

Abstract
Small leucine rich repeat proteoglycans (SLRPs) are a group of active components of the extracellular matrix in all tissues. SLRPs bind to collagens and regulate collagen fibril growth and fibril organization. SLRPs also interact with various cytokines and extracellular compounds, which lead to various biological functions such cell adhesion and signaling, proliferation, and differentiation. Mutations in SLRP genes are associated with human diseases. Now crystal structures of five SLRPs are available. We describe some features of amino acid sequence and structures of SLRPs. We also review ligand interactions and then discuss the interaction surfaces. Furthermore, we map mutations associated with human diseases and discuss possible effects on structures by the mutations.
Keywords: Super motif, Aromatic or methionine–aromatic interaction, Collagen binding, Interaction surface, Mutations
Introduction
Small leucine rich repeat proteoglycans (SLRPs) are a group of active components of the extracellular matrix (ECM) in all tissues. Leucine rich repeats (LRRs) occur tandemly in SLRPs. SLRPs bind to various types of collagens (I, II, III, V, VI, IX, XII, XIV, and XVIII) and regulate collagen fibril growth and fibril organization (collagen fibrillogenesis) (Chen and Birk 2013; Hindson et al. 2019). SLRPs also interact with various cytokines including transforming growth factor-beta (TGF-β) and von Willebrand factor (vWF), and with extracellular compounds such as Toll-like receptors (TLR2 and TLR4) and epidermal growth factor receptor (EGFR) (Pietraszek-Gremplewicz et al. 2018). These interactions lead to various biological functions, including cell adhesion and signaling, proliferation, and differentiation. Mutations in SLRP genes are associated with human diseases (Matsushima et al. 2019).
SLRPs contain LRR domains flanked by clusters of cysteine residues at both N- and C-termini which are called as LRRNT and LRRCT or LRRCE, respectively; LRRCE is a capping motif containing “ear” LRR (Park et al. 2008). In addition to these domains, there are low complexity sequences such a poly-Asp in asporin at the extreme ends. SLRPs are composed of eighteen members in human genome and are divided into five classes (Fig. 1) (Iozzo and Schaefer 2015). SLRPs undergo post translational modifications: glycosylation with glycosaminoglycan (GAG) including chondroitin sulfate, dermatan sulfate or keratan sulfate and N- or O- linked oligosaccharides, and tyrosine sulphation (Zappia et al. 2020). Their core proteins have molecular weights between 36 and 77 kDa. Class I identifies five members of biglycan (BGN), decorin (DCN), asporin (ASPN), extracellular matrix 2 (ECM2), and extracellular matrix X (ECMX). Class II containing five members are divided into 3 subgroups. Subgroup A consists of fibromodulin (FMOD) and lumican (LUM), subgroup B includes keratocan (KERA) and proline and arginine rich end leucine rich protein (PRELP), and subgroup C is composed of osteomodulin/osteoadherin (OMD). Class III is composed of osteoglycin/mimecan (OGN), epiphycan (EPN), and opticin (OPTC). Class IV SLRPs include chondroadherin (CHAD), chondroadherin-like protein (CHADL), nyctalopin (NYX), and tsukushi (TSK). Class V identifies podocan (PODN) and podocan like 1 (PODN1). Several SLRPs (asporin, PRELP, opticin, nyctalopin, and tsukushi) do not carry GAG side chains; ECM2 and ECMX has no data on GAG or glycosylation (Zappia et al. 2020). CHADL has two LRR domains which are highly similar to that in CHAD, indicating that CHADL was created by a duplication of CHAD.
Fig. 1.

The class and the repeating unit length (RUL) of individual LRRs in each protein of human SLRPs. Each box represents one structural element. Shaded areas indicate the LRR class; light yellow indicate Bacterial LRR, light blue Typical LRR. The numerical values indicate RUL of individual LRRs. N-box, conserved N-terminal disulfide bonded cap (LRRNT); E-box, ear repeat; C-box, C-terminal disulfide bonded cap (LRRCT). This figure is a modification of the one by McEwan et al (2006)
LRRs occur in tandem (Kobe and Deisenhofer 1994; Bella et al. 2008; Matsushima and Kretsinger 2016). Typical LRRs are 20–29 amino acid long. Some LRRs contain longer repeats or inversely shorter ones (Matsushima et al. 2021). Individual LRRs are separated into a hallmark motif, highly conserved segment (HCS) with the consensus of LxxLxLxxNxL, LxxLxLxxNxxL, LxxLxLxxxL and a variable segment (VS). In LRRs short β-strands (at positions 3–5) in HCS form a parallel β-sheet, which produce a super helix arrangement (called a solenoid structure). The VS part forms various secondary structures such α-helix, 3(10)-helix, polyproline II, and β-turns. At least eleven LRR classes including Bacterial, Typical, and SDS22-like have been recognized (Kobe and Kajava 2001; Matsushima et al. 2010; Kajava et al. 2008). Known structures indicate that all highly conserved hydrophobic residues such as Leu, Ile, Val, and Phe within LRR domains contribute to the hydrophobic core (Matsushima and Kretsinger 2016). The side chain of the conserved Asn at position 9 in HCS forms a hydrogen bond network in LRRs (called Asn ladder) (Kobe and Deisenhofer 1994). The LRR solenoid structure is divided in four regions of a concave (inside) surface, ascending loops, a convex (outside) surface, and descending loops (Matsushima et al. 2019). Protein, ligand interactions in LRR domains occur on the concave surface, the ascending surface, the descending surface, the capping regions, and the combinations thereof.
Several excellent reviews on SLRPs have been published (McEwan et al. 2006; Chen and Birk 2013; Iozzo and Schaefer 2015; Zappia et al. 2020; Pang et al. 2020; Zeng-Brouwers et al. 2020). We try to avoid gilding refined gold. We describe some features of amino acid sequence and structures of SLRPs. Next, we review ligand interactions and then discuss protein–ligand interfaces. Finally, we map all mutations associated with human diseases and discuss possible effects on LRR structures by the mutations.
Sequence features
Types and super motif of LRRs
Crystal structures of LRR domains in biglycan and decorin from bos taurus, and fibromodulin, osteomodulin, and chondroadherin from humans have been determined (Scott et al. 2004, 2006; Ramisch et al. 2017; Paracuellos et al. 2017; Tashima et al. 2018). The difference between the consensus sequences of respective LRR classes provides various secondary structures in the VS parts (Batkhishig et al. 2018, 2020, 2021). To get a deeper understanding of sequence—secondary structure correlations in LRRs we performed the following procedures. First, we assigned the secondary structures of the five known structures by four programs (Chebrek et al. 2014). Next, we identified a total 237 LRR units in human 18 SLRPs. Third, we divided by repeating unit length (RUL) and estimated respective consensus sequences. Finally, we could divide the 237 LRRs into three groups that are Bacterial LRR, Typical LRR, and others; Bacterial LRR are first identified in bacterial proteins such as YopM protein from Yersina pestis and Ipa4 and Ipa7 proteins from Shigell flexneri, while Typical LRR with RUL = 24 is the most abundant class at that time (Kajava 1998).
Sixty-one LRRs with RUL = 20 (17 times) and RUL = 21 (44) are Bacterial type, because the former N-subtype has the consensus of LxxLxLxxNxI xxLPxLPxx and the latter T-subtype has the consensus LxxLxLxxNxI xxLPxxLPxx (Batkhishig et al. 2018). The N-subtype VS adopts polyproline II (PPII) consisting of three to five residues, while the T-subtype VS prefers PPII conformation. The C-terminal side in the VS parts adopt β-turns, as well as the ones in the other LRRs (Fig. 2). Bacterial N-subtype LRR is characterized by a super secondary structure consisting of PPII with four, five, or six residues and a type I β-turn (Batkhishig et al. 2018).
Fig. 2.
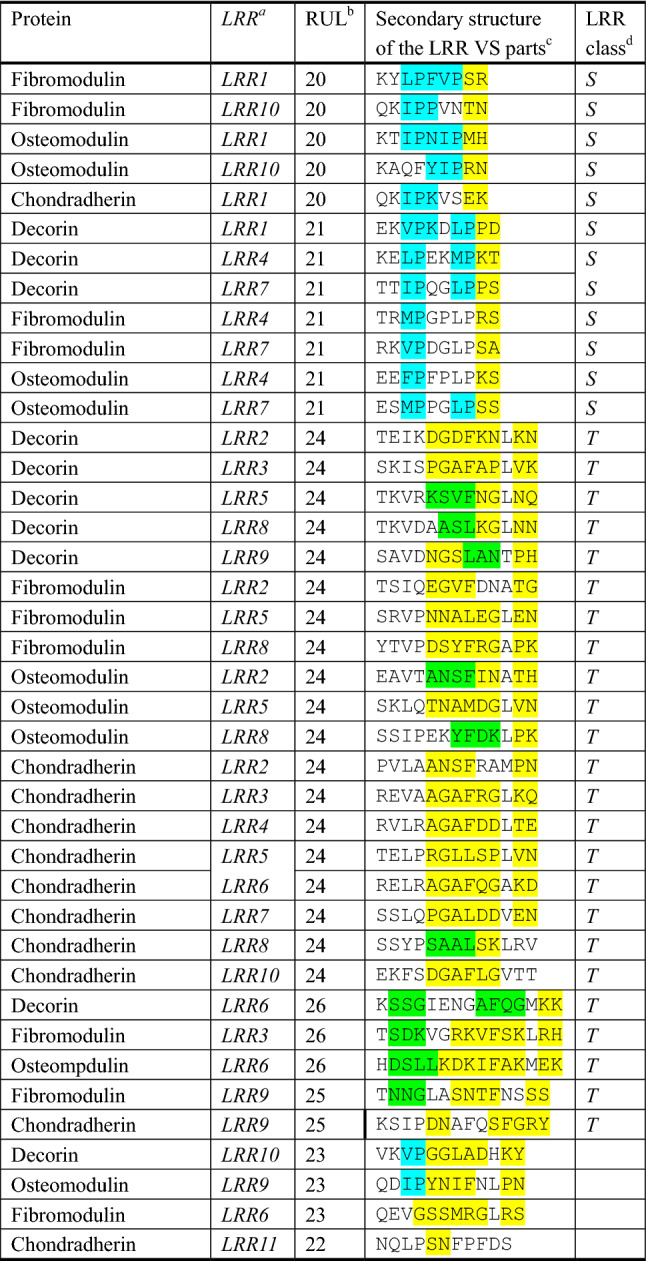
Secondary structures of the variable segment (VS) part in individual LRRs in the known structures of five SLRPs. aThe position number of LRR units. bThe repeating unit length of LRRs. cShaded areas indicate the secondary structure elements; light blue indicate polyproline II helix conformations, yellow b-turns in which the first residue and the last residues show residue i + 1 and i + 2, respectively, green α-helices in which the first residue and the last residues show residues Nc + 1 and Cc − 1, respectively; Nc is the first helical residue and Cc is the last helical residue. d“S” is Bacterial LRR and “T” is Typical LRR. Bos taurus biglycan (PDB: ID: 2FT3); Bos taurus decorin (PDB ID: 1XKU); Human fibromodulin (PDB ID: 5MX0); Human osteomodulin (PDB ID: 5YQ5); Human chondradherin (PDB ID: 5LFN)
Eighty-seven LRRs with RUL = 24 are Typical type with consensus sequence of LxxLxLxxNxL xxLxxxxFxxLxx. Thirty-four LRRs with RUL = 26 are a Typical variant with the consensus of LxxLxLxxNxL xxxxLxxxxFxxLxx. In both the central part of VS adopts tandem b-turns or 3(10)-helix (Fig. 2). The insertion of two residues in the VS part of the RUL = 26 LRR produce a short 3(10)-helix. Three LRRs with RUL = 25 and HCV = 12 (LRR4 in nyctalopin and LRR9 and LRR10 in tsukushi) are also Typical type, because the VS consensus is completely consistent to the Typical one. The remaining fourteen LRRs with RUL = 25 likely belong to Typical type, because (L/V)xxxxF, which is highly conserved in Typical LRR VS, are conserved. The secondary structure elements are similar to those of Typical LRR with RUL = 26. Eight LRRs with RUL = 23 may belong to Typical type, because the VS prefer b-turns or 3(10)-helix.
Others are four LRRs with RUL = 22 and twenty ones with longer RULs. Two repeats with RUL = 22 (LRR5 in lumican and LRR11 in the second LRR domain of chondroadherin-like protein) appear SDS22-like type with the consensus of LxxLxLxxNxL xxLxxLxxLxx. The eleventh repeat (LRR11) in classes I and II and the eigth one (LRR8) in class III is longer with RUL = 31–37, which was referred as the “ear repeat” (Scott et al. 2004). The last LRR in class IV with long RUL constitutes an LRRCT. The last two LRRs in podocin like protein 1 likely form a C-terminal capping structure through a disulfide bond.
In summary, one of the sequence features is the presence of super motifs consisting of Bacterial LRR (S) and Typical one (T) in classes I, II, III and IV (Fig. 1), as seen in the FLRT family (Matsushima et al. 2000; Matsushima and Kamiya 2000). The first nine LRRs in classes I and II SLRPs adopt almost three tandem repeats of the STT super motif. Podocan contain complete seven tandem repeats of STT, while podocin-like contains five STT repeats. Class III contains (STT)(ST). Class IV has no super motif.
Cysteine capping motifs
LRRNT typically contains four cysteines in a CxmCxCxnC pattern with n and m being variable number (Fig. 3). There are also two cysteines in ECM2, five in chondroaderin-like protein (in the second LRR domain), and seven in nyctalopin. The known structures of five SLRPs indicate that typical LRRNT forms a disulfide knot between the first LRR repeat (LRR1) and a β-hairpin, as first found in the decorin structure (Scott et al. 2004). This capping motif structure does not form a separate domain, but integrates seamlessly into the LRR architecture (McEwan et al. 2006). Two disulfide bonds are formed between the first and third cysteines, and between the second and fourth cysteines; they are C64–C70 and C68–C77 in biglycan, C55–C61 and C59–C68 in decorin; C76–C82 and C80-C92 in fibromodulin, C62–C68 and C66–C78 in osteomodulin; and C23–C29 and C27–C38 in chondroadherin. A single β-strand (β0) consists of four residues (three residues in only fibromodulin) and is the only one antiparallel to the rest. A β-turn is mostly formed between the first and second cysteines. The β0 strand contains the second and third cysteines, and the strand of LRR1 contains the fourth cysteine that is located on position 6 of the LRR1 HCV; positions 6 is occupied by the conserved hydrophobic residues such a leucine in the rest repeat. Significantly, the internal spacing between the third and fourth cysteines provides a difference in the β0–β1 loop. The β0–β1 loop contains only one β-turn in class I SLRPs (biglycan and decorin), which was also observed in the FLRT3 structure. In contrast, it contains two b-turns in class II SLRPs (fibromodulin and osteomodulin) and chondroadherin. In nyctalopin and tsukusi, there are more residues between conserved cysteines, which will produce longer β0–β1 loop. The seven cysteine clusters in nyctalopin will have one additional disulfide bond (McEwan et al. 2006). LRRNT shields the hydrophobic core of the first LRR, which prevents aggregation.
Fig. 3.

Amino acid sequence alignment of the LRRNT motif and the secondary structures identified from known structures of five SLRPs and FLRT3. aChondroadherin like protein (1) and (2) indicate the first and the second LRR domains, respectively. Shaded areas indicate the secondary structure elements; light blue indicate polyproline II helix conformations, yellow b-turns in which the first residue and the last residues show residue i + 1 and i + 2, respectively, pink b-strands. Cysteine residues are shown in white text over light blue, pink, or black background. Underlined residues “A”, “M”, and “E” are “S”, “L”, and “D” in human biglycan and decorin, respectively. Bos taurus biglycan (PDB ID: 2FT3); Bos taurus decorin (PDB ID: 1XKU); Human fibromodulin (PDB ID: 5MX0); Human osteomodulin (PDB ID: 5YQ5); Human chondradherin (PDB ID: 5LFN); FLRT3 (PDB ID: 6JBU)
Class I, II, and III SLRPs have a specific C-terminal capping motif with two cysteines containing the ear repeat (named LRRCE) (Park et al. 2008). This LRRCE motif encompasses the ear repeat, which is extended laterally, the last LRR following it, and the final b-strand closing the domain (Park et al. 2008). The last two LRRs is connected by a single disulfide bond; the crystal structures confirm the presence of this disulfide bond (C285–C318 in biglycan; C284–C317 in decorin, C334–C367 in fibromodulin, C321–C353 in osteomodulin). The cap structure forms parallel stacking of three β-strands (β11-, β12- and β13-strands) which integrates seamlessly into the LRR fold, as in the case of LRRNT. Conserved hydrophobic residues (I325 in biglycan, V324 in decorin, and I374 in fibromodulin, I229 in osteomodulin) in the final β-strand (β13) participate in the hydrophobic core the domain. Aromatic/methionine- aromatic interactions also occur in F284–F316 (biglycan), F283–F315 (decorin), F328–F333 (fibromodulin), and M320–F354 (osteomodulin) (Burley and Petsko 1985; Pal and Chakrabarti 2001).
LRRCT contains normally four cysteines having a CxCxmCxnC pattern in chondroadherin, chondradherin-like protein (the first and second LRR domains), and nyctalopin with one additional cysteine, as well as in many LRR proteins such as FLRT3, nogo receptor, and GPIBb; podocan does not have any C-terminal capping motif. The crystal structure of chondroadherin indicates that the disulfide bond connectivity is the same as that of LRRNT with the four cysteines; two sulfide bonds (C304–C326 and C306–C346) are formed. The last LRR contains a common sequence of NPWxCxCx4Lx2WL. Characteristic of this LRRCT motif is a short 3(10)-helix and a contiguous 10–13 residue α-helix on its convex side. The helical secondary structures cover the left-over hydrophobic surface of the LRR domain to prevent aggregation. Finally, both LRRNT and LRRCT would contribute to the stability of the entire domain.
Low complexity sequences
Several SLRPs have low complexity sequences flanking the LRR domains on the extreme N- an/or C-terminal sides (Kalamajski and Oldberg 2010). They are poly-Asp in asporin, Arg/Pro-rich amino acids in PRELP, Glu/Pro-rich amino acids in epiphycan, Thr/Ser-rich amino acids in opticin on the N-terminal regions, while on the C-terminal regions, Lys/Arg-rich amino acids in chondroadherin-like protein and poly-Glu in podocan. In chondroadherin-like protein, a linker connecting the two LRR domains consist of Glu/Pro-rich amino acids. In ECM2, Glu-rich amino acids intermediates between vWFC and the LRR domain. These low complexity regions may be regarded as intrinsically disordered regions (van der Lee et al. 2014). The N-terminal poly-Asp region in asporin binds calcium and regulates hydroxyapatite formation (Kalamajski et al. 2009). The basic N-terminal Arg/Pro-rich region in PRELP binds heparin and heparan sulfate (Bengtsson et al. 2000).
Tyrosine clusters
Class II SLRPs (fibromodulin, lumican, and osteomodulin) undergo sulphation in tyrosine clusters in the N-terminal regions; in addition, for osteomodulin, two adjacent tyrosines in the C-terminal region are sulphated. The remaining class II and III SLRPs also harbor sulfotyrosines in the N-terminal regions (Jensen and Karring 2020). Residues 73–130 in fibromodulin and 54–124 in osteomodulin on the extreme N-terminal sides are not visible in the electron density maps of the crystal structures, indicating that these regions are disordered; all nine tryosines in fibromodulin was substituted by serine in the molecule used for the crystal analysis (Praracuellos et al. 2017; Tashima et al. 2018). An LRR protein, platelet glycoprotein Ibα (GPIbα) binds α-thrombin. The interaction is enhanced by sulfotyrosine of Tyr278 at the C-terminus of GPIbα (Zarpellon et al. 2011). The tyrosine sulfate-rich domains of both fibromodulin and osteomodulin bind basic cluster motifs that are shared by various heparin-binding proteins and growth factors (Tillgren et al. 2009). The sulfotyrosines of fibromodulin and osteomodulin might be directly involved in the binding to their proteins.
Solenoid structures of five SLRPs
HELFIT analysis
Most solenoid structures of LRRs fold into a right-handed or left-handed super helix. The repeat number of LRRs in SLRPs ranges from 8 to 22 (Fig. 1). To get geometrical features of the LRR solenoid structures, we calculated the helix parameters by the HELFIIT analysis (Enkhbayar et al. 2014). A helix may be characterized by helix axis, helix pitch (P), helix radius (R), number of residues per turn (N), and handedness. HELFIT computes these parameters which also yield the rise per repeat unit/residue (Δz = P/N) and the rotation per repeat unit/residue in the helix (ΔΦ = 360°/N). The HELFIT analysis requires only four data points: the coordinates of the α-carbon (Cα) of residue. In LRRs, the Cα coordinates of the consensus leucine residue at position 4 in HCS, which is located in the center of individual β-strands, are used. The five SLRPs are represented by a right-handed helix. The helix parameters range over: P = 24.2 → 80.6 Å, N = 29.2 → 37.7 units/turn, R = 24.1 → 28.7 Å, Δz = 0.97 → 2.10 Å, and ΔΦ = 9.4° → 12.4°; p = 0.04 → 0.17 Å. Plots of 2⋅R⋅sin (ΔΦ/2) versus Δz is well fit by a circle with radius D.
| 1 |
The above equation is a circle equation and yields D = 5.09 ± 0.03 Å which corresponds to the inter-strand distance allowing the formation of hydrogen bonds. This equation is established in all known structures with a very few exceptions (Enkhbayar et al. 2014). The helical parameters of the five SLRPs seem to be more comparable to those of Typical LRR domains (for example, Drosophila Toll) than Bacterial LRR domains (IpaH9.8).
Multiple aromatic/methionine–aromatic interactions
The known five structures indicate that aromatic–aromatic and methionine (Met)–aromatic interactions frequently occur. These are noncovalent interactions stabilizing protein structure (Burley and Petsko 1985; Pal and Chakrabarti 2001). Examples are F74–F98 in biglycan, F72–F96 and F141–F167 in decorin, F125–F151 and F111–F137 in osteomodulin, and F71–F95–F119 in chondroradherin. The phenylalanines are a consensus hydrophobic residue at position 19 in the RUL = 24 typical LRR or position 21 in the RUL = 25 Typical LRR. The benzyl groups form stacks of Phe side chain. The Phe’s of consecutive units is called the Phe spine, which was first recognized in nogo receptor (He et al 2003). A big aromatic–aromatic interaction is observed. It is F242–F239–F364–F287–F289–W314 in chondroadherin. Others are F284–F316 in biglycan, F264–F290 in fibromodulin, Y274–F277–F296 in osteomodulin, and F259–F264 in chondroadherin. Met–aromatic interaction occurs in F72–F96–M119–F141–F167 and F141–M170–F167 in decorin, F156–M182–F208 in osteomodulin, in which more than one aromatic residue interact with one Met in a "bridging" motif of the general form aromatic-Met-aromatic (Weber and Warren 2019). Others are F328–F333–M360 in fibromodulin, M227–Y252–F253 and M320–F354 in osteomodulin. In summary, aromatic/Met–aromatic interactions is a significant factor to stabilize the entire LRR domains in SLRPs.
Ligand interactions
Collagen—binding
Decorin binds collagen I via sequence SYIRIADTNIT in LRR7, which is supportive of a concave surface binding (Kalmajski et al. 2007). The collagen-binding sites are mainly located in LRR4 and LRR5 (Sevensson et al. 1995). The deletion of M176–K201 (LRR6) and the mutation E180K (in LRR6) drastically interfered with the binding to reconstituted collagen I fibrils, indicating that an electrostatic interaction is a critical factor for the binding (Kresse et al. 1997). Decorin and asporin compete for binding to collagen via LRR10 to LRR12 (Kalmajski et al. 2009). Fibromodulin and lumican bind to same region on collagen I (Svensson et al. 2000). Fibromodulin interact with collagen cross-linking sites (Kalamajski et al. 2016). Fibromodulin binds collagen I via residues (E354 and K356) in LRR12 (Kalamajski and Oldberg 2007). Fibromodulin and lumican bind to collagen via a more proximal region located between LRR6 and LRR8 (Kalamajski and Oldberg 2009). In osteomodulin, residues E284 and E303 (in LRR 12 and LRR13, respectively) mediate collagen I-binding (Tashima et al. 2018). Taken together, collagen may span across 7–8 LRRs in these SLRPs. The collagen binding site is presumably mapped on the concave surface, the ascending loops or the combinations thereof of the LRR domains (Islam et al. 2013).
Chondroadherin binds to a sequence of GAOGPSGFQGLOGPOGPO (O is hydroxyproline) in collagen (Paracuellos et al. 2017). A GFx in the six triples is in common with several other collagen-binding proteins including OSCAR (Zhou et al. 2016). Histidines and aromatic residues are markedly concentrated on the concave surface concave surface of chondroadherin, as well as fibromodulin (Paracuellos et al. 2017). Taken together, phenylalanine in the GFX motif might participate in the collagen-binding interactions with chondroadherin through histidine–aromatic interaction and/or aromatic–aromatic interaction, while the interactions with osteomodulin is dominated by weak electrostatic forces (in particular by E284 and E303) and controlled by entropic factors (Tashima et al. 2018).
TGF—binding
Biglycan, decorin, asporin, fibromodulin, and tsukushi bind TGF-b (Yamaguchi and Ruoslahti 1988; Yamaguchi et al. 1990; Nakajima et al. 2007; Hildebrand et al. 1994; Niimori et al. 2012), as does an LRR protein, GARP (glycoprotein A repetitions predominant) with 22 LRRs (Lienart et al. 2018). Decorin fragment Leu155-Val260 (LRR5 to LRR9) interacts with TGF-β (Schoherr et al. 1998), while asporin fragment His159-Asn205 (LRR4 to LRR6) mediate its interaction with TGFβ1 (Kou et al. 2010). The binding footprint of TGF-β1 to GARP comprises the ascending and convex surfaces (LRR4 to LRR11). The five SLRPs directly might interact with TGF-β via the ascending and convex surfaces. In contrast, a modeling study indicates that TGF-β interacts with biglycan on its concave surface (Cho et al. 2016).
LRR proteins—binding
Some SLRPs bind other LRR proteins. Biglycan binds to TLR2, TLR4, and CD14 (Schaefer et al. 2005; Roedig et al. 2019). Lumican interacts with TLR4 (Wu et al. 2007). Decorin binds EGFR (Santra et al. 2002). EGFR and IGF-1 (insulin-like growth factor 1 receptor) contain five non-canonical LRRs in the L1 and L2 domains (Miyashita et al. 2014). A part of the LRR2 VS (H394-I402) in the L2 domain is essential for decorin and EGF binding. Decorin also binds and suppresses the IGF-IR (Morcavallo et al. 2014; Schonherr et al. 2005; Iozzo et al. 2011).
Other ligands
In addition to the above ligands, SLRPs belonging to classes I, II, and III interact with many other ligands including vWF (decorin) and matrix metalloproteinase-14 (MMP-14) (lumican) (Guidetti et al. 2004; Pietraszek-Gremplewicz et al. 2018). The glycosaminoglycan side chain of decorin mediates and the degree of sulfation regulates this interaction with vWF. The concave face of the LRR β-sheet in the GPIbα-VWF complex is involved in protein, ligand interactions (Sadler 2002). Similarly, the catalytic domain of MMP-14 may directly interact with the concave surface of the lumican LRR domain.
Classes IV and V SLRPs also interact with various ligands. Nyctalopin is located on the surface of photoreceptor-to-ON bipolar cell synapse in the retina (Gregg et al. 2007). Nyctalopin interacts directly with transient receptor potential cation channel subfamily member 1 (TRPM1) (Pearring, et al. 2011). Tsukushi is expressed in the primitive streak and Hensen’s node (Ahmad et al. 2018; Ohta et al. 2006). Tsukushi (TSK) binds to nodal/Vg1/TGF-β1, BMP4/chordin, Delta, FGF8, Frizzled4 and CCN2/CTGF (Ohta et al. 2004, 2011, 2019). Structural studies of the complexes remain.
Human diseases associated with mutations
Mutations in genes encoding SLRPs proteins are associated with human diseases (Matsushima et al. 2019). Exonic mutations (including missense mutations, nonsense mutations, insertions, deletions, frame shift mutations, stop codon/nonsense mutations, and silent mutations) and intronic mutations have been identified in the SLRP genes. Here we focus the exonic mutations on the LRR domains.
Spondyloepimetaphyseal Dysplasia, X-linked (SEMDX), and Meester-Loeys Syndrome (MRLS) associated with the BGN mutations
SEMDX is an X-linked recessive bone disease that impairs bone growth; it occurs only in males. It is characterized by anomalies of the spine and the epiphyses and metaphyses of the long bones. SEMDX is caused by BGN mutations (K147E, and G259V) (Cho et al. 2016). MRLS is a connective tissue disorder that is characterized by thoracic aortic aneurysms and dissections. The BGN mutations of G80S, Q303P, and Trp2* cause MRLS (Meester et al. 2017). The four missense mutations occur residues (in the HCS part) on the concave or ascending surfaces of the LRR domains.
Congential Stromal Corneal Dystrophy (CSCD) and Congenital Hereditary Stromal Dystrophy (CHSD) associated with the DCN mutations
CSCD is an inherited eye disorder that is characterized by numerous opaque flaky or feathery areas of clouding in the stroma (Bredrup et al. 2005). Mutations in DCN have been identified in families with CSCD (Bredrup et al. 2005; Rodahl et al. 2006; Kim et al. 2011; Chen et al. 2011; Jing et al. 2014). Five mutations result in the loss of the C-terminal 33 to 45 amino acids that is involved in the LRRCE motif. A truncated form lacking the 33 amino acids is retained in an unfolded protein response (Chen et al. 2013).
The DCN mutation may be linked to CHSD. CHSD is a rare autosomal dominant disorder that manifests as bilateral neonatal corneal opacification. Minute stromal opacity of the cornea gradually decreases in vision. The C346G mutation was proposed as a mild form of CHSD (Lee et al. 2012). This mutation eliminates interlocking disulfide bonds in the LRRCE motif.
Autosomal recessive cornea plana (CNA2) associated with the KERA mutations
CNA2 is characterized by a flattened corneal surface leading to a decrease in refraction, reduced visual activity, strong hyperopia, widened limbus zone, opacities in the corneal parenchyma, and marked arcus senilis (often detected at an early age) (Forsius et al. 1998). Mutations in KERA encoding keratocan cause CNA2 (Lehmann et al. 2001; Pellegata et al. 2000; Ebenezer et al. 2005; Liskova et al. 2007; Roos et al. 2015; Kumari et al. 2016; Dudakova et al. 2018; Khan 2018; Khan et al. 2006; Khan and Kambouris 2004; Dudakova et al. 2014).
Position 9 in the HCS consensus is usually occupied by Asn of which the side chain forms a network of H-bonds between the backbone carboxy and amide group of the neighboring repeats (asparagine ladder). Residues N31, N131, and N247 correspond to position 9. Thus, the missense mutations of N81A, N31D, N247S likely break the H-bonds. Position 1 and 11 in the HCS consensus is highly occupied by hydrophobic residues. T215 and I247 are located on their positions, as the sequence of the LRR8 HCS is 215-TMQLFLDNNSI-247. Threonine clearly show lower hydrophobicity than Leu, Ile or Val. However, the crystal structure of the lamprey variable lymphocyte receptor C (VLRC) with seven LRRs reveals that Thr69 (located at position 1 in the LRR1 HCS) is completely buried in the inner side of the LRR domain (Kanda et al. 2014). The charged residue substitution of the T215K mutation disrupt the hydrophobic core, that probably induces large structural changes and a subsequent mis-folding and/or an aberrant aggregation. The I225T mutation perturbs the overall folding of the LRR domain and then may induce mis-folding. P70 (in LRR1) and P208 (in LRR7) are located in the Bacterial VS part. The substitutions to Leu and Arg (P70L and P208R) perturb the preference of polyproline II conformation. Three nonsense mutations (Q174*, R279*, and R313*) could not preserve the solenoid structure. F125del will break a short b-strand in LRR4. The frameshift mutation of C343fs*26 may bring out a large change of the LRRCE structure.
Night Blindness, Congenital Stationary (CSNB) associated with the NYX mutations
CSNB is a non-progressive retinal disorder characterized by impaired night vision and predominantly by abnormal function of the rod system. There are type 1A (CSNB1A) and type 1F (CSNB1F) (Tsang and Sharma 2018, review). CSNB1A is caused by the mutations of nyctalopin (Bech-Hansen et al. 2000; Pusch et al. 2000; Zhou et al. 2015; Leroy et al. 2009; Dai et al. 2015; Pradhan et al. 2011; Wang et al. 2012; Xiao et al. 2006; Zeitz et al. 2005; Sui et al. 2008; Ivanova et al. 2019) as well as CSNB1F by those of LRIT3 (Zeitz et al. 2013, 2015; Dan et al. 2017), CACNA1F, TRPM1 (Abdelkader et al. 2018), RPM1 (AlTalbishi et al. 2019), and GNAT1 (Marmor and Zeitz 2018).
Nyctalopin contains thirteen LRRs of which most is Typical type, flanked by LRRNT and LRRCT. Many mutations have been mapped to the LRR domain (Matsushima et al. 2019). Eighteen mutations are located on highly conserved residues (at positions 1, 4, 6, and 11) in the HCS part (V46G, A64E, L91Q, L98P, L117P, L117Q, L123Q, L142P, L142R, L161R, L184P, A187K, L213Q, L232P, L235P, L280F, L285P, and L307P) and at “F” and “L” position in the “Typical” VS part (I101T and F298S). Among these mutations the charged residue substitutions (A64E, L142R, and L142R) disrupt the hydrophobic core in the LRR domain, as seen in the KERA mutation. The mutations of V46G, L91Q, I101T, L117Q, L123Q, L213Q, and F298S disturb largely hydrophobic core, while the I101T and L280F mutations induce its smaller disturbance. The remaining mutations are the substitutions to proline (L98P, L112P, L117P, L142P, L184P, L232P, L235P, L285P, and L307P).
Four mutations at Asn ladder position (N72H, N216S, N264K, and N312S) disturb a network of H-bonds. Nine mutations (C31S, C31Y, V46G, R50P, P57P, W346C, L347P, G370V, and C386R) in the LRRNT and LRRCT structures disturb sulfide bridges, folding, and stability. The mutations of A143P mutation (at position 6 in the HCS) and T258P (at position 3) disturb H-bond between β-strand that form the convex surface. The P57T, P151L and P175R mutations in the VS part change polyproline II conformation. The A177M mutation changes the conformation of plausible tandem β-turns in the VS region. The five mutations of Arg (R50P, R94P, R162P, R173P, and R257P) on the convex surface or on the ascending loop may impair interaction with a ligand such a TRPM1.
Indels (insertion and deletion) are observed. The insertions are the mutations of L155LSVPERLL (an addition of seven residues), R207RLLR and R209RCLR (an addition of three residues). These insertions probably form a bulge but keep a solenoid structure. The deletions are R29-A36del, E114-A118Adel, and A243-P246del which induce destructive structural change. The deletion of one residue of conserved hydrophobic residue occurs I101del which destabilize and then induce a structural change of the LRR domain. Nonsense mutations (Q299x) and frameshifts (H95s and D286Tfs) are also observed.
High Myopia associated with the mutations of LUM, FMOD, PRELP, OPTC, and NYX
If an eye requires -6.0 diopters or more of lens correction, it is usually considered to have high myopia (Cai et al. 2019). The mutations in genes coding opticin, lumican, fibromodilin, PRELP, and nyctalopin are associated with high myopia (Wang et al. 2009; Majava et al. 2007; Acharya et al. 2007; Yip et al. 2013; Zhang et al. 2007; Zhou et al. 2015).
The OPTC mutations of G329S and R330H occur at the C-terminus of the protein. The crystal structures of biglycan, decorin, and fibromodulin reveal that I325 (biglycan), V324 (decorin), and I374 (fibromodulin) participate in the hydrophobic cores (Scott et al. 2004, 2006; Praracuellos et al. 2017). Sequence alignment indicates that F331 in opticin corresponds to their residues. Thus, the G329 and R330H mutations disrupt the LRRCE structure. The NYX mutation of C48W disrupt disulfide bond in the LRRNT structure. The mutations of Arg (R191Q and R209P in NYX, R324P in FMOD, and R146L, R229H and R325W in OPTC may impair interaction with a ligand, as seen in the NYX mutations associated with CSNB. Similarly, the mutations of P46L at the N-terminal Tyr-rich region of fibromodulin, G33R at the N-terminal Arg/Pro-rich region of PRELP (PRELP), N348H at the LRRCT of PRELP also might prevent ligand-interaction. The mutations of L112P (NYX), M157V (PRELP), and L199P (LUM) disturb the hydrophobic core in the LRR domain. The mutations of A177T (NYX), G147D (FMOD), and T177R, P267L, and L268P (OPT) change β-turn conformation in the VS region. The mutations of P46L at the N-terminal Tyr-rich region of fibromodulin, G33R at the N-terminal Arg/Pro-rich region of PRELP (PRELP), N348H at the LRRCT motif of PRELP may impair interaction with a ligand. Furthermore, nonsense mutations (L134x and L270x in OPT) and frameshift mutation (E41fs*100) are also observed.
Cancer or Carcinoma associated with BGN
Cancer is a group of diseases that involve abnormal increases in the number of cells, with the potential to invade or spread to other parts of the body. Carcinoma are cancers derived from epithelial cells. Mutations in genes encoding various LRR proteins are associated with cancer or carcinoma. They include breast cancer in biglycan, as seen in NLRP14, densin-180, colorectal cancer in FBXL2, LRRC47, Slitrk4, and ISLR, papillary cancer in TSHR, lung cancer in MXRA5, multiple self-healing palmoplantar carcinoma (MSPC) in NLRP1, renal cell carcinoma in FBXL12, ovarian serous carcinoma in trk-A, lung adenocarcinoma in trk-B, gastric adenocarcinoma in trk-C and insulin receptor, and colorectal adenocarcinoma in ErbB4 (Matsushima et al. 2019); MSPC is an autosomal dominant disease that is characterized by recurrent keratoacanthomas in palmoplantar skin as well as in conjunctival and corneal epithelia (Zhong et al. 2016).
The BGN mutations of R266T and K288N is in breast cancer. Residues R266 and K288 on the ascending loops are exposed to solvent (Sjoblom et al. 2006). We infer that these mutations are gain-of-function.
Concluding remarks
We described some features of amino acid sequence and structures of SLRPs. The secondary structure assignment supports the presence of the super motifs consisting of Bacterial and Typical LRRs. The known five structures indicate that aromatic/methionine–aromatic interaction to stabilize the entire LRR domains frequently occur. We reviewed the ligand interactions of SLRPs. The concave surface of LRR domains is in contact with some ligands. In addition, other surfaces presumably participate in the interactions. We mapped all mutations associated with human diseases and discussed possible effects on LRR structures by the mutations.
Abbreviations
- Cα
α-Carbon of residue
- CNA2
Autosomal recessive cornea plana
- BGN
Biglycan
- CHAD
Chondroadherin
- CHADL
Chondroadherin-like protein
- CSCD
Congential Stromal Corneal Dystrophy
- CHSD
Congenital Hereditary Stromal Dystrophy
- DCN
Decorin
- LRRCE
Ear-containing LRR C-terminal capping motif
- EGFR
Epidermal growth factor receptor
- ECM
Extracellular matrix
- ECMX
Extracellular matrix X
- FMOD
Fibromodulin
- FLRT
Fibronectin-leucine-rich transmembrane protein
- GAG
Glycosaminoglycan
- P
Helix pitch
- R
Helix radius
- LUM
Lumican
- KERA
Keratocan
- LRR
Leucine rich repeat
- LRRCT
LRR C-terminal capping motif
- HCS
LRR highly conserved segment
- LRRNT
LRR N-terminal capping motif
- VS
LRR variable segment
- MMP-14
Matrix metalloproteinase-14
- MRLS
Meester-Loeys Syndrome
- CSNB
Night Blindness, Congenital Stationary
- N
Number of residues per turn in helix
- NYX
Nyctalopin
- OPTC
Opticin
- RUL
Repeating unit length of LRR
- PPII
Polyproline II helix
- GPIb
Platelet glycoprotein Ib
- PRELP
Proline and arginine rich end leucine rich protein
- Δz
Rise per repeat unit/residue in helix
- ΔΦ
Rotation per repeat unit/residue in helix
- SLRP
Small leucine rich repeat proteoglycan
- SEMDX
Spondyloepimetaphyseal Dysplasia, X-linked
- TLR
Toll-like receptor
- TGF
Transforming growth factor
- TSK
Tsukushi
- TRPM1
Transient receptor potential cation channel subfamily member 1
- vWF
Von Willebrand factor
Funding
None.
Declarations
Conflict of interest
The authors declare that they have no competing financial interests or personal relationships that could have appeared to influence the work in this paper.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Abdelkader E, AlHilali S, Neuhaus C, Bergmann C, AlMurshed T, Schatz P. Congenital stationary night blindness associated with morning glory disc malformation: a novel hemizygous mutation in CACNA1F. Ophthalmic Genet. 2018;39:659–661. doi: 10.1080/13816810.2018.1498526. [DOI] [PubMed] [Google Scholar]
- Acharya M, Mookherjee S, Bhattacharjee A, Thakur SK, Bandyopadhyay AK, Sen A, Chakrabarti S, Ray K. Evaluation of the OPTC gene in primary open angle glaucoma: functional significance of a silent change. BMC Mol Biol. 2007;8:21. doi: 10.1186/1471-2199-8-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad SAI, Anam MB, Ito N, Ohta K. Involvement of Tsukushi in diverse developmental processes. J Cell Commun Signal. 2018;12:205–210. doi: 10.1007/s12079-018-0452-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AlTalbishi A, Zelinger L, Zeitz C, Hendler K, Namburi P, Audo I, Sheffer R, Yahalom C, Khateb S, Banin E, Sharon D. RPM1 Mutations are the most common cause of autosomal recessive congenital stationary night blindness (CSNB) in the Palestinian and Israeli populations. Sci Rep. 2019;9:12047. doi: 10.1038/s41598-019-46811-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batkhishig D, Bilguun K, Enkhbayar P, Miyashita H, Kretsinger RH, Matsushima N. Super secondary structure consisting of a polyproline II helix and a β-turn in leucine rich repeats in bacterial type III secretion system effectors. Protein J. 2018;37:223–236. doi: 10.1007/s10930-018-9767-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batkhishig D, Enkhbayar P, Kretsinger RH, Matsushima N. A strong correlation between consensus sequences and unique super secondary structures in leucine rich repeats. Proteins. 2020;88:840–852. doi: 10.1002/prot.25876. [DOI] [PubMed] [Google Scholar]
- Batkhishig D, Enkhbayar P, Kretsinger RH, Matsushima N. A crucial residue in the hydrophobic core of the solenoid structure of leucine rich repeats. Biochim Biophys Acta Proteins Proteom. 2021;1869:140631. doi: 10.1016/j.bbapap.2021.140631. [DOI] [PubMed] [Google Scholar]
- Bech-Hansen NT, Naylor MJ, Maybaum TA, Sparkes RL, Koop B, Birch DG, Bergen AA, Prinsen CF, Polomeno RC, Gal A, Drack AV, Musarella MA, Jacobson SG, Young RS, Weleber RG. Mutations in NYX, encoding the leucine-rich proteoglycan nyctalopin, cause X-linked complete congenital stationary night blindness. Nat Genet. 2000;26:319–323. doi: 10.1038/81619. [DOI] [PubMed] [Google Scholar]
- Bella J, Hindle L, McEwan PA, Lovell SC. The leucine-rich repeat structure. Cell Mol Life Sci. 2008;65:2307–2333. doi: 10.1007/s00018-008-8019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtsson E, Aspberg A, Heinegard D, Sommarin Y, Spillmann D. The amino-terminal part of PRELP binds to heparin and heparan sulfate. J Biol Chem. 2000;275:40695–40702. doi: 10.1074/jbc.M007917200. [DOI] [PubMed] [Google Scholar]
- Bredrup C, Knappskog PM, Majewski J, Rødahl E, Boman H. Congenital stromal dystrophy of the cornea caused by a mutation in the decorin gene. Investig Ophthalmol Vis Sci. 2005;46:420–426. doi: 10.1167/iovs.04-0804. [DOI] [PubMed] [Google Scholar]
- Burley SK, Petsko GA. Aromatic-aromatic interaction: a mechanism of protein structure stabilization. Science. 1985;229:23–28. doi: 10.1126/science.3892686. [DOI] [PubMed] [Google Scholar]
- Cai XB, Shen SR, Chen DF, Zhang Q, Jin ZB. An overview of myopia genetics. Exp Eye Res. 2019;188:107778. doi: 10.1016/j.exer.2019.107778. [DOI] [PubMed] [Google Scholar]
- Chebrek C, Leonard S, de Brevern AG, Gelly JC. PolyprOnline: polyproline helix II and secondary structure assignment database. Database. 2014;2009:1–14. doi: 10.1093/database/bau102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Birk DE. The regulatory roles of small leucine-rich proteoglycans in extracellular assembly. FEBS J. 2013;280:2120–2137. doi: 10.1111/febs.12136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Sun M, Meng X, Iozzo RV, Kao WW, Birk DE. Pathophysiological mechanisms of autosomal dominant congenital stromal corneal dystrophy: C-terminal-truncated decorin results in abnormal matrix assembly and altered expression of small leucine-rich proteoglycans. Am J Pathol. 2011;179:2409–2419. doi: 10.1016/j.ajpath.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Sun M, Iozzo RV, Kao WW, Birk DE. Intracellularly-retained decorin lacking the C-terminal ear repeat causes ER stress: a cell-based etiological mechanism for congenital stromal corneal dystrophy. Am J Pathol. 2013;183:247–256. doi: 10.1016/j.ajpath.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SY, Bae JS, Kim NKD, Forzano F, Girisha KM, Baldo C, Faravelli F, Cho TJ, Kim D, Lee KY, Ikegawa S, Shim JS, Ko AR, Miyake N, Nishimura G, Superti-Furga A, Spranger J, Kim OH, Park WY, Jin DK. BGN mutations in X-linked spondyloepimetaphyseal dysplasia. Am J Hum Genet. 2016;98:1243–1248. doi: 10.1016/j.ajhg.2016.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai S, Ying M, Wang K, Wang L, Han R, Hao P, Li N. Two novel NYX gene mutations in the Chinese families with X-linked congenital stationary night blindness. Sci Rep. 2015;5:12679. doi: 10.1038/srep12679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan H, Song X, Li J, Xing Y, Li T. Mutation screening of the LRIT3, CABP4, and GPR179 genes in Chinese patients with Schubert-Bornschein congenital stationary night blindness. Ophthalmic Genet. 2017;38:206–210. doi: 10.1080/13816810.2016.1193876. [DOI] [PubMed] [Google Scholar]
- Dudakova L, Palos M, Hardcastle AJ, Liskova P. Corneal endothelial findings in a Czech patient with compound heterozygous mutations in KERA. Ophthalmic Genet. 2014;35:252–254. doi: 10.3109/13816810.2013.811272. [DOI] [PubMed] [Google Scholar]
- Dudakova L, Vercruyssen JHJ, Balikova I, Postolache L, Leroy BP, Skalicka P, Liskova P. Analysis of KERA in four families with cornea plana. Acta Ophthalmol. 2018;96(1):e87–e91. doi: 10.1111/aos.13484. [DOI] [PubMed] [Google Scholar]
- Ebenezer ND, Patel CB, Hariprasad SM, Chen LL, Patel RJ, Hardcastle AJ, Allen RC. Clinical and molecular characterization of a family with autosomal ecessive cornea plana. Arch Ophthalmol. 2005;123:1248–1253. doi: 10.1001/archopht.123.9.1248. [DOI] [PubMed] [Google Scholar]
- Enkhbayar P, Miyashita H, Kretsinger RH, Matsushima N. Helical parameters and correlations of tandem leucine rich repeats in proteins. J Proteomics Bioinform. 2014;7:139–150. [Google Scholar]
- Forsius H, Damsten M, Eriksson AW, Fellman J, Lindh S, Tahvanainen E. Autosomal recessive cornea plana. A clinical and genetic study of 78 cases in Finland. Acta Ophthalmol Scand. 1998;76:196–203. doi: 10.1034/j.1600-0420.1998.760215.x. [DOI] [PubMed] [Google Scholar]
- Gregg RG, Kamermans M, Klooster J, Lukasiewicz PD, Peachey NS, Vessey KA, McCall MA. Nyctalopin expression in retinal bipolar cells restores visual function in a mouse model of complete X-linked congenital stationary night blindness. J Neurophysiol. 2007;98:3023–3033. doi: 10.1152/jn.00608.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidetti GF, Bartolini B, Bernardi B, Tira ME, Berndt MC, Balduini C, Torti M. Binding of von Willebrand factor to the small proteoglycan decorin. FEBS Lett. 2004;574:95–100. doi: 10.1016/j.febslet.2004.08.011. [DOI] [PubMed] [Google Scholar]
- He XL, Bazan JF, McDermott G, Park JB, Wang K, Tessier-Lavigne M, He Z, Garcia KC. Structure of the Nogo receptor ectodomain: a recognition module implicated in myelin inhibition. Neuron. 2003;38:177–185. doi: 10.1016/s0896-6273(03)00232-0. [DOI] [PubMed] [Google Scholar]
- Hildebrand A, Romarís M, Rasmussen LM, Heinegard D, Twardzik DR, Border WA, Ruoslahti E (1994) Interaction of the small interstitial proteoglycans biglycan, decorin and fibromodulin with transforming growth factor beta. Biochem J 302:527–534. 10.1042/bj3020527 [DOI] [PMC free article] [PubMed]
- Hindson VJ, Gallagher JT, Halfter W, Bishop PN (2019) Opticin binds to heparan and chondroitin sulfate proteoglycans. Arch Biochem Biophys 672:108053. 10.1167/iovs.05-0883 [DOI] [PubMed]
- Iozzo RV, Schaefer L. Proteoglycan form and function: a comprehensive nomenclature of proteoglycans. Matrix Biol. 2015;42:11–55. doi: 10.1016/j.matbio.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iozzo RV, Buraschi S, Genua M, Xu SQ, Solomides CC, Peiper SC, Gomella LG, Owens RC, Morrione A. Decorin antagonizes IGF receptor I (IGF-IR) function by interfering with IGF-IR activity and attenuating downstream signaling. J Biol Chem. 2011;286:34712–43421. doi: 10.1074/jbc.M111.262766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam M, Gor J, Perkins SJ, Ishikawa Y, Bächinger HP, Erhard Hohenester E. The concave face of decorin mediates reversible dimerization and collagen binding. J Biol Chem. 2013;288:35526–35533. doi: 10.1074/jbc.M113.504530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova ME, Zolnikova IV, Gorgisheli KV, Atarshchikov DS, Ghosh P, Barh D. Novel frameshift mutation in NYX gene in a Russian family with complete congenital stationary night blindness. Ophthalmic Genet. 2019;40:558–563. doi: 10.1080/13816810.2019.1698617. [DOI] [PubMed] [Google Scholar]
- Jensen MM, Karring H. The origins and developments of sulfation-prone tyrosine-rich and acidic N- and C-terminal extensions of class ll and lll small leucine-rich repeat proteins shed light on connective tissue evolution in vertebrates. BMC Evol Biol. 2020;20:73. doi: 10.1186/s12862-020-01634-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing Y, Kumar PR, Zhu L, Edward DP, Tao S, Wang L, Chuck R, Zhang C. Novel decorin mutation in a Chinese family with congenital stromal corneal dystrophy. Cornea. 2014;33:288–293. doi: 10.1097/ICO.0000000000000055. [DOI] [PubMed] [Google Scholar]
- Kajava AV. Structural diversity of leucine-rich repeat proteins. J Mol Biol. 1998;277:519–527. doi: 10.1006/jmbi.1998.1643. [DOI] [PubMed] [Google Scholar]
- Kajava AV, Anisimova M, Peeters N. Origin and evolution of GALA-LRR, a new member of the CC-LRR subfamily: From plants to bacteria? PLoS ONE. 2008;3:e1694. doi: 10.1371/journal.pone.0001694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalamajski S, Oldberg A. Fibromodulin binds collagen type I via Glu-353 and Lys-355 in leucine-rich repeat 11. J Biol Chem. 2007;282:26740–26745. doi: 10.1074/jbc.M704026200. [DOI] [PubMed] [Google Scholar]
- Kalamajski S, Oldberg A. Homologous sequence in lumican and fibromodulin leucine-rich repeat 5–7 competes for collagen binding. J Biol Chem. 2009;284:534–539. doi: 10.1074/jbc.M805721200. [DOI] [PubMed] [Google Scholar]
- Kalamajski S, Oldberg A. The role of small leucine-rich proteoglycans in collagen fibrillogenesis. Matrix Biol. 2010;29:248–253. doi: 10.1016/j.matbio.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Kalamajski S, Aspberg A, Oldberg A. The decorin sequence SYIRIADTNIT binds collagen type I. J Biol Chem. 2007;282:16062–16067. doi: 10.1074/jbc.M700073200. [DOI] [PubMed] [Google Scholar]
- Kalamajski S, Aspberg A, Lindblom K, Heinegård D, Oldberg A. Asporin competes with decorin for collagen binding, binds calcium and promotes osteoblast collagen mineralization. Biochem J. 2009;423(1):53–59. doi: 10.1042/BJ20090542. [DOI] [PubMed] [Google Scholar]
- Kalamajski S, Bihan D, Bonna A, Rubin K, Farndale RW. Fibromodulin interacts with collagen cross-linking sites and activates lysyl oxidase. J Biol Chem. 2016;291:7951–7960. doi: 10.1074/jbc.M115.693408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda R, Sutoh Y, Kasamatsu J, Maenaka K, Kasahara M, Ose T. Crystal structure of the lamprey variable lymphocyte receptor C reveals an unusual feature in its N-terminal capping module. PLoS ONE. 2014;9:e85875. doi: 10.1371/journal.pone.0085875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan AO. Corneal ectasia in a boy with homozygous KERA mutation. Ophthalmic Genet. 2018;39:141–143. doi: 10.1080/13816810.2017.1350724. [DOI] [PubMed] [Google Scholar]
- Khan A, Kambouris M. A novel KERA mutation associated with autosomal recessive cornea plana. Ophthalmic Genet. 2004;25:147–152. doi: 10.1080/13816810490514397. [DOI] [PubMed] [Google Scholar]
- Khan AO, Aldahmesh M, Meyer B. Recessive cornea plana in the Kingdom of Saudi Arabia. Ophthalmology. 2006;113:1773–1778. doi: 10.1016/j.ophtha.[CNA2]. [DOI] [PubMed] [Google Scholar]
- Kim JH, Ko JM, Lee I, Kim JY, Kim MJ, Tchah H. A novel mutation of the decorin gene identified in a Korean family with congenital hereditary stromal dystrophy. Cornea. 2011;30:1473–1477. doi: 10.1097/ICO.0b013e3182137788. [DOI] [PubMed] [Google Scholar]
- Kobe B, Deisenhofer J. The leucine-rich repeat: a versatile binding motif. Trends Biochem Sci. 1994;19:415–421. doi: 10.1016/0968-0004(94)90090-6. [DOI] [PubMed] [Google Scholar]
- Kobe B, Kajava AV. The leucine-rich repeat as a protein recognition motif. Curr Opin Struct Biol. 2001;11:725–732. doi: 10.1016/s0959-440x(01)00266-4. [DOI] [PubMed] [Google Scholar]
- Kou I, Nakajima M, Ikegawa S (2010) Binding characteristics of the osteoarthritis-associated protein asporin. J Bone Miner Metab 28:395–402. 10.1007/s00774-009-0145-8 [DOI] [PubMed]
- Kresse H, Liszio C, Schönherr E, Fisher LW. Critical role of glutamate in a central leucine-rich repeat of decorin for interaction with type I collagen. J Biol Chem. 1997;272:18404–18410. doi: 10.1074/jbc.272.29. [DOI] [PubMed] [Google Scholar]
- Kumari D, Tiwari A, Choudhury M, Kumar A, Rao A, Dixit M. A novel KERA mutation in a case of autosomal recessive cornea plana with primary angle-closure glaucoma. J Glaucoma. 2016;25:e106–109. doi: 10.1097/IJG.0000000000000258. [DOI] [PubMed] [Google Scholar]
- Lee JH, Ki CS, Chung ES, Chung TY. A novel decorin gene mutation in congenital hereditary stromal dystrophy: a Korean family. Korean J Ophthalmol. 2012;6:301–305. doi: 10.3341/kjo.2012.26.4.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann OJ, El-ashry MF, Ebenezer ND, Ocaka L, Francis PJ, Wilkie SE, Patel RJ, Ficker L, Jordan T, Khaw PT, Bhattacharya SS. A novel keratocan mutation causing autosomal recessive cornea plana. Investig Ophthalmol Vis Sci. 2001;42:3118–3122. [PubMed] [Google Scholar]
- Leroy BP, Budde BS, Wittmer M, De Baere E, Berger W, Zeitz C. A common NYX mutation in Flemish patients with X linked CSNB. Br J Ophthalmol. 2009;93:692–696. doi: 10.1136/bjo.2008.143727. [DOI] [PubMed] [Google Scholar]
- Liénart S, Merceron R, Vanderaa C, Lambert F, Colau D, Stockis J, van der Woning B, De Haard H, Saunders M, Coulie PG, Savvides SN, Lucas S. Structural basis of latent TGF-β1 presentation and activation by GARP on human regulatory T cells. Science. 2018;362:952–956. doi: 10.1126/science.aau2909. [DOI] [PubMed] [Google Scholar]
- Liskova P, Hysi PG, Williams D, Ainsworth JR, Shah S, de la Chapelle A, Tuft SJ, Bhattacharya SS. Study of p.N247S KERA mutation in a British family with cornea plana. Mol Vis. 2007;13:1339–1347. [PubMed] [Google Scholar]
- Majava M, Bishop PN, Hagg P, Scott PG, Rice A, Inglehearn C, Hammond CJ, Spector TD, Ala-Kokko L, Mannikko M. Novel mutations in the small leucine-rich repeat protein/proteoglycan (SLRP) genes in high myopia. Hum Mutat. 2007;28:336–344. doi: 10.1002/humu.20444. [DOI] [PubMed] [Google Scholar]
- Marmor MF, Zeitz C. Riggs-type dominant congenital stationary night blindness: ERG findings, a new GNAT1 mutation and a systemic association. Doc Ophthalmol. 2018;137:57–62. doi: 10.1007/s10633-018-9651-0. [DOI] [PubMed] [Google Scholar]
- Matsushima N, Kamiya M. Super-motifs of leucine-rich repeats (LRRs) proteins. Genom Inf. 2000;11:343–345. [Google Scholar]
- Matsushima N, Kretsinger R. Leicine rich repeats: sequences, structures, ligand-interactions, and evolution. Saarbrücken: LAMBERT Academic Publishing; 2016. [Google Scholar]
- Matsushima N, Ohyanagi T, Tanaka T, Kretsinger RH. Super-motifs and evolution of tandem leucine-rich repeats within the small proteoglycans—biglycan, decorin, lumican, fibromodulin, PRELP, keratocan, osteoadherin, epiphycan, and osteoglycin. Proteins. 2000;38:210–225. doi: 10.1002/(sici)1097-0134(20000201)38:2<210::aid-prot9>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- Matsushima N, Miyashita H, Mikami T, Kuroki Y. A nested leucine rich repeat (LRR) domain: the precursor of LRRs is a ten or eleven residue motif. BMC Microbiol. 2010;10:235. doi: 10.1186/1471-2180-10-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima N, Takatsuka S, Miyashita H, Tamaki S, Kretsinger RH. Leucine rich repeat proteins: Sequences, mutations, structures and diseases. Protein Pept Lett. 2019;26:108–131. doi: 10.2174/0929866526666181208170027. [DOI] [PubMed] [Google Scholar]
- Matsushima N, Miyashita H, Tamaki S, Kretsinger RH. Shrinking of repeating unit length in leucine-rich repeats from double-stranded DNA viruses. Arch Virol. 2021;166:43–64. doi: 10.1007/s00705-020-04820-2. [DOI] [PubMed] [Google Scholar]
- McEwan PA, Scott PG, Bishop PN, Bella J. Structural correlations in the family of small leucine-rich repeat proteins and proteoglycans. J Struct Biol. 2006;155:294–305. doi: 10.1016/j.jsb.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Meester JA, Vandeweyer G, Pintelon I, Lammens M, van Hoorick L, de Belder S, Waitzman K, Young L, Markham LW, Vogt J, Richer J, Beauchesne LM, Unger S, Superti-Furga A, Prsa M, Dhillon R, Reyniers E, Dietz HC, Wuyts W, Mortier G, Verstraeten A, van Laer L, Loeys BL. Loss-of-function mutations in the X-linked biglycan gene cause a severe syndromic form of thoracic aortic aneurysms and dissections. Genet Med. 2017;19:386–395. doi: 10.1038/gim.2016.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita H, Kretsinger, RH, and Matsushima N. (2014) Comparative structural analysis of the extracellular regions of the insulin and epidermal growth factor receptors whose L1 and L2 domains have non-canonical, leucine rich repeats. Enliven: Bioinformatics 1:1–9.
- Morcavallo A, Buraschi S, Xu SQ, Belfiore A, Schaefer L, Iozzo RV, Morrione A. Decorin differentially modulates the activity of insulin receptor isoform A ligands. Matrix Biol. 2014;35:82–90. doi: 10.1016/j.matbio.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima M, Kizawa H, Saitoh M, Kou I, Miyazono K, Ikegawa S. Mechanisms for asporin function and regulation in articular cartilage. J Biol Chem. 2007;282:32185–32192. doi: 10.1074/jbc.M700522200. [DOI] [PubMed] [Google Scholar]
- Niimori D, Kawano R, Felemban A, Niimori-Kita K, Tanaka H, Ihn H, Ohta K. Tsukushi controls the hair cycle by regulating TGF-b1 signaling. Dev Biol. 2012;372:81–87. doi: 10.1016/j.ydbio.2012.08.030. [DOI] [PubMed] [Google Scholar]
- Ohta K, Lupo G, Kuriyama S, Keynes R, Holt CE, Harris WA, Tanaka H, Ohnuma S. Tsukushi functions as an organizer inducer by inhibition of BMP activity in cooperation with chordin. Dev Cell. 2004;7:347–358. doi: 10.1016/j.devcel.2004.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta K, Kuriyama S, Okafuji T, Gejima R, Ohnuma S, Tanaka H. Tsukushi cooperates with VG1 to induce primitive streak and Hensen's node formation in the chick embryo. Development. 2006;133:3777–3786. doi: 10.1242/dev.02579. [DOI] [PubMed] [Google Scholar]
- Ohta K, Ito A, Kuriyama S, Lupo G, Kosaka M, Ohnuma S, Nakagawa S, Tanaka H. Tsukushi functions as a Wnt signaling inhibitor by competing with Wnt2b for binding to transmembrane protein Frizzled4. Proc Natl Acad Sci USA. 2011;108:14962–14967. doi: 10.1073/pnas.1100513108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta K, Aoyama E, Ahmad SAI, Ito N, Anam MB, Kubota S, Takigawa M. CCN2/CTGF binds the small leucine rich proteoglycan protein Tsukushi. J Cell Commun Signal. 2019;13:113–118. doi: 10.1007/s12079-018-0487-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal D, Chakrabarti P. Non-hydrogen bond interactions involving the methionine sulfur atom. J Biomol Struct Dyn. 2001;19(115):128. doi: 10.1080/07391102.2001.10506725. [DOI] [PubMed] [Google Scholar]
- Pang X, Dong N, Zheng Z. Small leucine-rich proteoglycans in skin wound healing. Front Pharmacol. 2020;10:1649. doi: 10.3389/fphar.2019.01649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paracuellos P, Kalamajski S, Bonna A, Bihan D, Farndale RW, Erhard Hohenester E. Structural and functional analysis of two small leucine-rich repeat proteoglycans, fibromodulin and chondroadherin. Matrix Biol. 2017;63:106–116. doi: 10.1016/j.matbio.2017.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Huxley-Jones J, Ray J, Boot-Handford P, Bishop PN, Attwood TK, Bella J. LRRCE: a leucine-rich repeat cysteine capping motif unique to the chordate lineage. BMC Genom. 2008;9:599. doi: 10.1186/1471-2164-9-599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearring JN, Bojang P, Jr, Shen Y, Koike C, Furukawa T, Nawy S, Gregg RG. A role for nyctalopin, a small leucine-rich repeat protein, in localizing the TRP melastatin 1 channel to retinal depolarizing bipolar cell dendrites. J Neurosci. 2011;31:10060–10066. doi: 10.1523/JNEUROSCI.1014-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegata NS, Dieguez-Lucena JL, Joensuu T, Lau S, Montgomery KT, Krahe R, Kivela T, Kucherlapati R, Forsius H, de la Chapelle A. Mutations in KERA, encoding keratocan, cause cornea plana. Nat Genet. 2000;25:91–95. doi: 10.1038/75664. [DOI] [PubMed] [Google Scholar]
- Pietraszek-Gremplewicz K, Karamanou K, Niang A, Dauchez M, Belloy N, Maquart FX, Baud S, Brézillon S. Small leucine-rich proteoglycans and matrix metalloproteinase-14: key partners? Matrix Biol. 2018;75–76:271–285. doi: 10.1016/j.matbio.2017.12.006. [DOI] [PubMed] [Google Scholar]
- Pradhan M, Sharp D, Mora J, Wittmer M, Berger W, Vincent A. A novel NYX mutation associated with X-linked congenital stationary night blindness in a New Zealand family. J Clin Exp Oohthamol. 2011;2:1–4. [Google Scholar]
- Pusch CM, Zeitz C, Brandau O, Pesch K, Achatz H, Feil S, Scharfe C, Maurer J, Jacobi FK, Pinckers A, Andreasson S, Hardcastle A, Wissinger B, Berger W, Meindl A. The complete form of X-linked congenital stationary night blindness is caused by mutations in a gene encoding a leucine-rich repeat protein. Nat Genet. 2000;26:324–327. doi: 10.1038/81627. [DOI] [PubMed] [Google Scholar]
- Ramisch S, Pramhed A, Tillgren V, Aspberg A, Logan DT. Crystal structure of human chondroadherin: solving a difficult molecular-replacement problem using de novo models. Acta Crystallogr D Struct Biol. 2017;73:53–63. doi: 10.1107/S205979831601980X. [DOI] [PubMed] [Google Scholar]
- Rodahl E, Ginderdeuren RV, Knappskog PM, Bredrup C, Boman H. A second decorin frame shift mutation in a family with congenital stromal corneal dystrophy. Am J Ophthalmol. 2006;142:520–521. doi: 10.1016/j.ajo.2006.03.064. [DOI] [PubMed] [Google Scholar]
- Roedig H, Nastase MV, Frey H, Moreth K, Zeng-Brouwers J, Poluzzi C, Hsieh LT, Brandts C, Fulda S, Wygrecka M, Schaefer L. Biglycan is a new high-affinity ligand for CD14 in macrophages. Matrix Biol. 2019;77:4–22. doi: 10.1016/j.matbio.2018.05.006. [DOI] [PubMed] [Google Scholar]
- Roos L, Bertelsen B, Harris P, Bygum A, Jensen H, Gronskov K, Tumer Z. Case report: a novel KERA mutation associated with cornea plana and its predicted effect on protein function. BMC Med Genet. 2015;16:40. doi: 10.1186/s12881-015-0179-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadler JE. Biomedicine. Contact-how platelets touch von Willebrand factor. Science. 2002;297:1128–1129. doi: 10.1126/science. [DOI] [PubMed] [Google Scholar]
- Santra M, Reed CC, Iozzo RV. Decorin binds to a narrow region of the epidermal growth factor (EGF) receptor, partially overlapping but distinct from the EGF-binding epitope. J Biol Chem. 2002;277:35671–35681. doi: 10.1074/jbc.M205317200. [DOI] [PubMed] [Google Scholar]
- Schaefer L, Babelova A, Kiss E, Hausser HJ, Baliova M, Krzyzankova M, Marsche G, Young MF, Mihalik D, Götte M, Malle E, Schaefer RM, Gröne HJ. The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J Clin Investig. 2005;115:2223–2233. doi: 10.1172/JCI23755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönherr E, Broszat M, Brandan E, Bruckner P, Kresse H. Decorin core protein fragment Leu155-Val260 interacts with TGF-beta but does not compete for decorin binding to type I collagen. Arch Biochem Biophys. 1998;355:241–248. doi: 10.1006/abbi.1998.0720. [DOI] [PubMed] [Google Scholar]
- Schönherr E, Sunderkötter C, Iozzo RV, Schaefer L. Decorin, a novel player in the insulin-like growth factor system. J Biol Chem. 2005;280:15767–15772. doi: 10.1074/jbc.M500451200. [DOI] [PubMed] [Google Scholar]
- Scott PG, McEwan PA, Dodd CM, Bergmann EM, Bishop PN, Bella J. Crystal structure of the dimeric protein core of decorin, the archetypal small leucine-rich repeat proteoglycan. Proc Natl Acad Sci USA. 2004;101:15633–15638. doi: 10.1073/pnas.0402976101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott PG, Dodd CM, Bergmann EM. Crystal structure of the biglycan dimer core protein. J Biol Chem. 2006;281:13324–13332. doi: 10.1074/jbc.M513470200. [DOI] [PubMed] [Google Scholar]
- Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, Meeh P, Markowitz SD, Willis J, Dawson D, Willson JK, Gazdar AF, Hartigan J, Wu L, Liu C, Parmigiani G, Park BH, Bachman KE, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- Sui R, Li F, Zhao J, Jiang R. Clinical and genetic characterization of a Chinese family with CSNB1. Adv Exp Med Biol. 2008;613:245–252. doi: 10.1007/978-0-387-74904-4_28. [DOI] [PubMed] [Google Scholar]
- Svensson L, Heinegård D, Oldberg A. Decorin-binding sites for collagen type I are mainly located in leucine-rich repeats 4–5. J Biol Chem. 1995;270:20712–20716. doi: 10.1074/jbc.270.35.20712. [DOI] [PubMed] [Google Scholar]
- Svensson L, Närlid I, Oldberg A. Fibromodulin and lumican bind to the same region on collagen type I fibrils. FEBS Lett. 2000;470:178–182. doi: 10.1016/s0014-5793(00)01314-4. [DOI] [PubMed] [Google Scholar]
- Tashima T, Nagatoishi S, Caaveiro JMM, Nakakido M, Sagara H, Kusano-Arai O, Iwanari H, Mimuro H, Hamakubo T, Ohnuma SI, Tsumoto K. Molecular basis for governing the morphology of type-I collagen fibrils by Osteomodulin. Commun Biol. 2018;1:33–33. doi: 10.1038/s42003-018-0038-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillgren V, Onnerfjord P, Haglund L, Heinegård D. The tyrosine sulfate-rich domains of the LRR proteins fibromodulin and osteoadherin bind motifs of basic clusters in a variety of heparin-binding proteins, including bioactive factors. J Biol Chem. 2009;284:28543–28553. doi: 10.1074/jbc.M109.047076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang SH, Sharma T. Congenital stationary night blindness. Adv Exp Med Biol. 2018;1085:61–64. doi: 10.1007/978-3-319-95046-4_13. [DOI] [PubMed] [Google Scholar]
- van der Lee R, Buljan M, Lang B, Weatheritt RJ, Daughdrill GW, Dunker AK, Fuxreiter M, Gough J, Gsponer J, Jones DT, Kim PM, Kriwacki RW, Oldfield CJ, Pappu RV, Tompa P, Uversky VN, Wright PE, Babu MM. Classification of intrinsically disordered regions and proteins. Chem Rev. 2014;114:6589–6631. doi: 10.1021/cr400525m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Li S, Xiao X, Guo X, Zhang Q. An evaluation of OPTC and EPYC as candidate genes for high myopia. Mol Vis. 2009;15:2045–2049. [PMC free article] [PubMed] [Google Scholar]
- Wang Q, GaoY LS, Guo X, Zhang Q. Mutation screening of TRPM1, GRM6, NYX and CACNA1F genes in patients with congenital stationary night blindness. Int J Mol Med. 2012;30:521–526. doi: 10.3892/ijmm.2012.1039. [DOI] [PubMed] [Google Scholar]
- Weber DS, Warren JJ (2019) The interaction between methionine and two aromatic amino acids is an abundant and multifunctional motif in proteins. Arch Biochem Biophys 672:108053. 10.1016/j.abb.2019.07.018 [DOI] [PubMed]
- Wu F, Vij N, Roberts L, Lopez-Briones S, Joyce S, Chakravarti S. A novel role of the lumican core protein in bacterial lipopolysaccharide-induced innate immune response. J Biol Chem. 2007;282:26409–26417. doi: 10.1074/jbc.M702402200. [DOI] [PubMed] [Google Scholar]
- Xiao X, Jia X, Guo X, Li S, Yang Z, Zhang Q. CSNB1 in Chinese families associated with novel mutations in NYX. J Hum Genet. 2006;51:634–640. doi: 10.1007/s10038-006-0406-5. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Ruoslahti E. Expression of human proteoglycan in Chinese hamster ovary cells inhibits cell proliferation. Nature. 1988;336:244–246. doi: 10.1038/336244a0. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Mann DM, Ruoslahti E. Negative regulation of transforming growth factor-beta by the proteoglycan decorin. Nature. 1990;346:281–284. doi: 10.1038/346281a0. [DOI] [PubMed] [Google Scholar]
- Yip SP, Li CC, Yiu WC, Hung WH, Lam WW, Lai MC, Ng PW, Fung WY, Chu PH, Jiang B, Chan HH, Yap MK. A novel missense mutation in the NYX gene associated with high myopia. Ophthalmic Physiol Opt. 2013;33:346–353. doi: 10.1111/opo.12036. [DOI] [PubMed] [Google Scholar]
- Zappia J, Joiret M, Sanchez C, Lambert C, Geris L, Muller M, Henrotin Y. From translation to protein degradation as mechanisms for regulating biological functions: a review on the SLRP family in skeletal tissues. Biomolecules. 2020;10:80. doi: 10.3390/biom10010080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarpellon A, Celikel R, Roberts JR, McClintock RA, Mendolicchio GL, Moore KL, Jing H, Varughese KI, Ruggeri ZM. Binding of alpha-thrombin to surface-anchored platelet glycoprotein Ib(alpha) sulfotyrosines through a two-site mechanism involving exosite I. Proc Natl Acad Sci USA. 2011;108:8628–8633. doi: 10.1073/pnas.1017042108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitz C, Minotti R, Feil S, Matyas G, Cremers FP, Hoyng CB, Berger W. Novel mutations in CACNA1F and NYX in Dutch families with X-linked congenital stationary night blindness. Mol Vis. 2005;11:179–183. [PubMed] [Google Scholar]
- Zeitz C, Robson AG, Audo I. Congenital stationary night blindness: an analysis and update of genotype-phenotype correlations and pathogenic mechanisms. Prog Retin Eye Res. 2015;45:58–110. doi: 10.1016/j.preteyeres.2014.09.001. [DOI] [PubMed] [Google Scholar]
- Zeitz C, Jacobson SG, Hamel CP, Bujakowska K, Neuille M, Orhan E, Zanlonghi X, Lancelot ME, Michiels C, Schwartz SB, Bocquet B, Antonio A, Audier C, Letexier M, Saraiva JP, Luu TD, Sennlaub F, Nguyen H, Poch O, Dollfus H, Lecompte O, Kohl S, Sahel JA, Bhattacharya SS, Audo I. Whole-exome sequencing identifies LRIT3 mutations as a cause of autosomal-recessive complete congenital stationary night blindness. Am J Hum Genet. 2013;92:67–75. doi: 10.1016/j.ajhg.2012.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng-Brouwers J, Pandey S, Trebicka J, Wygrecka M, Schaefer L. Communications via the small leucine-rich proteoglycans: molecular specificity in inflammation and autoimmune diseases. J Histochem Cytochem. 2020;68:887–906. doi: 10.1369/0022155420930303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Xiao X, Li S, Jia X, Yang Z, Huang S, Caruso RC, Guan T, Sergeev Y, Guo X, Hejtmancik JF. Mutations in NYX of individuals with high myopia, but without night blindness. Mol Vis. 2007;13:330–336. [PMC free article] [PubMed] [Google Scholar]
- Zhong FL, Mamai O, Sborgi L, Boussofara L, Hopkins R, Robinson K, Szeverenyi I, Takeichi T, Balaji R, Lau A, Tye H, Roy K, Bonnard C, Ahl PJ, Jones LA, Baker PJ, Lacina L, Otsuka A, Fournie PR, Malecaze F, Lane EB, Akiyama M, Kabashima K, Connolly JE, Masters SL, Soler VJ, Omar SS, McGrath JA, Nedelcu R, Gribaa M, Denguezli M, Saad A, Hiller S, Reversade B (2016) Germline NLRP1 mutations cause skin inflammatory and cancer susceptibility syndromes via inflammasome activation. Cell 167:187–202. e17. 10.1016/j.cell.2016.09.001 [DOI] [PubMed]
- Zhou L, Li T, Song X, Li Y, Li H, Dan H. NYX mutations in four families with high myopia with or without CSNB1. Mol Vis. 2015;21:213–223. [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Hinerman JM, Blaszczyk M, Miller JL, Conrady DG, Barrow AD, Chirgadze DY, Bihan D, Farndale RW, Herr AB. Structural basis for collagen recognition by the immune receptor OSCAR. Blood. 2016;127(5):529–537. doi: 10.1182/blood-2015-08-667055. [DOI] [PMC free article] [PubMed] [Google Scholar]