

SUMMARY
Deconvolution of regulatory mechanisms that drive transcriptional programs in cancer cells is key to understanding tumor biology. Herein, we present matched transcriptome (scRNA-seq) and chromatin accessibility profiles (scATAC-seq) at single-cell resolution from human ovarian and endometrial tumors processed immediately following surgical resection. This dataset reveals the complex cellular heterogeneity of these tumors and enabled us to quantitatively link variation in chromatin accessibility to gene expression. We show that malignant cells acquire previously unannotated regulatory elements to drive hallmark cancer pathways. Moreover, malignant cells from within the same patients show substantial variation in chromatin accessibility linked to transcriptional output, highlighting the importance of intratumoral heterogeneity. Finally, we infer the malignant cell type-specific activity of transcription factors. By defining the regulatory logic of cancer cells, this work reveals an important reliance on oncogenic regulatory elements and highlights the ability of matched scRNAseq/scATACseq to uncover clinically relevant mechanisms of tumorigenesis in gynecologic cancers.
Keywords: Single-Cell Genomics, scRNA-seq, scATAC-seq, Endometrial Cancer, Ovarian Cancer, Gastro-Intestinal Stromal Tumors, Intratumoral Heterogeneity, Enhancer Elements
eTOC blurb
Regner & Wisniewska et al. present an integrated analysis of single-cell transcriptomics and chromatin accessibility data to define the regulatory logic of malignant cell states in human gynecologic cancers. They identify thousands of salient cancer-specific distal regulatory elements and uncover differential transcription factor activity that drives intratumor heterogeneity.
Graphical Abstract
INTRODUCTION
Dynamic interactions between various types of malignant and non-malignant cells in solid tumors contributeto a range of biological phenomena, from cancer progression to therapeutic response. Single-cell genomic technologies refined our ability to interrogate the underlying cellular heterogeneity of tumors, but most efforts to date have been limited to transcriptomics via single-cell RNA-seq (scRNA-seq) (Patel et al., 2014, Lambrechts et al., 2018, Slyper et al., 2020, Davidson et al., 2020, Kim et al., 2020, Cochrane et al., 2020). While initial reports have been transformative, it is evident that non-coding regions of the genome, containing regulatory elements (e.g. cis-acting distal enhancer elements), contribute profoundly to tumor biology (Corces et al., 2018). These regulatory elements are often rewired and repurposed by cancer cells to drive oncogenic transcription (Roadmap Epigenomics et al., 2015, Mansour et al., 2014, Zhang et al., 2016, Roe et al., 2017, Corces et al., 2018). Thus, a deeper understanding of the regulatory logic of cancer cells will provide novel insights into the molecular underpinnings of tumor biology and heterogeneity.
Advancements in the assay for transposase-accessible chromatin at the single cell level (scATAC-seq) enable robust profiling of the chromatin accessibility landscape, unveiling layers of gene regulation including cis-regulatory elements (Buenrostro et al., 2015, Cusanovich et al., 2015). Together, scRNA-seq and scATAC-seq offer unprecedented resolution to reveal complex epigenetic events underlying tumor biology and give potential for the discovery of pathways governing tumorigenesis going beyond the standard taxonomic identification of cell types.
Few cancer datasets with matched scRNA-seq and scATAC-seq exist and none have been reported for human gynecologic tumors (Granja et al., 2019). Ovarian cancer (OC) and Endometrial cancer (EC) represent two of the deadliest cancers among women (Siegel et al., 2018). This is partly due to the aggressive nature of these cancers, lack of targeted therapies, and often late-stage of diagnosis. Of note, OC portends a poor prognosis and, although less common than breast cancer, it is three times more lethal (Siegel et al., 2018). EC is the 6th most frequently diagnosed cancer in women globally and is one of few cancers that is rising in mortality (Lortet-Tieulent et al., 2018, Society, 2016, Henley et al., 2018). The Cancer Genome Atlas (TCGA) consortium has proposed molecular subtypes for these cancers, but these stratification systems fail to account for cell type composition and malignant cell heterogeneity within tumors (Cancer Genome Atlas Research, 2011, Cancer Genome Atlas Research et al., 2013). We posit that cell populations within and between patient tumors are delineated by noncoding regulatory elements that drive oncogene expression conferring enhanced proliferation, drug resistance, and/or survival.
Herein, we present a catalog of matched scRNA-seq and scATAC-seq data for 11 human gynecologic tumors (Table 1, Table S1). This dataset, encompassing over 170,000 single cells, is of broad utility to the fields of single-cell genomics and cancer biology. By analyzing these tumors with matched scRNA-seq and scATAC-seq, we uncover clinically relevant non-coding mechanisms for intratumoral heterogeneity and pathogenesis of EC and OC. We also infer the activity of transcription factors (TFs) that interact with malignant cell type-specific regulatory elements and prioritize TFs based on predicted druggability (Tym et al., 2016, Mitsopoulos et al., 2020, Malladi et al., 2020).
Table 1. Abbreviated clinical data and single-cell metadata for each patient tumor.
The last two columns reflect the number of cells obtained post QC and in parentheses the total number of cells estimated by Cell Ranger. Asterisks in the Tumor site column denote a metastatic event. Race column abbreviations: African American (AA), Caucasian (CAU), Asian (AS). Extended clinical data for each patient (de-identified) can be found in Table S1.
| Patient | Cancer type | Tumor site | Histology | Stage | Age | Race | BMI | scATAC-seq cells | scRNA-seq cells |
|---|---|---|---|---|---|---|---|---|---|
| Patient 1 | Endometrial | Endometrium | Endometrioid | IA | 70 | AA | 39.89 | 6,348 (6,649) | 5,279 (5,697) |
| Patient 2 | Endometrial | Endometrium | Endometrioid | IA | 70 | CAU | 30.50 | 7,248 (6,658) | 7,277 (7,963) |
| Patient 3 | Endometrial | Endometrium | Endometrioid | IA | 70 | CAU | 38.55 | 4,165 (7,241) | 4,974 (6,054) |
| Patient 4 | Endometrial | Endometrium | Endometrioid | IA | 49 | CAU | 55.29 | 7,597 (7,917) | 7,413 (8,110) |
| Patient 5 | Endometrial | Endometrium | Endometrioid | IA | 62 | CAU | 49.44 | 6,797 (7,881) | 7,291 (8,403) |
| Patient 6 | Endometrial | Ovary*** | Serous | IIIA | 74 | CAU | 29.94 | 6,643 (2,351) | 6,866 (8,009) |
| Patient 7 | Ovarian | Ovary | Endometrioid | IA | 76 | CAU | 34.80 | 5,924 (7,107) | 6,454 (8,295) |
| Patient 8 | Ovarian | Ovary | HGSOC | IIB | 61 | CAU | 22.13 | 8,014 (7,898) | 7,454 (8,181) |
| Patient 9 | Ovarian | Ovary | HGSOC | IIIC | 59 | AS | 22.37 | 9,670 (9,942) | 6,192 (6,939) |
| Patient 10 | Ovarian | Ovary | Carcinosarcoma | IVB | 69 | CAU | 23.72 | 4,439 (8,977) | 7,663 (8,984) |
| Patient 11 | Gastric | Ovary*** | GIST | IV | 59 | CAU | 33.96 | 7,776 (11,066) | 8,660 (10,094) |
RESULTS
Matched scRNA-seq and scATAC-seq of human gynecologic tumors
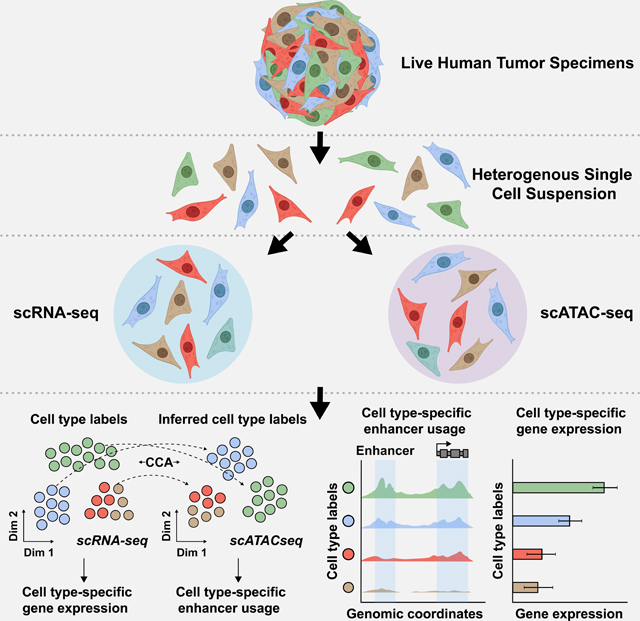
Eleven, treatment naïve, patients underwent debulking surgery with curative intent to remove tumors found either in the endometrium or ovary (Table 1, Table S1). Following surgical resection, each tumor was dissociated into a suspension of live cells and prepped for lipid droplet-based scRNA-seq and scATAC-seq (Figure 1A and STAR Methods). Tumor specimens where never frozen or fixed in any way, enabling high levels of cell viability and robust sequencing coverage in single cells. All tumors were primary tumors except for Patient 6, diagnosed as an EC that metastasized to the ovary, and Patient 11, diagnosed as a gastro-intestinal stromal tumor (GIST) that metastasized to the ovary. After quality control and doublet removal for each patient dataset (STAR Methods), we obtained 75,523 cells profiled by scRNA-seq and 74,621 cells profiled by scATAC-seq.
Figure 1. Overview of matched scRNA-seq and scATAC-seq workflow for patient tumors.

A) Cartoon showing patient tumor workflow. The female reproductive system cartoons, top, were created with BioRender.com.
B) UMAP plot all scRNA-seq cells color-coded by cell type across 11 patient tumors (left). UMAP plot of all scATAC-seq cells color-coded by inferred cell type across 11 patient tumors (right).Color shades denote subclusters within each cell type.
C) UMAP plot of scRNA-seq cells (left) and scATAC-seq cells (right) as shown in panel B but color-coded by patient of origin.
D) Stacked bar charts showing contribution of each patient to each subcluster in scRNA-seq (left) and to each inferred cell type subcluster in scATAC-seq (right).
To analyze scRNA-seq cells from the entire cohort, we performed principal component analysis (PCA) using the top 2,000 most variably expressed genes across all 75,523 cells. Cells were classified into transcriptionally-distinct clusters with graph-based clustering using the top 50 principal components (PCs) and visualized using a Uniform Manifold Approximation and Projection (UMAP) plot. This revealed that clusters could be annotated to known cell types (Aran et al., 2019) (Figure 1B [left], Figure S1A, Table S2, and STAR Methods) and batch effects were not a major confounder (Figure 1C, left). To identify malignant clusters across the entire cohort, we used clinical biomarker gene expression and inferred copy number amplification/deletion events (Figures S2-S4). We used expression of FDA approved biomarkers, MUC16/CA125 and WFDC2/HE4, to identify EC and OC cancer clusters (Duffy et al., 2005, Sturgeon et al., 2008, Hellström et al., 2003, Li et al., 2009, Dong et al., 2017). Expression of KIT/CD117 was used to identify GIST cancer clusters (Sarlomo-Rikala et al., 1998). Inferred copy number variation was used to help identify OC and GIST, but not EC since the disease rarely exhibits copy number variation (Berger et al., 2018).
To analyze scATAC-seq cells from the entire cohort, we created a matrix of contiguous genomic tiles, across the genome, in which we quantified fragment counts. We performed iterative latent semantic indexing on the top 25,000 most variable genomic tiles (Cusanovich et al., 2015, Satpathy et al., 2019, Granja et al., 2021). To assign cell type cluster labels from matching scRNA-seq data to scATAC-seq cells, we used the Seurat v3 cross-modality integration approach (constrained to cells of the same patient tumor) (Figure 1B [right], Figure S1, Table S3, and STAR Methods) (Stuart et al., 2019). This revealed scATAC-seq cells that clustered mainly by cell type and not by patient, highlighting the quality of the dataset (Figure 1C, right).
Overall, we found ten general cell types in the entire cohort with 36 subclusters present in both modalities. Although these subclusters vary in size, immune subclusters contain roughly equal proportions of cells across all patients, while malignant and fibroblast subclusters remain highly patient-specific (Figure 1D, Figures S5-S6). This is partly reflected by the uniqueness of each inferred CNV profile from each tumor (Figures S2-S3). Our observations are consistent with previous scRNA-seq reports in OC (Izar et al., 2020), lung cancer (Lambrechts et al., 2018), and nasopharyngeal cancer (Chen et al., 2020). These patterns likely reflect biological overlap of non-malignant cells across all patients and highlight the unique, and possibly tractable, biological features of malignant cells within each tumor.
Systematic discovery of cancer-specific distal regulatory elements (dREs) in human gynecologic cancers
We next explored the chromatin landscape to identify distal regulatory elements that could help explain distinct biological states of these malignant cells. To identify putative regulatory elements across all scATAC-seq cells, we first carried out peak calling within each cell type subcluster and used an iterative overlap peak merging procedure to generate a peak-by-cell matrix (Zhang et al., 2008, Granja et al., 2021, Liu, 2014, Corces et al., 2018). In order to link variation in chromatin accessibility to differences in gene expression, we executed a large-scale peak-to-gene linkage analysis and developed a robust empirical false discovery rate (eFDR) procedure for determining statistically significant peak-to-gene associations in single-cell data (STAR Methods) (Granja et al., 2021, Storey and Tibshirani, 2003).
Briefly, we aggregated the sparse peak counts within groups of similar scATAC-seq cells, identified via k-nearest neighbors, to generate more informative metacell observations for our peak-to-gene correlation analysis. We then used the scATAC-seq metacells (i.e. aggregates of similar cells) to compute the correlation between accessibility of every peak and expression of every gene in cis, imputed for each scATAC-seq cell (STAR Methods). This peak-to-gene correlation analysis resulted in 2,748,906 peak-to-gene combinations in cis (Figure 2A [top], Figure S7A [top]). To estimate the eFDR, we selected a raw p-value threshold of 1e-12 and recorded the number of observed peak-to-gene associations with a raw p-value ≤ 1e-12 (see STAR Methods). The peak-to-gene correlation analysis was repeated 100 times under the permuted null condition where, for each permutation, we shuffled scATAC-seq metacell labels to break the link between peak accessibility and gene expression (Figure 2A [bottom], Figure S7A [bottom]). For every permutation, there was less correlation between peak-to-gene pairs compared to observed data and the raw p-value distribution was near uniform. The eFDR was then calculated by dividing the median number of null peak-to-gene associations with a raw pvalue ≤ 1e-12 by the number of observed associations with a raw p-value ≤ 1e-12. These data highlight the genuine biological relationships between peak accessibility and gene expression in the observed data (Figure 2A, Figure S7, and STAR Methods).
Figure 2. Systematic in silico identification of cancer-specific distal regulatory elements.

A) Cartoon showing peak-to-gene correlation analysis with an eFDR (top).Histograms of correlation values and raw p-values for n=2,748,906 peak-to-gene link tests (middle) and peak-to-gene link tests under the null condition (bottom). Dashed red lines represent the alpha threshold or raw p-value cutoff of 1e-12 for calling statistically significant peak-to-gene links.
B) Row-scaled heatmaps of statistically significant distal peak-to-gene links. Each row represents expression of a gene (left) correlated to accessibility of a distal peak (right). Cancer-enriched k-means clusters are marked in red. Distal peaks participating in cancer-enriched k-means groups are used in the overlap analysis presented in panel C.
C) Venn diagram showing the number of cancer-specific distal peaks (orange) after overlapping the genomic coordinates of cancer-enriched distal peaks with the genomic coordinates of normal ovarian surface epithelium enhancer elements, normal fallopian tube enhancer elements, and all ENCODE regulatory element annotations (gray).
D) Bar charts comparing proportion of distal peaks per number of linked genes between cancer-specific (orange) and normal (gray) distal peak groups (left).Bar chart comparing mean number of linked genes per distal peak between cancer-specific (orange) and normal (gray) distal peak groups (right). Asterisks denote a statistically significant difference (Wilcoxon Rank Sum test). Error bars represent ±1 S.E.M.
E) Browser track showing the accessibility profile at the RHEB locus across all malignant subclusters (orange) and select non-malignant subclusters (gray) (left). Putative cancer-specific dREs for RHEB are highlighted by light blue shadows. Matching scRNA-seq expression of RHEB is shown for each subcluster (middle). Asterisks denote a statistically significant difference in gene expression between cells in the 3-Ovarian cancer subcluster and all remaining subclusters (average logFC > 1.0 & Bonferroni-corrected p-value <0.01, Wilcoxon Rank Sum test). Relative expression of mTOR pathway members is shown in the box plot (right). Asterisks denote statistically significant differences in mTOR pathway expression across all subclusters (Kruskal-Wallis test, p-value <0.01). Known regulatory element annotations, as used in panel C, are shown below the browser track. Peak-to-gene loops show the correlation value between peak accessibility and RHEB expression (bottom).
F) Kaplan-Meier survival curve based on progression-free survival for 614 OC patients stratified by high and low RHEBexpression.
The peak-to-gene correlation analysis revealed 345,791 statistically significant peak-togene links (p-value ≤ 1e-12 with eFDR=0.00014) (Data S1). To identify positive regulatory effects (i.e. positive correlation between peak accessibility and gene expression), we focused on peak-to-gene links with a correlation ≥ 0.45 (n=133,811). Most of these peak-to-gene links involved intronic peaks (50.2%) and distal peaks (28.3%). Promoter and exonic peak-to-gene links were lowest among this set (11.3% and 10.2%, respectively) (Figure S7D). To unveil distal regulatory mechanisms active within these gynecologic tumors, we proceeded with the 37,833 distal peak-to-gene links in our downstream analyses (Data S1). We further categorized peak-to-gene links into 36 k-means clusters and observed highly consistent patterns between inferred gene expression and linked peak accessibility (Figure 2B). We refer to these linked distal peaks as putative distal regulatory elements (dREs). The majority of identified dREs are annotated by the Encyclopedia of DNA Elements Consortium (ENCODE), providing support for our computational approach and suggesting they are bona fide regulatory elements (Consortium, 2012, Consortium et al., 2020).
To identify dREs specific to cancer cells across all patients, we extracted distal peaks from cancer-enriched k-means groups and carried out a genomic interval overlap analysis with epigenomic profiles from non-cancer tissues (Figure 2C, Figure S8A-E). We overlapped the genomic coordinates of our 14,043 cancer-enriched distal peaks with putative enhancer elements (defined by H3K27ac) active in cell lines derived from normal ovarian surface epithelium and normal fallopian tube secretory epithelium tissue (Coetzee et al., 2015). We also screened against all existing ENCODE regulatory elements (Consortium et al., 2020). The overlap analysis revealed 3,688 distal peaks that are not present in normal ovarian surface epithelium, normal fallopian tube secretory epithelium, nor the ENCODE database. Thus, these 3,688 distal peaks, participating in 5,827 peak-to-gene links, represent cancer-specific dREs (Data S1). The remaining distal peaks (n=22,166) represent regulatory elements that are active in normal tissue.
To further characterize cancer-specific dREs, we quantified the linked target genes per distal peak in both cancer-specific and normal peak groups. Strikingly, the cancer-specific peaks link to more genes (mean=1.58) compared to the non-malignant peaks (mean=1.44) (Wilcoxon Rank Sum test, p-value=1.6e-05) (Figure 2D, Figure S8F-I). Previous studies have proposed similar estimates of the number of putative target genes per dRE and we anticipate this difference to be magnified in a larger group of patients (Mills et al., 2020, Moore et al., 2020, Corces et al., 2018).
We found many salient instances of cancer-specific dREs linked to upregulated genes in malignant cell populations measured by scRNA-seq (Data S1). For example, the hallmark mTOR pathway regulator RHEB is significantly upregulated in the subcluster labeled as 3-Ovarian cancer, that comes from Patient 7 diagnosed with endometrioid OC (Figure 2E, Table 1, Table S1) (Yang et al., 2017). This subcluster of malignant cells also shows positive enrichment for the mTOR pathway gene signature (Liberzon et al., 2015) (see STAR Methods) (Kruskal-Wallis test, p-value <0.01). We found strong chromatin accessibility signal at the RHEB promoter across all malignant populations, but we highlight the marked increases in accessibility of four cancer-specific dREs enriched in the 3-Ovarian cancer subcluster (Figure 2E). Together, this offers a possible mechanism for mTOR pathway dysregulation through oncogenic dREs enriched in malignant cells of endometrioid OC. Indeed, high RHEB expression is prognostic of worse outcome in OC patients (Figure 2F and Table S4) (Gyorffy et al., 2012).
Our eFDR peak-to-gene linkage and genomic interval overlap analyses revealed additional putative cancer-specific dREs for clinical biomarkers CA125 andCD117 in EC/OC and GIST, respectively (Data S1). These genes are also predictive of poor survival in OC and gastric cancer, respectively (Table S4). Together with our findings for RHEB, this suggests that molecular rewiring of dREs play critical roles in the pathogenesis of gynecologic malignancies and have important clinical implications (Gyorffy et al., 2012, Szasz et al., 2016).
To transition from the full cohort analysis into cancer-type specific analyses, and identify even finer transcriptomic and epigenomic differences, we performed pseudo-bulk clustering analysis (Kimes et al., 2017) (STAR Methods). This analysis revealed two groups of patient tumors that were conserved across data types: Patients 1–5 (endometrioid endometrial cancer (EEC)) and Patients 8 & 9 (high-grade serous ovarian cancer (HGSOC)). These groupings reflect the original histological classifications in Table 1. Interestingly, tumors from Patient 6 and Patient 10 are more similar to the HGSOC tumors in terms of pseudo-bulk RNA-seq, but are more similar to EEC tumors in terms of pseudo-bulk ATAC-seq (Figure S9).
Cancer-specific regulatory mechanisms in Endometrioid Endometrial Cancer
EC is the most common gynecologic malignancy in the United States and the endometrioid histologic type accounts for a majority of cases (Siegel et al., 2021, Ritterhouse and Howitt, 2016). To analyze the EEC patient cohort, we merged all cells from Patients 1–5, resulting in 32,234 cells profiled by scRNA-seq and 32,155 cells profiled by scATAC-seq (STAR Methods). We found that cells clustered mainly by cell type and not by patient, suggesting batch effects were not a major confounder (Figure 3A-B, Figure S10). Overall, we observed eight general cell types across Patients 1–5 with 29 subclusters in scRNA-seq and 28 subclusters in scATAC-seq. In scATAC-seq, the 20-Fibroblast subcluster had only 10 cells and was therefore removed from downstream analysis. We next screened for malignant subclusters using the EC biomarkers MUC16/CA125 and WFDC2/HE4 (Figure S11) (Dong et al., 2017, Li et al., 2009). Again, we observed that fibroblast/stromal and EC subclusters were highly patient-specific (Figure 3C, Figure S10). We also highlight that four subclusters are almost entirely formed by cells coming from Patient 3 (6-,14-,15- and 21-Endometrial cancer), suggesting a high degree of intratumoral heterogeneity within this tumor.
Figure 3. A cancer-specific distal regulatory element helps drive IMPA2 expression within the Endometroid Endometrial Cancer patient cohort.

A) UMAP plot of scRNA-seq cells color-coded by cell types found in Patients 1–5 (left). UMAP plot of scATAC-seq cells color-coded by inferred cell type across Patients 1–5 (right).
B) UMAP plot of scRNA-seq cells as shown in panel A but color-coded by patient of origin (left). UMAP plot of scATAC-seq cells as shown in panel A but color-coded by patient of origin (right).
C) Stacked bar charts showing contribution of each patient to each subcluster.
D) Row-scaled heatmaps of statistically significant distal peak-to-gene links where each row represents expression of a gene (left) correlated to accessibility of a distal peak (right). Select k-means clusters containing IMPA2 are marked in red text.
E) Browser track showing the accessibility profile at the IMPA2 locus across all cell type subclusters (left). Subclusters are color-coded either malignant (orange) or non-malignant (gray). Putative cancer-specific dRE of IMPA2 is highlighted by the light blue shadow. Matching scRNA-seq expression of IMPA2 is shown for all subclusters (right). Asterisks denote a statistically significant difference in gene expression between cells in marked subclusters when aggregated (average logFC = 0.23 & Bonferroni-corrected p-value <0.01, Wilcoxon Rank Sum test). Known regulatory element annotations for normal ovarian surface epithelium, normal fallopian tube, and ENCODE, are shown below the browser track. Peak-to-gene loops show the correlation value between peak accessibility and IMPA2 expression (bottom).
F) Kaplan–Meier survival curve based on recurrence-free survival for 422 Uterine Corpus Endometrial Carcinoma (UCEC) patients stratified by high and low IMPA2 expression.
Next, we wanted to better understand transcriptional differences between these EEC subclusters and if any patterns could be explained by variation in chromatin accessibility. We performed the cancer-specific peak-to-gene linkage analysis in the EEC cohort and identified 324,626 peak-to-gene links (p-value ≤ 1e-12 with eFDR = 5.5e-5), of which 34,231 were distal with a correlation ≥ 0.45 (Data S1, Figure 3D). Comparison to normal reference epigenomic profiles identified 1,943 putative cancer-specific distal peaks forming 2,950 cancer-specific peak-to-gene links (Data S1) (Consortium et al., 2020, Coetzee et al., 2015). Interestingly, we observe the same increase in number of genes linked to cancer-specific peaks relative to normal peaks for the EEC patient cohort (Wilcoxon Rank Sum test, p-value=4.23e-05).
To evaluate if these dREs were shared across EEC patients, we repeated the peak-to-gene linkage analysis for each patient individually using the same set of peaks from the full EEC analysis (Figure S12A). We asked what proportion of the 34,231 dREs, or peak-gene pairs, were recoverable in each patient. The patient-specific analyses from Patients 1–5 recovered 49.68%, 52.03%, 40.91%, 62.17% and 52.32% of the original EEC dREs, respectively (Figure S12B). Moreover, we found that 17.23% of the original EEC dREs were recovered in every patient-specific analysis. Thus, multiple patients participate in these putative regulatory relationships.
Next, we wanted to investigate the extent to which cancer-specific dREs are rewired in malignant cell populations relative to normal cell populations of the EEC cohort. We repeated our peak-to-gene linkage analysis for malignant and non-malignant fractions of the EEC cohort independently and assessed how many cancer-specific dREs were recovered in each fraction (Figure 3C, Figure S13). We identified 27,738 dREs in the malignant-specific analysis and 34,172 dREs in the non-malignant analysis (Figure S13B top). The malignant-specific analysis recovered more of the 2,950 cancer-specific dREs than the non-malignant analysis (47.5% versus 6.3%, respectively) (Figure S13B, bottom). These data suggest that the distal regulatory landscape is rewired in malignancy relative to normal cell states.
We then identified three clear examples of cancer-specific dREs that explain upregulated gene expression in malignant populations relative to normal cell populations in the EEC cohort. For example, there is increased IMPA2 expression in the malignant fraction of the EEC cohort and increased chromatin accessibility of a cancer-specific dRE within the IMAP2 locus (Figure 3E). IMPA2 encodes the inositol monophosphatase 2 protein involved in phosphatidylinositol signaling. While few works have reported a role for IMPA2 in cancer, high IMPA2 expression is predictive of poor survival in Uterine Corpus Endometrioid Carcinoma (UCEC) patients (Figure 3F, Table S4) (Zhang et al., 2020, Nagy et al., 2021, Ohnishi et al., 2007). We also found three clear cancer-specific dREs linked to increased SOX9 expression in the malignant fraction of the EEC cohort (Data S1). Since high SOX9 expression portends a worse outcome for UCEC patients and SOX9 has been implicated in formation of endometrial hyperplastic lesions in EC, these data may offer insights into non-coding mechanisms behind carcinogenesis of the endometrium (Table S4) (Saegusa et al., 2012, Gonzalez et al., 2016, Nagy et al., 2021). Finally, we note that CD24 is highly expressed in the malignant fraction of the EEC cohort, and we highlight three cancer-specific dREs linked to CD24 expression (Data S1). CD24 is reported to be an effective differentiator between endometrial hyperplastic lesions and EC (Nagy et al., 2021, Kim et al., 2009). Additionally, increased CD24 expression offers resistance to chemotherapeutic agents and facilitates immune escape from macrophage phagocytosis in endometrial carcinoma cells (Lin et al., 2021, Pandey et al., 2010). These clinically relevant oncogenic dREs are just a snapshot of the altered regulatory landscape in EEC. We have tabulated all significant cancer-specific dRE-gene interactions in Data S1.
Cancer cell populations of High-Grade Serous Ovarian Cancer acquire cancer-specific dREs for genes involved in drug resistance
HGSOC is the most common histologic type of OC and is characterized by high copy number alterations and few driver mutations, which is thought to account for the clinical aggressiveness of this disease (Coward et al., 2015, Macintyre et al., 2018). To analyze the HGSOC patient cohort, we merged all cells from Patients 8 & 9, resulting in 13,646 cells profiled by scRNA-seq and 17,677 cells profiled by scATAC-seq (STAR Methods). Overall, we observed six general cell types across Patients 8 & 9 with 24 subclusters in scRNA-seq and 19 subclusters in scATAC-seq. In scATAC-seq, five cell type subclusters had less than 30 cells and were therefore removed from downstream analysis. (Figure 4A-B, Figure S14). We identified malignant subclusters using inferred CNV events and expression of the OC biomarkers MUC16/CA125 and WFDC2/HE4 (Figure S15) (Li et al., 2009, Duffy et al., 2005, Hellström et al., 2003, Sturgeon et al., 2008). Again, we observed that the fibroblast/stromal and OC subclusters are highly patient-specific, reflecting the biological uniqueness of malignant and fibroblast populations from each patient tumor as partly supported by their distinct inferred CNV profiles (Figure S3 and Figure S14). Of note, Patient 9 has four malignant subclusters suggesting a high degree of intratumoral heterogeneity within this tumor (Figure S14).
Figure 4. Malignant populations of the High-Grade Serous Ovarian Cancer patient cohort acquire novel enhancer-like elements that drive LAPTM4B expression.

A) UMAP plot of scRNA-seq cells color-coded by cell types found in Patients 8 and 9 (left). UMAP plot of scATAC-seq cells color-coded by inferred cell type across Patients 8 and 9 (right).
B) UMAP plot of scRNA-seq cells as seen in panel A but color-coded by patient of origin (left). UMAP plot of scATAC-seq cells as seen in panel A but color-coded by patient of origin (right).
C) Row-scaled heatmaps of statistically significant distal peak-to-gene links where each row represents expression of a gene (left) correlated to accessibility of a distal peak (right). Select k-means clusters containing LAPTM4B are marked in red text.
D) Browser track showing the accessibility profile at the LAPTM4B locus across all subclusters (left). Subclusters are color-coded either malignant (orange) or non-malignant (gray). Putative dREs of LAPTM4B are highlighted by light blue shadows. Matching scRNA-seq expression of LAPTM4B is shown in the box plot (right) for all subclusters. Asterisks denote a statistically significant difference in gene expression between cells in marked subclusters when aggregated (average logFC = 1.77 & Bonferroni-corrected p-value <0.01, Wilcoxon Rank Sum test). Known regulatory element annotations for normal ovarian surface epithelium, normal fallopian tube, and ENCODE, are shown below the browser track. Peak-to-gene loops show the correlation value between peak accessibility and LAPTM4B expression (bottom).
E) Kaplan-Meier survival curve based on overall survival for 1,656 OC patients stratified by high and low LAPTM4B expression.
F) Summary cartoon and table of Find Individual Motif Occurrences (FIMO) predictions within Enhancer 2, Enhancer 4 and LAPTM4B promoter (top, middle, bottom, respectively). Matching scRNA-seq TF expression in the malignant fraction of Patient 9 is shown in the box plots (right).
To understand the regulatory landscape of these subclusters, we carried out the peak-to-gene linkage analysis to identify putative cancer-specific dREs driving the transcriptional profiles of malignant populations. This analysis identified 486,293 statistically significant (p-value ≤ 1e-12 with eFDR = 2.1e-06) peak-to-gene links, of which 62,087 were distal with a correlation ≥ 0.45 (Data S1, Figure 4C). The genomic interval overlap analysis identified 5,202 putative cancer-specific distal peaks forming 11,134 cancer-specific peak-to-gene links (Data S1) (Consortium et al., 2020, Coetzee et al., 2015). Overall, cancer-specific peaks linked to more genes on average relative to the normal peaks for the HGSOC cohort (Wilcoxon Rank Sum test, p-value=6.6e-12). We again investigated the extent to which the cancer-specific dREs are rewired in malignant cell populations of the HGSOC cohort and found that a malignant-specific analysis recovered more of the 11,134 cancer-specific dREs than the non-malignant analysis (63.6% versus 3.9%, respectively) (Figure S16).
Of the 11,134 cancer-specific dREs in the HGSOC cohort, we highlight two examples of cancer-specific gene regulation in the malignant fraction. PI3, encoding peptidase inhibitor 3 (Elafin protein), is highly expressed in the malignant fraction and its upregulation can be explained by four cancer-specific dREs (Data S1). Not only is PI3 predictive of poor survival in serous ovarian cancer patients, it is implicated in OC chemoresistance and confers OC cells a proliferative advantage through activation of MEK-ERK signaling (Table S4) (Gyorffy et al., 2012, Labidi-Galy et al., 2015, Clauss et al., 2010, Wei et al., 2012, Williams et al., 2005).
We also highlight two cancer-specific dREs that were strongly associated with increased LAPTM4B expression in the malignant fraction of the HGSOC patient cohort (Figure 4D). LAPTM4B is predictive of poor survival in OC patients and has been reported as a potent facilitator of chemotherapeutic drug efflux as well as PI3K/AKT signaling (Figure 4E, Table S4) (Li et al., 2010, Tan et al., 2015, Gyorffy et al., 2012). We labeled LAPTM4B cancer-specific dREs as Enhancer 2 (Enh2) and Enhancer 4 (Enh4), and we note that there are three additional dREs annotated within this locus (Enhancer 1, 3, and 5). To interrogate TF occupancy at these dREs, we performed Find Individual Motif Occurrences (FIMO) analysis for each putative enhancer region using the Patient 9 DNA sequence after accounting for single-nucleotide variants in the malignant fraction (subclusters 0-,7-,11-,16-Ovarian cancer) of Patient 9 (Figure 4F and STAR Methods) (Bailey et al., 2015, Grant et al., 2011, Bailey et al., 2009). Interestingly, cells from the Patient 9 malignant fraction harbor a SNP (rs10955131) within Enhancer 2, but we are unable to determine if this mutation is somatically acquired as we did not achieve sufficient read depth in normal immune cells at this particular genomic region to perform variant calling (Figure S17). We observed statistically significant TF motif matches within each putative enhancer region and further ranked them by scRNA-seq TF expression within the Patient 9 malignant fraction (Figure 4F and Table S5). Of note, we found YY1 motifs within Enhancer 2, Enhancer 4 and the LAPTM4B promoter region, suggesting these cancer-specific enhancers participate in active enhancer-promoter connections within malignant cells of Patient 9 (Weintraub et al., 2017).
Functional validation of LAPTM4B enhancers and predicted TF regulators
To further validate our dRE identification pipeline, we conducted experiments to confirm these dREs and TFs as bona fide enhancers of LAPTM4B expression. First, we used dCas9-KRAB-mediated CRISPR interference assays, in the HGSOC cell line OVCAR3, to inhibit the most highly active cancer-specific dRE (Enhancer 2) and lineage-specific dRE (Enhancer 3) in the LAPTM4B locus (Figure 5A-C and STAR Methods) (Fulco et al., 2016, Larson et al., 2013, Gilbert et al., 2013, Qi et al., 2013). OVCAR3 cells stably expressing dCas9-KRAB were transfected with single guide RNAs (sgRNAs) targeting Enhancer 2 and Enhancer 3 to induce local chromatin repression (Figure 5B and STAR Methods). We then measured the consequences on gene expression and found that LAPTM4B was significantly reduced when targeting Enhancer 2 and Enhancer 3 (Figure 5D). Thus, we conclude that Enhancer 2 and Enhancer 3 are bona-fide enhancers of LAPTM4B, providing support for the remaining dREs identified throughout this study.
Figure 5. Functional validation of cancer-specific LAPTM4B regulatory model in high-grade serous ovarian cancer cells.

A) Browser track showing the accessibility profile at the LAPTM4B locus, as in Fig. 4D, but between malignant (orange) and non-malignant (gray) fractions of the HGSOC patient cohort. Coverage is normalized by sequencing depth as well as reads in TSS regions. Known regulatory element annotations for normal ovarian surface epithelium, normal fallopian tube, and ENCODE, are shown below the browser track.
B) Cartoon of dCas9-KRAB mediated CRISPR interference.
C) Western blot of OVCAR3 cells stably expressing dCas9-KRAB.
D) RT-qPCR results showing expression of LAPTM4B after dCas9-KRAB mediated repression of Enhancer 2 and Enhancer 3. Expression is shown as fold change relative to ACTB expression.
E) Cartoon depicting inferred TF-mediated enhancer-promoter connections.
F) RT-qPCR results of LAPTM4B expression after siRNA-mediated knockdown of GAPDH and predicted TF regulators: YY1, CEBPD, and KLF6. Expression is shown as fold change relative to ACTB expression.
G) RT-qPCR results of expression of TF regulators after siRNA knockdown. Expression is shown as fold change relative to ACTB expression.
H) RT-qPCR results of expression of GAPDH after siRNA-mediated knockdown of GAPDH and TF regulators. Expression is shown as fold change relative to ACTB expression. Data in D, F, G, and H shown as mean ± S.E.M.; *p< 0.05, **p< 0.01, ***p< 0.001, one-tailed Welch’s t-test.
We next validated predicted TF regulators of LAPTM4B via RNAi-mediated knockdown in OVCAR3 cells (Figure 5E). We measured the expression of LAPTM4B after knockdown of each predicted TF regulator: YY1, CEBPD, and KLF6. Indeed, we observed a statistically significant decrease in LAPTM4B expression when targeting YY1, CEBPD, and KLF6, but not when targeting the negative control, GAPDH (Figure 5E-H). Thus, YY1, CEBPD, and KLF6 are bona-fide TF regulators of LAPTM4B and provide confidence for our TF predictions (Figure 5E).
Linking dREs to transcription factor activity in human gynecologic malignancies
After identifying dREs that may play critical roles in cancer progression, we interrogated trans-acting factors present at these dREs across the entire dataset to better understand the regulatory logic of these tumors. We adapted our published method called Total Functional Score of Enhancer Elements (TFSEE) to predict which TFs are enriched at active dREs (enhancer-like elements) within malignant cell types (Figure 6A, STAR Methods) (Malladi et al., 2020, Franco et al., 2018). By adapting this method to matched scRNA-seq and scATAC-seq, TFSEE allows for concurrent assessment of TF expression, enhancer activity, enhancer location, and TFs present at enhancers. Across the full patient cohort, there were 11 malignant cell type subclusters chosen for TFSEE analysis based on patient specificity, inferred CNV events, and/or cancer biomarker expression patterns (Figure S18). We conducted the TFSEE analysis and observed that the malignant cell types tend to cluster by patient and by cancer type (Figure 6B). To further prioritize enriched TFs across active enhancer elements, we highlighted each TF by its predicted druggability status (binary) as determined by the canSAR database through structure-based and ligand-based assessments (Tym et al., 2016, Mitsopoulos et al., 2020).
Figure 6. Functional scoring of cell type-specific enhancer activity and their cognate transcription factors helps prioritize potential therapeutic targets across gynecologic malignancies.

A) Cartoon of matrix operations performed in the Total Functional Score of Enhancer Elements (TFSEE) method. Only malignant cell type clusters with 100% patient specificity were chosen for TFSEE analysis.
B) Unsupervised hierarchical clustering heatmap of cell type normalized TFSEE scores (n=102 TFs across active enhancers). Each row of the heatmap represents TF activity across cell type-specific enhancers enriched in each column. Predicted druggability status for each TF is marked with druggable/not druggable according to the canSAR database.
C) Rank-ordered plot showing the difference in scaled TFSEE score for each TF between subclone 1 (orange) and subclone 2 (blue) of the Patient 6 tumor representing serous EC. Each point represents a TF and is colored by predicted druggability status. Notable TFs enriched in either condition (subclone 1/subclone 2) are labeled in light blue regions of the plot.
D) Rank-ordered plot showing the difference in scaled TFSEE score for each TF between carcinoma (pink) and sarcoma (green) fractions of the Patient 10 tumor representing carcinosarcoma OC. Each point represents a TF and is colored by predicted druggability status. Notable TFs enriched in either condition (sarcoma/carcinoma) are labeled in light blue regions of the plot.
To exemplify the utility of TFSEE with single-cell data, we investigated intratumoral heterogeneity of two patients with rare histological subtypes. For Patient 6, diagnosed as EC of serous histology that metastasized to the ovary, there were two distinct tumor subclones (19- and 34-Endometrial cancer) highlighted by their distinct CNV profiles (Figure 6C, Figure S2, Table 1, Table S1). We visualized the differences in TF activity between these two subclones and observed several notable TFs enriched in each subclone (Figure 6C). Of note, we found MAFB to be enriched in the 19-Endometrial cancer subclone of the Patient 6 tumor relative to the 34-Endometrial cancer subclone. Moreover, MAFB is predicted to be druggable by ligand-based assessment according to the canSAR database (Mitsopoulos et al., 2020, Tym et al., 2016). We also observed STAT1 is enriched in the 34-Endometrial cancer subclone of the Patient 6 tumor (Mitsopoulos et al., 2020, Tym et al., 2016). These differences in TF activity may provide valuable insight into intratumoral heterogeneity of serous EC.
We also chose to investigate the two histopathological fractions (16- and 17-Ovarian cancer) of the Patient 10 tumor diagnosed as an ovarian carcinosarcoma (Table 1, Table S1). While these two histopathological fractions have similar inferred CNV profiles, a pseudo-bulk gene-set variation analysis (GSVA) across all malignant cell types revealed a higher enrichment of epithelial-to-mesenchymal transition (EMT) and Invasion gene signatures within the 16-Ovarian cancer subcluster (Figures S3 and S18). This suggests the 16-Ovarian cancer subcluster represents the sarcoma fraction while the 17-Ovarian cancer subcluster represents the carcinoma fraction. These fraction identity assignments are also supported by clustering of 16-Ovarian cancer with the GIST subclusters, 0-/27-GIST, and clustering of 17-Ovarian cancer with the HGSOC subclusters, 9-/10-Ovarian cancer (Figure 6B). To uncover differences in TF activity between the carcinoma fraction (17-Ovarian cancer) and sarcoma fraction (16-Ovarian cancer) of the Patient 10 tumor, we visualized the differences in scaled TFSEE score and identified a number of TFs enriched in each fraction. ZEB1 was enriched in the sarcoma fraction relative to carcinoma fraction (Figure 6D) (Mitsopoulos et al., 2020, Tym et al., 2016). This result is in line with ZEB1’s role in EMT and repression of epithelial-specific genes (Sánchez-Tilló et al., 2011, Watanabe et al., 2019). We also observed the epithelial-specific transcription factor ELF3 enriched in the carcinoma fraction relative to the sarcoma fraction (Figure 6D) (Sengez et al., 2019, Brembeck et al., 2000). These distinct TF activity profiles, along with the shared inferred CNV events between the histopathological fractions of the ovarian carcinosarcoma, may help researchers and clinicians better understand the etiologyof gynecologic carcinosarcomas (Barker and Scott, 2020, Kostov et al., 2020).
Our TFSEE analysis allowed us to make additional comparisons of serous versus endometrioid OC, serous versus endometrioid EC, and GIST versus serous OC (Figure S19). In each case, we identify important TF regulators enriched in either histologic type. Of note, we observed RARG enriched in serous OC relative to endometrioid OC, MAFB enriched in serous EC relative to endometrioid EC, and ZEB1 enriched in GIST relative to serous OC (Figure S19B-D). Overall, our TFSEE analysis is a novel framework in single-cell genomics that reveals robust inferences of TF activity coupled to TF expression. This strategy attempts to lower the false positive rate of motif-based TF predictions by enriching for TFs with non-zero expression and giving lower weight to TFs with zero or negligible expression. In some instances, some TFs can still be functional without being actively transcribed. Therefore, we chose to explore an alternate version of the TFSEE analysis that is agnostic to TF expression by omitting the last element-wise multiplication with the TF expression matrix and found similar results (Figure S20).
DISCUSSION
To date, the standard of care for OC and EC is a combination of surgery, chemotherapy, and radiation. Despite these aggressive treatments, most women with advanced stage EC and OC will succumb to their disease, highlighting the need to develop better targeted therapies. Our work represents a valuable multi-omic resource that charts the transcriptional and regulatory landscape of gynecologic tumors at single-cell resolution. Deconvolution of this dataset identified novel mechanisms that facilitate tumorigenesis and prioritized potential avenues for therapeutic intervention that were hidden using bulk genomic approaches. We also shed light on non-coding regulatory mechanisms for a number of clinically relevant biomarkers and major playersinvolved in cancer pathogenesis (Yang et al., 2017, Duffy et al., 2005, Dong et al., 2017, Sturgeon et al., 2008, Sarlomo-Rikala et al., 1998). Moreover, we anticipate that this dataset will help inspire novel therapeutic treatment strategies in EC and/or OC by serving as a reference for 1) clinicians in understanding intratumoral heterogeneity, 2) hypothesis generation in cancer biology, 3) cell type annotation in future single-cell datasets, and 4) the development of novel bioinformatic methods.
We reiterate four important findings from analyzing this single-cell dataset. First, we demonstrated that cancer cells acquire de novo non-coding dREs that modulate hallmark cancer pathways, including mTOR signaling, in a cancer-specific manner (Figures 2–5, Data S1). This is consistent with recent clinical trials testing mTOR inhibitors in combination therapy for ovarian cancer patients (Das et al., 2017, Westin, 2014, Banerji, 2014). From this, we speculate that the mTOR-enriched Patient 7 may benefit from an mTOR inhibitor treatment, although further investigation is needed. Nonetheless, these data demonstrate important non-coding mechanisms for how cancer cells may acquire aggressive phenotypes due to changes in chromatin accessibility and TF occupancy.
Moreover, cancer-specific dREs identified in each analysis cohort linked to more target genes on average compared to the lineage-specific dREs (Figure 2D). Based on our data, we anticipate this trend to be even greater across a larger group of patient tumors and posit that salient cancer-specific dREs carry a higher ‘regulatory load’ relative to dREs active in normal tissues. This could be explained by alterations in topologically associating domain boundaries and higher order chromatin structure, but this warrants further investigation (Akdemir et al., 2020).
Next, malignant populations within and between patient tumors show substantial heterogeneity in chromatin accessibility linked to transcriptional output (Figures 1–6). This poses a challenging obstacle in EC and OC treatment, and highlights the importance of intratumoral heterogeneity and the growing need for more single-cell datasets of solid tumors, especially in response to chemotherapy. The extent to which malignant cell populations can be described as distinct ‘cell types’ or ‘cell states’ remains elusive and inspires further study into temporally regulated oncogenic regulatory elements and lineage tracing of malignant cell populations (Clevers et al., 2017).
Lastly, our methodology to infer differential TF activity between populations of malignant cells reveals another complex layer of gene regulation that is repurposed in cancer cells (Figure 6 and Figures S19-S20). Our TFSEE analysis is a powerful tool that facilitates integration of scRNA-seq and scATAC-seq datasets to interrogate complex mechanisms of gene regulation. This helps prioritize TFs for follow up investigation and could help inspire novel therapeutic avenues in gynecologic malignancies. As a whole, this resource showcases important principles of gene regulation and tumor biology determined through single-cell multi-omic data.
Limitations of study
We recognize the true richness of the dataset cannot be exemplified here in full, and that there are some limitations associated with our approach. First, scRNA-seq and scATAC-seq libraries were prepared for each tumor by independent sampling of the cell suspension generated for each tumor. While Seurat v3 allows for robust alignment of cell types across datasets, there are methods for profiling the transcriptome and chromatin landscape within the same cell (Cao et al., 2018, Chen et al., 2019, Ma et al., 2020). However, these methods have yet to become widely accessible and come with their own set of technical nuances. Secondly, the number of cell type subclusters identified in the scRNA-seq data is dependent on user-defined parameters such as number of PCs and clustering resolution (Xu and Su, 2015, Stuart et al., 2019). While we did not explore all possible parameter sets, we note that characterizing cell type composition of each tumor was not the main focus of our study. Therefore, there may be even more complexity in these single-cell data. Thirdly, we realize that our Kaplan-Meier survival analyses were derived from bulk measurements in contrast to our single-cell data. Finally, we acknowledge that our study was limited by a small number of patients with a mix of histotypes which could affect the generalizability of our resource. However, we note that our requirement for treatment-naïve tumors prevented us from being more selective in regard to tumor histology. All patient specimens presented are treatment-naïve tumors, which are difficult to procure since the standard of care for HGSOC is shifting towards neo-adjuvant treatment. Nonetheless, these data and the analyses described herein represent a true baseline for these cancers, serving as a foundation for defining the regulatory logic of malignant cells at single-cell resolution.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Hector L. Franco (hfranco@med.unc.edu).
Materials availability
Plasmids generated in this study are available upon request.
Data and code availability
Processed single-cell RNA-seq data and single-cell ATAC-seq have been deposited at GEO(https://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE173682 and are publicly available as of the date of publication. Raw data (10x FASTQs) will be available with controlled access via dbGAP under the accession number phs002340.v1.p1 (https://www.ncbi.nlm.nih.gov/gap/).
All original code has been deposited on the Zenodo platform (DOI: 10.5281/zenodo.5546110) and is publicly available at the Github repository scENDO_scOVAR_2020 (https://github.com/RegnerM2015/scENDO_scOVAR_2020).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact (hfranco@med.unc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Patient Samples and Tumor Dissociation
Eleven, treatment naïve, Ovarian and Endometrial cancer patients were enrolled in the ‘Genomics of Ovarian and Endometrial Cancers’ study at the UNC Cancer Hospital (IRB Protocol 18–3198) and underwent debulking surgery with curative intent to remove their tumors (Table 1, Table S1). Tumor specimens were sectioned for pathology review and the remaining tissues were de-identified and collected for this study through UNC’s Tissue Procurement Facility. To minimize the time elapsed between the surgical removal of tumor tissue and processing for single-cell genomics, we established an efficient pipeline between the medical professionals (surgeon/clinical research coordinator/clinical pathologist), the coordinating team (project managers/pathology technician) and our labs’ research technicians before procedure day. The tumor specimens were never frozen or fixed in any way, and transported immediately after surgical resection to the lab on ice in media containing DMEM/F12 media (Gibco) + 1% Penicillin/Streptomycin (Corning). Before dissociation, tumor samples were weighed. Tissue mass varied between 0.5 g and 4.68 g. Tumor specimens were then minced using two razor blades and digested overnight in 20–30 mL DMEM/F12 + 5% FBS, 15mM HEPES (Gibco), 1x Glutamax (Gibco), 1x Collagenase/Hyaluronidase (Stem Cell Technologies, 07912), 1% Penicillin/Streptomycin (Corning), and 0.48 μg/mL Hydrocortisone (Stem Cell Technologies, 74144) on a stir plate at 37C and 180 rpm. For ovarian tumors, Gentle Collagenase/Hyaluronidase (Stem Cell Technologies, 07919) was used instead of Collagenase/Hyaluronidase. After digestion, tumor cells were washed twice with cold PBS + 2% FBS and 10mM HEPES (PBS-HF) and centrifuged at 1200 rpm for 5 min at room temperature. To remove red blood cells, the cell pellet was treated with 4 or 8 mL cold Ammonium Chloride Solution (Stem Cell Technologies, 07850) with 1 or 2 mL PBS-HF (ratio 1:4), respectively, for 1 minute, then centrifuged at 1200 rpm for 5 min. The amount of Ammonium Chloride Solution added was based on the size of the cell pellet and visual assessment of pink or red color present in the pellet. This step was repeated a second time if the pellet still exhibited a pink or red color after initial treatment. To further dissociate the cells, pellets were resuspended in 1–2 mL 0.05% Trypsin-EDTA (Gibco) and the suspension was gently pipetted up and down for 1 min. After 1 min, trypsin was inactivated by adding 10mL PBS-HF solution. The suspension was then centrifuged at 1200rpm for 5 min. If cell suspensions were clumpy, cells were resuspended with 1–2 mL Dispase (Stem Cell Technologies, 07923) and 200 μL 1mg/mL DNase I (Stem Cell Technologies, 07900) for 1 min, then inactivated with 10 mL PBS-HF. If the Dispase step was not necessary, cells were treated with DNase I during the trypsinization step. Cells were again centrifuged at 1200 rpm for 5 min, then washed in 10 mL PBS-HF and filtered through a 100μm cell strainer. A final centrifugation step was done at 1200 rpm for 5 min. The cell pellet was resuspended in DMEM/F12 + 5% FBS using a volume based on the final pellet size and filtered using a 40μm cell strainer. Single-cell suspension concentration and cell viability was measured by adding 10 μL 0.4% Trypan Blue to 10 μL cell suspension and measuring with the Countess II Automated Cell Counter (Thermo Fisher, AMQAX1000). We aimed for cell viability above 60% for the cells to be used for single-cell sequencing. Cell viability varied between 64% and 94% across all samples, with the majority of tumor suspensions being over 70% viable.
Cell Culture
OVCAR3 and HEK-293T cell lines were obtained from ATCC. OVCAR3 cells were grown in RPMI media (Gibco, 11875–093) supplemented with 10% FBS (Sigma) and 1% penicillin/streptomycin (Corning). HEK-293T cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco, 11995065) supplemented with 10% FBS and 1% penicillin/streptomycin. OVCAR3-dCas9-KRAB-blast (OVCAR3-KRAB) cells were grown in RPMI media supplemented with 10% FBS, 1% penicillin/streptomycin and 1 μg/mL blasticidin (Corning, 30100RB) after selection. All cell cultures were incubated at 37 °C in 5% CO2. Before use, OVCAR3 cells were authenticated with Short Tandem Repeat profiling through ATCC. All cell lines were tested for mycoplasma.
METHOD DETAILS
Single-cell Sequencing
To continue with scRNA-seq, the cell suspension was diluted to 1200 cells/μL. 10,000 cells were used to prepare scRNA-seq libraries using the following 10x Genomics Single Cell 3’ kits: Chromium Single Cell 3’ GEM, Library & Gel Bead Kit v3 (PN-1000075), Chromium Chip B Single Cell Kit (PN-10000153), and Chromium i7 Multiplex Kit (PN-120262) following the manufacturer’s protocol.
To continue with scATAC-seq, 500,000 cells were used in nuclei isolation following the Nuclei Isolation for Single Cell ATAC Sequencing protocol from 10x Genomics. For the lysis step, cells were lysed for 4 min. For the resuspension step, nuclei were resuspended in 50 μL 1x Nuclei Buffer. Nuclei were counted by adding 10 μL 0.4% Trypan Blue to 10 μL nuclei suspension and counted with the Countess II Automated Cell Counter. 10,000 nuclei were then used in library preparation using the following 10x Genomics Single Cell ATAC Kits: Chromium Single Cell ATAC Library & Gel Bead Kit v1 (PN-1000110), Chromium Chip E Single Cell ATAC Kit (PN-1000082), and Chromium i7 Multiplex Kit N, Set A (PN-1000084) following the manufacturer’s protocol. All libraries were sequenced using the 10X Genomics suggested sequencing parameters on an Illumina NextSeq 500 instrument.
Engineering OVCAR3-dCas9-KRAB cells
Lentivirus containing the Lenti-dCas9-KRAB-blast vector(Xie et al., 2017) (Addgene #89567) was packaged in HEK-293T cells. HEK-293T cells were seeded in a T75 flask and transfected with the following plasmids: 6.67 μg Lenti-dCas9-KRAB-blast, 5 μg psPAX2 (gift from Didier Trono, Addgene #12260), and 3.33 μg PMD2G (gift from Didier Trono, Addgene #12259) using Fugene 6 (Promega, E2691) following the manufacturer’s protocol. The lentivirus containing supernatant was harvested 48–72 hours after transfection and lentivirus was concentrated using Lenti-X Concentrator (Takara, 631231) following the manufacturer’s protocol. OVCAR3 cells were seeded in a six-well plate at 50,000 cells/well and transduced with the harvested lentivirus in RPMI media with 10% FBS and 10 μg/mL polybrene (Millipore, TR1003G). Transduced cells were incubated with lentivirus for 72 hours, then placed in RPMI selection media with 3 μg/mL blasticidin for 7 days. Batch selected OVCAR3-KRAB cells were validated by western blot. For western blot analysis, cells were lysed using the following lysis buffer: 50 mM Tris HCl (pH 8), 0.5 M NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS and 1x protease inhibitor. The primary antibodies used for Western blotting were as follows: Anti-beta Tubulin Loading Control (Abcam, ab6046), Anti-Cas9 Antibody (7A9–3A3) (Santa Cruz Biotechnology, sc-517386). The β-tubulin antibody was used at a 1:1500 dilution in 5% BSA in TBST with overnight incubation at 4°C. The Cas9 antibody was used at a 1:1500 dilution in 5% BSA in TBST with overnight incubation at 4°C. The secondary antibodies used for Western blotting were as follows: Donkey anti-rabbit IgG, Whole Ab, HRP-conjugated (GE Healthcare, NA934) and Donkey anti-Mouse IgG (H+L), HRP-conjugated (Thermo Fisher Scientific, PA1–28748). Secondary antibodies were used at a 1:5000 dilution in 5% BSA in TBST.
sgRNA Design and Vector Cloning
sgRNAs targeting Enhancer 2 and Enhancer 3 were designed using the CRISPOR web tool(Concordet and Haeussler, 2018). Two sgRNAs targeting unique regions of each enhancer were designed to be transfected together. The negative control sgRNA (sgScramble) used was previously published(Lawhorn et al., 2014). The sgRNA cloning vector pX-sgRNA-eGFP-MI is a modified version of pSpCas9(BB)-2A-Puro (pX459) v2.0(Ran et al., 2013) (Addgene #62988). Cas9 was removed from pX459 and replaced with eGFP to allow for visualization of sgRNA expression. To improve sgRNA stability and optimize for assembly with dCas9, the sgRNA stem-loop was extended and modified with an A-U base pair flip(Chen et al., 2013). sgRNA vector cloning was done following the protocol from Feng Zheng’s group(Ran et al., 2013). Briefly, sgRNA oligonucleotides were ordered from Integrated DNA Technologies (IDT). Oligonucleotides were duplexed with the following reaction: 10 μM sgRNA forward oligo, 10 μM sgRNA reverse oligo, 10 U T4 polynucleotide kinase (NEB, M0201L), and 1x T4 ligation buffer under the following conditions: 37°C for 30 minutes, 95°C for 5 minutes, then ramp down to 25°C at 5°C/minute. Duplexed sgRNAs were diluted 1:100, then 2 μL of this dilution was used in a ligation reaction with 100 ng pX-sgRNA-eGFP-MI linearized with BbsI-HF (NEB, R3539S). The ligation product was transformed into Subcloning Efficiency DH5alpha Competent Cells (Invitrogen, 18265017) following the manufacturer’s protocol. Each completed sgRNA vector was verified by Sanger sequencing using the human U6 promoter sequencing primer (GGC-CTA-TTT-CCC-ATG-ATT-CC). sgRNA oligonucleotide sequences can be found in Table S6.
CRISPRi
OVCAR3-KRAB cells were seeded in 6-well plates at 200,000 cells/well using antibiotic-free RPMI media supplemented with 10% FBS. After 24 hours, cells were transfected with a total of 1.5 μg sgRNA vector per well using Fugene 6 (Promega, E2691) following the manufacturer’s protocol. For the negative control well (Scramble), a single sgRNA vector was transfected. For wells targeting Enhancer 2 and Enhancer 3, two unique sgRNAs were co-transfected in each well. 72 hours after transfection, cells were visualized for GFP expression to ensure good transfection efficiency. Cells were then washed with 1x PBS and RNA was extracted using the Zymo Quick-RNA Miniprep Kit (Zymo, R1055) with on-column DNaseI treatment. The experiment was conducted three times to ensure reproducibility.
RNAi
OVCAR3 cells were seeded in 6-well plates at 150,000 cells per well in antibiotic-free RPMI media. After 24 hours, cells were transfected with 40 nM of siRNA (siGENOME SMARTpool, Dharmacon) using 3 μL RNAiMAX (Invitrogen, 13778075) following the manufacturer’s protocol. After 48 hours, wells were washed with 1x PBS and RNA was extracted using the Zymo Quick-RNA Miniprep Kit (Zymo, R1055) with on-column DNaseI treatment. The experiment was conducted three times to ensure reproducibility. The siRNA sequences can be found in Table S7.
RT-qPCR
RNA extracted from CRISPRi and RNAi experiments was treated with the Turbo DNA-free Kit (Invitrogen, AM1907) following the manufacturer’s protocol to ensure removal of all genomic DNA. Next, 2 μg of RNA was reverse-transcribed using the iScript cDNA Synthesis Kit (BioRad, 1708891) following the manufacturer’s protocol. The resulting cDNA was analyzed by qPCR with SYBR Green using the QuantStudio 6 Flex System (Applied Biosystems) and the primers listed below. mRNA expression was normalized to ACTB using the 2-ΔΔCT method. All experiments were conducted three times to ensure reproducibility. Results are shown as the mean fold change ± S.E.M. Statistical analysis was conducted with the GraphPad Prism 9.0.0 software using Welch’s one-tailed t-test. Statistical significance is indicated by *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001. Primer oligonucleotide sequences can be found in Table S8.
Single-cell RNA-seq Quantification and Quality Control (QC)
Raw andfiltered feature-barcode matrices for each patient tumor sample were generated using 10x Genomics Cell Ranger. For each patient tumor sample, the filtered feature-barcode matrix was then converted into a Seurat object using the Seurat R package (Stuart et al., 2019, Team, 2020). To enrich for high quality cells in each patient dataset, QC and doublet removal were performed for each patient dataset individually. First, outlier cells were defined in each of the following metrics: log(UMI counts) (>2 MADs, low end), log(number of genes expressed) (>2 MADs, low end) and log(percent mitochondrial read count +1) (>2 MADs, high end)(McCarthy et al., 2017). Only non-outlier cells meeting all three criteria were kept for doublet detection. Note that for the two lowest viability samples, collected from Patients 2 & 7, we had to manually set these QC thresholds. To reduce the false positive rate in doublet calling, only cells marked as doublets by both DoubletDecon(DePasquale et al., 2019) and DoubletFinder(McGinnis et al., 2019) were removed from downstream analysis. After QC and doublet removal for each patient dataset, the individual patient datasets were combined using Seurat’s merge() to form each patient cohort presented in this study (All patients, endometrioid endometrial cancer (EEC), high-grade serous ovarian cancer (HGSOC)).
Single-cell RNA-seq normalization, feature selection and clustering
Gene expression matrices were normalized using Seurat’s NormalizeData() with the normalization method set to “LogNormalize.” Feature selection was performed with Seurat’s FindVariableFeatures()with the selection method set to “vst” and the number of top variable features set to 2,000. Before principal component analysis (PCA), we scaled the expression for all genes in the dataset using Seurat’s ScaleData(). We opted not to regress out UMI counts per cell because either 1) PC1 was not correlated to UMI counts per cell or 2) evidence of biological variation was found in PC1 based on the number of inferred CNVs and cell type gene signature enrichment. We opted not to regress out percent mitochondrial read count per cell because it could represent meaningful biological variation as increased metabolic activity is a hallmark feature of cancer cells. The top 2,000 most variable genes were summarized by PCA into 50 principal components (PCs) and the cells were visualized in a two-dimensional UMAP embedding using Seurat’s RunUMAP() with all 50 PCs, as suggested by the results of Seurat’s JackStraw() (data not shown). To identify groups of transcriptionally distinct cells, graph-based Louvain clustering was performed using Seurat’s FindNeighbors() with all 50 PCs and Seurat’s FindClusters() with a resolution of 0.7. scRNA-seq UMAP plots were generated in R(Team, 2020) using ggplot2(Wickham, 2016).
Inference of copy number variation (CNV) from single-cell RNA-seq
For each patient tumor sample,putative copy number events were inferred for each cell cluster using the R package inferCNV(Tickle, 2019). To determine which cell clusters would serve as a normal background, each cell was scored for enrichment in the ESTIMATE immune gene signature(Yoshihara et al., 2013) and in the PanglaoDB(Franzen et al., 2019) plasma cell gene signature using Seurat’s AddModuleScore(). Cell clusters having a median enrichment score >0.1 in either of these gene signatures were deemed as normal immune cell types and were used as a normal background for inferCNV. The remaining cell clusters, representing the remaining cellular fraction of the tumor, were specified in inferCNV annotations file to infer CNVs at the level of these clusters. The standard inferCNV algorithm was invoked with infercnv::run() with cutoff set to “0.1”, denoise set to “TRUE”, scale_data set to “TRUE” and HMM set to “TRUE”. The default i6 Hidden Markov Model (HMM) was used to predict CNV levels based on a six-state CNV model ranging from complete loss to >2 copies. The Bayesian Network Latent Mixture Model was used to estimate the posterior probability of each CNV level at each predicted CNV region. CNV regions with a posterior probability of a normal diploid state <0.05 were deemed as putative CNV events and were further used to justify the CNV status of each cluster (and thus the CNV status for each cell). The inferred CNVs determined individually for each patient dataset were retained after combining multiple patient datasets into the different patient cohort datasets. Box plots showing the number of inferred CNV events in each cell type subcluster were generated in R(Team, 2020) using ggplot2(Wickham, 2016).
Single-cell RNA-seq cell type annotation
Cell type annotation was performed using a combination of 1) reference-based annotation with the R package SingleR(Aran et al., 2019) and 2) gene signature enrichment with Seurat’s AddModuleScore(). After QC, doublet removal, and dimension reduction for each patient dataset, single cells were annotated to known cell types using SingleR with a reference scRNA-seq dataset. Datasets for Patients 1–5 were annotated based on a reference scRNA-seq dataset from the human endometrium(Wang et al., 2020). Datasets forPatients 6–11 were annotated based on a reference scRNA-seq dataset from a human ovarian tumor (sample ID: HTAPP-624-SMP-3212)(Slyper et al., 2020). The individual patient datasets were then combined using Seurat’s merge() to form each patient cohort presented in this study and subsequently reprocessed according to the normalization, feature selection and clustering methods described previously. The resulting clusters in each patient cohort dataset were annotated based on the majority cell type label within each cluster. Finally, SingleR cell type annotations were verified by calculating single cell enrichment scores for cell type gene signatures from PangladoDB(Franzen et al., 2019) using Seurat’s AddModuleScore(). The cell type annotations for each cluster were then modified to include the cluster number identity hyphened with the cell type identity. To identify malignant cell clusters, MUC16/CA125 and WFDC2/HE4 expression levels were used to identify EC and OC (Duffy et al., 2005, Sturgeon et al., 2008, Hellström et al., 2003, Li et al., 2009, Dong et al., 2017) and KIT/CD117 expression level was used to identify GIST(Sarlomo-Rikala et al., 1998). A cluster was deemed malignant if it had inferCNV events and/or statistically significant increased expression (Wilcoxon Rank Sum test, Bonferroni-corrected p-value <0.01) of any of these markers relative to the predicted non-malignant fraction (Figure S4, Figure S11, Figure S15). These criteria defined the final cell type subcluster identities for scRNA-seq that were used in label transferring to the matching scATAC-seq data.
Calculating enrichment of gene signatures in single-cell RNA-seq
Single-cell gene signature enrichment was calculated using Seurat’s AddModuleScore() with the search parameter set to “TRUE” to find aliases for gene names. Gene signature enrichment for pseudo-bulk clusters was performed using the R package GSVA(Hanzelmann et al., 2013). To generate pseudo-bulk transcriptome profiles for each cluster as shown in Figure S18, raw gene counts were summed across all cells in each cluster. The resulting matrix of genes by n clusters was then used as input into GSVA with the method argument set to “gsva” and the kcdf argument set to “Poisson.” Gene signature enrichment violin plots and/or boxplots were generated in R(Team, 2020) using ggplot2(Wickham, 2016).
Single-cell ATAC-seq quality control (QC)
For each patient tumor sample, a list of unique ATAC-seq fragments with associated barcodeswas generated using 10x Genomics Cell Ranger ATAC. The list of unique fragments per barcode for each patient tumor sample was read into the R package ArchR(Granja et al., 2021) to perform quality control and doublet removal for each patient dataset individually. To enrich for cellular barcodes, we took advantage of the bimodal distributions in log10(TSS enrichement+1) and in log10(number of unique fragments) characterizing two different populations of barcodes (cellular and non-cellular). Barcode cutoff thresholds for log10(TSS enrichement+1) and log10(number of unique fragments) were estimated using a Gaussian Mixture Model (GMM) for each metric, as implemented in the R package mclust(Scrucca et al., 2016). Only barcodes above these estimated thresholds in both metrics were kept as cellular barcodes for doublet detection. Note that for our lowest viability samples, collected from Patients 2 & 7, we manually set these QC thresholds. Doublet enrichment scores were calculated for cellular barcodes using ArchR’s addDoubletScores() with the knnMethod set to “UMAP.” Cellular barcodes with doublet enrichment scores >1 were marked as potential doublets and subsequently removed based on the filterRatio parameter of ArchR’s filterDoublets() function.
Single-cell ATAC-seq quantification, feature selection and integration with single-cell RNA-seq
We opted not to use the peak-barcode matrices generated by Cell Ranger ATAC because these peaks were called in a pooled/bulk setting (i.e. using all fragments captured by the assay in such a way that is agnostic to barcode identity). This would effectively drown out the signal from rare cell types present in the dataset. Therefore, we used the R package ArchR(Granja et al., 2021) to construct an initial feature matrix of 500 bp genomic tiles across all cells in each patient cohort. To reduce dimensions of the genomic tile features, we adopted the iterative latent semantic indexing(Cusanovich et al., 2015, Satpathy et al., 2019, Granja et al., 2019) (LSI) procedure implemented in the ArchR R package(Granja et al., 2021). Briefly, this procedure performs term frequency-inverse document frequency (TF-IDF) normalization to upweight more informative features followed by an initial LSI reduction on the top accessible tiles. Graph-based Louvain clustering is used to identify low resolution clusters in which feature counts are summed across all cells in each cluster to identify the top 25,000 most variable features across clusters. This procedure was iterated once more by inputting the top 25,000 most variable tiles from iteration 1 as the top accessible tiles in iteration 2. The iterative LSI procedure computed 50 LSI dimensions that were then collapsed further into a two dimensional UMAP embedding using ArchR’s addUMAP() with the same UMAP parameters used in Seurat’s RunUMAP(). LSI dimensions that were correlated with sequencing depth (>0.75, Pearson correlation) were not included in downstream analysis. scATAC-seq UMAP plots were generated in R(Team, 2020) using ggplot2(Wickham, 2016).
Before transferring labels from scRNA-seq to scATAC-seq, gene activity scores were inferred in scATAC-seq using ArchR’s addGeneScoreMatrix(). Briefly, this method uses the following features to estimate gene activity: 1) fragment counts mapping to the gene body, 2) an exponential weighting function to give higher weights to fragment counts closer to the gene and lower weights to fragment counts father away from the gene, and 3) gene boundaries to prevent the contribution of fragments from other genes. Seurat’s CCA implementation(Stuart et al., 2019) in FindTransferAnchors() and TransferData() was used to assign each of the scATAC-seq cells a cell type subcluster identity from the matching scRNA-seq data and an associated label prediction score. This label transferring procedure was constrained to only align cells of the same patient dataset (e.g. Patient 1 scATAC-seq cells were assigned only to cell type subclusters represented by Patient 1 scRNA-seq cells). All scATAC-seq cells were included in UMAP visualization and in calculating patient contribution per cluster, but only scATAC-seq cells with a label prediction score >0.5 were included in downstream analyses. Also, only inferred cell type subclusters with >10 cells were included in downstream analysis to ensure enough cells for peak calling in each cluster. This criterion was raised to >30 cells for the HGSOC patient cohort analysis. After scATAC-seq cells received a cell type subcluster label, pseudo-bulk replicates were generated for each inferred cell type subcluster in the R package ArchR(Granja et al., 2021) and pseudo-bulk peak calling was performed within each inferred cell type subcluster using MACS2(Zhang et al., 2008, Liu, 2014). ArchR’s default iterative overlap procedure was used to merge all peak calls into a single peak by barcode matrix across all cellular barcodes in each patient cohort dataset. Genomic browser tracks displaying the pseudo-bulk ATAC-seq coverage patterns within cell types were generated using ArchR’s plotBrowserTrack() function(Granja et al., 2021).
Differential gene expression and differential peak accessibility
Differential gene expression analysis in scRNA-seq was performed using Seurat’s FindAllMarkers() with the min.pct set to “0.25” and only.pos set to “FALSE”. This procedure identifies differentially expressed genes (DEGs) between two groups of cells using a Wilcoxon Rank Sum test. Unless otherwise noted in figure legends, DEGs were identified for each cell cluster by comparing the expression values of genes across all cells in a cluster (group 1) relative to the expression values for all remaining cells in the dataset (group 2). We chose a stringent Bonferroni-corrected p-value threshold of 0.01 for determining differentially expressed genes after multiple testing. For some cases, we pooled together malignant clusters to form group 1 and compared against non-malignant clusters to form group 2. For these special cases, we set the min.pct parameter to zero. Differential peak accessibility analysis in scATAC-seq was performed using ArchR’s getMarkerFeatures() with the bias argument set to include both “TSSEnrichment” and “log10(number of fragments)”. This procedure identifies differentially accessibility peaks (DEPs) between two groups of cells using a Wilcoxon Rank Sum test. DEPs were identified for each cell cluster by comparing the accessibility values of peaks across all cells in a cluster (group 1) relative to the accessibility values for a group of background cells matched for TSS enrichment and read depth (group 2). We chose a stringent Benjamini-Hochberg corrected p-value threshold of 0.01 for determining differentially accessible peaks (Log2FC >= 1.25) after multiple testing, and used these thresholds for determining distal marker peaks for the Total Functional Score of Enhancer Elements (TFSEE) analysis (Figure 6, Figure S19-S20).
Kaplan-Meier (KM) survival curves
All KM plots and hazard ratio statistics for each gene were generated using the Kaplan Meier Plotter web tool(Gyorffy et al., 2012, Nagy et al., 2018, Szasz et al., 2016) available at https://kmplot.com/analysis/. Detailed metadata for each KM analysis, such as datasets used, filtering criteria, etc., are listed in Table S4.
To determine the expression cutoff for stratifying patients into high versus low groups, we used the auto select best cutoff option. Briefly, this method involves computing all possible cutoff values between the lower and upper quartiles and choosing the KM plot result with the maximum difference between the p-value and hazard ratio.
Pseudo-bulk clustering of patient tumors
To create a pseudo-bulk transcriptome profile for each patient tumor sample as shown in Figure S9, the raw feature barcode matrix generated by 10x Genomics Cell Ranger (v3.1.0) was collapsed into a single profile by row summing the raw counts across all barcodes (cellular and non-cellular). Only genes expressed across all patient samples were kept for downstream analysis due to a lack of replicates to distinguish biological zeros from technical zeros. The resulting matrix of 19,914 genes by 11 patients was transformed with the regularized logarithm transformation in the DESeq2(Love et al., 2014) R package to stabilize variance and to account for differences in library size between patients. The top 5% most variable genes were chosen for unsupervised hierarchical clustering and principal component analysis (PCA). Hierarchical clustering, with complete linkage and 1-Pearson correlation as the distance metric, was performed in the R package sigclust2(Kimes et al., 2017) to assess statistical significance of splitting. Dendrograms were generated by invoking sigclust2::shc() with the alpha set to 0.05 and n_min set to 8. The R package ComplexHeatmap(Gu et al., 2016) was used to generate the heatmap of the top 5% most variable genes across 11 patients using the custom dendrogram generated by sigclust2. The PCA plot of 11 patient tumors based on the top 5% most variable genes was generated using DESeq2’s plotPCA().
To create a pseudo-bulk chromatin accessibility profile for each patient tumor sample as shown in Figure S9, the position sorted bam file generated by 10x Genomics Cell Ranger ATAC (v 1.2.0) was inputted into the R package csaw(Lun and Smyth, 2016) to quantify ATAC fragments into 200 bp contiguous genomic tiles. The read parameters were set using csaw’s readParam() with minq set to “20”, pe set to “both”, dedup set to “TRUE”, max.frag set to “500”, and discard to set to a Granges object listing hg38 blacklist regions. The 200 bp genomic tile matrix was constructed using csaw’s windowCounts() with ext set to “100”, width set to “200”, and bin set to “TRUE”. Only genomic tiles accessible across all patient samples were kept for downstream analysis due to a lack of replicates to distinguish biological zeros from technical zeros. The resulting matrix of 6,052,083 genomic tiles by 11 patients was transformed with the regularized logarithm transformation in the DESeq2(Love et al., 2014) R package to stabilize variance and to account for differences in library size between patients. The top 5% most variable genomic tiles were chosen for unsupervised hierarchical clustering and principal component analysis (PCA). Hierarchical clustering, with complete linkage and 1-Pearson correlation as the distance metric, was performed in the R package sigclust2(Kimes et al., 2017) to assess statistical significance of splitting. Dendrograms were generated by invoking sigclust2::shc() with the alpha set to 0.05 and n_min set to 8. The R package ComplexHeatmap(Gu et al., 2016) was used to generate the heatmap of 3,000 randomly sampled features out of the top 5% most variable genomic tiles across 11 patients using the custom dendrogram generated by sigclust2. The PCA plot of 11 patient tumors based on the top 5% most variable genomic tiles was generated using DESeq2’s plotPCA().
Peak-to-gene correlation analysis with empirically derived FDR (eFDR)
Peak-to-gene correlation analysis was performed to identify putative regulatory relationships by correlating peak accessibility to imputed gene expression across scATAC-seq metacells. This procedure was invoked by ArchR’s addPeak2GeneLinks() with reducedDims set to “IterativeLSI” and dimsToUse set to “1:50”. Gene expression in scATAC-seq was imputed after the Seurat label transfer procedure. This procedure calculated imputed gene expression values by multiplying the scRNA-seq expression values by the anchor weights matrix defining the association between each scATAC-seq cell and each anchor. Next, low-overlapping aggregates of scATAC-seq cells were generated via a k-nearest neighbor procedure in the LSI space to reduce noise and to ensure robust correlations in the features. Aggregates with >80% overlap with any other aggregate were removed to reduce to bias. This procedure resulted in approximately 500 aggregates of scATAC-seq cells which were used to correlate the accessibility of every peak to the imputed expression of every gene on the same chromosome using an implementation of fast feature correlations in C++ using the Rcpp package implemented by the ArchR(Granja et al., 2021) R package.
To assess statistical significance of the peak-to-gene correlations as shown in Figure S7, we developed an elaborate empirical FDR (eFDR) procedure to help screen for robust peak-to-gene associations(Storey and Tibshirani, 2003). To estimate the eFDR, the number of observed peak-to-gene associations with a raw p-value ≤ 1e-12 was first recorded. The peak-to-gene correlation analysis was then repeated 100 times under the permuted null condition where, for each permutation, the scATAC-seq metacell labels were shuffled for the peak data only to break the link between peak accessibility and gene expression. To calculate the eFDR, the median number of null peak-to-gene associations with a raw p-value ≤ 1e-12 across all 100 permutations was divided by the number of observed peak-to-gene associations with a raw p-value ≤ 1e-12. This entire procedure was conducted for each patient analysis cohort (full cohort, EEC, and HGSOC) based on the peak matrices generated for each patient analysis. The initial raw p-value threshold of 1e-12 was chosen over the first-quartile of the observed p-value distribution because in two out of three analysis cohorts, the 1e-12 raw p-value threshold offered a preferable (lower) eFDR relative to the first-quartile approach.
To compute the distribution of the number peaks per gene and vice versa as shown in Figures 2D and S8, a peak-to-gene metadata table was first created where each row contained a peak name, or set of genomic coordinates, and a corresponding gene name. The distribution of the number peaks per gene was computed by tallying the number of unique gene names. The distribution of the number genes per peak was computed by tallying the number of unique peak names.
To identify patient-specific and malignant cell type-specific peak-to-gene correlations, as shown in Figures S12, S13, and S16, the scATAC-seq ArchR dataset was subsetted accordingly to only include patient or malignant cell type barcodes of interest before re-computing the peak-to-gene links.
Genomic coordinate overlap analysis with normal epigenome profiles
To identify putative cancer-specific distal regulatory elements (dREs) within each patient analysis cohort as demonstrated in Figure S8, the genomic coordinates of the distal peaks participating in the cancer-enriched peak-to-gene links were overlapped with a set of normal epigenome profiles.
H3K27ac ChIP-seq peaks of ovarian surface epithelium cell lines iOSE4 and iOSE11 were downloaded from GSE68104. The hg19 genomic coordinates from iOSE4 rep1, iOSE4 rep2, iOSE11 rep1, and iOSE11 rep2 were merged into one combined peak set using the reduce() function from the GenomicRanges R package(Coetzee et al., 2015, Lawrence et al., 2013). After liftOver from hg19 to hg38, this combined peak set served as the normal reference enhancer profile for ovarian surface epithelium(Maintainer, 2020). H3K27ac ChIP-seq peaks of fallopian tube secretory epithelial cell lines iFTSEC33 and iFTSEC246 were downloaded from GSE68104. The hg19 genomic coordinates from iFTSEC33 rep1, iFTSEC33 rep2, iFTSEC246 rep1, and iFTSEC246 rep2 were merged into one combined peak set using the reduce() function from the GenomicRanges R package(Coetzee et al., 2015, Lawrence et al., 2013). After liftOver from hg19 to hg38, this combined peak set served as the normal reference enhancer profile for fallopian tube secretory epithelium(Maintainer, 2020). The last normal reference epigenome profile was supplied by the full list of Candidate cis-Regulatory Elements by ENCODE (ENCODE cCREs) in hg38 (Consortium et al., 2020).
findOverlapsOfPeaks() from the ChIPpeakAnno R package was used to find overlaps between the cancer-enriched peaks and the normal reference epigenome profiles(Zhu et al., 2010). Genomic coordinate overlap between features was defined as a minimum of 1 bp overlap. The cancer-enriched peak coordinates that did not overlap with any of the normal reference epigenome profiles were deemed cancer-specific peaks.
Predicting transcription factor occupancy at select putative enhancer regions in High-Grade Serous OC (HGSOC)
The sequences of the select putative enhancers in the malignant fraction of Patient 9, as shown in Figure 4D, were extracted with bedtools(Quinlan and Hall, 2010) getfasta() after accounting for single-nucleotide variants relative to the hg38 reference genome. Single-nucleotide variants in the malignant fraction were called using bcftools(Danecek and McCarthy, 2017) mpileup followed by bcftools(Danecek and McCarthy, 2017) consensus with a bam file containing fragments only from cellular barcodes present in in the Patient 9 malignant fraction. This malignant-specific bam file was generated using Cell Ranger’s bamslice. The putative enhancer sequences were inputted into Find Individual Motif Occurrences (FIMO) (Bailey et al., 2015) motif scanning with the --bgfile parameter set to “motif-file” and with a motif database supplied by JASPAR2020 (Fornes et al., 2020). The FIMO output listing matching motif occurrences was filtered for matches with a q-value < 0.10. This list of statistically significant motif matches was further ranked by TF expression in the malignant fraction of Patient 9 calculated by summing the normalized TF counts across all cells in the malignant fraction. TF expression box plots were generated in R(Team, 2020) using ggplot2(Wickham, 2016).
Total Functional Score of Enhancer Elements (TFSEE)
TFSEE analysis, as presented in Figure 6, was performed to identify transcription factors (TFs) enriched at active distal regulatory elements (dREs) for each malignant cell type(Malladi et al., 2020) (Franco et al., 2018). Referring back to the entire patient cohort, 11 out of 36 cell type subclusters were chosen for TFSEE analysis based on patient specificity, inferred copy number events and malignant cell type identity (Figure 1D, Figure S18). Only malignant cell type clusters with 100% patient specificity were chosen for the TFSEE analysis.
To generate the dRE or enhancer activity matrix, statistically significant dREs identified in the peak-to-gene linkage analysis (Pearson correlation >0.45, p-value <= 1e-12) were set intersected with a list of differentially accessible peaks enriched (Benjamini-Hochberg corrected p-value <= 0.01 & log2FC >= 1.25) in each of the malignant cell type groups. Pseudo-bulk enhancer activity profiles were generated by row summing the counts across all cells in each malignant cell type. Only enhancer regions that were accessible across all malignant cell types were included in the analysis due to a lack of replicates to distinguish biological zeros from technical zeros. The resulting matrix of enhancers by malignant cell types was transformed with the regularized logarithm transformation in the DESeq2(Love et al., 2014) R package to stabilize variance and to account for differences in library size between malignant cell type groups. Post-transformation, the enhancer activity matrix was scaled from 0 to 1 (cell type-wise) prior to the TFSEE matrix operations.
To generate the TF motif prediction matrix, motif search and matching were performed with MEME and TOMTOM, respectively using MEME suite of programs(Bailey et al., 2009, Bailey et al., 2015). The sequences of the enhancers in each malignant cell type were extracted with bedtools(Quinlan and Hall, 2010) getfasta() using the hg38 reference genome. The enhancer sequences were then inputted into MEME motif searching using the following flags: - dna, -mod zoops, -nmotifs 15, -minw 8, -maxw 15, and -revcomp. The MEME outputs were inputted into TOMTOM motif matching using the flags -evalue and -thresh 10 with a motif database supplied by JASPAR2020(Fornes et al., 2020). The outputs of MEME and TOMTOM were parsed using a custom Python script written by the original authors (Malladi et al., 2020) of TFSEE to generate a matrix of TF motif prediction scores (https://git.biohpc.swmed.edu/gcrb/tfsee). This motif prediction score matrix was scaled from 0 to 1 (enhancer-wise) prior to the TFSEE matrix operations.
To generate the TF expression matrix, pseudo-bulk gene expression profiles were generated by row summing the gene counts across all cells in each malignant cell type. Only genes that were expressed across all malignant cell types were included in the analysis due to a lack of replicates to distinguish biological zeros from technical zeros. The resulting matrix of genes by malignant cell types was transformed with the regularized logarithm transformation in the DESeq2(Love et al., 2014) R package to stabilize variance and to account for differences in library size between malignant cell type groups. Post-transformation, the gene expression matrix was subsetted to TFs identified in the motif prediction analysis and then scaled from 0 to 1 (cell type-wise) prior to the TFSEE matrix operations.
The enhancer activity matrix was multiplied with the TF motif prediction matrix to form an intermediate matrix product. This matrix product was element-wise multiplied with the TF expression matrix to form the final TFSEE matrix used in downstream analysis (Figure 6A). Heatmaps of the final TFSEE matrix were generated in R(Team, 2020) using ComplexHeatmap(Gu et al., 2016, Wickham, 2016).
The rank order frequency distribution plots were generated by computing the difference in scaled TFSEE score between two conditions or malignant cell types of interest. If multiple malignant cell types were represented in a condition, the average TFSEE score profile was computed to form one observation for that condition group in the difference calculation. Rank order plots were generated in R(Team, 2020) using ggplot2(Wickham, 2016).
QUANTIFICATION AND STATISTICAL ANALYSIS
For computational analyses, statistical details can be found in the corresponding figure legends and in the publicly available Github repository (https://github.com/RegnerM2015/scENDO_scOVAR_2020). Most of the computational analyses and statistical tests were performed in R version 4.0.3 (Team, 2020). Statistical significance for correlation, Wilcoxon-Rank Sum, and Kruskal-Wallis tests were defined as a p-value < 0.01 unless otherwise indicated in the figure legends or method details section. The remaining statistical analyses were performed through the Unix command line interface with the Cell Ranger software or the MEME suite of tools (Grant et al., 2011, Bailey et al., 2009, Bailey et al., 2015). Statistical significance for Cell Ranger related analyses can be described further here: https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger. Statistically significant motif matches identified by the FIMO software were defined as a Benjamini-Hochberg corrected p-value (i.e. q-value) < 0.10.
For RT-qPCR, statistical details of experiments can be found in the corresponding figure legends. Results are shown as the mean fold change (n=3) ± S.E.M. (n = number of biological replicates). Statistical analysis was conducted with the GraphPad Prism 9.0.0 software using Welch’s one-tailed t-test. Statistical significance is indicated by *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001.
Supplementary Material
Table S1. Extended clinical data and library information for 11 gynecologic tumor specimens, Related to Table 1. (Table_S1_clinical_data.xlsx)
Table S2. scRNA-seq barcode metadata, clustering, and cell type annotations, Related to Figures 1, 3, and 4.(Table_S2_scRNA_metadata.xlsx)
Table S3. scATAC-seq barcode metadata, clustering, and inferred cell type annotations, Related to Figures 1, 3, and 4. (Table_S3_scATAC_metadata.xlsx)
Table S4. Kaplan-Meier data summary and associated metadata with directions to reproduce the analyses on kmplot.com, Related to STAR Methods. (Table_S4_KM_metadata.xlsx)
Table S5. FIMO transcription factor motif scanning results for the LAPTM4B enhancers 1–5 and promoter in high-grade serous ovarian cancer, Related to Figure 4. (Table_S5_ranked_FIMO_results.xlsx)
There are three sets of files for each cohort of patients in this study: 1) statistically significant peak-to-gene links with all peak types and no correlation thresholding, 2) statistically significant distal peak-to-gene links with correlation >= 0.45, and 3) statistically significant cancer-specific distal peak-to-gene links with correlation >= 0.45. (Data_S1_Peak_to_Gene_Links.tsv.gz)
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | |
|---|---|---|---|
| Antibodies | |||
| Anti-beta Tubulin antibody – Loading Control | Abcam | Cat#ab6046; RRID: AB_2210370 | |
| Anti-Cas9 Antibody (7A9–3A3) | Santa Cruz Biotechnology | Cat#sc-517386; RRID: AB_2800509 | |
| Donkey anti-rabbit IgG, Whole Ab, HRP-conjugated | GE Healthcare | Cat#NA934, RRID: AB_772206 | |
| Donkey anti-Mouse IgG (H+L), HRP-conjugated | Thermo Fisher Scientific | Cat#PA1–28748, RRID: AB_10982166 | |
| Bacterial and virus strains | |||
| Subcloning Efficiency DH5alpha Competent Cells | Invitrogen | Cat#18265017 | |
| Chemicals, peptides, and recombinant proteins | |||
| FuGENE 6 | Promega | Cat#E2691 | |
| Collagenase/Hyaluronidase | Stemcell Technologies | Cat#07912 | |
| Gentle Collagenase/Hyaluronidase | Stemcell Technologies | Cat#07919 | |
| Hydrocortisone | Stemcell Technologies | Cat#74144 | |
| Dispase | Stemcell Technologies | Cat#07923 | |
| DNase I | Stemcell Technologies | Cat#07900 | |
| Blasticidin | Corning | Cat#30100RB | |
| Lenti-X Concentrator | Takara | Cat#631231 | |
| Polybrene | Millipore | Cat#TR1003G | |
| RNAiMAX | Invitrogen | Cat#13778075 | |
| Critical commercial assays | |||
| Chromium Single Cell 3’ GEM, Library & Gel Bead Kit v3 | 10x Genomics | Cat#PN-1000075 | |
| Chromium Single Cell ATAC Library & Gel Bead Kit v1 | 10x Genomics | Cat#PN-1000110 | |
| Chromium Chip B Single Cell Kit | 10x Genomics | Cat#PN-10000153 | |
| Chromium i7 Multiplex Kit | 10x Genomics | Cat#PN-120262 | |
| Chromium Chip E Single Cell ATAC Kit | 10x Genomics | Cat#PN-1000082 | |
| Chromium i7 Multiplex Kit N, Set A | 10x Genomics | Cat#PN-1000084 | |
| Quick-RNA Miniprep Kit | Zymo | Cat#R1055 | |
| Turbo DNA-free Kit | Invitrogen | Cat#AM1907 | |
| iScript cDNA Synthesis Kit | BioRad | Cat#1708891 | |
| Deposited data | |||
| scRNA-seq (processed data) | This Paper | GSE173682 | |
| scATAC-seq (processed data) | This Paper | GSE173682 | |
| scRNA-seq (raw data) | This Paper | phs002340.v1.p1 | |
| scATAC-seq (raw data) | This Paper | phs002340.v1.p1 | |
| Normal ovarian epithelial H3K27ac ChIP-seq peaks | Coetzee et. al., 2015 | GSE68104 | |
| Normal fallopian tube H3K27ac ChIP-seq peaks | Coetzee et. al., 2015 | GSE68104 | |
| Experimental models: Cell lines | |||
| Human: NIH:OVCAR-3 [OVCAR3] | ATCC | Cat#HTB-161, RRID: CVCL_0465 | |
| Human: HEK-293T | ATCC | Cat#CRL-3216, RRID: CVCL_0063 | |
| Experimental models: Organisms/strains | |||
| Human patients consented to participation in ‘Genomics of Ovarian and Endometrial Cancers’ study at the UNC Cancer Hospital (IRB Protocol 18–3198) | This Paper | Table 1, Table S1 | |
| Oligonucleotides | |||
| See Table S6, Table S7 and Table S8 | |||
| Recombinant DNA | |||
| Lenti-dCas9-KRAB-blast vector | Xie et al., 2017 | Addgene #89567 | |
| psPAX2 | Gift from Didier Trono | Addgene #12260 | |
| pMD2.G | Gift from Didier Trono | Addgene #12259 | |
| pSpCas9(BB)-2A-Puro (pX459) v2.0 | Ran et al., 2013 | Addgene #62988 | |
| pX-sgRNA-eGFP-MI | This paper | n/a | |
| Software and algorithms | |||
| Code used to analyze data presented in this paper | This Paper | 10.5281/zenodo.5546110 | |
| Prism (v9.0.0) | GraphPad | www.graphpad.com | |
| R (v4.0.2 or v4.0.3) | The R Project for Statistical Computing | https://www.r-project.org/ | |
| Seurat (v3.2.0 or v3.2.1) | Stuart et al., 2019 | https://satijalab.org/seurat/index.html | |
| ArchR (v0.9.5) | Granja et al., 2021 | https://www.archrproject.com/ | |
| mclust (v5.4.6 or v5.4.7) | Scrucca et al., 2016 | https://cran.r-project.org/web/packages/mclust/index.html | |
| scater (v1.17.5 or v1.18.6) | McCarthy et al., 2017 | https://bioconductor.org/packages/release/bioc/html/scater.html | |
| DESeq2 (v1.29.13 or v1.30.1) | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html | |
| inferCNV (v1.4.0 or v1.6.0) | Tickle, 2019 | http://www.bioconductor.org/packages/release/bioc/html/infercnv.html | |
| DoubletDecon (v1.1.5 or v1.1.6) | DePasquale et al., 2019 | https://github.com/EDePasquale/DoubletDecon | |
| DoubletFinder (v2.0.3) | McGinnis et al., 2019 | https://github.com/chris-mcginnis-ucsf/DoubletFinder | |
| GSVA (v1.36.1 or v1.36.2) | Hanzelmann et al., 2013 | http://bioconductor.org/packages/release/bioc/html/GSVA.html | |
| ggplot2 (v3.3.2 or v3.3.3) | Wickham, 2016 | https://cran.r-project.org/web/packages/ggplot2/index.html | |
| ComplexHeatmap (v2.4.3 or v2.6.2) | Gu et al., 2016 | https://jokergoo.github.io/ComplexHeatmap-reference/book/ | |
| Cell Ranger (v3.1.0) | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/installation | |
| Cell Ranger ATAC (v1.2.0) | 10x Genomics | https://support.10xgenomics.com/single-cell-atac/software/pipelines/latest/installation | |
| MEME suite (v4.12.0) | Bailey et al, 2009 | https://meme-suite.org/meme/index.html | |
| Python (v3.6.10) | Python Software Foundation | https://www.python.org/ | |
| Biopython (v1.78) | Python tools for computational biology | https://biopython.org / | |
| scikit-learn (v0.23.2) | Machine Learning in Python | https://scikit-learn.org/stable/ | |
| scipy (v1.5.2) | Fundamental Algorithms for Scientific Computing in Python | https://www.scipy.org/ | |
Highlights.
First matched scRNA-seq and scATAC-seq dataset of human gynecologic tumors
Rewiring of chromatin accessibility linked to transcriptional output in cancer cells
Identification of cancer-specific and clinically relevant distal regulatory elements
Differential transcription factor activity drives intratumor heterogeneity
ACKNOWLEDGEMENTS
We thank all patients and their families. We thank the UNC Tissue Procurement Facility and UNC Translational Genomics Core Facility for helping us acquire tumor specimens and sequence genomic libraries. We thank Michele Hayward at the Office of Genomics research for help in navigating the IRB and data submission process. We thank Dr. Yuchao Jiang for helpful discussion on statistical considerations needed for single-cell analysis. We thank Dr. Katie Hoadley and Dr. Steve Marron for insights into statistical considerations regarding pseudo-bulk clustering of patient tumors. We thank Olivia Brown in the UNC School of Medicine for helpful discussion on the clinical interpretation of our single-cell analysis. Finally, we thank members of the Franco Lab for their helpful comments and discussions.
This work was supported by grants from the NIH/National Cancer Institute (5-P50-CA058223-25), the Susan G. Komen Breast Cancer Research Foundation (CCR19608601), and the V Foundation for Cancer Research (V2019-015) to H.L.F. Additional support was provided by the She Rocks Foundation to V.B.J.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- AKDEMIR KC, LE VT, CHANDRAN S, LI Y, VERHAAK RG, BEROUKHIM R, CAMPBELL PJ, CHIN L, DIXON JR & FUTREAL PA 2020. Disruption of chromatin folding domains by somatic genomic rearrangements in human cancer. Nature genetics, 52, 294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARAN D, LOONEY AP, LIU L, WU E, FONG V, HSU A, CHAK S, NAIKAWADI RP, WOLTERS PJ, ABATE AR, BUTTE AJ & BHATTACHARYA M 2019. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol, 20, 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAILEY TL, BODEN M, BUSKE FA, FRITH M, GRANT CE, CLEMENTI L, REN J, LI WW & NOBLE WS 2009. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res, 37, W202–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAILEY TL, JOHNSON J, GRANT CE & NOBLE WS 2015. The MEME Suite. Nucleic Acids Res, 43, W39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BANERJI U 2014. A Phase I Trial of the Combination of AZD2014 and Weekly Paclitaxel [Online]. Available: https://ClinicalTrials.gov/show/NCT02193633 [Accessed].
- BARKER HE & SCOTT CL Genomics of gynaecological carcinosarcomas and future treatment options. Seminars in cancer biology, 2020. Elsevier, 110–120. [DOI] [PubMed] [Google Scholar]
- BERGER AC, KORKUT A, KANCHI RS, HEGDE AM, LENOIR W, LIU W, LIU Y, FAN H, SHEN H & RAVIKUMAR V 2018. A comprehensive pan-cancer molecular study of gynecologic and breast cancers. Cancer cell, 33, 690–705. e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BREMBECK FH, OPITZ OG, LIBERMANN TA & RUSTGI AK 2000. Dual function of the epithelial specific ets transcription factor, ELF3, in modulating differentiation. Oncogene, 19, 1941–1949. [DOI] [PubMed] [Google Scholar]
- BUENROSTRO JD, WU B, LITZENBURGER UM, RUFF D, GONZALES ML, SNYDER MP, CHANG HY & GREENLEAF WJ 2015. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature, 523, 486–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CANCER GENOME ATLAS RESEARCH, N. 2011. Integrated genomic analyses of ovarian carcinoma. Nature, 474, 609–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CANCER GENOME ATLAS RESEARCH, N., KANDOTH C, SCHULTZ N, CHERNIACK AD, AKBANI R, LIU Y, SHEN H, ROBERTSON AG, PASHTAN I, SHEN R, BENZ CC, YAU C, LAIRD PW, DING L, ZHANG W, MILLS GB, KUCHERLAPATI R, MARDIS ER & LEVINE DA 2013. Integrated genomic characterization of endometrial carcinoma. Nature, 497, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAO J, CUSANOVICH DA, RAMANI V, AGHAMIRZAIE D, PLINER HA, HILL AJ, DAZA RM, MCFALINE-FIGUEROA JL, PACKER JS, CHRISTIANSEN L, STEEMERS FJ, ADEY AC, TRAPNELL C & SHENDURE J 2018. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science, 361, 1380–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN B, GILBERT LA, CIMINI BA, SCHNITZBAUER J, ZHANG W, LI GW, PARK J, BLACKBURN EH, WEISSMAN JS, QI LS & HUANG B 2013. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell, 155, 1479–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN S, LAKE BB & ZHANG K 2019. High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nat Biotechnol, 37, 1452–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN Y-P, YIN J-H, LI W-F, LI H-J, CHEN D-P, ZHANG C-J, LV J-W, WANG Y-Q, LI X-M & LI J-Y 2020. Single-cell transcriptomics reveals regulators underlying immune cell diversity and immune subtypes associated with prognosis in nasopharyngeal carcinoma. Cell research, 30, 1024–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLAUSS A, NG V, LIU J, PIAO H, RUSSO M, VENA N, SHENG Q, HIRSCH MS, BONOME T & MATULONIS U 2010. Overexpression of elafin in ovarian carcinoma is driven by genomic gains and activation of the nuclear factor κB pathway and is associated with poor overall survival. Neoplasia, 12, 161-IN15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLEVERS H, RAFELSKI S, ELOWITZ M, KLEIN A, SHENDURE J, TRAPNELL C, LEIN E, LUNDBERG E, UHLEN M & MARTINEZ-ARIAS A 2017. What is your conceptual definition of “cell type” in the context of a mature organism? Cell Systems, 4, 255–259. [DOI] [PubMed] [Google Scholar]
- COCHRANE DR, CAMPBELL KR, GREENING K, HO GC, HOPKINS J, BUI M, DOUGLAS JM, SHARLANDJIEVA V, MUNZUR AD, LAI D, DEGROOD M, GIBBARD EW, LEUNG S, BOYD N, CHENG AS, CHOW C, LIM JL, FARNELL DA, KOMMOSS S, KOMMOSS F, ROTH A, HOANG L, MCALPINE JN, SHAH SP & HUNTSMAN DG 2020. Single cell transcriptomes of normal endometrial derived organoids uncover novel cell type markers and cryptic differentiation of primary tumours. J Pathol, 252, 201–214. [DOI] [PubMed] [Google Scholar]
- COETZEE SG, SHEN HC, HAZELETT DJ, LAWRENSON K, KUCHENBAECKER K, TYRER J, RHIE SK, LEVANON K, KARST A & DRAPKIN R 2015. Cell-type-specific enrichment of risk-associated regulatory elements at ovarian cancer susceptibility loci. Human molecular genetics, 24, 3595–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CONCORDET JP & HAEUSSLER M 2018. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res, 46, W242–W245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CONSORTIUM EP 2012. An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CONSORTIUM EP, MOORE JE, PURCARO MJ, PRATT HE, EPSTEIN CB, SHORESH N, ADRIAN J, KAWLI T, DAVIS CA, DOBIN A, KAUL R, HALOW J, VAN NOSTRAND EL, FREESE P, GORKIN DU, SHEN Y, HE Y, MACKIEWICZ M, PAULI-BEHN F, WILLIAMS BA, MORTAZAVI A, KELLER CA, ZHANG XO, ELHAJJAJY SI, HUEY J, DICKEL DE, SNETKOVA V, WEI X, WANG X, RIVERA-MULIA JC, ROZOWSKY J, ZHANG J, CHHETRI SB, ZHANG J, VICTORSEN A, WHITE KP, VISEL A, YEO GW, BURGE CB, LECUYER E, GILBERT DM, DEKKER J, RINN J, MENDENHALL EM, ECKER JR, KELLIS M, KLEIN RJ, NOBLE WS, KUNDAJE A, GUIGO R, FARNHAM PJ, CHERRY JM, MYERS RM, REN B, GRAVELEY BR, GERSTEIN MB, PENNACCHIO LA, SNYDER MP, BERNSTEIN BE, WOLD B, HARDISON RC, GINGERAS TR, STAMATOYANNOPOULOS JA & WENG Z 2020. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature, 583, 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CORCES MR, GRANJA JM, SHAMS S, LOUIE BH, SEOANE JA, ZHOU W, SILVA TC, GROENEVELD C, WONG CK, CHO SW, SATPATHY AT, MUMBACH MR, HOADLEY KA, ROBERTSON AG, SHEFFIELD NC, FELAU I, CASTRO MAA, BERMAN BP, STAUDT LM, ZENKLUSEN JC, LAIRD PW, CURTIS C, CANCER GENOME ATLAS ANALYSIS N, GREENLEAF WJ & CHANG HY 2018. The chromatin accessibility landscape of primary human cancers. Science, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COWARD JI, MIDDLETON K & MURPHY F 2015. New perspectives on targeted therapy in ovarian cancer. Int J Womens Health, 7, 189–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CUSANOVICH DA, DAZA R, ADEY A, PLINER HA, CHRISTIANSEN L, GUNDERSON KL, STEEMERS FJ, TRAPNELL C & SHENDURE J 2015. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science, 348, 910–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DANECEK P & MCCARTHY SA 2017. BCFtools/csq: haplotype-aware variant consequences. Bioinformatics, 33, 2037–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAS A, REIS F, MAEJIMA Y, CAI Z & REN J 2017. mTOR Signaling in Cardiometabolic Disease, Cancer, and Aging. Oxid Med Cell Longev, 2017, 6018675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAVIDSON S, EFREMOVA M, RIEDEL A, MAHATA B, PRAMANIK J, HUUHTANEN J, KAR G, VENTO-TORMO R, HAGAI T, CHEN X, HANIFFA MA, SHIELDS JD & TEICHMANN SA 2020. Single-Cell RNA Sequencing Reveals a Dynamic Stromal Niche That Supports Tumor Growth. Cell Rep, 31, 107628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEPASQUALE EAK, SCHNELL DJ, VAN CAMP PJ, VALIENTE-ALANDI I, BLAXALL BC, GRIMES HL, SINGH H & SALOMONIS N 2019. DoubletDecon: Deconvoluting Doublets from Single-Cell RNA-Sequencing Data. Cell Rep, 29, 1718–1727 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DONG C, LIU P & LI C 2017. Value of HE4 combined with cancer antigen 125 in the diagnosis of endometrial cancer. Pakistan journal of medical sciences, 33, 1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUFFY M, BONFRER J, KULPA J, RUSTIN G, SOLETORMOS G, TORRE G, TUXEN M & ZWIRNER M 2005. CA125 in ovarian cancer: European Group on Tumor Markers guidelines for clinical use. International Journal of Gynecologic Cancer, 15. [DOI] [PubMed] [Google Scholar]
- FORNES O, CASTRO-MONDRAGON JA, KHAN A, VAN DER LEE R, ZHANG X, RICHMOND PA, MODI BP, CORREARD S, GHEORGHE M, BARANAŠIĆ D, SANTANA-GARCIA W, TAN G, CHÈNEBY J, BALLESTER B, PARCY F, SANDELIN A, LENHARD B, WASSERMAN WW & MATHELIER A 2020. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res, 48, D87–D92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FRANCO HL, NAGARI A, MALLADI VS, LI W, XI Y, RICHARDSON D, ALLTON KL, TANAKA K, LI J, MURAKAMI S, KEYOMARSI K, BEDFORD MT, SHI X, BARTON MC, DENT SYR & KRAUS WL 2018. Enhancer transcription reveals subtype-specific gene expression programs controlling breast cancer pathogenesis. Genome Res, 28, 159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FRANZEN O, GAN LM & BJORKEGREN JLM 2019. PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database (Oxford), 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FULCO CP, MUNSCHAUER M, ANYOHA R, MUNSON G, GROSSMAN SR, PEREZ EM, KANE M, CLEARY B, LANDER ES & ENGREITZ JM 2016. Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science, 354, 769–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GEISTLINGER L, OH S, RAMOS M, SCHIFFER L, LARUE RS, HENZLER CM, MUNRO SA, DAUGHTERS C, NELSON AC, WINTERHOFF BJ, CHANG Z, TALUKDAR S, SHETTY M, MULLANY SA, MORGAN M, PARMIGIANI G, BIRRER MJ, QIN LX, RIESTER M, STARR TK & WALDRON L 2020. Multiomic Analysis of Subtype Evolution and Heterogeneity in High-Grade Serous Ovarian Carcinoma. Cancer Res, 80, 4335–4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GILBERT LA, LARSON MH, MORSUT L, LIU Z, BRAR GA, TORRES SE, STERN-GINOSSAR N, BRANDMAN O, WHITEHEAD EH, DOUDNA JA, LIM WA, WEISSMAN JS & QI LS 2013. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell, 154, 442–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GONZALEZ G, MEHRA S, WANG Y, AKIYAMA H & BEHRINGER RR 2016. Sox9 overexpression in uterine epithelia induces endometrial gland hyperplasia. Differentiation, 92, 204–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRANJA JM, CORCES MR, PIERCE SE, BAGDATLI ST, CHOUDHRY H, CHANG HY & GREENLEAF WJ 2021. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat Genet, 53, 403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRANJA JM, KLEMM S, MCGINNIS LM, KATHIRIA AS, MEZGER A, CORCES MR, PARKS B, GARS E, LIEDTKE M, ZHENG GXY, CHANG HY, MAJETI R & GREENLEAF WJ 2019. Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia. Nat Biotechnol, 37, 1458–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRANT CE, BAILEY TL & NOBLE WS 2011. FIMO: scanning for occurrences of a given motif. Bioinformatics, 27, 1017–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GU Z, EILS R & SCHLESNER M 2016. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics, 32, 2847–9. [DOI] [PubMed] [Google Scholar]
- GYORFFY B, LANCZKY A & SZALLASI Z 2012. Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocr Relat Cancer, 19, 197–208. [DOI] [PubMed] [Google Scholar]
- HANZELMANN S, CASTELO R & GUINNEY J 2013. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics, 14, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HELLSTRÖM I, RAYCRAFT J, HAYDEN-LEDBETTER M, LEDBETTER JA, SCHUMMER M, MCINTOSH M, DRESCHER C, URBAN N & HELLSTRÖM KE 2003. The HE4 (WFDC2) protein is a biomarker for ovarian carcinoma. Cancer research, 63, 3695–3700. [PubMed] [Google Scholar]
- HENLEY SJ, MILLER JW, DOWLING NF, BENARD VB & RICHARDSON LC 2018. Uterine cancer incidence and mortality—United States, 1999–2016. Morbidity and Mortality Weekly Report, 67, 1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IZAR B, TIROSH I, STOVER EH, WAKIRO I, CUOCO MS, ALTER I, RODMAN C, LEESON R, SU MJ, SHAH P, IWANICKI M, WALKER SR, KANODIA A, MELMS JC, MEI S, LIN JR, PORTER CBM, SLYPER M, WALDMAN J, JERBY-ARNON L, ASHENBERG O, BRINKER TJ, MILLS C, ROGAVA M, VIGNEAU S, SORGER PK, GARRAWAY LA, KONSTANTINOPOULOS PA, LIU JF, MATULONIS U, JOHNSON BE, ROZENBLATT-ROSEN O, ROTEM A & REGEV A 2020. A single-cell landscape of high-grade serous ovarian cancer. Nat Med, 26, 1271–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIM KH, CHOI JS, CHOI Y-L, SHIN YK, LEE H-C, SEONG IO, KIM BK, CHAE SW & KIM S-H 2009. Enhanced CD24 expression in endometrial carcinoma and its expression pattern in normal and hyperplastic endometrium. Histology and histopathology. [DOI] [PubMed] [Google Scholar]
- KIM N, KIM HK, LEE K, HONG Y, CHO JH, CHOI JW, LEE JI, SUH YL, KU BM, EUM HH, CHOI S, CHOI YL, JOUNG JG, PARK WY, JUNG HA, SUN JM, LEE SH, AHN JS, PARK K, AHN MJ & LEE HO 2020. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat Commun, 11, 2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIMES PK, LIU Y, NEIL HAYES D & MARRON JS 2017. Statistical significance for hierarchical clustering. Biometrics, 73, 811–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOSTOV S, KORNOVSKI Y, IVANOVA Y, DZHENKOV D, STOYANOV G, STOILOV S, SLAVCHEV S, TRENDAFILOVA E & YORDANOV A 2020. Ovarian Carcinosarcoma with Retroperitoneal Para-Aortic Lymph Node Dissemination Followed by an Unusual Postoperative Complication: A Case Report with a Brief Literature Review. Diagnostics, 10, 1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LABIDI-GALY SI, CLAUSS A, NG V, DURAISAMY S, ELIAS KM, PIAO H-Y, BILAL E, DAVIDOWITZ RA, LU Y & BADALIAN-VERY G 2015. Elafin drives poor outcome in high-grade serous ovarian cancers and basal-like breast tumors. Oncogene, 34, 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAMBRECHTS D, WAUTERS E, BOECKX B, AIBAR S, NITTNER D, BURTON O, BASSEZ A, DECALUWE H, PIRCHER A, VAN DEN EYNDE K, WEYNAND B, VERBEKEN E, DE LEYN P, LISTON A, VANSTEENKISTE J, CARMELIET P, AERTS S & THIENPONT B 2018. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med, 24, 1277–1289. [DOI] [PubMed] [Google Scholar]
- LARSON MH, GILBERT LA, WANG X, LIM WA, WEISSMAN JS & QI LS 2013. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat Protoc, 8, 2180–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAWHORN IE, FERREIRA JP & WANG CL 2014. Evaluation of sgRNA target sites for CRISPR-mediated repression of TP53. PLoS One, 9, e113232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAWRENCE M, HUBER W, PAGES H, ABOYOUN P, CARLSON M, GENTLEMAN R, MORGAN MT & CAREY VJ 2013. Software for computing and annotating genomic ranges. PLoS computational biology, 9, e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI J, DOWDY S, TIPTON T, PODRATZ K, LU W-G, XIE X & JIANG S-W 2009. HE4 as a biomarker for ovarian and endometrial cancer management. Expert review of molecular diagnostics, 9, 555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI L, WEI XH, PAN YP, LI HC, YANG H, HE QH, PANG Y, SHAN Y, XIONG FX, SHAO GZ & ZHOU RL 2010. LAPTM4B: a novel cancer-associated gene motivates multidrug resistance through efflux and activating PI3K/AKT signaling. Oncogene, 29, 5785–95. [DOI] [PubMed] [Google Scholar]
- LIBERZON A, BIRGER C, THORVALDSDOTTIR H, GHANDI M, MESIROV JP & TAMAYO P 2015. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst, 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIN C-Y, TSAI C-L, CHAO A, LEE L-Y, CHEN W-C, TANG Y-H, CHAO A-S & LAI C-H 2021. Nucleophosmin/B23 promotes endometrial cancer cell escape from macrophage phagocytosis by increasing CD24 expression. Journal of Molecular Medicine, 1–13. [DOI] [PubMed] [Google Scholar]
- LIU T 2014. Use model-based Analysis of ChIP-Seq (MACS) to analyze short reads generated by sequencing protein-DNA interactions in embryonic stem cells. Methods Mol Biol, 1150, 81–95. [DOI] [PubMed] [Google Scholar]
- LORTET-TIEULENT J, FERLAY J, BRAY F & JEMAL A 2018. International Patterns and Trends in Endometrial Cancer Incidence, 1978–2013. J Natl Cancer Inst, 110, 354–361. [DOI] [PubMed] [Google Scholar]
- LOVE MI, HUBER W & ANDERS S 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol, 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUN AT & SMYTH GK 2016. csaw: a Bioconductor package for differential binding analysis of ChIP-seq data using sliding windows. Nucleic Acids Res, 44, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MA S, ZHANG B, LAFAVE LM, EARL AS, CHIANG Z, HU Y, DING J, BRACK A, KARTHA VK, TAY T, LAW T, LAREAU C, HSU YC, REGEV A & BUENROSTRO JD 2020. Chromatin Potential Identified by Shared Single-Cell Profiling of RNA and Chromatin. Cell, 183, 1103–1116 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MACINTYRE G, GORANOVA TE, DE SILVA D, ENNIS D, PISKORZ AM, ELDRIDGE M, SIE D, LEWSLEY LA, HANIF A, WILSON C, DOWSON S, GLASSPOOL RM, LOCKLEY M, BROCKBANK E, MONTES A, WALTHER A, SUNDAR S, EDMONDSON R, HALL GD, CLAMP A, GOURLEY C, HALL M, FOTOPOULOU C, GABRA H, PAUL J, SUPERNAT A, MILLAN D, HOYLE A, BRYSON G, NOURSE C, MINCARELLI L, SANCHEZ LN, YLSTRA B, JIMENEZLINAN M, MOORE L, HOFMANN O, MARKOWETZ F, MCNEISH IA & BRENTON JD 2018. Copy number signatures and mutational processes in ovarian carcinoma. Nat Genet, 50, 1262–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAINTAINER BP 2020. liftOver: Changing genomic coordinate systems with rtracklayer:: liftOver. R package version, 1. [Google Scholar]
- MALLADI VS, NAGARI A, FRANCO HL & KRAUS WL 2020. Total Functional Score of Enhancer Elements Identifies Lineage-Specific Enhancers That Drive Differentiation of Pancreatic Cells. Bioinform Biol Insights, 14, 1177932220938063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MANSOUR MR, ABRAHAM BJ, ANDERS L, BEREZOVSKAYA A, GUTIERREZ A, DURBIN AD, ETCHIN J, LAWTON L, SALLAN SE, SILVERMAN LB, LOH ML, HUNGER SP, SANDA T, YOUNG RA & LOOK AT 2014. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science, 346, 1373–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCCARTHY DJ, CAMPBELL KR, LUN AT & WILLS QF 2017. Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics, 33,1179–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCGINNIS CS, MURROW LM & GARTNER ZJ 2019. DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst, 8, 329–337.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MILLS C, MURUGANUJAN A, EBERT D, MARCONETT CN, LEWINGER JP, THOMAS PD & MI H 2020. PEREGRINE: A genome-wide prediction of enhancer to gene relationships supported by experimental evidence. PloS one, 15, e0243791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MITSOPOULOS C, DI MICCO P, FERNANDEZ EV, DOLCIAMI D, HOLT E, MICA IL, COKER EA, TYM JE, CAMPBELL J, CHE KH, OZER B, KANNAS C, ANTOLIN AA, WORKMAN P & ALLAZIKANI B 2020. canSAR: update to the cancer translational research and drug discovery knowledgebase. Nucleic Acids Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOORE JE, PRATT HE, PURCARO MJ & WENG Z 2020. A curated benchmark of enhancer-gene interactions for evaluating enhancer-target gene prediction methods. Genome biology, 21, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAGY A, LANCZKY A, MENYHART O & GYORFFY B 2018. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci Rep, 8, 9227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAGY Á, MUNKÁCSY G & GYŐRFFY B 2021. Pancancer survival analysis of cancer hallmark genes. Sci Rep, 11, 6047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OHNISHI T, OHBA H, SEO K-C, IM J, SATO Y, IWAYAMA Y, FURUICHI T, CHUNG S-K & YOSHIKAWA T 2007. Spatial expression patterns and biochemical properties distinguish a second myo-inositol monophosphatase IMPA2 from IMPA1. Journal of Biological Chemistry, 282, 637–646. [DOI] [PubMed] [Google Scholar]
- OLBRECHT S, BUSSCHAERT P, QIAN J, VANDERSTICHELE A, LOVERIX L, VAN GORP T, VAN NIEUWENHUYSEN E, HAN S, VAN DEN BROECK A & COOSEMANS A 2021. High-grade serous tubo-ovarian cancer refined with single-cell RNA sequencing: specific cell subtypes influence survival and determine molecular subtype classification. Genome Medicine, 13, 1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PANDEY V, JUNG Y, KANG J, STEINER M, QIAN P-X, BANERJEE A, MITCHELL MD, WU Z-S, ZHU T & LIU D-X 2010. Artemin reduces sensitivity to doxorubicin and paclitaxel in endometrial carcinoma cells through specific regulation of CD24. Translational oncology, 3, 218-IN5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PATEL AP, TIROSH I, TROMBETTA JJ, SHALEK AK, GILLESPIE SM, WAKIMOTO H, CAHILL DP, NAHED BV, CURRY WT, MARTUZA RL, LOUIS DN, ROZENBLATT-ROSEN O, SUVA ML, REGEV A & BERNSTEIN BE 2014. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science, 344, 1396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QI LS, LARSON MH, GILBERT LA, DOUDNA JA, WEISSMAN JS, ARKIN AP & LIM WA 2013. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell, 152, 1173–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUINLAN AR & HALL IM 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics, 26, 841–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAN FA, HSU PD, WRIGHT J, AGARWALA V, SCOTT DA & ZHANG F 2013. Genome engineering using the CRISPR-Cas9 system. Nat Protoc, 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RITTERHOUSE LL & HOWITT BE 2016. Molecular pathology: predictive, prognostic, and diagnostic markers in uterine tumors. Surgical pathology clinics, 9, 405–426. [DOI] [PubMed] [Google Scholar]
- ROADMAP EPIGENOMICS C, KUNDAJE A, MEULEMAN W, ERNST J, BILENKY M, YEN A, HERAVI-MOUSSAVI A, KHERADPOUR P, ZHANG Z, WANG J, ZILLER MJ, AMIN V, WHITAKER JW, SCHULTZ MD, WARD LD, SARKAR A, QUON G, SANDSTROM RS, EATON ML, WU YC, PFENNING AR, WANG X, CLAUSSNITZER M, LIU Y, COARFA C, HARRIS RA, SHORESH N, EPSTEIN CB, GJONESKA E, LEUNG D, XIE W, HAWKINS RD, LISTER R, HONG C, GASCARD P, MUNGALL AJ, MOORE R, CHUAH E, TAM A, CANFIELD TK, HANSEN RS, KAUL R, SABO PJ, BANSAL MS, CARLES A, DIXON JR, FARH KH, FEIZI S, KARLIC R, KIM AR, KULKARNI A, LI D, LOWDON R, ELLIOTT G, MERCER TR, NEPH SJ, ONUCHIC V, POLAK P, RAJAGOPAL N, RAY P, SALLARI RC, SIEBENTHALL KT, SINNOTT-ARMSTRONG NA, STEVENS M, THURMAN RE, WU J, ZHANG B, ZHOU X, BEAUDET AE, BOYER LA, DE JAGER PL, FARNHAM PJ, FISHER SJ, HAUSSLER D, JONES SJ, LI W, MARRA MA, MCMANUS MT, SUNYAEV S, THOMSON JA, TLSTY TD, TSAI LH, WANG W, WATERLAND RA, ZHANG MQ, CHADWICK LH, BERNSTEIN BE, COSTELLO JF, ECKER JR, HIRST M, MEISSNER A, MILOSAVLJEVIC A, REN B, STAMATOYANNOPOULOS JA, WANG T & KELLIS M 2015. Integrative analysis of 111 reference human epigenomes. Nature, 518, 317–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROE JS, HWANG CI, SOMERVILLE TDD, MILAZZO JP, LEE EJ, DA SILVA B, MAIORINO L, TIRIAC H, YOUNG CM, MIYABAYASHI K, FILIPPINI D, CREIGHTON B, BURKHART RA, BUSCAGLIA JM, KIM EJ, GREM JL, LAZENBY AJ, GRUNKEMEYER JA, HOLLINGSWORTH MA, GRANDGENETT PM, EGEBLAD M, PARK Y, TUVESON DA & VAKOC CR 2017. Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell, 170, 875–888 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAEGUSA M, HASHIMURA M, SUZUKI E, YOSHIDA T & KUWATA T 2012. Transcriptional up-regulation of Sox9 by NF-κB in endometrial carcinoma cells, modulating cell proliferation through alteration in the p14ARF/p53/p21WAF1 pathway. The American journal of pathology, 181, 684–692. [DOI] [PubMed] [Google Scholar]
- SARLOMO-RIKALA M, KOVATICH AJ, BARUSEVICIUS A & MIETTINEN M 1998. CD117: a sensitive marker for gastrointestinal stromal tumors that is more specific than CD34. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc, 11, 728–734. [PubMed] [Google Scholar]
- SATPATHY AT, GRANJA JM, YOST KE, QI Y, MESCHI F, MCDERMOTT GP, OLSEN BN, MUMBACH MR, PIERCE SE, CORCES MR, SHAH P, BELL JC, JHUTTY D, NEMEC CM, WANG J, WANG L, YIN Y, GIRESI PG, CHANG ALS, ZHENG GXY, GREENLEAF WJ & CHANG HY 2019. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat Biotechnol, 37, 925–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCRUCCA L, FOP M, MURPHY TB & RAFTERY AE 2016. mclust 5: Clustering, Classification and Density Estimation Using Gaussian Finite Mixture Models. R J, 8, 289–317. [PMC free article] [PubMed] [Google Scholar]
- SENGEZ B, AYGÜN I, SHEHWANA H, TOYRAN N, TERCAN AVCI S, KONU O, STEMMLER MP & ALOTAIBI H 2019. The transcription factor Elf3 is essential for a successful mesenchymal to epithelial transition. Cells, 8, 858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIEGEL RL, MILLER KD, FUCHS HE & JEMAL A 2021. Cancer statistics, 2021. CA: a cancer journal for clinicians, 71, 7–33. [DOI] [PubMed] [Google Scholar]
- SIEGEL RL, MILLER KD & JEMAL A 2018. Cancer statistics, 2018. CA Cancer J Clin, 68, 7–30. [DOI] [PubMed] [Google Scholar]
- SLYPER M, PORTER CBM, ASHENBERG O, WALDMAN J, DROKHLYANSKY E, WAKIRO I, SMILLIE C, SMITH-ROSARIO G, WU J, DIONNE D, VIGNEAU S, JANE-VALBUENA J, TICKLE TL, NAPOLITANO S, SU MJ, PATEL AG, KARLSTROM A, GRITSCH S, NOMURA M, WAGHRAY A, GOHIL SH, TSANKOV AM, JERBY-ARNON L, COHEN O, KLUGHAMMER J, ROSEN Y, GOULD J, NGUYEN L, HOFREE M, TRAMONTOZZI PJ, LI B, WU CJ, IZAR B, HAQ R, HODI FS, YOON CH, HATA AN, BAKER SJ, SUVA ML, BUENO R, STOVER EH, CLAY MR, DYER MA, COLLINS NB, MATULONIS UA, WAGLE N, JOHNSON BE, ROTEM A, ROZENBLATT-ROSEN O & REGEV A 2020. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat Med, 26, 792–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOCIETY AC 2016. Cancer facts & figures. American Cancer Society. [Google Scholar]
- STOREY JD & TIBSHIRANI R 2003. Statistical significance for genomewide studies. Proceedings of the National Academy of Sciences, 100, 9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STUART T, BUTLER A, HOFFMAN P, HAFEMEISTER C, PAPALEXI E, MAUCK WM 3RD, HAO Y, STOECKIUS M, SMIBERT P & SATIJA R 2019. Comprehensive Integration of Single-Cell Data. Cell, 177, 1888–1902 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STURGEON CM, DUFFY MJ, STENMAN U-H, LILJA H, BRUNNER N, CHAN DW, BABAIAN R, BAST JR RC, DOWELL B & ESTEVA FJ 2008. National Academy of Clinical Biochemistry laboratory medicine practice guidelines for use of tumor markers in testicular, prostate, colorectal, breast, and ovarian cancers. Oxford University Press. [DOI] [PubMed] [Google Scholar]
- SZASZ AM, LANCZKY A, NAGY A, FORSTER S, HARK K, GREEN JE, BOUSSIOUTAS A, BUSUTTIL R, SZABO A & GYORFFY B 2016. Cross-validation of survival associated biomarkers in gastric cancer using transcriptomic data of 1,065 patients. Oncotarget, 7, 49322–49333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SÁNCHEZ-TILLÓ E, SILES L, DE BARRIOS O, CUATRECASAS M, VAQUERO EC, CASTELLS A & POSTIGO A 2011. Expanding roles of ZEB factors in tumorigenesis and tumor progression. American journal of cancer research, 1, 897. [PMC free article] [PubMed] [Google Scholar]
- TAN X, SUN Y, THAPA N, LIAO Y, HEDMAN AC & ANDERSON RA 2015. LAPTM4B is a PtdIns(4,5)P2 effector that regulates EGFR signaling, lysosomal sorting, and degradation. EMBO J, 34, 475–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TEAM RC 2020. R: A Language and Environment for Statistical Computing. [Google Scholar]
- TICKLE TI, GEORGESCU C, BROWN M & HAAS B 2019. inferCNV of the Trinity CTAT Project [Online]. Available: https://github.com/broadinstitute/inferCNV [Accessed].
- TYM JE, MITSOPOULOS C, COKER EA, RAZAZ P, SCHIERZ AC, ANTOLIN AA & AL-LAZIKANI B 2016. canSAR: an updated cancer research and drug discovery knowledgebase. Nucleic Acids Res, 44, D938–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG W, VILELLA F, ALAMA P, MORENO I, MIGNARDI M, ISAKOVA A, PAN W, SIMON C. & QUAKE SR 2020. Single-cell transcriptomic atlas of the human endometrium during the menstrual cycle. Nat Med, 26, 1644–1653. [DOI] [PubMed] [Google Scholar]
- WATANABE K, PANCHY N, NOGUCHI S, SUZUKI H. & HONG T. 2019. Combinatorial perturbation analysis reveals divergent regulations of mesenchymal genes during epithelial-to-mesenchymal transition. NPJ systems biology and applications, 5, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEI H, HELLSTRÖM KE & HELLSTRÖM I. 2012. Elafin selectively regulates the sensitivity of ovarian cancer cells to genotoxic drug-induced apoptosis. Gynecologic oncology, 125, 727–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEINTRAUB AS, LI CH, ZAMUDIO AV, SIGOVA AA, HANNETT NM, DAY DS, ABRAHAM BJ, COHEN MA, NABET B, BUCKLEY DL, GUO YE, HNISZ D, JAENISCH R, BRADNER JE, GRAY NS & YOUNG RA 2017. YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell, 171, 1573–1588.e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WESTIN SN 2014. mTORC1/2 Inhibitor AZD2014 or the Oral AKT Inhibitor AZD5363 for Recurrent Endometrial and Ovarian [Online]. Available: https://ClinicalTrials.gov/show/NCT02208375 [Accessed].
- WICKHAM H. 2016. ggplot2: Elegant Graphics for Data Analysis, Springer-Verlag; New York. [Google Scholar]
- WILLIAMS J, LUCAS PC, GRIFFITH KA, CHOI M, FOGOROS S, HU YY & LIU JR 2005. Expression of Bcl-xL in ovarian carcinoma is associated with chemoresistance and recurrent disease. Gynecologic oncology, 96, 287–295. [DOI] [PubMed] [Google Scholar]
- XIE S, DUAN J, LI B, ZHOU P. & HON GC 2017. Multiplexed Engineering and Analysis of Combinatorial Enhancer Activity in Single Cells. Mol Cell, 66, 285–299.e5. [DOI] [PubMed] [Google Scholar]
- XU C. & SU Z. 2015. Identification of cell types from single-cell transcriptomes using a novel clustering method. Bioinformatics, 31, 1974–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG H, JIANG X, LI B, YANG HJ, MILLER M, YANG A, DHAR A. & PAVLETICH NP 2017. Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature, 552, 368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YOSHIHARA K, SHAHMORADGOLI M, MARTINEZ E, VEGESNA R, KIM H, TORRES-GARCIA W, TREVINO V, SHEN H, LAIRD PW, LEVINE DA, CARTER SL, GETZ G, STEMKE-HALE K, MILLS GB & VERHAAK RG 2013. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun, 4, 2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG K, LIU L, WANG M, YANG M, LI X, XIA X, TIAN J, TAN S. & LUO L. 2020. A novel function of IMPA2, plays a tumor-promoting role in cervical cancer. Cell death & disease, 11, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG X, CHOI PS, FRANCIS JM, IMIELINSKI M, WATANABE H, CHERNIACK AD & MEYERSON M. 2016. Identification of focally amplified lineage-specific super-enhancers in human epithelial cancers. Nat Genet, 48, 176–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG Y, LIU T, MEYER CA, EECKHOUTE J, JOHNSON DS, BERNSTEIN BE, NUSBAUM C, MYERS RM, BROWN M, LI W. & LIU XS 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol, 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHU LJ, GAZIN C, LAWSON ND, PAGÈS H, LIN SM, LAPOINTE DS & GREEN MR 2010. ChIPpeakAnno: a Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC bioinformatics, 11, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Extended clinical data and library information for 11 gynecologic tumor specimens, Related to Table 1. (Table_S1_clinical_data.xlsx)
Table S2. scRNA-seq barcode metadata, clustering, and cell type annotations, Related to Figures 1, 3, and 4.(Table_S2_scRNA_metadata.xlsx)
Table S3. scATAC-seq barcode metadata, clustering, and inferred cell type annotations, Related to Figures 1, 3, and 4. (Table_S3_scATAC_metadata.xlsx)
Table S4. Kaplan-Meier data summary and associated metadata with directions to reproduce the analyses on kmplot.com, Related to STAR Methods. (Table_S4_KM_metadata.xlsx)
Table S5. FIMO transcription factor motif scanning results for the LAPTM4B enhancers 1–5 and promoter in high-grade serous ovarian cancer, Related to Figure 4. (Table_S5_ranked_FIMO_results.xlsx)
There are three sets of files for each cohort of patients in this study: 1) statistically significant peak-to-gene links with all peak types and no correlation thresholding, 2) statistically significant distal peak-to-gene links with correlation >= 0.45, and 3) statistically significant cancer-specific distal peak-to-gene links with correlation >= 0.45. (Data_S1_Peak_to_Gene_Links.tsv.gz)
Data Availability Statement
Processed single-cell RNA-seq data and single-cell ATAC-seq have been deposited at GEO(https://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE173682 and are publicly available as of the date of publication. Raw data (10x FASTQs) will be available with controlled access via dbGAP under the accession number phs002340.v1.p1 (https://www.ncbi.nlm.nih.gov/gap/).
All original code has been deposited on the Zenodo platform (DOI: 10.5281/zenodo.5546110) and is publicly available at the Github repository scENDO_scOVAR_2020 (https://github.com/RegnerM2015/scENDO_scOVAR_2020).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact (hfranco@med.unc.edu).