

Abstract
High lipoprotein(a) [Lp(a)] levels are associated with the development of atherosclerotic cardiovascular disease (ASCVD) and with calcific aortic valve stenosis (CAVS) both observationally and causally from human genetic studies. The mechanisms are not well characterized but likely involve its role as a carrier of oxidized phospholipids (OxPLs), which are known to be increased in pro-inflammatory states, to induce pro-inflammatory changes in monocytes leading to plaque instability, and to impair vascular endothelial cell function, a driver of acute and recurrent ischemic events. In addition, Lp(a) itself has prothrombotic activity. Current lipid lowering strategies do not sufficiently lower Lp(a) serum levels. Lp(a)-specific lowering drugs, targeting apolipoprotein(a) synthesis, lower Lp(a) by up to 90% and are being evaluated in ongoing clinical outcome trials. This review summaries the current knowledge on the associations of Lp(a) with ASCVD and CAVS, the current role of Lp(a) assessment in the clinical setting, as well as emerging Lp(a) specific lowering therapies.
Lipoprotein (a) [Lp(a)] was first described by Kåre Berg in 1963 (1). Half a century after its discovery, it is well established that increased levels of Lp(a) confer an increased risk for cardiovascular disease (CVD). However, a standard management approach regarding Lp(a) in the clinical setting is not yet identified, mainly due to the absence of Lp(a)-specific lowering therapies. With the plethora of effective available low-density lipoprotein cholesterol (LDL-C) lowering therapies, there is a growing interest in Lp(a), which possibly represents a significant contributor to the atherosclerotic risk that persists despite optimal LDL-C lowering and other primary and secondary prevention atherosclerotic cardiovascular disease (ASCVD) therapies. This review highlights the associations of Lp(a) with CVD, the current role of Lp(a) in the clinical setting, as well as emerging Lp(a) specific lowering therapies.
Lipoprotein(a) structure
Initial reports of Lp(a)’s molecular weight and composition varied, possibly due to different purification methods used by different laboratories (2, 3). Lp(a) consists of an LDL-like core lipoprotein molecule, containing apolipoprotein B (apo-B), to which a glycoprotein of variable molecular weight, apolipoprotein (a), is covalently bound via a cysteine-cysteine disulfide bond (3). Apo(a) has been formerly termed Lipoprotein(a) antigen.
Apolipoprotein (a) is a large glycoprotein that exhibits size heterogeneity among individuals. The LPA gene, which codes for the protein, is located on chromosome 6q26. Sequencing of cloned human apolipoprotein cDNA showed that the LPA gene resembles the human plasminogen gene (4). Human plasminogen is a serine protease proenzyme that is converted to its active form, plasmin, via the action of tissue plasminogen activator, urokinase, factors XIa, XIIa and kalikrein. Plasminogen consists of five domains, named kringles (K-I, K-II, K-III, K-IV and K-V) and one protease domain at the end. Kringles are loop like structures that are stabilized by three internal disulfide bridges and were named after Scandinavian pretzels due to shape resemblance (5). (Figure 1)
Figure 1.
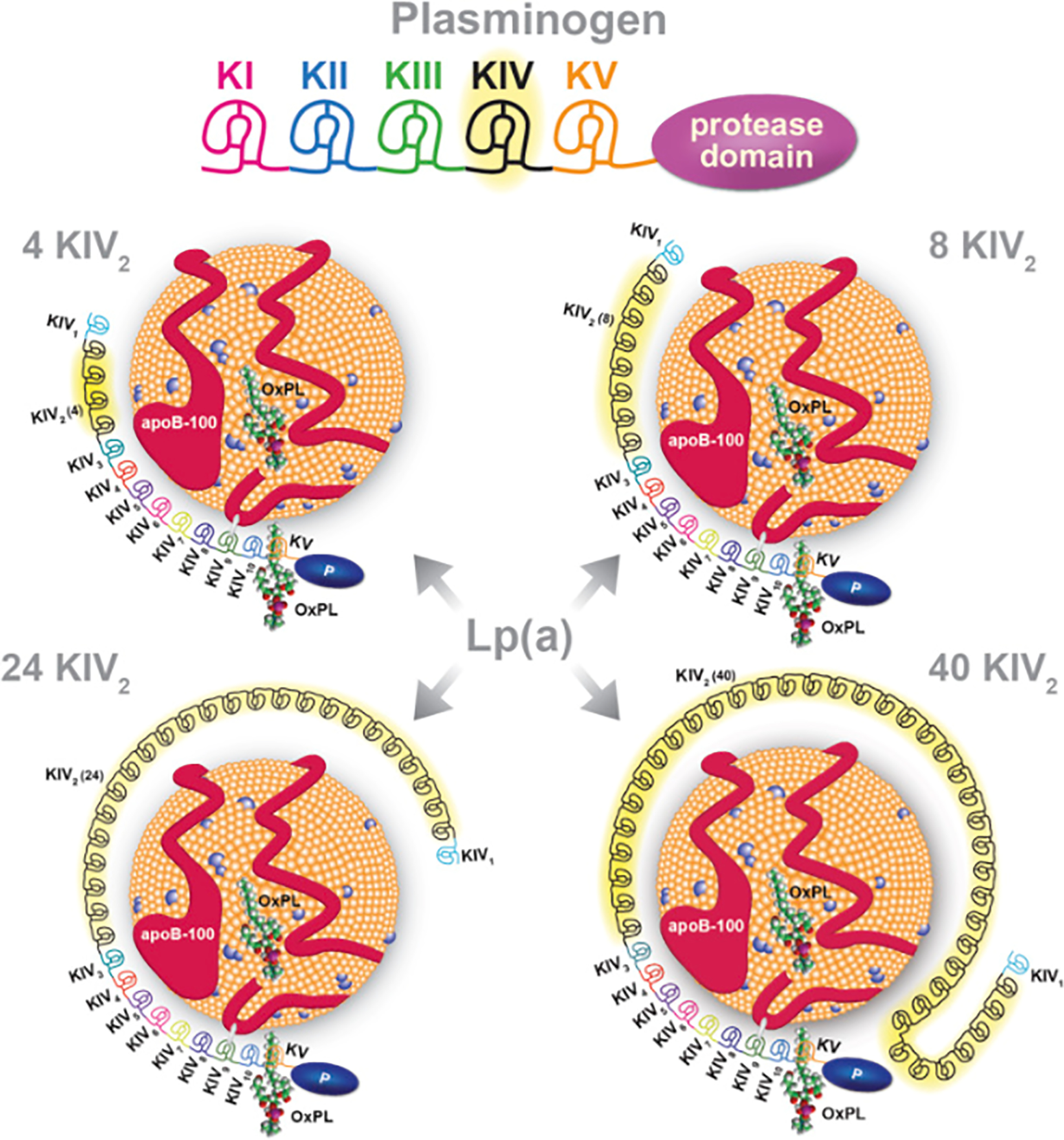
Lipoprotein [Lp(a)] is composed of apolipoprotein B-100 (apoB-100) covalently bound to apolipoprotein (a) [apo(a)], which is derived from Kringle IV (KIV) and KV, and the protease domain of plasminogen. Plasminogen has 1 copy each of KI to KV and an active protease domain. Apo(a) contains 10 subtypes of KIV repeats, composed of 1 copy each of KIV1, multiple copies of KIV2, and 1 copy of KIV310, KV, and an inactive protease-like (P) domain. In these examples, apo(a) isoforms of 4, 8, 24, and 40 KIV2 repeats are shown, representing 13, 17, 33, and 49 total KIV repeats. Oxidized phospholipids (OxPL), represented here by 1-palmitoyl-2-oxovaleroyl-sn-glycero-3- phosphocholine (POVPC), are present covalently bound to apo(a), and also dissolved in the lipid phase of apoB-100 (11). Reproduced with permission from “A Test in Context: Lipoprotein(a). Diagnosis, Prognosis, Controversies, and Emerging Therapies”(11).
In contrast, apolipoprotein (a) consists of a single Kringle V and 10 different types of Kringle IV derived from amino acid substitutions: Kringle-IV 1–10, with K-IV one and three to 10 present in one copy and Kringle IV-2 existing in multiple copies (ranging from two to >40 copies). The copy number variation (CNV) of Kringle IV-2 results in the size polymorphism of the apolipoprotein (a) molecule. The disulfide bond between apo(a) and apo-B is formed between two cysteine molecules, one being the only free cysteine residue within the apo(a) molecule, located in KIV-9, and the second one within the C-terminal region of apoB (6–8). Within populations, the copy number variations of Kringle IV-2 also presents with great heterogeneity, thus making it a very useful trace of genetic heritability (9–11). Similar to plasminogen, there is a protease at the end; however, this protease cannot be activated by tissue plasminogen activator, urokinase or streptokinase due to a single substitution mutation (serine for arginine) at the position where plasminogen is normally cleaved to be converted to active plasmin. Kringles IV 3–10, Kringle V and the protease domain are invariable (10). The above characteristics of apolipoprotein (a) reinforce the hypothesis that the LPA gene arose from gene duplication of the human plasminogen gene and subsequent deletions of the Kringle I, II and III exons (4).
Finally, another key structural component of Lp(a) are oxidized phospholipids (OxPLs) which are either carried by the apoB-100 component of the LDL-like molecule or covalently bound to the apo(a) molecule (11). In vitro studies show that OxPLs only bind to the KIV10 molecule when the lysine residue of this position is intact (12).
Synthesis and Catabolism of Lp(a)
Synthesis:
Changes of genetic apolipoprotein phenotypes caused by liver transplantation provided evidence that apolipoprotein (a) is produced in the liver (13). The exact location of apolipoprotein (a) and apo-B binding, leading to the assembly of Lp(a) in vivo, has not been described to date. In vitro studies in cultured HepG2 cells, hepatic cells of transgenic mice, and baboon hepatocytes demonstrate that assembly of Lp(a) most likely occurs extracellularly, at the hepatocyte surface or in the plasma (space of Disse, hepatocyte-blood interface surface) (9, 14–17). At the same time, studies in human hepatocytes, plasmid transfected HepG2 cells, and in vivo turnover studies indicate possible intracellular hepatocyte assembly of lipoprotein(a) (18–20). The exact location of synthesis is still uncertain.
The LPA gene genetically determines the variance in Lp(a) plasma concentrations, ranging from 36% in the PROCARDIS consortium (21) to 70%−90% in genome wide association studies, with larger apo(a) isoforms associated with lower concentrations of Lp(a) (22, 23). On a cellular level, apo(a), as a secretory protein, undergoes post translational modifications in the endoplasmic reticulum (ER). This process is dependent on apo(a)’s size, with modification of larger apo(a) isoforms taking more time. This leads to a slower production rate of larger apo(a) molecules per unit of time, and thus lower Lp(a) plasma concentrations (7).
Catabolism:
Lipoprotein (a) catabolism is not yet well defined. Murine studies demonstrate that Lp(a) is cleared primarily in the liver, and to a smaller extent, by the kidney (24, 25). On a molecular level, the specific receptor that facilitates Lp(a) clearance or its specific target on the LP(a) has not been precisely identified. Five main receptors were identified in the Lp(a) clearance process, i.e. lipoprotein receptors, toll-like and scavenger receptors, lectins, and plasminogen receptors. Apolipoprotein (a), apo-B and the OxPLs carried in the Lp(a) molecule, have all been identified as ligands for receptors involved in Lp(a) catabolism. The variety of receptors as well as their respective ligands and animal models used for their discovery were exceptionally reviewed by McCormick & Schneider (26).
Lp(a) and cardiovascular disease (CVD)
There are several mechanisms by which Lipoprotein (a) is believed to cause cardiovascular disease. The first, similar to all apoB containing lipoproteins, is an interaction with proteoglycans on the arterial wall leading to subendothelial deposition and triggering an inflammatory immune response involving T-lymphocytes and macrophages. This response results in endothelial injury and dysfunction and subsequent atherosclerotic plaque formation (27, 28).
However, it is believed that the pivotal mechanism by which Lp(a) contributes to ASCVD is through its role as a carrier of OxPLs, which are associated with several cellular mechanisms promoting atherosclerosis (29). In fact, more than 85% of serum OxPLs are bound to Lp(a) (30). A strong correlation of the oxidized phospholipids: apoB-100 ratio and Lp(a) lipoprotein was demonstrated by Tsimikas et al. (31), reinforcing the notion that Lp(a) is the main carrier of OxPLs. Moreover, the OxPL:apoB ratio was strongly associated with both the extent and the amount of atherosclerotic coronary artery disease, independent of other risk factors except for Lp(a), suggesting that OxPL are the main atherogenic component of the Lp(a) molecule (32). Results from the Dallas Heart Study also showed the important role of Lp(a) carrying OxPLs as compared to other apo-B lipoproteins, by a log-linear relationship between the OxPLs/ApoB ratio and Lp(a) above an Lp(a) threshold of ~30 nmol/L (33). For reference, this value is slightly below the median Lp(a) level in the adult U.S. population (40 nmol/L) (34). A recent in vivo study reinforced this concept: 18- Fluorodeoxyglucose PET scans showed that patients with high levels of Lp(a) had increased reuptake of the isotope in the aorta and the carotids, suggesting higher levels of inflammation, and increased mononuclear cell trafficking to the vessel walls. In vitro studies using samples from the same patients revealed increased monocyte transmigration capacity and pro-inflammatory cytokine production. These effects were annulled by OxPL antibodies, suggesting a strong causal role for OxPL as responsible for the pro-inflammatory effects (35). Finally, OxPLs can potentially activate macrophages via interaction with the CD36 receptor (36).
Because of apolipoprotein (a)’s resemblance to plasminogen, but without the ability to be activated to plasmin, it is believed that high levels of lipoprotein(a) inhibit the natural conversion of plasminogen to plasmin via competitive inhibition, thus causing a pro-thrombotic milieu. Although in vitro experiments support this hypothesis (37), in vivo studies fail to confirm the results (38, 39).
Several studies have established Lp(a) as an important risk factor for coronary heart disease (CHD). In a recent meta-analysis of 36 cohort studies, pooling 126,634 individuals, there was a 16% and 10% relative increase in CHD events and stroke, respectively, for each standard deviation increase in Lp(a) (40). Similarly, in the Copenhagen City Heart Study, participants with Lp(a) levels above the 90th percentile and 95th percentile had a 1.9- and 2.6-fold increased risk of myocardial infarction (MI) over a 16-year follow-up period, respectively, when compared to individuals with Lp(a) levels < 5 mg/dL (22nd percentile) (41). In addition, in the Copenhagen General Population Study, patients with familial hypercholesterolemia (FH) and Lp(a) levels >50 mg/dL or Lp(a) < 50 mg/dL, had a 5.3- and 3.2- fold increase in the hazard for myocardial infarction, compared to patients without FH and LP(a) levels of ≤ 50 mg/dL. These findings suggest that measuring Lp(a) is beneficial in all patients with FH, providing valuable information to further stratify patients with FH and identify those with the highest risk for MI (42).
In addition, in the PROCAM study, participants with Lp(a) ≥ 20 mg/dL had an increased risk for coronary events compared to those with lower levels, especially if they had an LDL cholesterol ≥ 4.1 nmol/L (2.6-fold increase), HDL cholesterol ≤ 0.9 nmol/L (8.3-fold increase), hypertension (3.2-fold increase) or belonged in the two highest quartiles of global cardiovascular risk (2.7-fold increase) (43).
These associations are also replicated by genome wide association and Mendelian randomization studies, showing evidence of causality between Lp(a) and CVD (21, 44–46).
Lp(a) and Calcific Aortic Valve Disease (CAVD)
Clinical studies demonstrate a significant association between Lp(a) levels and calcific aortic valve disease CAVD and genome wide and Mendelian randomization studies support a causal association. In the Copenhagen General Population cohort, Lp(a) levels in the 90th - 95th percentile carried a two-fold increase and levels above the 95th percentile almost a three-fold increase in the hazard of developing CAVD, compared to participants with Lp(a) levels under 5mg/dL (33d percentile) (47). Subsequent analysis of the same population showed that OxPLs bound to apoB (OxPL-apoB) levels above the 90th and 95th percentiles were associated with a two- and 3.4-fold increase in the odds for CAVD respectively, compared to levels below the 34th percentile, suggesting that OxPL carried in Lp(a) may be a mediator of CAVD progression (47). The same association of high Lp(a) levels and CAVD was reported in patients with asymptomatic FH, further supporting the notion that measurement of Lp(a) in this subpopulation is of critical importance (48). In genome wide association studies, the most frequent single nucleotide polymorphism associated with higher levels of Lp(a) and CAVD is rs10455872, with studies reporting a two-fold or greater increase in the odds of developing aortic stenosis in homozygous carriers, compared to those not carrying this nucleotide polymorphism (45, 49, 50).
In addition to these observations, a study of 220 patients with mild-to-moderate CAVD showed that patients within the 3rd tertile of Lp(a) (>58.5 mg/dl) and OxPL-apoB (>5.50 nM) concentrations had more rapid disease progression, independently of other CAVD risk factors. Moreover, these patients also had higher rates of aortic valve replacement (51). A study by Zheng et al, presented similar conclusions in terms of disease progression and need for aortic valve replacement and further showed that patients in the top Lp(a) tertile had increased valvular calcification activity on baseline 18F-sodium fluoride (18F-NaF) positron emission tomography (PET) and increased aortic valve computed tomography calcium score (52).
Lp(a) in acute coronary syndromes
A study by Tsimikas et al. reported an acute rise in Lp(a) levels of 100% to 150% above baseline in eight patients with an acute MI, which persisted for up to 120 days, before returning to baseline thereafter (31). In the same study, this increase was also associated with a simultaneous acute rise of OxPL levels, by up to 54% at discharge, remaining at 36% above baseline at 30 days (31). Though limited by a small sample size, these data emphasize the critical role of Lp(a) as a carrier of OxPLs, which is likely potentiated in ACS. A similar relationship of acute increases in Lp(a) and OxPLs also occurs 6 to 24 hours following percutaneous coronary interventions (PCI) in patients with stable angina (53).
The importance of Lp(a) in the pathophysiology of ACS may be even more pronounced in younger individuals, particularly in those <45 years old, in whom elevated Lp(a) levels (> 120 nmol/L, 80th percentile) are associated with a 3-fold increased risk of MI (54, 55). In addition, studies consistently demonstrate that the age at which patients present with a first MI is inversely related to Lp(a) levels (54, 55). This likely reflects the importance of other, traditional atherosclerotic risk factors in older individuals in contrast to a more important role of Lp(a) in younger individuals. This is not surprising, given that Lp(a) levels are highly genetically determined, as previously stated. This implies lifelong exposure to an important risk factor as opposed to other traditional risk factors which typically have a later in life onset.
Lowering Lipoprotein(a)
Lifestyle factors:
Even though Lp(a) levels are mostly genetically determined by the LPA gene, lifestyle factors may also play a role. Most evidence however comes from small studies or anecdotal data, with controlled trials missing. In addition, one caveat with dietary trials is the potential confounding of one intervention (e.g. caloric intake restriction) by a concomitant change (e.g. lower fat intake), making it challenging to establish causality. Interestingly, a low fat, high carbohydrate content diet significantly increases Lipoprotein(a) and OxPL-apoB compared to a high fat, low carbohydrate diet (56). In addition, a recent study found that weight loss following caloric restriction led to a decrease in LDL-C levels with a simultaneous increase in Lp(a) levels, in obese individuals with, and those without, diabetes (57). Interestingly, there was no significant rise in Lp(a) levels in patients who underwent bariatric surgery, despite significant weight loss (57). The paradoxical Lp(a) increase may be a consequence of the LDL-C lowering effect of the diets. The decrease of LDL-C requires a new buffer for OxPL in the circulation, possibly stimulating the liver to produce more Lp(a).
Existing lipid lowering therapies:
Statins
Until recently, the effect of statins on Lp(a) was controversial. In a recent meta-analysis by Tsimikas et al. including 5256 patients from six randomized trials (3 placebo controlled statin trials and 3 trials comparing different statins), statins increased Lp(a) levels by approximately 20%, despite lowering atherogenic non-HDL lipoprotein concentrations substantially and reducing overall cardiovascular risk (58).
Importantly, the risk attributed to elevated Lp(a) levels persists in statin-treated individuals and in those with low LDL-C levels. Another meta-analysis of 30,000 patients from randomized trials of statin therapy suggests an even stronger association of Lp(a) levels and ASCVD risk in patients with a statin as compared to those treated with placebo (59). This may be partially related to the increase in Lp(a) levels with statin therapy. The mechanism by which statins increase Lp(a) levels is not completely understood. In addition, when HepG2 cells were treated with atorvastatin for 24h, there was a 1.5-fold increase in LPA mRNA expression, indicating that statins may cause an increase in apolipoprotein (a) expression and thus an increase in serum Lp(a) (58).
Cholesterol Ester Transferase Protein (CETP) Inhibitors and Niacin
In the ACCELERATE trial, Evacetrapib, a CETP inhibitor, decreased Lp(a) levels by about 20%, in addition to decreasing LDL-C and increasing HDL-C, without, however, any clinical cardiovascular benefit (60).
In general, niacin also decreases Lp(a) levels by approximately 20%−30% (61). In addition, Yeang et al reported a decrease in both OxPL and Lp(a) levels with niacin monotherapy whereas, there was an increase in both, after 24 weeks of simvastatin and ezetimibe (62). In the AIM-HIGH trial, niacin achieved favorable lipoprotein changes, decreasing Lp(a), however, these changes were not associated with a clinical cardiovascular benefit (63).
There are a few reasons why these outcome-driven randomized trials of a CETP inhibitor and niacin failed to show a clinical benefit. Initially, these trials were not specifically designed to evaluate patients with high Lp(a), as indicated by median Lp(a) levels of 30 to 60 nmol/L in their respective trials. It is believed that the lack of benefit of Lp(a) reduction in these studies may have been due to:
(1) the therapies didn’t lower Lp(a) to a level sufficient to achieve a clinical effect. This argument is also supported by Mendelian randomization studies which indicate that in order to achieve a CHD risk reduction by Lp(a) lowering that is equivalent to lowering LDL-C by 1 mmol/L (38.7 mg/dL), Lp(a) would have to be reduced by 27 to 42 nmol/L (65 to 100 mg/dL) (45, 64).
(2) the baseline Lp(a) was too low to achieve the absolute reduction in Lp(a) levels required to observe a clinical benefit. However, in the AIM-HIGH study, even in patients with Lp(a) levels > 125 nmol/ L who achieved adequate Lp(a) reduction (39% and 64% for the 75th and 90th Lp(a) percentiles respectively), there was no reduction in the event rate.
Proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors
PCSK9 inhibitors lower Lp(a) by approximately 20%−30%, in addition to a 50%−60% reduction in LDL-C (65–70). In the FOURIER study of patients with clinically evident ASCVD, evolocumab reduced the composite endpoint of CHD death, MI or urgent revascularization by 15% over a median follow-up of 2.2 years as compared to placebo (67). A sub-analysis of the data demonstrated that those patients with baseline Lp(a) levels above the median of 37 nmol/L experienced a 23% reduction in the composite endpoint, as compared to a 7% reduction for those patients with baseline Lp(a) levels below the median, despite similar LDL-C reductions in the two groups (69). Similarly, in the ODYSSEY OUTCOMES study of patients with a recent acute coronary syndrome (1 to 12 months prior to enrolment), alirocumab reduced the composite endpoint of CHD death, MI, ischemic stroke or unstable angina (UA) requiring hospitalization by 15% over a median follow up of 2.8 years (68), with a subsequent analysis showing a median 5 mg/dl decrease in Lp(a), which independently reduced the risk for MACE (70). In both studies, greatest absolute Lp(a) reductions were observed for patients in the highest quartile of baseline Lp(a) values. The data from these two studies suggest two conclusions: first, at least part of the benefit of PCSK9-inhibition is mediated through a reduction in Lp(a) levels; and second, similar to LDL-C, the magnitude of benefit from Lp(a) reduction is likely related to the absolute, not relative, reduction in Lp(a) levels.
In general, the mechanisms by which the aforementioned lipid lowering treatments affect Lp(a) are not well understood. In fact, one would assume that HMG-CoA reductase inhibition would result in the same downstream upregulation of LDL-R, in a similar manner with PCSK-9 inhibition, leading to a decrease of Lp(a) via a common mechanism. This paradox of statins increasing Lp(a) while PCK9 inhibitors decrease LP(a) levels may be due to (1) a potency dependent threshold in order to observe an Lp(a) lowering effect through the LDL-R pathway; (2) the Lp(a) increasing effect of statins is mediated by other mechanisms (e.g. increase in LPA expression); (3) the Lp(a) lowering effect of PCSK9 inhibitors is independent of the PCSK9 pathway. The latter is supported by the fact that serum PCSK9 levels do not correlate with the levels of serum Lp(a) or differ between Lp(a) tertiles (71). Finally, in the HPS2-THRIVE study, the Lp(a) lowering effect of niacin-laropiprant was related to the apo(a) isoform size of Lp(a) (72). Again, the mechanism responsible for this association is not well understood.
Emerging Therapies
Two major ongoing clinical trials are investigating Lp(a)- specific lowering medications. The HORIZON phase III trial is investigating the impact of Lp(a) lowering with an antisense oligonucleotide (ASO), TQJ230, on major cardiovascular events in patients with established cardiovascular disease (NCT03887520). TQJ230 binds directly to the mRNA and inhibits transcription to apolipoprotein(a) in the hepatocyte (73). Results from the Phase I and Phase II trials of TQJ230, formerly mentioned as ISIS-APO(a)Rx and AKCEA-APO(a)-LRx, demonstrated a dose-dependent reduction of up to 90% in Lp(a) and associated OxPLs without significant safety concerns (74–76).
A phase II study is also being conducted to evaluate the efficacy, safety, and tolerability of a small interfering RNA (siRNA) molecule, AMG 890, in subjects with elevated Lp(a) levels (NCT04270760). AMG 890 is conjugated to trivalent N-acetylgalactosamine (GalNAc) which enhances its hepatocellular uptake. Upon uptake, AMG 890 binds to the mRNA transcribed from the LPA gene, blocking its translation to apolipoprotein (a).
Lp(a) in the current clinical setting. What are the challenges?
Even though Lp(a) is established as an independent cardiovascular risk factor and abnormally high levels are present in 20% of the general population it is still not measured routinely in clinical practice. In addition, an ICD-10 code of “Elevated Lp(a) level” was only established in June of 2018.
One of the biggest challenges in the clinical setting with lipoprotein (a) is the method of measurement. Most methods used to quantify Lp(a) are immunoassays using polyclonal antibodies to target the apo(a) molecule. However, this method is subject to miscalculation due to the heterogeneity of the apo(a) molecule both between individuals but also within the same individual due to heterozygosity of the apo(a) gene. In addition, mass concentration reporting in (mg/dL) is highly influenced by the number of Kringle form repeats and can lead to overestimation with big isoforms (77). The effect of the number of apo(a) Kringle domains on Lp(a) measurement and its implications for clinical testing were reported by Marcovina et al. (78, 79). It is for these reasons that the National Heart, Lung, and Blood Institute (NHLBI) suggested measuring Lp(a) in nmol/L, switching from the most commonly used total mass assay which reports Lp(a) in mg/dL (80). Finally, converting mg/dL to nmol/ L with an estimate of 2– 2.5 multiplication factor should be avoided, due to uncertainty of the molecular weight of apo(a), leading to frequent miscalculations (80).
The 2010 European Atherosclerosis Society (EAS) guidelines (81) recommended a one-time measurement in all patients at intermediate or high risk of CVD or CHD, presenting with:
premature cardiovascular disease
familial hypercholesterolemia
a family history of premature cardiovascular disease and/or elevated Lp(a)
recurrent CVD despite statin therapy
≥ 3% 10-year risk of fatal CVD
≥ 10% 10-year risk of fatal and/or non-fatal CHD according to AHA guidelines
Nine years later, the European Society of Cardiology guidelines for the management of dyslipidemias (82) recommended measuring Lp(a) levels at least once to identify patients with Lp(a) levels above 430 nmol/L, which confers a very high risk for atherosclerotic cardiovascular disease, one similar to that of heterozygotic FH. In addition, this one-time measurement of Lp(a) is recommended to further classify patients whose risk levels are borderline between moderate and high for ASCVD. Since Lp(a) levels are primarily genetically determined, the one-time testing can easily identify individuals at high risk, without the need for repeat measurement in those with normal values. With emerging Lp(a) therapies it is crucial to identify those at high risk.
Conclusion:
In conclusion, even though it is well established that high Lp(a) levels are associated with the development and progression of ASCVD and CAVD, Lp(a) has been an underappreciated lipoprotein, with minimal testing in the clinical setting, mainly due to the lack of potential therapeutic interventions in patients with elevated levels (83).
On a molecular level, many factors are not understood regarding its metabolism and catabolism as well the mechanisms responsible for its association with atherosclerotic disease. In the clinical setting, establishing a universal measurement method and increasing physician awareness of Lp(a)’s importance as a risk factor so appropriate patients can be widely tested is key.
At the same time, it is crucial to explore the role of Lp(a) and OxPL during the peri- and early post-acute coronary syndrome (ACS) period given the current lack of extensive reports and investigate whether Lp(a)-specific lowering interventions at this time have a role in the management of ACS, potentially improving short-and long-term clinical outcomes. The presence and timing of percutaneous coronary intervention (PCI) should also be considered as possible confounders in future ACS Lp(a) lowering trials due to the association of the procedure with rises in Lp(a).
The ongoing TQJ230 Horizon trial will study whether Lp(a) lowering will provide a clinical benefit in patients with established cardiovascular disease. As the field moves forward with additional clinical outcome studies, it will be key to identify populations most likely to benefit from reducing Lp(a). In view of the results from prior studies, future trials might focus on patients with elevated baseline Lp(a) so as to achieve meaningful absolute reductions in Lp(a) levels. In addition, participants should already be optimally treated for other cardiovascular risk factors, with achieved guideline targets or maximum tolerated treatment regimens, to reduce potential confounding by differences in concurrent therapies. In addition to the baseline Lp(a) levels, the clinical context should also be considered. It may be that patients with acute coronary syndromes (ACS), presenting with acute rises in Lp(a) and OxPL levels, will derive a more significant benefit from Lp(a) lowering than do those with stable ischemic heart disease (SIHD).
If these Lp(a)-specific lowering drugs achieve a significant clinical benefit, and with a 20% prevalence of elevated Lp(a) levels in the general population, and a probable higher prevalence in those with known disease, a dedicated Lp(a) outpatient clinic effort, similar to that of the FH clinic, may be a valuable addition to the outpatient management of patients with, and at increased risk for, atherosclerotic and aortic valve disease.
Footnotes
The final publication is available at https://www.sciencedirect.com/science/article/pii/S1109966620302165?via%3Dihub
REFERENCES:
- 1.Berg K A NEW SERUM TYPE SYSTEM IN MAN--THE LP SYSTEM. Acta Pathol Microbiol Scand 1963;59:369–382. [DOI] [PubMed] [Google Scholar]
- 2.Gaubatz JW, Heideman C, Gotto AM, Morrisett JD, Dahlen GH. Human plasma lipoprotein [a]. Structural properties. J. Biol. Chem. 1983;258:4582–4589. [PubMed] [Google Scholar]
- 3.Utermann G, Weber W. Protein composition of Lp(a) lipoprotein from human plasma. FEBS Letters 1983;154:357–361. [DOI] [PubMed] [Google Scholar]
- 4.McLean JW, Tomlinson JE, Kuang W-J, et al. cDNA sequence of human apolipoprotein(a) is homologous to plasminogen. Nature 1987;330:132–137. [DOI] [PubMed] [Google Scholar]
- 5.Utermann G The mysteries of lipoprotein(a). Science 1989;246:904–910. [DOI] [PubMed] [Google Scholar]
- 6.Callow MJ, Rubin EM. Site-specific mutagenesis demonstrates that cysteine 4326 of apolipoprotein B is required for covalent linkage with apolipoprotein (a) in vivo. J. Biol. Chem. 1995;270:23914–23917. [DOI] [PubMed] [Google Scholar]
- 7.Dieplinger H, Utermann G. The seventh myth of lipoprotein(a): where and how is it assembled? Current Opinion in Lipidology 1999;10:275–284. [DOI] [PubMed] [Google Scholar]
- 8.Koschinsky ML, Côté GP, Gabel B, van der Hoek YY. Identification of the cysteine residue in apolipoprotein(a) that mediates extracellular coupling with apolipoprotein B-100. J. Biol. Chem. 1993;268:19819–19825. [PubMed] [Google Scholar]
- 9.Kronenberg F, Utermann G. Lipoprotein(a): resurrected by genetics. J. Intern. Med. 2013;273:6–30. [DOI] [PubMed] [Google Scholar]
- 10.Haibach C, Kraft HG, Köchl S, Abe A, Utermann G. The number of kringle IV repeats 3–10 is invariable in the human apo(a) gene. Gene 1998;208:253–258. [DOI] [PubMed] [Google Scholar]
- 11.Tsimikas S A Test in Context: Lipoprotein(a): Diagnosis, Prognosis, Controversies, and Emerging Therapies. J. Am. Coll. Cardiol. 2017;69:692–711. [DOI] [PubMed] [Google Scholar]
- 12.Leibundgut G, Scipione C, Yin H, et al. Determinants of binding of oxidized phospholipids on apolipoprotein (a) and lipoprotein (a)1. J Lipid Res 2013;54:2815–2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kraft HG, Menzel HJ, Hoppichler F, Vogel W, Utermann G. Changes of genetic apolipoprotein phenotypes caused by liver transplantation. Implications for apolipoprotein synthesis. J Clin Invest 1989;83:137–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lobentanz EM, Krasznai K, Gruber A, et al. Intracellular metabolism of human apolipoprotein(a) in stably transfected Hep G2 cells. Biochemistry 1998;37:5417–5425. [DOI] [PubMed] [Google Scholar]
- 15.White AL, Lanford RE. Cell surface assembly of lipoprotein(a) in primary cultures of baboon hepatocytes. J. Biol. Chem. 1994;269:28716–28723. [PubMed] [Google Scholar]
- 16.White AL, Rainwater DL, Hixson JE, Estlack LE, Lanford RE. Intracellular processing of apo(a) in primary baboon hepatocytes. Chem. Phys. Lipids 1994;67–68:123–133. [DOI] [PubMed] [Google Scholar]
- 17.White AL. Biogenesis of Lp(a) in transgenic mouse hepatocytes. Clin. Genet. 1997;52:326–337. [DOI] [PubMed] [Google Scholar]
- 18.Edelstein C, Davidson NO, Scanu AM. Oleate stimulates the formation of triglyceride-rich particles containing apoB100-apo(a) in long-term primary cultures of human hepatocytes. Chem. Phys. Lipids 1994;67–68:135–143. [DOI] [PubMed] [Google Scholar]
- 19.Bonen DK, Hausman AM, Hadjiagapiou C, Skarosi SF, Davidson NO. Expression of a recombinant apolipoprotein(a) in HepG2 cells. Evidence for intracellular assembly of lipoprotein(a). J. Biol. Chem. 1997;272:5659–5667. [DOI] [PubMed] [Google Scholar]
- 20.Su W, Campos H, Judge H, Walsh BW, Sacks FM. Metabolism of Apo(a) and ApoB100 of lipoprotein(a) in women: effect of postmenopausal estrogen replacement. J. Clin. Endocrinol. Metab. 1998;83:3267–3276. [DOI] [PubMed] [Google Scholar]
- 21.Clarke R, Peden JF, Hopewell JC, et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N. Engl. J. Med. 2009;361:2518–2528. [DOI] [PubMed] [Google Scholar]
- 22.Boerwinkle E, Leffert CC, Lin J, Lackner C, Chiesa G, Hobbs HH. Apolipoprotein(a) gene accounts for greater than 90% of the variation in plasma lipoprotein(a) concentrations. J. Clin. Invest. 1992;90:52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmidt K, Noureen A, Kronenberg F, Utermann G. Structure, function, and genetics of lipoprotein (a). J. Lipid Res. 2016;57:1339–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hrzenjak A, Frank S, Wo X, Zhou Y, Van Berkel T, Kostner GM. Galactose-specific asialoglycoprotein receptor is involved in lipoprotein (a) catabolism. Biochem J 2003;376:765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cain WJ, Millar JS, Himebauch AS, et al. Lipoprotein [a] is cleared from the plasma primarily by the liver in a process mediated by apolipoprotein [a]. J. Lipid Res. 2005;46:2681–2691. [DOI] [PubMed] [Google Scholar]
- 26.McCormick SPA, Schneider WJ. Lipoprotein(a) catabolism: a case of multiple receptors. Pathology 2019;51:155–164. [DOI] [PubMed] [Google Scholar]
- 27.Tabas I, Williams KJ, Borén J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation 2007;116:1832–1844. [DOI] [PubMed] [Google Scholar]
- 28.Borén J, Williams KJ. The central role of arterial retention of cholesterol-rich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: a triumph of simplicity. Curr. Opin. Lipidol. 2016;27:473–483. [DOI] [PubMed] [Google Scholar]
- 29.Berliner JA, Leitinger N, Tsimikas S. The role of oxidized phospholipids in atherosclerosis. J Lipid Res 2009;50:S207–S212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bergmark C, Dewan A, Orsoni A, et al. A novel function of lipoprotein [a] as a preferential carrier of oxidized phospholipids in human plasma. J. Lipid Res. 2008;49:2230–2239. [DOI] [PubMed] [Google Scholar]
- 31.Tsimikas S, Bergmark C, Beyer RW, et al. Temporal increases in plasma markers of oxidized low-density lipoprotein strongly reflect the presence of acute coronary syndromes. Journal of the American College of Cardiology 2003;41:360–370. [DOI] [PubMed] [Google Scholar]
- 32.Tsimikas S, McConnell JP, Witztum JL. Oxidized Phospholipids, Lp(a) Lipoprotein, and Coronary Artery Disease. The New England Journal of Medicine 2005:12. [DOI] [PubMed] [Google Scholar]
- 33.Tsimikas S, Clopton P, Brilakis ES, et al. Relationship of Oxidized Phospholipids on Apolipoprotein B-100 Particles to Race/Ethnicity, Apolipoprotein(a) Isoform Size, and Cardiovascular Risk Factors: Results From the Dallas Heart Study. Circulation 2009;119:1711–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Varvel S, McConnell JP, Tsimikas S. Prevalence of Elevated Lp(a) Mass Levels and Patient Thresholds in 532 359 Patients in the United States. Arterioscler Thromb Vasc Biol. 2016;36:2239–2245. [DOI] [PubMed] [Google Scholar]
- 35.van der Valk FM, Bekkering S, Kroon J, et al. Oxidized Phospholipids on Lipoprotein(a) Elicit Arterial Wall Inflammation and an Inflammatory Monocyte Response in Humans. Circulation 2016;134:611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Greenberg ME, Sun M, Zhang R, Febbraio M, Silverstein R, Hazen SL. Oxidized phosphatidylserine–CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J Exp Med 2006;203:2613–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caplice NM, Panetta C, Peterson TE, et al. Lipoprotein (a) binds and inactivates tissue factor pathway inhibitor: a novel link between lipoproteins and thrombosis. Blood 2001;98:2980–2987. [DOI] [PubMed] [Google Scholar]
- 38.Kamstrup PR, Tybjærg--Hansen A, Nordestgaard BG. Genetic Evidence That Lipoprotein(a) Associates With Atherosclerotic Stenosis Rather Than Venous Thrombosis. Arterioscler Thromb Vasc Biol. 2012;32:1732–1741. [DOI] [PubMed] [Google Scholar]
- 39.Lippi G, Bassi A, Brocco G, Manzato F, Marini M, Guidi G. Lipoprotein(a) concentration is not associated with venous thromboembolism in a case control study. Haematologica 1999;84:726–729. [PubMed] [Google Scholar]
- 40.Emerging Risk Factors Collaboration, Erqou S, Kaptoge S, et al. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA 2009;302:412–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kamstrup PR, Tybjærg-Hansen A, Steffensen R, Nordestgaard BG. Genetically Elevated Lipoprotein(a) and Increased Risk of Myocardial Infarction. JAMA 2009;301:2331–2339. [DOI] [PubMed] [Google Scholar]
- 42.Langsted A, Kamstrup PR, Benn M, Tybjærg-Hansen A, Nordestgaard BG. High lipoprotein(a) as a possible cause of clinical familial hypercholesterolaemia: a prospective cohort study. The Lancet Diabetes & Endocrinology 2016;4:577–587. [DOI] [PubMed] [Google Scholar]
- 43.von Eckardstein A, Schulte H, Cullen P, Assmann G. Lipoprotein(a) further increases the risk of coronary events in men with high global cardiovascular risk. Journal of the American College of Cardiology 2001;37:434–439. [DOI] [PubMed] [Google Scholar]
- 44.Anon. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet 2013;45:25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burgess S, Ference BA, Staley JR, et al. Association of LPA Variants With Risk of Coronary Disease and the Implications for Lipoprotein(a)-Lowering Therapies: A Mendelian Randomization Analysis. JAMA Cardiol 2018;3:619–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kamstrup PR, Tybjærg-Hansen A, Nordestgaard BG. Extreme Lipoprotein(a) Levels and Improved Cardiovascular Risk Prediction. J Am Coll Cardiol 2013;61:1146–1156. [DOI] [PubMed] [Google Scholar]
- 47.Kamstrup PR, Hung M-Y, Witztum JL, Tsimikas S, Nordestgaard BG. Oxidized Phospholipids and Risk of Calcific Aortic Valve Disease - The Copenhagen General Population Study. Arterioscler Thromb Vasc Biol 2017;37:1570–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vongpromek R, Bos S, Kate G-JR ten, et al. Lipoprotein(a) levels are associated with aortic valve calcification in asymptomatic patients with familial hypercholesterolaemia. Journal of Internal Medicine 2015;278:166–173. [DOI] [PubMed] [Google Scholar]
- 49.Arsenault Benoit J, Matthijs Boekholdt S., Marie-Pierre Dubé, et al. Lipoprotein(a) Levels, Genotype, and Incident Aortic Valve Stenosis. Circulation: Cardiovascular Genetics 2014;7:304–310. [DOI] [PubMed] [Google Scholar]
- 50.Thanassoulis G, Campbell CY, Owens DS, et al. Genetic Associations with Valvular Calcification and Aortic Stenosis. New England Journal of Medicine 2013;368:503–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Capoulade R, Chan KL, Yeang C, et al. Oxidized Phospholipids, Lipoprotein(a), and Progression of Calcific Aortic Valve Stenosis. Journal of the American College of Cardiology 2015;66:1236–1246. [DOI] [PubMed] [Google Scholar]
- 52.Zheng KH, Tsimikas S, Pawade T, et al. Lipoprotein(a) and Oxidized Phospholipids Promote Valve Calcification in Patients With Aortic Stenosis. J Am Coll Cardiol 2019;73:2150–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsimikas S, Lau HK, Han K-R, et al. Percutaneous Coronary Intervention Results in Acute Increases in Oxidized Phospholipids and Lipoprotein(a): Short-Term and Long-Term Immunologic Responses to Oxidized Low-Density Lipoprotein. Circulation 2004;109:3164–3170. [DOI] [PubMed] [Google Scholar]
- 54.Rallidis LS, Pavlakis G, Foscolou A, et al. High levels of lipoprotein (a) and premature acute coronary syndrome. Atherosclerosis 2018;269:29–34. [DOI] [PubMed] [Google Scholar]
- 55.Isser HS, Puri VK, Narain VS, Saran RK, Dwivedi SK, Singh S. Lipoprotein (a) and lipid levels in young patients with myocardial infarction and their first-degree relatives. Indian Heart J 2001;53:463–466. [PubMed] [Google Scholar]
- 56.Faghihnia N, Tsimikas S, Miller ER, Witztum JL, Krauss RM. Changes in lipoprotein(a), oxidized phospholipids, and LDL subclasses with a low-fat high-carbohydrate diet. J. Lipid Res. 2010;51:3324–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Berk KA, Yahya R, Verhoeven AJM, et al. Effect of diet-induced weight loss on lipoprotein(a) levels in obese individuals with and without type 2 diabetes. Diabetologia 2017;60:989–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tsimikas S, Gordts PLSM, Nora C, Yeang C, Witztum JL. Statin therapy increases lipoprotein(a) levels. European Heart Journal 2019:ehz310. [DOI] [PubMed] [Google Scholar]
- 59.Willeit P, Ridker PM, Nestel PJ, et al. Baseline and on-statin treatment lipoprotein(a) levels for prediction of cardiovascular events: individual patient-data meta-analysis of statin outcome trials. The Lancet 2018;392:1311–1320. [DOI] [PubMed] [Google Scholar]
- 60.Lincoff AM, Nicholls SJ, Riesmeyer JS, et al. Evacetrapib and Cardiovascular Outcomes in High-Risk Vascular Disease. New England Journal of Medicine 2017;376:1933–1942. [DOI] [PubMed] [Google Scholar]
- 61.Sahebkar A, Reiner Ž, Simental-Mendía LE, Ferretti G, Cicero AFG. Effect of extended-release niacin on plasma lipoprotein(a) levels: A systematic review and meta-analysis of randomized placebo-controlled trials. Metabolism - Clinical and Experimental 2016;65:1664–1678. [DOI] [PubMed] [Google Scholar]
- 62.Yeang C, Hung M-Y, Byun Y-S, et al. Effect of therapeutic interventions on oxidized phospholipids on apolipoprotein B100 and lipoprotein(a). Journal of Clinical Lipidology 2016;10:594–603. [DOI] [PubMed] [Google Scholar]
- 63.Albers JJ, Slee A, O’Brien KD, et al. Relationship of Apolipoproteins A-1 and B, and Lipoprotein(a) to Cardiovascular Outcomes: The AIM-HIGH Trial (Atherothrombosis Intervention in Metabolic Syndrome With Low HDL/High Triglyceride and Impact on Global Health Outcomes). Journal of the American College of Cardiology 2013;62:1575–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lamina C, Kronenberg F. Estimation of the Required Lipoprotein(a)-Lowering Therapeutic Effect Size for Reduction in Coronary Heart Disease Outcomes: A Mendelian Randomization Analysis. JAMA Cardiol 2019;4:575–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gaudet D, Kereiakes DJ, McKenney JM, et al. Effect of Alirocumab, a Monoclonal Proprotein Convertase Subtilisin/Kexin 9 Antibody, on Lipoprotein(a) Concentrations (a Pooled Analysis of 150 mg Every Two Weeks Dosing from Phase 2 Trials). American Journal of Cardiology 2014;114:711–715. [DOI] [PubMed] [Google Scholar]
- 66.Raal FJ, Giugliano RP, Sabatine MS, et al. Reduction in lipoprotein(a) with PCSK9 monoclonal antibody evolocumab (AMG 145): a pooled analysis of more than 1,300 patients in 4 phase II trials. J. Am. Coll. Cardiol. 2014;63:1278–1288. [DOI] [PubMed] [Google Scholar]
- 67.Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. New England Journal of Medicine 2017;376:1713–1722. [DOI] [PubMed] [Google Scholar]
- 68.Schwartz GG, Steg PG, Szarek M, et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. New England Journal of Medicine 2018;379:2097–2107. [DOI] [PubMed] [Google Scholar]
- 69.O’Donoghue Michelle L, Fazio Sergio, Giugliano Robert P., et al. Lipoprotein(a), PCSK9 Inhibition, and Cardiovascular Risk. Circulation 2019;139:1483–1492. [DOI] [PubMed] [Google Scholar]
- 70.Bittner VA, Szarek M, Aylward PE, et al. Effect of Alirocumab on Lipoprotein(a) and Cardiovascular Risk After Acute Coronary Syndrome. J. Am. Coll. Cardiol. 2020;75:133–144. [DOI] [PubMed] [Google Scholar]
- 71.Yang S-H, Li S, Zhang Y, et al. Analysis of the association between plasma PCSK9 and Lp(a) in Han Chinese. J. Endocrinol. Invest. 2016;39:875–883. [DOI] [PubMed] [Google Scholar]
- 72.Sarah Parish, Hopewell Jemma C., Hill Michael R., et al. Impact of Apolipoprotein(a) Isoform Size on Lipoprotein(a) Lowering in the HPS2-THRIVE Study. Circulation: Genomic and Precision Medicine 2018;11:e001696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Graham MJ, Viney N, Crooke RM, Tsimikas S. Antisense inhibition of apolipoprotein (a) to lower plasma lipoprotein (a) levels in humans. J. Lipid Res. 2016;57:340–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tsimikas S, Viney NJ, Hughes SG, et al. Antisense therapy targeting apolipoprotein(a): a randomised, double-blind, placebo-controlled phase 1 study. The Lancet 2015;386:1472–1483. [DOI] [PubMed] [Google Scholar]
- 75.Viney NJ, van Capelleveen JC, Geary RS, et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet 2016;388:2239–2253. [DOI] [PubMed] [Google Scholar]
- 76.Tsimikas S, Karwatowska-Prokopczuk E, Gouni-Berthold I, et al. Lipoprotein(a) Reduction in Persons with Cardiovascular Disease. New England Journal of Medicine 2020. Available at: 10.1056/NEJMoa1905239. Accessed July 4, 2020. [DOI] [PubMed]
- 77.Kronenberg F, Tsimikas S. The challenges of measuring Lp(a): A fight against Hydra? Atherosclerosis 2019;289:181–183. [DOI] [PubMed] [Google Scholar]
- 78.Marcovina SM, Albers JJ, Gabel B, Koschinsky ML, Gaur VP. Effect of the number of apolipoprotein(a) kringle 4 domains on immunochemical measurements of lipoprotein(a). Clin. Chem. 1995;41:246–255. [PubMed] [Google Scholar]
- 79.Marcovina SM, Albers JJ. Lipoprotein (a) measurements for clinical application. J. Lipid Res. 2016;57:526–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tsimikas S, Fazio S, Ferdinand KC, et al. NHLBI Working Group Recommendations to Reduce Lipoprotein(a)-Mediated Risk of Cardiovascular Disease and Aortic Stenosis. Journal of the American College of Cardiology 2018;71:177–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nordestgaard BG, Chapman MJ, Ray K, et al. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur. Heart J. 2010;31:2844–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mach F, Baigent C, Catapano AL, et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. European Heart Journal 2020;41:111–188. [DOI] [PubMed] [Google Scholar]
- 83.Tsimikas S, Hall JL. Lipoprotein(a) as a potential causal genetic risk factor of cardiovascular disease: a rationale for increased efforts to understand its pathophysiology and develop targeted therapies. J. Am. Coll. Cardiol. 2012;60:716–721. [DOI] [PubMed] [Google Scholar]