

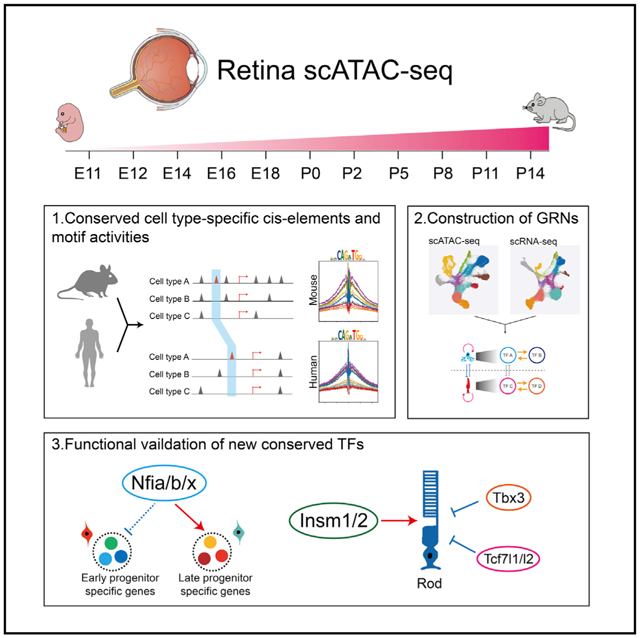
SUMMARY
Gene regulatory networks (GRNs), consisting of transcription factors and their target sites, control neurogenesis and cell-fate specification in the developing central nervous system. In this study, we use integrated single-cell RNA and single-cell ATAC sequencing (scATAC-seq) analysis in developing mouse and human retina to identify multiple interconnected, evolutionarily conserved GRNs composed of cell-type-specific transcription factors that both activate genes within their own network and inhibit genes in other networks. These GRNs control temporal patterning in primary progenitors, regulate transition from primary to neurogenic progenitors, and drive specification of each major retinal cell type. We confirm that NFI transcription factors selectively activate expression of genes promoting late-stage temporal identity in primary retinal progenitors and identify other transcription factors that regulate rod photoreceptor specification in postnatal retina. This study inventories cis- and trans-acting factors that control retinal development and can guide cell-based therapies aimed at replacing retinal neurons lost to disease.
Graphical Abstract
In brief
Using integrated analysis of gene expression and chromatin accessibility at the single-cell level, Lyu et al. identify evolutionarily conserved gene regulatory networks that regulate temporal patterning, neurogenesis, and cell-fate specification for all major cell types in the developing mouse and human retina and functionally validate these predictions.
INTRODUCTION
The central nervous system (CNS) consists of many distinct cell types, which are generated in discrete though often overlapping temporal windows (Holguera and Desplan, 2018; Oberst et al., 2019; Paridaen and Huttner, 2014). In both vertebrates and invertebrates, temporal patterning is controlled intrinsically by dynamically regulated expression of transcription factors (TFs), which in turn regulate the ability of neural progenitors to proliferate and generate specific cell types (Cayouette et al., 2003; Doe, 2017; Rossi et al., 2021; Thor, 2017). Multiple individual TFs control temporal patterning in both Drosophila (Bayraktar and Doe, 2013; Erclik et al., 2017; Konstantinides et al., 2021) and mammalian (Sagner et al., 2020; Telley et al., 2019) neural progenitors, and large-scale gene expression analysis of developing CNS has identified many other dynamically expressed TFs (Carter et al., 2018; La Manno et al., 2021; Tiklová et al., 2019). However, the high cell type diversity in developing CNS (Zeng and Sanes, 2017) has hindered identification of the genomic targets of these transcription factors, the networks into which they are organized, and the mechanisms by which they control temporal transitions and neurogenesis.
Unlike most brain regions, the retina is a tractable system for identifying molecular mechanisms controlling temporal patterning and neurogenesis. The retina is composed of seven major cell types, whose birth order and molecular properties are well characterized. Retinal ganglion cells, cone photoreceptors, horizontal cells, and GABAergic amacrine cells (ACs) are specified during early stages of neurogenesis prior to embryonic day (E)18, although non-GABAergic ACs, bipolar cells (BCs), Müller glia (MG), and most rod photoreceptors are specified at later ages (Bassett and Wallace, 2012; Cepko, 2014; Young, 1985a). Much effort has been directed toward identifying factors controlling retinal cell identity (Malin and Desplan, 2021; Sanes and Zipursky, 2010). Some transcription factors, such as Otx2, which promotes photoreceptor and BC fate while repressing AC specification, act as master regulators of retinal cell-fate specification (Ghinia Tegla et al., 2020; Nishida et al., 2003). Several recent single-cell RNA sequencing (scRNA-seq) studies have profiled gene expression in mouse, human, and zebrafish retinas across neurogenesis (Clark et al., 2019; Cowan et al., 2020; Lu et al., 2020; Xu et al., 2020). These have identified multiple TFs that are selectively expressed in early- or late-stage retinal progenitor cells (RPCs). Genetic analysis has shown that several of these TFs are required for generation of individual retinal cell types (Clark et al., 2019; Elliott et al., 2008; Javed et al., 2020; Liu et al., 2020; Mattar et al., 2015).
Despite these advances, the detailed mechanisms by which retinal cell-fate specification is regulated remain largely unknown. Gene expression data alone cannot identify direct regulatory relationships between TFs and target genes. Likewise, it is not clear how TFs selectively expressed in early- or late-stage RPCs regulate temporal identity. Although RPCs appear to commit to specific cell fates during terminal mitosis (Cepko, 2014), the molecular mechanisms that control this process are also unknown. The organization of the gene regulatory networks (GRNs) that control retinal neurogenesis and cell-fate specification remains unexplored at the single-cell level.
To address this, we generated chromatin accessibility profiles of developing mouse retina using sinlge cell assay for transposon accessible chromatin (scATAC)-seq. We identified cis-regulatory elements and putative TF binding sites from scATAC-seq data and integrated these with existing, age-matched scRNA-seq data from mouse (Clark et al., 2019) and newly generated scRNA-seq and scATAC-seq data from developing human retina (Thomas et al., 2021) to identify evolutionarily conserved GRNs that control developmental transitions and cell-fate specification. Cell-type-specific TFs activate expression of other TFs within these GRNs, although often also inhibiting (or more rarely activating) expression of TFs in other networks. We identified GRNs specific to neuroepithelial-like cells, early- and late-stage primary and neurogenic RPCs, and all major retinal neuronal and glial cell types. By modeling regulatory relationships among TFs in cell-specific GRNs, we can make and validate predictions about their function. For instance, we show that the nuclear factor I (NFI) factors Nfia/b/x, which promote specification of late-born retinal cell types (Clark et al., 2019), directly activate expression of TFs selectively expressed in late-stage RPCs and MG. We also identify activators (Insm1/2) and inhibitors (Tbx3 and Tcf7l1/2) of rod photoreceptor specification and differentiation. This resource provides a roadmap for the research community to identify gene regulatory networks that control retinal development.
RESULTS
scATAC-seq profiling of developing mouse retina
To profile dynamic changes in chromatin accessibility across retinogenesis, we conducted scATAC-seq analysis using the 10× Genomics Chromium platform on dissociated cell nuclei obtained from whole mouse retina at 11 time points: E11, E12, E14, E16, and E18, as well as postnatal day (P) 0, P2, P5, P8, P11, and P14 (Figure 1A), profiling a total of 108,975 cells (Figure S1A). The size distribution and position of accessible DNA sequences relative to transcriptional start sites (TSSs) was consistent among each sample (Figure S1B), demonstrating overall high-quality data. High overall correlations are seen between age-matched scATAC-seq and bulk ATAC-seq RPC samples at E11 (r = 0.82) and P2 (r = 0.94), although lower correlations were seen between age-mismatched E11 and P2 samples (Figure S1C; Zibetti et al., 2019). scATAC-seq analysis detected peaks seen in bulk ATAC-seq data and which reflected temporal differences in gene expression, as shown for the basic-helix-loop-helix (bHLH) factor Hes5, which is enriched in late-stage RPCs (Hojo et al., 2000; Figure S1D).
Figure 1. Overview of the study.

(A) Schematic summary of the study. scATAC-seq of the mouse whole retinas was performed at 11 different time points. Cell types and cell-type-specific accessible chromatin regions were identified through dimensional reduction and clustering analysis. By integrating age-matched scRNA-seq data with our data, we reconstructed gene regulatory networks (GRNs) using the IReNA v2 analytic pipeline and identified candidate regulators controlling temporal patterning and cell-fate specification during the retinal development.
(B) Combined UMAP projection of all mouse retinal cells profiled using scATAC-seq (top) and scRNA-seq data (bottom). Each point (cell) is colored by cell type (left) and age (right).
(C) Examples of mRNA levels and chromatin accessibility for selected cell-type-specific genes.
(D) The relative abundance of retinal cell types is similar between age-matched scATAC-seq (left panel) and scRNA-seq (right panel). Bar plots show fraction of cells (y axis) at each time point of each cell type (x axis). ACs, amacrine cells; BCs, bipolar cells; HCs, horizontal cells; MG, Müller glia; NGs, neurogenic progenitor cells; RGCs, retinal ganglion cells; RPCs, retinal progenitor cells.
Clustering and UMAP analysis was then performed on data obtained from each time point to identify individual cell types, which were annotated based on differential accessibility of a panel of well-characterized, cell-type-specific genes (Mendeley dataset). UMAP analysis of scATAC-seq data showed broad similarity to age-matched scRNA-seq data (Figure 1B). Several features were observed from this analysis. First, as reported using scRNA-seq analysis (Clark et al., 2019), a clear distinction was seen between early-stage neuroepithelial cells (which we call RPCs stage 1) and both early-stage and late-stage primary RPCs (RPCs stage 2 and 3, respectively), with MG arising directly from late-stage primary RPCs. Distinct populations of early- and late-stage neurogenic RPCs were also detected, which RNA velocity analysis indicated arose from early- and late-stage primary RPCs, respectively (Melsted et al., 2021; Figures 1B and 1C). Second, four major trajectories of differentiating neurons were seen: retinal ganglion cells (RGCs); ACs and horizontal cells (HCs); rod and cone photoreceptors; and BCs, respectively (Figures 1B and S1). Third, the generation of each retinal cell type and their relative abundance was similar between the two datasets (Figure 1D; Mendeley dataset). Neuroepithelial cells (RPCs stage 1) dominated E11 and E12 samples, although RGCs, cones, and ACs and HCs were detected by E14. A rapid transition between early- and late-stage primary and neurogenic RPCs was seen between E16 and E18, coinciding with a dramatic reduction in relative abundance of RGCs, as seen with scRNA-seq analysis (Clark et al., 2019). Likewise, late-born BCs and MG were first seen at P5 (Figure 1D). High overall correlation was seen between scATAC-seq and scRNA-seq profiles of individual cell types (Figure S1E).
A previous study conducted bulk ATAC-seq and chromatin immunoprecipitation (ChIP)-seq analysis of chromatin modifications in whole mouse retina across development (Aldiri et al., 2017). We investigated whether open chromatin regions (OCRs) detected using scATAC-seq matched genomic annotations defined by hidden Markov modeling (HMM) from age-matched bulk retinal samples (Aldiri et al., 2017). Although HMM analysis showed that most genomic regions lacked predicted regulatory potential at all ages (Aldiri et al., 2017), most OCRs (identified in P0 and P14 retina using aggregated scATAC-seq data) overlapped with regions identified as active promoters or enhancers (as defined using HMM analysis of bulk ChIP-seq and ATAC-seq data; Figure S1F). A comparison between OCRs present in specific retinal cell types and age-matched HMM data revealed that the most abundant cell types showed the strongest predicted regulatory potential, with OCRs in primary and neurogenic RPCs showing high regulatory potential at P0, as did rod photoreceptor OCRs at P14 (Figure S1F). However, even in rarer cell types–such as RGCs at P0 or MG at P14–at least one-third of all OCRs were predicted to show regulatory potential. These reflect overall changes in retinal cell composition and highlight the importance of the higher resolution analysis provided by scATAC-seq data. Genes specific to specific mature retinal cell types–such as Aqp4 in MG, Tfap2b in ACs, Opn1sw in cones, Rho in rods, and Cabp5 in BCs–showed expected cell-type-specific patterns of chromatin accessibility at P14 (Figure S1G).
Analysis of dynamic chromatin accessibility and TF activity during mouse retinal development
Next, we analyzed scATAC-seq data to identify cell-type-specific cis-regulatory elements (Figure 2A; Mendeley dataset). Patterns of chromatin accessibility of well-characterized, cell-type-specific genes generally correlate with their mRNA expression (Figure S2A), although some transcription factors expressed in neurogenic RPCs, such as Atoh7 and Olig2, also showed high levels of accessibility in early-born neurons. All major retinal cell types showed many specific peaks of chromatin accessibility, with MG (12,757 peaks) and RGCs (8,576 peaks) showing the most and early neurogenic RPCs showing the fewest peaks (1,497). Next, we identified changes in gene expression associated with these cell-type-specific accessibility regions. For each cell-type-specific accessible peak, we calculated the peak-gene correlation and identified genes whose expression was positively correlated with regions of chromatin accessibility, identifying potential cell-type-specific enhancers and promoters. In total, we identified 11,203 corresponding genes for all cell types (Figure 2B; Mendeley dataset). These include Nrl and Nr2e3 in rods, Mlc1 and Aqp4 in MG, and Sfrp2 and Foxp1 in stage 1+2 RPCs. Gene Ontology (GO) analysis reveals that genes associated with progenitor-specific differentially accessible regions (DARs) are enriched for cell cycle regulation; genes associated with rod- and cone-specific DARs for phototransduction; and genes associated with neurogenic RPC-specific DARs for regulation of development (Figure 2C; Mendeley dataset).
Figure 2. Single-cell regulatory landscape of mouse retinal development.

(A) Heatmap of cell-type-specific peaks. The numbers of cell-type-specific peaks are indicated at left. Cell types are shown at the bottom.
(B) Heatmaps of the expression level of positively correlated genes. Cell types are shown at the bottom of the plot.
(C) Representative genes along with GO enrichment for each cluster. The x axis indicates the −log10(p value) of the GO term.
(D) Heatmap of the chromVAR Z score for cell-type-specific motifs. The number of motifs in each cell type is indicated at left. Cell types are indicated at the bottom. Representative motif logos are shown on the right.
(E) Examples of TF footprint profiles for Tfap2a, Pou4f2, Otx2, and Nfix in indicated scATAC-seq clusters. Tn5 insertion tracks are shown below.
We next analyzed the activity of TFs that could interact with the DARs. We measured the gain or loss of global chromatin accessibility in DARs containing individual TF motifs by using chromVAR software (Figure 2D; Mendeley dataset; Schep et al., 2017). Many TFs showed some degree of cell type specificity, with the number of cell-type-specific motifs ranging from 641 in MG to 41 in BCs (Mendeley dataset). We further validated chromVAR scores by using footprinting analysis for known TF markers selectively expressed in specific retinal cell types. These include Tfap2a motifs in ACs and HCs; Pou4f2 motifs in RGCs; Otx2 motifs in photoreceptors; and Nfix motifs in MG (Figures 2E and S2B). Integrated scRNA-seq and scATAC-seq analysis can thus identify targets of known cell-type-specific TFs in mice.
Comparison of mouse and human scATAC-seq data reveals evolutionary-conserved regulatory elements and motif activities
To identify evolutionarily conserved regulatory elements and TFs controlling retinal neurogenesis and cell-fate specification, we compared our mouse data to scATAC-seq and scRNA-seq data obtained from whole human fetal retinas at six developmental time points, ranging from 7.5 to 19 gestational weeks (Thomas et al., 2021). As in the mouse, UMAP analysis identified each major cell type (Figures S3A and S3B; Mendeley dataset) and resembled an aggregate UMAP plot of scRNA-seq analysis of developing human retina (Figure S3C). We next identified evolutionarily conserved cell-type-specific regulatory elements for all major retinal cell types. 3%–15% of these elements were conserved between mouse and human, with RGCs showing the highest and cones and MG the lowest conservation (Figure 3A). This may in part reflect oversampling of early time points in the human data, as these samples are enriched for RGCs and have few MG (Mendeley dataset). The low conservation of cone-specific elements is consistent with the finding that cones are the most transcriptionally divergent retinal cell type between mice and humans (Lu et al., 2020). Overall, 8.3% of mouse peaks and 6.4% of human peaks were evolutionarily conserved (Figure 3B; Mendeley dataset). No enrichment was seen in evolutionarily conserved peaks relative to all peaks in either species, with the exception of evolutionarily conserved human peaks showing greater enrichment for TSSs (4.9% versus 2.1%; p < 2.2e–16).
Figure 3. Conserved single-cell regulatory landscape in mouse and human retinal development.

(A) Heatmap of evolutionary conserved cell-type-specific peaks. Numbers of peaks are indicated at left. Cell types are shown at the bottom. Representative conserved and positively correlated genes are shown on the right.
(B) Pie chart depicts percentage of total and conserved peaks from mouse (top) and human (bottom). TES, transactional end site; TSS, transcriptional start site.
(C) Heatmap of the chromVAR Z score of the conserved cell-type-specific motifs from mouse (left) and human (right). The number of motifs is indicated at left. Cluster identities are indicated at the bottom. Representative motif logos are shown on the right.
(D) TF footprint profile of Neurod1 and Sox9 from selected mouse and human retinal cell types.
To analyze the evolutionary conservation of trans-acting factors regulating cell-type-specific gene expression, we compared cell-type-specific active motifs in mouse and human cell types based on chromVAR score (Schep et al., 2017). As expected, we observed a higher percentage of conserved active motifs of cell-type-specific regulatory elements, with numbers of conserved cell-type-specific motifs ranging from 122/161 in cones to 50/641 in MG. Cell-type-specific active motifs include well-characterized TFs, such as Sox9, Neurod1, Pou4f2, Tfap2a, and Crx (Figure 3C; Mendeley dataset). Similar developmental patterns of TF footprinting were seen for many of these TFs, as illustrated by Neurod1 in late neurogenic RPCs and Sox9 in stage 3 RPCs and MG (Figure 3D; Mendeley dataset). Using the same analytic approach applied to mouse retina, we can also identify targets of known cell-type-specific TFs in developing human retina.
GRNs controlling temporal patterning of retinal progenitors
Because the generation of all retinal cell types is ultimately controlled by the dynamic temporal patterning of primary RPCs during neurogenesis (Cepko, 2014; Zechner et al., 2020), we next set out to identify the GRNs that control this process. To identify key TFs controlling temporal patterning, we first constructed these GRNs by integrating scRNA-seq and scATAC-seq data using a modified form of the Integrated Regulatory Network Analysis (IReNA) analysis pipeline (Hoang et al., 2020; Figures S4A and S4B). We then extracted predicted regulatory relationships among stage-specific TFs, identifying positive feedback loops of co-expressed TFs used to maintain stage-specific identity and negative feedback loops used to ensure mutually exclusive expression of TFs specific to different stages. We further filtered TFs based on their regulatory strength in cell-specific GRNs and whether their expression pattern was conserved between mouse and human retina (Figures 4A and S4A).
Figure 4. Model of GRNs controlling temporal patterning of retinal progenitors.

(A) Schematic of the analytic pipeline used to identify TFs controlling retinal development. The role of feedback loops (double positive and double negative) in controlling transitions between cell states and during the retinal-cell-fate specification is shown in the Waddington epigenetic landscape model.
(B) UMAPs of retinal progenitors from scRNA-seq (left) and scATAC-seq (right). Cells are colored by pseudotime and cell type.
(C) A model for transitions of primary retinal progenitors and MG during E11-P8.
(D) Heatmaps show expression of cell-type-specific DEGs (left) and their correlated accessible regions (CARs) (right) across pseudotime. The left bar indicates cell types (RPCs S1–S3 and MG) and the classes of CARs (TSS, positively correlated and negatively correlated).
(E) Genome track visualization of the Hes1 locus. Each track represents the aggregated scATAC-seq signals across the RPC-MG trajectory. Inferred links of Hes1-associated CARs (correlated accessible regions) are shown at top. Expression level of Hes1 measured by scRNA-seq across the RPC-MG trajectory is shown at right.
(F) Full network diagram on the left showing TF pairs linked by reciprocal positive or negative regulatory relationships during the RPC-MG transition. Each node represents an individual cell-type-specific TF. Each edge represents a statistically significant feedback relation between TF pairs. Simplified intermodular regulatory networks of retinal progenitors are shown on the right. Colored nodes represent specific cell types. Connections indicate statistically significant regulations among modules. The width of connections indicates their regulatory enrichment fold.
(G) The top 10 TFs predicted to activate expression of genes specific to stage 3 RPCs, as inferred from IReNA v2 analysis (left). Bar plots show expression levels of these TFs in mouse and human stage 3 RPCs progenitors (right).
We focused on four major cell states, including stage 1–3 RPCs and MG, to study GRNs controlling temporal patterning of neurogenesis. We first performed pseudotime analysis for both scATAC-seq and scRNA-seq data from primary progenitors and MG, as previously described (Hoang et al., 2020; Lu et al., 2020; Figure 4B). Our scRNA-seq and scATAC-seq data suggest that many primary RPCs progressively transition between stages 1, 2, and 3 before eventually becoming MG (Figure 4C). Analysis of scRNA-seq identified differentially expressed genes (DEGs) during each of these stages, with Foxp1 and Sfrp2 enriched in stage 1 RPCs, Fgf15 and Pou3f1 in stage 2, Ascl1 in stage 3, and Nfia/b/x and Hes5 in MG (Figure 4D), matching reported results (Clark et al., 2019). scATAC-seq data were then used to identify correlated accessible regions (CARs) associated with these DEGs (Figure 4D; Mendeley dataset). CARs include the accessible peaks near TSSs and distal accessible peaks (regions <100 kb from the TSS) either positively or negatively correlated with the DEGs (Figure S4B; Mendeley dataset). Dynamic regulation of both positive and negative correlated elements can be clearly seen in the case of Hes1, which is most highly expressed in S3 RPCs and MG (Figure 4E). At the Hes1 locus, one distal and two proximal positively correlated elements show increased accessibility across pseudotime, although three negatively correlated elements show decreased accessibility. Accessibility at the TSS, however, does not change (Figure 4E).
We next inferred patterns of TF binding by integrating TF expression patterns identified using scRNA-seq with footprinting in CARs identified by scATAC-seq (Figure S4B). This allowed us to identify TF-TF regulatory relationships among each of the four cell states (Figure 4F; Mendeley dataset). Many state-specific TFs were connected in positive-feedback loops that may maintain expression of state-specific TFs, while also repressing TFs specific to other cell states. Each cell state possessed a self-activating GRN, with the stage 1 and 3 RPC- and MG-specific networks predicted to be strong and the stage 2 RPC network relatively weak. GRNs specific to stage 1 and 2 RPCs showed both positive and negative regulatory relationships between one another, with positive regulatory relationships slightly dominating. A similar situation was seen with stage 3 RPCs and MG, although positive regulatory relationships between these networks were relatively stronger. Strong negative regulatory relationships were seen between stage 1 and 2 RPCs and stage 3 RPCs and MG, respectively (Figure 4F).
Nfia/b/x promote late-stage RPC temporal identity
We next sought to predict which TFs play an essential role in controlling RPC and MG temporal identity by integrating multiple data, which included gene regulatory relationships, gene expression specificity, and evolutionary conservation of gene expression patterns (Mendeley dataset). NFI family members were among the top TFs predicted to activate expression of TFs specific to stage 3 RPCs and MG (Figure 4G). Previous genetic analysis suggests that NFI factors Nfia, Nfib, and Nfix might both control temporal identity in retinal progenitors and formation of late-born retinal cell types (Clark et al., 2019). However, it is not known whether Nfia/b/x are necessary to activate expression of genes specific to late-stage RPCs. Likewise, although NFIA/B/X overexpression in late-stage RPCs promotes cell cycle exit and generation of late-born cell types (Clark et al., 2019), it is not known whether their misexpression in early-stage RPCs confers late-stage temporal identity. To address these questions, we used ex vivo electroporation to overexpress human homologs of NFIA/B/X in E14 retina and profiled changes in gene expression and chromatin accessibility in fluorescence-activated cell sorting (FACS)-isolated electroporated cells at E16 and P0 using scRNA-seq and scATAC-seq. We performed similar analysis in P2 and P14 Nfia/b/x conditional knockout (cKO) retina (Figures 5A and S5A).
Figure 5. Nfia/b/x promote late-stage temporal identity in retinal progenitors.

(A) Overview of experimental design for characterizing Nfia/b/x function in retinal progenitors.
(B) Boxplots showing changes in the Nfia/b/x motif enrichment in retinal progenitors following overexpression or knockout of Nfia/b/x. Bars are colored by genotype.
(C) Bar plots showing the fraction of each retinal cell type by ages and genotypes (top: scRNA-seq data; bottom: scATAC-seq data).
(D) Immunostaining showing fewer RGCs and more photoreceptors at P0 following NFIA/B/X overexpression at E14 retinal explants. The fractions of RGCs and photoreceptors are shown on the right.Error bars indicate standard deviation. n> 5 retinas/group. Scale bars represent 20 μm.
(E) Dot plot showing gene set enrichment results for DEGs enriched in early- and late-stage RPCs and MG following overexpression or knockout of Nfia/b/x.
(F) Dot plot showing peak set enrichment results for DARs enriched in early- and late-stage RPCs and MG following overexpression or knockout of Nfia/b/x.
(G) Venn diagrams showing overlap between direct Nfia/b/x binding regions identified using ChIP-seq and cell-type-specific DARs. The p value on top of each Venn diagram indicates the significance of their overlap using the hypergeometric test.
(H) Summary of Nfia/b/x action during the transition from early- to late-stage RPCs.
scATAC-seq analysis showed that NFIA/B/X overexpression induced increased chromatin accessibility at sequences containing the consensus NFI motif, as reported in non-neuronal cells (Denny et al., 2016), although loss of function of Nfia/b/x reduced accessibility at these sites (Figure 5B). Relative to retinas electroporated with a control GFP plasmid, E16 retinas overexpressing NFIA/B/X showed fewer RGCs and more RPCs when analyzed by both scRNA-seq and scATAC-seq (Figure 5C). At P0, scRNA-seq analysis and immunohistochemistry show that NFIA/B/X-overexpressing retinas contain more primary RPCs and photoreceptors and fewer RGCs compared to the control (Figures 5C and 5D). scATAC-seq analysis of P2 Nfia/b/x cKO retinas showed more RPCs and fewer rod photoreceptors and AC and HC neurons (Figure 5C). In P14 Nfia/b/x cKO retina, more RPCs were detected, although bipolar neurons were absent (Figure 5C).
To determine whether gain or loss of function of Nfia/b/x altered the expression of genes and patterns of chromatin accessibility specific to the three RPC states or MG, we performed gene set enrichment analysis (GSEA) and peak set enrichment analysis (PSEA). Overexpression of NFIA/B/X upregulated MG-enriched genes in primary RPCs by E16 (Figures 5E and S5B; Mendeley dataset). By P0, this effect was more pronounced, with stage 3 RPC-enriched genes also upregulated. Furthermore, stage 1 RPC-enriched genes were downregulated relative to GFP controls (Figures 5E and S5B). The opposite pattern was seen in Nfia/b/x cKO retina, with stage 3 RPC and MG-enriched genes downregulated and stage 2 RPC-enriched genes upregulated at P2. By P14, downregulation of MG-enriched genes and upregulation of stage 1 and stage 2 RPC-enriched genes was more prominent (Figures 5E and S5B). Changes in patterns of chromatin accessibility matched those of gene expression, with NFIA/B/X overexpression inducing RPCs to adopt a state resembling MG and the loss of function inducing a state that resembled stage 1 and 2 RPCs (Figures 5F and S5C). Motif analysis indicated that NFIA/B/X motifs were enriched in DARs upregulated following NFIA/B/X overexpression and downregulated in Nfia/b/x cKO retina (Figure S5C). These data show that NFI factors directly regulate temporal patterning in RPCs.
To identify genes directly regulated by Nfia/b/x, we performed ChIP-seq analysis on P2 wild-type retina using antibodies that recognize all three NFI factors (Mendeley dataset). We identified 13,680 Nfia/b/x ChIP-seq peaks (Figure S5D), most of which (83.9%) are located in open chromatin (Figure S5E). These peaks are mostly in intergenic and intronic regions. NFI factor binding sites are enriched in stage 3 RPC-specific accessible regions (Figure S5F). We then asked whether changes in RPC identity are directly mediated by NFIA/B/X. In E16 NFIA/B/X-overexpressing primary RPCs, 61% (425/702) of DARs specific to stage 3 RPCs and/or MG were directly bound by NFIA/B/X, but only 4/22 of DARs specific to stage 1 and/or 2 RPCs were directly bound (Figure 5G). In P2 Nfia/b/x cKO retina, 51% (869/1,689) of DARs specific to stage 3 RPCs and/or MG overlapped with NFIA/B/X ChIP-seq peaks, in contrast to only 13% (48/371) DARs specific to stage 1 and/or 2 RPCs. Similar pattern was seen in P14 Nfia/b/x cKO retina (Figure 5G). This result suggests that DARs specific to stage 3 RPCs and/or MG could be directly regulated by the binding of Nfia/b/x at these regions and was further confirmed by the comparison of temporal identity-associated DEGs and Nfia/b/x ChIP-seq peaks (Figure S5G). In summary, our analysis suggests that NFI factors alter the chromatin accessible regions specific to late progenitor-specific genes by direct binding to these regions and in turn activates expression of stage 3 RPCs and/or MG genes and represses the expression of early progenitor-specific genes. By this mechanism, NFI factors control temporal identity in retinal progenitors and formation of late-born retinal cell types.
GRNs that control specification of retinal neurons
We applied the same approach to identify GRNs controlling neurogenesis and the specification of individual types of neurons. To do this, we generated three combined datasets, corresponding to early, intermediate, and late stages of retinal neurogenesis, identifying DEGs and CARs that are selectively active as cells adopt different identities (Mendeley dataset). The early dataset consisted of E14 and E16 and included all early-stage neurogenic progenitors, as well as differentiating RGCs, cone photoreceptors, and early-born ACs and HCs (Figure 6A). The intermediate dataset consisted of E18, P0, and P2 and included late-stage neurogenic progenitors, as well as differentiating rod photoreceptors and late-born ACs (Figure S6A). The late dataset consisted of P5 and P8 and included late-stage neurogenic progenitors and differentiating rods and BCs (Figure S6E).
Figure 6. Model of GRNs controlling specification of retinal neuronal cell types.

(A) A diagram showing development of early-born retina cell types (left). UMAPs of scRNA-seq and scATAC-seq data from E14–E16 retina (right) are shown. Color indicates cell type.
(B) UMAPs showing trajectories constructed from scRNA-seq and scATAC-seq at E14–E16. Color indicates pseudotime state.
(C) Heatmaps showing expression of cell-type-specific DEGs (left) and the accessibility of their corresponding CARs (right) along differentiation trajectories. The top bars are colored by pseudotime state for each trajectory. The left bar indicates cell type and the classes of CARs.
(D) Networks showing feedback relationships between TF pairs at E14–E16. Each node represents an individual cell-type-specific TF. Each edge represents a positive- or negative-feedback regulatory relationship between TF pairs.
(E–G) Simplified intermodular GRNs of RPCs and neurons at different stages (E, early-stage; F, intermediate-stage; G, late-stage). Colored nodes represent cell types. Connections indicate statistically significant regulatory relationships among GRNs specific to each cell type. The width of connections indicates their regulatory enrichment fold.
We then used pseudotime analysis to analyze four major developmental trajectories in the aggregated early development dataset. Specifically, these were (1) the transitions from stage 2 primary RPCs to early neurogenic RPCs, (2) from early neurogenic RPCs to RGCs, (3) from early neurogenic RPCs to GABAergic ACs and HCs, and (4) from early neurogenic RPCs to cone photoreceptors (Figure 6B). To identify GRNs controlling these transitions, we identified both DEGs and CARs for each developmental trajectory (Figure 6C) and inferred putative regulatory relationships among cell-specific TFs (Figures 6D and 6E), as previously done for primary RPCs and MG. A similar analysis was performed for intermediate stages of neurogenesis, when glycinergic and non-GABAergic, non-glycinergic (nGnG) ACs and rods are generated (Figures S6B-S6D), and late stages of neurogenesis, when rods and BCs are generated (Figures S6F-S6H). This identified many TFs predicted to selectively activate or repress genes specific to individual cell types (Mendeley dataset).
GRNs controlling neuronal cell-fate specification showed many similarities with GRNs controlling state changes in primary RPCs (Figure 4F). Cell type identity is maintained by strong positive regulatory relationships among cell-type-specific TFs (Figures 6E-6G; Mendeley dataset). Regulatory relationships among different cell types often contain both positive and negative components. GRNs specific to primary RPCs and all neuronal subtypes are connected by many regulatory relationships, which are almost all negative. In contrast, GRNs specific to most neuronal cell types are generally connected by fewer negative regulatory relationships, and positive regulatory relationships predominantly connect GRNs of some transcriptionally similar cell types, such as RGC, AC, and HC. Regulatory relationships between neurogenic and primary RPCs are more dynamic. It is weakly positive at early stages of neurogenesis, weakly negative during intermediate stages, and strongly positive at late stages. This shift may reflect the fact that a rapid increase in the relative fraction of neurogenic RPCs relative to primary RPCs occurs during late neurogenesis (Clark et al., 2019). The regulatory relationship between neurogenic RPCs and neuronal networks is likewise dynamic. During early stages of neurogenesis, neurogenic RPC networks strongly inhibit RGC networks, weakly inhibit horizontal and early-born amacrine networks, and also weakly activate cone networks (Figure 6E). At intermediate stages, they weakly inhibit late-born amacrine networks and also weakly inhibit rod networks (Figure 6F). At late stages, they strongly inhibit rod networks but activate bipolar networks (Figure 6G). Notably, these regulations roughly correspond to the order in which these neuronal subtypes are generated during retinal development, with RGCs formed first and BCs last (Cepko et al., 1996).
Identification of TFs that control neurogenesis and cell-fate specification in postnatal retina
Our GRN analysis predicts that many TFs will act as either positive or negative regulators of neurogenesis and/or cell-fate specification. Many of these predicted regulatory relationships have been previously validated using genetic analysis (Figure S7A; Mendeley dataset). Considering TFs with the highest number of predicted regulatory relationships active in E18–P2 retina, for instance, we find that Otx2, Crx, Prdm1, Rax, Rorb, Nrl, and Nr2e3 are all predicted to activate expression of rod-specific genes (Akhmedov et al., 2000; Brzezinski et al., 2010; Furukawa et al., 1997; Irie et al., 2015; Jia et al., 2009; Mears et al., 2001; Nishida et al., 2003); Pax6, Tfap2a, and Tfap2b are predicted to repress photoreceptor specification (Jin et al., 2015; Remez et al., 2017); and Zfp36l1/2, Nfia, Hes1/5, Sox2/8/9, and Lhx2 are predicted to both promote RPC maintenance and inhibit rod differentiation (Bosze et al., 2020; Clark et al., 2019; Marquardt et al., 2001; de Melo et al., 2016; Muto et al., 2009; Roy et al., 2013; Taranova et al., 2006; Wall et al., 2009; Wu et al., 2020). Knockdown of Nfib, which is predicted to be one of the top activators of rod-specific genes, has also been shown to reduce rod-specific gene expression in human organoid cultures (Xie et al., 2020). Given our success in predicting the function of these TFs, we conducted gain- and loss-of-function analysis via electroporation for several previously uncharacterized candidate TFs on retinal explants. We analyzed the resulting phenotypes using scRNA-seq and immunohistochemistry to determine whether other TFs with high numbers of regulatory relationships showed predicted phenotypes (Figure 7A). We analyzed five different TFs: Insm1; Insm2; Tcf7l1; Tcf7l2; and Tbx3. Insm1 and Insm2 are predicted to activate genes specific to neurogenic progenitors and rod photoreceptors. In contrast, Tcf7l1/2 are predicted to inhibit neurogenesis and promote a stage 3 primary RPC and MG identity. Lastly, Tbx3 is predicted to inhibit rod specification while promoting amacrine formation (Figures S7A-S7C).
Figure 7. Identification of TFs controlling cell-fate specification in postnatal retina.

(A) A schematic diagram for gain- and loss-of-function analysis of candidate TFs in postnatal mouse retina explants.
(B) Bar plots showing the fraction of each cell type at P5 as measured by scRNA-seq analysis of FACS-isolated GFP-positive cells for each treatment condition.
(C and D) Immunohistochemistry and quantification of MG (SOX9 positive) and photoreceptors (GFP positive in the ONL layer) in P11 retina explants in control and overexpression of INSM1, INSM2, TCF7L1/2, and TBX3. Arrowheads indicate SOX9/GFP double-positive cells. Error bars indicate standard deviation. **p < 0.05; ***p < 0.001; n > 7 retinas/group. Each dot represents a retinal explant. INL, inner nuclear layer; ONL, outer nuclear layer; OS, outer segment. Scale bar represents 30 μm.
(E) GSEA of DEGs from each cell type in each experiment. GSEA was performed with the cell-type-specific gene sets obtained from the combined scRNA-seq data (E11–P8). Only significant enrichment results (p < 0.05) are shown in the dot plot. Each dot was colored by NES and sized by −log(p value). The x axis indicates the cell type where DEGs are calculated. The y axis indicates the specific gene sets used in the analysis.
(F) Examples of GSEA results from (E). Heatmaps show DEGs used in the GSEA analysis, with DEGs ranked by log2 fold change, as shown in the middle panel. The right annotation shows the distribution of significantly enriched gene sets among the DEGs. Representative cell-type-specific genes are also labeled.
(G) Summary of observed phenotypes.
Overexpression of either Insm1 or Insm2 at P0 led to a dramatic reduction in the relative fraction of primary RPC and MG cells and an increase in the fraction of amacrine cells and cone photoreceptors at P5, as measured by scRNA-seq analysis of FACS-isolated, GFP-positive electroporated cells (Figure 7B). Insm2 overexpression also led to a modest increase in the fraction of rods. Immunohistochemical analysis of P11 retinas shows that Insm1 or Insm2 overexpression significantly increases the fraction of GFP+ cells in the photoreceptor layer and leads to a corresponding decrease in the MG fraction (Figures 7C, 7D, and S7E). Both Insm1 and Insm2 strongly activate expression of rod-specific genes in all other cell types (Figure 7E). Insm1 and Insm2 both accelerate the normal developmental increase in the expression of rod-specific genes in rod precursors, such as Gngt1, Sag, and Rho (Figure 7F). In contrast, somatic CRISPR-mediated loss of function of Insm1/2 reduced the relative fraction of rods and increased the fraction of primary and neurogenic RPCs at P5 and also reduced expression of rod-specific genes, such as Nrl, in rod precursors (Figures 7B, 7E, and S7F; Mendeley dataset). However, no statistically significant changes in cell composition were seen following loss of function of Insm1/2 at P11 (data not shown). Although insm1a has been reported to be required for rod differentiation in zebrafish (Forbes-Osborne et al., 2013), our data show that Insm1/2 promote retinal neurogenesis, rod photoreceptor specification, and rod-specific gene expression.
Tcf7l1/2 are expressed in RPC and MG and are predicted to inhibit rod specification while maintaining RPC identity (Figures S7A and S7B). scRNA-seq analysis of P5 GFP-positive cells that overexpress TCF7L1/2 revealed a substantial increase in the fraction of stage 3 RPC and MG cells, with a corresponding reduction in the fraction of rods (Figure 7B). Immunohistochemical analysis of P11 retinas overexpressing TCF7L1/2 leads to a reduction in relative fraction of rod photoreceptors and BCs and a corresponding increase in the fraction of MG (Figures 7C, 7D, and S7E). In rod precursors, Tcf7l1/2 inhibited expression of genes enriched in mature rods, such as Gngt1 and Pdc, as well as TFs that promote rod specification, such as Prdm1 (Figures 7E and 7F; Mendeley dataset).
Finally, Tbx3 is expressed in ACs and MG and is predicted to inhibit rod photoreceptor specification (Figures S7A and S7C). scRNA-seq analysis of P5 GFP-positive cells revealed that Tbx3 overexpression led to a reduction in rod photoreceptors at P5, along with a corresponding increase in the fraction of not only ACs but also other inner retinal cell types, including stage 3 RPC and MG and BCs (Figure 7B). Immunohistochemical analysis of retinas overexpressing Tbx3 revealed changes in cell composition are maintained at P11 (Figures 7C and S7E). Tbx3 overexpression also reduced expression of Neurod1 in neurogenic progenitors, which promotes rod differentiation (Akagi et al., 2004; Figures 7E and 7F; Mendeley dataset). These results, which are summarized in Figure 7G, validate our predicted GRNs and demonstrate that multiplexed scRNA-seq analysis can be scaled to analyze the function of major transcriptional regulators of retinal development.
DISCUSSION
This study provides a comprehensive picture of the cellular-level landscape of dynamic chromatin accessibility over the full course of retinal development and provides both a map of both candidate cis-regulatory elements and TF binding patterns. By integrating scRNA-seq data with scATAC-seq data, we have reconstructed transcriptional regulatory networks that control all aspects of retinal development. We observe similarities between the retinal and other systems in both general mechanisms and in specific genes that control this process. Our work fills an important gap in our understanding of how temporal patterning is controlled. Although several recent studies have used scATAC-seq to identify active TF motifs in the developing brain (Domcke et al., 2020; Kim et al., 2021; Sarropoulos et al., 2021), only one recent study has systematically integrated these data to identify GRNs controlling neurogenesis and specification of major cell types (Di Bella et al., 2021). Moreover, although previous studies have used ATAC-seq, ChIP-seq, and HiC analysis to profile changes in chromatin accessibility, conformation, and covalent modification during retinal development, the information in these data has been limited by high cellular heterogeneity (Aldiri et al., 2017; Norrie et al., 2019; Xie et al., 2020). Our scATAC-seq data allow us to visualize developmentally dynamic changes in chromatin accessibility within each major retinal cell type. Furthermore, direct comparison of stage-matched mouse and human data identifies conservation of cell-type-specific patterns of both gene expression and chromatin accessibility and allows efficient identification of both evolutionary-conserved and species-specific components of GRNs that control retinal neurogenesis and cell-fate specification.
Integrated scRNA-seq and scATAC-seq analysis reveals the redundant and complex patterns of gene regulatory relationships maintaining each cellular state and ensures developmental processes remain consistent and robust in the face of a variety of environmental perturbations. This may explain the observation that genetic disruption of individual cis-regulatory elements typically results in only developmental phenotypes that are either modest or only alter expression of a subset of the cell-type-specific TFs regulated by these genes (Chan et al., 2020; Ghiasvand et al., 2011). GRNs controlling retinal development are parallel, redundant, and complex. Much like in Drosophila (Doe, 2017; Rossi et al., 2021), cell states are maintained by networks of TFs that activate expression of TFs within cell-type-specific GRNs but often also repress expression of TFs in GRNs specific to other cell states, potentially mediating rapid and irreversible transitions between different stable transcriptional states. Regulatory relationships among individual cell-type-specific GRNs are temporally dynamic, often containing a mixture of positive- and negative-feedback loops, and reflect developmental changes in the timing of retinal neurogenesis. For instance, although GRNs of primary RPCs show only weakly positive or negative regulation of GRNs specific to neurogenic RPCs between E14 and P2, they strongly activate neurogenic RPC-specific GRNs at P5 and P8. This may reflect the increase seen in the relative fraction of terminal neurogenic divisions at these ages (Cepko, 2014), as well as the increased fraction of neurogenic RPCs relative to primary RPCs (Clark et al., 2019). Likewise, neurogenic RPC-specific GRNs most strongly activate expression of TFs in GRNs specific to the neuronal subtypes that are generated latest at each stage: cones at E14–E16, rods at E18–P2, and bipolar cells at P5–P8 (Young, 1985b). Identifying the mechanisms that control these changes in the organization of cell-type-specific GRNs awaits further analysis.
Transitions between cell states are driven by changes in both gene expression and chromatin accessibility. In some cases, TFs act to alter chromatin accessibility at regulatory sites associated with stage-specific genes, prior to initiation of changes in gene expression (Ma et al., 2020). This is seen with Nfia/b/x, which are enriched in late-stage RPCs and MG. Nfia/b/x overexpression triggers increased accessibility at regulatory sites associated with genes expressed in late-stage RPCs and MG, although loss of function of Nfia/b/x produces the opposite effect. This leads to activation of expression of these genes and, indirectly, to repression of genes specific to early-stage RPCs, inhibiting generation of early-born cell types, such as RGCs, and promoting rod photoreceptor specification. This establishes NFI factors as bona fide regulators of temporal patterning in RPCs and identifies the mechanism by which they regulate changes in retinal progenitor competence.
This study serves as a broadly useful resource for the community for further functional characterization of GRNs that control retinal neurogenesis and cell-fate specification. Furthermore, it may help facilitate and improve strategies for reprogramming of endogenous MG and/or directed differentiation of embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) to replace neurons lost due to blinding diseases (Javed and Cayouette, 2017; Lahne et al., 2020; Miltner and La Torre, 2019). Sequential expression of TFs that promote formation of early- or late-stage neurogenic RPCs, followed by TFs that drive specification of rods, cones, or RGCs, could provide a robust approach to generate these neurons for therapeutic purposes.
Limitations of the study
Other than rod photoreceptors, all major classes of retinal neurons are present in multiple molecularly distinct subtypes in the mouse and/or human retina (Masland, 2012). Like the major retinal cell types, individual neuronal subtypes are generated during different temporal intervals (Cepko, 2014). Although in principle, the approach outlined here should be able to identify GRNs controlling retinal neuronal subtype specification, this dataset is not large enough to accomplish this. Profiling much larger numbers of retinal cells, however, may make it possible to identify these GRNs.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Seth Blackshaw (sblack@jhmi.edu).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and code availability
All mouse and human scRNA-seq and scATAC-seq data can be accessed at GEO accession numbers GSE181251 and GSE183684 (Thomas et al., 2021). Supplemental datasets listed in Mendeley dataset are available at https://doi.org/10.17632/nrstc3xhwb.1.
Code for IReNA v2 pipeline is available at https://github.com/Pinlyu3/IReNA-v2. Interactive displays of all scRNA-seq and scATAC-seq data can be accessed through the St Jude Cloud Visualization Community at https://viz.stjude.cloud/blackshaw-lab/visualization/scatac-seq-analysis-of-mouse-retinal-development~92.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
CD1 mice were purchased from Charles River Laboratories. All experimental procedures were pre-approved by the Institutional Animal Care and Use Committee (IACUC) of the Johns Hopkins University School of Medicine. Mouse embryos or pups at different time points of retinal development (E11, E12, E14, E16, E18, P0, P2, P5, P8, P11 and P14) were used for this study. Chx10-Cre-EGFP;Nfiafl/fl;Nfibfl/fl;Nfixfl/fl mice were generated as described previously (Clark et al., 2019).
Human
The Seattle Children’s Hospital (SCH) Institutional Review Board reviewed and approved all tissue procurement procedures. Experiments were performed in accordance with SCH ethical and legal guidelines. Developing human retinas were obtained from the Birth Defects Research Laboratory at the University of Washington, with ethics board approval and maternal written consent obtained before specimen collection. Developmental age postconception, sex, and postmortem interval prior to collection are as follows for each donor: Sample 1:53 days, female, 7hr 10 min. Sample 2:59 days, male, 7hr30 min. Sample 3: 74 days, male, 6hr 12. Sample 4: 78 days, male, 5hr 39 min. Sample 5:113 days, female, 7hr 25 min. Sample 3: 132 days, male, 4hr 16 min.
METHOD DETAILS
Retinal cell dissociation
Mice were euthanized, and eyes were removed and incubated in ice-cold PBS. For embryonic stages, embryos were first removed from the uterus, placed in iced-cold PBS and decapitated. Whole embryo heads were then transferred to a new Petri dish containing iced-cold PBS, where the skin layer surrounding the eyes were removed to expose the eyeballs. Retinas were then micro-dissected from the eyeballs, and retinal cells were dissociated using Papain Dissociation System as described previously (Hoang et al., 2020). Each sample contains a minimum of 4 retinas from 4 animals, regardless of sex. Dissociated cells were resuspended in ice-cold PBS containing 0.04% bovine serum albumin (BSA). Cell count and viability were assessed by Trypan blue staining.
Ex vivo retinal electroporation and fluorescence-activated cell sorting (FACS)
Retinas from CD1 mouse embryos at day 14 (E14) and postnatal day 0 (P0) were used for ex vivo electroporation as described previously (de Melo and Blackshaw, 2011). For overexpression studies, pCAGIG was used as a control, while pCAGIG-based plasmids encoding full-length ORFs were used for overexpression (see Key resources table). For analysis of NFIA/B/X and TCF7L1/2 function, equal molar amounts of each plasmid were combined prior to electroporation.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-GFP | ThermoFisher | A-16455, RRID:AB_221570 |
| Rabbit anti-GFP | Abcam | ab290, RRID:AB_303395 |
| Chicken anti-GFP | ThermoFisher | A10262, RRID:AB_2534023 |
| Goat anti-GFP | Rockland Antibodies | 600-101-215, RRID: AB_218182 |
| Mouse anti-TFAP2A | DSHB | 3B5, RRID:AB_528084 |
| Rabbit anti-RBPMS | GeneTex | GTX118619, RRID:AB_10720427 |
| Goat anti-OTX2 | R&D Systems | AF1979, RRID:AB_2157172 |
| Rabbit anti-SOX9 | MiliporeSigma | AB5535, RRID:AB_2239761 |
| Rabbit anti-RBPMS | Proteintech | 15187-1-AP, RRID:AB_2238431 |
| Mouse anti-CRX | Abnova | H00001406-M02, RRID:AB_606098 |
| Sheep anti-VSX2 | ExAlpha | X1180P, RRID:AB_2314191 |
| Donkey anti-Sheep 568 | Invitrogen | A21099, RRID:AB_2535753 |
| Donkey anti-Mouse 647 | Invitrogen | A31571, RRID:AB_162542 |
| Donkey anti-Goat 488 | Invitrogen | A11055, RRID:AB_2534102 |
| Donkey anti-Rabbit 568 | Invitrogen | A10042, RRID:AB_2534017 |
| Donkey anti-Rabbit 488 | Invitrogen | A21206, RRID:AB_2535792 |
| Donkey anti-Mouse 568 | Invitrogen | A21206, RRID:AB_2535792 |
| Donkey anti-Sheep 647 | Invitrogen | A21448, RRID:AB_1500712 |
| Anti-NFIA | Sigma-Aldrich | HPA006111, RRID:AB_1854422 |
| Anti-NFIB | Active Motif | 39091, RRID:AB_2314934 |
| Anti-NFIX | Sigma-Aldrich | SAB1401263, RRID:AB_10608433 |
| Mouse IgG | Diagenode | C15400001, RRID:AB_2722553 |
| Rabbit IgG | Diagenode | C15410206, RRID:AB_2722554 |
| Critical commercial assays | ||
| 10x scRNaseq 3′ v3.1 | 10X Genomics | 1000268 |
| 10x scATACseq v1.1 | 10X Genomics | 1000175 |
| PrecisionX Multiplex gRNA Cloning Kit | System Biosciences | CAS9-GRNA-KIT |
| GeneArt Genomic Cleavage Detection Kit | ThermoFisher | A24372 |
| truChIP chromatin shearing kit | Covaris | 520154 |
| iDeal ChIP-seq kit for transcription factors | Diagenode | C01010170 |
| Deposited data | ||
| All mouse scRNA-seq and scATAC-seq data | GEO | GSE181251 |
| All human scRNA-Seq and scATAC-Seq data | GEO | GSE183684 |
| Datasets 1-14 | Mendeley Data | https://doi.org/10.17632/nrstc3xhwb.1 |
| Interactive web portal for mouse scATAC-seqdata | St Jude’s Children’s Hospital | https://viz.stjude.cloud/blackshaw-lab/visualization/scatac-seq-analysis-of-mouse-retinal-development~92 |
| Experimental models: organisms/strains | ||
| CD-1 mice | Charles River Labs | N/A |
| Tg(Chx10-EGFP/Cre/-ALPP)2Clc mice | Dr. Connie Cepko | (Rowan and Cepko, 2004) |
| Nfialox/lox;Nfib lox/lox;Nfixlox/lox mice | Dr. Dr. Richard Gronostajski | (Clark et al., 2019) |
| Oligonucleotides | ||
| Insm1 gRNA1 | IDT DNA | AGTCCACGCCCGTGTCCTAC |
| Insm1 gRNA2 | IDT DNA | CGCTCCCGCCGAGCTCAAGA |
| Insm2 gRNA1 | IDT DNA | GGTGACCACGTCCCCGGTGC |
| Insm2 gRNA2 | IDT DNA | AGACTGGGGCACCCTTACCG |
| Recombinant DNA | ||
| Full-length NFIA ORF | ThermoFisher | IOH12791 |
| Full-length NFIB ORF | ThermoFisher | IOH3202 |
| Full-length NFIX ORF | ThermoFisher | BC117115 |
| Full-length INSM1 ORF | Genecopoeia | GC-F0042 |
| Full-length INSM2 ORF | Genecopoeia | GC-Z3358 |
| Full-length TBX3 ORF | ThermoFisher | IOH12474 |
| Full-length TCF7L1 ORF | Genecopoeia | GW-C0075 |
| Full-length TCF7L2 ORF | ThermoFisher | IOH21979 |
| Software and algorithms | ||
| Cell Ranger | 10X Genomics | Version 4.0.0 and 6.0.1 |
| Cell Ranger ATAC | 10X Genomics | Version 1.2.0 |
| Seurat | https://github.com/satijalab/seurat | Version 3.1.2 |
| Signac | https://github.com/timoast/signac/ | Version 0.1.6 |
| ArchR | https://github.com/GreenleafLab/ArchR | Version 0.9.5 |
| Slingshot | https://github.com/kstreet13/slingshot | Version 1.4.0 |
| Motifmatchr | https://github.com/GreenleafLab/motifmatchr | Version 1.8.0 |
| TOBIAS | https://github.com/loosolab/TOBIAS | Version 0.12.10 |
| Harmony | https://github.com/immunogenomics/harmony | Version 1.0 |
| MAGIC | https://github.com/KrishnaswamyLab/MAGIC | Version 2.0.3 |
| chromVAR | https://github.com/GreenleafLab/chromVAR | Version 1.4.1 |
| MAnorm | https://github.com/shao-lab/MAnorm | Version 1.0 |
| Trimmomatic | https://github.com/usadellab/Trimmomatic | Version 0.38 |
| bowtie2 | https://github.com/BenLangmead/bowtie2 | Version 2.3.5 |
| SAMtools | https://github.com/samtools/samtools | Version 1.9 |
| Picard | https://github.com/broadinstitute/picard | Version 2.18.2 |
| MACS2 | https://github.com/macs3-project/MACS | Version 2.1.2 |
| ComplexHeatmap | https://github.com/jokergoo/ComplexHeatmap | Version 2.0.0 |
| Fgsea | https://github.com/ctlab/fgsea | Version 1.12.0 |
| Rtracklayer | https://bioconductor.org/packages/release/bioc/html/rtracklayer.html | Version 1.46.0 |
| CHOPCHOP | https://chopchop.cbu.uib.no/ | N/A |
| GraphPad Prism | GraphPad Software | N/A |
| ImageJ/Fiji | https://imagej.net/software/fiji/ | N/A |
| Cytoscape | Cytoscape Consortium | Version 3.7.2 |
| BioRender | https://biorender.com/ | N/A |
| TargetScanHuman | http://www.targetscan.org/vert_72/ | Release 7.2, March 2018 |
For somatic CRISPR-mediated gene knockout, the CBh promoter of Cas9-P2A-GFP plasmid (Addgene #48138) was replaced by pCAG promoter (pCAGIG, Addgene #11159) to allow for more robust Cas9-P2A-GFP expression in retinal explants. Dual gRNAs targeting two different exon regions were cloned into a single Cas9 plasmid using PrecisionX Multiplex gRNA Cloning Kit with U6 and H1 promoters. gRNAs were designed using the CHOPCHOP tool. For combined Insm1/2 knockout, equal molar amounts of each gRNA-Cas9 plasmid were mixed prior to electroporation. Retinal cells were dissociated from explants for fluorescence-activated cell sorting (FACS) as described previously (Hoang et al., 2020). GFP+ cells were collected in ice-cold PBS with 10% heat-inactivated fetal bovine serum (FBS). To determine CRISPR-mediated knockout efficiency, genomic DNA was extracted from GFP+ cells from Insm1/2 knockout and empty Cas9 control, and subjected for PCR and digestion using GeneArt Genomic Cleavage Detection Kit.
Immunohistochemistry
Explants used for immunohistochemical analyses were cultured to P0 or P11 equivalent (6 or 11 days in vitro), fixed in 4% paraformaldehyde in PBS, and processed through sucrose gradients before mounting in OCT compound, cryosectioning (15 μm sections), and immunohistochemical analyses. Stained slides of retinal explant sections were imaged using a Zeiss LSM 800 confocal microscope. For each immunostaining condition, 2-3 single-plane confocal images per retinal explant were counted, with counts aggregated across individual explants. Individual data points shown in Figures 5D and 7C represent cell counts obtained from individual explants.
Single Cell RNA-seq library construction and sequencing
ScRNA-seq analysis was performed on dissociated retinal cells using 10x Genomics. Whole retina was used in all cases except for all overexpression and somatic CRISPR analysis, where FACS-isolated GFP-positive electroporated cells were analyzed. Briefly, dissociated retinal cells (~10,000 cells per sample) were loaded into a 10x Genomics Chromium Single Cell system using Chromium Single Cell 3′ Reagents Kits v3.1 (10X Genomics, Pleasanton, CA). scRNA libraries were generated by following the manufacturer’s instructions. Libraries were pooled and sequenced on Illumina NextSeq 500 or NovaSeq 6000. Sequencing reads were processed through the Cell Ranger 3.1 pipeline (10x Genomics) using default parameters.
Single Cell ATAC-seq library construction and sequencing
ScATAC-seq was performed using the 10x Genomic single cell ATAC reagent v1.1 kit following the manufacturer’s instruction. Briefly, dissociated cells were centrifuged at 300xg for 5 min at 4°C. Cell pellet was resuspended in 100 μL of Lysis buffer, mixed 10x by pipetting and incubated on ice for 3 min. Wash buffer (1 ml) was added to the lysed cells, and cell nuclei were centrifuged at 500xg for 5 min at 4°C. Nuclei pellet was re-suspended in 250 μL of 1x Nuclei buffer. Cell nuclei were then counted using Trypan blue. Re-suspended cell nuclei (10-15k) were used for transposition and loaded into the 10x Genomics Chromium Single Cell system. Libraries were amplified with 10 PCR cycles and were sequenced on Illumina NextSeq or NovaSeq with ~200 million reads per library. Sequencing data were processed through the Cell Ranger ATAC 1.1.0 pipeline (10x Genomics) using default parameters.
ChIP-seq
Freshly dissected P2 retinas were homogenized and cross-linked for 10 minutes using 1% formaldehyde (ThermoFisher Scientific Cat# 28906) on a tube rotator at room temperature. Glycine was added to a final concentration of 0.125 M to quench the cross-linking reaction and washed three times with ice-cold PBS with cOmplete protease inhibitors (Millipore Sigma Cat# 11836170001). The cells were then prepared for sonication using the truChIP chromatin shearing kit (Covaris Cat# 520154). Briefly, cells were lysed at 4C on a tube rotator for 10 minutes using the 1X lysis buffer B. Intact nuclei were then collected by centrifugation at 1700xg for 5 minutes and washed with 1X wash buffer C before being resuspended in 1ml of 1X shearing buffer D3. Nuclei were then transferred to 1mL milliTUBE with AFA fiber (Covaris Cat# 520130) and sonicated using the E220 focused-ultrasonicator (Covaris Cat# 500239). Chromatin immunoprecipitation was then performed on the sheared DNA using the iDeal ChIP-seq kit for transcription factors (Diagenode Cat# C01010170). One percent of total chromatin was set aside to be used as an input control. Antibodies against targets used for chromatin immunoprecipitation are NFI (NFIA, NFIB, NFIX), and IgG. Briefly, equal volume of sheared chromatin was incubated overnight with 3 μg of antibody in iC1b buffer with protease inhibitors and BSA and washed DiaMag Protein A-coated magnetic beads (Diagenode Cat# C03010020-220) on a tube rotator at 4°C overnight. The magnetic beads were then washed sequentially with wash buffers iW1, iW2, iW3 and iW4. DNA was then de-crosslinked and eluted for 4 hours at 65C before being purified using IPure beads (Diagenode Cat# C03010014). The purified DNA was then subjected to sequencing library preparation or qPCR analysis. Libraries were prepared from 5ng of DNA using the Ovation Ultralow System V2 (Tecan Genomics Cat# 0344NB-32) and sequenced on the Illumina NextSeq500.
QUANTIFICATION AND STATISTICAL ANALYSIS
Single-cell ATAC-seq analysis
Preprocessing
The Cell Ranger (Zheng et al., 2017) ATAC pipeline was used to process the raw sequencing data for mapping, de-duplication and identification of Tn5 cut sites. We first convert BCL files to fastq format with the function ‘cellranger-atac mkfastq’. Then, we mapped the fastq files to the mm10 genome (refdata-cellranger-atac-GRCh38-1.2.0) with the function ‘cellranger-atac count’. This function outputs the aligned, barcoded, and Tn5 insertion corrected fragment files, which were used for all downstream analysis.
Filtering cells by TSS enrichment, unique fragments, nucleosome banding and doublet score
The ArchR package(Granja et al., 2021) was used to process the fragment files. We calculated the TSS enrichment, unique fragments and nucleosome banding for each cell with the function ‘createArrowFiles’’. Then we kept the high-quality cells with the following criteria: 1) The number of unique nuclear fragments > 1000. 2) TSS enrichment score > 10. 3) nucleosome banding score < 4. We next identified potential doublets with the function ‘addDoubletScores’, and removed doublets using ‘filterDoublets’’ with the following parameters: cutEnrich = 2, cutScore = −Inf, and filterRatio = 2. Finally, we filtered the fragment files according to the cells we retained. These cleaned fragment files were used for all downstream analysis.
Generating union peaks
Union peaks were generated for all the samples as described by Satpathy et al. (2019). We first constructed 2.5kb tiled windows across the mm10 genome and computed a cell-by-window sparse matrix by counting Tn5 insertion (from cleaned fragment files) overlaps for each cell. Next, we binarized the cell-by-window matrix and created a Seurat object for each sample with the Signac (Stuart et al., 2020) package. Then we performed dimension reduction and clustering analysis using the functions ‘RunTFIDF’, ‘RunSVD’, ‘FindNeighbors’ and ‘FindClusters’ with 2-50 dimensions and 0.3 resolution. Next, we call peaks for each identified cluster in each sample using MACS2 (Zhang et al., 2008) software with the following parameters: ‘-shift −75–extsize 150–nomodel–callsummits–nolambda–keep-dup all −q 0.05’. We further extended the peak summits on both sides to a final width of 500 bp, and filtered these fixed-width peaks if they overlapped with mm10 v2 blacklist regions (https://github.com/Boyle-Lab/Blacklist/blob/master/lists/mm10blacklist.v2.bed.gz). Finally, we kept the top 120,000 fixed-width peaks for each cluster in each sample according to their −log10(q-value), and then merged them to the final union peak sets using the ‘reduce’ function from the GenomicRanges (Lawrence et al., 2013) package.
LSI clustering, visualization, and identification of cell types
For each sample, the cell-by-peak matrix was generated by the union peak sets, and was binarized and inputted to the Signac pipeline. Then we performed dimension reduction, clustering and UMAP analysis using the standard Signac workflow.
To annotate cell types corresponding to each cluster, we used existing mouse and human scRNA-seq data (Clark et al., 2019; Thomas et al., 2021) to interpret our scATAC-seq cell types using the CCA (canonical correlation analysis) integration method in the Seurat package. First, we downloaded the mouse scRNA-seq data (“https://github.com/gofflab/developing_mouse_retina_scRNASeq”) and converted them to Seurat objects. Second, for each scATAC-seq sample, we calculate the ‘gene activity’ profile for each cell using the function ‘CreateGeneActivityMatrix’. Finally, for each age-matched sample pair from scATAC-seq and scRNA-seq data, we identified anchors between them using the function ‘transfer.anchors’, and we used the ‘TransferData’ to obtain the cell type prediction results for each cell. We further filtered out cells with a prediction score < 0.5, and annotated each cluster according to their predicted cell types.
Integration of E11-P14 single-cell ATAC-seq data
To Integrate and visualize all the cells from the scATAC-seq data (E11 to P14), we used the following 3 steps:
Filtering cell types. To better identify differences in chromatin accessibility during retinal development, we removed the cells which are not annotated as retinal cells in each time point before integration. We kept the following cell types: RPCs, Neurogenic, RGC, AC/HC, Cone, Rod, BC and MG. The total cell-by-peak matrix is filtered according to the retinal cells and used in the downstream analysis.
Selecting variable peaks. To remove the potential batch effect, we selected the variable peaks separately in each sample. Because scATAC-seq data are very sparse, we aimed to aggregate similar cells to create a more dense cell-by-peaks matrix to facilitate variable peaks calling. First, based on the UMAP embedding, we used the kNN approach to find the 100 nearest cells for each individual cell. Next we aggregate raw counts for each cell by its corresponding 100 nearest cells to create a new cell-by-peaks aggregate matrix. We then identified the variable peaks based on the new matrix using Seurat pipelines: ‘NormalizeData’ and ‘’FindVariableFeatures’ (selection.method = “mvp”). Finally, we combined all the variable peaks from each sample into a master variable feature set, which was used in the downstream dimension reduction and clustering procedure.
LSI clustering and visualization. First, we binarized the filtered cell-by-peak matrix from Step 1 and performed the TF-IDF normalization. Then we used the master variable feature set from Step 2 to perform the dimension reduction with ‘RunSVD.’ Next, we used the 2nd-20th dimensions to identify clusters with a resolution of 1, and calculated the UMAP coordinates for visualization. Finally, we plotted the 3D UMAP of all retinal cells with the plotly graphing library in Python.
ChromVAR and footprint analysis
We used the chromVAR (Schep et al., 2017) R package to infer global TF activity in each cell. First, we fed the raw cell-by-peak matrix into chromVAR and to correct for GC bias with the mm10 reference genome. Next, we generated a TF z-score matrix with the mouse TF Motif database (TransFac2018) using the function ‘computeDeviations’. The z-score for each cell was used to generate the heat-map and visualization using previously calculated UMAP coordinates.
To analyze and plot TF footprints in different retinal cell types, we used the same methods described in Corces et al. (2018). First, we predicted the TF binding sites with the TF PWM matrix and the identified accessibility region with the function ‘matchmotifs’ in the motifmatchr R package. Second, for each motif, we generated 3 tables: 1) Table1: An aggregated observed 6-bp hexamer table in ± 250bp region relative to the motif centers. 2) Table 2: An aggregated expected 6-bp hexamer table from the mm10 genome. 3) Table 3: An observed Tn5 insertion signal table around the ± 250bp relative to the motif centers. Next, we obtain the O/E 6-bp hexamer table by dividing the two hexamer tables (Table 4 = Table 1/Table2). We then normalized the signal using the O/E 6-bp hexamer table (Table 5 = Table 3/Table 4) to get the final Tn5 bias-corrected signal.
Single-cell RNA-seq analysis
We processed raw scRNA-seq data with the Cell Ranger software for formatting reads, demultiplexing samples, genomic alignment, and generating the cell-by-gene count matrix. The ‘cellranger mkfastq’ function was used to convert BCL files to fastq files. The ‘cellranger count’ function was used to process fastq files for each sample with the mm10 mouse reference index provided by 10x Genomics. The cell-by-gene count matrix is the final output from the Cell Ranger pipeline. We used the cell-by-gene count matrix for all downstream analysis.
We applied the Seurat (Stuart et al., 2019) package to create Seurat objects for each sample with the cell-by-gene count matrix with the function ‘CreateSeuratObject’ (min.cells = 3, min.features = 200). After visual checking the violin plot of the total counts for each cell, we filtered out cells with nCount_RNA < 800 or nCount_RNA > 8000. We further filtered out the cells with a mitochondrial fraction 10%. Next, we used Scrublet (Wolock et al., 2019) to identify and remove doublets with default parameters. After performing dimensional reduction and clustering analysis through the standard Seurat pipeline, we annotated the cell types for each cluster according to the well-known markers. We filtered out non-neuroretinal cells, such as microglia and astrocytes for each sample. The cleaned data were used for downstream integration and DEG analysis.
Trajectory inference and pseudotime analysis
We applied Slingshot (Street et al., 2018) software to infer pseudotime based on the UMAP coordinates. We first filtered the UMAP coordinates matrix by keeping the cells involved in the developmental process we plan to investigate. Then we ran Slingshot with the filtered UMAP coordinates matrix for each observed trajectory separately (Figure 4: RPCS1 to MG, Figures 6 and S6: RPC to NG, NG to RGC, NG to AC/HC, NG to Cone, NG to Rod, NG to BC) with the function ‘getLineages‘ and ‘getCurves‘. Then, we applied the ‘slingPseudotime‘ function to calculate pseudotime state for each cell. Finally, we merged the pseudotime to 20-50 bins for each trajectory, and calculated the average gene expression or average accessibility level for each bin.
Identification of cell-type specific genes, peaks and motif activities
We calculated the cell-type specific genes across all the retina cells with the mouse scRNaseq data. The function ‘FindMarkers‘ in Seurat package were used to identify the marker genes for each cell type with the following parameters: min.pct = 0.1, logfc.threshold = 1, only.pos = TRUE, p-adjust < 0.01. These gene sets are used in the downstream gene set enrichment analysis (GSEA).
We calculated the cell-type specific peaks across all the retina cells for mouse and human scATACseq respectively. The function ‘getMarkerFeatures’ in ArchR package were used to calculate the enrichment of peaks for each cell type with the following parameters: normBy = ‘nFrags’, bias = c(“TSSEnrichment,””log10(nFrags)”) and testMethod = “wilcoxon.” We then further filtered the results to get the final specific peak sets with the function “getMarkers”: cutOff = “FDR ≤ 0.01 & Log2FC >= 1.5.” These peak sets are used in the downstream peak set enrichment analysis (PSEA).
The cell-type-specific motifs were identified based on the chromVAR results. First, we identify the significant enriched motif for each cell type. We converted the chromVAR deviation matrix to a Seurat object. Then we added the cell type information to the Seurat object and measured the enriched motif for each cell type by the function “FindAllMarkers” with following parameters: only.pos = TRUE, test.use = ‘LR’, and p value < 0.01. Second, we further filtered the significant motifs according to their average Z-score and their ranks among the retina cell types. We only kept the motifs for each cell type if they 1) show average chromVAR Z-score 1, and 2) average chromVAR Z-score are the highest or the second highest among all the cell types.
Identification of conserved peaks between mouse and human
We compared cell-type specific peaks between mouse (mm10) and human (hg38) using the rtracklayer package in R. we converted mouse peak region from mm10 assembly to hg38 assembly with the function ‘liftOver’. We then identified the overlapping peak pairs between mouse converted peaks and human peaks with the function ‘findOverlaps’. We also calculated the overlap ratio for each pair as: Overlap ratio = width(Mouse converted peak) / width(Overlapping region). Finally, we identified the pairs of peaks as conserved peak pairs if their overlap ratio > 0.5.
Constructing gene regulatory networks by integrating scRNA-seq and scATAC-seq data
To infer cell-type-specific GRNs from scRNA-seq and scATAC-seq data, we modified IReNA pipeline to IReNA v2 (Figures S4A and S4B), which contains the following main modules:
1. Selecting candidate genes
The DEGs were used as candidate genes for GRNs construction. For each developmental process which we aim to study in mouse and human, we identified the enriched genes for each cell type using the function ‘FindMarkers’ in Seurat. In constructing the GRNs of progenitors transition, the following parameters of ‘FindMarkers’ were used: min.pct = 0.05, logfc.threshold = 0.20, only.pos = TRUE, p-adjust < 0.01. In constructing GRNs regulating neurogenesis, the following parameters of ‘FindMarkers’ were used: min.pct = 0.1, logfc.threshold = 0.25, only.pos = TRUE and p-adjust < 0.01.
2. Identifying significant peak-to-gene links
We used the ArchR package to identify the significant peak-to-gene links. First, we integrated the age-matched scRNA-seq and scATAC-seq data for each time point using unconstrained Integration method with the function ‘addGeneIntegrationMatrix’. Then, using the function ‘addPeak2GeneLinks’, we calculated the correlation between accessibility peak intensity and gene expression. Finally, we identified the significant peak-to-gene links with the following cutoff: abs(correlation) > 0.2 and fdr < 1e-6.
3. Identifying the potential cis-regulatory elements for each candidate gene
We identified potential cis-regulatory elements for each candidate gene based on their location and the peak-to-gene links from Step 2. We first classified all peaks into three categories according to their genomic location related to their potential target genes: 1) Promoter. 2) Gene body. 3) Intergenic. For the peaks in the promoter region,we treated all of them as correlated accessible chromatin regions (CARs) of their overlapping target genes. For the peaks in the gene body region, we defined them as CARs of their overlapping genes if they met the following criteria: 1) the distance between the peak and the TSS of its overlapping gene is < 100kb. 2) the links between the peak and its overlapping gene are significant. For the peaks in the intergenic region, we first find their target genes and construct the peak-gene pairs if the target genes’ TSS are located within the upstream 100kb or downstream 100 kb of the intergenic peaks. Then we keep the peak-gene pairs if their peak-to-gene links are significant in step 2. These peaks were identified as CARs of their gene pairs.
4. Predicting cell-type specific TFs binding in cis-regulatory elements
With the cis-regulatory elements identified in step 3, we next predicted the TF binding in these elements for each cell type with the PWMs extracted from TRANSFAC database. First, we searching the motifs in all the cis-regulatory elements with the function ‘match-Motifs (p.cutoff = 5e-05)’ from the motifmatchr package. Then we filtered these motif regions according to their footprint score and their corresponding TF’s expression for each cell type.
To calculate the footprint score for each motif region in each cell type, we re-grouped the insertion fragments based on their origin of cell type and converted these cell-type-specific fragments into bam files using a custom script. Then we fed the bam files to TOBIAS software and obtained the bias-corrected Tn5 signal (log2(obs/exp)) with the default parameters except: ATACorrect–read_shift 0 0. Next, we calculated footprint scores including NC, NL and NR for each motif’s binding region. NC indicated the average bias-corrected Tn5 signal in the center of the motif. NL and NR indicated the average bias-corrected Tn5 signal in the left and right flanking regions of the motif, respectively. The flanking region is triple the size of the center region. We kept the motifs with the following criteria: NC < −0.1 and NL > 0.1 and NR > 0.1.
We further removed the motifs binding region for each cell type if the expression level of their corresponding TFs are not enriched in that cell type (from step 1).
5. Calculating gene-gene correlation
We calculated the expression correlations between all the expressed genes at the single-cell level. First, we extracted the cell-by-matrix from Seurat objects and filtered out the non-expressed genes in the matrix. Then we applied the MAGIC software to impute missing values and recover the gene interactions with the cell-by-gene matrix. The output matrix from MAGIC was used to calculate gene-gene correlation using the function ‘cor’ in R. To identify the significant gene-gene correlations, we ranked all the gene-gene correlations (~1X10e8). The top 2.5% correlations were treated as significant positive correlations (p < 0.025) and the bottom 2.5% correlations were treated as significant negative correlations (p < 0.025).
6. Constructing gene regulatory networks
By integrating data from Step 1-Step 5, We constructed cell-type specific GRNs with the following procedure:
We first obtained the peak-target links from Step 3, and cell-type specific TF-peak links from Step 4. We then merged these 2 types of links to the cell-type specific TF-peak-target relationships. Next, we classified these TF-peak-target relationships into activation or repression relationships based on the sign of the expression correlation between TF and target from Step 5. The significant positive/negative correlated TF-targets were selected as the activating/repressive regulatory relationships, respectively.
Finally, we removed all the duplicated TF-target regulatory relationships for each cell type and merged them to the final GRNs which were used for the downstream analysis.
7. Identifying and visualizing feedback TF pairs
With the GRNs constructed in the previous steps, we searched for TF pairs connected by either positive or negative feedback regulatory relationships. The TF pairs that activated each other were identified as ‘double positive’ pairs and the TF pairs repressed each other were identified as ‘double negative’ pairs. We visualized these feedback TFs pairs using Cytoscape software.
Constructing inter-module regulatory networks
To study the global regulatory relationships among retinal cell types in each development stage, we calculated the regulatory significance and regulatory tendency between each pair of cell types using a previously described approach (Hoang et al., 2020). We first filtered the TFs and targets in the GRNs, we only kept the highly enriched TFs and targets for each cell type if their log2 fold change were > 0.5. Then for each pair of cell types, we counted the regulations from cell type A to cell type B and calculated the significance of positive or negative regulations using the hypergeometric test respectively. We set p value < 0.01 as a cutoff to determine the significant regulations. To further quantify which positive / negative regulatory relationships are present between cell type A and cell type B, we calculate the regulatory enrichment fold. We first normalized positive or negative regulations by the total number of positive or negative regulations in that stage, then calculated regulatory enrichment fold as: abs(log2(Rpos/Rneg)), where Rpos indicate the normalized positive regulations and Rneg indicate the normalized negative regulations.
ChIP-seq data analysis
After removing adaptors with Trimmomatic (Bolger et al., 2014), we mapped the cleaned fastq files to mm10 genome using bowtie2 (Langmead and Salzberg, 2012). We next filtered low quality reads with SAMtools (Li et al., 2009)(MAPQ < 10), and removed PCR duplicates using Picard tools (http://broadinstitute.github.io/picard/). For NFI ChIP-seq data, we used IgG and Input samples as control, and used MACS2 (Zhang et al., 2008) to call peaks with the default parameters except: −q 0.01. Finally, we identified 13,680 NFI binding peaks and used them in the downstream analysis.
Integration analysis of single-cell RNA-seq or single-cell ATAC-seq data between control and treatment samples
We applied the CCA method from Seurat to integrate the scRNA-seq data from different genotypes at the same age (Figures S5 and S7). Briefly, we first processed the cleaned seurat objects generated before using the functions ‘NormalizeData’, and ‘FindVariableFeatures’. Next, we integrated all the Seurat objects from different genotypes using the ‘FindIntegrationAnchors’ and ‘IntegrateData’ function with the options: dim.use = 1:50. Finally, we used the integrated dimensions to perform clustering analysis, and calculated UMAP coordinates for visualization.
We applied the Harmony (Korsunsky et al., 2019) package to integrate the scATAC-seq data from different genotypes at the same age (Figure S5). Briefly, we first merged the cell-by-peak matrix from the same age, then inputted the cell-by-peak matrix into the Signac analysis pipeline. We normalized and obtained a low-dimensional representation of the cell-by-peak matrix using the functions ‘FindTopFeatures’, ‘RunTFIDF’ and ‘RunSVD’. Next, we integrated all the cells from different genotypes (Ctrl versus NFI Overexpress or Ctrl versus NFI TKO) using the ‘RunHarmony’ function with the options: dim.use = 2:50, group.by.vars = ‘genotypes’, reduction = ‘lsi’ and project.dim = FALSE. Finally, we used the harmony dimensions to perform clustering analysis, and calculated UMAP coordinates for visualization.
Inferring Nfia/b/x targets in progenitors
We predicted NFI target genes by integrating the information obtained from scRNA-seq, scATAC-seq and ChIP-seq analysis with the following steps (Figure S5G):
1. Identify DEGs resulting from NFIA/B/X overexpression and Nfia/b/x conditional knockout.
We performed the differential gene expression analysis between control RPC/MG cells and Nfia/b/x TKO or NFIA/B/X overexpressing RPC/MG cells (Figure S5B) using the function ‘FindMarkers’ with the options: min.pct = 0.1, logfc.threshold = 0.25. Then we selected the DEGs with adjusted p value < 0.01.
2. Identify DARs following NFIA/B/X overexpression or Nfia/b/x conditional knockout.
To explore which ATAC regions are changed following NFIA/B/X overexpression or Nfia/b/x conditional knockout (Figure S5C), we applied the MAnorm (Shao et al., 2012) algorithm to perform the differential peak analysis. First, we selected cells in the ‘RPC’ and ‘MG’ cluster and then separated these cells according to their genotypes (control, NFIA/B/X overexpression or Nfia/b/x conditional knockout). Next, we aggregated the cells in the same condition by summing their count signals for each peak, and created a new condition-by-peak count matrix, and fed this into the MAnorm pipeline. We performed the MAnorm test and identified the differential peaks using the cutoff: LOG_p > 5, abs (M_value_rescaled) > 0.5 and A_value_scaled > 4.
3. Predict NFIA/B/X regulatory targets in RPC/MG
We predicted the NFIA/B/X regulatory targets by integrating the information from DEGs, DARs, Nfia/b/x ChIP-seq peaks and peak-to-gene links (that showed dynamic regulation across the RPC-MG developmental process) for each experiment. (Figures 5G and S5G)
First, we identified the Nfia/b/x-binding DARs by overlapping the Nfia/b/x ChIP-seq peaks with the DARs upon NFIA/B/X overexpression or Nfia/b/x conditional knockout. For each Nfia/b/x-binding DAR, we then predicted their target genes using the same method described in the RPC-MG GRNs construction: For the Nfia/b/x-binding DARs in the TSS region, their overlapping genes are considered as their regulatory target if these genes are also DEGs. For the Nfia/b/x-binding DARs in the gene body region, their overlapping genes are considered as their regulatory target if they meet: 1) The distance between the DARs and the TSS of its target gene is < 100kb. 2) The link between the DARs and its target gene is significant. 3) Their target genes are DEGs. For the Nfia/b/x-binding DARs in the intergenic region, all the genes are considered as target genes if they meet: 1) The distance between the DARs and the TSS of its target gene is < 100kb. 2) The links between the DARs and its target gene are significant. 3) Their target genes are DEGs.
Differential gene expression analysis with single-cell RNaseq data
To identify DEGs resulting from Insm1,Insm2,Tbx3,Tcf7l1/l2 overexpression and Insm1/2 CRISPR-mediated knockdown for each cell type (Figures 7E and 7F), we used the Wilcoxon Rank Sum Test with the function ‘FindMarkers’ from the Seurat package with the options: min.pct = 0.1, logfc.threshold = 0.25. Then we selected DEGs with adjusted p value < 0.01.
Gene/peaks set enrichment analysis
We performed Gene/peak set enrichment analysis using the fgsea package in R with the default parameters (Korotkevich et al., 2021). The significant DEGs and DARs were ranked based on their log2 fold change (treatment/control). The retina cell-type-specific gene sets and peak sets we used in the GSEA / PSEA were generated using all retinal cells as mentioned before.
GO term analysis
To understand what biological functions are enriched in the gene set we are interested, we applied Gorilla (Eden et al., 2009) algorithm to calculate the enriched Gene Ontology terms for our gene sets with the default parameters (P value threshold = 0.001, ontology = ‘Process’).
Supplementary Material
Highlights.
Single-cell profile of chromatin accessibility changes during mouse retinal development
Identification of conserved gene regulatory networks controlling retinal development
NFI factors directly active genes promoting late-stage retinal progenitor identity
Identification of regulators of rod photoreceptor specification
ACKNOWLEDGMENTS
The authors would like to thank Richard Gronostajski for providing Nfia/b/x conditional mice, Linda Orzolek and Tyler Creamer at the Hopkins Transcriptomics and Deep Sequencing Core for use of the 10× Genomics Chromium controller and library sequencing, and J. Nathans, H. Jaspers, M. Cayouette, A. Javed, L. Goff, J. Kebschull, W. Yap, and members of the Blackshaw lab for comments on the manuscript. This work was supported by the NIH National Eye Institute grants R01EY020560 and U01EY027267 to S.B., EY001765 in support of flow sorting, R01EY029548 and P30EY001765 to J.Q., R00EY027844 to B.S.C., R01EY028584 to T.J.C., P30 EY002687 in support of confocal imaging, and 2T32EY007143 to C.P.S. S.B. is supported by a Stein Innovation Award from Research to Prevent Blindness. T.J.C. is supported by a grant from the Lowy Medical Research Institute, and B.S.C. is supported by an unrestricted grant to the Washington University Department of Ophthalmology and Visual Sciences and a Career Development Award from Research to Prevent Blindness.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109994.
DECLARATION OF INTERESTS
S.B. co-founded and is a shareholder of CDI Labs, LLC. S.B. is also a consultant for Third Rock Ventures, LLC.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper received support from a program designed to increase minority representation in science. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
SUPPORTING CITATIONS
The following references appear in the supplemental information: Stein-O’Brien et al. (2019).
REFERENCES
- Akagi T, Inoue T, Miyoshi G, Bessho Y, Takahashi M, Lee JE, Guillemot F, and Kageyama R (2004). Requirement of multiple basic helix-loop-helix genes for retinal neuronal subtype specification. J. Biol. Chem 279, 28492–28498. [DOI] [PubMed] [Google Scholar]
- Akhmedov NB, Piriev NI, Chang B, Rapoport AL, Hawes NL, Nishina PM, Nusinowitz S, Heckenlively JR, Roderick TH, Kozak CA, et al. (2000). A deletion in a photoreceptor-specific nuclear receptor mRNA causes retinal degeneration in the rd7 mouse. Proc. Natl. Acad. Sci. USA 97, 5551–5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldiri I, Xu B, Wang L, Chen X, Hiler D, Griffiths L, Valentine M, Shirinifard A, Thiagarajan S, Sablauer A, et al. ; St. Jude Children’s Research Hospital–Washington University Pediatric Cancer Genome Project (2017). The dynamic epigenetic landscape of the retina during development, reprogramming, and tumorigenesis. Neuron 94, 550–568.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett EA, and Wallace VA (2012). Cell fate determination in the vertebrate retina. Trends Neurosci 35, 565–573. [DOI] [PubMed] [Google Scholar]
- Bayraktar OA, and Doe CQ (2013). Combinatorial temporal patterning in progenitors expands neural diversity. Nature 498, 449–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosze B, Moon M-S, Kageyama R, and Brown NL (2020). Simultaneous requirements for Hes1 in retinal neurogenesis and optic cup-stalk boundary maintenance. J. Neurosci 40, 1501–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzezinski JA 4th, Lamba DA, and Reh TA (2010). Blimp1 controls photoreceptor versus bipolar cell fate choice during retinal development. Development 137, 619–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter RA, Bihannic L, Rosencrance C, Hadley JL, Tong Y, Phoenix TN, Natarajan S, Easton J, Northcott PA, and Gawad C (2018). A single-cell transcriptional atlas of the developing murine cerebellum. Curr. Biol 28, 2910–2920.e2. [DOI] [PubMed] [Google Scholar]
- Cayouette M, Barres BA, and Raff M (2003). Importance of intrinsic mechanisms in cell fate decisions in the developing rat retina. Neuron 40, 897–904. [DOI] [PubMed] [Google Scholar]
- Cepko C (2014). Intrinsically different retinal progenitor cells produce specific types of progeny. Nat. Rev. Neurosci 15, 615–627. [DOI] [PubMed] [Google Scholar]
- Cepko CL, Austin CP, Yang X, Alexiades M, and Ezzeddine D (1996). Cell fate determination in the vertebrate retina. Proc. Natl. Acad. Sci. USA 93, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CSY, Lonfat N, Zhao R, Davis AE, Li L, Wu M-R, Lin C-H, Ji Z, Cepko CL, and Wang S (2020). Cell type- and stage-specific expression of Otx2 is regulated by multiple transcription factors and cis-regulatory modules in the retina. Development 147, dev187922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark BS, Stein-O’Brien GL, Shiau F, Cannon GH, Davis-Marcisak E, Sherman T, Santiago CP, Hoang TV, Rajaii F, James-Esposito RE, et al. (2019). Single-cell RNA-seq analysis of retinal development identifies NFI factors as regulating mitotic exit and late-born cell specification. Neuron 102, 1111–1126.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Granja JM, Shams S, Louie BH, Seoane JA, Zhou W, Silva TC, Groeneveld C, Wong CK, Cho SW, et al. ; Cancer Genome Atlas Analysis Network (2018). The chromatin accessibility landscape of primary human cancers. Science 362, eaav1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan CS, Renner M, De Gennaro M, Gross-Scherf B, Goldblum D, Hou Y, Munz M, Rodrigues TM, Krol J, Szikra T, et al. (2020). Cell types of the human retina and its organoids at single-cell resolution. Cell 182, 1623–1640.e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Melo J, and Blackshaw S (2011). In vivo electroporation of developing mouse retina. J. Vis. Exp, 2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Melo J, Zibetti C, Clark BS, Hwang W, Miranda-Angulo AL, Qian J, and Blackshaw S (2016). Lhx2 is an essential factor for retinal gliogenesis and Notch signaling. J. Neurosci 36, 2391–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denny SK, Yang D, Chuang C-H, Brady JJ, Lim JS, Grüner BM, Chiou S-H, Schep AN, Baral J, Hamard C, et al. (2016). Nfib promotes metastasis through a widespread increase in chromatin accessibility. Cell 166, 328–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Bella DJ, Habibi E, Stickels RR, Scalia G, Brown J, Yadollahpour P, Yang SM, Abbate C, Biancalani T, Macosko EZ, et al. (2021). Molecular logic of cellular diversification in the mouse cerebral cortex. Nature 595, 554–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doe CQ (2017). Temporal patterning in the Drosophila CNS. Annu. Rev. Cell Dev. Biol 33, 219–240. [DOI] [PubMed] [Google Scholar]
- Domcke S, Hill AJ, Daza RM, Cao J, O’Day DR, Pliner HA, Aldinger KA, Pokholok D, Zhang F, Milbank JH, et al. (2020). A human cell atlas of fetal chromatin accessibility. Science 370, eaba7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, and Yakhini Z (2009). GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott J, Jolicoeur C, Ramamurthy V, and Cayouette M (2008). Ikaros confers early temporal competence to mouse retinal progenitor cells. Neuron 60, 26–39. [DOI] [PubMed] [Google Scholar]
- Erclik T, Li X, Courgeon M, Bertet C, Chen Z, Baumert R, Ng J, Koo C, Arain U, Behnia R, et al. (2017). Integration of temporal and spatial patterning generates neural diversity. Nature 541, 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes-Osborne MA, Wilson SG, and Morris AC (2013). Insulinoma-associated 1a (Insm1a) is required for photoreceptor differentiation in the zebrafish retina. Dev. Biol 380, 157–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa T, Morrow EM, and Cepko CL (1997). Crx, a novel otx-like homeobox gene, shows photoreceptor-specific expression and regulates photoreceptor differentiation. Cell 91, 531–541. [DOI] [PubMed] [Google Scholar]
- Ghiasvand NM, Rudolph DD, Mashayekhi M, Brzezinski JA 4th, Goldman D, and Glaser T (2011). Deletion of a remote enhancer near ATOH7 disrupts retinal neurogenesis, causing NCRNA disease. Nat. Neurosci 14, 578–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghinia Tegla MG, Buenaventura DF, Kim DY, Thakurdin C, Gonzalez KC, and Emerson MM (2020). OTX2 represses sister cell fate choices in the developing retina to promote photoreceptor specification. eLife 9, e54279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granja JM, Ryan Corces M, Pierce SE, Tansu Bagdatli S, Choudhry H, Chang HY, and Greenleaf WJ (2021). ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet 53, 403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang T, Wang J, Boyd P, Wang F, Santiago C, Jiang L, Yoo S, Lahne M, Todd LJ, Jia M, et al. (2020). Gene regulatory networks controlling vertebrate retinal regeneration. Science 370, eabb8598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hojo M, Ohtsuka T, Hashimoto N, Gradwohl G, Guillemot F, and Kageyama R (2000). Glial cell fate specification modulated by the bHLH gene Hes5 in mouse retina. Development 127, 2515–2522. [DOI] [PubMed] [Google Scholar]
- Holguera I, and Desplan C (2018). Neuronal specification in space and time. Science 362, 176–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie S, Sanuki R, Muranishi Y, Kato K, Chaya T, and Furukawa T (2015). Rax homeoprotein regulates photoreceptor cell maturation and survival in association with Crx in the postnatal mouse retina. Mol. Cell. Biol 35, 2583–2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javed A, and Cayouette M (2017). Temporal progression of retinal progenitor cell identity: implications in cell replacement therapies. Front. Neural Circuits 11, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javed A, Mattar P, Lu S, Kruczek K, Kloc M, Gonzalez-Cordero A, Bremner R, Ali RR, and Cayouette M (2020). Pou2f1 and Pou2f2 cooperate to control the timing of cone photoreceptor production in the developing mouse retina. Development 147, dev188730. [DOI] [PubMed] [Google Scholar]
- Jia L, Oh ECT, Ng L, Srinivas M, Brooks M, Swaroop A, and Forrest D (2009). Retinoid-related orphan nuclear receptor RORbeta is an early-acting factor in rod photoreceptor development. Proc. Natl. Acad. Sci. USA 106, 17534–17539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Jiang H, Xiao D, Zou M, Zhu J, and Xiang M (2015). Tfap2a and 2b act downstream of Ptf1a to promote amacrine cell differentiation during retinogenesis. Mol. Brain 8, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SS, Jagadeesh K, Dey KK, Shen AZ, Raychaudhuri S, Kellis M, and Price AL (2021). Leveraging single-cell ATAC-seq to identify disease-critical fetal and adult brain cell types. bioRxiv. 10.1101/2021.05.20.445067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinides N, Rossi AM, Escobar A, Dudragne L, Chen Y-C, Tran T, Jaimes AM, Özel MN, Simon F, Shao Z, et al. (2021). A comprehensive series of temporal transcription factors in the fly visual system. bioRxiv. 10.1101/2021.06.13.448242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, and Sergushichev A (2021). Fast gene set enrichment analysis. bioRxiv 10.1101/060012. [DOI] [Google Scholar]
- Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh P-R, and Raychaudhuri S (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahne M, Nagashima M, Hyde DR, and Hitchcock PF (2020). Reprogramming Müller glia to regenerate retinal neurons. Annu. Rev. Vis. Sci 6, 171–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, and Carey VJ (2013). Software for computing and annotating genomic ranges. PLoS Comput. Biol 9, e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Liu X, Li S, Huang X, Qian H, Jin K, and Xiang M (2020). Foxn4 is a temporal identity factor conferring mid/late-early retinal competence and involved in retinal synaptogenesis. Proc. Natl. Acad. Sci. USA 117, 5016–5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Shiau F, Yi W, Lu S, Wu Q, Pearson JD, Kallman A, Zhong S, Hoang T, Zuo Z, et al. (2020). Single-cell analysis of human retina identifies evolutionarily conserved and species-specific mechanisms controlling development. Dev. Cell 53, 473–491.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Zhang B, LaFave LM, Earl AS, Chiang Z, Hu Y, Ding J, Brack A, Kartha VK, Tay T, et al. (2020). Chromatin potential identified by shared single-cell profiling of RNA and chromatin. Cell 183, 1103–1116.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malin J, and Desplan C (2021). Neural specification, targeting, and circuit formation during visual system assembly. Proc. Natl. Acad. Sci. USA 118, e2101823118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manno G, Siletti K, Furlan A, Gyllborg D, Vinsland E, Albiach AM, Langseth CM, Khven I, Lederer AR, Dratva LM, et al. (2021). Molecular architecture of the developing mouse brain. Nature 596, 92–96. [DOI] [PubMed] [Google Scholar]
- Marquardt T, Ashery-Padan R, Andrejewski N, Scardigli R, Guillemot F, and Gruss P (2001). Pax6 is required for the multipotent state of retinal progenitor cells. Cell 105, 43–55. [DOI] [PubMed] [Google Scholar]
- Masland RH (2012). The neuronal organization of the retina. Neuron 76, 266–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattar P, Ericson J, Blackshaw S, and Cayouette M (2015). A conserved regulatory logic controls temporal identity in mouse neural progenitors. Neuron 85, 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mears AJ, Kondo M, Swain PK, Takada Y, Bush RA, Saunders TL, Sieving PA, and Swaroop A (2001). Nrl is required for rod photoreceptor development. Nat. Genet 29, 447–452. [DOI] [PubMed] [Google Scholar]
- Melsted P, Booeshaghi AS, Liu L, Gao F, Lu L, Min KHJ, da Veiga Beltrame E, Hjörleifsson KE, Gehring J, and Pachter L (2021). Modular, efficient and constant-memory single-cell RNA-seq preprocessing. Nat. Biotechnol 39, 813–818. [DOI] [PubMed] [Google Scholar]
- Miltner AM, and La Torre A (2019). Retinal ganglion cell replacement: current status and challenges ahead. Dev. Dyn 248, 118–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muto A, Iida A, Satoh S, and Watanabe S (2009). The group E Sox genes Sox8 and Sox9 are regulated by Notch signaling and are required for Müller glial cell development in mouse retina. Exp. Eye Res 89, 549–558. [DOI] [PubMed] [Google Scholar]
- Nishida A, Furukawa A, Koike C, Tano Y, Aizawa S, Matsuo I, and Furukawa T (2003). Otx2 homeobox gene controls retinal photoreceptor cell fate and pineal gland development. Nat. Neurosci 6, 1255–1263. [DOI] [PubMed] [Google Scholar]
- Norrie JL, Lupo MS, Xu B, Al Diri I, Valentine M, Putnam D, Griffiths L, Zhang J, Johnson D, Easton J, et al. (2019). Nucleome dynamics during retinal development. Neuron 104, 512–528.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberst P, Agirman G, and Jabaudon D (2019). Principles of progenitor temporal patterning in the developing invertebrate and vertebrate nervous system. Curr. Opin. Neurobiol 56, 185–193. [DOI] [PubMed] [Google Scholar]
- Paridaen JTML, and Huttner WB (2014). Neurogenesis during development of the vertebrate central nervous system. EMBO Rep 15, 351–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remez LA, Onishi A, Menuchin-Lasowski Y, Biran A, Blackshaw S, Wahlin KJ, Zack DJ, and Ashery-Padan R (2017). Pax6 is essential for the generation of late-born retinal neurons and for inhibition of photoreceptor-fate during late stages of retinogenesis. Dev. Biol 432, 140–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi AM, Jafari S, and Desplan C (2021). Integrated patterning programs during Drosophila development generate the diversity of neurons and control their mature properties. Annu. Rev. Neurosci 44, 153–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan S, and Cepko CL (2004). Genetic analysis of the homeodomain transcription factor Chx10 in the retina using a novel multifunctional BAC transgenic mouse reporter. Dev. Biol 271, 388–402. [DOI] [PubMed] [Google Scholar]
- Roy A, de Melo J, Chaturvedi D, Thein T, Cabrera-Socorro A, Houart C, Meyer G, Blackshaw S, and Tole S (2013). LHX2 is necessary for the maintenance of optic identity and for the progression of optic morphogenesis. J. Neurosci 33, 6877–6884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagner A, Zhang I, Watson T, Lazaro J, Melchionda M, and Briscoe J (2020). Temporal patterning of the central nervous system by a shared transcription factor code. bioRxiv. 10.1101/2020.11.10.376491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, and Zipursky SL (2010). Design principles of insect and vertebrate visual systems. Neuron 66, 15–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarropoulos I, Sepp M, Frömel R, Leiss K, Trost N, Leushkin E, Okonechnikov K, Joshi P, Giere P, Kutscher LM, et al. (2021). Developmental and evolutionary dynamics of cis-regulatory elements in mouse cerebellar cells. Science 373, eabg4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpathy AT, Granja JM, Yost KE, Qi Y, Meschi F, McDermott GP, Olsen BN, Mumbach MR, Pierce SE, Corces MR, et al. (2019). Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol 37, 925–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep AN, Wu B, Buenrostro JD, and Greenleaf WJ (2017). chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z, Zhang Y, Yuan G-C, Orkin SH, and Waxman DJ (2012). MAnorm: a robust model for quantitative comparison of ChIP-seq data sets. Genome Biol 13, R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein-O’Brien GL, Clark BS, Sherman T, Zibetti C, Hu Q, Sealfon R, Liu S, Qian J, Colantuoni C, Blackshaw S, et al. (2019). Decomposing cell identity for transfer learning across cellular measurements, platforms, tissues, and species. Cell Syst 8, 395–411.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N, Purdom E, and Dudoit S (2018). Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Srivastava A, Lareau C, and Satija R (2020). Multimodal single-cell chromatin analysis with Signac. bioRxiv. 10.1101/2020.11.09.373613. [DOI] [Google Scholar]
- Taranova OV, Magness ST, Fagan BM, Wu Y, Surzenko N, Hutton SR, and Pevny LH (2006). SOX2 is a dose-dependent regulator of retinal neural progenitor competence. Genes Dev 20, 1187–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telley L, Agirman G, Prados J, Amberg N, Fièvre S, Oberst P, Bartolini G, Vitali I, Cadilhac C, Hippenmeyer S, et al. (2019). Temporal patterning of apical progenitors and their daughter neurons in the developing neocortex. Science 364, eaav2522. [DOI] [PubMed] [Google Scholar]
- Thomas ED, Timms AE, Giles S, Harkins-Perry S, Lyu P, Hoang T, Qian J, Jackson V, Bahlo M, Blackshaw S, et al. (2021). Multi-omic analysis of developing human retina and organoids reveals cell-specific cis-regulatory elements and mechanisms of non-coding genetic disease risk. bioRxiv. 10.1101/2021.07.31.454254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thor S (2017). Nervous system development: temporal patterning of large neural lineages. Curr. Biol 27, R392–R394. [DOI] [PubMed] [Google Scholar]
- Tiklová K, Björklund ÅK, Lahti L, Fiorenzano A, Nolbrant S, Gillberg L, Volakakis N, Yokota C, Hilscher MM, Hauling T, et al. (2019). Single-cell RNA sequencing reveals midbrain dopamine neuron diversity emerging during mouse brain development. Nat. Commun 10, 581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall DS, Mears AJ, McNeill B, Mazerolle C, Thurig S, Wang Y, Kageyama R, and Wallace VA (2009). Progenitor cell proliferation in the retina is dependent on Notch-independent Sonic hedgehog/Hes1 activity. J. Cell Biol 184, 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolock SL, Lopez R, and Klein AM (2019). Scrublet: computational identification of cell doublets in single-cell transcriptomic data. Cell Syst 8, 281–291.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F, Kaczynski T, Matheson LS, Liu T, Wang J, Turner M, and Mu X (2020). Zfp36l1 and Zfp36l2 balances proliferation and differentiation in the developing retina. bioRxiv. 10.1101/2020.12.15.422926. [DOI] [Google Scholar]
- Xie H, Zhang W, Zhang M, Akhtar T, Li Y, Yi W, Sun X, Zuo Z,Wei M, Fang X, et al. (2020). Chromatin accessibility analysis reveals regulatory dynamics of developing human retina and hiPSC-derived retinal organoids. Sci. Adv 6, eaay5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Tang X, Jin M, Zhang H, Du L, Yu S, and He J (2020). Unifying developmental programs for embryonic and postembryonic neurogenesis in the zebrafish retina. Development 147, dev185660. [DOI] [PubMed] [Google Scholar]
- Young RW (1985a). Cell differentiation in the retina of the mouse. Anat. Rec 212, 199–205. [DOI] [PubMed] [Google Scholar]
- Young RW (1985b). Cell proliferation during postnatal development of the retina in the mouse. Brain Res 353, 229–239. [DOI] [PubMed] [Google Scholar]
- Zechner C, Nerli E, and Norden C (2020). Stochasticity and determinism in cell fate decisions. Development 147, dev181495. [DOI] [PubMed] [Google Scholar]
- Zeng H, and Sanes JR (2017). Neuronal cell-type classification: challenges, opportunities and the path forward. Nat. Rev. Neurosci 18, 530–546. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et al. (2017). Massively parallel digital transcriptional profiling of single cells. Nat. Commun 8, 14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zibetti C, Liu S, Wan J, Qian J, and Blackshaw S (2019). Epigenomic profiling of retinal progenitors reveals LHX2 is required for developmental regulation of open chromatin. Commun. Biol 2, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All mouse and human scRNA-seq and scATAC-seq data can be accessed at GEO accession numbers GSE181251 and GSE183684 (Thomas et al., 2021). Supplemental datasets listed in Mendeley dataset are available at https://doi.org/10.17632/nrstc3xhwb.1.
Code for IReNA v2 pipeline is available at https://github.com/Pinlyu3/IReNA-v2. Interactive displays of all scRNA-seq and scATAC-seq data can be accessed through the St Jude Cloud Visualization Community at https://viz.stjude.cloud/blackshaw-lab/visualization/scatac-seq-analysis-of-mouse-retinal-development~92.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.