

Abstract
Exposure to carcinogenic alkylating agents, oxidizing agents, and ionizing radiation modulates transcript levels for over one third of Saccharomyces cerevisiae's 6,200 genes. Computational analysis delineates groups of coregulated genes whose upstream regions bear known and novel regulatory sequence motifs. One group of coregulated genes contain a number of DNA excision repair genes (including the MAG1 3-methyladenine DNA glycosylase gene) and a large selection of protein degradation genes. Moreover, transcription of these genes is modulated by the proteasome-associated protein Rpn4, most likely via its binding to MAG1 upstream repressor sequence 2-like elements, that turn out to be almost identical to the recently identified proteasome-associated control element (G. Mannhaupt, R. Schnall, V. Karpov, I. Vetter, and H. Feldmann, FEBS Lett. 450:27–34, 1999). We have identified a large number of genes whose transcription is influenced by Rpn4p.
Biological processes depend upon the structural integrity of the molecules that comprise living organisms. The structural integrity of the genome is particularly important because molecular alterations in the genetic material, usually DNA, can lead to permanent inheritable changes, i.e., mutations. However, the structural integrity of other cellular molecules, such as proteins, RNA, carbohydrates, and lipids, is also important, because the precise three-dimensional shape and the detailed chemistry of these molecules orchestrate the biochemical processes vital for life. Most biomolecules are inherently reactive, and as such their structural integrity is constantly challenged by reactive chemical and physical agents in the environment. It should therefore come as no surprise that all cells can sense and respond to unfavorable molecular alterations. Indeed, it is well known that cells sense and respond to damaged DNA and proteins, and such responses are exemplified by the SOS and heat shock responses that have been well characterized in Escherichia coli and other organisms (11, 12, 28).
Here we explore the transcriptional response of Saccharomyces cerevisiae to a wide range of chemical and physical damaging agents. Specifically, we explore how transcript levels for every S. cerevisiae gene and open reading frame (ORF) respond when cellular molecules are damaged by a selection of environmentally and clinically relevant chemical and physical carcinogens. The global transcriptional response of this budding yeast to these damaging agents turns out to be far more extensive than anticipated. However, computational analysis of almost 200,000 data points reveals patterns in the data that allow us to define novel regulatory networks. We find that the responses of S. cerevisiae to each of six damaging agents are markedly different and that, for at least one agent, the response is dramatically affected by the cell's position in the cell cycle at the time of exposure. Computational clustering of the data and subsequent searching for common sequence motifs in promoter regions reveal nine such motifs, only five of which have known binding factors. Furthermore, we find that a large number of protein degradation genes and a selection of base excision and nucleotide excision DNA repair genes are linked in a transcriptional regulon controlled by Rpn4p, a proteasome-associated protein (22).
MATERIALS AND METHODS
Strains, media, growth conditions, and damaging-agent exposure.
Log-phase S. cerevisiae strain DBY747 (MATa his3-Δ1 leu2-3,112 ura3-52 trp1-289a gal2 can1 CUP1s) or BY4740 and its Δrpn4 derivative (MATa ura3Δ0 lys2Δ0 leu2Δ0 Δrpn4) was grown to a density of 5 × 106 cells per ml in 1% yeast extract–2% peptone–2% glucose. Cultures were split in two, and methyl methanesulfonate (MMS) was added to 0.1% to one half. Incubation was continued for 10, 30, or 60 min. For one experiment, MMS was added to 0.05, 0.1 or 0.2% and incubated for 1 h. For the other agents, log-phase cells were grown to a density of 5 × 106 cells per ml in 1% yeast extract–2% peptone–2% glucose. Cultures were split, and N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) (6.7 or 27 μg/ml), mitomycin C (MMC) (2 μg/ml), 4-nitroquinoline n-oxide (4NQO) (2 or 8 μg/ml), 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) (200 μM), or tert-butyl hydroperoxide (t-BuOOH) (5 mM) was added, and incubation was continued for 60 min. For cell synrochony, log-phase cells were arrested in G1 by α-factor (3 μM for 120 min), in S phase by hydroxyurea (0.1 M for 210 min), and in G2 by nocodazole (15 μg/ml for 120 min). Stationary-phase cells were harvested after 3 days of growth (5 × 108 cells/ml). Arrested cells were confirmed by microscopy and by fluorescence-activated cell sorting analysis (data not shown) and split in two, and 0.1% MMS was added to half the cultures for 1 h before RNA isolation.
GeneChip hybridizations.
RNA isolation and purification and cRNA labeling were done as described (17). Hybridizations with a set of four oligonucleotide arrays (GeneChip Ye6100 arrays; Affymetrix, Santa Clara, Calif.) containing probes for 6,218 yeast ORFs were done at 45°C for 16 h with constant mixing in 200 μl of MES buffer (100 mM MES [morpholimeethanesulfonic acid], 1 M Na+, 20 mM EDTA, 0.01% Tween 20) with 10 μg of labeled cRNA. After hybridization, arrays were washed in nonstringent wash A buffer (6× SSPE [1× SSPE is 0.18 M NaCl, 10 mM NaH2 PO4, and 1 mM EDTA (pH 7.7)], 0.01% Tween 20, 0.005% antifoam, 25°C) followed by stringent wash B buffer (100 mM MES, 0.1 M Na+, 0.01% Tween 20, 50°C). Arrays were then stained with strepavidin-phycoerythrin (30 min, 25°C; Molecular Probes), followed by rinsing with wash A buffer. Arrays were stained with R-phycoerythrin-streptavidin (10 μg/ml; Molecular Probes) in 100 mM MES–1.0 M Na+–0.01% Tween 20 at 25°C. All washes were automated on a fluidics station (Affymetrix). Arrays were scanned using a specialized confocal laser scanning microscope (Hewlett Packard or Molecular Dynamics) and analyzed using the GeneChip analysis suite, version 3.1. All arrays were scaled so that the average of the average intensity difference of the perfect match probes minus the mismatch probes was 300. This scaling allowed all the arrays to be directly compared with each other. Integrity of the sample was determined by measuring the intensity of probes derived from both the 3′ and 5′ ends of actin and TATA-binding protein. The signal from probes corresponding to the 5′ end was not less then twofold of the intensity of probes derived from the 3′ end of the gene in any sample. These measurements suggest that the mRNA is not more degraded in the treated samples than in the controls.
Determination of fold change.
Three control untreated log-phase samples were hybridized to three different sets of GeneChip arrays. A baseline value was determined by averaging the hybridization intensity from the three control experiments. Each gene has approximately 20 pairs of oligonucleotide probes; within each pair, one is a perfect match to the gene and one has a mismatch. Hybridization intensity was determined by calculating the average intensity difference between the perfect match signal and the mismatch signal across the 20 pairs of probes. Fold changes were calculated by dividing the average intensity difference values from experimental samples by the baseline values. Note that in our original report we arbitrarily chose fourfold as the cutoff for induction and threefold as the cutoff for repression (17). Given an improved algorithm for calculating hybridization intensities, we now adopt threefold as the cutoff for both induction and repression. Accordingly, the number of genes categorized as responsive in this study has increased compared to our previous study.
Cluster analysis.
All genes showing a change of 3.0-fold or more in at least one experimental condition were included in the analysis. Three control untreated experiments were performed. The baseline intensity value was calculated as the average of the three average difference values. For each treatment, the fold change was determined by dividing the average intensity difference from the experimental sample by the baseline intensity. The average difference was determined using GeneChip analysis suite 3.1 software (Affymetrix). Although some genes have less than the ideal number of probes, they were still included in the analysis. Cluster analysis was performed using one of three methods: Euclidean distances (34), hierarchical (10), or self-organized maps (SOMs) (33). For Euclidean distance measurements, log-transformed fold changes was arbitrarily clustered into groups of genes having similar expression profiles. Hierarchical clustering was done as described (10). The third method was based on SOMs using Genecluster 1.0 (33). A. filter was used to eliminate genes which had a relative change of less than 3.0-fold and whose expression level were less than 60 across all treatments. Expression levels were normalized to have a mean of 0 and a variance of 1, which forced genes to be grouped based on the shape of their expression pattern rather than on their absolute values (33). The number of clusters was chosen to give the largest number of fundamentally different patterns.
On the arrays, many genes are represented by more than one set of probes. In order to accurately determine the distribution of functional categories present, only one set of probes was used for each gene. When more than one set was present, sets containing less than the ideal number of probes were eliminated. In other cases the probe set with the higher signal was included.
Statistical calculation.
To determine if fold changes were statistically significant between the triplicate experiments, a Student t test was performed in Microsoft Excel using a two-tailed distribution. To determine if SOMs produced distinct clusters, correlation coefficients were determined from the mean of each group. Correlation coefficients p(x,y) were determined using Microsoft Excel with the formula
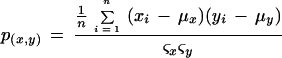 |
where x,y represents any pair of clusters, xi or yi equals the value of treatment i in cluster x or y, μx and μy equal the means of the normalized intensities across the 26 treatments in clusters x and y, and ςx and ςy equal the standard deviations of the normalized intensities across the 26 treatments in clusters x and y, respectively.
Only four groups showed significant similarity. They included clusters 1 and 4 (0.86), clusters 14 and 17 (0.83), clusters 17 and 15 (0.78), and clusters 9 and 12 (0.79). Hypergeometric distribution [P(x)] was used to determine the chance probability of observing the number of genes of a particular function category or with a particular upstream motif within each cluster, as described (34); calculations were determined using the formula
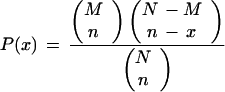 |
where x is the number of genes in a functional category or with a motif in a defined cluster, n is the total number of genes in the cluster, M is the total number of genes in a functional category or with a particular motif in the dataset, and N is the total number of genes in the dataset.
RESULTS AND DISCUSSION
Reproducibility of transcriptional profiling by oligonucleotide DNA microarray analysis.
We previously used Affymetrix GeneChip oligonucleotide arrays to characterize the global transcriptional response of S. cerevisiae upon exposure to a mildly toxic dose of a monofunctional SN2 alkylating agent, MMS (17). MMS is typical of a large class of reactive chemicals present in the air we breathe and the food we eat, as well as being representative of some normal cellular metabolites (23). To our surprise, transcript levels for roughly 400 of S. cerevisiae's ∼6,200 genes were responsive to MMS exposure; ∼300 genes were induced by 4- to 250-fold, and ∼100 genes were repressed by 3- to 18-fold (17). Before undertaking a much larger study, we assessed the reproducibility of the transcriptional responsiveness measured by GeneChip analysis. Six separate S. cerevisiae cultures were grown to mid-log phase; three were exposed to 0.1% MMS for 60 min, and three remained untreated. cRNA prepared from each culture was hybridized to the GeneChip arrays as described above (17, 38). Figure 1a displays the range of hybridization intensities obtained for a representative selection of 69 MMS-inducible and 31 MMS-repressible genes; note that in virtually no instance do the error bars (representing standard deviations) for treated and untreated come close to overlapping. Figures 1b and c display individual hybridization intensities for the 693 genes whose transcripts changed by threefold or more upon MMS treatment (the identities of these genes and the raw data can be found at www.hsph.harvard.edu/geneexpression.) Individual values for the three untreated cultures are plotted against their averages in Fig. 1b, and individual values for the three MMS-treated cultures are plotted against their averages in Fig. 1c; this provides a graphic representation of the variation between experiments, viewing the untreated and treated groups separately. Figure 1d displays the average hybridization intensities for the 693 responsive genes, but here the average values for the MMS-treated cultures are plotted against those for the untreated cultures to provide a graphic representation of where the 693 responsive genes fall in the 3- to 217-fold range of MMS responsiveness. The data in Fig. 1 confirm that, in our hands, transcript levels measured by GeneChip analysis are highly reproducible and show that transcript levels observed in MMS-treated cells are likewise reproducible. Furthermore, the changes in transcript level are also reproducible, and 648 of the 693 responsive genes (94%) showed a statistically significant change at a 90% confidence level. We are therefore confident that our estimate of such a surprisingly large number of MMS-responsive genes is reliable.
FIG. 1.

Reproducibility of mRNA profiling by GeneChip analysis. Hybridization intensities for transcripts from three untreated log-phase samples and three 0.1% MMS-treated samples. (a) Hybridization intensities for 100 MMS-responsive genes; symbols represent basal transcript intensity, averaged from three untreated cultures, ± standard deviation. Green and red symbols represent values for the three MMS-treated cultures, with green for induced and red for repressed. (b) Intensity of hybridization signals for 693 MMS-responsive genes from three independent untreated cultures versus the average of their intensity values. (c) Intensity of hybridization signals for 693 MMS-responsive genes from three independent MMS-treated cultures versus the average of their intensity values. In panels b and c, red lines represent twofold changes from the average. (d) Average intensity value of the untreated cultures versus the average intensity value for the MMS-treated cultures for the 693 MMS-responsive genes. Red lines represent 2-fold, 5-fold, and 10-fold differences in transcript levels.
Having established that mRNA profiling using the Affymetrix oligonucleotide chips was reproducible, we adopted an experimental strategy first suggested by Eisen et al. (10), that it is much more informative to establish mRNA profiles for a wide variety of conditions than to make repeat observations on identical conditions. We therefore committed our available resources to monitoring changes in transcript levels induced by numerous different MMS exposures and induced by roughly equitoxic exposures to numerous carcinogens. Note that by using a 90% confidence level to determine significance, we may increase the number of false-positive results while increasing the number of responsive genes. We chose to be more inclusive with our data, relying on the clustering algorithms to determine interesting patterns that would be unaffected by a few false-positive results.
Kinetics of the MMS-induced transcriptional response.
The collection of MMS-responsive S. cerevisiae genes observed after 60 min of exposure to 0.1% MMS (17) (Fig. 1) represents a simple snapshot of the transcriptional response of this eukaryote to alkylation damage. This raises the possibility that if one could monitor transcriptional responses as a continuum, even more genes might be counted as MMS responsive. In an attempt to gain insight into this continuum, we monitored transcriptionally responsive genes as a function of time in 0.1% MMS and as a function of MMS dose. Figures 2a and b provide a diagrammatic representation of the results (the identities of the genes and the raw data can be found at www.hsph.harvard /geneexpression). Represented in Fig. 2a and b are genes whose transcript levels either increased (green) or decreased (red) by threefold or more for at least one of the treatments. In addition, the responsive genes are clustered into groups that show similar kinetics using the hierarchical clustering program developed by Eisen et al. (10). It is immediately apparent that many more than 400 genes are MMS responsive, and the total number of genes represented in Fig. 2a and b is 969 and 1,863, respectively. The set of transcriptionally responsive genes is quite different at early versus late times and at low versus high alkylation levels. For the temporal response, there appear to be distinct groups of early-, middle-, and late-responsive genes, clusters IV, III, and II, respectively, in Fig. 2a. In addition, the response of several sets of genes appears to be transient, in that their responsiveness is seen at only one or two time points (e.g., clusters I, V, and VI). For the dose response (Fig. 2b), it is clear that with increasing alkylation levels, the number of responsive genes as well as the degree of responsiveness increases in a cumulative way. At the highest dose, we monitored 1,426 responsive genes, with 999 upregulated and 427 downregulated; this represents over 20% of the S. cerevisiae genome. Why such a large fraction of the yeast genome should be MMS responsive is discussed below.
FIG. 2.
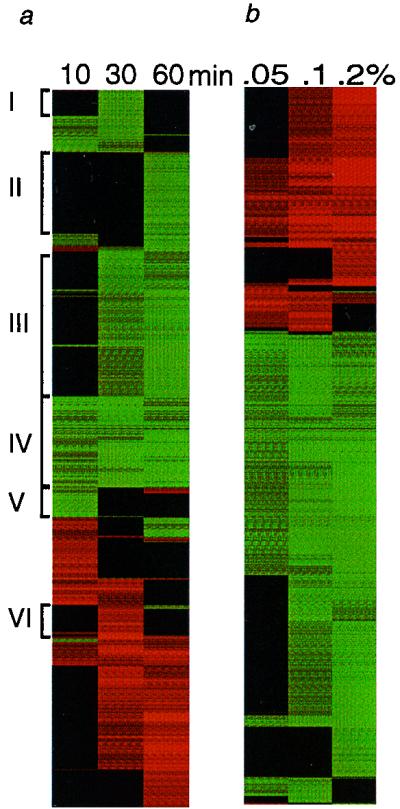
Expression patterns with different MMS exposures. (a) Cells were exposed to 0.1% MMS for the indicated times. The 969 genes that showed a transcript level change of threefold or higher were grouped by similar patterns of expression using hierarchical clustering and visualized graphically using colors to represent the direction and extent of change. Increases in mRNA expression are represented by shades of green, and decreases are shown by shades of red. (b) Cells were exposed to the indicated MMS dose for 60 min. The expression pattern for 1,863 responsive genes is shown as described above. The results in panels a and b were derived independently using different datasets.
MMS-induced transcriptional response as a function of cell cycle position.
As eukaryotic cells move through the cell cycle, specific sets of genes are transcriptionally activated and inactivated, although transcript levels for the vast majority of genes do not change (4, 32). Moreover, responses to DNA-damaging agents are known to vary throughout the cell cycle; e.g., G1 cells that experience DNA damage activate a G1/S checkpoint, those in S phase activate an S-phase delay, and those in G2 or M activate a G2/M checkpoint (16, 21, 30, 37). For these reasons we monitored how S. cerevisiae responds to MMS-induced alkylation damage as a function of cell cycle. (Note that in our initial dataset [17] very few of the MMS-responsive genes turned out to be cell cycle regulated genes.) Cells arrested in G1, S, G2, or stationary phase were exposed to 0.1% MMS for 60 min; the MMS-induced transcriptional profiles for each synchronized population are diagrammed in Fig. 3 and presented numerically at www.hsph.harvard.edu/geneexpression. Cell cycle stage had a profound effect on the MMS-induced transcriptional profiles. Numerous genes appear to be cell cycle specific in that they were only scored as weakly responsive or nonresponsive in MMS-treated log-phase cultures, but were scored as clearly responsive in a synchronized culture. Among them, 199 genes appear responsive only if cells experience damage in G1 (clusters II, V, and VI); 84 genes are only responsive in S phase (clusters IV and VII); 94 are only responsive in G2 (clusters III and IX); and 229 are only responsive in stationary phase (clusters I, VIII, and X). Fewer than 20% of these 614 genes were previously shown to have cell cycle-dependent expression (4, 32).
FIG. 3.

Effect of cell cycle on MMS-induced mRNA profiles. (a) Cells were arrested in G1 with α-factor, in S with hydroxyurea, or in G2 with nocodazole or allowed to grow to stationary (stat) phase. Transcript levels in each population were measured with and without 0.1% MMS exposure for 60 minutes. The data were divided into 40 clusters using a K-means algorithm (34). The centroids of the 40 clusters were grouped by hierarchical clustering to group clusters with similar profiles together. These 1,876 genes were responsive by more than 3.0-fold. (b) Hierarchical clustering (10) of transcript changes in MMS-treated log-phase cells, untreated stationary-phase cells (compared to untreated log-phase cells), and MMS-treated stationary-phase cells. Genes with more than a three- fold change in any one condition are shown. Fold changes are presented as described above. Clusters I to X represent cell cycle-specific responsive clusters.
It turns out that a large fraction of the genes that are responsive to MMS in log-phase cycling cells are also responsive to simply being held in stationary phase, independent of MMS exposure. This is shown clearly in Fig. 3b, where the transcript level changes for MMS-treated log-phase cells and those for stationary-phase versus log-phase cells are reclustered and shown alongside each other. The MMS exposure (0.1% for 60 min) to some extent appears to mimic the arrest of cells in stationary phase, at least in terms of transcriptional profile. At first it seemed that fewer genes are MMS responsive in stationary-phase cells than in cells in other parts of the cell cycle (Fig. 3a and b). However, this may be explained by the fact that 335 transcripts that ordinarily respond to MMS are already up- or downregulated in stationary-phase cells prior to MMS exposure (Fig. 3b, clusters I and II, respectively) and respond no further upon alkylation exposure. There appears to be an overlap of responsive genes by two different stressful conditions, MMS exposure and stationary growth. This may reflect a general stress response pathway, although we do not yet know whether these are a primary, secondary or tertiary response to stress.
Transcriptional responses to other damaging agents.
One of our major goals is to understand exactly how cells respond to a range of carcinogenic alkylating agents representative of those present in our environment and those used in the cancer clinic. We therefore set out to compare the transcriptional response of S. cerevisiae to various candidate alkylating agents, including the SN2 alkylating agent MMS, the SN1 alkylating agent MNNG and the chemotherapeutic alkylating agent BCNU. In addition, we wished to determine which aspects of the responses are alkylating agent specific, and so for comparison we determined the transcriptional profile of cells exposed to three other types of damaging agent: γ-irradiation, 4NQO, and the oxidizing agent t-BuOOH (12). Cells were exposed to roughly equitoxic doses of each agent, as measured by colony formation, and the resulting profiles are shown in Fig. 4 (shown numerically at www.hsph.harvard.edu/geneexpression). Doses were relatively nontoxic, resulting in 75 to 100% survival. Very few genes were responsive to all of the agents; indeed, among the hundreds of responsive genes, the transcript levels for only 12 turned out to be induced by all treatments, and transcripts for only 9 were repressed by all treatments. These 21 genes do not include any DNA repair genes, and since they were determined independently of clustering, they are detailed at http://www.hsph.harvard.edu/geneexpression for easy access. Furthermore, there were surprisingly extensive differences between the transcriptional profiles induced by each of the six damaging agents. Even the closely related methylating agents MMS and MNNG induce quite distinct transcriptional profiles at roughly equitoxic doses (Fig. 4, lane 4 versus 6). At a higher MNNG dose (24% survival), the profile begins to overlap more with the MMS-induced profile, although each profile still remains quite distinct (lane 4 versus 7). It is also notable that the profiles produced by equitoxic exposure to γ-rays and the oxidizing agent t-BuOOH are dramatically different. Since it has been estimated that following ionizing radiation, ∼65% of the damage to DNA occurs by base oxidation and only ∼35% occurs directly by ionization (12, 36), one might have expected more overlap.
FIG. 4.
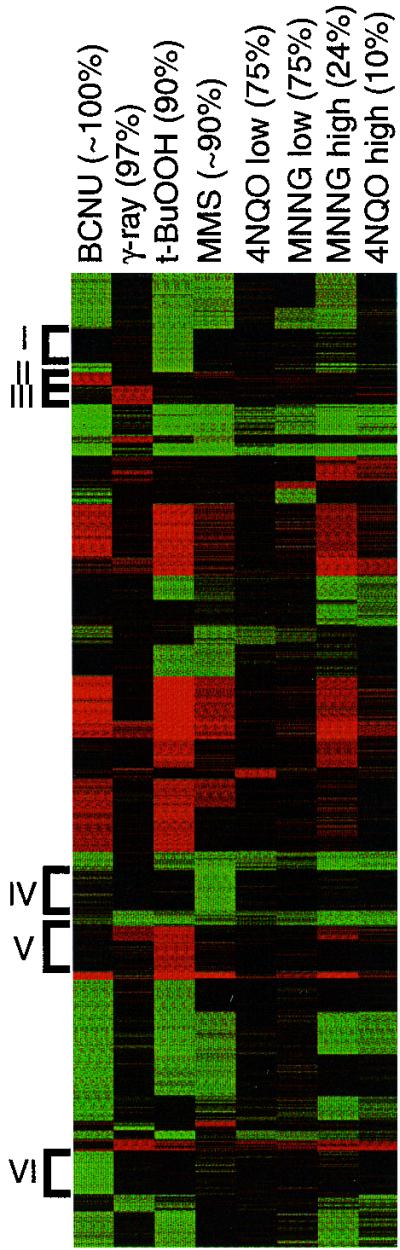
Global transcriptional profiles in response to different damaging agents. Log-phase cells were exposed to the indicated agents; exposure was limited to 1 h and resulted in the percent survival (as determined by colony-forming ability) indicated in parentheses. Expression profiles for 2,324 responsive genes were grouped by a K-means clustering algorithm into 30 clusters and are represented by colors as described in the legend to Fig. 2. Six clusters containing members whose transcript levels appear to be responsive to only a particular agent are indicated. Treatments were as follows: 200 μM BCNU, 30-kilorad γ-ray, 5 mM t-BuOOH, 0.1% MMS, 4NQO at 2 (low) or 8 (high) μg/ml, and MNNG 8 at (low) or 27 (high) μg/ml. Clusters I to IV represent DNA-damaging agent-specific clusters.
In addition, there appear to be groups genes that are specifically responsive to each damaging agent (clusters I to VI), and these may turn out to represent unique signatures for each agent. It should be noted once again that these profiles represent snapshots of transcriptional responses, and upon further kinetic analysis, genes that appear to be agent specific in Fig. 4 may turn up in response to the other agents. In fact, only 30% of the genes that appear to be agent specific in Fig. 4 (excluding the MMS-specific cluster IV) can be found among the numerous MMS-induced profiles described in this study. However, a more extensive kinetic analysis will be needed to establish if there are certain responsive genes that are truly specific for a particular agent or class of agent. Finally, it is clear from Fig. 4 that the volume of genes responding to exposure to a damaging agent is not a good predictor of toxicity. For example, the most toxic treatment (4NQO, producing 10% survival) influenced the expression of far fewer genes than did the least toxic treatment (BCNU, producing 100% survival).
Over one third of S. cerevisiae genes respond to cellular damage.
Taken as a whole, this study shows that damaging cells by physical and chemical carcinogens elicits significant changes in transcript level for more than 2,500 of S. cerevisiae's ∼6,200 genes. These transcriptionally responsive genes can be categorized by functional category (as defined by the S. cerevisiae genome database [1]) and are summarized in Table A at http: //www.harvard.edu/geneexpression. The number of induced genes is listed in green, the number of repressed genes is listed in red, and each number is linked to its corresponding list of genes and to a numerical representation of transcript levels and fold induction values. By far the largest category of responsive genes is genes of unknown function, and the next largest categories include those for protein and mRNA metabolism. Surprisingly, DNA repair, DNA replication, and cell cycle progression genes are only modestly represented in the dataset.
SOMs of the responsive genes in 26 transcriptional profiles.
A powerful computational method for seeking meaningful patterns in large datasets can now be applied to transcriptional profiling data (33). The organization of data into SOMs places genes into clusters that behave similarly across multiple conditions. Using this algorithm, we organized 26 transcriptional profiles into 18 such SOMs (Fig. 5A); the 26 conditions are listed in the figure legend, and at the website each box links to a list of the genes in that particular SOM. Note that for 3,600 genes, either the transcript levels did not change significantly for any of the 26 treatments or they were expressed at very low levels and were eliminated from the dataset; the remaining 2,610 genes are apportioned to the 18 SOMs. For this analysis, transcript levels are compared across all 26 conditions, and clusters are created based on whether or not the transcript levels go up or down; the analysis does not weigh the actual fold differences in transcript levels, but instead notes the trend. Put another way, genes whose transcript levels change by up to 10-fold in any one of the 26 conditions may be clustered with those that change up to 100-fold, provided the up and down trend is similar across all 26 conditions. Such computational organization of transcriptionally responsive genes is designed to cluster together genes that respond to some of the same signal transduction events, and thus genes whose expression may be controlled by the same regulatory proteins. In other words, it is hoped that such clustering will identify individual regulons and their regulators.
FIG. 5.



SOMs of transcript levels for 2,610 genes that change by threefold or more across 26 exposure conditions. The 26 exposure conditions are as follows: untreated no. 1, untreated no. 2, untreated no. 3, 30 kilorads of irradiation, G1 arrest, 0.1% MMS for 10 min, stationary-phase growth plus 0.1% MMS for 60 min, stationary-phase growth, G1 phase arrest plus 0.1% MMS for 60 min, 0.1% MMS for 30 min, 0.2% MMS for 60 min, S-phase arrest plus 0.1% MMS for 60 min, G2 phase arrest plus 0.1% MMS for 60 min, 0.1% MMS for 60 min no. 1, 0.1% MMS for 60 min no. 2, 0.05% MMS for 60 min, 0.1% MMS for 60 min no. 3, 5 mM t-BuOOH, 200 μM BCNU, 4NQO at 8 μg/ml, MNNG at 27 μg/ml, MNNG at 6.7 μg/ml, S-phase arrest, 4NQO at 2 μg/ml, G2 arrest, and MMC at 2 μg/ml. (A) A total of 2,610 genes were grouped into 18 clusters as described in the text, and the average expression profile for the genes in each cluster is represented; the number of genes in each cluster is indicated. The cluster containing the MAG1 gene is highlighted. The 18 clusters are numbered left to right and top to bottom. (B) Data from SOMs are converted to color representation as described in the legend to Fig. 3, and fold changes are compared to the average transcript levels in three untreated cultures. Lane labels indicate cell cycle phase (G1, G2, S, or stationary [STAT]), agent, concentration, and time (in minutes). The treatments are clustered based on hierarchical methods to group treatments that produce similar profiles together, and their correlation is represented by the dendogram. (C) Distribution of genes containing a MAG1 URS2-like element among the 18 clusters.
Figure 5B presents a diagrammatic representation of the 18 SOMs, but this time the fold changes in transcript levels are presented as they relate to the average transcript levels observed in the three untreated log-phase cultures (see Fig. 1). The individual transcriptional profiles are also arranged so that those that are most similar to each other lie next to each other, and the extent of their relatedness is indicated by the dendogram above the figure. Accordingly, almost all of the MMS-induced profiles lie together, with the exception that the untreated stationary-phase profile sorts with this group, as mentioned above (Fig. 3b). Note that the profiles induced by the two agents that, like MMS, are strong inducers of protein degradation and amino acid metabolism genes (BCNU and t-BuOOH; see Table A at www.hsph.harvard.edu/geneexpression) sort next to the MMS-induced profiles. Again, it is surprising that the profile induced upon exposure to the oxidizing agent t-BuOOH sorts so far away from that induced by an equitoxic γ-radiation dose, given that γ-irradiation-induced toxicity is thought to derive in large part from a flux of oxidative damage (12, 36).
The SOMs produced a distinct organization of genes. Of the 162 possible pairwise comparisons of the patterns within each cluster (Fig. 5A), only four showed significant similarity (with a correlation coefficient greater than 0.75), while the vast majority did not. This indicates that for the most part, this analysis divided the responsive genes into distinctive groups. For some of the SOMs derived from this diverse array of damage-inducible profiles, it is quite apparent that genes encoding functionally related proteins become grouped together. As examples, SOM1 (Fig. 5A and B) contains 130 of the 212 responsive protein synthesis genes; SOM3 contains 47 of the 62 responsive genes involved in energy metabolism; SOM5 contains 13 of the 19 responsive genes involved in mating; and SOM13 contains 29 of the 96 responsive amino acid metabolism genes. Such grouping of functionally related genes agrees well with the results of Eisen et al. (10), who first proposed that clustering the combined data from transcriptional profiles generated by a large number of treatments would allow genes to be sorted into functional groups.
Identification of an MMS-responsive regulon that includes the MAG1 3-methyladenine DNA glycosylase gene and protein degradation genes.
Several S. cerevisiae DNA repair and DNA metabolism genes have long been known to be induced upon MMS exposure, among them the MAG1 3-methyladenine DNA glycosylase gene, known for its important role in base excision repair and in alkylation resistance (2, 3, 31, 39). Indeed, it was the fact that MMS-responsive genes like MAG1 are important for protecting cells against carcinogenic alkylating agents that prompted us to seek the identity of all MMS-responsive S. cerevisiae genes, on the premise that some of these genes may also be important for alkylation resistance. We were therefore particularly interested to determine which genes cluster with MAG1 across the 26 conditions shown in Fig. 5. MAG1 turns out to cluster with 213 other genes in cluster 14, as highlighted in Fig. 5A and detailed in Table 1 (and presented numerically at the website). To our surprise, the largest category of known genes to cluster with MAG1 were the protein degradation genes, and only four other DNA repair genes were present in the cluster. In our initial report (17), we noted that a large fraction of protein degradation genes were induced by MMS, along with an equally high fraction of amino acid metabolism genes. We inferred from these data that MMS exposure might signal the induction of a program to eliminate and replace alkylated proteins. Here, SOM analysis indicates a correlation between the regulation of MAG1 and nearly 50% of the responsive protein degradation genes (most amino acid metabolism genes cluster elsewhere). This observation led us to search for common regulatory motifs upstream of MAG1, upstream of the protein degradation genes, and upstream of the other genes in cluster 14.
TABLE 1.
Genes and ORFs coregulated with MAG1a
| Category | No. of genes |
|---|---|
| Protein degradation | 33 |
| Protien folding | 8 |
| Amino acid metabolism | 7 |
| Transcription | 7 |
| Signaling | 6 |
| DNA repair | 5 |
| Secretion | 5 |
| Cytoskeleton | 4 |
| mRNA metabolism | 4 |
| Stress response | 4 |
| Others | 38 |
| Unknown | 93 |
| Total | 214 |
Categories are based on the Saccharomyces cerevesiae database (1) and yeast protein database (6). Only categories with more than three members are included. For a complete list of individual genes, see www.hsph.harvard.edu /geneexpression
Several years ago we identified an upstream repressor sequence (URS), called URS2, in the MAG1 promoter region with the sequence GGTGGCGA (31, 39). Using the AlignACE and ScanACE programs developed by Roth et al. (25), we now find that sequence motifs similar to the MAG1 URS2 can be found upstream of 56 of the 214 genes in cluster 14 and that 33 of these 56 genes are protein degradation and ubiquitination related. In total, 68 responsive genes are involved in protein degradation, and almost 50% (33) are found in this cluster. In order to show the significance of this finding, Fig. 5C displays the distribution of genes with MAG1 URS2-like elements among the 18 SOMs; clearly this element is overrepresented (P = <10−300) in cluster 14 containing the MAG1 and protein degradation genes.
We and others have pointed out that sequence motifs similar to the MAG1 URS2 element are found upstream of over a dozen DNA repair and metabolism genes (27, 31, 39). These elements have been referred to as damage repair consensus elements (27), and many but not all genes bearing such elements are damage responsive. More recently, a similar putative regulatory sequence was identified for numerous genes encoding proteins involved in ubiquitin-mediated protein degradation; this was named the proteasome-associated control element (PACE) (22). It is now clear that damage repair consensus elements can be separated into two different sequence motif groups, one of which is indistinguishable from the PACE sequence motif group and which includes the MAG1 URS2 element (P. Estep G. Church, unpublished data). A protein that binds specifically to the PACE sequence motif was identified by one-hybrid analysis as Rpn4 (22), a protein thought to be associated with proteasomes (13, 14). It appears that Rpn4p binds the PACE sequence to serve as a transcriptional activator (22).
Here we characterize an rpn4 deletion strain for its ability to induce MAG1 transcript levels in response to MMS. Figure 6a shows the dramatic loss of MAG1 MMS inducibility in the rpn4 deletion strain, and as a result the rpn4 deletion strain turns out to be MMS sensitive, although not as sensitive as a mag1 deletion strain (Fig. 6b). That the MAG1 URS2 element behaves as a repressor binding site (31) does not necessarily exclude Rpn4p's behaving as an activator at this site; our current model predicts that Rpn4p and a putative repressor compete for binding at the GGTGGCGA sequence. We also monitored the MMS inducibility of two other genes that contain the MAG1 URS2 sequence motif. The loss of Rpn4p caused a dramatic loss of inducibility for the RAD23 nucleotide excision repair gene and attenuated the inducibility of the PRE2 proteasome subunit gene. Rpn4p thus influences the regulation of genes in at least three different pathways, namely, base excision repair, nucleotide excision repair, and protein degradation. Note that two other MMS-inducible genes, neither of which localized to cluster 14, are totally unaffected by the absence of the Rpn4 transcriptional activator (Fig. 6a).
FIG. 6.

Characterization of an rpn4 deletion strain. (a) Northern analysis of wild-type (WT) and Δrpn4 cells treated with 0.1% MMS for the indicated times. Blots were probed for expression with MAG1-, RAD23-, PRE2-, RNR3-, and SNZ1-derived probes. (b) Colony-forming ability of wild-type, Δmag1, and Δrpn4 cells was measured after 1 h of exposure to 0.1, 0.2 and or 0.3% MMS. (c) SOM clustering of transcript changes in a Δrpn4 deletion strain versus wild (WT), in wild-type cells treated with 0.1% MMS, and in Δrpn4 cells treated with. 0.1% MMS. (d) Relative transcript levels of the 44 genes containing a MAG1 URS2-like sequence in wild-type (WT) and Δrpn4 cells with and without 0.1% MMS for 1 h. Black lines show 21 genes from cluster 3, blue lines show 12 genes from cluster 6, and orange lines show 11 genes from other clusters.
Rpn4p influences the basal and damage-responsive expression of many genes.
We find that S. cerevisiae transcriptional profiles change dramatically in the absence of Rpn4p, both with and without MMS exposure. Lane 1 in Fig. 6c displays transcript level changes in the untreated rpn4 deletion strain compared to its untreated wild-type parent. A total of 350 genes are downregulated, suggesting that Rpn4p affects transcriptional activation, and an even larger group of genes, 389, are upregulated, suggesting that Rpn4p affects transcriptional repression. Lanes 2 and 3 in Fig. 6c depict MMS-responsive genes in wild-type and rpn4-deleted cells, respectively, treated with 0.1% MMS for 60 min. Extensive differences between the two profiles are quite apparent, and both the upregulation and downregulation of transcripts are affected by the loss of Rpn4. The data in Fig. 6c were organized into 12 SOMs, and the multiple effects of losing the Rpn4 regulatory protein can be summarized as follows. (i) A total of 230 genes that are not MMS responsive in wild-type cells become susceptible to repression by MMS (cluster 2) and 85 become susceptible to induction by MMS (cluster 5); (ii) 461 genes become refractory to MMS induction (clusters 3, 8, and 10); (iii) 333 genes become more sensitive to MMS repression (clusters 4 and 7) and 455 become more sensitive to MMS induction (clusters 9 and 12); and (iv) 660 genes show little difference in their response to MMS despite the fact that their basal-level expression changed in rpn4-deleted cells (clusters 1, 6, and 11).
For the group of 213 genes that clustered with MAG1 (cluster 14, Fig. 5A), 56 had an upstream MAG1 URS2-1ike sequence; 44 of these 56 appear in the profiles shown in Fig. 6c because their expression was affected by Rpn4p. The relative expression of all 44 genes is shown in Fig. 6d, and the genes are grouped into three categories based on their distribution in Fig. 6c. Shown in black are 21 genes from cluster 3 (which contains MAG 1), in blue are 12 genes from cluster 6, and in orange are the remaining 11 genes from four separate clusters. The following conclusions can be made: on average, their basal expression is lower in the rpn4 deletion strain than in the wild-type strain, and on average, the absence of Rpn4p renders these genes less MMS inducible. Presumably the constellation of other transcription factors at each promoter determines how Rpn4p influences transcription.
Finally, two DNA nucleotide excision repair genes, RAD23 and SSL2, display another intriguing link to the ubiquitin-mediated proteasome degradation pathway. First, RAD23 and SSL2 are coregulated with MAG1 and protein degradation genes. Second, Rad23p has an N-terminal ubiquitin-like domain that interacts with the 26S proteasome and a C-terminal domain that interacts with Rad4p (29). The Rad23p-Rad4p complex in turn interacts with TFIIH (which contains Ssl2p), the transcription initiation factor known to be required for nucleotide excision repair (15). Thus, just as regulation of RAD23, SSL2, and the proteasome genes is transcriptionally linked, their products are physically linked via protein-protein interactions. Moreover, recent in vitro and in vivo evidence demonstrates that such protein interactions are important for optimal nucleotide excision repair activity (26). Since the transcription of three DNA glycosylase genes (MAG1, NTG1, and NTG2) is also coregulated with proteasome genes, it is tempting to speculate that optimal base excision repair is connected in a similar way to proteasome function.
Identification of several known and several putative control elements upstream of damage-responsive genes.
The promoter regions for the genes in each of the 18 clusters identified by SOMs in Fig. 5A were analyzed by the AlignACE program (25) for common sequence motifs, and Table 2 lists the consensus sequence for each motif with a MAP score of >10. This score is an internal metric used to determine the significance of an alignment. AlignACE searches in unaligned sequences for conserved DNA motifs and scores each motif based on the alignment and on the frequency of occurrences in intergenic regions. Motifs were considered significant if their MAP score was greater than 10 and if their distribution was significantly enriched in a particular cluster. Nine significant sequence motifs were identified, of which five are bound by known factors.
TABLE 2.
Upstream promotor sequence motifs
| Clustera | %b | Motifc | Binding factor | MAP scored | Putative functione |
|---|---|---|---|---|---|
| 1 | 45 | ATGTAYGGRTG | Rap1p | 150 | Ribosomal (G, BF) |
| 1 | 11 | YYNCCNGGCRNNA | Unknown | 10 | Ribosomal (G) |
| 2, 4 | 25, 26 | GNGATGAGMTf | Unknown | 82 | RNA metabolism (G) |
| 3 | 10 | GCCAATCAG | HAP CCAAT-binding complex | 23 | Mitochrondrial (G, BF) |
| 3 | 10 | YTGGGCRGC | Unknown | 13 | Mitochrondrial (G) |
| 4 | 15 | TYCGTNNNWNGTGA | Abf1p | 18 | RNA metabolism (G)/translation (BF) |
| 5 | 16 | TGTTTCARA | Ste12p | 33 | Mating/cell cycle (G, BF) |
| 13 | 7.5 | GCMCTGCGMK | Unknown | 11 | Unknown |
| 14 | 25 | GGTGGCAA | Rpn4p | 73 | Protein degradation (G, BF) |
Cluster numbers are as in Fig. 5.
Percentage of genes in that particular cluster which contain the motif within 600 bp upstream of the transcription start site.
Only motifs with a MAP score greater then 10 and whose motif is confined to a single cluster are included (25). Y is C or T; R is A or G; N is A, C, G, or T; W is A or T; M is A or C; and K is G or T.
MAP score is a statistical value of how “good” a particular motif is and is based on the alignment and the frequency of occurrence in noncoding sequences.
Function of the majority of the genes in the cluster containing the motif (G) or genes known to be controlled by the indicated binding factor (BF).
This consequence sequence has been identified upstream of many RNA polymerase A and C subunit genes and has been referred to as the PAC box (8).
Five of the sequence motifs have known binding factors, namely, Rpn4p (discussed above), Rap1p (19), Hap2/3/4/5p (24), Abf1p (7), and Ste12p (40). Rap1p regulates ribosomal protein gene transcription (19), and accordingly Rap1 binding sites were found upstream of 45% of the genes in cluster 1 (Fig. 5A), where most of the ribosomal protein genes sort. Indeed, a consensus Rap1p binding sequence was recently determined by a systematic search of all upstream ribosomal protein transcription start sites (18) that turns out to be identical to the motif identified here by the blind AlignACE search of our dataset. The HAP2/3/4/5p binding complex is important for the transcription of many mitochondrial proteins (20); 10% of the genes in cluster 3 contain a HAP2/3/4/5p binding site, more than half of which turn out to be involved in mitochondrial functions, including ATP synthesis, oxidative phosphorylation, and respiration. Abf1p binds to a sequence motif present in replication origins, promoters of rRNA genes, and other genes involved in translation and glycoylsis and at mating type silencing sequences (6); cluster 4 contains a concentration of RNA metabolism genes and translation genes. Ste12p is a transcription factor for yeast mating genes and associated cell cycle regulation genes (40), and these sites are found in 16% of the cluster 5 genes; most of the mating genes sort to cluster 5. The factors that bind the remaining four motifs (if any) remain to be identified.
Concluding comments.
Exploring transcriptional profiles is inherently descriptive. For S. cerevisiae, most of the transcriptional profiling carried out to date describes changes that occur upon specific alterations in growth conditions or upon specific alterations in genotype (4, 5, 9, 17, 18, 32, 35). Here we describe changes in transcriptional profiles that take place when cells are exposed to a reactive chemical or physical agent, such that virtually every molecule in the cell is at risk of being altered in some way. In retrospect, we should perhaps not have been surprised by the fact that over one third of S. cerevisiae's entire gene repertoire can respond to the deluge of damage. Nevertheless, the results were surprising, and they challenge us to determine what roles, if any, such a myriad of transcriptional changes play in protecting cells against inevitable exposures to carcinogenic agents.
One way to make sense of global transcriptional responses is to break them down into smaller components, by identifying individual regulons and their regulators. Ultimately, manipulating each regulon to alter the response component by component should help to reveal their in vivo roles. Despite the complexity and the sheer volume of information contained in global transcriptional profiles, elegant computational methods can unveil patterns in the data. In this study, these patterns led us to genetically define a novel MMS-responsive regulon that is controlled, at least in part, by the proteasome-associated protein Rpn4p. Moreover, the Rpn4p binding site was only one of nine sequence motifs identified upstream of the damage-responsive genes. Among the remaining eight motifs, four are known to be bound by previously characterized transcription factors, and four warrant further investigation. In this way, it may be ultimately possible to systematically study each component of the complex transcriptional response of eukaryotes to carcinogenic agents. Perhaps more important will be subsequent determination of the relative importance of each component in protecting against the cytotoxic, mutagenic, and thus carcinogenic effects of the kinds of damaging agents used in this study.
It is important to note that the transcriptional profiles from all the diverse exposures were required in order to generate the SOMs that link the proteasome and the DNA repair genes. If some of the profiles are omitted, the apparent connection between protein degradation and DNA repair is lost. This underscores the power of combining the information from numerous diverse treatments in order to generate informative patterns in the data.
The presentation of enormous datasets associated with transcriptional profiling in conventional publications must of necessity be limited to describing patterns and trends in the data, rather than discussing the identity of every transcript whose expression is affected. Such patterns and trends hold the promise of identifying novel biological pathways, elucidating how pathways are regulated, assigning to genes of unknown function a known or probable function, and ultimately (in conjunction with proteomics and other emerging techniques) elucidating how all the molecular components of cells integrate to make a living organism. However, in order to understand the final integrated picture, the identity of each gene whose changing expression produces the patterns and trends must ultimately be considered. It is therefore important that the information be scrutinized by experts in many different areas of molecular biology. Here we have inspected our dataset from the perspective of DNA repair in general and DNA alkylation repair in particular. We hope that others will inspect our data (at http://www.hsph.harvard.edu/geneexpression) from the perspective of their own highly specialized areas of expertise.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grant RO1 CA5502 to L.D.S. and ONR grant N00014-97-1-0865 to G.M.C. S.A.J. was supported by National Institutes of Health training grant CA09078 and NRSA grant CA81744. The Affymetrix academic user program was supported in part by National Institutes of Health grant PO1-HG0132. L.S. was a Burroughs Wellcome Toxicology Scholar.
REFERENCES
- 1.Ball C A, Dolinski K, Dwight S S, Harris M A, Issel-Tarver L, Kasarskis A, Scafe C R, Sherlock G, Binkley G, Jin H, Kaloper M, Orr S D, Schroeder M, Weng S, Zhu Y, Botstein D, Cherry J M. Integrating functional genomic information into the Saccharomyces genome database. Nucleic Acids Res. 2000;28:77–80. doi: 10.1093/nar/28.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen J, Derfler B, Samson L. Saccharomyces cerevisiae 3-methyladenine DNA glycosylase has homology to the AlkA glycosylase of E. coli and is induced in response to DNA alkylation damage. EMBO J. 1990;9:4569–4575. doi: 10.1002/j.1460-2075.1990.tb07910.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen J, Samson L. Induction of S. cerevisiae MAG 3-methyladenine DNA glycosylase transcript levels in response to DNA damage. Nucleic Acids Res. 1991;19:6427–6432. doi: 10.1093/nar/19.23.6427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cho R J, Campbell M J, Winzeler E A, Steinmetz L, Conway A, Wodicka L, Wolfsberg T G, Gabrielian A E, Landsman D, Lockhart D J, Davis R W. A genome-wide transcriptional analysis of the mitotic cell cycle. Mol Cell. 1998;2:65–73. doi: 10.1016/s1097-2765(00)80114-8. [DOI] [PubMed] [Google Scholar]
- 5.Chu S, DeRisi J, Elsen M, Mulholland J, Botstein D, Brown P O, Herskowitz I. The transcriptional program of sporulation in budding yeast. Science. 1998;282:699–705. doi: 10.1126/science.282.5389.699. [DOI] [PubMed] [Google Scholar]
- 6.Costanzo M C, Hogan J D, Cusick M E, Davis B P, Fancher A M, Hodges P E, Kondu P, Lengieza C, Lew-Smith J E, Lingner C, Roberg-Perez K J, Tillberg M, Brooks J E, Garrels J I. The yeast proteome database (YPD) and Caenorhabditis elegans proteome database (WormPD): comprehensive resources for the organization and comparison of model organism protein information. Nucleic Acids Res. 2000;28:73–76. doi: 10.1093/nar/28.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Della Seta F, Ciafre S A, Marck C, Santoro B, Presutti C, Sentenac A, Bozzoni I. The ABF1 factor is the transcriptional activator of the L2 ribosomal protein genes in Saccharomyces cerevisiae. Mol Cell Biol. 1990;10:2437–2441. doi: 10.1128/mcb.10.5.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dequard-Chablat M, Riva M, Carles C, Sentenac A. RPC19, the gene for a subunit common to yeast RNA polymerases A (I) and C (III) J Biol Chem. 1991;266:15300–15307. [PubMed] [Google Scholar]
- 9.DeRisi J L, Iyer V R, Brown P O. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- 10.Eisen M B, Spellman P T, Brown P O, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elledge S J. Cell cycle checkpoints: preventing an identity crisis. Science. 1996;274:1664–1672. doi: 10.1126/science.274.5293.1664. [DOI] [PubMed] [Google Scholar]
- 12.Friedberg E C, Walker G C, Siede W. DNA repair and mutagenesis. Washington, D.C.: ASM Press; 1995. [Google Scholar]
- 13.Fujimuro M, Tanaka K, Yokosawa H, Toh-e A. Son1p is a component of the 26S proteasome of the yeast Saccharomyces cerevisiae. FEBS Lett. 1998;423:149–154. doi: 10.1016/s0014-5793(98)00084-2. [DOI] [PubMed] [Google Scholar]
- 14.Glickman M H, Rubin D M, Fried V A, Finley D. The regulatory particle of the Saccharomyces cerevisiae proteasome. Mol Cell Biol. 1998;18:3149–3162. doi: 10.1128/mcb.18.6.3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guzder S N, Habraken Y, Sung P, Prakash L, Prakash S. Reconstitution of yeast nucleotide excision repair with purified Rad proteins, replication protein A, and transcription factor TFIIH. J Biol Chem. 1995;270:12973–12976. doi: 10.1074/jbc.270.22.12973. [DOI] [PubMed] [Google Scholar]
- 16.Hartwell L H, Weinert T A. Checkpoints: controls that ensure the order of cell cycle events. Science. 1989;246:629–634. doi: 10.1126/science.2683079. [DOI] [PubMed] [Google Scholar]
- 17.Jelinsky S A, Samson L D. Global response of Saccharomyces cerevisiae to an alkylating agent. Proc Natl Acad Sci USA. 1999;96:1486–1491. doi: 10.1073/pnas.96.4.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lascaris R F, Mager W H, Planta R J. DNA-binding requirements of the yeast protein Rap1p as selected in silico from ribosomal protein gene promoter sequences. Bioinformatics. 1999;15:267–277. doi: 10.1093/bioinformatics/15.4.267. [DOI] [PubMed] [Google Scholar]
- 19.Li B, Nierras C R, Warner J R. Transcriptional elements involved in the repression of ribosomal protein synthesis. Mol Cell Biol. 1999;19:5393–5404. doi: 10.1128/mcb.19.8.5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Z, Butow R A. A transcriptional switch in the expression of yeast tricarboxylic acid cycle genes in response to a reduction or loss of respiratory function. Mol Cell Biol. 1999;19:6720–6728. doi: 10.1128/mcb.19.10.6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lowndes N F, Murguia J R. Sensing and responding to DNA damage. Curr Opin Genet Dev. 2000;10:17–25. doi: 10.1016/s0959-437x(99)00050-7. [DOI] [PubMed] [Google Scholar]
- 22.Mannhaupt G, Schnall R, Karpov V, Vetter I, Feldmann H. Rpn4p acts as a transcription factor by binding to PACE, a nonamer box found upstream of 26S proteasomal and other genes in yeast. FEBS Lett. 1999;450:27–34. doi: 10.1016/s0014-5793(99)00467-6. [DOI] [PubMed] [Google Scholar]
- 23.Marnett L J, Burcham P C. Endogenous DNA adducts: potential and paradox. Chem Res Toxicol. 1993;6:771–785. doi: 10.1021/tx00036a005. [DOI] [PubMed] [Google Scholar]
- 24.McNabb D S, Xing Y, Guarente L. Cloning of yeast HAP5: a novel subunit of a heterotrimeric complex required for CCAAT binding. Genes Dev. 1995;9:47–58. doi: 10.1101/gad.9.1.47. [DOI] [PubMed] [Google Scholar]
- 25.Roth F P, Hughes J D, Estep P W, Church G M. Finding DNA regulatory motifs within unaligned noncoding sequences clustered by whole-genome mRNA quantitation. Nat Biotechnol. 1998;16:939–945. doi: 10.1038/nbt1098-939. [DOI] [PubMed] [Google Scholar]
- 26.Russell S J, Reed S H, Huang W, Friedberg E C, Johnston S A. The 19S regulatory complex of the proteasome functions independently of proteolysis in nucleotide excision repair. Mol Cell. 1999;3:687–695. doi: 10.1016/s1097-2765(01)80001-0. [DOI] [PubMed] [Google Scholar]
- 27.Sancar G B, Ferris R, Smith F W, Vandeberg B. Promoter elements of the PHR1 gene of Saccharomyces cerevisiae and their roles in the response to DNA damage. Nucleic Acids Res. 1995;23:4320–4328. doi: 10.1093/nar/23.21.4320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Santoro M G. Heat shock factors and the control of the stress response. Biochem Pharmacol. 2000;59:55–63. doi: 10.1016/s0006-2952(99)00299-3. [DOI] [PubMed] [Google Scholar]
- 29.Schauber C, Chen L, Tongaonkar P, Vega I, Lambertson D, Potts W, Madura K. Rad23 links DNA repair to the ubiquitin/proteasome pathway. Nature. 1998;391:715–718. doi: 10.1038/35661. [DOI] [PubMed] [Google Scholar]
- 30.Shackelford R E, Kaufmann W K, Paules R S. Cell cycle control, checkpoint mechanisms, and genotoxic stress. Environ Health Perspect. 1999;107(Suppl. 1):5–24. doi: 10.1289/ehp.99107s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh K K, Samson L. Replication protein A binds to regulatory elements in yeast DNA repair and DNA metabolism genes. Proc Natl Acad Sci USA. 1995;92:4907–4911. doi: 10.1073/pnas.92.11.4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spellman P T, Sherlock G, Zhang M Q, lyer V R, Anders K, Eisen M B, Brown P O, Botstein D, Futcher B. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell. 1998;9:3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tamayo P, Slonim D, Mesirov J, Zhu Q, Kitareewan S, Dmitrovsky E, Lander E S, Golub T R. Interpreting patterns of gene expression with self-organizing maps: methods and application to hematopoietic differentiation. Proc Natl Acad Sci USA. 1999;96:2907–2912. doi: 10.1073/pnas.96.6.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tavazoie S, Hughes J D, Campbell M J, Cho R J, Church G M. Systematic determination of genetic network architecture. Nat Genet. 1999;22:281–285. doi: 10.1038/10343. [DOI] [PubMed] [Google Scholar]
- 35.ter Linde J J, Liang H, Davis R W, Steensma H Y, van Dijken J P, Pronk J T. Genome-wide transcriptional analysis of aerobic and anaerobic chemostat cultures of Saccharomyces cerevisiae. J Bacteriol. 1999;181:7409–7413. doi: 10.1128/jb.181.24.7409-7413.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ward J F. DNA damage produced by ionizing radiation in mammalian cells: identities, mechanisms of formation, and reparability. Prog Nucleic Acid Res Mol Biol. 1988;35:95–125. doi: 10.1016/s0079-6603(08)60611-x. [DOI] [PubMed] [Google Scholar]
- 37.Weinert T A, Hartwell L H. The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae. Science. 1988;241:317–322. doi: 10.1126/science.3291120. [DOI] [PubMed] [Google Scholar]
- 38.Wodicka L, Dong H, Mittmann M, Ho M H, Lockhart D J. Genome-wide expression monitoring in Saccharomyces cerevisiae. Nat Biotechnol. 1997;15:1359–1367. doi: 10.1038/nbt1297-1359. [DOI] [PubMed] [Google Scholar]
- 39.Xiao W, Singh K K, Chen B, Samson L. A common element involved in transcriptional regulation of two DNA alkylation repair genes (MAG and MGT1) of Saccharomyces cerevisiae. Mol Cell Biol. 1993;13:7213–7221. doi: 10.1128/mcb.13.12.7213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yuan Y O, Stroke I L, Fields S. Coupling of cell identity to signal response in yeast: interaction between the alpha 1 and STE12 proteins. Genes Dev. 1993;7:1584–1597. doi: 10.1101/gad.7.8.1584. [DOI] [PubMed] [Google Scholar]