

Abstract
Metal-catalysed cross-couplings are a mainstay of organic synthesis and are widely used for the formation of C–C bonds, particularly in the production of unsaturated scaffolds1. However, alkyl cross-couplings using native sp3-hybridized functional groups such as alcohols remain relatively underdeveloped2. In particular, a robust and general method for the direct deoxygenative coupling of alcohols would have major implications for the field of organic synthesis. A general method for the direct deoxygenative cross-coupling of free alcohols must overcome several challenges, most notably the in situ cleavage of strong C–O bonds3, but would allow access to the vast collection of commercially available, structurally diverse alcohols as coupling partners4. We report herein a metallaphotoredox-based cross-coupling platform in which free alcohols are activated in situ by N-heterocyclic carbene salts for carbon–carbon bond formation with aryl halide coupling partners. This method is mild, robust, selective and most importantly, capable of accommodating a wide range of primary, secondary and tertiary alcohols as well as pharmaceutically relevant aryl and heteroaryl bromides and chlorides. The power of the transformation has been demonstrated in a number of complex settings, including the late-stage functionalization of Taxol and a modular synthesis of Januvia, an antidiabetic medication. This technology represents a general strategy for the merger of in situ alcohol activation with transition metal catalysis.
Over the past half century, advances in transition metal-catalysed cross-coupling have revolutionized the field of synthetic chemistry, enabling the rapid diversification of simple, abundant starting materials as well as the late-stage modification of highly complex molecular architectures1,5. Synthetic chemists today can select from a wide range of cross-coupling methods to gain easy access to unsaturated scaffolds from sp2-hybridized coupling partners6. However, significant limitations remain with respect to the activation and coupling of sp3-hybridized substrates2. Despite notable recent advances in the use of bench-stable sp3-hybridized coupling partners, such as carboxylic acids7 and alkyl halides8,9, the most abundant and versatile alkyl source—the alcohol—remains underdeveloped10 (Fig. 1a).
Fig. 1 |. Direct deoxygenative arylation of alcohols.
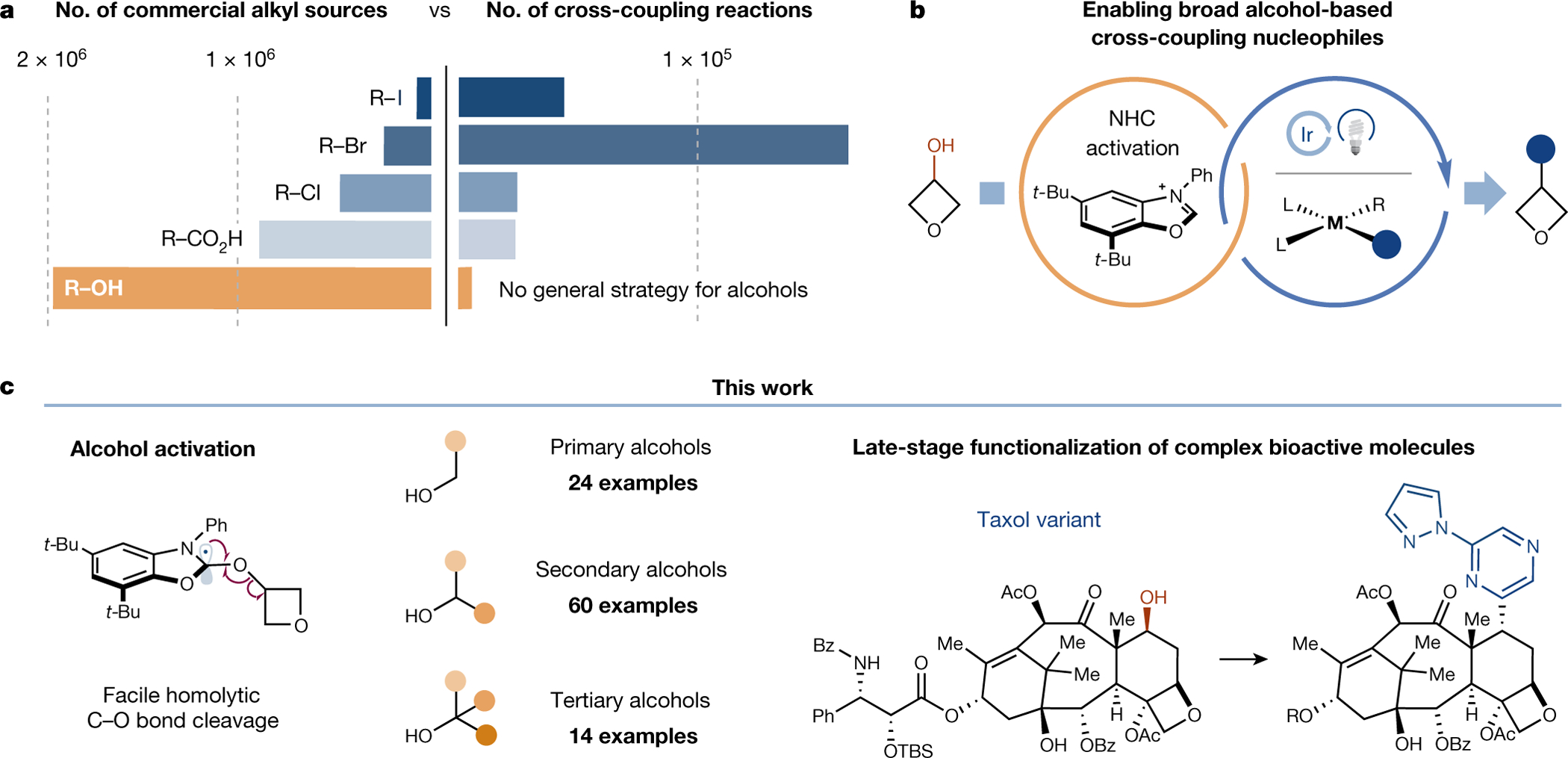
a, Alcohols are the most widely available alkyl fragment; however, there is no general strategy for the use of alcohols as C(sp3) fragments in cross-coupling reactions. b, A general strategy for the cross-coupling of alcohols is enabled by the merger of NHC-mediated alcohol activation, photoredox catalysis, and nickel catalysis. c, The success of alcohol deoxygenative cross-coupling relies on the facile NHC-mediated C–O bond homolytic cleavage. This method is amenable to a wide range of primary, secondary and tertiary alcohols. This activation mode can be used for the late-stage deoxygenative arylation of drugs, such as Taxol. NHC, nitrogen-heterocyclic carbene; Ac, acetyl; Bz, benzoyl; Me, methyl; Ph, phenyl; t-Bu, tert-butyl; TBS, tert-butyldimethylsilyl.
The alcohol is a well-established ‘native’ functional group, remarkably widely represented in commercially available sources and pharmaceutically relevant molecules11. A general method by which to accomplish direct deoxygenative alcohol cross-coupling would allow unparalleled entry into a vast new collection of diverse substrates. To date, however, no general strategy for the direct cross-coupling of alcohols has been reported. Attempts to achieve alcohol cross-coupling have been beset by both scope limitations and significant issues of substrate generality10,12–14, often arising from the difficult C(sp3)–OH cleavage step. Although kinetically facile homolytic deoxygenation methods are well-known, these methods often rely on main group elements, such as phosphorus15 and sulfur16 that cannot be generically merged with transition metal catalysis17. As an alternative, the alcohol coupling partner may be pre-activated prior to introduction to the cross-coupling reaction, although this process requires additional chemical steps and purifications18–20. We recognized that, to truly harness the potential of alcohol substrates for cross-couplings, it would be necessary to conceive of a new alcohol activation strategy that would allow direct deoxygenative coupling of a diverse array of sp3-hybridized alcohols. Ideally, this new activation mode should: (1) require no separate alcohol pre-activation step, (2) be amenable to all classes of alkyl alcohol substrates, (3) be readily adaptable across diverse transition-metal-based cross-coupling platforms, and (4) exhibit exceptional levels of functional group tolerance. To demonstrate this conceptually novel disconnection as a generic platform for metal-mediated transformations, we have elected to perform deoxygenative arylation with pharmaceutically relevant aryl halides as coupling partners.
From the outset, we recognized that a fundamental challenge would involve the in situ alcohol activation prior to C(sp3)–O bond cleavage. To accommodate a diverse range of chemical complexity and substrate C–O bond strengths, this activation step would require a significant thermodynamic driving force, as well as a kinetically facile, exothermic C(sp3)–O bond homolysis step. Along these lines, we took note of reports that N-heterocyclic carbenes (NHCs) undergo reversible condensation with the polar O–H bond21. We envisioned that subsequent oxidation of the electron-rich NHC–alcohol adduct should provide an exothermic pathway for the formation of a benign aromatic byproduct, thereby providing a strong driving force for C(sp3)–O homolytic bond cleavage. Based on recent investigations into α-amino C–H bond homolysis by our group22 and others23, we anticipated using photoredox catalysis to selectively generate the open-shell NHC–alcohol intermediate via sequential single electron transfer (SET) and proton transfer (PT) events. Photoredox activation would also provide a general platform to interface the oxidatively generated carbon-centred radicals with a variety of transition metal-catalysed transformations (Fig. 1b). As an important design criteria, we hoped to exploit the NHC–alcohol motif as an efficient quencher of excited state photocatalysts, given the ultra-fast rate at which anilinic systems are known to undergo SET with established photocatalysts22. Beyond rapid C–O bond homolysis, such a system would further allow chemoselective oxidation of the NHC adduct in the presence of prototypical medicinal chemistry moities such as carboxylates and tertiary amines, thereby ensuring broad functional group tolerance. On this basis, we herein report the development of a nickel metallaphotoredox-catalysed deoxygenative arylation of alcohols with aryl halide electrophiles (Fig. 1c).
The proposed mechanism for the deoxygenative arylation is outlined in Fig. 2a. An alcohol substrate (1) condenses with a benzoxazolium salt (2) to form a NHC–alcohol adduct (3) under mild, basic conditions. Excitation of photocatalyst [Ir(ppy)2(dtbbpy)](PF6) (4, ppy = 2-phenylpyridine, dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine) under blue light is known to generate the long-lived triplet-excited state IrIII complex (5, with a lifetime, τ, of 1.3 μs)24. This excited-state Ir complex (E1/2red[IrIII*/IrII] = +0.66 V versus SCE) can readily oxidize the anilinic nitrogen atom in 3 via a single electron transfer mechanism22. The C–H bond adjacent to the resulting nitrogen radical cation intermediate (7), now significantly weakened and more acidic (pKα ≈ 10)25, can be deprotonated by a suitable base to yield an α-amino radical (8). This unique carbon-centred radical 8, located adjacent to three heteroatoms, should undergo rapid β-scission to give carbamate 926,27, and deoxygenated alkyl radical 10. Importantly, formation of this aromatized carbamate byproduct (9), possessing a strong C=O double bond, is anticipated to provide a universal thermodynamic driving force for alcohol C–O bond homolysis. In the nickel catalytic cycle, the Ni(0) species 12, generated from a Ni(ii) precatalyst via two sequential SET events with reduced photocatalyst 6 (E1/2red[IrIII/IrII] = −1.51 V versus SCE)23, is expected to undergo facile oxidative addition into an aryl bromide (13) to form an aryl Ni(ii) species (14). Trapping of the alcohol-derived radical species 10 by 14 should yield the key Ni(iii) species 15. Finally, reductive elimination from the Ni(iii) metal centre forges the requisite C–C bond, delivering the deoxygenative arylation product 16 and expelling the Ni(i) intermediate 11, thereby simultaneously completing both the photoredox and the nickel catalytic cycle.
Fig. 2 |. Proposed mechanism and nitrogen-heterocyclic carbene evaluation for deoxygenative arylation.
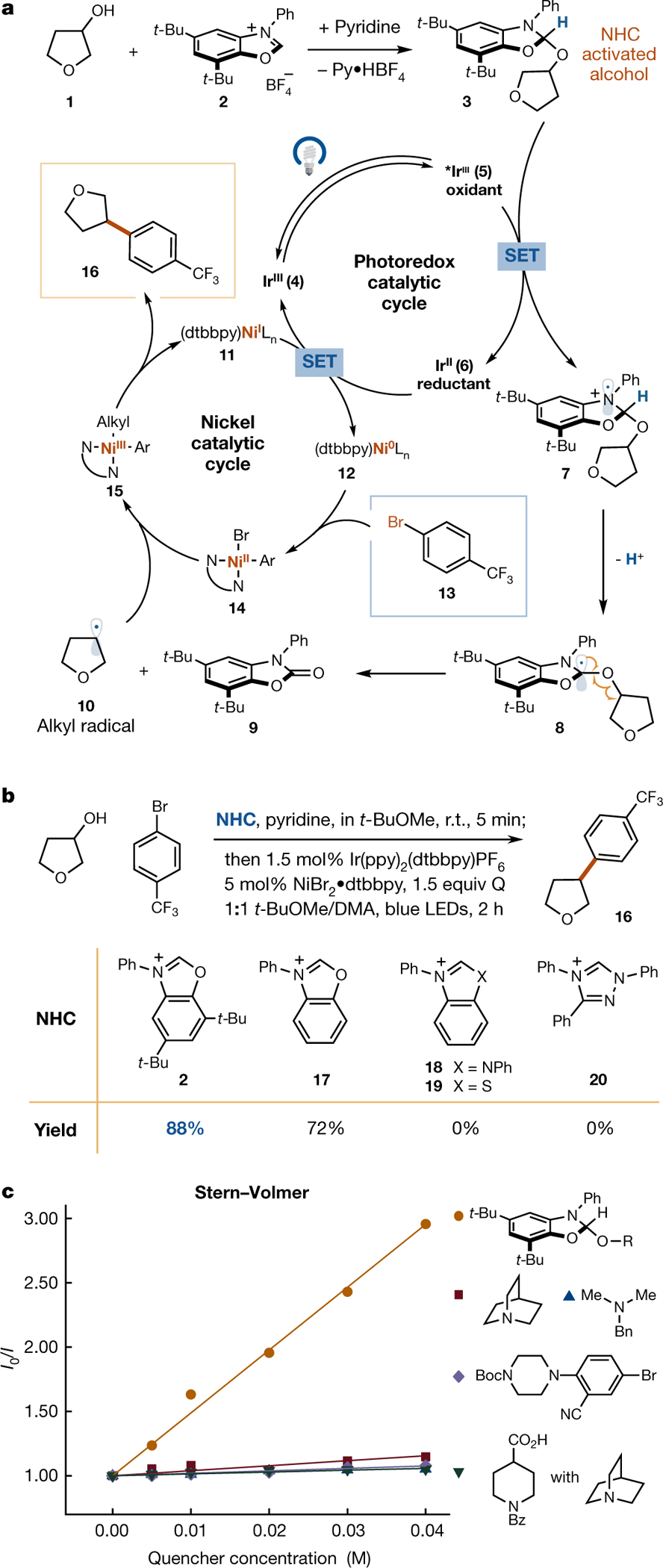
a, The starting alcohol 1 is converted to adduct 3 in the presence of NHC salt 2 and base. The deoxygenative radical 10 is generated from 3 upon sequential electron–proton transfer and followed by facile β-scission, which can be captured by Ni-aryl species 14 to yield the arylated product 16. b, Evaluation of N-heterocyclic carbene salts for deoxygenative arylation. For detailed optimization, see Supplementary Information. c, Stern–Volmer quenching comparison of NHC adduct 3 and other readily oxidizable functional groups. Py•HBF4, pyridinium tetrafluoroborate; Q, quinuclidine; t-BuOMe, methyl tert-butyl ether; DMA, dimethylacetamide; R, 3-tetrahydrofuranyl.
With this working hypothesis in hand, we first sought to identify suitable NHC salts. One major challenge is the propensity of the NHC–alcohol adduct to transfer a free NHC ligand to the metal centre and release the alcohol, owing to the reversible, dynamic nature of the NHC–alcohol bond28. Upon extensive screening of NHC salts, we found a highly electron-deficient precursor N-aryl benzoxazolium salt (17)29 to be an effective activating agent, delivering the desired deoxygenative coupling product (16) in 72% yield. In contrast, other common NHC salts—such as benzimidazolium (18)30, benzothiazolium (19), and triazolium salts (20)31 failed to produce the desired product (Fig. 2b). For a detailed rationale of this unique NHC skeleton preference, see Supplementary Figs. 7, 8. Modifications to the benzoxazolium backbone revealed modified NHC salt 2 to be highly effective at activating alcohol 1 for cross-coupling. Based on previous studies, the benzoxazolium-based free carbene represents the most electron-deficient NHC synthesized to date32. We hypothesize that the electron-deficient nature of the NHC significantly decreases the tendency of the NHC-alcohol adduct (3) to dissociate. Indeed, we did not observe 3 undergo dynamic exchange with other alcohols or metal catalysts under reaction conditions, which is opposite to the reported behaviour of more electron-rich NHC-alcohol adducts21,28. More surprisingly, the benzoxazolium salt 2 readily condensed with alcohols in less than 5 minutes with high efficiency in ethereal solvents. The resulting solution of NHC-alcohol adduct 3 can be directly mixed with the aryl halide, 1.5 mol% Ir(ppy)2(dtbbpy)PF6, and 5 mol% Ni(dtbbpy)Br2 as catalysts, 1.5 equivalents of quinuclidine as base, without isolation or purification of compound 3. Upon 450 nm blue light irradiation of the crude reaction mixture, the desired adduct 16 was obtained in 88% yield. As an important design component, we were satisfied to find that NHC-alcohol adduct 3 does indeed quench the excited state photocatalyst 5 significantly faster than a number of medicinal chemistry functionalities that can be susceptible to SET, including carboxylates and tertiary amines (Fig. 2c). As can be discerned vide infra, this design feature was critical to enabling the remarkable level of generality observed for this deoxygenative coupling. Furthermore, we note that the benzoxazolium salt (2) is readily synthesized in two steps on 150 g scale without any work-up or purification, and is now available commercially.
With an operationally simple protocol in hand, we first explored the scope of the deoxygenative arylation with respect to the alcohol component (Fig. 3). We were delighted to find that this activation mode was competent in delivering a diverse array of adducts arising from both stabilized and unstabilized radical species. For example, high-energy deuteromethyl and trifluoroethyl radicals were generated from deuteromethanol (21) and trifluoroethanol (22), respectively, to give arylated product in good yields. This technology is also compatible with alcohols bearing chiral β-substituents, including a free alcohol, amide, and enolizable ester, directly yielding chiral arylated products with 100% enantiospecificity (23–25). Importantly, a high degree of chemo- and regio-selectivity was observed in this reaction, as the NHC 2 preferentially activated the less-hindered primary alcohol in the presence of both a carbamate N–H bond (24) and a secondary benzylic alcohol (25).
Fig. 3 |. Alcohol scope for deoxygenative arylation.

Primary, secondary and tertiary alcohols can be used as alkyl coupling partners in the arylation. All yields are isolated unless otherwise noted. Experiments typically run with 1.0 equivalent of aryl halide, 1.7 equivalent of alcohol and 1.6 equivalent of NHC salts on 0.5 mmol scale. *See Supplementary Information for experimental details. §Ni(TMHD)2 and methyl 4-bromobenzoate are used as the catalyst and aryl halide coupling partner, respectively. Boc, tert-butyloxycarbonyl; Bn, benzyl; Et, ethyl; PMB, 4-methoxybenzyl; Piv, pivaloyl; Ts, 4-toluenesulfonyl; Ni(TMHD)2, nickel(ii) bis(2,2,6,6-tetramethyl-3,5-heptanedionate).
We next examined a series of secondary alcohol coupling partners. As shown in Fig. 3 variety of cyclic alcohols, ranging in size from three- to seven-membered rings, underwent deoxygenative arylation in good to excellent yields. Viable motifs in this transformation include the cyclopropyl group (33), oxetane (34), azetidine (35), pyrrolidine (37, 38), indane (39), menthol (40), piperidine (41) and diazacycloheptane (44). Alcohols located on pharmaceutically relevant strained bridged bicycles, such as (48–50), gave arylated products with good diastereo-selectivity. Finally, acyclic secondary alcohols, such as 45 and the bulky pinacolyl alcohol (46), were successfully coupled. We note that this method has a reactivity profile orthogonal to common sp2–sp3 coupling protocols, as arylation occurs exclusively at the alcohol site (42, 43, 48) while secondary alkyl bromides and carboxylic acid are left untouched.
We next probed whether tertiary alcohol substrates could be employed to generate quaternary carbon centres. We were pleased to find that, in the presence of a N-4-(trifluoromethyl)phenyl benzoxazolium salt activating agent, a variety of tertiary alcohols served as viable coupling partners in the deoxygenative arylation; see Supplementary Fig. 7 for details. Thus, cyclopropyl (51), oxetane (52) and azetidine (53) tertiary alcohols gave the desired quaternary carbon product without any detectable regioisomers. An alcohol positioned at a [1.1.1] bicyclo bridge head (54) underwent homolysis to form a high-energy radical species en route to the desired arylation product. Moreover, arylation with the C2 tertiary radical of [1.1.1]bicyclopentane (55) was achieved for the first time. For more hindered and electron-rich tertiary radicals such as trialkyl acyclic (56–57), cyclic (58–61), or fused-ring bridgehead variant (62), we found that Ni(TMHD)2 proved to be more effective to form the quaternary carbon centres. In contrast, Ni(dtbbpy) Br2 catalyst gave only trace product with tertiary radicals, consistent with the reported ligand-induced change of mechanism for this C–C bond-forming step.33
Saccharides represent an important class of biological molecules, and the development of versatile methods by which to directly functionalize these alcohols is of particular interest. As shown in Fig. 3, the hemiacetal at the anomeric carbon of commercially available pyranose (66) and furanoses (63 and 64) could be activated to provide the deoxygenatively coupled products with excellent diastereoselectivity. Similarly, chiral alcohols on the ribose core (65) and hindered fructopyranose (67) were activated to generate the corresponding coupled products.
We next sought to evaluate the scope of the aryl halide coupling partner employing Boc-protected 3-hydroxy piperidine as the standard alcohol coupling partner. It is worth noting that this alcohol cannot be converted to the corresponding alkyl halide via a conventional Appel reaction.34 As shown in Extended Data Fig. 1, aryl bromides with different electronic properties (68, 69) or bulky ortho-substituents (74, 75) gave desired products in generally good yields. Importantly, many medicinally relevant functional groups were well-tolerated, including: primary sulfonamide (70), aryl boronic pinacol ester (71), tertiary amine (72), primary benzyl amine (73) and free benzylic alcohols (76). We were pleased to observe that challenging five-membered heterocyclic bromides (93–106) generally participate effectively in this coupling. Moreover, we found that our standard conditions are also amenable to the coupling of electron-deficient aryl chlorides (107–109) and heteroaryl chlorides (110–115). Notably, the transformation can be used for the late-stage functionalization of the chloride-containing drug molecules Zomepirac (108) and Etoricoxib (115).
To further demonstrate the synthetic value of this new versatile technology, we sought to achieve sequential double-deoxygenative arylations of C2-symmetric diols (Fig. 4a). The condensation of NHC with 1,2-diol substrates was found to be highly mono-selective, ultimately yielding mono-arylated adduct (121–123) with high regio- and diastereo-selectivity. These monoarylated products were then employed as starting materials en route to bis-arylated products, which were isolated with excellent diastereo-selectivity in moderate to good yields (126–128). When commercially available chiral diol 116 was employed, the enantiopurity was quantitively transferred to the bis-arylated product (121). Similarly, commercially available C2-symmetric chiral 1,4-diols (119 and 120) were readily diversified into complex arylated products (129–130) with excellent diastereo-selectivity. This chirality transfer technology is anticipated to provide a valuable new bond disconnection strategy for chiral pool synthesis.
Fig. 4 |. Chirality transfer from chiral diol and late-stage drug molecule functionalization.
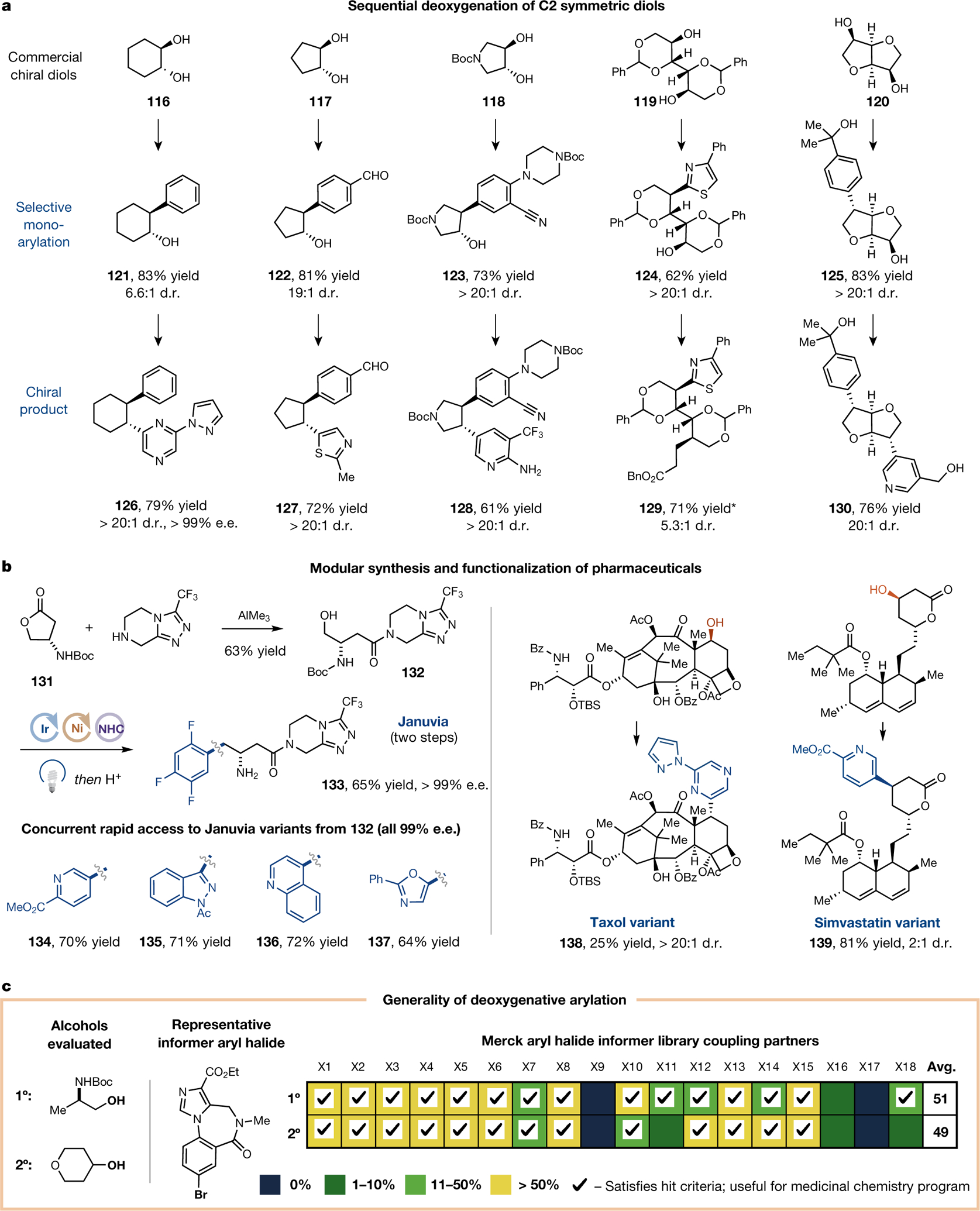
a, Double deoxygenative functionalization of commercially available C2-symmetric diols permits transfer of chirality to bis-arylated products via diastereocontrol. *Benzyl arylate was used instead of nickel catalyst. b, This protocol can also be applied to a modular synthesis of Januvia and its variants, as well as to the late-stage functionalization of pharmaceutical variants, such as Taxol and Simvastatin. All yields are isolated; see Supplementary Information for exact conditions. c, To demonstrate the generality of the deoxygenative arylation, primary and secondary alcohols were evaluated with 18 different complex halides. Among 36 distinct combinations of coupling partners, 32 reactions successfully gave the desired product. Yields were obtained by UPLC analysis; see Supplementary Information for details.
We next sought to demonstrate the robustness of the coupling reaction in the context of complex, drug-like molecules (Fig. 4b). Toward this end, the blockbuster antidiabetic drug Januvia was synthesized in two steps from commercially available starting materials. Under standard deoxygenative arylation conditions, the alcohol 132 was converted to enantiopure Januvia (133) with 65% yield. Four Januvia variants (134–137) were similarly prepared in straightforward fashion from intermediate 132. To demonstrate the expansive utility of this cross-coupling strategy, we selectively functionalized the alcohol on the high-density core of the anticancer drug Taxol (138). Moreover, deoxygenative arylation of the lipid-lowering drug simvastatin proceeded in good yield (139).
To further demonstrate the generality of the aryl halide scope and its potential value in medicinal chemistry campaigns, we tested a Merck aryl-halide informer library, containing 18 different halides, using representative primary and secondary alcohols (Fig. 4c)35. Of the 36 reactions performed, 28 gave synthetically useful yields for a medicinal chemistry program (0.1 mmol scale). With our protocol, an average yield of 50% was observed across the entire Merck halide informer library. This result represents the highest level of reaction efficiency to date for any sp2–sp3 coupling technology benchmarked by the informer library,36 highlighting the power and versatility of this deoxygenative transformation for complex substrates.
Extended Data
Extended Data Fig. 1 |. Aryl halide scope for deoxygenative arylation.
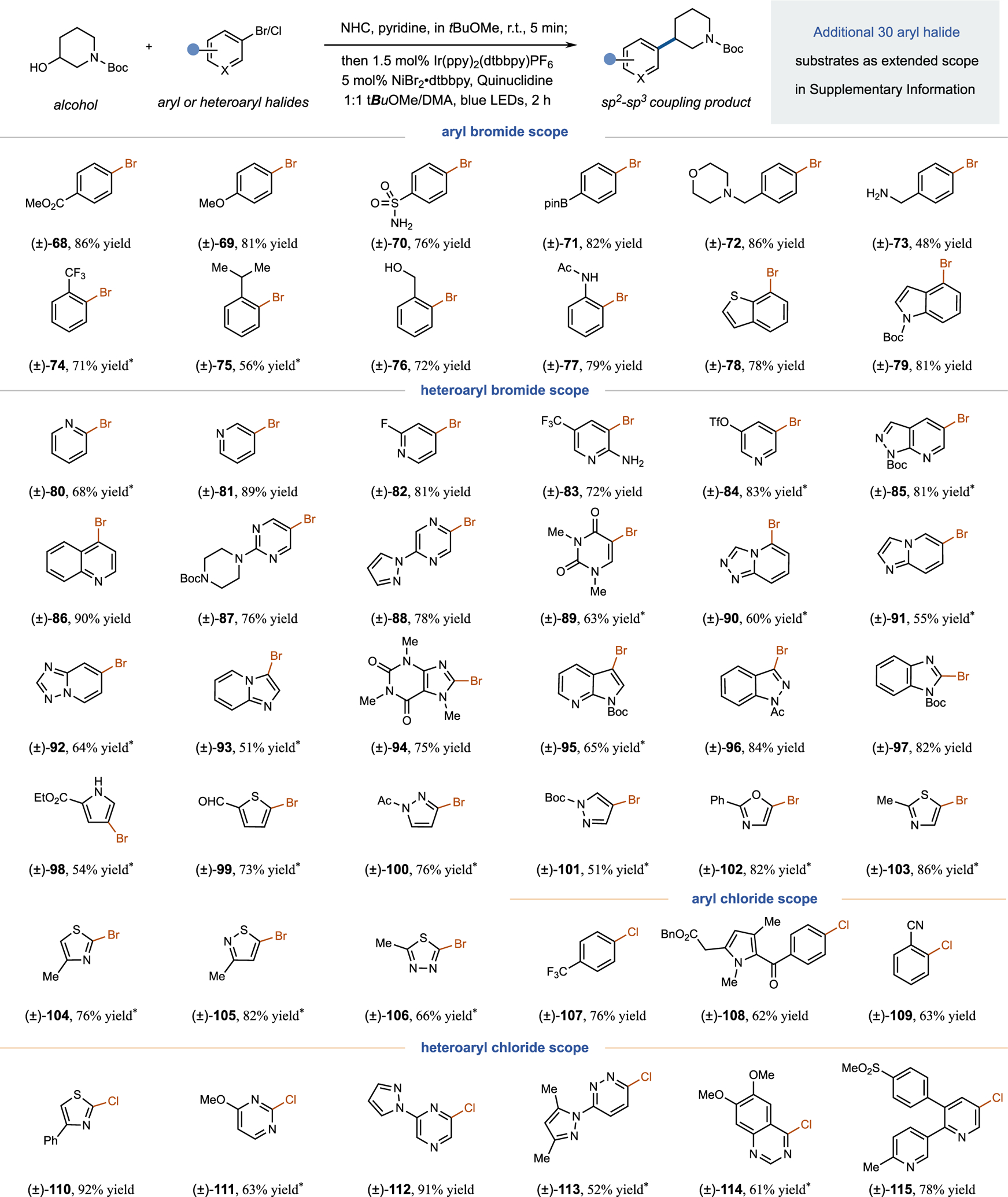
Both (hetero)aryl bromides and chlorides can be utilized under same reaction conditions. All yields are isolated. Experiments typically run with 1.0 equivalent of aryl halide, 1.7 equivalent of alcohol and 1.6 equivalent of NHC on 0.5 mmol scale. *See Supplementary Information for experimental details.
Supplementary Material
Acknowledgements
Research reported in this publication was supported by the NIH National Institute of General Medical Sciences (R35 GM134897-02) and gifts from Merck, Bristol-Myers Squibb, Eli Lilly, and Janssen Research and Development LLC. The authors thank C. Liu and R. Lambert for assistance in preparing this manuscript.
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-021-03920-6.
Competing interests The authors declare no competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41586-021-03920-6.
Data availability
The data supporting the findings of this study are available within the paper and its Supplementary Information.
References
- 1.Corbet J-P & Mignani G Selected patented cross-coupling reaction technologies. Chem. Rev 106, 2651–2710 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Choi J & Fu GC Transition metal–catalyzed alkyl-alkyl bond formation: another dimension in cross-coupling chemistry. Science 356, eaaf7230 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herrmann JM & König B Reductive deoxygenation of alcohols: catalytic methods beyond Barton–McCombie deoxygenation. Eur. J. Org. Chem 2013, 7017–7027 (2013). [Google Scholar]
- 4.Blakemore DC et al. Organic synthesis provides opportunities to transform drug discovery. Nat. Chem 10, 383–394 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Ruiz-Castillo P & Buchwald SL Applications of palladium-catalyzed C–N cross-coupling reactions. Chem. Rev 116, 12564–12649 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walters WP, Green J, Weiss JR & Murcko MA What do medicinal chemists actually make? a 50-year retrospective. J. Med. Chem 54, 6405–6416 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Zuo Z et al. Merging photoredox with nickel catalysis: coupling of α-carboxyl sp3-carbons with aryl halides. Science 345, 437–440 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Everson DA, Jones BA & Weix DJ Replacing conventional carbon nucleophiles with electrophiles: nickel-catalyzed reductive alkylation of aryl bromides and chlorides. J. Am. Chem. Soc 134, 6146–6159 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sakai HA, Liu W, Le C & MacMillan DWC Cross-electrophile coupling of unactivated alkyl chlorides. J. Am. Chem. Soc 142, 11691–11697 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suga T & Ukaji Y Nickel-catalyzed cross-electrophile coupling between benzyl alcohols and aryl halides assisted by titanium co-reductant. Org. Lett 20, 7846–7850 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Ertl P & Schuhmann T A systematic cheminformatics analysis of functional groups occurring in natural products. J. Nat. Prod 82, 1258–1263 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Jia X-G, Guo P, Duan J & Shu X-Z Dual nickel and Lewis acid catalysis for cross-electrophile coupling: the allylation of aryl halides with allylic alcohols. Chem. Sci 9, 640–645 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo P et al. Dynamic kinetic cross-electrophile arylation of benzyl alcohols by nickel catalysis. J. Am. Chem. Soc 143, 513–523 (2021). [DOI] [PubMed] [Google Scholar]
- 14.Li Z et al. Electrochemically enabled, nickel-catalyzed dehydroxylative cross-coupling of alcohols with aryl halides. J. Am. Chem. Soc 143, 3536–3543 (2021). [DOI] [PubMed] [Google Scholar]
- 15.Barton DHR & McCombie SW A new method for the deoxygenationof secondary alcohols. J. Chem. Soc. Perkin Trans 1 16, 1574–1585. (1975). [Google Scholar]
- 16.Zhang L & Koreeda M Radical deoxygenation of hydroxyl groups via phosphites. J. Am. Chem. Soc 126, 13190–13191 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Vara BA, Patel NR & Molander GA O-benzyl xanthate esters under Ni/photoredox dual catalysis: selective radical generation and Csp3 –Csp2 cross-coupling. ACS Catal 7, 3955–3959 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang C-T et al. Copper-catalyzed cross-coupling of nonactivated secondary alkyl halides and tosylates with secondary alkyl grignard reagents. J. Am. Chem. Soc 134, 11124–11127 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Zhang X & MacMillan DWC Alcohols as latent coupling fragments for metallaphotoredox catalysis: sp3 –sp2 cross-coupling of oxalates with aryl halides. J. Am. Chem. Soc 138, 13862–13865 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ye Y, Chen H, Sessler JL & Gong H Zn-mediated fragmentation of tertiary alkyl oxalates enabling formation of alkylated and arylated quaternary carbon centers. J. Am. Chem. Soc 141, 820–824 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Coulembier O et al. Alcohol adducts of N-heterocyclic carbenes: latent catalysts for the thermally-controlled living polymerization of cyclic esters. Macromolecules 39, 5617–5628 (2006). [Google Scholar]
- 22.McNally A, Prier CK & MacMillan DWC Discovery of an α-amino C–H arylation reaction using the strategy of accelerated serendipity. Science 334, 1114–1117 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Joe CL & Doyle AG Direct acylation of C(sp3)−H bonds enabled by nickel and photoredox catalysis. Angew. Chem. Int. Ed 55, 4040–4043 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Slinker JD et al. Efficient yellow electroluminescence from a single layer of a cyclometalated iridium complex. J. Am. Chem. Soc 126, 2763–2767 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Dinnocenzo JP & Banach TE Deprotonation of tertiary amine cation radicals. A direct experimental approach. J. Am. Chem. Soc 111, 8646–8653 (1989). [Google Scholar]
- 26.Kochi JK Chemistry of alkoxy radicals: cleavage reactions. J. Am. Chem. Soc 84, 1193–1197 (1962). [Google Scholar]
- 27.Zhao L, Zhang C, Zhuo L, Zhang Y & Ying JY Imidazolium salts: a mild reducing and antioxidative reagent. J. Am. Chem. Soc 130, 12586–12587 (2008). [DOI] [PubMed] [Google Scholar]
- 28.Trnka TM et al. Synthesis and activity of ruthenium alkylidene complexes coordinated with phosphine and N-heterocyclic carbene ligands. J. Am. Chem. Soc 125, 2546–2558 (2003). [DOI] [PubMed] [Google Scholar]
- 29.Bellemin-Laponnaz S Synthesis of N,O-heterocyclic carbene and coordination to rhodium(I) and copper(I). Polyhedron 29, 30–33 (2010). [Google Scholar]
- 30.Sladojevich F, Arlow SI, Tang P & Ritter T Late-stage deoxyfluorination of alcohols with PhenoFluor. J. Am. Chem. Soc 135, 2470–2473 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kato T, Matsuoka S & Suzuki M N-heterocyclic carbene-mediated redox condensation of alcohols. Chem. Commun 52, 8569–8572 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Gusev DG Electronic and steric parameters of 76 N-heterocyclic carbenes in Ni(CO)3(NHC). Organometallics 28, 6458–6461 (2009). [Google Scholar]
- 33.Yuan M, Song Z, Badir SO, Molander GA & Gutierrez O On the nature of C(sp3)–C(sp2) bond formation in nickel-catalyzed tertiary radical cross-couplings: a case study of Ni/photoredox catalytic cross-coupling of alkyl radicals and aryl halides. J. Am. Chem. Soc 142, 7225–7234 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gonnard L, Guérinot A & Cossy J Cobalt-catalyzed cross-coupling of 3- and 4-iodopiperidines with grignard reagents. Chem. Eur. J 21, 12797–12803 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Kutchukian PS et al. Chemistry informer libraries: a chemoinformatics enabled approach to evaluate and advance synthetic methods. Chem. Sci 7, 2604–2613 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang R et al. Profiling and application of photoredox C(sp3)–C(sp2) cross-coupling in medicinal chemistry. ACS Med. Chem. Lett 9, 773–777 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of this study are available within the paper and its Supplementary Information.