

Abstract
Thiol dioxygenases (TDOs) are a class of metalloenzymes that oxidize various thiol-containing substrates to their corresponding sulfinic acids. Originally established by X-ray crystallography for cysteine dioxygenase (CDO), all TDOs are believed to contain a 3-histidine facial triad that coordinates the necessary Fe(II) cofactor. However, very little additional information is available for cysteamine dioxygenase (ADO), the only other mammalian TDO besides CDO. Previous spectroscopic characterizations revealed that ADO likely binds substrate cysteamine in a monodentate fashion, while a mass spectrometry study provided evidence that a thioether crosslink can form between Cys206 and Tyr208 (mouse ADO numbering). In the present study we have used electronic absorption and electron paramagnetic resonance (EPR) spectroscopies to investigate the species formed upon incubation of Fe(III)ADO with sulfhydryl-containing substrates and the superoxide surrogates azide and cyanide. Our data reveal that azide is unable to coordinate to cysteamine-bound Fe(III)ADO, suggesting that the Fe(III) center lacks an open coordination site or azide competes with cysteamine for the same binding site. Alternatively, cyanide binds to either cysteamine- or Cys-bound Fe(III)ADO to yield a low-spin (S = 1/2) EPR signal that is distinct from that observed for cyanide/Cys-bound Fe(III)CDO, revealing differences in the active site pockets between ADO and CDO. Finally, EPR spectra obtained for cyanide/cysteamine adducts of wild-type Fe(III)ADO and its Tyr208Phe variant are superimposable, implying that either an insignificant fraction of as-isolated wild-type enzyme is crosslinked or that formation of the thioether bond has minimal effects on the electronic structure of the iron cofactor.
Keywords: Thiol dioxygenases, cysteamine dioxygenase, cysteine dioxygenase, non-heme iron enzymes, electron paramagnetic resonance
Graphical Abstract
Introduction
Thiol dioxygenases (TDOs) are a family of non-heme Fe-dependent enzymes that oxidize thiol substrates to their corresponding sulfinic acids via the incorporation of both oxygen atoms from dioxygen. Currently, five classes of TDOs are known: cysteine dioxygenase (CDO); 3-mercaptopropionate dioxygenase (MDO); mercaptosuccinate dioxygenase (MSDO); plant cysteine oxidase (PCO); and cysteamine dioxygenase (ADO). Each TDO class has a unique substrate specificity.
CDO provided the founding structure for the TDO family [1], which is part of the cupin superfamily. The structure revealed that CDO binds its Fe(II) cofactor via a 3 histidine (3-His) facial triad as opposed to the 2-His-1-carboxylate metal binding triad found in most Fe(II)-dependent oxygenases [2, 3]. Indeed, this 3-His binding motif is apparently rare, as only a small number of cupin-fold enzymes feature this Fe binding motif, including CDO [1], MDO [4], and PCO [5]. Furthermore, CDO contains an unusual thioether linkage between Cys93 and a Tyr157 (Mus musculus CDO [MmCDO] numbering) in the secondary sphere of the active site that increases activity by properly positioning the substrate and suppressing water coordination to the substrate- bound Fe(II) site [6–8].
CDO oxidizes L-cysteine (Cys) to cysteine sulfinic acid (CSA) [9]. In subsequent steps, CSA is further metabolized into hypotaurine and taurine [10–13]. Though Cys is essential for life, an elevated level off Cys has potentially devastating neurological effects and has been linked to Alzheimer’s and Parkinson’s diseases [14–16]. Hence, tight regulation of intracellular Cys levels is vital. Hypotaurine can also be biosynthesized from the oxidation of cysteamine (2-aminoethanethiol, 2-AET), a reaction catalyzed by ADO [17]. The gene encoding ADO drew attention when researchers noticed a discrepancy between the high levels of taurine and low levels of cdo gene expression in certain tissues. Thus, ADO was postulated to be responsible for taurine production in certain tissues, most notably the brain.
Similar to CDO, the ADO active site features a 3-His facial triad that coordinates the Fe(II) ion, as judged on the basis of sequence alignment [17], site-directed mutagenesis [17, 18], and spectroscopic studies [18, 19]. In addition, also similar to CDO, the existence of a Cys-Tyr crosslink has been established in Homo sapiens (Hs) ADO via incorporation of an unnatural amino acid, 3,5,-difluoro-tyrosine, in conjunction with mass spectrometry and NMR experiments [20]. Interestingly, the positions of the residues contributing to the Cys-Tyr crosslink within the amino acid primary sequence are strikingly different between ADO and CDO (Cys206-Tyr208 in MmADO vs Cys93-Tyr157 in MmCDO). Beyond these conserved features, ADO and CDO have low sequence identity (~15%) and use different binding modes for their thiol substrate (monodentate thiol in ADO versus bidentate thiol and amino in CDO [18, 19, 21]), raising the question of whether there are also differences in the binding of O2 to the Fe(II) center and the nature of reaction intermediates, such as the putative Fe(III)-superoxide species.
Intriguingly, recent studies revealed that ADO might also carry out an essential posttranslational modification similar to that reported for PCO [22], which initiates an N-degron pathway in plants [23, 24]. Under oxic conditions, PCO is able to oxidize the N-terminal Cys of a peptide substrate. The N-terminal CSA then serves as a substrate for arginyl tRNA transferase ATE1, thus marking the arginylated peptide for ubiquitination and proteosomal degradation. Under anoxia, PCO is unable to carry out the N-terminal oxidation and the peptide persists, allowing initiation of a hypoxic response to submergence or waterlogging in plants. Similar to this regulatory role for PCO, it has recently been observed that natriuretic polypeptides such as RGS4 and RGS5 are putative ADO substrates [25]. These peptides serve as negative regulators of G protein signaling that controls cardiovascular function [26].
Direct trapping of O2-derived intermediates in the catalytic cycles of TDOs has not yet been achieved. To address this issue, O2 and superoxide surrogates that do not support turnover have been productively used to probe the nature of the active site and make deductions about possible reaction intermediates [27–31]. As azide has similar frontier orbitals and the same charge as superoxide (O2•−), this anion has frequently been used to prepare models of putative Fe(III)-superoxide intermediates that are amenable to a wide range of spectroscopic techniques [27, 28, 30]. In the case of CDO, azide does not coordinate directly to the Fe(III) center of Cys-bound enzyme but instead occupies a pre-binding site in the enzyme [27]. Alternatively, treatment of Cys-bound Fe(III)CDO with excess cyanide, another widely used O2•− surrogate, causes the high-spin (S = 5/2) electron paramagnetic resonance (EPR) signal to convert to a low-spin (S = 1/2) EPR signal, demonstrating the direct coordination of cyanide to the Fe(III) ion to presumably form a six-coordinate adduct where the thiol and amino groups of Cys, cyanide, and the 3-His motif make up the iron coordination sphere. Interestingly, the cyano/Cys-Fe(III)CDO complex not only provides a model of a putative reaction intermediate, but also displays EPR parameters that are sensitive to changes in the secondary coordination sphere of CDO, such as the formation of the Cys-Tyr crosslink [32].
In the present study, we have used electronic absorption (Abs) and EPR spectroscopies to characterize the complexes that are formed upon incubation of Fe(III)ADO with 2-AET or thiol-containing derivatives and azide or cyanide. As ADO appears to feature a similar 3-His metal-binding site as CDO, it is reasonable to assume that oxidation of 2-AET by this enzyme also proceeds via transient formation of an Fe(III)-superoxo species. As such, our results obtained for Fe(III)ADO incubated with 2-AET (analogues) and O2•− surrogates permit a detailed assessment of the geometric and electronic properties of potential ADO reaction intermediates. Similarities and differences between the azide and cyanide complexes of substrate-bound CDO and ADO are discussed, and inferences are made as to the influence of the unique secondary sphere of ADO on substrate specificity. Additionally, the effect of the putative crosslink between residues Cys206 and Tyr208 on the positioning of substrate (analogues) is assessed via EPR studies of WT and Y208F ADO incubated with cyanide and 2-AET.
Materials and methods
Recombinant gene expression and protein purification.
Gene expression and protein purification of wild-type (WT) Mus musculus (Mm) ADO were conducted in the same manner as described elsewhere [18]. In brief, the ado gene was expressed in Escherichia coli Rosetta 2(DE3) cells through induction with isopropyl-β-D-thiogalactopyranoside (IPTG). ADO was purified using an immobilized metal affinity chromatography column and a gel filtration column. After each column, fractions were pooled on the basis of purity visualized via stain-free sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Once enzyme was eluted from the final column, activity was verified via qualitative thin-layer chromatography as described elsewhere [18].
The Tyr208Phe mutation was introduced in MmADO using primers 5’-GACTGCCACTTCTACCGCGTTG-3’ and 5’-CAACGCGGTAGAAGTGGCAGTC-3’ purchased from Integrated DNA Technology. The cloned gene sequence was confirmed at the University of Wisconsin-Madison Biotechnology Center. Variant ADO was prepared as described elsewhere with minor adjustments [18]. E. coli Rosetta 2(DE3) cells were transformed with the mutant DNA and grown at 37 °C in 4 L of LB medium (containing 10 g of tryptone, 10 g of NaCl, and 5 g of yeast extract per liter). Gene overexpression was induced by adding IPTG to a final concentration of 140 μM along with ferrous ammonium sulfate to a final concentration of 80 μM.
Tyr208Phe ADO was purified using immobilized metal affinity chromatography with a TALON Co-resin column. Protein-containing fractions were pooled according to purity by stain-free SDS-PAGE. Activity was confirmed qualitatively with thin layer chromatography, as described elsewhere [18].
Sample preparation.
The Fe(II) and Fe(III) contents of ADO were determined through a colorimetric assay using the iron chelator tripyridyl triazine and an ε595 of 22.1 mM−1 cm−1 [33]. The assay was performed in the absence and presence of a reductant to determine the proportion of Fe(II) versus Fe(III) initially present. Purified ADO was typically 70–80% Fe-loaded, with the majority (~85%) of metalated sites containing Fe(II). As iron-loading of the ADO active sites was incomplete, all concentrations cited herein refer to the Fe-bound fraction, not that of total protein.
All samples were prepared aerobically in 20 mM Tris, pH 8.0 with 250 mM NaCl buffer. Oxidation to Fe(III)ADO was accomplished via a 30-minute reaction of as-isolated ADO (~0.4 mM) with a 3-fold molar excess of potassium hexachloroiridate(IV), followed by buffer exchange via centrifugation to remove any remaining oxidant [34]. The desired protein complexes were obtained by incubating 0.4 mM Fe(III)ADO with substrate 2-AET, 3-mercaptopropanoic acid (3-MPA), or Cys for 2 min and then with a 200-fold molar excess (80 mM) or 10-fold molar excess (4 mM) of azide or cyanide, respectively. A recent study by Wang et al. revealed that upon incubation with substrate, Fe(III)ADO is reduced to Fe(II)ADO within 20 min [19]. Thus, to avoid reduction, all samples were frozen within 5 min.
Spectroscopy.
Abs spectra were recorded at room temperature with a double-beam Varian Cary 4 Bio spectrophotometer set to a spectral bandwidth of 0.5 nm. X-band EPR data were collected using a Bruker ELEXSYS E500 spectrometer. The sample temperature was maintained at 20 K by an Oxford ESR 900 continuous flow liquid He cryostat regulated by an Oxford ITC-503S temperature controller. All EPR spectra were obtained using the following experimental parameters: frequency = 9.386 GHz; microwave power = 12.62 mW; modulation amplitude = 3 G; modulation frequency = 100 kHz. These parameters were kept constant to allow for a direct comparison of the relative contributions from different (i.e., low-spin versus high-spin) species to the different samples investigated. EPR spectral fits for the low-spin (S = 1/2) species were performed using EasySpin Version 5.2.25 [35].
Results
The electronic Abs spectrum of Fe(III)ADO presented in Figure 1A, solid line lacks any discernible features in the UV/Vis spectral region. When incubated with 2-AET or Cys, Fe(III)ADO turns light blue in color due to the appearance of a broad band centered at ~690 nm (λmax of 693 and 692 nm for 2-AET-bound and Cys-bound Fe(III)ADO, respectively, Figure 1B and 1D, solid line). Analogous features in the Abs spectra of Fe(III)CDO [36] and oxidized superoxide reductase [30], a non-heme iron enzyme with a comparable active site structure, have been assigned as SCys→Fe(III) charge transfer (CT) transitions on the basis of spectroscopic and computational studies. Addition of 3-MPA to Fe(III)ADO causes the appearance of a dominant Abs feature with a λmax of 647 nm, also suggestive of S→Fe(III) charge transfer (Figure 1C, solid line). However, the unique peak position of this feature provides the evidence that the electronic structure of the 3-MPA-bound ADO complex is distinct from that of the 2-AET-bound ADO complex.
Figure 1.
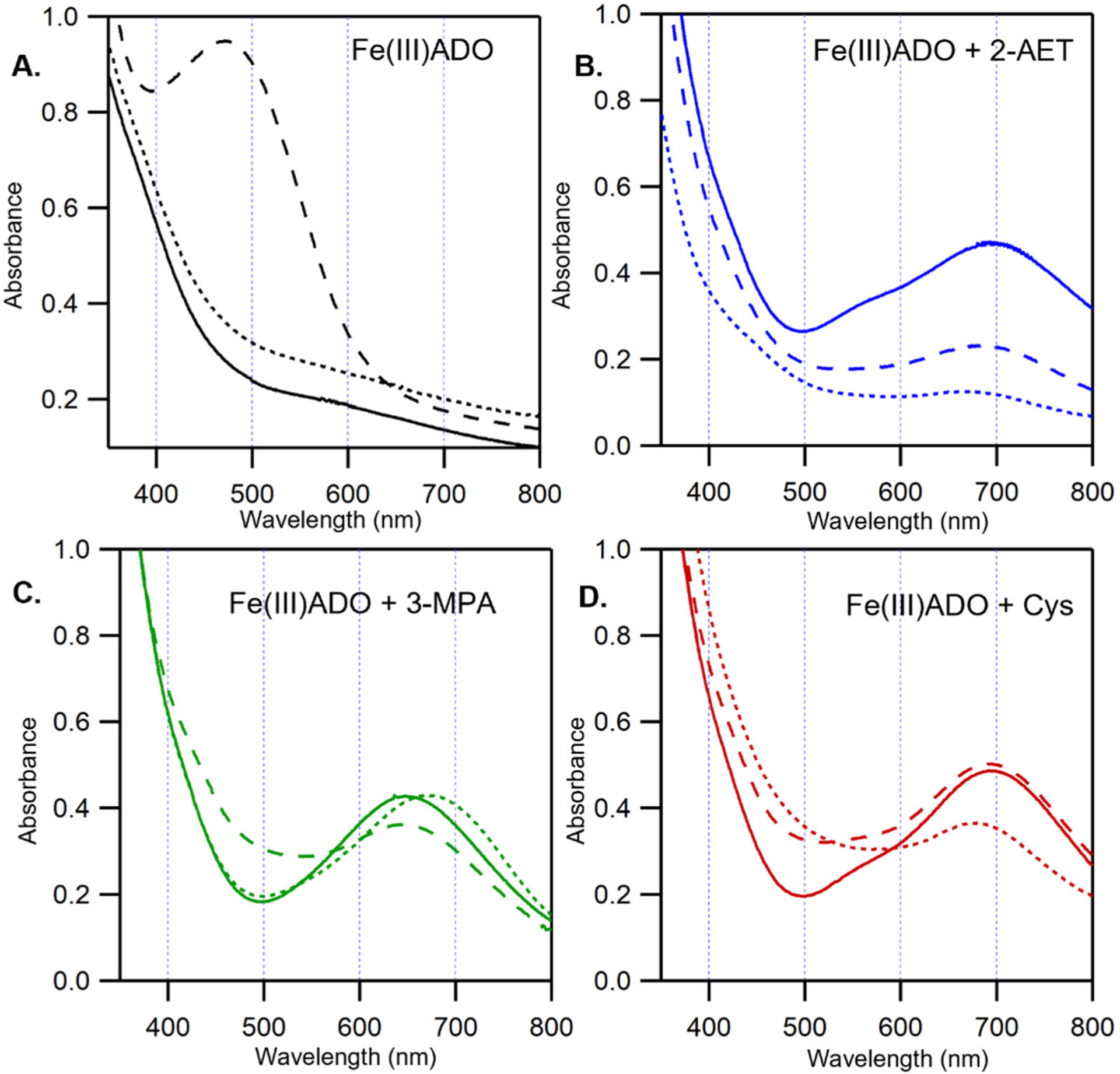
Room-temperature UV/Vis spectra of 0.4 mM Fe(III)ADO in the absence and presence of substrate (analogues) and O2•− surrogates. A) Fe(III)ADO, B) Fe(III)ADO incubated with a 5-fold excess (2 mM) of 2-AET, C) Fe(III)ADO incubated with a 5-fold excess (2 mM) of 3-MPA, and D) Fe(III)ADO incubated with a 10-fold excess (4 mM) of Cys. Spectra were collected either in the absence of an O2•− surrogate (solid lines), the presence of a 200-fold excess (80 mM) of azide (long dashes), or a 10-fold excess (4 mM) of cyanide (short dashes).
Azide-Fe(III)ADO adduct.
Upon incubation with azide, the color of Fe(III)ADO changes from pale yellow to an orange-pink. This change in color is accompanied by the appearance of a new Abs feature with a λmax of 472 nm (Figure 1A, dashed line). A similar feature in the absorption spectrum of Fe(III) superoxide dismutase incubated with azide was previously assigned as an N3−→Fe(III) CT transition on the basis of resonance Raman spectroscopic studies [37]. Likewise, CDO has been reported to form an inner-sphere azide-Fe(III)CDO complex on the basis of Abs, EPR, and MCD spectroscopic studies [27]. Thus, like these other metalloenzymes, azide coordinates directly to the Fe(III) center of ADO in the absence of substrate. The Fe(III)ADO incubated with azide also displays a rhombic, high-spin (S = 5/2) EPR signal that closely resembles that of as-isolated ADO (Figure 2B and 2A, respectively), suggesting that azide binds to the Fe(III)ADO active site by replacing a ligand with comparable donor strength, likely a solvent-derived hydroxide.
Figure 2.
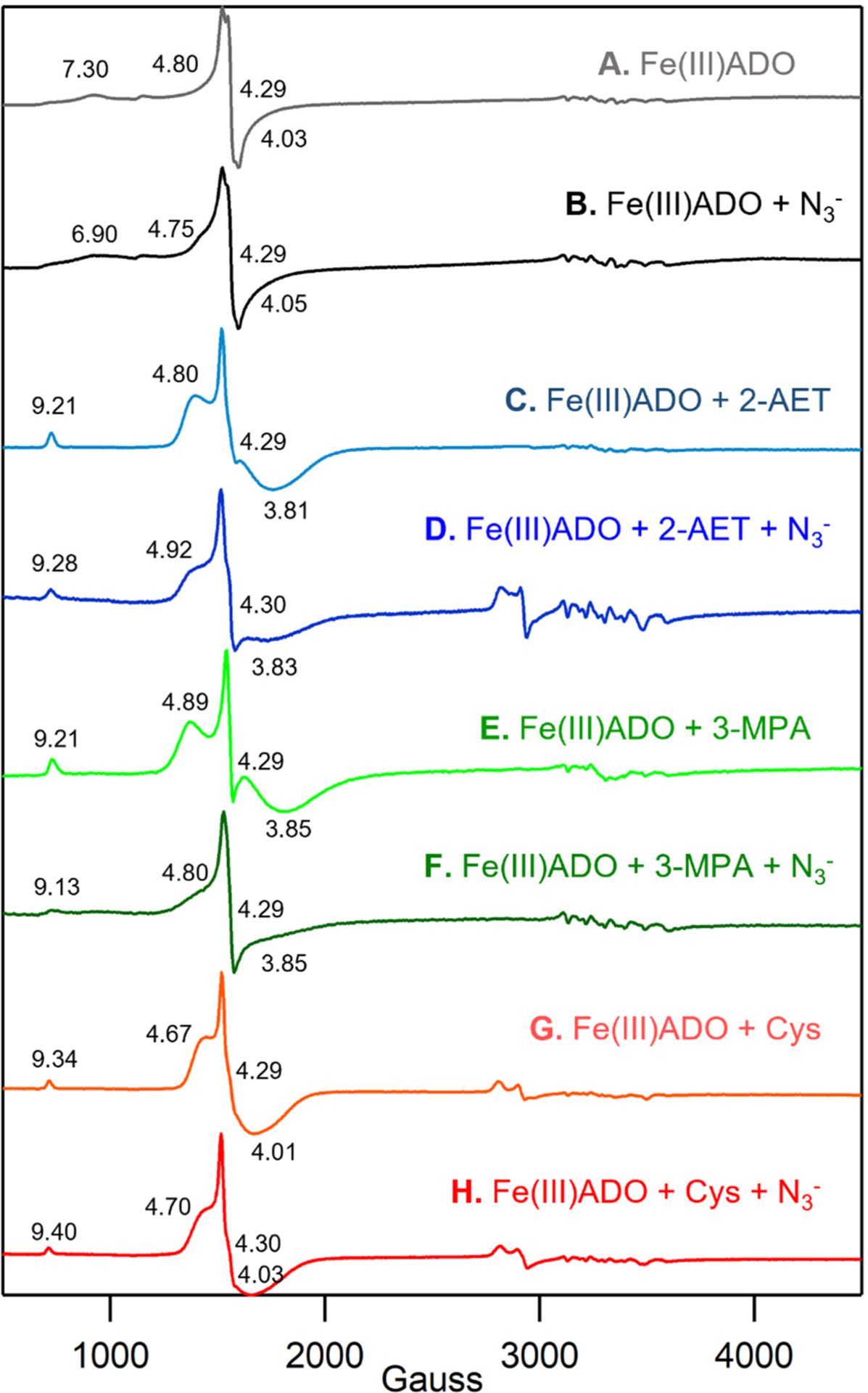
X-band EPR spectra at 20 K of 0.4 mM Fe(III)ADO in the absence and presence of various substrate analogues and/or O2•− surrogates. A) Fe(III)ADO, B) Fe(III)ADO incubated with a 200-fold excess (80 mM) of azide, C) Fe(III)ADO incubated with a 5-fold excess (2 mM) of 2-AET, D) Fe(III)ADO incubated with a 5-fold excess (2 mM) of 2-AET and a 200-fold excess (80 mM) of azide, E) Fe(III)ADO incubated with a 10-fold excess (4 mM) of 3-MPA, F) Fe(III)ADO incubated with a 10-fold excess (4 mM) of 3-MPA and a 200-fold excess (80 mM) of azide, G) Fe(III)ADO incubated with a 10-fold excess (4 mM) of Cys, and H) Fe(III)ADO incubated with a 5-fold excess (2 mM) of Cys and a 200-fold excess (80 mM) of azide. Effective g values are provided for the S = 5/2 species. The hyperfine structure in the 3000–3700 cm−1 Gauss region is due to a minor Mn(II) impurity.
Addition of azide to Fe(III)ADO incubated with substrate 2-AET causes a minor (~9 nm) blue shift of the λmax of the visible Abs feature from 693 nm to 684 nm (Figure 1B, dashed line) and a slight increase in the rhombicity of the EPR signal (Figure 2D) compared to that displayed by 2-AET-bound Fe(III)ADO in the absence of azide (Figure 2C) [18]. The relatively small magnitude of the spectral perturbations caused by the addition of azide to 2-AET-bound Fe(III)ADO indicates that this O2•− surrogate binds near, but not directly to the Fe(III) center in the majority of active sites. However, the EPR spectrum of Fe(III)ADO incubated with 2-AET and azide additionally contains a minor low-spin (S = 1/2) signal in the high field region. A similar, weak low-spin EPR signal was previously reported for Fe(III)CDO treated with Cys and azide. In the case of Fe(III)CDO, this S = 1/2 EPR signal was attributed to a minor fraction of Fe(III) centers that form an inner-sphere complex with azide, while the appearance of a slightly perturbed Abs spectrum and retention of the dominant S = 5/2 EPR signal was interpreted to indicate that azide binds in the vicinity of, rather than to, the majority of Fe(III) centers [27]. The low affinity of Cys-bound Fe(III)CDO for azide was suggested to be due to the presence of a sixth ligand, most likely a hydroxide ion, that saturates the Fe(III) coordination sphere. By analogy, we propose that the Fe(III) center in 2-AET-bound Fe(III)ADO is also six-coordinate, precluding the direct coordination of azide to the vast majority of active sites.
As evidenced by the appearance of a prominent Abs band at 647 nm, the thiol group of 3-MPA can coordinate to Fe(III) in ADO (Figure 1C, solid line). Addition of azide to 3-MPA-bound Fe(III)ADO causes only a slight blue-shift of the λmax of the prominent Abs band in the visible spectral region to 645 nm (Figure 1C, dashed line), along with a small increase in rhombicity of the S = 5/2 EPR signal (Figure 2E). Importantly, no low-spin EPR signal characteristic of the azide and thiol-bound enzyme is present. The similarity of λmax and the EPR spectra of 3-MPA- bound ADO in the absence and presence of azide and lack of a signal in the high field region of the EPR spectrum indicate that azide does not directly bind to the Fe(III) ion when 3-MPA is present (note: the small changes in the Abs spectrum observed upon incubation of 3-MPA-bound Fe(III)ADO with azide could be due to a change in ionic strength or binding of azide near the active site without coordinating the iron).
As with 3-MPA, the Abs spectra collected for Cys-bound Fe(III)ADO in the absence and presence of azide (Figure 1D, dashed line) are nearly identical, suggesting that this O2•− surrogate does not directly coordinate to the Fe(III) center in the majority of active sites. While the EPR spectrum of Fe(III)ADO incubated with Cys and azide contains a minor low-spin (S = 1/2) signal (Figure 2F, dashed line), seemingly suggesting that azide directly coordinates to a small subset of Cys-bound Fe(III)ADO active sites, the same low-spin signal is also observed for Cys-bound ADO in the absence of azide [18]. In fact, these spectra overlay perfectly, leading us to conclude that azide minimally perturbs the active site of Cys-bound Fe(III)ADO.
Cyanide-Fe(III)ADO adduct.
The Abs and EPR spectra of Fe(III)ADO collected in the absence and presence of cyanide are nearly identical (Figure 1, dotted line, and 3A), indicating that this O2•− surrogate does not coordinate to substrate-free Fe(III)ADO. In contrast, addition of cyanide to 2-AET-bound Fe(III)ADO causes the appearance of a dominant low-spin (S = 1/2) EPR signal (g = 2.30, 2.20, 1.96; Figure 3B) and a small but noticeable change to the Abs spectrum, signifying direct coordination of the cyanide ion to the Fe(III) center in this species. A similar requirement for the presence of substrate to enable binding of cyanide to the Fe(III) ion has been reported for Fe(III)CDO [32].
Figure 3.
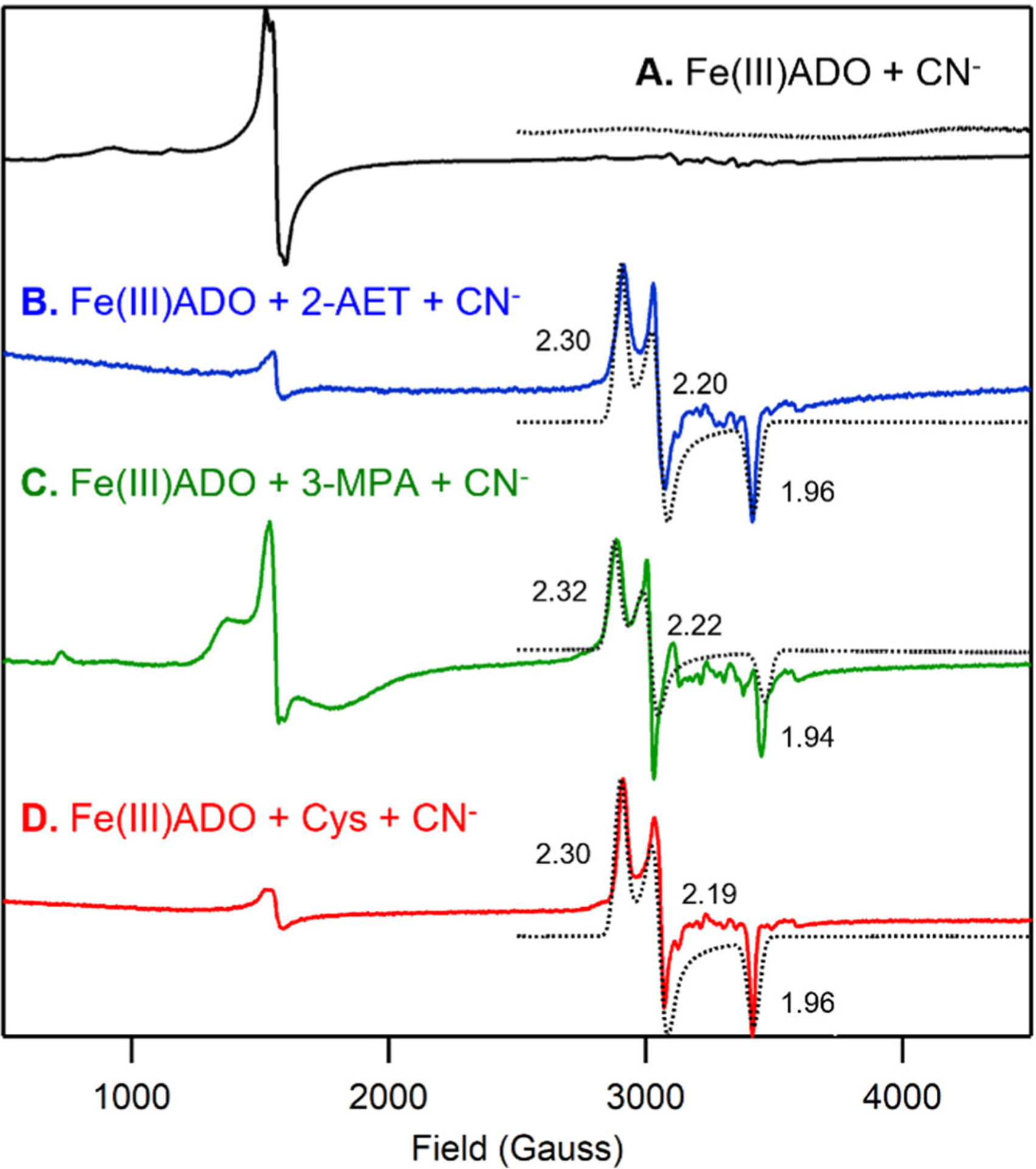
X-band EPR spectra at 20 K of 0.4 mM Fe(III)ADO incubated with a 10-fold excess (4 mM) of cyanide in the absence of substrate (A) and in the presence of a 15-fold excess (6 mM) of 2-AET (B), a 5-fold (2 mM) excess of 3-MPA (C), or a 10-fold excess (4 mM) of Cys (D). g values are provided for the S = 1/2 species. The hyperfine structure in the 3000–3700 cm−1 Gauss region is due to a minor Mn(II) impurity.
Incubation of 3-MPA-bound Fe(III)ADO with cyanide leads to a 25 nm red-shift of the prominent Abs feature at ~650 nm (Figure 1C, dotted line) and the appearance of an identical low-spin EPR signal as observed for 2-AET-bound Fe(III)ADO in the presence of cyanide (Figure 3C vs 3B), again signaling formation of an Fe(III)-cyano-substrate complex. However, the relative intensity of the residual high-spin (S = 5/2) EPR signal (Figure 3C) is much higher in the EPR spectrum of cyanide-treated 3-MPA-bound Fe(III)ADO, indicating that cyanide coordinates to a smaller fraction of Fe(III)ADO in the presence of the substrate analogue 3-MPA.
The Abs and EPR spectra of Cys-bound Fe(III)ADO in the presence of cyanide (Fig 1D, dotted line and Figure 3D) are nearly identical to those obtained for 2-AET-bound Fe(III)ADO incubated with cyanide (Fig 1D, dotted line and Figure 3D). These data reveal that the active sites of 2-AET- and Cys-bound Fe(III)ADO have comparable affinities for and form similar inner-sphere complexes with cyanide. Even with this similarity, ADO turnover of Cys is very slow [17], suggesting that a step in the catalytic cycle after Cys and O2 binding to Fe(II)ADO is perturbed.
Impact of crosslink on spectral properties.
Recently, Liu and coworkers demonstrated that a Cys-Tyr crosslink can be formed in human ADO (involving Cys206 and Tyr208 using MmADO numbering scheme), by genetically incorporating an unnatural amino acid, 3,5-difluoro-tyrosine, specifically into Tyr208 and using mass spectrometry and 19F NMR spectroscopy [20]. Although in the absence of a crystal structure the position of the crosslink relative to the active site remains unknown, the authors noted that crosslink formation required the presence of cysteamine and O2, suggesting that it resides in close proximity to the Fe center. In support of this hypothesis, the catalytic activity (determined via O2 uptake studies) was found to be reduced by a factor of ~4 in the Tyr208Ala ADO variant.
To assess whether a crosslink has any noticeable effect on the interaction between substrate 2-AET and the Fe(III)ADO active site, we prepared the Tyr208Phe ADO variant, which prevents the formation of the thioether crosslink. Notably, the EPR spectra obtained for cyanide/2-AET adducts of WT and Y208F Fe(III)ADO (Figure 4) are superimposable, suggesting that formation of the Cys206–Tyr208 crosslink has negligible effects on the geometric and electronic properties of the ADO active site. Given the differences in primary sequence positions of residues involved, it is perhaps not surprising that Cys-Tyr thioether bond formation would affect the substrate/active site interactions differently in ADO and CDO. However, because only a small fraction of as-isolated WT ADO appears to be crosslinked based on mass spectrometry analysis [20], it is possible that the sample of cyanide/2-AET-bound WT Fe(III)ADO used to collect the EPR data in Figure 4 contained an insignificant fraction of crosslinked enzyme. Also, the modest decrease in catalytic activity displayed by ADO in response to the non-conservative Tyr208Ala substitution variant could be due to more global structural changes rather than the presence of a substantial fraction of crosslinked WT enzyme under physiologically relevant conditions.
Figure 4.
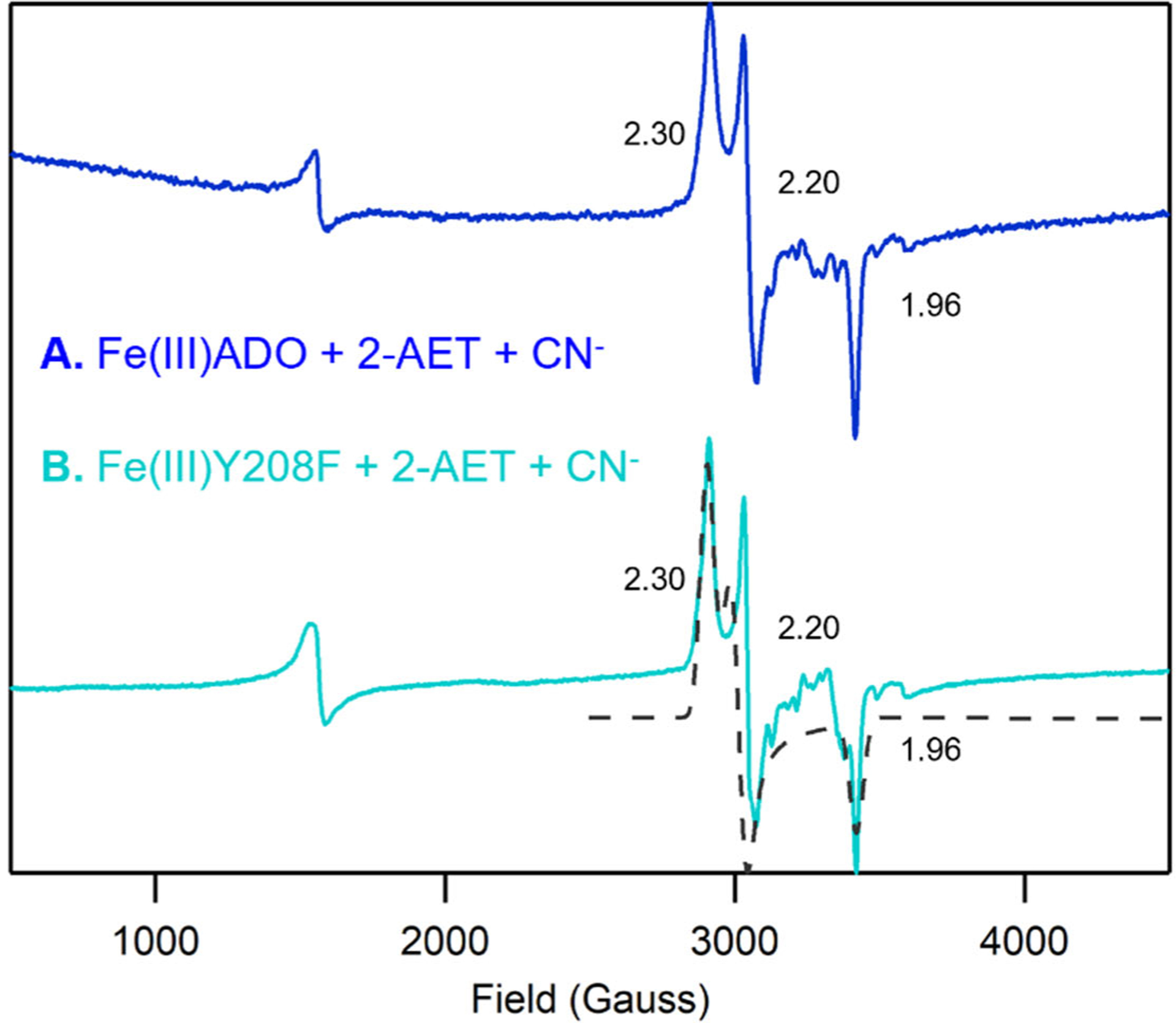
X-band EPR spectra at 20 K of (A) 0.4 mM Fe(III)ADO incubated with a 10-fold excess (4 mM) of cyanide in the presence of a 15-fold excess (6 mM) of 2-AET (B) 0.4 mM Y208F Fe(III)ADO incubated with a 10-fold excess (4 mM) of cyanide in the presence of a 15-fold excess (6 mM) of 2-AET. g values are provided for the S = 1/2 species. The hyperfine structure in the 3000–3700 cm−1 Gauss region is due to a minor Mn(II) impurity.
Discussion
Despite the significant role that ADO plays in mammalian sulfur metabolism, little is known about its active site structure. Although sequence homology modeling could, in principle, be used to predict the three-dimensional structure of ADO and guide site-directed mutagenesis studies, this approach is hampered by the fact that ADO and other crystallographically characterized TDOs display low sequence identity (e.g., mouse ADO and CDO share only 14% sequence identity). In particular, it is difficult to assess the influence of secondary sphere residues on the nature of the substrate/active site interactions from homology models. Thus, in this study, we have used Abs and EPR spectroscopy to investigate the binding of superoxide surrogates to Fe(III)ADO in the absence and presence of substrate and substrate analogues.
Azide coordinates to the iron center of Fe(III)ADO in the absence of substrate.
When Fe(III)ADO is incubated with azide, an intense Abs feature appears at 472 nm, providing evidence for direct coordination of this O2•− surrogate to the active site of Fe(III)ADO in the absence of substrate (analogue). The distinct rhombic, high-spin (S = 5/2) signal in the corresponding EPR spectrum indicates that azide likely coordinates to the active site by replacing a ligand with similar donor strength, presumably a solvent-derived hydroxide. Similar spectral changes were noted when substrate-free Fe(III)CDO was incubated with azide [27]. A combined spectroscopic and computational analysis of the resulting azide adduct of Fe(III)CDO were consistent with a single azide bound to the Fe(III) center to yield a five-coordinate complex or an azide and a water bound to give six-coordinate complex. Our results indicate that azide-bound Fe(III)ADO also contains a five-coordinate or six-coordinate Fe(III) center possessing hydroxide and azide ligands.
Azide binding to Fe(III)ADO is suppressed in the presence of substrate (analogues).
The EPR data of azide/2-AET-bound Fe(III)ADO demonstrate that azide binds directly to only a minor fraction of Fe(III)ADO active sites complexed with 2-AET, suggesting that the Fe(III) center of 2-AET-bound Fe(III)ADO is either coordinatively saturated or otherwise inaccessible in the presence of substrate. The presence of a six-coordinate Fe(III) center has also been proposed for Cys-bound Fe(III)CDO, with a hydroxide ion completing a distorted octahedral coordination environment of the active site Fe(III) ion, and azide occupying an outer-sphere pocket without binding to iron [27]. Unlike Fe(III)CDO, however, Fe(III)ADO appears to bind its 2-AET substrate in a monodentate fashion via coordination of only the thiolate moiety [18, 38]. Thus, we hypothesize that the Fe(III) ion of substrate-bound Fe(III)ADO resides in a distorted octahedral coordination environment composed of the 3-His facial triad, the 2-AET terminal thiolate, and two hydroxide ions.
While a small fraction of 2-AET-bound Fe(III)ADO active sites form inner-sphere complexes with azide as judged by the presence of a low-spin EPR signal, azide is unable to bind to the 3-MPA-bound or Cys-bound Fe(III)ADO active site. This difference likely stems from the fact that the ADO active site has evolved to promote monodentate binding of the 2-AET substrate (and potentially N-terminal Cys peptides), which causes the non-coordinating, negatively-charged carboxylate groups in 3-MPA- and Cys-bound Fe(III)ADO to repel anionic O2•− surrogates away from the active site. In CDO, an analogous electrostatic repulsion between the substrate carboxylate group and azide is alleviated by the formation of a salt bridge between the Cys carboxylate and the Arg60 outer-sphere residue. The prediction that ADO cannot form a salt bridge that stabilizes substrate binding is consistent with the observation that this enzyme is unable produce cysteine sulfinic acid from Cys [17].
Effect of outer-sphere residues on the cyanide/2-AET-Fe(III)ADO adduct.
The EPR g-values of cyanide/Cys-bound Fe(III)CDO have been found to be sensitive to changes in the outer coordination sphere, specifically the absence and presence of the Cys93-Tyr157 crosslink [29, 31]. A spectroscopically validated computational analysis of this adduct revealed that in the absence of the crosslink, the Fe–C–N unit is nearly linear. Upon crosslink formation, the Fe–C–N unit adopts a more bent configuration and the repositioning of Tyr157 perturbs the network of hydrogen bonds between the side chain of Arg60, the phenol of Tyr157, and the substrate Cys carboxylate, causing a lengthening of the Fe–S bond by 0.02 Å. Although seemingly minor, these structural changes have a measurable effect on the electronic and spectroscopic properties of the enzyme active site.
To correlate changes in EPR g-values with specific geometric perturbations, Fiedler and coworkers performed CASSCF/NEVPT2 ab initio calculations on active site models of non-crosslinked and crosslinked cyanide/Cys-bound Fe(III)CDO as well as synthetic mimics [29]. These computations indicated that a lengthening of the Fe–S bond causes an overall increase in the spread of g-values and a decrease in the intermediate g-value. Hence, the significantly smaller g spread displayed by cyanide/2-AET-bound Fe(III)ADO (2.30, 2.20, 1.96) relative to those reported for non-crosslinked and crosslinked cyanide/Cys-bound Fe(III)CDO (2.34, 2.21, 1.95 versus 2.38, 2.23, 1.94, respectively) implies that the cyanide/2-AET-bound Fe(III)ADO complex possesses the shortest Fe-S bond of these species. An even smaller g spread was found for a synthetic cyanide/Cys-bound Fe(III)CDO mimic (2.20, 2.16, 1.99), which lacks any outer-sphere interactions involving the carboxylate group of the substrate Cys analogue and thus possesses a particularly short Fe-S bond. Collectively, these results suggest that the amine group of 2-AET in substrate-bound Fe(III)ADO engages in some, but considerably weaker interactions with secondary sphere residues than does the carboxylate group of Cys with Arg60 in substrate-bound Fe(III)CDO. This finding is consistent with the much narrower substrate scope displayed by CDO compared to ADO (i.e., while CDO is only capable of turning over L-Cys efficiently [14], ADO has been shown to oxidize 2-AET [17] as well as the amino-terminal Cys residue of a much larger polypeptide from RGS5 [22]).
Conclusion
The spectroscopic data obtained in this study provide new insights into the nature of the species that are formed upon incubation of Fe(III)ADO with substrate/substrate analogues and O2•− surrogate, which serve as models of putative reaction intermediates. Azide is only able to coordinate to the Fe(III) center of substrate-free ADO, likely indicating that cysteamine-bound Fe(III)ADO lacks an open coordination site. An analogous inhibitory effect of substrate on azide binding was previously reported for Fe(III)CDO, where the presence of a six-coordinate Fe(III) center with azide occupying an outer-sphere pocket was established on the basis of spectroscopic, computational, and X-ray crystallographic studies. Conversely, the O2•− surrogate cyanide requires coordination of substrate cysteamine (analogues) before it can bind to the Fe(III)ADO active site, again paralleling the behavior of Fe(III)CDO. A comparison of the EPR g-values obtained for cyanide/cysteamine-bound Fe(III)ADO and those reported for cyanide/Cys-bound Fe(III)CDO reveals that the interaction of the thiol substrate with outer-sphere residues is considerably weaker in ADO than in CDO. Lastly, elimination of the putative Cys206–Tyr208 crosslink via Tyr208Phe substitution has negligible effects on the EPR signal of the cyanide/cysteamine adduct, indicating that either an insignificant fraction of as-isolated wild-type enzyme contains the crosslink or that formation of the thioether bond has minimal effects on the electronic structure of the substrate-bound active site.
Supplementary Material
Acknowledgement
The authors would like to thank Prof. Martha H. Stipanuk for sharing the original mouse ADO plasmid used to begin this work.
Funding
The authors are grateful for financial support from the National Institute of General Medical Sciences of the National Institutes of Health (Grant GM117120 to T.C.B.) and the Department of Biochemistry and the Office of the Vice Chancellor for Research and Graduate Education (to B.G.F.). This research was conducted in part while R.L.F. was supported by the National Institute of General Medical Sciences of the National Institutes of Health (Grant T32GM008505).
Footnotes
References
- 1.McCoy JG, Bailey LJ, Bitto E, et al. (2006) Structure and mechanism of mouse cysteine dioxygenase. Proc Natl Acad Sci 103:3084–3089. 10.1073/pnas.0509262103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Straganz GD, Nidetzky B (2006) Variations of the 2-His-1-carboxylate theme in mononuclear non-heme Fe II oxygenases. ChemBioChem 7:1536–1548. 10.1002/cbic.200600152 [DOI] [PubMed] [Google Scholar]
- 3.Koehntop KD, Emerson JP, Que L (2005) The 2-His-1-carboxylate facial triad: A versatile platform for dioxygen activation by mononuclear non-heme iron(II) enzymes. J Biol Inorg Chem 10:87–93. 10.1007/s00775-005-0624-x [DOI] [PubMed] [Google Scholar]
- 4.Tchesnokov EP, Fellner M, Siakkou E, et al. (2015) The cysteine dioxygenase homologue from Pseudomonas aeruginosa is a 3-mercaptopropionate dioxygenase. J Biol Chem 290:24424–24437. 10.1074/jbc.M114.635672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.White MD, Carbonare LD, Puerta ML, et al. (2020) Structures of Arabidopsis thaliana oxygen-sensing plant cysteine oxidases 4 and 5 enable targeted manipulation of their activity. Proc Natl Acad Sci U S A 117:23140–23147. 10.1073/pnas.2000206117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davies CG, Fellner M, Tchesnokov EP, et al. (2014) The Cys-Tyr cross-link of cysteine dioxygenase changes the optimal ph of the reaction without a structural change. Biochemistry 53:7961–7968. 10.1021/bi501277a [DOI] [PubMed] [Google Scholar]
- 7.Aloi S, Davies CG, Karplus PA, et al. (2019) Substrate Specificity in Thiol Dioxygenases. Biochemistry 2398–2407. 10.1021/acs.biochem.9b00079 [DOI] [PubMed] [Google Scholar]
- 8.Driggers CM, Kean KM, Hirschberger LL, et al. (2016) Structure-Based Insights into the Role of the Cys–Tyr Crosslink and Inhibitor Recognition by Mammalian Cysteine Dioxygenase. J Mol Biol 428:3999–4012. 10.1016/j.jmb.2016.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamaguchi K, Hosokawa Y (1987) Cysteine Dioxygenase. Methods Enzymol 143:395–403. 10.1016/0076-6879(87)43069-3 [DOI] [PubMed] [Google Scholar]
- 10.Huxtable RJ (1992) Physiological Actions of Taurine. Physiol Rev 72:101–163 [DOI] [PubMed] [Google Scholar]
- 11.Schaffer SW, Ju Jong C, KC R, Azuma J (2010) Physiological roles of taurine in heart and muscle. J Biomed Sci 17:101–163. 10.1186/1423-0127-17-S1-S2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu Y-J, Arneja AS, Tappia PS, Dhalla NS (2008) The potential health benefits of taurine in cardiovascular disease. Exp Clin Cardiol 13:57–65 [PMC free article] [PubMed] [Google Scholar]
- 13.Stipanuk MH (1986) Metabolism of sulfur-containing amino acids. Annu Rev Nutr 6:179–209. 10.1146/annurev.nu.06.070186.001143 [DOI] [PubMed] [Google Scholar]
- 14.Simmons CR, Hirschberger LL, Machi MS, Stipanuk MH (2006) Expression, purification, and kinetic characterization of recombinant rat cysteine dioxygenase, a non-heme metalloenzyme necessary for regulation of cellular cysteine levels. Protein Expr Purif 47:74–81. 10.1016/j.pep.2005.10.025 [DOI] [PubMed] [Google Scholar]
- 15.Heafield MT, Fearn S, Steventon GB, et al. (1990) Plasma cysteine and sulphate levels in patients with motor neurone, Parkinson’s and Alzheimer’s disease. Neurosci Lett 110:216–220. 10.1016/0304-3940(90)90814-P [DOI] [PubMed] [Google Scholar]
- 16.Slivka A, Cohen G (1993) Brain ischemia markedly elevates levels of the neurotoxic amino acid, cysteine. Brain Res 608:33–37. 10.1016/0006-8993(93)90770-N [DOI] [PubMed] [Google Scholar]
- 17.Dominy JE, Simmons CR, Hirschberger LL, et al. (2007) Discovery and characterization of a second mammalian thiol dioxygenase, cysteamine dioxygenase. J Biol Chem 282:25189–25198. 10.1074/jbc.M703089200 [DOI] [PubMed] [Google Scholar]
- 18.Fernandez RL, Dillon SL, Stipanuk MH, et al. (2020) Spectroscopic Investigation of Cysteamine Dioxygenase. Biochemistry 59:2450–2458. 10.1021/acs.biochem.0c00267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y, Davis I, Chan Y, et al. (2020) Characterization of the nonheme iron center of cysteamine dioxygenase and its interaction with substrates. J Biol Chem 295:11789–11802. 10.1074/jbc.ra120.013915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y, Griffith WP, Li J, et al. (2018) Cofactor Biogenesis in Cysteamine Dioxygenase: C–F Bond Cleavage with Genetically Incorporated Unnatural Tyrosine. Angew Chemie Int Ed 57:8149–8153. 10.1002/anie.201803907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ye S, Wu X, Wei L, et al. (2007) An insight into the mechanism of human cysteine dioxygenase: Key roles of the thioether-bonded tyrosine-cysteine cofactor. J Biol Chem 282:3391–3402. 10.1074/jbc.M609337200 [DOI] [PubMed] [Google Scholar]
- 22.Masson N, Keeley TP, Giuntoli B, et al. (2019) Conserved N-terminal cysteine dioxygenases transduce responses to hypoxia in animals and plants. Science (80-) 365:65–69. 10.1126/science.aaw0112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.White MD, Klecker M, Hopkinson RJ, et al. (2017) Plant cysteine oxidases are dioxygenases that directly enable arginyl transferase-catalysed arginylation of N-end rule targets. Nat Commun 8:. 10.1038/ncomms14690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van V, Smith AT (2020) ATE1-Mediated Post-Translational Arginylation Is an Essential Regulator of Eukaryotic Cellular Homeostasis. ACS Chem Biol. 10.1021/acschembio.0c00677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee MJ, Tasaki T, Moroi K, et al. (2005) RGS4 and RGS5 are in vivo of the N-end rule pathway. Proc Natl Acad Sci U S A 102:15030–15035. 10.1073/pnas.0507533102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tamirisa P, Blumer KJ, Muslin AJ (1999) RGS4 inhibits G-protein signaling in cardiomyocytes. Circulation 99:441–447. 10.1161/01.CIR.99.3.441 [DOI] [PubMed] [Google Scholar]
- 27.Blaesi EJ, Fox BG, Brunold TC (2014) Spectroscopic and Computational Investigation of Iron(III) Cysteine Dioxygenase: Implications for the Nature of the Putative Superoxo-Fe(III) Intermediate. Biochemistry 53:5759–5770. 10.1021/bi500767x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blaesi EJ, Fox BG, Brunold TC (2015) Spectroscopic and computational investigation of the H155A variant of cysteine dioxygenase: Geometric and electronic consequences of a third-sphere amino acid substitution. Biochemistry 54:2874–2884. 10.1021/acs.biochem.5b00171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fischer AA, Miller JR, Jodts RJ, et al. (2019) Spectroscopic and Computational Comparisons of Thiolate-Ligated Ferric Nonheme Complexes to Cysteine Dioxygenase: Second-Sphere Effects on Substrate (Analogue) Positioning. Inorg Chem 58:16487–16499. 10.1021/acs.inorgchem.9b02432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clay MD, Jenney FE, Hagedoorn PL, et al. (2002) Spectroscopic studies of Pyrococcus furiosus superoxide reductase: Implications for active-site structures and the catalytic mechanism. J Am Chem Soc 124:788–805. 10.1021/ja016889g [DOI] [PubMed] [Google Scholar]
- 31.Pierce BS, Gardner JD, Bailey LJ, et al. (2007) Characterization of the nitrosyl adduct of substrate-bound mouse cysteine dioxygenase by electron paramagnetic resonance: Electronic structure of the active site and mechanistic implications. Biochemistry 46:8569–8578. 10.1021/bi700662d [DOI] [PubMed] [Google Scholar]
- 32.Li W, Blaesi EJ, Pecore MD, et al. (2013) Second-sphere interactions between the C93- Y157 cross-link and the substrate-bound Fe site influence the O2 coupling efficiency in mouse cysteine dioxygenase. Biochemistry 52:9104–9119. 10.1021/bi4010232 [DOI] [PubMed] [Google Scholar]
- 33.Fischer DS, Price DC (1964) a Simple Serum Iron Method Using the New Sensitive Chromogen Tripyridyl-S-Triazine. Clin Chem 10:21–31 [PubMed] [Google Scholar]
- 34.Crawford JA, Li W, Pierce BS (2011) Single turnover of substrate-bound ferric cysteine dioxygenase with superoxide anion: Enzymatic reactivation, product formation, and a transient intermediate. Biochemistry 50:10241–10253. 10.1021/bi2011724 [DOI] [PubMed] [Google Scholar]
- 35.Stoll S, Schweiger A (2006) EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J Magn Reson 178:42–55. 10.1016/j.jmr.2005.08.013 [DOI] [PubMed] [Google Scholar]
- 36.Gardner JD, Pierce BS, Fox BG, Brunold TC (2010) Spectroscopic and computational characterization of substrate-bound mouse cysteine dioxygenase: Nature of the ferrous and ferric cysteine adducts and mechanistic implications. Biochemistry 49:6033–6041. 10.1021/bi100189h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xie J, Yikilmaz E, Miller AF, Brunold TC (2002) Second-sphere contributions to substrate- analogue binding in iron(III) superoxide dismutase. J Am Chem Soc 124:3769–3774. 10.1021/ja016254h [DOI] [PubMed] [Google Scholar]
- 38.Wang Y, Davis I, Chan Y, et al. (2020) Characterization of the nonheme iron center of cysteamine dioxygenase and its interaction with substrates. J Biol Chem 295:11789–11802. 10.1074/jbc.ra120.013915 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.