragments using a 1/8’ microtip on a Q500 sonicator (Qsonica) for 15 minutes (10 seconds on, 20 seconds off). The sample tube was held suspended in a −20 °C 80% ethanol bath to prevent sample heating during sonication. Cell debris was then pelleted and the supernatant retained for ChIP. For each ChIP reaction, 30 μL Protein G Dynabeads (Invitrogen) were blocked (PBS + 0.5% BSA), prebound with 10 μL anti-V5 antibody (SV5-Pk1, BioRad Cat# MCA1360G) or 10 μL anti-FLAG antibody (M2, SIGMA Cat# F1804) and washed once with PBS before incubation with supernatant (4 °C, overnight). Dynabeads were then washed (5 minutes per wash) twice in FA lysis buffer, twice in high-salt FA lysis buffer (50 mM HepesKOH pH 8.0, 500 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 1 mM PMSF), twice in ChIP wash buffer (10 mM TrisHCl pH 7.5, 0.25 M LiCl, 0.5% NP-40, 0.5% sodium deoxycholate, 1 mM EDTA, 1 mM PMSF) and once in TE wash buffer (10 mM TrisHCl pH 7.5, 1 mM EDTA, 50 mM NaCl). DNA was eluted in ChIP elution buffer (50 mM TrisHCl pH 7.5, 10 mM EDTA, 1% SDS) at 65 °C for 15-20 minutes. Eluted DNA was incubated to reverse crosslinks (65 °C, 5 hours), before treatment with RNAse A (37 °C, 1 hour) and then Proteinase K (65 °C, 2 hours). DNA was purified using the ChIP DNA Clean & Concentrator kit (Zymo Research). Indexed sequencing libraries were prepared using the NEBNext Ultra II DNA Library Prep kit for Illumina (NEB, # E7645) and pooled before paired-end (2x150 bp) Illumina sequencing (Genewiz, NJ).
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA-seq data processing
Because some samples analyzed in this study contained S. cerevisiae as well as reference spike-in S. pombe RNA, a combined S. cerevisiae and S. pombe genome file was created using the sacCer3 and ASM294v2 versions of the respective genomes, and a combined transcriptome annotation was created using the S. pombe gene models available from PomBase (Lock et al., 2019) and an S. cerevisiae set of gene models updated using transcript-end mapping data as previously described (Shipony et al., 2020). For the purposes of RNA-seq data quality evaluation and genome browser track generation, reads were aligned against the combined genome and annotated set of splice junctions using the STAR aligner (version 2.5.3a; settings: --limitSjdbInsertNsj 10000000 --outFilterMultimapNmax 50 --outFilterMismatchNmax 999 --outFilterMismatchNoverReadLmax 0.04 --alignIntronMin 10 --alignIntronMax 1000000 --alignMatesGapMax 1000000 --alignSJoverhangMin 8 --alignSJDBoverhangMin 1 --sjdbScore 1 --twopassMode Basic --twopass1readsN −1) (Dobin et al., 2013). Read mapping statistics and genome browser tracks were generated using custom Python scripts. For quantification purposes, reads were aligned as 2x50mers in transcriptome space against an index generated from the combined annotation described above using Bowtie (Langmead et al. 2009; version 1.0.1; settings: -e 200 -a -X 1000). Alignments were then quantified using express (version 1.5.1) (Roberts and Pachter, 2013) before effective read count values and TPM (Transcripts Per Million transcripts) were then separated for each genome and renormalized TPMs were calculated with respect to the total reads for S. cerevisiae.
Differential expression analysis by DESeq2 was performed using technical replicates to compare RNA-seq data from different size bins in the experiment shown in Figures 1D-F and S1 (Love et al., 2014). To calculate the total amount of transcription during the G1 arrest/release RNA-seq time course experiment (Fig. 2C-D & S2), TPM values were normalized to the mean for each experiment and the Area Under the Curve (AUC) for TPM as a function of time was calculated for each time course using the R function auc(type = "spline") from the MESS package.
For the analysis of cell cycle gene expression dynamics in these experiments (Fig. S2F-G) cell-cycle genes (n=240) were taken as the overlap in high confidence cell-cycle regulated genes defined in both de Lichtenberg et al., (2005) and Spellman et al., (1998)
Classification of sub-scaling and super-scaling transcripts
To classify transcripts whose expression sub-scales with cell size, we analyzed data from two experiments: (1) the RNA-seq experiment on size-sorted populations of cells (Fig. 1D-F and S1) and (2) the elutriation, G1 arrest/release RNA-seq time course experiment (Fig. 2A-D and S2). Two biological replicates of each experiment were performed. Sub-scaling genes were classified as genes that passed the following criteria in both biological replicates of each experiment:
(1) At least one pair-wise comparison between the four-size bins has a false-discovery rate (FDR) adjusted p-value <0.01, a TPM fold-change > 1.5, and bin 1 TPM > bin 4 TPM.
(2) Small cells’ TPM Area Under Curve (AUC) > medium cells’ TPM AUC > large cells’ TPM AUC and TPM AUC fold-change > 1.3. See above (RNA-seq data processing) for details of the AUC calculation.
To classify transcripts whose expression sub-scales with cell size we applied the same criteria as for sub- scaling genes but instead (1) bin 1 PM < bin 4 TPM and (2) Small cells’ TPM Area Under Curve (AUC) < medium cells’ TPM AUC < large cells’ TPM AUC.
Genes identified by the above criteria are listed in Table S2 – list of sub- and super-scaling genes (related to Fig. 3).
GO term enrichment
GO term enrichment (Fig. 3A&3F) was performed using the GOrilla GO analysis tool (Eden et al., 2009). Enrichment of size-independent gene transcripts was performed versus a background set of all genes that had a TPM value > 0 in all RNA-seq samples.
ChIP-seq analysis
Demultipexed fastq files were mapped to the sacCer3 assembly of the S. cerevisiae genome as 2×36mers using Bowtie (v.1.0.1) (Langmead et al., 2009) with the following settings: -v 2 -k 2 -m 1 --best --strata. Duplicate reads were removed using picard-tools (v.1.99). Peaks were called using MACS2 (v.2.1.0) (Feng et al., 2012) with the following settings: -g 12000000-f BAMPE. RPM (Reads Per Million) normalized read coverage genome browser tracks were generated using custom-written python scripts. Coverage tracks show the region +/− 50bp around the midPoint of each mapped fragment.
Gene deletion collection screen by microarray
We analyzed the correlation between RNA levels and cell size in 1,484 gene deletion stains using published microarray data and cell size measurements of these strains (Hoose et al., 2012; Jorgensen et al., 2002; Kemmeren et al., 2014; O'Duibhir et al., 2014; Ohya et al., 2005; Soifer and Barkai, 2014). Gene expression changes relative to wild-type were from (O'Duibhir et al., 2014), where we used the dataset transformed to correct for effects of slow growth. The same trends were observed in the uncorrected dataset. For each gene we then analyzed the correlation between the relative fold-change of its expression in a given deletion with the size of that deletion strain across all the deletion strains for which both data were available. Pearson correlation coefficients were calculated using the R function cor (Fig. 3E & S5D).
GFP fusion collection screen by flow cytometry
To examine the size-dependence of histone protein’s expression, we analyzed a genome-wide dataset of flow cytometry-based GFP intensity measurements (Parts et al., 2014), where each measurement is from a single well containing two strains both expressing the same protein C-terminally fused to GFP (Fig. 4A-C). One strain is in the BY4741 background (replicate 1) and the other in the RM11 background (replicate 2). Cells were grown in low fluorescence media containing 2% glucose and measured using an BD LSRII flow cytometer as described in Parts et al., 2014. Cells were separated into budded and unbudded populations on the basis of the side scatter width (SSC-W). Co-cultured strains of each background were separated on the basis of HTB2-mCherry intensity (RM low, BY high). Size was defined by the area of the side scatter signal (SSC-A). We used the lowest expressed gene from each plate as a background for that plate, thereby controlling for plate-to-plate variation in measurements. To calculate the background, we fitted a linear function to SSC-A and total GFP fluorescence for these low-expressing cells (python function polyfit(matplotlib)). We then subtracted the fit for these lowest expressing cells from the GFP intensity for all other cells. Strains with noisy signals (i.e., their mean expression is less than the standard deviation) and cells with saturated signals (mean expression is greater than 200000) were excluded. SSC-A and background subtracted GFP intensity were then normalized to the mean and a linear function was then fitted (python function polyfit(matplotlib)). The slope of this function was used as a measurement for a protein’s size-dependence.
Cell cycle and protein partitioning modeling
The cell cycle was modeled as reported in (Chandler-Brown et al., 2017). We simulated the entire cell cycle, where cells grew and divided according to measured growth and cell cycle transition rates. This accounts for cell-to-cell variability. To examine the role of protein partitioning in the overall scaling of protein expression, we simulated the synthesis of a constitutively expressed protein (p) in each cell (Fig. 5D & S7B). Within the model, protein synthesis and partitioning properties were varied as follows. Protein synthesis was modelled as either scaling in proportion to cell size or constant independent of cell size and protein partitioning was modeled as either volume-proportional partitioning at cytokinesis ( and ) or partitioned in the manner empirically measured for Whi5 . We note that the partitioning ratio of ~1.4 corresponds to approximately 50% of Whi5 molecules being partitioned by volume and 50% being partitioned by amount. Cells were simulated until a steady-state distribution was achieved and all cells at the last time-point were plotted.
Supplementary Material
Table S2 – List of sub- and super-scaling genes. Related to Figure 3.
Highlights.
A transcriptional mechanism uncouples Whi5 synthesis from cell size
Histones are the major class of sub-scaling mRNAs in addition to WHI5
Chromatin-based partitioning ensures Whi5 sub-scaling is inherited during division
Disruption of Whi5 sub-scaling weakens G1 size homeostasis
Acknowledgments.
We would like to thank Bruce Futcher and Yuping Chen for invaluable advice on centrifugal elutriation, as well as Andreas Cuny, Fabian Rudolf and Sahand Rahi for advice on improving our image analysis pipeline. We thank Fabian Rudolf sharing the TETpr constructs, Gabriel Neurohr for sending strain A17896, Chris You for assistance with the ChIP-seq experiments and Jon Turner for help in optimizing the smFISH protocol. We thank Christine Jacobs-Wagner and Helena Cantwell for comments on the manuscript and members of the Skotheim laboratory for feedback throughout the project. This work was supported by the NIH (GM134858), the HHMI-Simons (JMS, Faculty Scholars Program). MPS was supported by a Simons Foundation Fellowship of the Life Sciences Research Foundation and an EMBO Long-Term Postdoctoral Fellowship. KMS was supported by the Human Frontier Science Program (Postdoctoral Fellowship and Career Development Award). JK was supported by a Stanford MPTP T32 training grant.
Footnotes
Declaration of interests. The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Andrews BJ, and Herskowitz I (1989). Identification of a DNA binding factor involved in cell-cycle control of the yeast HO gene. Cell 57, 21–29. [DOI] [PubMed] [Google Scholar]
- Azizoğlu A, Brent R, and Rudolf F (2020). A precisely adjustable, variation-suppressed eukaryotic transcriptional controller to enable genetic discovery. bioRvix 2019.12.12.874461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber F, Amir A, and Murray AW (2020). Cell-size regulation in budding yeast does not depend on linear accumulation of Whi5. Proc Natl Acad Sci U S A 117, 14243–14250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler-Brown D, Schmoller KM, Winetraub Y, and Skotheim JM (2017). The Adder Phenomenon Emerges from Independent Control of Pre- and Post-Start Phases of the Budding Yeast Cell Cycle. Curr Biol 27, 2774–2783 e2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhao G, Zahumensky J, Honey S, and Futcher B (2020). Differential Scaling of Gene Expression with Cell Size May Explain Size Control in Budding Yeast. Mol Cell 78, 359–370 e356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia W, Somers WG, and Wang H (2008). Drosophila neuroblast asymmetric divisions: cell cycle regulators, asymmetric protein localization, and tumorigenesis. J Cell Biol 180, 267–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claude KL, Bureik D, Chatzitheodoridou D, Adarska P, Singh A, and Schmoller KM (2021). Transcription coordinates histone amounts and genome content. Nat Commun 12, 4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, Nishikawa JL, Tang X, Millman JS, Schub O, Breitkreuz K, Dewar D, Rupes I, Andrews B, and Tyers M (2004). CDK activity antagonizes Whi5, an inhibitor of G1/S transcription in yeast. Cell 117, 899–913. [DOI] [PubMed] [Google Scholar]
- Creanor J, and Mitchison JM (1982). Patterns of protein synthesis during the cell cycle of the fission yeast Schizosaccharomyces pombe. J Cell Sci 58, 263–285. [DOI] [PubMed] [Google Scholar]
- Crissman HA, and Steinkamp JA (1973). Rapid, simultaneous measurement of DNA, protein, and cell volume in single cells from large mammalian cell populations. J Cell Biol 59, 766–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross SL, and Smith MM (1988). Comparison of the structure and cell cycle expression of mRNAs encoded by two histone H3-H4 loci in Saccharomyces cerevisiae. Mol Cell Biol 8, 945–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin RA, McDonald WH, Kalashnikova TI, Yates J 3rd, and Wittenberg C (2004). Cln3 activates G1-specific transcription via phosphorylation of the SBF bound repressor Whi5. Cell 117, 887–898. [DOI] [PubMed] [Google Scholar]
- de Lichtenberg U, Jensen LJ, Fausboll A, Jensen TS, Bork P, and Brunak S (2005). Comparison of computational methods for the identification of cell cycle-regulated genes. Bioinformatics 21, 1164–1171. [DOI] [PubMed] [Google Scholar]
- Di Talia S, Skotheim JM, Bean JM, Siggia ED, and Cross FR (2007). The effects of molecular noise and size control on variability in the budding yeast cell cycle. Nature 448, 947–951. [DOI] [PubMed] [Google Scholar]
- Dietler N, Minder M, Gligorovski V, Economou AM, Joly D, Sadeghi A, Chan CHM, Kozinski M, Weigert M, Bitbol AF, et al. (2020). A convolutional neural network segments yeast microscopy images with high accuracy. Nat Commun 11, 5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doncic A, Eser U, Atay O, and Skotheim JM (2013). An algorithm to automate yeast segmentation and tracking. PLoS One 8, e57970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doncic A, Falleur-Fettig M, and Skotheim JM (2011). Distinct interactions select and maintain a specific cell fate. Mol Cell 43, 528–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsey S, Tollis S, Cheng J, Black L, Notley S, Tyers M, and Royer CA (2018). G1/S Transcription Factor Copy Number Is a Growth-Dependent Determinant of Cell Cycle Commitment in Yeast. Cell Syst 6, 539–554 e511. [DOI] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, and Yakhini Z (2009). GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott SG (1983). Coordination of growth with cell division: regulation of synthesis of RNA during the cell cycle of the fission yeast Schizosaccharomyces pombe. Mol Gen Genet 192, 204–211. [DOI] [PubMed] [Google Scholar]
- Elliott SG, and McLaughlin CS (1979). Regulation of RNA synthesis in yeast. III. Synthesis during the cell cycle. Mol Gen Genet 169, 237–243. [DOI] [PubMed] [Google Scholar]
- Elliott SG, Warner JR, and McLaughlin CS (1979). Synthesis of ribosomal proteins during the cell cycle of the yeast Saccharomyces cerevisiae. J Bacteriol 137, 1048–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eser U, Falleur-Fettig M, Johnson A, and Skotheim JM (2011). Commitment to a cellular transition precedes genome-wide transcriptional change. Mol Cell 43, 515–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Liu T, Qin B, Zhang Y, and Liu XS (2012). Identifying ChIP-seq enrichment using MACS. Nat Protoc 7, 1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrezuelo F, Colomina N, Futcher B, and Aldea M (2010). The transcriptional network activated by Cln3 cyclin at the G1-to-S transition of the yeast cell cycle. Genome Biol 11, R67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser RS, and Nurse P (1978). Novel cell cycle control of RNA synthesis in yeast. Nature 271, 726–730. [DOI] [PubMed] [Google Scholar]
- Fraser RS, and Nurse P (1979). Altered patterns of ribonucleic acid synthesis during the cell cycle: a mechanism compensating for variation in gene concentration. J Cell Sci 35, 25–40. [DOI] [PubMed] [Google Scholar]
- Gunjan A, and Verreault A (2003). A Rad53 kinase-dependent surveillance mechanism that regulates histone protein levels in S. cerevisiae. Cell 115, 537–549. [DOI] [PubMed] [Google Scholar]
- Heldt FS, Lunstone R, Tyson JJ, and Novak B (2018). Dilution and titration of cell-cycle regulators may control cell size in budding yeast. PLoS Comput Biol 14, e1006548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoose SA, Rawlings JA, Kelly MM, Leitch MC, Ababneh QO, Robles JP, Taylor D, Hoover EM, Hailu B, McEnery KA, et al. (2012). A systematic analysis of cell cycle regulators in yeast reveals that most factors act independently of cell size to control initiation of division. PLoS Genet 8, e1002590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, Petela N, Kurze A, Chan KL, Chapard C, and Nasmyth K (2015). Biological chromodynamics: a general method for measuring protein occupancy across the genome by calibrating ChIP-seq. Nucleic Acids Res 43, e132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston GC, Pringle JR, and Hartwell LH (1977). Coordination of growth with cell division in the yeast Saccharomyces cerevisiae. Exp Cell Res 105, 79–98. [DOI] [PubMed] [Google Scholar]
- Jorgensen P, Edgington NP, Schneider BL, Rupes I, Tyers M, and Futcher B (2007). The size of the nucleus increases as yeast cells grow. Mol Biol Cell 18, 3523–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen P, Nishikawa JL, Breitkreutz BJ, and Tyers M (2002). Systematic identification of pathways that couple cell growth and division in yeast. Science 297, 395–400. [DOI] [PubMed] [Google Scholar]
- Keifenheim D, Sun XM, D'Souza E, Ohira MJ, Magner M, Mayhew MB, Marguerat S, and Rhind N (2017). Size-Dependent Expression of the Mitotic Activator Cdc25 Suggests a Mechanism of Size Control in Fission Yeast. Curr Biol 27, 1491–1497 e1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemmeren P, Sameith K, van de Pasch LA, Benschop JJ, Lenstra TL, Margaritis T, O'Duibhir E, Apweiler E, van Wageningen S, Ko CW, et al. (2014). Large-scale genetic perturbations reveal regulatory networks and an abundance of gene-specific repressors. Cell 157, 740–752. [DOI] [PubMed] [Google Scholar]
- Koch C, Moll T, Neuberg M, Ahorn H, and Nasmyth K (1993). A role for the transcription factors Mbp1 and Swi4 in progression from G1 to S phase. Science 261, 1551–1557. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litsios A, Huberts D, Terpstra HM, Guerra P, Schmidt A, Buczak K, Papagiannakis A, Rovetta M, Hekelaar J, Hubmann G, et al. (2019). Differential scaling between G1 protein production and cell size dynamics promotes commitment to the cell division cycle in budding yeast. Nat Cell Biol 21, 1382–1392. [DOI] [PubMed] [Google Scholar]
- Lock A, Rutherford K, Harris MA, Hayles J, Oliver SG, Bahler J, and Wood V (2019). PomBase 2018: user-driven reimplementation of the fission yeast database provides rapid and intuitive access to diverse, interconnected information. Nucleic Acids Res 47, D821–D827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord PG, and Wheals AE (1981). Variability in individual cell cycles of Saccharomyces cerevisiae. J Cell Sci 50, 361–376. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucena R, Alcaide-Gavilan M, Schubert K, He M, Domnauer MG, Marquer C, Klose C, Surma MA, and Kellogg DR (2018). Cell Size and Growth Rate Are Modulated by TORC2-Dependent Signals. Curr Biol 28, 196–210 e194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marguerat S, and Bahler J (2012). Coordinating genome expression with cell size. Trends Genet 28, 560–565. [DOI] [PubMed] [Google Scholar]
- Marguerat S, Schmidt A, Codlin S, Chen W, Aebersold R, and Bahler J (2012). Quantitative analysis of fission yeast transcriptomes and proteomes in proliferating and quiescent cells. Cell 151, 671–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateus C, and Avery SV (2000). Destabilized green fluorescent protein for monitoring dynamic changes in yeast gene expression with flow cytometry. Yeast 16, 1313–1323. [DOI] [PubMed] [Google Scholar]
- Moran L, Norris D, and Osley MA (1990). A yeast H2A-H2B promoter can be regulated by changes in histone gene copy number. Genes Dev 4, 752–763. [DOI] [PubMed] [Google Scholar]
- Nasmyth K, and Dirick L (1991). The role of SWI4 and SWI6 in the activity of G1 cyclins in yeast. Cell 66, 995–1013. [DOI] [PubMed] [Google Scholar]
- Neumann FR, and Nurse P (2007). Nuclear size control in fission yeast. J Cell Biol 179, 593–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neurohr GE, Terry RL, Lengefeld J, Bonney M, Brittingham GP, Moretto F, Miettinen TP, Vaites LP, Soares LM, Paulo JA, et al. (2019). Excessive Cell Growth Causes Cytoplasm Dilution And Contributes to Senescence. Cell 176, 1083–1097 e1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris D, and Osley MA (1987). The two gene pairs encoding H2A and H2B play different roles in the Saccharomyces cerevisiae life cycle. Mol Cell Biol 7, 3473–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Duibhir E, Lijnzaad P, Benschop JJ, Lenstra TL, van Leenen D, Groot Koerkamp MJ, Margaritis T, Brok MO, Kemmeren P, and Holstege FC (2014). Cell cycle population effects in perturbation studies. Mol Syst Biol 10, 732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohya Y, Sese J, Yukawa M, Sano F, Nakatani Y, Saito TL, Saka A, Fukuda T, Ishihara S, Oka S, et al. (2005). High-dimensional and large-scale phenotyping of yeast mutants. Proc Natl Acad Sci U S A 102, 19015–19020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padovan-Merhar O, Nair GP, Biaesch AG, Mayer A, Scarfone S, Foley SW, Wu AR, Churchman LS, Singh A, and Raj A (2015). Single mammalian cells compensate for differences in cellular volume and DNA copy number through independent global transcriptional mechanisms. Mol Cell 58, 339–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parts L, Liu YC, Tekkedil MM, Steinmetz LM, Caudy AA, Fraser AG, Boone C, Andrews BJ, and Rosebrock AP (2014). Heritability and genetic basis of protein level variation in an outbred population. Genome Res 24, 1363–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pramila T, Wu W, Miles S, Noble WS, and Breeden LL (2006). The Forkhead transcription factor Hcm1 regulates chromosome segregation genes and fills the S-phase gap in the transcriptional circuitry of the cell cycle. Genes Dev 20, 2266–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y, Jiang J, Liu X, Wei P, Yang X, and Tang C (2019). Cell Cycle Inhibitor Whi5 Records Environmental Information to Coordinate Growth and Division in Yeast. Cell Rep 29, 987–994 e985. [DOI] [PubMed] [Google Scholar]
- Raj A, and Tyagi S (2010). Detection of individual endogenous RNA transcripts in situ using multiple singly labeled probes. Methods Enzymol 472, 365–386. [DOI] [PubMed] [Google Scholar]
- Roberts A, and Pachter L (2013). Streaming fragment assignment for real-time analysis of sequencing experiments. Nat Methods 10, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmoller KM, Lanz MC, Kim J, Koivomagi M, Qu Y, Tang C, Kukhtevich IV, Schneider R, Rudolf F, Moreno DF, et al. (2020). Whi5 is diluted and protein synthesis does not dramatically increase in pre-Start G1. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmoller KM, Turner JJ, Koivomagi M, and Skotheim JM (2015). Dilution of the cell cycle inhibitor Whi5 controls budding-yeast cell size. Nature 526, 268–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipony Z, Marinov GK, Swaffer MP, Sinnott-Armstrong NA, Skotheim JM, Kundaje A, and Greenleaf WJ (2020). Long-range single-molecule mapping of chromatin accessibility in eukaryotes. Nat Methods. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skotheim JM, Di Talia S, Siggia ED, and Cross FR (2008). Positive feedback of G1 cyclins ensures coherent cell cycle entry. Nature 454, 291–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soifer I, and Barkai N (2014). Systematic identification of cell size regulators in budding yeast. Mol Syst Biol 10, 761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Anders K, Eisen MB, Brown PO, Botstein D, and Futcher B (1998). Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell 9, 3273–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston JE, Schierenberg E, White JG, and Thomson JN (1983). The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol 100, 64–119. [DOI] [PubMed] [Google Scholar]
- Sun XM, Bowman A, Priestman M, Bertaux F, Martinez-Segura A, Tang W, Whilding C, Dormann D, Shahrezaei V, and Marguerat S (2020). Size-Dependent Increase in RNA Polymerase II Initiation Rates Mediates Gene Expression Scaling with Cell Size. Curr Biol 30, 1217–1230 e1217. [DOI] [PubMed] [Google Scholar]
- Travesa A, Kalashnikova TI, de Bruin RA, Cass SR, Chahwan C, Lee DE, Lowndes NF, and Wittenberg C (2013). Repression of G1/S transcription is mediated via interaction of the GTB motifs of Nrm1 and Whi5 with Swi6. Mol Cell Biol 33, 1476–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trcek T, Chao JA, Larson DR, Park HY, Zenklusen D, Shenoy SM, and Singer RH (2012). Single-mRNA counting using fluorescent in situ hybridization in budding yeast. Nat Protoc 7, 408–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JJ, Ewald JC, and Skotheim JM (2012). Cell size control in yeast. Curr Biol 22, R350–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tutucci E, Vera M, Biswas J, Garcia J, Parker R, and Singer RH (2018). An improved MS2 system for accurate reporting of the mRNA life cycle. Nat Methods 15, 81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youk H, Raj A, and van Oudenaarden A (2010). Imaging single mRNA molecules in yeast. Methods Enzymol 470, 429–446. [DOI] [PubMed] [Google Scholar]
- Zenklusen D, Larson DR, and Singer RH (2008). Single-RNA counting reveals alternative modes of gene expression in yeast. Nat Struct Mol Biol 15, 1263–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhurinsky J, Leonhard K, Watt S, Marguerat S, Bahler J, and Nurse P (2010). A coordinated global control over cellular transcription. Curr Biol 20, 2010–2015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2 – List of sub- and super-scaling genes. Related to Figure 3.